

Abstract
J Clin Hypertens (Greenwich). 2012;14:198–205. ©2012 Wiley Periodicals, Inc.
Under resting conditions the arterial vasculature exists in a vasoconstricted state referred to as vascular tone. Physiological dilatation in response to increased flow, a function of normal endothelium is necessary to maintain normal blood pressure. Endothelial dysfunction in vascular smooth muscle cells thus results in loss of normal vasorelaxant function and the inability of arteries to appropriately dilate in response to increased blood flow in either a systemic or regional vascular bed, resulting in increased blood pressure, a sequence that may represent a common pathway to hypertension. Normal vasorelaxation is mediated by a number of endothelial systems including nitric oxide (NO), prostaglandins (PGI2 and PGE2), and a family of endothelial‐derived hyperpolarizing factors (EDHF). In response to hemodynamic shear stress, endothelium continuously releases NO, EDHF, and PGI2 to provide vasodilatation. EDHF, not a single molecule but rather a group of molecules that includes epoxyeicosatrienoic acids, hydrogen peroxide, carbon monoxide, hydrogen sulfide, C‐natriuretic peptide, and K+ itself, causes vasodilatation by activation of vascular smooth muscle cell K+ channels, resulting in hyperpolarization and thus vasorelaxation. The understanding and effective management of blood pressure requires an understanding of both physiologic and pathophysiologic regulation of vascular tone. This review describes molecular mechanisms underlying normal endothelial regulation and pathological states, such as increased oxidative stress, which cause loss of vasorelaxation. Possible pharmacological interventions to restore normal function are suggested.
The biomarker par excellence of hypertension is chronic elevation of systemic arterial blood pressure (BP) above normal. When BP is abnormally and chronically elevated, it is often assumed that either the cardiac output is too high or abnormal vasoconstriction has increased peripheral vascular resistance. 1 , 2 However, the arterial vasculature basally exists in a vasoconstricted state, known as “vascular tone.” 1 , 2 Thus, the inability of the blood vessels to dilate in response to increased flow may result in an increase in BP. Inability to appropriately vasodilate may occur on a systemic or a regional basis.
Vasodilation in response to increased flow is the function of the normal endothelium through the secretion of nitric oxide (NO), prostacyclin (PGI2), and endothelial‐derived hyperpolarizing factors (EDHFs). The loss of normal endothelial function results in impaired vasodilation and increased BP and may be a common pathway to the disease hypertension.
Below we describe endothelial systems that mediate vasodilation, ie, NO, EDHFs, and prostaglandins, that have been linked to experimental increases in BP. Also discussed are mechanisms that induce endothelial dysfunction and thus may be of importance in the pathogenesis of hypertension, eg, oxidative stress and diabetes mellitus.
Mechanisms Associated With Endothelial‐Mediated Vasodilation
The Figure illustrates the various mechanisms involved in endothelial‐mediated vasodilation. The reader is encouraged to refer frequently to the Figure to fully appreciate the integrated nature of the response.
Figure 1.
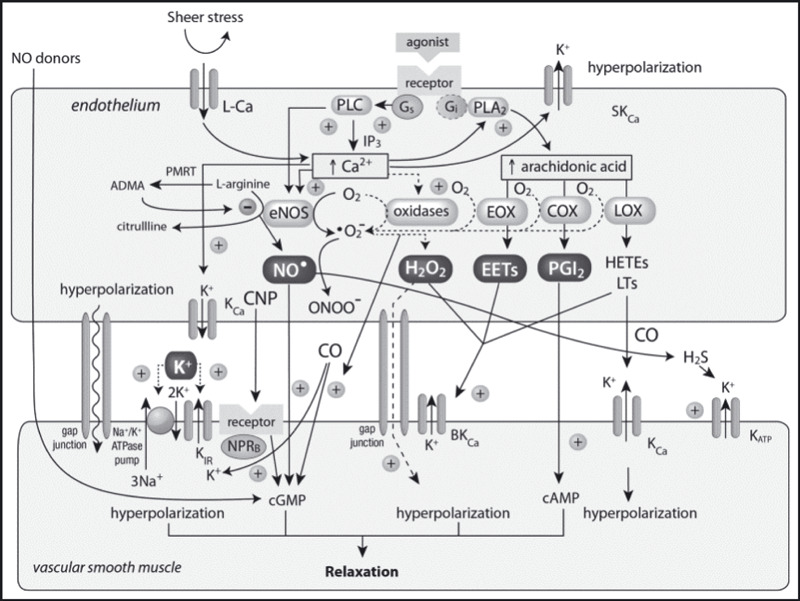
Vascular smooth muscle relaxation results from cellular hyperpolarization due to a variety of mechanisms that ultimately cause increases in cyclic guanosine monophosphate (cGMP), cyclic adenosine monophosphate (cAMP), or transmembrane K+ and Na+ transport that may occur via gap junctions, ion pumps, direct receptor or channel activation, or direct action of nitric oxide (NO) donors. An array of endothelial‐derived hyperpolarizing factors contributes to the hyperpolarization. ADMA indicates asymmetric dimethylarginine; CaM, calmodulin; COX, cyclooxygenase; CNP, C‐type natriuretic peptide; EET, epoxyeicosatetraenoic acid; eNOS, endothelial nitric oxide synthase; EOX, epoxygenase; HETE, hydroxyleicosa‐tetraenoic acid; KCa, Ca2+‐activated K+ channels; KIR, inward rectifier; KATP, ATP‐dependent; LCa, L‐type Ca channel; LOX, lipoxygenase; LT, leukotriene; PLA2, phospholipase A2; NPRB, natriuretic peptide receptor subtype B; PLC, phospholipase C; PRMT, protein arginine N‐methyltransferases. (+), stimulates; (↑), increases. Adapted from Félétou et al 21 and Miura et al. 44
Nitric Oxide
The principal regulator of endothelial vasodilator function through NO is vascular shear. Shear is the frictional force exerted on the vascular wall secondary to the flow of blood. These forces open calcium channels on endothelial cells, thus promoting the calcium‐dependent activation of endothelial NO synthase (eNOS), which, in turn, induces the release of NO. NO then diffuses to the underlying vascular smooth muscle, where it activates soluble guanylate cyclase, causing an increase in cyclic guanosine monophosphate (cGMP) and smooth muscle relaxation. Constitutive NO synthase (NOS) can be competitively inhibited by guanidine‐substituted analogues of l‐arginine, such as N G‐monomethyl‐l‐arginine (L‐NMMA). 3 It is not surprising that administration of these substances to experimental animal results in large increases in systemic arterial BP.
