

Abstract
Coronary heart disease (CHD) is one of the leading causes of heart-associated deaths worldwide. This study aimed to investigate the mechanism by which microRNA-363-3p (miR-363-3p) regulates endothelial injury induced by inflammatory responses in CHD. The expression patterns of miR-363-3p, NADPH oxidase 4 (NOX4), and p38 MAPK/p-p38 MAPK were examined in an established atherosclerosis (AS) model in C57BL/6 mice and in isolated coronary arterial endothelial cells (CAECs) after gain- or loss-of-function experiments. We also measured the levels of inflammatory factors (IL-6, ICAM-1, IL-10 and IL-1β), hydrogen peroxide (H2O2), and catalase (CAT) activity, followed by detection of cell viability and apoptosis. In AS, miR-363-3p was downregulated and NOX4 was upregulated, while miR-363-3p was identified as targeting NOX4 and negatively regulating its expression. The AS progression was reduced in NOX4 knockout mice. Furthermore, miR-363-3p resulted in a decreased inflammatory response, oxidative stress, and cell apoptosis in CAECs while augmenting their viability via blockade of the p38 MAPK signaling pathway. Overall, miR-363-3p hampers the NOX4-dependent p38 MAPK axis to attenuate apoptosis, oxidative stress injury, and the inflammatory reaction in CAECs, thus protecting CAECs against CHD. This finding suggests the miR-363-3p-dependent NOX4 p38 MAPK axis as a promising therapeutic target for CHD.
Keywords: microRNA-363-3p, NADPH oxidase 4, p38 MAPK, coronary heart disease, endothelial cell
INTRODUCTION
Coronary heart disease (CHD) is a highly prevalent cardiovascular disease primarily caused by atherosclerosis (AS), due to the passive accumulation of cholesterol and the proliferation of inflammatory macrophages in blood vessel walls [1]. As an active inflammatory disease, AS is characterized by inflammation of the arterial wall, which is mediated by an array of thrombotic factors, growth factors, cytokines, and vasoactive molecules [2]. Endothelial cells are vital for the maintenance of the functional integrity of the vascular wall and nonthrombogenic blood-tissue reactions and serve as modulators of immune responses [3]. Furthermore, endothelial dysfunction is a primary contributing factor to several cardiovascular diseases [4, 5]. Indeed, there is a clear correlation between the atherosclerotic burden and various other risk factors due to the extent of endothelial dysfunction [3]. Due to a persistent lack of clinically approved therapeutic options for ameliorating endothelial cell inflammation, there is an urgent need for the development of novel approaches derived from insights into the underlying pathophysiological mechanisms.
MicroRNAs (miRs) are noncoding nucleotide sequences that modulate gene expression, thus potentially altering endothelial cell phenotypes and vascular smooth muscle function in physiological and disease contexts through translational repression and/or effects on posttranscriptional degradation [6]. miRs can function as direct or indirect modulators at posttranscriptional levels in vascular biology, such that miRs with specific effects on vascular remodeling have emerged as promising targets in treatment [7]. Other reports have also identified vital roles for miRs in the pathogenesis of AS and CHD [8, 9]. Interestingly, extensive miR expression profiling and functional knockdown experiments in human embryonic stem cell-derived cardiomyocytes have identified miR-363 as a possible upstream negative regulator of the left ventricular specification transcription factor HAND1 [10, 11]. miR-363-3p is a 22 bp linear RNA sequence (AATTGCACGGTATCCATCTGTA) (https://www.ncbi.nlm.nih.gov/nuccore/KT921595.1). Moreover, miR-363-3p has been reported to exert neuroprotective effects against ischemic stroke in females [12]. miR-363-3p can influence the angiogenic characteristics of endothelial cells, regulate the expression of angiogenesis-related factors, and control the interaction between hematopoietic cells and endothelial cells [13]. The Targetscan website (http://www.targetscan.org) has predicted that NADPH oxidase 4 (NOX4) is a target of miR-363-3p. The NOX4 gene encodes an oxidoreductase that can facilitate the conversion of molecular oxygen into different reactive oxygen species (ROS), with diverse consequences on the biology of endothelial cells as well as on blood pressure [14]. Overexpression of NOX4 has been observed in several pulmonary and cardiovascular diseases, including AS, pulmonary hypertension, and pulmonary fibrosis [15, 16]. Moreover, the downregulation of NOX4 expression induced by dihydrotanshinone I (a natural compound extensively used for treating cardiovascular diseases) may impede atherosclerotic plaque formation and reduce oxidative stress in atherosclerotic mice [17]. Atorvastatin has demonstrated its inhibitory role in the homocysteine-induced oxidative stress and apoptosis in endothelial progenitor cells via the inactivation of the NOX4-dependent p38 mitogen-activated protein kinase (MAPK) signaling pathway [18]. An existing study has illustrated the relationship between the pathogenesis of cardiovascular diseases and the p38 MAPK signaling pathway [19], the inhibition of which is considered to be a therapeutic target for vascular remodeling [20]. On the basis of the aforementioned literature, we hypothesized that miR-363-3p may inhibit endothelial cell inflammation-induced injuries by targeting NOX4 via modulation of the p38 MAPK signaling pathway. To test this hypothesis, we examined the effects of miR-363-3p-mediated inhibition of NOX4 and the subsequent modulation of the p38 MAPK signaling pathway on the inflammatory injury of endothelial cells following CHD.
RESULTS
Increased blood lipid and calcium levels in AS mice are reduced by a NOX4 knockout
Blood lipid and calcium levels of mice in each group were quantified using an Abbott AerosetTM biochemical analyzer and are summarized in Table 1. In contrast to the levels observed in the normal mouse group, the serum levels of total cholesterol (TC), triglyceride (TG), low-density lipoprotein cholesterol (LDLC) and Ca2+ were increased in the AS group (p < 0.05), whereas there was no significant difference in the serum level of high-density lipoprotein cholesterol (HDLC) (p > 0.05). However, the aforementioned indicators did not differ among the normal (normal mice), N-NOX4-/- (NOX4 knockout mice), and AS-NOX4-/- (NOX4 knockout mice following AS induction) groups (p > 0.05). These results suggested that the elevated blood lipid and calcium levels in AS mice could be prevented by the NOX4 knockdown.
Table 1. Levels of TC, TG, LDLC, HDLC and Ca2+ were increased in AS mice and could be reduced by NOX4 knockout (mmol/L).
| Group | TC | TG | LDLC | HDLC | Ca2+ |
| Normal | 4.41 ± 0.34 | 1.21 ± 0.39 | 1.13 ± 0.29 | 2.26 ± 0.24 | 2.39 ± 0.38 |
| AS | 16.87 ± 1.39* | 4.22 ± 0.57* | 5.82 ± 0.99* | 2.61 ± 0.31 | 3.57 ± 0.67* |
| N-NOX4-/- | 5.64±0.31 | 1.53 ± 0.31 | 1.21 ± 0.11 | 2.21 ± 0.20 | 2.41 ± 0.22 |
| AS-NOX4-/- | 7.62 ± 0.27 | 2.22 ± 0.57 | 2.71 ± 0.11 | 2.26 ± 0.21 | 2.71 ± 0.25 |
Note: NOX4, NADPH oxidase 4; AS, atherosclerosis; TC, total cholesterol; TG, triglyceride; LDLC, low density lipoprotein cholesterol; HDLC, high density lipoprotein cholesterol; the data were analyzed by one-way ANOVA with Tukey's test; n = 10; * p < 0.05 vs. the normal group.
