

Abstract
J Clin Hypertens (Greenwich). 2012; 14:365–371. ©2012 Wiley Periodicals, Inc.
The concept of developmental origins of adult disease derives from both epidemiologic and basic sciences. This brief review considers the impact of the intrauterine milieu, intrauterine growth retardation, premature birth, and infant feeding on later hypertension and kidney disease.
The concept of developmental origins of adult diseases derives from several different types of observations. The thought that there were critical or “sensitive” periods of development originated more than 50 years ago and was put forward by Widdowson and McCance, 1 who demonstrated that malnutrition led to distinct and different effects, depending on when the life cycle malnutrition occurred. In the 1980s, epidemiologic data in which an association was observed between birth weight, as well as placental size, and subsequent cardiovascular disease led to the understanding that the perinatal environment was important in the risk of future disease. 2 , 3 , 4 The implications of such epidemiologic observations were brought to prominence by the work of David Barker and colleagues, and the data mesh well with the hypothesis put forth nearly concomitantly by Brenner and colleagues, 5 who hypothesized that the final number of nephrons, which is fixed by term birth in humans, might explain subsequent hypertension and chronic kidney disease. In the decades since the work in this area began, the data have shifted from phenomenology to a current emphasis on seeking mechanisms for these observations. In this brief review, we examine the information available to explain the pathogenesis and mechanisms of what has been widely termed perinatal programming or developmental origins of health and disease. In 2001, Bateson 6 suggested that a better term for the subtle changes that occur in the perinatal period but have long‐ranging effects might be phenotypic induction, a label that may better combine the various forces at work—both environmental and epigenetic—that lead to time‐spanning changes in the individual. This paper considers the impact of the intrauterine milieu, intrauterine growth retardation, premature birth, and infant feeding on later hypertension and renal disease.
Prenatal Stress and the Intrauterine Milieu
A number of perinatal perturbations have been associated with subsequent hypertension—maternal malnutrition, exposures to medications or toxins during pregnancy, and dietary deficiencies. 7 , 8 , 9 , 10 The influence of dietary alterations and stress on the developing fetus or immature (premature) newborn may be very mild, changing the developmental sequence very subtly, yet permanently (Table and Figure). Such changes may include alterations in the distribution of cell types within an organ or differences in gene expression within given cells within the organ. The effects of stress may vary, depending on when the stress occurs. For example, embryos of dams exposed to a low‐protein diet only within the preimplantation period had decreased inner cell mass in the early blastocyst and, in the mid to late blastocyst, a decreased number of cells were found in the trophoectoderm. 11 While the mechanism is unclear, such decreased cell numbers may lead to long‐term changes in the developing organism. It is thought that increases in maternal glucocorticoid levels via stress may transduce some of the changes observed in the offspring. The placenta normally has 11‐beta‐hydroxysteroid dehydrogenase type 1, which inactivates maternal glucocorticoids. During stress, placentas express less of this enzyme, and the fetus is exposed to higher glucocorticoid levels as a result (summarized in Baum 9 ). It has been demonstrated that glucocorticoids at critical windows lead to decreased nephrogenesis, at least in the rat, likely by inhibition of branching morphogenesis and, ultimately, resulting in a relatively low nephron number. 12 , 13
Table TABLE.
Stressors and Developmental Origins of Health and Disease
| Timing of Stressor | Exposure Type | Effects in Models | Observations in Humans |
|---|---|---|---|
| Preimplantation | Malnutrition/low‐protein diet | Cell deficiencies in blastocysts | – |
| Placental | Placental insufficiency, multiple pregnancies | Models of placental insufficiency associated with intrauterine growth restriction | Infants with intrauterine growth restriction often have small placentas; placental size correlated with birth weight and with future cardiovascular disease in initial Barker studies |
| Intrauterine | Hormones (folate, glucocorticoids) Methylated compounds | Impaired glucose tolerance test/diabetes mellitus reduced β‐cell mass Obesity; impaired nephrogenesis | Impaired glucose tolerance test/diabetes mellitus reduced β‐cell mass Obesity; impaired nephrogenesis |
| Protein‐calorie malnutrition; Hyperglycemia High glucocorticoids Renin angiotensin system alterations Inflammation Free fatty acids | Increased type 1 angiotensin II receptor expression; cardiorenal abnormalities, vascular structural changes; hypertension | Low nephron number, vascular structural changes; hypertension | |
| Perinatal | Malnutrition Overnutrition Toxic substances Hypoxia Oxidative stress Trace metal deficiencies | Epigenetic changes observed Future cardiorenal syndrome Future metabolic syndrome | Future cardiovascular disease Future metabolic syndrome |
Figure FIGURE.