In response to hemodynamic shear stress, the endothelium continuously releases NO, EDHF, and prostaglandins and up‐regulates the gene that expresses NOS; NO provides vasodilation. 4 This mechanotransduction is truly remarkable with realization that shear stress is in the range of 10 to 20 dynes/cm2 whereas BP, measured in mm Hg, is orders of magnitude greater (1 mm Hg=1333.22 dynes/cm2).
The production of NO is catalyzed by NOS, which convert the amino acid l‐arginine to l‐citrulline and NO. 5 , 6 NOSs are members of a family of cytochrome P450–like reductases linked to a nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme. 7 , 8 Modulation of NO production is also provided by asymmetric dimethylarginine (ADMA), an endogenous inhibitor of NOS. ADMA concentrations are a reflection of oxidative stress. 9 , 10
NO plays an important role in regulating systemic vascular resistance, arterial relaxation, and distensibility. Collectively, these actions reduce cardiac hemodynamic load, thereby reducing myocardial hypertrophy and left ventricular dysfunction and protecting target organs. 11 , 12 , 13 , 14 , 15 Thus, NO plays a major role in maintaining and regulating BP. 16 , 17 , 18 , 19 , 20
Endothelial‐Derived Hyperpolarizing Factor(s)
The recognition of another endothelial vasodilatory mechanism resulting from K+ channel activation led to the concept of an “endothelial‐derived hyperpolarizing factor,” or EDHF. This topic has recently been extensively reviewed. 21 , 22 , 23 While initially thought to be a single substance, it is now apparent that there is no “universal” EDHF. Regional differences in vascular function reflect the complex mechanisms of EDHF, with 2 primary mechanistic pathways implicated: myoendothelial gap junctions mediating the spread of endothelial cell hyperpolarization and/or small signaling molecules to the smooth muscle and diffusible mediators released from the endothelium. What has become apparent is that there are a number of molecules, including at least K+ ions, arachidonic acid metabolites (epoxyeicosatrienoic acids), hydrogen peroxide (H2O2), carbon monoxide (CO), hydrogen sulfide (H2S), and C‐natriuretic peptide, that cause vasodilation by hyperpolarization and hence relaxation of vascular smooth muscle by activating different K+ channels. Furthermore, there exists a separate pathway that produces relaxation of smooth muscle vascular cells by increasing endothelial cell intracellular Ca2+ concentration, resulting in the opening of K+ channels. 21
Potassium Channels
Three subtypes of Ca2+‐activated K+ channels have been identified in the vascular wall, characterized by large (BKCa), intermediate (IKCa or KCa3.1 isoform), and small (SKCa or KCa2.3 isoform) conductance. IKCA and SKCA, especially the SK3 α subunit, are expressed in endothelial cells, but with very specific cellular and subcellular locations.
Following SKCa activation, smooth muscle hyperpolarization is preferentially evoked by electrical coupling through myoendothelial gap junctions, whereas following IKCa activation, K+ efflux can activate smooth muscle Kir2.1 and/or Na+/K+ATPase. 21 , 22 The in vivo relevance of these endothelial KCa has been demonstrated in a mutant mouse model with deletion of genes encoding for either or both endothelial SK3 (SKCa, KCa2.3) and IK1 (IKCa, KCa3.1). K+ channels in EDHF responses are severely impaired and arterial BPs are elevated. 24 , 25 Combined IK1/SK3 deficiency in IK1(−/−)/SK3(T/T) mice abolished endothelial KCa currents and impaired acetylcholine‐induced smooth muscle hyperpolarization and EDHF‐mediated dilation in conduit arteries and in resistance arterioles in vivo. IK1 deficiency had a severe impact on acetylcholine‐induced EDHF‐mediated vasodilation, whereas SK3 deficiency impaired NO‐mediated dilation to acetylcholine and to shear stress stimulation. As a consequence, SK3/IK1‐deficient mice exhibited an elevated arterial BP, which was most prominent during physical activity. Overexpression of SK3 in IK1(−/−)/SK3(T/T) mice partially restored EDHF‐ and NO‐mediated vasodilation and lowered elevated BP. The IK1‐opener SKA‐31 enhanced EDHF‐mediated vasodilation and lowered BP in SK3‐deficient IK1(+/+)/SK3(T/T) mice to normotensive levels. 25 Furthermore, pharmacologic opening of endothelial KCa3.1/KCa2.3 channels stimulates endothelium‐derived hyperpolarizing factor–mediated arteriolar dilation and lowers BP. 24
C‐Natriuretic Peptide
C‐natriuretic peptide (CNP) has been demonstrated to cause hyperpolarization and relaxation of vascular smooth muscle cells, including those of human forearm resistance vessels. This response results from activation of the B subtype of the naturetic peptide receptor located on vascular smooth muscle. Receptor activation, in turn, leads to stimulation of particulate guanylate cyclase, thus increasing cGMP and subsequent opening of BKCa and KATP channels. 26 , 27 However, it is most likely that the physiologic effects of CNP are directed more toward preventing smooth muscle proliferation, leukocyte recruitment, and platelet reactivity. As such, endothelium‐derived CNP is likely to exert a strong antiatherogenic influence on blood vessel walls preventing smooth muscle proliferation and thus exerting antiatherogenic activity. 28 , 29
Gasotransmitters
In addition to NO, CO and H2S are also water‐soluble low molecular weight molecules that easily cross lipid membranes and diffuse homogeneously from synthetic site to target. This novel family of endogenous gaseous transmitters has been termed “gasotransmitters.” Functionally, H2S has been implicated in the induction of hippocampal long‐term potentiation, brain development, and BP regulation. By acting specifically on KATP channels, H2S can hyperpolarize cell membranes, relax smooth muscle cells, or decrease neuronal excitability. 30 The physiologic cardiovascular effects of H2S (inflammatory and antioxidant properties, vasodilation, and a decrease in BP) have been linked to the activation of cystathionine beta‐synthase by calcium‐calmodulin; H2S is produced and released by endothelial cells, in a Ca2+‐dependent manner following neurohumoral stimulation and evokes hyperpolarization and relaxation of vascular smooth muscle cells by activating KATP. 30 , 31 , 32 H2S production from vascular tissues appears to be enhanced by NO. 33 Genetic deletion of cystathionine beta‐synthase (cystathionine gamma‐lyase) in mice markedly reduces H2S levels in the serum, heart, aorta, and other tissues, resulting in pronounced hypertension and diminished endothelium‐dependent vasorelaxation. Such observations support the concept that H2S is a physiologic vasodilator and regulator of BP. 34
The majority of endogenous CO is catalyzed by inducible (HO‐1) and constitutive (HO‐2) heme oxygenases. The release of CO by vascular cells may modulate blood flow and fluidity by inhibiting vasomotor tone, smooth muscle cell proliferation, and platelet aggregation. CO may also maintain the integrity of the vessel wall by directly blocking vascular cell apoptosis and by inhibiting the release of proapoptotic inflammatory cytokines from the vessel wall. These effects of CO are mediated via multiple pathways, including activation of soluble guanylate cyclase, potassium channels, p38 mitogen‐activated protein kinase, or inhibition of cytochrome P450. 35 , 36
In a chronic renovascular hypertension model, hypertension, cardiac hypertrophy, acute renal failure, and acute mortality induced by one kidney‐one clip surgery were more severe in HO‐1‐null mice. In contrast, mice with cardiac‐specific overexpression of HO‐1 had an improvement in cardiac function, smaller myocardial infarctions, and reduced inflammatory and oxidative damage after coronary artery ligation and reperfusion. 37 Systolic BP of spontaneously hypertensive rats (SHRs) was normalized and this normalization maintained for 9 months after hemin infusion; HO‐1 expression, HO activity, soluble guanylyl cyclase expression, and cGMP content increased, but phosphodiesterase‐5 expression was downregulated in the mesenteric arteries. The infusion also reversed SHR‐featured arterial eutrophic inward remodeling and decreased expression levels of vascular endothelial growth factor. 38 Taken together, these studies suggest that an absence of HO‐1 has detrimental consequences, whereas overexpression of HO‐1 plays a protective role in hypoperfusion and ischemia/reperfusion injury.