miR-363-3p expression is downregulated and NOX4 mRNA expression is upregulated in AS mice, and NOX4 knockout decreases the CD34 level in endothelial cells and aortic plaque formation in AS mice
CD34 is the surface marker of mature endothelial cells, and CD34 immunostaining in coronary artery sections can reflect the integrity of the vascular endothelium [21]. Therefore, we undertook immunohistochemical staining to detect the level of the coronary aorta-specific marker, CD34. As depicted in Figure 1A, the level of CD34 was increased in the endothelial cells in the AS and the AS-NOX4-/- groups when compared to the normal group but was reduced in the AS-NOX4-/- group compared to the AS group (p < 0.05), suggesting that the loss of NOX4 might attenuate endothelial cell injury caused by AS. Movat’s pentachrome staining was used to stain the nuclei and elastic fibers in black, the proteoglycans in blue, the collagen fibers and the reticular fibers in yellow, the muscles in red, and the fibrin and the red cells in bright red, thereby demonstrating the specific areas of artery trunk lesions in the head and arm of the mice in each group (Figure 1B, Table 2). The plaque area, external elastic membrane area and plaque area/vascular area were increased in the AS group compared with the normal group (p < 0.05). There were no significant differences in the plaque area, external elastic membrane area, or the plaque area/vascular area among the normal, N-NOX4-/- and AS-NOX4-/- groups (p > 0.05). Overall, these results showed that AS may lead to an increased level of CD34 in endothelial cells and enhance aortic plaque formation in mice, which could be rescued by NOX4 knockout.miR-363-3p expression and the mRNA expression of NOX4 in coronary artery tissues in the normal, AS, N-NOX4-/- and AS-NOX4-/- groups, as detected by reverse transcription quantitative polymerase chain reaction (RT-qPCR), are shown in Figure 1C. miR-363-3p expression was downregulated, while the NOX4 mRNA expression was upregulated in the coronary artery tissues in the AS group compared to the normal group (p < 0.05). There were no significant differences regarding the miR-363-3p expression in coronary artery tissues among the normal, N-NOX4-/- and AS-NOX4-/- groups. In addition, NOX4 mRNA expression was absent in the N-NOX4-/- and AS-NOX4-/- groups (p > 0.05). The aforementioned results indicated poor miR-363-3p expression and an amplified NOX4 expression in AS mice.
Figure 1.

miR-363-3p and CD34 expressions are downregulated and NOX4 mRNA expression is upregulated in AS mice and the formation of aortic plaques in AS mice could be prevented by NOX4 knockout. (A) quantification of positive expression of CD34 protein in mice; (B) quantification of the aortic plaque area; (C) the expression of miR-363-3p and NOX4 mRNA as detected by RT-qPCR; The data were analyzed by one-way ANOVA with Tukey's test; n = 10; * p < 0.05 vs. the normal group; # p < 0.05 vs. the AS group; AS, atherosclerosis; NOX4, NADPH oxidase 4.
Table 2. The aortic plaque formation in AS mice could be prevented by NOX4 knockout.
| Group | Plaque area (× 103 um2) | Plaque area/Vascular area |
| Normal | 201.54 ± 15.27 | 0.46 ± 0.05 |
| AS | 542.43 ± 17.69* | 0.72 ± 0.08* |
| N-NOX4-/- | 191.76 ± 12.85 | 0.53 ± 0.04 |
| AS-NOX4-/- | 208.46 ± 20.33 | 0.49 ± 0.07 |
Note: The data were analyzed by one-way ANOVA with Tukey's test; n = 10; * p < 0.05 vs. the normal group; AS, atherosclerosis; NOX4, NADPH oxidase 4.
NOX4 knockout decreases phosphorylated (p)-p38 MAPK/p38 MAPK in the coronary artery tissues of AS mice
Figure 2A, 2B depicts the protein expression of NOX4 and p38 MAPK and the extent of p38 MAPK phosphorylation in the coronary artery tissues of the normal, AS, N-NOX4-/- and AS-NOX4-/- groups, as detected by Western blot analysis. The protein expression of NOX4 and the ratio of p-p38 MAPK/p38 MAPK were upregulated in coronary artery tissues of the AS group compared to the normal group (p < 0.05). No protein expression of NOX4 was observed in the coronary artery tissues of the N-NOX4-/- and AS-NOX4-/- groups (p > 0.05), whereas the ratio of p-p38 MAPK/p38 MAPK was decreased compared with the AS group. These results suggested that the increased p-p38 MAPK/p38 MAPK by AS could be inhibited by the NOX4 knockout.
Figure 2.

The protein expression of NOX4 and ratio of p-p38 MAPK/p38 MAPK are upregulated in coronary artery tissues of AS mice. (A) The protein expressions of NOX4, p38 MAPK and p-p38 MAPK were detected by Western blot; (B) Statistical comparison of NOX4, p38 MAPK and p-p38 MAPK protein expressions among four groups; The data were analyzed by one-way ANOVA with Tukey's test; n = 10; * p < 0.05 vs. the normal group; # p < 0.05 vs. the AS group; AS, atherosclerosis; NOX4, NADPH oxidase 4.
Differential expression of miR-363-3p, NOX4, and p38 MAPK in homocysteine (HCY)-induced coronary arterial endothelial cells (CAECs)
We isolated primary CAECs from normal mice and used a goat antibody to CD34 for immunohistochemical detection, which clearly demonstrated brown staining in the cytoplasm of the positive staining group but no brown staining in the negative control (NC) group. Immunohistochemical staining with a rabbit antibody to factor VIII was negative in the positive- and negative-staining groups, thereby confirming that the isolated cells were indeed endothelial cells. These results are shown in Figure 3A, 3B. Next, we investigated the role of the miR-363-3p/NOX4/p38 MAPK axis in CAECs. HCY was incubated with the isolated CAECs for 24 h for CHD induction. The results of the RT-qPCR showed a downregulated miR-363-3p expression and an upregulated mRNA expression of NOX4 in the CAECs in the HCY group in comparison to the control group (p < 0.05, Figure 3C). Moreover, as illustrated in Figure 3D, the protein expression of NOX4 and the ratio of p-p38 MAPK/p38 MAPK were augmented in the CAECs in the HCY group compared to the control group (p < 0.05). These findings suggested a reduced miR-363-3p and elevated NOX4 and p38 MAPK in HCY-induced CAECs.
Figure 3.

Reduced miR-363-3p expression and abundant NOX4 and p38 MAPK expressions in HCY-induced CAECs. (A) positive and negative staining for goat antibody to CD34 (scale bar = 25 μm); (B) positive and negative staining for rabbit antibody to Factor VIII (scale bar = 25 μm); (C) miR-363-3p expression and NOX4 mRNA expression were determined by RT-qPCR in HCY-induced CAECs; (D) Representative Western blots of NOX4, p38 MAPK and p-p38 MAPK proteins and their quantitation in HCY-induced CAECs, normalized by β-actin. The data were analyzed by paired t-test. n = 3. * p < 0.05 vs. the control group; CAECs, coronary arterial endothelial cells; SP, streptavidin-peroxidase.
NOX4 is a target gene of miR-363-3p
The target relationship between miR-363-3p and NOX4 was predicted using the website available at http://www.targetscan.org (Figure 4A). Dual luciferase reporter assay was further conducted to validate the predicted relationship, which showed a reduced luciferase activity in the NOX4-3’untranslated region (3’UTR)-wild type (WT) (p < 0.05) but no significant difference in the luciferase activity of the NOX4-3’UTR-mutant (MUT) in the CAECs and the HEK-293 cells transfected with miR-363-3p mimic (p > 0.05) (Figure 4B). Therefore, miR-363-3p could target NOX4 and subsequently downregulated its expression in CAECs and HEK-293 cells.
Figure 4.

miR-363-3p binds to the 3’UTR NOX4 mRNA and inhibits its expression in CAECs and HEK-293 cells. (A) putative miR-363-3p binding sites in the 3’UTR of NOX4 mRNA in the online website (http://www.targetscan.org); (B) miR-363-3p binding with the 3’UTR of NOX4 mRNA in CAECs and HEK-293 cells confirmed by dual luciferase reporter assay. Comparisons between two groups were performed by unpaired t-test, and t-test was adopted for pairwise comparisons. n = 3; * p < 0.05 vs. the mimic NC group; miR, microRNA; NOX4, NADPH oxidase 4; 3’UTR, 3’ untranslated region.
Selection of an optimal small interfering RNA (siRNA) against NOX4 in CAECs
RT-qPCR was conducted for the detection of the mRNA expression of NOX4 to select the siRNA plasmid with the optimal silencing rate. In comparison with the NC group, the mRNA expression of NOX4 was lower in the siRNA NOX4-1, siRNA NOX4-2 and siRNA NOX4-3 groups (p < 0.05), with the siRNA NOX4-1 group showing the highest silencing efficiency (p < 0.05). No significant difference was observed between the NC group and the normal group (p > 0.05) (Table 3). Hence, siRNA NOX4-1 was selected for subsequent experimentation.
Table 3. The siRNA NOX4-1 group exhibited a highest silencing efficiency.
| Group | NOX4 mRNA | Silencing rate (%) |
| siRNA NOX4-1 | 0.246 ± 0.033*# | 75.79 ± 3.25 |
| siRNA NOX4-2 | 0.467 ± 0.011*#& | 33.87 ± 1.75 |
| siRNA NOX4-3 | 0.492 ± 0.023*#& | 31.15 ± 2.33 |
| Negative control | 0.816 ± 0.012 | 3.43 ± 1.24 |
| Blank control | 0.845 ± 0.042 |
Note: The data were analyzed by one-way ANOVA with Tukey's test; n = 3; * p < 0.05 vs. the negative control group; # p < 0.05 vs. the blank control group; & p < 0.05 vs. the siRNA NOX4-1 group; NOX4, NADPH oxidase 4.