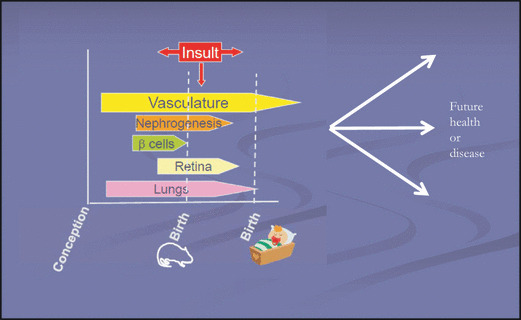
Perinatal stressors, growth, and cardiorenal endowment. This figure shows the length during which certain organ systems of interest develop, as well as potential timing of insults. Depending on whether there are intrauterine or perinatal challenges will inform the potential trajectories of health and disease throughout subsequent development and in adult life.
Normal Kidney Development
The antecedents of the human kidney form in three successive waves: the pronephros, the mesonephros, and the metanephros. The initial functioning of nephrons is present by about 9 weeks of gestation, and no new nephrons are formed after 34 to 36 weeks (after 7–10 days postnatal in rats). Several hundred genes are involved in orchestrating renal development. 14 , 15
The vasculature begins to develop early in the first trimester, but within the kidney, vasculogenesis continues as nephrons form. Major vessels are visible by the start of the third week of fetal life in the human, and the heart is formed by the 9th week. In the human embryo, vasculogenesis begins by about day 18, at which point cells from the splanchnopleuric mesoderm differentiate to become endothelial progenitors and, then, embryonic endothelial cells, which begin to form vessel networks. Subsequently, periendothelial smooth muscle cells, essentially for the formation of actual vessels, also appear. Vasculogenesis continues as organs develop. 14 , 15
Nephrogenesis and renal vascularization are intimately linked. Both glomerular and tubular epithelia are induced by the ureteric bud, which branches. Around the ureteric bud tip, there is a condensation of mesenchymal cells, which ultimately develop into glomeruli. 14 , 15 Concomitantly, the vasculature develops, and if coordination is not synchronized, the kidneys are markedly abnormal. The process is well‐traced in the mouse and some other mammalian models, but, for obvious reasons, is not well studied in humans.
Insults During Ongoing Nephrogenesis
Animal models and ex vivo studies indicate that intrauterine insults such as those imposed simply by a maternal low‐protein diet, may lead to decreased nephron number. There appear to be specific “windows,” at least in rodent models, during which exposure to stress has maximal effects. For example, the timing of the administration of a maternal low‐protein diet and also maternal glucocorticoid administration greatly impacts its effect. In rats, the provision of a low‐protein diet to pregnant dams during the second half of gestation but not during the first half results in a nephron deficit in the offspring. The administration of glucocorticoids at E13–15 leads to hypertension in rats, but not later or earlier. 14 , 15
The strategies that have been employed to examine the impact of challenges to the maternal‐fetal environment have included total caloric restriction, as well as low‐protein diet, high‐fat diet, vitamin A and D deficiencies, high‐salt diet, ethanol exposure, trace metal deficiencies, and glucocorticoid exposure or interruption to endogenous glucocorticoids. 16 , 17
Vasculature and Intrauterine Events
Both experimental models and epidemiologic data suggest that intrauterine events can affect both vasculogenesis and endothelial function, as well as vascular wall structure. Both endothelium‐independent and ‐dependent vasodilatation (well‐recognized precursors of hypertension and atherosclerosis) may be impaired in low‐birth weight infants and may persist to adult life. 18 , 19 , 20 , 21 , 22 Some differences in vascular reactivity are evident, even in the first week of life in small‐for‐gestational‐age neonates. 19 At 9 years of age, children who were low–birth‐weight neonates had evidence of aberrant response to acetylcholine but normal response to the endothelium‐independent vasodilator sodium nitroprusside. Experimental data also indicate exaggerated vasoconstrictor response to angiotensin II in such offspring. 23
There is experimental evidence of decreased vascular arborization during development that leads to microvascular rarefaction in the fetus or neonate exposed to adverse conditions. 24 , 25 , 26 Microvascular rarefaction (which comprises reduced density of arterioles and capillaries) is associated with hypertension. 27 Capillary and microvascular rarefaction can increase peripheral vascular resistance and are generally considered a consequence rather than a cause of hypertension. 28 However, experimental data have shown capillary rarefaction in major sites of peripheral resistance in newborn but not in fetal rats exposed to a maternal restricted protein diet that is prior to blood pressure elevation, 25 , 26 suggesting that microvascular rarefaction can be a primary event in the development of hypertension associated with earlier fetal stress. It can therefore be postulated that the increase in blood pressure could at least be, in part, an adaptation mechanism allowing sufficient capillary recruitment to maintain adequate perfusion to the peripheral tissues. 29 Reduced retinal vascularization and lower peripheral skin blood flow have been reported in young adults born at term but who had evidence of intrauterine growth restriction 30 , 31 or were born preterm (these persons were not diagnosed with formal retinopathy of prematurity during their neonatal period). 32