Hydrogen Peroxide
Endothelial cells express enzymes that produce reactive oxygen species (ROS) in response to various stimuli, and H2O2 is a potent relaxant of vascular smooth muscle and has been postulated to represent yet another endogenous HDHF, playing a key role in the control of resistance artery tone. H2O2 itself can mediate endothelium‐dependent relaxations in some vascular beds by potentiating Ca2+ release from endothelial stores, probably via redox modification of the InsP3 receptor with some contribution of gap junctions, leading to the opening of hyperpolarizing endothelial KCa channels and an electrotonically mediated relaxant response. Paradoxically, H2O2 is a potent vasoconstrictor if KCa channels are blocked. 39 , 40 , 41 , 42 , 43 , 44 , 45 Thus, the mechanism of H2O2‐induced hyperpolarization appears to be complex, depending on the blood vessel used and the type of vascular wall cells examined. 43 , 44 Arterioles from human right atrial appendages obtained at the time of cardiac surgery dilate and hyperpolarize in response to exogenous H2O2; dilation is reduced by catalase, providing evidence that shear stress induces endothelial release of H2O2 and may contribute to flow‐mediated dilatation in patients with heart disease. 45 H2O2 flow and agonist‐dependent relaxation has also been reported in endothelium‐stripped human mesenteric arteries. In this model, relaxations to bradykinin were markedly inhibited by catalase. 43
By using rabbit iliac artery rings, hydrogen peroxide, generated by pro‐oxidant effects of ascorbic acid and tetrahydrobiopterin, has been demonstrated to amplify IKCa‐ and SKCa‐driven hyperpolarization‐mediated relaxation by facilitating IP3‐mediated Ca2+ release from endothelial stores. 21 , 46
Oxidases
Oxidases, including cytochrome P‐450 epoxygenases, cyclooxygenases (COXs), lipoxygenases, and xanthine oxidases; mitochondrial respirator chain enzymes; and NADPH oxidases can also produce superoxide, which is dismutated by superoxide dismutase into H2O2. 43 , 45 , 47
Epoxyeicosatrienoic Acid
Endothelial epoxyeicosatrienoic acid (EET) hyperpolarized bovine coronary smooth muscle cells by activating BKCa (KCa1.1) channels. 48 Bradykinin induces hyperpolarization of endothelial cells; it also hyperpolarizes myocytes by a mechanism independent of endothelial cell hyperpolarization, which involves endothelial cell production of EETs (most likely 14,15‐ and/or 11,12‐EET), that, in turn, open endothelial IKCa and SKCa channels and also activate large‐conductance calcium‐sensitive K+ channels (BKCa) on the surrounding myocytes. 49 The 11,12‐EET causes relaxation by activating smooth muscle cell BKCa channels in human internal mammary artery. 50
Potassium
Lastly, K+ ions activate Na+/K+ pumps and inward rectifier K+ channels (Kir) to cause hyperpolarization. 23 , 51 , 52
Prostaglandins
The COX products PGI2 and PGE2 relax vascular smooth muscle. 53 , 54 , 55 PGI2 is continuously released into the circulation by the lungs to counter platelet aggregation from the release of thromboxane A2 (TxA2). 33 The PGI2/TxA2 ratio has been observed to be important; manipulation of this ratio with small doses of aspirin has similar beneficial effects as antithrombotic therapy, 33 , 56 but with little (if any) role in maintaining vasoreactivity in normotensive type 2 diabetic patients. 57 , 58 Synthesis of PGI2 is enhanced in the spontaneously hypertensive and Goldblatt hypertensive rat. Metabolism of PGE2, PGF2‐α, and PGI2 by prostaglandin 15‐hydroxydehydrogenase is impaired in genetic models. Responses to endothelium‐dependent vasodilators are impaired in acute and chronic animal models of hypertension. Production of relaxing factor by the endothelium is not inhibited, but rather the vascular smooth muscle fails to respond. Acute, severe hypertension potentiates the response to serotonin, presumably by attenuating the release or response to relaxing factor(s). In the aorta of the SHR, the endothelium releases a constricting factor in response to acetylcholine. Pulmonary arterial endothelium (and other vessels) releases a vasoconstrictor that is blocked by inhibitors of COX; this pressor factor may be TxA2. Certainly less, PGI2 is produced in acute and chronic models of hypertension. In severe hypertension the response to serotonin is potentiated by the attenuated response to relaxing factors; this response is blunted in the presence of COX inhibitors. 59 In patients with hypertension, production of vasoactive prostanoids is selectively impaired and may contribute to the increased systemic vascular resistance and increased incidence of thrombosis. 60 Recent animal studies have shown that PGI2 may, in fact, paradoxically induce vasoconstriction rather than vasodilatation in certain circumstances. In the aortic rings from SHR and aging Wistar Kyoto rats, the endothelium‐dependent contractions elicited by acetylcholine most likely involve the release of PGI2 with a concomitant contribution of PGH2. 61 , 62 Additional studies with rat aortic strips have indicated that PGI2 induces relaxation through a PGI2‐PGE1 receptor; however, higher concentrations of PGI2 act at the TxA2‐PGH2‐receptor to decrease PGI2‐induced relaxation. 63 , 64 , 65 Moreover, inhibition of NO with infusion of L‐NMMA has been shown to significantly increase BP and total peripheral artery resistance. 20
Linking Endothelial Dysfunction to Hypertension—Implications for Disease States
Decreased NO Production and Cardiovascular Effects
The cardiovascular effects of reduced NO bioactivity per se are suggested by data from studies in animals and humans. 66 , 67 , 68 , 69 For example, inhibition of NO with L‐NMMA increases arterial stiffness. 13 Further, in the pulmonary circulation of eNOS‐deficient mice, there is an increase in pulmonary artery pressure. 70 In patients with coronary heart disease, elevated C‐reactive protein level, an inflammatory marker, was shown to be significantly and independently associated with impaired NO bioavailability as measured by forearm blood flow (FBF) vasodilatory response to L‐NMMA. These findings further support the hypothesis that oxidative stress and inflammation are major causes of impaired NO. 71
Impaired NO and Hypertension
Endothelial dysfunction is often described in individuals with hypertension. 72 Multiple studies of flow‐mediated dilatation of the brachial artery, usually examined by venous occlusion plethysmography of FBF, have shown that patients with hypertension exhibit blunted arterial vasodilation in response to endothelium‐dependent vasodilators, such as Ach, while vasodilatory responses to endothelium‐independent vasodilators, such as sodium nitroprusside, are preserved.