MiR-363-3p negatively regulates NOX4 and blocks the p38 MAPK signaling pathway in CAECs
After uncovering the regulatory relationship between miR-363-3p and NOX4, we next aimed to elucidate the possible downstream mechanism of NOX4 in CAECs. First, we stably expressed miR-363-3p mimic, miR-363-3p inhibitor, and siNOX4, either separately or in combination, in CAECs and incubated the cells with HCY for 24 h for stress-induced CHD. Next, the expression of NOX4 in CAECs was determined by RT-qPCR (Figure 5), which showed that the mRNA expression of NOX4 was reduced in the miR-363-3p mimic group and the siNOX4 group, while it was higher in the miR-363-3p inhibitor group, compared to the NC group (p < 0.05). In comparison with the miR-363-3p inhibitor group, mRNA expression of NOX4 was reduced in the miR-363-3p inhibitor + siNOX4 group (p < 0.05). The mRNA expression of NOX4 was even lower in the miR-363-3p mimic + siNOX4 group than in the miR-363-3p mimic group (p < 0.05). These results indicated that miR-363-3p extensively suppressed the expression of NOX4 in CAECs.
Figure 5.
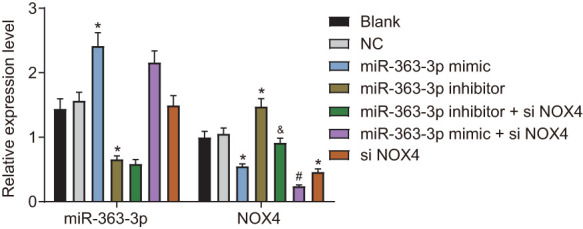
miR-363-3p downregulates the mRNA expression of NOX4 in CAECs. The data were analyzed by one-way ANOVA with Tukey's post hoc test; n = 3; * p < 0.05 vs. the NC group; # p < 0.05 vs. the miR-363-3p mimic group; & p < 0.05 vs. the miR-363-3p inhibitor group; CAECs, coronary artery endothelial cells; miR, microRNA; NOX4, NADPH oxidase 4; NC, negative control.
Western blot analysis (Figure 6A, 6B) showed a lower NOX4 protein expression and p-p38 MAPK/p38 MAPK ratio in the miR-363-3p mimic and the siNOX4 groups than in the NC group (p < 0.05). However, these markers were increased in the miR-363-3p inhibitor group compared to the NC group (p < 0.05). The protein expression of NOX4 and the ratio of p-p38 MAPK/p38 MAPK were lower in the miR-363-3p inhibitor + siNOX4 group than in the miR-363-3p inhibitor group (p < 0.05). In comparison with the miR-363-3p mimic group, the miR-363-3p mimic + siNOX4 group showed a lower NOX4 protein expression, along with a reduced ratio of p-p38 MAPK/p38 MAPK (p < 0.05). Taken together, these data uncovered that miR-363-3p downregulated NOX4 and blocked the activation of the p38 MAPK signaling pathway in CAECs.
Figure 6.

miR-363-3p downregulates protein expression of NOX4 and the ratio of p-p38 MAPK/p38 MAPK in CAECs. (A, B) Representative Western blots of NOX4, p38 MAPK and p-p38 MAPK proteins and their quantitation in CAECs, normalized by β-actin; the data were analyzed by one-way ANOVA with Tukey's post hoc test; n = 3; * p < 0.05 vs. the blank group; # p < 0.05 vs. the miR-363-3p mimic group; & p < 0.05 vs. the miR-363-3p inhibitor group; CAECs, coronary artery endothelial cells; miR, microRNA; NOX4, NADPH oxidase 4; NC, negative control.
miR-363-3p promotes CAEC viability by targeting NOX4
Figure 7 shows the viability of CAECs as measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. In comparison with the NC group, the cell viability was increased in the miR-363-3p mimic group, while it was lower in the miR-363-3p inhibitor group (p < 0.05). When compared to the miR-363-3p inhibitor group, the cell viability was increased in the miR-363-3p inhibitor + siNOX4 group (p < 0.05). These results highlighted the potential of miR-363-3p to increase the viability of CAECs by targeting NOX4.
Figure 7.
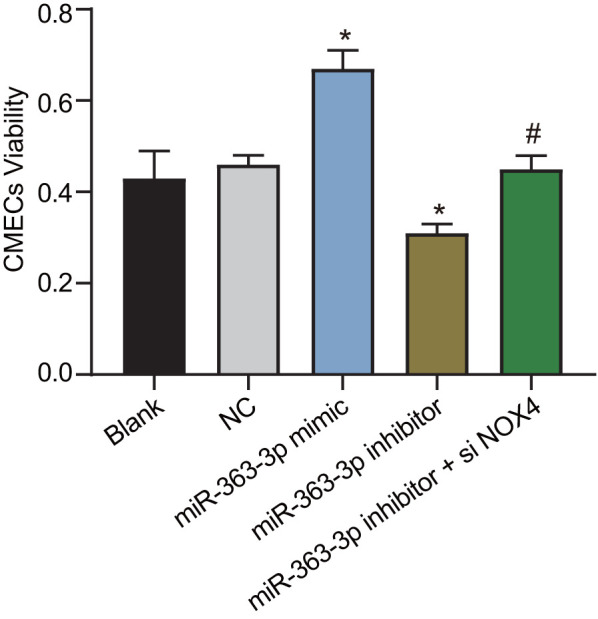
miR-363-3p promotes CAEC viability by targeting NOX4. The data were analyzed by one-way ANOVA with Tukey's post hoc test; n = 3; * p < 0.05 vs. the NC group; # p < 0.05 vs. the miR-363-3p inhibitor group; CAECs, coronary artery endothelial cells; miR, microRNA; NOX4, NADPH oxidase 4; NC, negative control.
miR-363-3p reduces the hydrogen peroxide (H2O2) level and catalase (CAT) activity in CAECs by targeting NOX4
H2O2 is an important active molecule that mediates oxidative stress-induced endothelial injury, and NOX4 is the major producer of H2O2 in living cells [22]. CAT is a kind of antioxidant enzyme widely distributed in almost all organisms, the main function of which is to catalyze H2O2 into water and O2 and to clear the H2O2 in the body to protect the cells from the toxicity of H2O2. The CAT level can also reflect the oxidative stress [23]. Therefore, we next examined the effect of miR-363-3p on the H2O2 level and the CAT activity in CAECs. In comparison with the NC group, the H2O2 level and CAT activity were decreased in the miR-363-3p mimic group, which were increased in the miR-363-3p inhibitor group (p < 0.05). The levels of H2O2 and CAT activity were much lower in the miR-363-3p inhibitor + siNOX4 group than in the miR-363-3p inhibitor group (p < 0.05) (Figure 8A, 8B). These results indicated that miR-363-3p could reduce the H2O2 levels and CAT activity in CAECs by targeting NOX4.
Figure 8.

miR-363-3p reduces H2O2 level and CAT activity in CAECs by targeting NOX4. (A) quantification of H2O2 level in CAECs; (B) quantification of CAT activity in CAECs; the data were analyzed by one-way ANOVA with Tukey's post hoc test; n = 3; * p < 0.05 vs. the blank group; # p < 0.05 vs. the miR-363-3p mimic group; CAECs, coronary artery endothelial cells; CAT, catalase; miR, microRNA; NOX4, NADPH oxidase 4; NC, negative control.
miR-363-3p inhibits apoptosis in CAECs by targeting NOX4
Hoechst 33258 staining and flow cytometry were employed to observe the apoptosis in CAECs, as shown in Figure 9A, 9B. No difference was evident in the apoptosis rate between the blank group and the NC group (p > 0.05). In comparison with the NC group, the apoptosis rate was decreased in the miR-363-3p mimic group and increased in the miR-363-3p inhibitor group (p < 0.05). When compared with the miR-363-3p inhibitor group, the apoptosis rate was attenuated in the miR-363-3p inhibitor + siNOX4 group (p < 0.05). According to these results, the apoptosis of CAECs could be repressed by miR-363-3p through inhibition of NOX4.
Figure 9.