Mechanisms underlying microvascular rarefaction associated with fetal and/or neonatal stress is incompletely understood. Experimentally, there appears to be an attenuated angiogenic potential early in life based on explants of aortic rings in pups exposed to a maternal restricted protein diet. 26 However, there have been studies indicating that a number of factors known to be participants in angiogenesis—AT1 receptor subtype; endothelial nitric oxide synthase, angiopoietin 1 and 2, the Tie 2 receptor, vascular endothelial growth factor (VEGF), VEGF F receptor‐2, and platelet‐derived growth factor. Controls were not affected. More recent data indicate that vascular progenitor cells could be affected by perinatal adverse conditions. Neonatal mice exposed to 10 days of hyperoxia have lower endothelial progenitor cells in the blood and lungs. 33 Furthermore, circulating endothelial progenitor cells from prematurely born children who were exposed to supplemental oxygen demonstrate impaired growth and proliferation when compared with endothelial progenitor cells taken from either premature infants who remained in room air or from term infants. 34
Low birth weight and premature birth are associated with increased arterial stiffness when tested later as adults, and also in children and adolescents who had such birth histories[ ]. 35 , 36 , 37 , 38 The proportion of elastin vs rigid collagen in the arterial wall is a major determinant of arterial stiffness, which is a key factor in increased systolic and pulse pressure in adults. 39 The main structural change in the artery wall associated with increased stiffness relates to relatively increased collagen and reduced elastin content. Elastin synthesis in vessels peaks in late gestation, decreases rapidly after birth, and is minimal in the adult aorta. 40 Experimental data indicate that extracellular matrix composition can be altered by fetal and/or neonatal stress and thereby contribute to resulting vascular and blood pressure changes. 41 , 42
Trajectory of Development After Perinatal Challenges
The inverse relationship between birth weight and blood pressure 43 , 44 is not present during the neonatal period or very early childhood, but it has been reported from the age of 4 and appears to increase with age. 45 Similarly in experimental models of developmental programming, animals are not born with hypertension but undergo an age‐dependent and earlier‐than‐expected increase in blood pressure. 25 , 46
Studies that consider gestational age (and therefore prematurity) in addition to low birth weight are relatively more recent. In a study of 430 Swedish men, aged 49 years, the authors estimated that every additional week of gestation achieved before birth was associated with a decrease in adult systolic blood pressure of 7.2 mm Hg. 47 Former preterm adolescents and young adults are also reported to have higher blood pressure. 48 Such impact of shortened gestation rather than or in addition to intrauterine growth restriction (IUGR) was noted by additional 43 , 49 but not by all studies. 50 , 51 Interestingly, in many studies, IUGR participants do not seem to differ from those who were born at an appropriate size for gestational age, within both the premature and term groups, suggesting that preterm birth per se plays an important role in the programming of later‐life cardiovascular function. 52
White and colleagues 53 looked at 31 case‐control and cohort studies that included data on more than 49,000 individual persons and data from more than 2 million persons in one record‐linkage report. Their analysis showed that the combined odds ratios for developing kidney disease supported a link between low birth weight and subsequent chronic kidney disease, although not all studies were consistent.
Mechanisms (Epigenetic and Post‐Genetic)
Growing evidence suggests that epigenetic modifications might explain many of the changes noted in perinatal programming. 54 , 55 , 56 Epigenetic changes consist of modifications to nucleic acids that do not change the sequence of base pairs. Such changes consist mainly of chemical reactions—5′‐methylation of cytosines in CpG dinucleotides, phosphorylation, ubiquitination of histones, ADP ribosylation, and other covalent modifications such as acetylation. 54 , 55 , 56 Noncoding RNAs may also influence and change the organization of chromatin. Of importance, epigenetic changes may change conformation and function of chromatin, modulating gene expression and binding of transcription activators and repressors. Methylation of DNA is associated with long‐term changes, while histone modifications lead to short‐term effects.