Oxidative Stress and Endothelial Dysfunction
Many of those mechanisms contributing to vascular smooth muscle relaxation and vasodilation are depicted in the Figure. The discussion of all possible mechanisms for robbing the endothelium of its ability to vasodilate is beyond the scope of this discussion. However, evidence is accumulating at an ever‐increasing rate that the neutralization of NO by oxidative stress may underlie the endothelial dysfunction that leads to hypertension and atherosclerosis.
The importance of NO in promoting endothelial homeostasis is demonstrated by the association between impaired NO bioactivity and endothelial dysfunction, which is characterized by the imbalance of endothelium‐derived vasoconstrictive and vasodilatory substances, with a shift toward greater vasoconstriction, inflammation, and thrombosis. 72
One proposed mechanism of impaired NO availability and endothelial dysfunction is oxidative stress. 73 , 74 Free radical molecules function normally as signals to modulate vascular tone. Oxidative stress exists when pro‐oxidant processes exceed the capacity of antioxidant mechanisms to maintain an appropriate balance. Oxidative stress is produced with increased production of ROS, including superoxide anion that is derived from xanthine oxidase, COX, and NADPH oxidase 75 , 76 enzyme systems. ROS react with NO forming peroxynitrite, thereby decreasing NO bioactivity. 74 , 77 Oxidative stress may cause a deficiency of l‐arginine and tetrahydrobiopterin (BH4). 77 , 78 BH4 is an essential cofactor of NOS, along with NADPH, Ca2+/calmodulin, and flavin nucleotides. 7 , 79 , 80 , 81 , 82 , 83 , 84 , 85 , 86 Reduced availability of BH4 causes Enos to produce superoxide instead of NO, a process known as NOS uncoupling; 87 , 88 this results in increased formation of peroxynitrite (PN), a highly reactive oxidant that can be highly toxic. 73 , 89 , 90 In certain circumstances, however, PN has been reported to have beneficial effects in the cardiovascular system; therefore, the biologic effects of PN can be paradoxical. 73 , 89 , 90 , 91 However, these apparently contradictory responses could well be due to the environment in which studies were conducted. 92 The results observed in nonsanguineous studies may be due to the conversion of PN into deleterious mediators with hydroxyl radical‐like activities, whereas in physiologic studies, the beneficial action of PN may be due to reaction of PN with plasma, red cell glutathione, or plasma cysteine and albumin, with the production of NO or an NO donor–like compound. 93 , 94 , 95 PN induces accumulation of cGMP in a glutathione‐dependent manner in endothelial and smooth muscle cells, and PN produces thiol‐dependent stimulation of purified guanylate cyclase. 96
Endothelial‐Mediated Vasodilation and the Cardiometabolic Syndrome (Glucose Metabolism and Insulin Resistance)
Clinical studies have shown that the arterial vasodilating effect of insulin in skeletal muscle, a primary mechanism of insulin sensitivity, is dependent on endothelial NO release. 97 , 98 Overall, NO appears necessary for regulation of insulin secretion, normal β‐cell function, and insulin sensitivity. 99
Insulin Resistance and Diabetes
Decreased NO bioactivity and endothelial dysfunction are associated with insulin resistance and diabetes mellitus. 19 , 100 , 101 Because the arterial vasodilating effect of insulin in skeletal muscle is dependent on endothelial NO release, NO impairment would, theoretically, reduce insulin sensitivity. Recent studies suggest that the use of biomarkers of oxidative and nitrosative stress may be the earliest manifestation for the presence of endothelial dysfunction in the development of diabetes, and that some of these studies suggest that this dysfunction may be due to reduced bioavailability of endothelial‐derived NO. 75 , 102 , 103 , 104 , 105 , 106 , 107 , 108 , 109 , 110 , 111 The observed dysfunction of the endothelial NOS/soluble guanylate cyclase/cGMP system is a common mechanism by which cardiovascular risk factors such as diabetes and hypertension mediate their deleterious effects on the vascular wall. 112 , 113 , 114 , 115 , 116 , 117 , 118 , 119 , 120
Conclusions
It is evident that endothelial‐dependent mechanisms play a major role in the regulation of blood flow and BP. There is abundant evidence that dysregulation of these mechanisms may be involved in the pathogenesis of arterial hypertension and the increase in BP that contributes to the target organ damage that ensues. One of the principal mechanisms by which the endothelium produces vasodilation is the production of NO. Interestingly, many pharmacologic agents currently used for the treatment of cardiovascular disease influence NO production, eg, statins, NO donors, antihypertensive drugs (CCBs, ACE inhibitors), use of stimulating sGC drugs, and the vasodilating β‐blocker, nebivolol. Furthermore, there is increasing evidence that manipulation of the various EDHF molecules may also have BP‐lowering effects. It is apparent that effective management of BP requires understanding of both physiologic and pathophysiologic regulation of vascular tone. The appropriate use of existing therapies and the development of new and novel approaches are necessarily based on such an understanding.