miR-363-3p attenuates apoptosis of CAECs by targeting NOX4. (A) Hoechst 33258 staining and quantification of apoptosis in CAECs (scale bar = 25 μm); (B) flow cytometry and quantification of apoptosis in CAECs; * p < 0.05 vs. the blank group; # p < 0.05 vs. the miR-363-3p inhibitor group; the data were analyzed by one-way ANOVA with Tukey's post hoc test; n = 3; CAECs, coronary artery endothelial cells; miR, microRNA; NOX4, NADPH oxidase 4; NC, negative control.
miR-363-3p increases tube formation of CAECs by targeting NOX4
The tube formation assay was performed to examine the effect of miR-363-3p on the tube formation of CAECs. As depicted in Figure 10A, 10B, the number of tubular structures was increased in the miR-363-3p mimic group and reduced in the miR-363-3p inhibitor group compared with the NC group (p < 0.05). In comparison with the miR-363-3p inhibitor group, the number of tubular structures formed in the miR-363-3p inhibitor + siNOX4 group was increased (p < 0.05). These results indicated that miR-363-3p stimulated tube formation in CAECs through inhibition of NOX4.
Figure 10.

miR-363-3p enhances tube formation of CAECs by targeting NOX4. (A) representative images of the tubular structures in CAECs (scale bar = 100 μm); (B) quantification of tubular structures in CAECs; the data were analyzed by one-way ANOVA with Tukey's test; n = 3; * p < 0.05 vs. the blank group; # p < 0.05 vs. the miR-363-3p mimic group; CAECs, coronary artery endothelial cells; miR, microRNA; NOX4, NADPH oxidase 4; NC, negative control.
miR-363-3p reduces the expression of inflammatory factors in CAECs by targeting NOX4
Enzyme-linked immunosorbent assay (ELISA) was conducted to observe the effects of miR-363-3p on inflammatory responses in CAECs. In comparison to the NC group, the secretion of interleukin (IL)-6, intercellular adhesion molecule-1 (ICAM-1), and IL-1β was lower in the miR-363-3p mimic group, accompanied by an increased IL-10 secretion (p < 0.05). In contrast, the miR-363-3p inhibitor group showed increased secretion of IL-6, ICAM-1, and IL-1β, as well as a lower secretion of IL-10 (p < 0.05). Relative to the miR-363-3p inhibitor group, the miR-363-3p inhibitor + siNOX4 group displayed reduced secretion of IL-6, ICAM-1 and IL-1β, along with elevated secretion of IL-10 (p < 0.05) (Table 4). The aforementioned data indicated that miR-363-3p could inhibit the expression of proinflammatory cytokines in CAECs through inhibition of NOX4.
Table 4. Levels of the IL-6, ICAM-1 and IL-1β were increased while IL-10 level was reduced in cells treated with miR-363-3p inhibitor (pg/mL).
| Group | IL-6 | ICAM-1 | IL-10 | IL-1β |
| Blank | 49.26 ± 5.02 | 24.57 ± 7.65 | 48.56 ± 11.05 | 89.14 ± 6.48 |
| Negative control | 51.64 ± 5.50 | 27.45 ± 8.19 | 50.43 ± 12.61 | 95.25 ± 4.64 |
| miR-363-3p mimics | 27.55 ± 3.31* | 12.34 ± 6.31* | 82.26 ± 16.53* | 45.88 ± 3.89* |
| miR-363-3p inhibitors | 89.56 ± 7.58* | 42.64 ± 13.02* | 25.96 ± 8.37* | 188.28 ± 5.31* |
| miR-363-3p inhibitors + si NOX4 | 53.61 ± 5.34# | 25.41 ± 6.18# | 46.13 ± 13.12# | 91.57 ± 4.25# |
Note: The data were analyzed by one-way ANOVA with Tukey's test; n = 3; * p < 0.05 vs. the blank group; # p < 0.05 compared with the miR-363-3p mimics group; IL-6, interleukin-6; ICAM-1, intercellular cell adhesion molecule-1; IL-10, interleukin-10; IL-1β, interleukin-1β; NOX4, NADPH oxidase 4.
DISCUSSION
This study was designed to investigate the effects of miR-363-3p on endothelial dysfunction and the inflammatory response in CHD based on a hypothesis involving molecular targeting of NOX4 via the p38 MAPK signaling pathway. Our findings suggested that miR-363-3p impeded the apoptosis in CAECs and the expression of pro-inflammatory cytokines in AS mice with CHD by inhibiting the NOX4-dependent p38 MAPK signaling pathway.
Our initial findings highlighted that AS mice had elevated serum levels of TC, TG, LDLC and calcium compared with the normal mice. In the clinic, an elevated level of LDLC is considered as a major risk factor for CHD, and reductions in the levels of TC, TG and LDLC are associated with a reduced morbidity and mortality of AS [24]. Consistent with our present results, Prendergast et al. has reported the presence of elevated calcium levels in endothelial cells prior to plaque development [25].
In the present study, we found a low expression of miR-363-3p but a high expression of NOX4 in both the coronary artery tissues of AS mice and the HCY-induced CAECs. miR-363-3p, a member of the miR-363 family, is derived from the miR-106a-363 cluster [26]. The miR-106a-363 cluster is a paralog of the extensively studied miR-17-92 cluster, which has been linked to the cardiovascular system [27, 28]. Previous studies also demonstrated that miR-363 is dysregulated in several cancers and is specifically downregulated in numerous cancers, such as head and neck squamous cell carcinoma and hepatocellular carcinoma [29, 30]. miR-363-3p suppresses metastasis and inhibits the epithelial-to-mesenchymal transition (EMT) in colorectal cancer by targeting Sox4 [31]. Interestingly, miR-363 was reported to negatively regulate HAND1, a vital left ventricular determining transcription factor, thereby functioning as a modulator in the generation of functional left-ventricular cardiomyocytes [11]. NOX4 has been found to be extensively expressed in H2O2-induced human coronary artery smooth muscle cells in AS [32]. There was a trend toward an increased expression of NOX4 in oxygen and glucose depletion/reoxygenation (OGD/R)-treated human brain vascular endothelial cells collected after AS-associated stroke [33]. Upregulation of NOX4 is evident in AS endothelial cells, thereby highlighting the vital functionality of the NOX4-containing NADPH oxidase [34].
miRNAs can modulate gene expression posttranscriptionally by interacting with the 3’UTR of specific target mRNAs [35]. In this study, a biological prediction website and a luciferase reporter assay identified that miR-363-3p bound to the 3’UTR of NOX4 mRNA and could negatively regulate its expression. A previous study revealed that miR-363-3p is involved in the metastasis of colorectal cancer by functioning as a tumor suppressor through the negative regulation of its target Sox4 gene [31]. MiR-363-3p can also function as a tumor suppressor in papillary thyroid carcinoma, where its suppressive effect is mediated by repression of PIK3CA [36]. The aforementioned studies collectively demonstrate that miR-363-3p can target different genes to affect the development of disease in several types of carcinomas.
NOX4, as a key cellular regulator of oxidative stress [37], is associated with the involvement of miR-363-3p in AS. H2O2 is considered as a key active molecule mediating the oxidative stress-induced endothelial injury in AS, and NOX4 has been found to be the major producer of H2O2 [22]. NOX4 knockdown has also been documented to elicit a decrease of H2O2 levels in human umbilical vein endothelial cells (HUVECs) treated with copper oxide nanoparticles (CuONPs), which normally evoke oxidative stress [38]. Treatment of cyclosporine A rats with CAT, an H2O2 scavenger, is capable of restoring NOX4 expression [39]. Endothelial dysfunction in the ascending aorta in a Marfan’s disease model has been prevented in mice following the knockdown of NOX4 [40]. Bao et al. confirmed that oxidative stress could influence p38 MAPK signaling and various downstream transcription factors, resulting in alterations in gene expression capable of impacting cell survival. Other reports have highlighted that these alterations involve reduced oxidative stress via NOX4 and regulation of p38 MAPK [18]. Another study reported a significant decrease in p38 MAPK mRNA at two days after transfection with the miRNA mimics [41]. Moreover, the multiMiR package in R showed that hsa-miR-363-3p was downregulated in gastric cancer and significantly enriched in the MAPK signaling pathway [42]. It is well known that MAPKs include ERK1/2, ERK5, p38 and JNK [43]. siRNA-mediated NOX4 silencing reduces ROS generation, p38 expression and ERK1/2 phosphorylation level in HUVECs, thus retarding TNF-α-induced oxidative stress and inflammation in AS [44]. In palmitic acid (PA)-induced murine hepatic AML12 cells, treatment with the JNK inhibitor SP600125 decreases NOX4 expression [45]. Inhibition of NOX4 and inactivation of JNK helped to mitigate the inflammatory response in fibroblast-like synoviocytes, exerting protective effects against the progression of rheumatoid arthritis [46]. Although there is no literature establishing a direct relationship between NOX4 and ERK5 (also named BMK1), in the aldosterone or high salt-induced hypertensive renal injury rat model, the expression of the NOX4 gene, along with ERK1/2, JNK and BMK1, is all increased, but the p38 MAPK activity was unchanged [47]. The aforementioned results revealed the close relationship between NOX4 and the MAPK members, along with their complex regulatory relationship. Thus, we cannot exclude the possibility of involvement of other signaling pathways in the pro-inflammatory effects of NOX4 in CAECs, given the very complex cellular microenvironments and the capacity of p38 MAPK to interact with other signaling pathways. The interaction between NOX4 and the p38 MAPK signaling pathway merits further in-depth analysis.