The term imprinting has multiple meanings. Genetic imprinting connotes alteration of a gene or its expression based on the observation that some genes will have differential expression that depends on maternal or paternal derivation. This expression is a function, in large part, of the epigenetic regulation of a gene. Generally, the methylation pattern of a gene is involved, with more methylation of imprinted regions, which renders them relatively less transcriptionally active. (Note that the term “imprinting” is also used in the field of developmental origins to describe a variety of metabolic changes that occur with intrauterine stresses that are transmitted to a fetus; this is an inexact term that has been used nearly interchangeably with programming and is not considered here.)
Epigenetic changes can and do occur at multiple points in the lifecycle. However, there are major epigenetic changes in the germline—when the germ cells move to the developing gonads and then during early embryonic life, when both maternal and paternal haploid genomes lose much of their DNA methylation and then undergo histone modification. The demethylation that occurs in the paternal genome is extensive, but it does not take place in genes that are paternally imprinted. 54 , 55 , 56 , 57 Some repetitive sequences and heterochromatin near some centromeres may not be demethylated. Thus, these processes erase much of the epigenetic information from the previous generation, although some sequences may remain.
A growing body of data suggests that epigenetic changes do occur with intrauterine stress. The low‐protein intake model of perinatal programming has been used to examine the role of epigenetic changes. Bogdarina and colleagues, 58 for example, examined the offspring of dams that were given low‐protein intake during gestation (8%) as compared with offspring of dams that were given normal protein intake (18%). The investigators examined adrenal expression of renin‐angiotensin system components and reported changes in the expression of genes of the RAS in the offspring at 1 day and at 12 weeks of life. They also reported that differences appeared to be due to demonstrable evidence of epigenetic changes. In particular, the AT1b receptor had a lower‐than‐normal amount of methylation. As in vitro, the AT1b gene is dependent on promoter methylation, the intrauterine effects of a low‐protein diet might permanently alter the gene expression of an important receptor for angiotensin II action.
In another series of experiments in a rat model, Vaiman and colleagues59 examined the level of IUGR‐induced deregulation of the transcriptome and noted that organs are differentially affected by insults such as a low‐protein diet. In general, organs that develop function late are affected more severely than are those that function early. For example, the kidney would be more markedly affected than the heart. In a recent study, Vaiman and colleagues examined offspring of normal protein dams (22%) vs those offspring of dams that had a low (9%) protein diet. They then examined the organ‐specific intensity of growth restriction, looked at clustering of the effects of organ response to stress, and analyzed specific gene groups. They examined epigenetic regulators, imprinted genes, and chromosome‐specific alterations of gene expression and concluded that many alterations would be permanent, affecting organ function well into adult life.
Other Factors
Among the many factors implicated in adverse perinatal conditions and developmental programming of hypertension, oxidative stress seems an important common denominator. Infants are exposed upon birth to relatively high concentrations of oxygen compared with intrauterine life. Indeed, under physiologic conditions the fetus is hypoxic compared with the adult. While human maternal arterial oxygen partial pressure is approximately 90 mm Hg, the highest arterial or venous oxygen partial pressure in the late gestation fetus rarely exceeds 30 mm Hg. In the fetal to neonatal transition, blood oxygen content and oxygen availability abruptly increase in the first few minutes after birth to adult values, eliciting the generation of a burst of oxygen‐free radicals. 60 , 61 , 62 Small‐for‐gestational age and premature infants have lower and less inducible antioxidant defenses because it is only during the last trimester of a normal pregnancy that antioxidant enzyme levels increase. 63 , 64 , 65 , 66 Therefore, the combination of immature antioxidant system to face this surge in oxygen partial pressure plus the need for therapy with oxygen supplementation because of lung immaturity, unstable lung dynamics, and sepsis altogether, can lead to significant oxidative stress in the immediate neonatal period. 67 , 68 , 69 In immature newborns, exposure to supplemental oxygen can halt microvessel growth in the lung and retina, leading to serious short‐term complications such as bronchopulmonary dysplasia and retinopathy of prematurity. 70 , 71 The possible long‐term consequences of early‐life vascular oxidative injury in susceptible individuals are only starting to be explored, and experimental data indicate a premature arrest in cell proliferation in developing organs with accelerated differentiation and oxidative DNA damage. 72 , 73
Management: What Can One Do to Ameliorate the Effect of Adverse Programming?
How do we identify those who are at risk for long‐term sequelae of an adverse intrauterine and perinatal experience? What do we know about managing them? Data from animal models suggest that progeny born to mothers who had a deficient diet during pregnancy remain normotensive and live longer if they remain on a restricted diet during postnatal life. Other studies demonstrate that rat pups born to dams that had uteroplacental insufficiency, owing to uterine vessel ligation, were “rescued” by cross‐fostering to normal mothers. There are data in humans that suggest that the pattern of weight gain after birth, if controlled, can lead to less hypertension and chronic kidney disease.