References
- 1. Furchgott R, Vanhoutte P. Endothelium‐derived relaxing and contracting factors. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- 2. Bevan JA, Henrion D. Pharmacological implications of the flow‐dependence of vascular smooth muscle tone. Annu Rev Pharmacol Toxicol. 1994;34:173–190. [DOI] [PubMed] [Google Scholar]
- 3. Sainz JM, Reche C, Rabano MA, et al. Effects of nitric oxide on aldosterone synthesis and nitric oxide synthase activity in glomerulosa cells from bovine adrenal gland. Endocrine. 2004;24:61–71. [DOI] [PubMed] [Google Scholar]
- 4. Gibbons GH. Endothelial function as a determinant of vascular function and structure: a new therapeutic target. Am J Cardiol. 1997;79:3–8. [DOI] [PubMed] [Google Scholar]
- 5. Moncada S. The L‐arginine: nitric oxide pathway, cellular transduction and immunological roles. Adv Second Messenger Phosphoprotein Res. 1993;28:97–99. [PubMed] [Google Scholar]
- 6. Cylwik D, Mogielnicki A, Buczko W. L‐arginine and cardiovascular system. Pharmacol Rep. 2005;57:14–22. [PubMed] [Google Scholar]
- 7. Mayer B, Heinzel B, Klatt P, et al. Nitric oxide synthase‐catalyzed activation of oxygen and reduction of cytochromes: reaction mechanisms and possible physiological implications. J Cardiovasc Pharmacol. 1992;20(Suppl 12):S54–S56. [DOI] [PubMed] [Google Scholar]
- 8. Smith GC, Tew DG, Wolf CR. Dissection of NADPH‐cytochrome P450 oxidoreductase into distinct functional domains. Proc Natl Acad Sci USA. 1994;91:8710–8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li J, Zhou Z, Jiang DJ, et al. Reduction of NO‐ and EDHF‐mediated vasodilatation in hypertension: role of asymmetric dimethylarginine. Clin Exp Hypertens. 2007;29:489–501. [DOI] [PubMed] [Google Scholar]
- 10. Boger RH. Asymmetric dimethylarginine (ADMA) and cardiovascular disease: insights from prospective clinical trials. Vasc Med. 2005;10(Suppl 1):S19–S25. [DOI] [PubMed] [Google Scholar]
- 11. Perticone F, Ceravolo R, Pujia A, et al. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation. 2001;104:191–196. [DOI] [PubMed] [Google Scholar]
- 12. Raij L. Workshop: hypertension and cardiovascular risk factors: role of the angiotensin II‐nitric oxide interaction. Hypertension. 2001;37:767–773. [DOI] [PubMed] [Google Scholar]
- 13. Wilkinson IB, Franklin SS, Cockcroft JR. Nitric oxide and the regulation of large artery stiffness: from physiology to pharmacology. Hypertension. 2004;44:112–116. [DOI] [PubMed] [Google Scholar]
- 14. Paulus WJ, Bronzwaer JG. Nitric oxide’s role in the heart: control of beating or breathing? Am J Physiol Heart Circ Physiol. 2004;287:H8–H13. [DOI] [PubMed] [Google Scholar]
- 15. Scherrer‐Crosbie M, Ullrich R, Bloch KD, et al. Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation. 2001;104:1286–1291. [DOI] [PubMed] [Google Scholar]
- 16. Spieker LE, Flammer AJ, Luscher TF. The vascular endothelium in hypertension. Handb Exp Pharmacol. 2006;(176 pt 2)::249–283. [DOI] [PubMed] [Google Scholar]
- 17. Cebeci SA, Kocaturk PA, Kavas GO. Hypertension: does impaired endothelium‐dependent relaxation affect superoxide scavenging? Biol Trace Elem Res. 2002;90:239–249. [DOI] [PubMed] [Google Scholar]
- 18. Taddei S, Virdis A, Ghiadoni L, et al. Endothelial dysfunction in hypertension. J Nephrol. 2000;13:205–210. [PubMed] [Google Scholar]
- 19. Fitzgerald SM, Brands MW. Nitric oxide may be required to prevent hypertension at the onset of diabetes. Am J Physiol Endocrinol Metab. 2000;279:E762–E768. [DOI] [PubMed] [Google Scholar]
- 20. Haynes WG, Noon JP, Walker BR, et al. Inhibition of nitric oxide synthesis increases blood pressure in healthy humans. J Hypertens. 1993;11:1375–1380. [DOI] [PubMed] [Google Scholar]
- 21. Félétou M, Vanhoutte PM. EDHF: an update. Clin Sci (Lond). 2009;117:139–155. [DOI] [PubMed] [Google Scholar]
- 22. Félétou M, Köhler R, Vanhoutte PM. Endothelium derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets. Current Rev Hypertens. 2010;12:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Edwards G, Feletou M, Weston AH. Endothelium‐derived hyperpolarizing factors and associated pathways: a synopsis. Pflugers Arch. 2010;459:863–879. [DOI] [PubMed] [Google Scholar]
- 24. Köhler R, Kaistha BP, Wulff H. Vascular Kca‐channels as therapeutic targets in hypertension and restenosis disease. Expert Opin Ther Targets. 2010;14:143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brähler S, Kaistha A, Schmidt VJ, et al. Genetic deficit of SK3 and IK1 channels disrupts the endothelium‐derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation. 2009;119:2323–2332. [DOI] [PubMed] [Google Scholar]
- 26. Wei CM, Hu S, Miller VM, et al. Vascular actions of C‐type natriuretic peptide in isolated porcine coronary arteries and coronary vascular smooth muscle cells. Biochem Biophys Res Commun. 1994;205:765–771. [DOI] [PubMed] [Google Scholar]
- 27. Honing ML, Smits P, Morrison PJ, et al. C‐type natriuretic peptide‐induced vasodilation is dependent on hyperpolarization in human forearm resistance vessels. Hypertension. 2001;37:1179–1183. [DOI] [PubMed] [Google Scholar]
- 28. Ahluwalia A, Hobbs AJ. Endothelium‐derived C‐type natriuretic peptide: more than just a hyperpolarizing factor. Trends Pharmacol Sci. 2005;26:162–167. [DOI] [PubMed] [Google Scholar]
- 29. Sandow SL, Tare M. C‐type natriuretic peptide: a new endothelium‐derived hyperpolarizing factor? Trends Pharmacol Sci. 2007;28:61–67. [DOI] [PubMed] [Google Scholar]
- 30. Wang R. Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–1798. [DOI] [PubMed] [Google Scholar]
- 31. Szabó C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. [DOI] [PubMed] [Google Scholar]