Furthermore, the present study demonstrated that miR-363-3p increased CAEC viability, tube formation, and IL-10 levels, as well as reducing apoptosis in the CAECs and the expression of the pro-inflammatory cytokines IL-6, ICAM-1, and IL-1β. These findings are indicative of the ability of miR-363-3p to suppress inflammation, thereby preventing endothelial cell injury. Inhibition or overexpression of certain miRs represents a promising therapeutic channel for the treatment of cardiovascular disorders [7]. Another study also found that reduced miR-363 expression resulted in a significantly decreased number of newly formed tubes among endothelial cells [13]. Furthermore, miR-363-3p suppresses the proliferation of human hepatocellular carcinoma cells by targeting S1PR1 or USP28 [48]. This phenomenon has also been evident in head and neck squamous cell carcinomas, where miR-363-3p inhibits the expression of the transmembrane glycoprotein podoplanin [29]. Cytokines, by functioning as modulators of immune responses and pro-inflammatory and anti-inflammatory stimulation, may play a significant role in the development of AS [49]. Although previous studies have characterized the relationship between miR-17-92 and immune responses [50], an understanding of the detailed mechanisms of how miRs affect specific cytokines still requires further investigation.
Overall, this study suggests that miR-363-3p confers cellular protection against inflammation-induced endothelial cell injury in an AS model by targeting NOX4 via inactivation of the p38 MAPK signaling pathway (Figure 11). The results of this study provide novel insights for the development of potential therapeutic strategies in the treatment of CHD. Further comprehensive experiments are required to expand our observations and to explore the mechanism of miR-363-3p-mediated downregulation of NOX4 and the relationships between miR-363-3p and specific inflammatory cytokines.
Figure 11.

A schematic for the function and mechanism of miR-363-3p in CHD. miR-363-3p targets NOX4 and disrupts the p38 MAPK/ signaling pathway, which consequently reduces the apoptosis of CAECs, oxidative stress injury and the expression of inflammatory factors in CAECs, thus protecting CAECs against CHD.
MATERIALS AND METHODS
Ethics statement
This study was conducted with the approval of the Ethics Committee of the Guizhou Provincial People’s Hospital and in strict accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Adequate measures were taken to minimize the numbers of animals and their suffering.
Study subjects
Twenty healthy male C57BL/6 mice (aged 3-4 months; weighing 25 ± 5 g) were acquired from the Peking University Health Science Center (Beijing, China). These mice were housed in plastic cages with a stainless-steel bottom, with five mice in each cage. Twenty healthy male NOX4-/- clean mice with a C57BL/6 genetic background were provided by the Peking University Health Science Center (Beijing, China). These mice were housed in the Laboratory of Clinical Medicine, which conformed to the grade 2 animal breeding standards. All mice were given ad libitum access to food and water.
Animal grouping and model establishment
The mice were assigned into the normal group (10 normal mice), the AS group (10 mice with AS), the N-NOX4-/- group (10 NOX4-/- normal mice) and the AS-NOX4-/- group (10 NOX4-/- mice with AS).
The AS mouse model was established in strict accordance with the procedure described by Fu et al. [51]. After a week of adaptive feeding, the mice in the normal and NOX4-/- groups were fed with conventional food and tap water, whereas mice in the AS and AS-NOX4-/- groups were fed with vitamin D3 powder at a concentration of 1.25 × 106 μg/kg along with a high-fat diet comprised of cholesterol (2%), sodium cholate (0.5%), lard (3%), propylthiouracil (0.2%) and basic food (94.3%). At the beginning of the experiment, the mice were given an intramuscular injection of vitamin D3 powder at a dose of 3 × 106 μg/kg in the right lower extremity. This injection protocol was repeated once every 30 days.
Collection of arterial blood and coronary artery specimens
Three months after model establishment, arterial blood and coronary artery specimens were obtained from mice in each group after overnight fasting. Under deep anesthesia via an intraperitoneal injection of sodium pentobarbital, the abdominal aorta was isolated and opened below the renal artery bifurcation. A puncture needle was then inserted, and an arterial blood sample was withdrawn using a syringe and centrifuged (3000 rpm) at 4° C for 5 min. Next, 1 mL of the separated serum was collected for determination of blood lipid and calcium levels. Additionally, a segment of the coronary artery (approximately 1 cm in length) was excised from the lower border of the coronary artery. The segment was fixed using 4% paraformaldehyde, embedded in paraffin and sliced into serial sections.
Detection of blood lipid and calcium levels
An Abbott AerosetTM biochemical analyzer (Abbott Laboratories, Abbott Park, IL, USA) was utilized to measure the serum levels of TC, TG, HDLC, LDLC and calcium in the mice.
Movat’s pentachromic staining
The paraffin-embedded coronary artery tissues were sectioned, dewaxed using xylene, and then treated with 5% sodium thiosulfate and 1% Alcian blue solution for 20 min. The sections were then heated at a high temperature in a microwave for 45 s in alkaline ethanol. Next, the sections were stained using Weigert hematoxylin solution, saffron/acid fuchsin solution and 5% phosphotungstic acid solution sequentially for 5 min each. Following the final staining regimen, the sections were transferred into 1% glacial acetic acid for 5 min. After conventional gradient ethanol dehydration, the sections were cleared using xylene, mounted using neutral gum and observed under a Leica optical microscope. The plaque area and vascular area were then measured and documented.
Immunohistochemical staining
The coronary artery tissues of mice in each group were resected and fixed in a conventional manner using 4% paraformaldehyde for 24 h, followed by dehydration with an ethanol series (80%, 90%, and 100%) and n-butanol. Next, the sections were immersed in a 60° C wax box, embedded, and then sliced into 5-μm serial sections. The sections were subsequently heated for 1 h at 60° C, dewaxed with xylene, rehydrated with gradient ethanol, and soaked in 3% H2O2 for 10 min, followed by high-pressure antigen retrieval for 90 s. The sections were then blocked with 100 μL of 5% bovine serum albumin (BSA) blocking solution and later incubated at 37° C for 30 min. The sections were incubated with 100 μL of the primary rabbit antibody against CD34 (1:1000, ab214035, Abcam, Cambridge, UK) at 4° C overnight. The sections were then incubated with the biotin-labeled secondary antibody, goat anti-rabbit working solution (1:100; HY90046, Shanghai HengYuan Biological Technology Co., Ltd., Shanghai, China), at 37° C for 30 min. After that, the sections were incubated in streptavidin-peroxidase solution (Beijing Zhongshan Biotechnology Co., Ltd., Beijing, China) at 37° C for 30 min and finally treated with diaminobenzidine (DAB) (Beijing Bioss Biotechnology Co., Ltd., Beijing, China) for development at room temperature. The sections were then soaked in hematoxylin for 5 min, rinsed in 1% hydrochloric acid for 4 s, and then immersed in tap water for 20 min to revert to the original blue color. The criterion for CD34 protein-positive cells was the appearance of a brown yellow color as observed by a microscopy. The Image-Pro Plus image analysis software (Media Cybernetics, Silver Springs, MD, USA) was used to detect the mean optical density (OD) values of the CD34-positive coloration in the coronary tissues of each group under high magnification microscopy, followed by a quantitative analysis.