Enhancing vasculogenesis postnatally can be promoted by exercise. Children randomized to daily school exercise lessons (vs twice a week for the control group) for 1 year improved their physical fitness and significantly increased the number of circulating progenitor cells. 74 Therefore, evidence so far indicate that common sense healthy life habits (limiting calories and regular exercise) can be particularly critical for the significant proportion of children and adults who were born with low birth weight or prematurely.
Unanswered Questions
At present, we do not have a method of assessing vascular and nephron endowment during early infancy in humans. Postmortem studies that assess nephron number strongly suggest that intrauterine events influence cardiorenal endowment, 75 , 76 , 77 , 78 but noninvasive means that could assess nephron number are still needed, as those infants with a nephron number deficit would likely be at high risk. Thus, practical means to distinguish whether all small‐for‐gestational age infants are at high risk would be of benefit. Whether fitting postnatal diet to endowment would impact later metabolic and cardiovascular diseases is unknown, although animal models suggest that such an approach would be of benefit.
Disclosures:
Dr Nuyt is supported by a Fonds de la Recherche en Sante′ du Quebec Senior Scholar Award, and is an investigator on two grants from the Canadian Institutes of Health Research. Dr Ingelfinger and Dr Nuyt have stated that they have no competing interests to disclose.
References
- 1. Widdowson EM, McCance RA. Some effects of accelerating growth. I. General somatic development. Proc R Soc Lond B Biol Sci. 1960;152:188–206. [DOI] [PubMed] [Google Scholar]
- 2. Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077–1081. [DOI] [PubMed] [Google Scholar]
- 3. Barker DJ, Osmond C, Golding J, et al. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barker D, Bull A, Osmond C, Simmonds S. Fetal and placental size and risk of hypertension in adult life. BMJ. 1990;301:259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens. 1988;1:335–347. [DOI] [PubMed] [Google Scholar]
- 6. Bateson P. Fetal experience and good adult design. Int J Epidemiol. 2001;30:928–934. [DOI] [PubMed] [Google Scholar]
- 7. Gluckman PD, Hanson MA. The Fetal Matrix: Evolution, Developmental and Disease. London, UK: Cambridge University Press; 2005. [Google Scholar]
- 8. Hoy WE, Ingelfinger JR, Hallan S, et al. The early development of the kidney and implications for future health. J Dev Orig Health Dis. 2010;1:216–233. [DOI] [PubMed] [Google Scholar]
- 9. Baum M. Role of the kidney in the prenatal and early postnatal programming of hypertension. Am J Physiol Renal Physiol. 2010;298: F235–F247. Epub 2009 Sep 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wells JCK. The concept of phenotypic induction (‘Programming’) and implications for growth. In Preedy VR, ed. Handbook of Growth and Growth Monitoring in Health and Disease. New York, NY: Springer; 2012: 13–25. [Google Scholar]
- 11. Kwong WY, Wild AE, Roberts P, et al. Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Dev Suppl. 2000;127:4195–4202. [DOI] [PubMed] [Google Scholar]
- 12. Ortiz LA, Quan A, Weinberg A, Baum M. Effect of prenatal dexamethasone on rat renal development. Kindey Int. 2001;59:1663–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Habib S, Gattineni J, Twombley K, Baum M. Evidence that prenatal programming of hypertension by dietary protein deprivation is mediated by fetal glucocorticoid exposure. Am J Hypertens. 2011;24(1): 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Woolf AS. Molecular bases of human kidney malformation. Pediatr Nephrol. 1997;11:373–376. [DOI] [PubMed] [Google Scholar]
- 15. Vise PD, Woolf AS, Bard JBL. The Kidney: From Normal Development to Congenital Disease. London, UK: Academic Press; 2003. [Google Scholar]
- 16. Moritz KM, Wintour EM, Black MJ, et al. Factors influencing mammalian kidney development: implications for health in adult life. Adv Anat Embryol Cell Biol. 2008;196:1–78. [DOI] [PubMed] [Google Scholar]
- 17. Nijland MJ, PW Nathanielsz. Developmental programming of the kidney. In: JP Newnham, MG Ross, eds. Early Life Origins of Human Health and Disease. Basel, Switzerland: Karger AG; 2009: 133–141 [Google Scholar]