- 32. Zhao W, Zhang J, Lu Y, et al. The vasorelaxant effect of H2S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moncada S, Vane JR. The role of prostacyclin in vascular tissue. Fed Proc. 1979;38:66–71. [PubMed] [Google Scholar]
- 34. Yang G, Wu L, Jiang B, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma‐lyase. Science. 2008;322:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Durante W. Carbon monoxide and bile pigments: surprising mediators of vascular function. Vasc Med. 2002;7:195–202. [DOI] [PubMed] [Google Scholar]
- 36. Wu L, Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev. 2005;57:585–630. [DOI] [PubMed] [Google Scholar]
- 37. Chen YH, Yet SF, Perrella MA. Role of heme oxygenase‐1 in the regulation of blood pressure and cardiac function. Exp Biol Med (Maywood). 2003;228:447–453. [DOI] [PubMed] [Google Scholar]
- 38. Wang R, Shamloul R, Wang X, et al. Sustained normalization of high blood pressure in spontaneously hypertensive rats by implanted hemin pump. Hypertension. 2006;48:685–692. [DOI] [PubMed] [Google Scholar]
- 39. Ellis A, Triggle CR. Endothelium‐derived reactive oxygen species: their relationship to endothelium‐dependent hyperpolarization and vascular tone. Can J Physiol Pharmacol. 2003;81:1013–1028. [DOI] [PubMed] [Google Scholar]
- 40. Lucchesi PA, Belmadani S, Matrougui K. Hydrogen peroxide acts as both vasodilator and vasoconstrictor in the control of perfused mouse mesenteric resistance arteries. J Hypertens. 2005;23:571–579. [DOI] [PubMed] [Google Scholar]
- 41. Edwards DH, Li Y, Griffith TM. Hydrogen peroxide potentiates the EDHF phenomenon by promoting endothelial Ca2+ mobilization. Arterioscler Thromb Vasc Biol. 2008;28:1774–1781. [DOI] [PubMed] [Google Scholar]
- 42. Matoba T, Shimokawa H, Nakashima M, et al. Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matoba T, Shimokawa H, Kubota H, et al. Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in human mesenteric arteries. Biochem Biophys Res Commun. 2002;290:909–913. [DOI] [PubMed] [Google Scholar]
- 44. Miura H, Bosnjak JJ, Ning G, et al. Role for hydrogen peroxide in flow‐induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–e40. [DOI] [PubMed] [Google Scholar]
- 45. Matoba T, Shimokawa H. Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in animals and humans. J Pharmacol Sci. 2003;92:1–6. [DOI] [PubMed] [Google Scholar]
- 46. Garry A, Edwards DH, Fallis IF, et al. Ascorbic acid and tetrahydrobiopterin potentiate the EDHF phenomenon by generating hydrogen peroxide. Cardiovasc Res. 2009;84:218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shimokawa H, Matoba T. Hydrogen peroxide as an endothelium‐derived hyperpolarizing factor. Pharmacol Res. 2004;49:543–549. [DOI] [PubMed] [Google Scholar]
- 48. Campbell WB, Gebremedhin D, Pratt PF, et al. Identification of epoxyeicosatrienoic acids as endothelium‐derived hyperpolarizing factors. Circ Res. 1996;78:415–423. [DOI] [PubMed] [Google Scholar]
- 49. Weston AH, Félétou M, Vanhoutte PM, et al. Bradykinin‐induced, endothelium‐dependent responses in porcine coronary arteries: involvement of potassium channel activation and epoxyeicosatrienoic acids. Br J Pharmacol. 2005;145:775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Archer SL, Gragasin FS, Wu X, et al. Endothelium‐derived hyperpolarizing factor in human internal mammary artery is 11,12‐epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation. 2003;107:769–776. [DOI] [PubMed] [Google Scholar]
- 51. Edwards G, Dora KA, Gardener MJ, et al. K+ is an endothelium‐derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. [DOI] [PubMed] [Google Scholar]
- 52. Edwards G, Thollon C, Gardener MJ, et al. Role of gap junctions and EETs in endothelium‐dependent hyperpolarization of porcine coronary artery. Br J Pharmacol. 2000;129:1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Greenberg S, Kadowitz PJ. Difference in prostaglandin modulation of arterial and venous smooth muscle responses to bradykinin and norepinephrine. Methods Find Exp Clin Pharmacol. 1982;4:7–24. [PubMed] [Google Scholar]
- 54. Chapple DJ, Dusting GJ, Hughes R, et al. Some direct and reflex cardiovascular actions of prostacyclin (PGI2) and prostaglandin E2 in anaesthetized dogs. Br J Pharmacol. 1980;68:437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lippton HL, Paustian PW, Mellion BT, et al. Cardiovascular actions of prostacyclin (PGI2) in the cat. Arch Int Pharmacodyn Ther. 1979;241:121–130. [PubMed] [Google Scholar]
- 56. de Kergommeaux BD, Ali M, McDonald JW. Effects of ASA on thromboxane and prostacyclin synthesis by rabbit aorta and pulmonary artery. Prostaglandins Leukot Med. 1983;11:225–231. [DOI] [PubMed] [Google Scholar]
- 57. Johnstone MT, Creager SJ, Scales KM, et al. Impaired endothelium‐dependent vasodilation in patients with insulin‐dependent diabetes mellitus. Circulation. 1993;88:2510–2516. [DOI] [PubMed] [Google Scholar]
- 58. Williams SB, Cusco JA, Roddy MA, et al. Impaired nitric oxide‐mediated vasodilation in patients with non‐insulin‐dependent diabetes mellitus. J Am Coll Cardiol. 1996;27:567–574. [DOI] [PubMed] [Google Scholar]
- 59. Peach MJ, Loeb AL. Changes in vascular endothelium and its function in systemic arterial hypertension. Am J Cardiol. 1987;60:110I–115I. [DOI] [PubMed] [Google Scholar]
- 60. Minuz P, Barrow SE, Cockcroft JR, et al. Prostacyclin and thromboxane biosynthesis in mild essential hypertension. Hypertension. 1990;15:469–474. [DOI] [PubMed] [Google Scholar]
- 61. Gluais P, Lonchampt M, Morrow JD, et al. Acetylcholine‐induced endothelium‐dependent contractions in the SHR aorta: the Janus face of prostacyclin. Br J Pharmacol. 2005;146:834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gluais P, Paysant J, Badier‐Commander C, et al. In SHR aorta, calcium ionophore A‐23187 releases prostacyclin and thromboxane A2 as endothelium‐derived contracting factors. Am J Physiol Heart Circ Physiol. 2006;291:H2255–H2264. [DOI] [PubMed] [Google Scholar]