Culture and identification of CAECs
The proximal coronary artery of normal mice was excised and soaked in a culture dish containing phosphate-buffered saline (PBS) and heparin for approximately 5 min to prevent any subsequent blood coagulation. The coronary artery was then rinsed 3 times with sterile PBS and soaked in 75% ethanol for 15 s. The artery tissues were sliced into 1 mm3 tissue blocks using a pair of ophthalmic scissors and detached for 6 min at 37° C using 3 mL of 0.2% type II collagenase. Subsequently, an equal volume of 0.25% trypsin was added, and the tissues were placed in a water bath at 37° C for 5 min, after which the detachment was terminated by the addition of 3 mL of Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). After the supernatant was discarded, the cells were resuspended in complete culture medium and incubated with 5% CO2 at 37° C. After 6 h, the medium was replaced, and the non-adherent cells were removed. When the remaining cells reached 80% confluence, the medium was discarded. Then, solutions of 0.25% trypsin and 0.02% ethylenediamine tetra acetic acid (EDTA) were added for complete detachment under gentle shaking, followed by microscopic observation. After contraction and conversion to a round shape by the majority of cells in the culture, a medium containing 10% FBS was added to terminate the detachment. The cells were then triturated using a pipette for detachment, and the cell suspension was centrifuged and resuspended in complete culture medium. After adjustment of the cell density to an appropriate level, the cells were maintained and passaged at 37° C with 5% CO2. The culture medium was substituted once every 3 days.
Streptavidin-peroxidase (SP) immunohistochemical staining was performed to identify the CAECs in mice; rabbit antibody to factor VIII and goat antibody to CD34 were selected as the primary antibodies. When the CAECs reached 80% confluence, they were detached by adding trypsin and then seeded onto coverslips in a 6-well plate. After being seeded with the CAECs, the coverslips were fixed using paraformaldehyde for 30 min and incubated with the specified diluted primary antibodies (PBS used as the NC) overnight at 4° C. The CAECs were then incubated with the goat anti-rabbit IgG at 37° C for 40 min. After that, the CAECs were developed with DAB, stained using hematoxylin for 1 min, differentiated using hydrochloric acid for 2 s, sealed with conventional resin and observed under an inverted microscope.
Plasmid construction of siRNA against NOX4
The NOX4 gene sequences were obtained from the GenBank. In all, 3 pairs of siRNA sequences were designed in strict accordance with the basic siRNA design principles provided online by Ambion Inc. (Austin, TX, USA). Furthermore, a BLAST analysis was then performed to determine the specificity of the siRNAs. The siRNAs against NOX4 were constructed and identified by the Shanghai Genechem Co., Ltd. (Shanghai, China). The gene sequences are shown in Table 5.
Table 5. The siRNA primer sequences.
| Gene | Sequence |
| siRNA NOX4-1 | F: 5'-CAUGCUGCUGCUGUUGCAUGUUUCA-3' R: 5'-CCCUCUGAUGUAAUGGAACUCCGUA-3' |
| siRNA NOX4-2 | F: 5′-GACCUGACUUUGUGAACAUTT-3′ R: 5′-AUGUUCACAAAAUCAGGUCTT-3′ |
| siRNA NOX4-3 | F: 5′-GCTGTGATAAUCACCUCUAC-3′ R: 5′-GATTGAGGUGAUUAUCACAGC-3′ |
| NOX4 | F: 5’-TAGCTGCCCACTTGGTGAACG-3’ R: 5’-TGTAACCATGAGGAACAATACCACC-3’ |
| Negative control siRNA | F: 5′-UUCUCCGAACGUGUCACGUTT-3′ R: 5′-ACGUGACACGUUCGGAGAATT-3′ |
Note: NOX4, NADPH oxidase 4; siRNA, small interfering RNA; F, forward; R, reverse.
Transfection of plasmids and selection of siRNAs
The CAECs were incubated in DMEM supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin at 37° C with 5% CO2 under a saturated air humidity. The cells were incubated in a six-well plate at a density of 2 × 105 cells/well. The cell confluence reached 90% after 24 h of incubation. Then, 4.0 μg of plasmid and 10 μL of LipofectamineTM 2000 reagents were added to each well for transfection. The cells were then divided into the siRNA NOX4-1 group (transfected with the siRNA NOX4-1 plasmid), the siRNA NOX4-2 group (transfected with the siRNA NOX4-2 plasmid), the siRNA NOX4-3 group (transfected with the siRNA NOX4-3 plasmid), NC group (exposed to liposomes only) and the blank control group (without treatment of siRNA or liposomes). The serum media were replaced 6 h post-transfection. After cell collection, the total RNA was extracted using a one-step method (Invitrogen, Inc., Carlsbad, CA, USA). NOX4 expression was determined by RT-qPCR. Ultraviolet (UV) analysis and formaldehyde gel electrophoresis were used to obtain high-quality RNA samples. The PCR primers were designed and synthesized by Invitrogen, Inc. (Carlsbad, CA, USA) (Table 6).
Table 6. The primer sequences for RT-qPCR.
| Gene | Primer sequence |
| miR-363-3p | F: 5'-AATTGCACGGTATCCATCTGTA-3' |
| R: 5'-TACAGATGGATACCGTGCAATT-3' | |
| miR-363-3p mimic | F: 5'-AATTGCACGGTATCCA-3' |
| R: 5'-GTGCAGGGTCCGAGGT-3' | |
| miR-363-3p inhibitor | F: 5'-UACAGAUGGAUACCGUGCAAUU-3' |
| β-actin | F: 5’-GTCAGGTCATCACTATCGGCAAT-3' |
| R: 5'-AGAGGTCTTTACGGATGTCAACGT-3' |
Note: miR-363-3p, microRNA-363-3p; F, forward; R, reverse; RT-qPCR, reverse transcription quantitative polymerase chain reaction.
Cell transfection and grouping
A CAEC model of CHD (which served as the HCY group) was established after incubation of the CAECs from normal mice for 24 h in 0.25 mmol/L HCY. The untreated CAECs were used as the control group. The DNA oligonucleotide sequence of the overexpressed RNA of the miR-363-3p was synthesized based on a target sequence that augmented the expression of miR-363-3p, from which a double-stranded DNA was generated. The double-stranded DNA was then ligated into the pGCSIL-GFP vector following digestion with the Age I and EcoRI restriction enzymes, after which the positive clones were selected and sequenced. Afterwards, the miR-363-3p mimic, the miR-363-3p inhibitor and a siRNA against NOX4 (siNOX4) were synthesized, and the liposomes were used to package the miR-363-3p mimic, miR-363-3p inhibitor and siNOX4 (Guangzhou RiboBio Co., Ltd., Guangzhou, China). After the CAECs at passage 3 reached 80% confluence, the diluted liposomes were added to the culture medium, followed by another regimen of 24-h incubation. The CAECs were divided into five groups: the blank group (CAECs without transfection), the NC group (CAECs transfected with the NC sequences), the miR-363-3p mimic group (CAECs transfected with the miR-363-3p mimic), the miR-363-3p inhibitor group (CAECs transfected with the miR-363-3p inhibitor) and the miR-363-3p inhibitor + siNOX4 group (CAECs transfected with the miR-363-3p inhibitor and siNOX4). After transfection, the CAECs were incubated with 0.25 mmol/l HCY for 24 h, after which the subsequent experiments were then carried out.
Dual-luciferase reporter gene assay
The binding of miR-363-3p to NOX4 was predicted based on a binding site in the NOX4 3’UTR identified using a bioinformatics website (http://www.targetscan.org). The NOX4 3’UTR, containing the binding site of miR-363-3p, was then synthesized. Next, a wild type NOX4 3’UTR (NOX4 3’UTR-WT) was constructed with the pmir-Glo plasmid (Supplementary Table 1). The primer sequence of the NOX4 3’UTR-WT was 5'-TAAAGAGGAACAAGTGCAATTTCTAAGACTTAGAAACTCAGCTGAATCAAACAGC-3', or 5'-ATGTGCAATGCTCATGTAGACTGAACTGTGGAGACATAAACTATGACATCATAGCAATGCTGTCTAGATTACAAAGAGTTTTCACAAAACACATACTACAAAAATAAAAATAAAAATAAAC CTCATAAAAAAAAACTTGGGCTTGTCCAGGCTTGATGTGCAATAC-3'. A mutant NOX4 3’UTR (NOX4 3’UTR-MUT) plasmid was also constructed with site-directed mutagenesis. Detection was performed in strict accordance with the manufacturer’s protocol provided in the plasmid extraction kit (Promega Corp., Madison, WI, USA). Afterwards, the CAECs in the logarithmic growth phase and the HEK-293 cells were seeded into a 96-well plate. Upon attaining 70% cell confluence, the cells were transfected using the LipofectamineTM 2000 reagent as outlined above. The NOX4 3’UTR-WT and miR-363-3p mimic plasmids were then cotransfected into the CAECs and the HEK-293 cells. NOX4-3’ UTR-WT + NC, NOX4-3’UTR-MUT + NC and NOX4-3’UTR-MUT + miR-363-3p mimic groups were prepared as controls and transfected into the CAECs and the HEK-293 cells, respectively. After 6 h of culture in an incubator, the transfected cells were cultured in medium containing 10% FBS for 48 h. A dual-luciferase reporter assay was performed to determine luciferase activity.