- 18. Goodfellow J, Bellamy MF, Gorman ST, et al. Endothelial function is impaired in fit young adults of low birth weight. Cardiovasc Res. 1998;40:600–606. [DOI] [PubMed] [Google Scholar]
- 19. Martin H, Hu J, Gennser G, Norman M. Impaired endothelial function and increased carotid stiffness in 9‐year‐old children with low birthweight. Circulation. 2000;102:2739–2744. [DOI] [PubMed] [Google Scholar]
- 20. Goh KL, Shore AC, Quinn M, Tooke JE. Impaired microvascular vasodilatory function in 3‐month‐old infants of low birth weight. Diabetes Care. 2001;24:1102–1107. [DOI] [PubMed] [Google Scholar]
- 21. Leeson CP, Kattenhorn M, Morley R, et al. Impact of low birth weight and cardiovascular risk factors on endothelial function in early adult life. Circulation. 2001;103:1264–1268. [DOI] [PubMed] [Google Scholar]
- 22. Franco MC, Christofalo DM, Sawaya AL, et al. Effects of low birth weight in 8‐ to 13‐year‐old children: implications in endothelial function and uric acid levels. Hypertension. 2006;48:45–50. [DOI] [PubMed] [Google Scholar]
- 23. Nuyt AM. Mechanisms underlying developmental programming of elevated blood pressure and vascular dysfunction: evidence from human studies and experimental animal models. Clin Sci. 2008;114:1–17. [DOI] [PubMed] [Google Scholar]
- 24. Norman M. Low birth weight and the developing vascular tree: a systematic review. Acta Paediatr. 2008;97:1165–1172. [DOI] [PubMed] [Google Scholar]
- 25. Yzydorczyk C, Comte B, Cambonie G, et al. Neonatal oxygen exposure in rats leads to cardiovascular and renal alterations in adulthood. Hypertension. 2008;52:889–895. [DOI] [PubMed] [Google Scholar]
- 26. Pladys P, Sennlaub F, Brault S, et al. Microvascular rarefaction and decreased angiogenesis in rats with fetal programming of hypertension associated with exposure to a low protein diet in utero. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1580–R1588. [DOI] [PubMed] [Google Scholar]
- 27. Boudier HA. Arteriolar and capillary remodelling in hypertension. Drugs. 1999, 58 Spec No 1 , 37–40. [PubMed] [Google Scholar]
- 28. le Noble FA, Stassen FR, Hacking WJ, Struijker Boudier HA. Angiogenesis and hypertension. J Hypertens. 1998;16:1563–1572. [DOI] [PubMed] [Google Scholar]
- 29. Greene AS, Tonellato PJ, Zhang Z, et al. Effect of microvascular rarefaction on tissue oxygen delivery in hypertension. Am J Physiol. 1992;262:H1486–H1493. [DOI] [PubMed] [Google Scholar]
- 30. Mitchell P, Liew G, Rochtchina E, et al. Evidence of arteriolar narrowing in low‐birth‐weight children. Circulation. 2008;118:518–524. [DOI] [PubMed] [Google Scholar]
- 31. Hellstrom A, Dahlgren J, Marsal K, Ley D. Abnormal retinal vascular morphology in young adults following intrauterine growth restriction. Pediatrics. 2004;113:e77–e80. [DOI] [PubMed] [Google Scholar]
- 32. Kistner A, Jacobson L, Jacobson SH, et al. Low gestational age associated with abnormal retinal vascularization and increased blood pressure in adult women. Pediatr Res. 2002;51:675–680. [DOI] [PubMed] [Google Scholar]
- 33. Balasubramaniam V, Mervis CF, Maxey AM, et al. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1073–L1084. [DOI] [PubMed] [Google Scholar]
- 34. Baker CD, Ryan SL, Ingram DA, et al. Endothelial colony‐forming cells from preterm infants are increased and more susceptible to hyperoxia. Am J Respir Crit Care Med. 2009;180:454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cheung YF, Wong KY, Lam BC, Tsoi NS. Relation of arterial stiffness with gestational age and birth weight. Arch Dis Child. 2004;89:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tauzin L, Risso F, Buffat C, et al. Vascular mechanisms in the developmental programming of cardio‐vascular disease. Pediatr Med Chir. 2005;27:18–23. [PubMed] [Google Scholar]
- 37. Rossi P, Tauzin L, Marchand E, et al. Respective roles of preterm birth and fetal growth restriction in blood pressure and arterial stiffness in adolescence. J Adolesc Health. 2011;48:520–522. [DOI] [PubMed] [Google Scholar]
- 38. McEniery CM, Bolton CE, Fawke J, et al. Cardiovascular consequences of extreme prematurity: the EPICure study. J Hypertens. 2011;29:1367–1373. [DOI] [PubMed] [Google Scholar]