- 63. Williams SP, Dorn GW 2nd, Rapoport RM. Prostaglandin I2 mediates contraction and relaxation of vascular smooth muscle. Am J Physiol. 1994;267:H796–H803. [DOI] [PubMed] [Google Scholar]
- 64. Panza JA, Quyyumi AA, Brush JE Jr, et al. Abnormal endothelium‐dependent vascular relaxation in patients with essential hypertension. N Engl J Med. 1990;323:22–27. [DOI] [PubMed] [Google Scholar]
- 65. Panza JA, Casino PR, Kilcoyne CM, et al. Role of endothelium‐derived nitric oxide in the abnormal endothelium‐dependent vascular relaxation of patients with essential hypertension. Circulation. 1993;87:1468–1474. [DOI] [PubMed] [Google Scholar]
- 66. Raij L. Nitric oxide in the pathogenesis of cardiac disease. J Clin Hypertens. 2006;8:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Giles TD. Aspects of nitric oxide in health and disease: a focus on hypertension and cardiovascular disease. J Clin Hypertens. 2006;8:2–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cooke JP. The pivotal role of nitric oxide for vascular health. Can J Cardiol. 2004;20(Suppl B):7B–15B. [PubMed] [Google Scholar]
- 69. Nakamura R, Egashira K, Arimura K, et al. Increased inactivation of nitric oxide is involved in impaired coronary flow reserve in heart failure. Am J Physiol Heart Circ Physiol. 2001;281:H2619–H2625. [DOI] [PubMed] [Google Scholar]
- 70. Champion HC, Bivalacqua TJ, Greenberg SS, et al. Adenoviral gene transfer of endothelial nitric‐oxide synthase (Enos) partially restores normal pulmonary arterial pressure in Enos‐deficient mice. Proc Natl Acad Sci USA. 2002;99:13248–13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fichtlscherer S, Breuer S, Schachinger V, et al. C‐reactive protein levels determine systemic nitric oxide bioavailability in patients with coronary artery disease. Eur Heart J. 2004;25:1412–1418. [DOI] [PubMed] [Google Scholar]
- 72. Brunner H, Cockcroft JR, Deanfield J, et al. Endothelial function and dysfunction. Part II: association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension. J Hypertens. 2005;23:233–246. [DOI] [PubMed] [Google Scholar]
- 73. Uppu RM, Nossaman BD, Greco AJ, et al. Cardiovascular effects of peroxynitrite. Clin Exp Pharmacol Physiol. 2007;34:933–937. [DOI] [PubMed] [Google Scholar]
- 74. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. [DOI] [PubMed] [Google Scholar]
- 75. Zalba G, Beaumont FJ, San JoseG, et al. Vascular NADH/NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension. 2000;35:1055–1061. [DOI] [PubMed] [Google Scholar]
- 76. Li JJ, Chen JL. Inflammation may be a bridge connecting hypertension and atherosclerosis. Med Hypotheses. 2005;64:925–929. [DOI] [PubMed] [Google Scholar]
- 77. Kawashima S, Yokoyama M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:998–1005. [DOI] [PubMed] [Google Scholar]
- 78. Kurowska EM. Nitric oxide therapies in vascular diseases. Curr Pharm Des. 2002;8:155–166. [DOI] [PubMed] [Google Scholar]
- 79. Wyatt AW, Steinert JR, Mann GE. Modulation of the L‐arginine/nitric oxide signalling pathway in vascular endothelial cells. Biochem Soc Symp. 2004;(71):143–156. [DOI] [PubMed] [Google Scholar]
- 80. Schmidt HH, Pollock JS, Nakane M, et al. Ca2+/calmodulin‐regulated nitric oxide synthases. Cell Calcium. 1992;13:427–434. [DOI] [PubMed] [Google Scholar]
- 81. Berka V, Yeh HC, Gao D, et al. Redox function of tetrahydrobiopterin and effect of L‐arginine on oxygen binding in endothelial nitric oxide synthase. Biochemistry. 2004;43:13137–13148. [DOI] [PubMed] [Google Scholar]
- 82. Perry JM, Marletta MA. Effects of transition metals on nitric oxide synthase catalysis. Proc Natl Acad Sci USA. 1998;95:11101–11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ortiz de Montellano PR, Nishida C, Rodriguez‐Crespo I, Gerber N. Nitric oxide synthase structure and electron transfer. Drug Metab Dispos. 1998;26:1185–1189. [PubMed] [Google Scholar]
- 84. Wang X, Hattori Y, Satoh H, et al. Tetrahydrobiopterin prevents endothelial dysfunction and restores adiponectin levels in rats. Eur J Pharmacol. 2007;555:48–53. [DOI] [PubMed] [Google Scholar]
- 85. Fukuda Y, Teragawa H, Matsuda K, et al. Tetrahydrobiopterin restores endothelial function of coronary arteries in patients with hypercholesterolaemia. Heart. 2002;87:264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ueda S, Matsuoka H, Miyazaki H, et al. Tetrahydrobiopterin restores endothelial function in long‐term smokers. J Am Coll Cardiol. 2000;35:71–75. [DOI] [PubMed] [Google Scholar]
- 87. Leopold JA, Loscalzo J. Organic nitrate tolerance and endothelial dysfunction: role of folate therapy. Minerva Cardioangiol. 2003;51:349–359. [PubMed] [Google Scholar]
- 88. Dixon LJ, Morgan DR, Hughes SM, et al. Functional consequences of endothelial nitric oxide synthase uncoupling in congestive cardiac failure. Circulation. 2003;107:1725–1728. [DOI] [PubMed] [Google Scholar]
- 89. Nossaman BD, Kadowitz PJ. Potential benefits of peroxynitrite. Open Pharmacol J. 2008;2:31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nossaman BD, Dabisch PA, Liles JT, et al. Peroxynitrite does not impair pulmonary and systemic vascular responses. J Appl Physiol. 2004;96:455–462. [DOI] [PubMed] [Google Scholar]
- 91. Nossaman BD, Bivalacqua TJ, Champion HC, et al. Analysis of vasodilator responses to peroxynitrite in the hindlimb vascular bed of the cat. J Cardiovasc Pharmacol. 2007;50:358–366. [DOI] [PubMed] [Google Scholar]