RT-qPCR
Total RNA extraction was performed using a one-step method by TRIzol reagent (Invitrogen, Inc., Carlsbad, CA, USA). The extracted total RNA was analyzed and verified using UV analysis and formaldehyde gel electrophoresis. Then, l μg of total RNA was reversely transcribed into complementary DNA (cDNA). The PCR primer sequences were designed and synthesized by Invitrogen, Inc. (Carlsbad, CA, USA) (Table 2). β-actin was used as an internal reference. The PCR amplification conditions were as follows: predenaturation at 94° C for 5 min, followed by 40 cycles of denaturation at 94° C for 40 s, annealing at 60° C for 40 s and extension at 72° C for 1 min, followed by a final extension at 72° C for 10 min. The PCR products were subjected to agarose gel electrophoresis, and the results were analyzed using the Opticon Monitor 3 software (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The data were analyzed by the 2-ΔΔCt method, as described by the following equation: ΔΔCt = [Ct (target gene) - Ct (reference gene)] (the experimental group) / [Ct (target gene) - Ct (reference gene)] (the control group). This experiment was repeated three times to obtain the mean value.
Western blot analysis
Total protein was extracted from tissues or cells with radio-immunoprecipitation assay (RIPA) lysis buffer containing phenylmethylsulfonyl fluoride (PMSF) (P0013C, Beyotime Institute of Biotechnology, Shanghai, China). The samples were then incubated on ice for 30 min and centrifuged at 8000 g for 10 min at 4° C to collect the supernatant. The protein concentrations were measured using a bicinchoninic acid (BCA) kit (Wuhan Boster Biological Technology Co., Ltd., Wuhan, Hubei, China). After the addition of loading buffer (30 μg/well), the proteins were boiled at 95° C for 10 min and then separated by 10% polyacrylamide gel electrophoresis (Wuhan Boster Biological Technology Co., Ltd., Wuhan, Hubei, China). Next, the proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane. Subsequently, the membrane was blocked using 5% BSA at room temperature for 1 h and then incubated at 4° C overnight with the primary rabbit anti-mouse antibodies to NOX4 (1:1000, ab195524), p38 MAPK (1:1500, ab170099) p-p38 MAPK (1:800, ab47363) and β-actin (1:2000, ab8227). The aforementioned antibodies were purchased from Abcam, Cambridge, UK. The following day, the membrane was rinsed three times with Tris-buffered saline-Tween 20 (TBST) (5 min per rinse) and incubated with the goat anti-rabbit secondary antibody at room temperature for 1 h. A chemiluminescence reagent was added to develop the membrane, with β-actin used as an internal reference. The bands were visualized using a Bio-Rad Gel Doc EZ imager (Bio-Rad, CA, USA), and the ImageJ software was used to analyze the grayscale value of the target bands. The experiment was repeated three times to obtain the mean value.
MTT assay
The CAECs at passage two or three were dispersed into a cell suspension and seeded in a 96-well plate at a density of 5 × 104 cells/well (6 parallel wells for each group). Upon attaining 80% cell confluence, the transfected cells were collected and incubated in 20 μL of MTT solution (Sigma-Aldrich Chemical Company, St Louis, MO, USA) at 37° C for 4 h. After removal of the MTT solution, the cells in each well were treated with 150 μL of dimethyl sulfoxide (Sigma-Aldrich Chemical Company, St Louis, MO, USA) in a shaker for 10 min. The OD of each well was measured at an excitation wavelength of 490 nm using a microplate reader. The average OD values were determined from the mean of three independent measurements. The cell viability was calculated according to the following equation: (OD the experimental group - OD the control group)/OD the control group.
H2O2 and CAT detection
Titanium sulfate colorimetry was used to detect the level of H2O2 in cells. Fe2+ was oxidized by H2O2 to produce Fe3+, and a purple product was formed with addition of xylenol orange in a specific solution to colorimetrically determine the concentration of H2O2 [52]. The cells were collected into centrifuge tubes, and the supernatant was then removed. A total of 1 × 106 cells were fully lysed using 100-200 μL lysis solution and then centrifuged at 12000 ×g for 3-5 min at 4° C, after which the H2O2 reagent was liquefied on ice or in an ice-bath. Cell culture medium was added to dilute the standard samples to variable concentrations of 1, 2, 5, 10, 20, 50, or 100 μM. The OD value at the excitation wavelength of 560 nm was measured, according to which a standard curve was plotted. Afterwards, 50 μL of standard sample was mixed with 100 μL of H2O2 reagent in each test well or tube, which was thoroughly mixed and then incubated for 30 min at room temperature (15-30° C). The level of H2O2 in the cells was eventually calculated relative to the standard curve.
When H2O2 is sufficient, CAT can catalyze H2O2 to generate H2O2 and O2, while the remaining H2O2 generates a red substance under the peroxidase catalysis [53]. A CAT detection kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China) was used to measure the CAT activity in the cells according to the manufacturer’s instructions of the CAT detection kit. The OD value at the excitation wavelength of 240 nm was measured using an ultraviolet spectrophotometer, and the CAT activity was calculated relative to a standard curve.
Hoechst 33258 staining
Cells in the logarithmic growth phase were isolated using 0.25% trypsin containing 0.02% EDTA. After centrifugation, the cells were resuspended in complete culture medium, followed by transfer of 500 μL of cell suspension into a 24-well plate at a density of 6 × 105 cells/well. The plate was incubated at 37° C with 5% CO2. The cells were fixed for 15 min using 4% paraformaldehyde. Subsequently, the cells were stained with Hoechst 33258 at 25° C for five min and dried conventionally. The cells were then observed in five randomly selected fields under an inverted fluorescence microscope. Reduced, dense and brightened cell nuclei were considered apoptotic cell nuclei. The number of positive cells was counted, and the apoptosis rate was calculated as the number of positive cells/total number of cells. The experiment was repeated three times independently to obtain the mean value.
Tube formation assay
Endothelial cells can form tubular structures. To observe this phenomenon, 5 mL Matrigel (#356230, BD Biosciences, San Jose, CA, USA), which was conventionally stored at -20° C, was thawed at 4° C overnight prior to experimentation. A micropipette (2 mL) and a 24-well plate were also precooled on ice. Then, 500 μL of Matrigel was added to each well and evenly distributed in the 24-well plate. The formation of air bubbles was prevented during this process. Matrigel was then solidified by incubation for 30 min in a 37° C incubator. After detachment, 500 μL of cell suspension (2 × 105 cells/mL) from each group was added to each well. The inoculation was repeated several times for each group. The plate was then incubated at 37° C, and the formation of tubes was observed under a microscope after 15 h of incubation. The number of vascular structures was calculated in three randomly selected fields of view in each well. The experiment was repeated three times independently to calculate the mean value.
ELISA
Cells in the logarithmic growth phase were detached using 0.25% trypsin-0.02% EDTA. After centrifugation, the cells were resuspended in complete culture medium and then subjected to incubation in a 24-well plate at a density of 6 × 105 cells/well at 37° C in a 5% CO2 atmosphere. After the cells adhered to the well, the cell supernatant was collected and stored at -20° C. The levels of ICAM-1 (ab2213, Abcam, Cambridge, UK), IL-6 (ab6672, Abcam, Cambridge, UK), IL-10 (ab100549, Abcam, Cambridge, UK), and IL-1β (ab214025, Abcam, Cambridge, UK) in the cell supernatant were detected in strict accordance with the provided instructions of the ELISA kit. The experiment was repeated three times to calculate the mean values.