- 39. O’Rourke MF, Staessen JA, Vlachopoulos C, et al. Clinical applications of arterial stiffness; definitions and reference values. Am J Hypertens. 2002;15:426–444. [DOI] [PubMed] [Google Scholar]
- 40. Berry CL, Looker T. An alteration in the chemical structure of the aortic wall induced by a finite period of growth inhibition. J Anat. 1973;114:83–94. [PMC free article] [PubMed] [Google Scholar]
- 41. Mivelaz Y, Yzydorczyk C, Barbier A, et al. Neonatal oxygen exposure leads to increased aortic wall stiffness in adult rats: a Doppler ultrasound study. J Dev Orig Health Dis. 2011;2:184–189. [DOI] [PubMed] [Google Scholar]
- 42. Khorram O, Momeni M, Ferrini M, et al. In utero undernutrition in rats induces increased vascular smooth muscle content in the offspring. Am J Obstet Gynecol. 2007;196:486–488. [DOI] [PubMed] [Google Scholar]
- 43. Leon DA, Lithell HO, Vagero D, et al. Reduced fetal growth rate and increased risk of death from ischaemic heart disease: cohort study of 15 000 Swedish men and women born 1915‐29. BMJ. 1998;317:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barker DJ, Martyn CN. The maternal and fetal origins of cardiovascular disease. J Epidemiol Community Health. 1992;46:8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Law CM, de SM, Osmond C, et al. Initiation of hypertension in utero and its amplification throughout life. BMJ. 1993;306:24–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roghair RD, Segar JL, Sharma RV, et al. Newborn lamb coronary artery reactivity is programmed by early gestation dexamethasone before the onset of systemic hypertension. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1169–R1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Siewert‐Delle A, Ljungman S. The impact of birth weight and gestational age on blood pressure in adult life: a population‐based study of 49‐year‐old men. Am J Hypertens. 1998;11:946–953. [DOI] [PubMed] [Google Scholar]
- 48. Hovi P, Andersson S, Eriksson JG, et al. Glucose regulation in young adults with very low birth weight. N Engl J Med. 2007;356:2053–2063. [DOI] [PubMed] [Google Scholar]
- 49. Gennser G, Rymark P, Isberg PE. Low birth weight and risk of high blood pressure in adulthood. Br Med J. (Clin Res Ed). 1988;296:1498–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Singhal A, Kattenhorn M, Cole TJ, et al. Preterm birth, vascular function, and risk factors for atherosclerosis. Lancet. 2001;358:1159–1160. [DOI] [PubMed] [Google Scholar]
- 51. Martyn CN, Barker DJ, Jespersen S, et al. Growth in utero, adult blood pressure, and arterial compliance. Br Heart J. 1995;73:116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kistner A, Celsi G, Vanpee M, Jacobson SH. Increased systolic daily ambulatory blood pressure in adult women born preterm. Pediatr Nephrol. 2005;20:232–233. [DOI] [PubMed] [Google Scholar]
- 53. White S, Perkovic V, Cass A, et al. Is low birth weight an antecedent of CKD in later life? A systematic review of observational studies. Am J Kidney Dis. 2009;54:248–261. Epub 2009 Apr 1. [DOI] [PubMed] [Google Scholar]
- 54. Burdge GC, Hanson MA, Slater‐Jefferies JL, Lillycrop KA. Epigenetic regulation of transcription: a mechanism for inducing variations in phenotype (fetal programming) by differences in nutrition during early life? Br J Nutr. 2007;97:1036–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dwivedi RS, Herman JG, McCaffrey TA, Raj DSC. Beyond genetics: epigenetic code in chronic kidney disease. Kidney Int. 2011;29:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Woroniecki R, Gaikwad AB, Susztak K. Fetal environment, epigenetics, and pediatric renal disease. Pediatr Nephrol. 2011;26:705–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mohtat D, Susztak K. Fine tuning gene expression: the epigenome. Semin Nephrol. 2010;30:468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bogdarina I, Welham S, King PJ, et al. Epigenetic modification of the renin‐angiotensin system in the fetal programming of hypertension Circ Res. 2007;100;520–526; originally published online Jan 25, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vaiman D, Gascoin‐Lachambre G, Boubred F, et al. The intensity of IUGR‐induced transriptome deregulations is inversely correlated with onset of organ function in a rat model. PLoS One. 2011;6: e21222Epub 2011 Jun 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Comporti M, Signorini C, Leoncini S, et al. Plasma F2‐isoprostanes are elevated in newborns and inversely correlated to gestational age. Free Radic Biol Med. 2004;37:724–732. [DOI] [PubMed] [Google Scholar]