- 92. Ronson RS, Thourani VH, Ma XL, et al. Peroxynitrite, the breakdown product of nitric oxide, is beneficial in blood cardioplegia but injurious in crystalloid cardioplegia. Circulation. 1999;100:II384–II391. [DOI] [PubMed] [Google Scholar]
- 93. Meister A. Glutathione‐ascorbic acid antioxidant system in animals. J Biol Chem. 1994;269:9397–9400. [PubMed] [Google Scholar]
- 94. Dowell FJ, Martin W. The effects of peroxynitrite on rat aorta: interaction with glucose and related substances. Eur J Pharmacol. 1997;338:43–53. [DOI] [PubMed] [Google Scholar]
- 95. Radi R, Beckman JS, Bush KM, et al. Peroxynitrite‐induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys. 1991;288:481–487. [DOI] [PubMed] [Google Scholar]
- 96. Mayer B, Schrammel A, Klatt P, et al. Peroxynitrite‐induced accumulation of cyclic GMP in endothelial cells and stimulation of purified soluble guanylyl cyclase. Dependence on glutathione and possible role of S‐nitrosation. J Biol Chem. 1995;270:17355–17360. [DOI] [PubMed] [Google Scholar]
- 97. Petrie JR, Ueda S, Webb DJ, et al. Endothelial nitric oxide production and insulin sensitivity. A physiological link with implications for pathogenesis of cardiovascular disease. Circulation. 1996;93:1331–1333. [DOI] [PubMed] [Google Scholar]
- 98. Scherrer U, Sartori C. Defective nitric oxide synthesis: a link between metabolic insulin resistance, sympathetic overactivity and cardiovascular morbidity. Eur J Endocrinol. 2000;142:315–323. [DOI] [PubMed] [Google Scholar]
- 99. Lajoix AD, Reggio H, Chardes T, et al. A neuronal isoform of nitric oxide synthase expressed in pancreatic beta‐cells controls insulin secretion. Diabetes. 2001;50:1311–1323. [DOI] [PubMed] [Google Scholar]
- 100. Cleland SJ, Petrie JR, Small M, et al. Insulin action is associated with endothelial function in hypertension and type 2 diabetes. Hypertension. 2000;35:507–511. [DOI] [PubMed] [Google Scholar]
- 101. Calver A, Collier J, Vallance P. Inhibition and stimulation of nitric oxide synthesis in the human forearm arterial bed of patients with insulin‐dependent diabetes. J Clin Invest. 1992;90:2548–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hink U, Li H, Mollnau H, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:E14–E22. [DOI] [PubMed] [Google Scholar]
- 103. Rask‐Madsen C, King GL. Mechanisms of disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46–56. [DOI] [PubMed] [Google Scholar]
- 104. Mather KJ, Lteif A, Steinberg HO, et al. Interactions between endothelin and nitric oxide in the regulation of vascular tone in obesity and diabetes. Diabetes. 2004;53:2060–2066. [DOI] [PubMed] [Google Scholar]
- 105. Steinberg HO, Baron AD. Vascular function, insulin resistance and fatty acids. Diabetologia. 2002;45:623–634. [DOI] [PubMed] [Google Scholar]
- 106. Nambi V, Ballantyne C. Role of biomarkers in developing new therapies for vascular disease. World J Surg. 2007;31:676–681. [DOI] [PubMed] [Google Scholar]
- 107. Tsukahara H. Biomarkers for oxidative stress: clinical application in pediatric medicine. Curr Med Chem. 2007;14:339–351. [DOI] [PubMed] [Google Scholar]
- 108. Zoppini G, Targher G, Zamboni C, et al. Effects of moderate‐intensity exercise training on plasma biomarkers of inflammation and endothelial dysfunction in older patients with type 2 diabetes. Nutr Metab Cardiovasc Dis. 2006;16:543–549. [DOI] [PubMed] [Google Scholar]
- 109. Matteucci E, Passerai S, Mariotti M, et al. Dietary habits and nutritional biomarkers in Italian type 1 diabetes families: evidence of unhealthy diet and combined‐vitamin‐deficient intakes. Eur J Clin Nutr. 2005;59:114–122. [DOI] [PubMed] [Google Scholar]
- 110. Ding H, Triggle CR. Endothelial cell dysfunction and the vascular complications associated with type 2 diabetes: assessing the health of the endothelium. Vasc Health Risk Manag. 2005;1:55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Miyata T, Kurokawa K. A detective story for biomedical footprints towards new therapeutic interventions in diabetic nephropathy. Intern Med. 2003;42:1165–1171. [PubMed] [Google Scholar]
- 112. Boger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “L‐Arginine Paradox” and acts as a novel cardiovascular risk factor. J Nutr. 2004;134:2842S–2847S. [DOI] [PubMed] [Google Scholar]
- 113. Siasos G, Tousoulis D, Antoniades C, et al. L‐Arginine, the substrate for NO synthesis: an alternative treatment for premature atherosclerosis? Int J Cardiol. 2007;116:300–308. [DOI] [PubMed] [Google Scholar]
- 114. Winer N, Sowers JR. Diabetes and arterial stiffening. Adv Cardiol. 2007;44:245–251. [DOI] [PubMed] [Google Scholar]
- 115. Shimasaki Y, Saito Y, Yoshimura M, et al. The effects of long‐term smoking on endothelial nitric oxide synthase mRNA expression in human platelets as detected with real‐time quantitative RT‐PCR. Clin Appl Thromb Hemost. 2007;13:43–51. [DOI] [PubMed] [Google Scholar]
- 116. Forgione MA, Loscalzo J. Oxidant stress as a critical determinant of endothelial function. Drug News Perspect. 2000;13:523–529. [DOI] [PubMed] [Google Scholar]
- 117. Cooke JP. Therapeutic interventions in endothelial dysfunction: endothelium as a target organ. Clin Cardiol. 1997;20:II‐45–51. [PubMed] [Google Scholar]
- 118. Signorello MG, Viviani GL, Armani U, et al. Homocysteine, reactive oxygen species and nitric oxide in type 2 diabetes mellitus. Thromb Res. 2007;120:607–613. [DOI] [PubMed] [Google Scholar]
- 119. Tawakol A, Omland T, Gerhard M, et al. Hyperhomocyst(e)inemia is associated with impaired endothelium‐dependent vasodilation in humans. Circulation. 1997;95:1119–1121. [DOI] [PubMed] [Google Scholar]
- 120. Natali A, Toschi E, Baldeweg S, et al. Clustering of insulin resistance with vascular dysfunction and low‐grade inflammation in type 2 diabetes. Diabetes. 2006;55:1133–1140. [DOI] [PubMed] [Google Scholar]