- 61. House JT, Schultetus RR, Gravenstein N. Continuous neonatal evaluation in the delivery room by pulse oximetry. J Clin Monit. 1987;3:96–100. [DOI] [PubMed] [Google Scholar]
- 62. Vento M, Asensi M, Sastre J, et al. Hyperoxemia caused by resuscitation with pure oxygen may alter intracellular redox status by increasing oxidized glutathione in asphyxiated newly born infants. Semin Perinatol. 2002;26:406–410. [DOI] [PubMed] [Google Scholar]
- 63. Jankov RP, Negus A, Tanswell AK. Antioxidants as therapy in the newborn: some words of caution. Pediatr Res. 2001;50:681–687. [DOI] [PubMed] [Google Scholar]
- 64. Asikainen TM, Heikkila P, Kaarteenaho‐Wiik R, et al. Cell‐specific expression of manganese superoxide dismutase protein in the lungs of patients with respiratory distress syndrome, chronic lung disease, or persistent pulmonary hypertension. Pediatr Pulmonol. 2001;32:193–200. [DOI] [PubMed] [Google Scholar]
- 65. Saugstad OD. Oxidative stress in the newborn – a 30‐year perspective. Biol Neonate. 2005;88:228–236. [DOI] [PubMed] [Google Scholar]
- 66. Ledo A, Arduini A, Asensi MA, et al. Human milk enhances antioxidant defenses against hydroxyl radical aggression in preterm infants. Am J Clin Nutr. 2009;89:210–215. [DOI] [PubMed] [Google Scholar]
- 67. Vento M, Moro M, Escrig R, et al. Preterm resuscitation with low oxygen causes less oxidative stress, inflammation, and chronic lung disease. Pediatrics. 2009;124(3):e439–e449. [DOI] [PubMed] [Google Scholar]
- 68. Vina J, Vento M, Garcia‐Sala F, et al. L‐cysteine and glutathione metabolism are impaired in premature infants due to cystathionase deficiency. Am J Clin Nutr. 1995;61:1067–1069. [DOI] [PubMed] [Google Scholar]
- 69. Asikainen TM, White CW. Pulmonary antioxidant defenses in the preterm newborn with respiratory distress and bronchopulmonary dysplasia in evolution: implications for antioxidant therapy. Antioxid Redox Signal. 2004;6:155–167. [DOI] [PubMed] [Google Scholar]
- 70. Saugstad OD. Optimal oxygenation at birth and in the neonatal period. Neonatology. 2007;91:319–322. [DOI] [PubMed] [Google Scholar]
- 71. Thebaud B, Ladha F, Michelakis ED, et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia‐induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation. 2005;112:2477–2486. [DOI] [PubMed] [Google Scholar]
- 72. Tarry‐Adkins JL, Martin‐Gronert MS, Chen JH, et al. Maternal diet influences DNA damage, aortic telomere length, oxidative stress, and antioxidant defense capacity in rats. Faseb J. 2008;22:2037–2044. [DOI] [PubMed] [Google Scholar]
- 73. Luyckx VA, Compston CA, Simmen T, Mueller TF. Accelerated senescence in kidneys of low‐birth‐weight rats after catch‐up growth. Am J Physiol Renal Physiol. 2009;297:F1697–F1705. [DOI] [PubMed] [Google Scholar]
- 74. Walther C, Gaede L, Adams V, et al. Effect of increased exercise in school children on physical fitness and endothelial progenitor cells: a prospective randomized trial. Circulation. 2009;120:2251–2259. [DOI] [PubMed] [Google Scholar]
- 75. Keller G, Zimmer G, Mall G, et al. Nephron number in patients with primary hypertension. N Engl J Med. 2003;348:101–108. [DOI] [PubMed] [Google Scholar]
- 76. Hughson MD, Gobe GC, Hoy WE, et al. Associations of glomerular number and birth weight with clinicopathological features of African Americans and whites. Am J Kidney Dis. 2008;52:18–28. [DOI] [PubMed] [Google Scholar]
- 77. Hinchliffe SA, Sargent PH, Howard CV, et al. Human intrauterine renal growth expressed in absolute number of glomeruli assessed by the disector method and Cavalieri principle. Lab Invest. 1991;64:777–784. [PubMed] [Google Scholar]
- 78. Dötsch J, Plank C, Amann K, Ingelfinger J. The implications of fetal programming of glomerular number and renal function. J Mol Med. 2009;87:841–848. Epub 2009 Aug 4. [DOI] [PubMed] [Google Scholar]