Flow cytometry
After transfection for 48 h, the cells were treated with 0.25% EDTA-free trypsin (YB15050057, Yubo Biotechnology, Shanghai, China). The cells were then collected in a flow tube and centrifuged, after which the supernatant was discarded. According to the protocols for the Annexin-V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (K201-100, Biovision, USA), the annexin-V-FITC, propidium iodide (PI), and 4-(2-hydroxyethyl)-1-piperazineëthanesulfonic acid (HEPES) buffer were formulated into the annexin-V-FITC/PI staining solution at a volume ratio of 1:2:50. Next, the cells were resuspended at a density of 1 × 106 cells per 100 μL of the staining solution. After incubation for 15 min at room temperature, the cells were treated with 1 mL of HEPES buffer (PB180325, Procell, Wuhan, China). With an excitation wavelength at 488 nm, a flow cytometer was employed to determine FITC fluorescence by a 525 nm bandpass filter and PI fluorescence by a 620 nm bandpass filter in order to detect cell apoptosis. The experiment was repeated three times [54].
Statistical analysis
Statistical analyses were performed using the SPSS 21.0 statistical software (IBM Corp., Armonk, NY, USA). Measurement data were presented as the mean ± standard deviation. The data following normal distribution between two groups were compared by an unpaired t-test, and a paired t-test was used for pairwise comparisons. One-way analysis of variance (ANOVA) was utilized for comparisons among multiple groups, followed by Tukey’s test with corrections for multiple comparisons. A value of p < 0.05 was considered to be statistically significant.
Supplementary Material
Footnotes
AUTHOR CONTRIBUTIONS: TZ, SNL, LHY and DKX designed the study. TZ and LHY collated the data, carried out data analyses and produced the initial draft of the manuscript. SNL and DKX contributed to drafting the manuscript. All authors have read and approved the final submitted manuscript.
CONFLICTS OF INTEREST: The authors report no conflicts of interest.
FUNDING: This work was supported by Science and Technology Foundation of Guizhou Provincial ([2019]1204).
REFERENCES
- 1.Horio E, Kadomatsu T, Miyata K, Arai Y, Hosokawa K, Doi Y, Ninomiya T, Horiguchi H, Endo M, Tabata M, Tazume H, Tian Z, Takahashi O, et al. Role of endothelial cell-derived angptl2 in vascular inflammation leading to endothelial dysfunction and atherosclerosis progression. Arterioscler Thromb Vasc Biol. 2014; 34:790–800. 10.1161/ATVBAHA.113.303116 [DOI] [PubMed] [Google Scholar]
- 2.Rodríguez-Saldaña J, Rodriguez-Flores M, Cantú-Brito C, Aguirre-Garcia J. A pathological study of the epidemiology of atherosclerosis in Mexico city. Cardiol Res Pract. 2014; 2014:264205. 10.1155/2014/264205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maier JA, Malpuech-Brugère C, Zimowska W, Rayssiguier Y, Mazur A. Low magnesium promotes endothelial cell dysfunction: implications for atherosclerosis, inflammation and thrombosis. Biochim Biophys Acta. 2004; 1689:13–21. 10.1016/j.bbadis.2004.01.002 [DOI] [PubMed] [Google Scholar]
- 4.Abedinzadeh N, Pedram B, Sadeghian Y, Nodushan SM, Gilasgar M, Darvish M, Mokarizadeh A. A histopathological analysis of the epidemiology of coronary atherosclerosis: an autopsy study. Diagn Pathol. 2015; 10:87. 10.1186/s13000-015-0324-y [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001; 104:545–56. 10.1016/s0092-8674(01)00241-0 [DOI] [PubMed] [Google Scholar]
- 6.Santulli G. microRNAs distinctively regulate vascular smooth muscle and endothelial cells: functional implications in angiogenesis, atherosclerosis, and in-stent restenosis. Adv Exp Med Biol. 2015; 887:53–77. 10.1007/978-3-319-22380-3_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santulli G. MicroRNAs and endothelial (Dys) function. J Cell Physiol. 2016; 231:1638–44. 10.1002/jcp.25276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo L, Chen B, Li S, Wei X, Liu T, Huang Y, Lin X. Plasma miR-10a: a potential biomarker for coronary artery disease. Dis Markers. 2016; 2016:3841927. 10.1155/2016/3841927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li S, Fan Q, He S, Tang T, Liao Y, Xie J. MicroRNA-21 negatively regulates treg cells through a TGF-β1/Smad-independent pathway in patients with coronary heart disease. Cell Physiol Biochem. 2015; 37:866–78. 10.1159/000430214 [DOI] [PubMed] [Google Scholar]
- 10.Gupta MK, Rao TN. Hearty miR-363 controls HAND1 in cardiac cell specification. Stem Cell Res Ther. 2014; 5:89. 10.1186/scrt478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagh V, Pomorski A, Wilschut KJ, Piombo S, Bernstein HS. MicroRNA-363 negatively regulates the left ventricular determining transcription factor HAND1 in human embryonic stem cell-derived cardiomyocytes. Stem Cell Res Ther. 2014; 5:75. 10.1186/scrt464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selvamani A, Sohrabji F. Mir363-3p improves ischemic stroke outcomes in female but not male rats. Neurochem Int. 2017; 107:168–81. 10.1016/j.neuint.2016.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Costa A, Afonso J, Osório C, Gomes AL, Caiado F, Valente J, Aguiar SI, Pinto F, Ramirez M, Dias S. miR-363-5p regulates endothelial cell properties and their communication with hematopoietic precursor cells. J Hematol Oncol. 2013; 6:87. 10.1186/1756-8722-6-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kriegel AJ, Baker MA, Liu Y, Liu P, Cowley AW Jr, Liang M. Endogenous microRNAs in human microvascular endothelial cells regulate mRNAs encoded by hypertension-related genes. Hypertension. 2015; 66:793–99. 10.1161/HYPERTENSIONAHA.115.05645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Hao B, Ma A, He J, Liu X, Chen J. The expression of NOX4 in smooth muscles of small airway correlates with the disease severity of COPD. Biomed Res Int. 2016; 2016:2891810. 10.1155/2016/2891810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vecchio D, Acquaviva A, Arezzini B, Tenor H, Martorana PA, Gardi C. Downregulation of NOX4 expression by roflumilast N-oxide reduces markers of fibrosis in lung fibroblasts. Mediators Inflamm. 2013; 2013:745984. 10.1155/2013/745984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao W, Li C, Gao H, Wu Q, Shi J, Chen X. Dihydrotanshinone I attenuates atherosclerosis in ApoE-deficient mice: role of NOX4/NF-κB mediated lectin-like oxidized LDL receptor-1 (LOX-1) of the endothelium. Front Pharmacol. 2016; 7:418. 10.3389/fphar.2016.00418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bao XM, Wu CF, Lu GP. Atorvastatin inhibits homocysteine-induced oxidative stress and apoptosis in endothelial progenitor cells involving Nox4 and p38MAPK. Atherosclerosis. 2010; 210:114–21. 10.1016/j.atherosclerosis.2009.11.032 [DOI] [PubMed] [Google Scholar]
- 19.Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007; 116:1413–23. 10.1161/CIRCULATIONAHA.106.679589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shirai H, Autieri M, Eguchi S. Small GTP-binding proteins and mitogen-activated protein kinases as promising therapeutic targets of vascular remodeling. Curr Opin Nephrol Hypertens. 2007; 16:111–15. 10.1097/MNH.0b013e3280148e4f [DOI] [PubMed] [Google Scholar]
- 21.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997; 275:964–67. 10.1126/science.275.5302.964 [DOI] [PubMed] [Google Scholar]
- 22.Langbein H, Brunssen C, Hofmann A, Cimalla P, Brux M, Bornstein SR, Deussen A, Koch E, Morawietz H. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J. 2016; 37:1753–61. 10.1093/eurheartj/ehv564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu P, Wu X, Khandelwal AR, Yu W, Xu Z, Chen L, Yang J, Weisbrod RM, Lee KS, Seta F, Hammock BD, Cohen RA, Zeng C, Tong X. Endothelial Nox4-based NADPH oxidase regulates atherosclerosis via soluble epoxide hydrolase. Biochim Biophys Acta Mol Basis Dis. 2017; 1863:1382–91. 10.1016/j.bbadis.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan A, Chandel AS, Ishaq F, Chhetri S, Rawat S, Malhotra D, Khan S, Rathore NS. Protective Role of Dietary Tocotrienols and Black Caraway Oil on Infection and Inflammation Induced Lipoprotein Oxidation in Animals. Der Pharmacia Sinica. 2011; 2:341–54. [Google Scholar]
- 25.Prendergast C, Quayle J, Burdyga T, Wray S. Atherosclerosis differentially affects calcium signalling in endothelial cells from aortic arch and thoracic aorta in apolipoprotein E knockout mice. Physiol Rep. 2014; 2:e12171. 10.14814/phy2.12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsuji S, Kawasaki Y, Furukawa S, Taniue K, Hayashi T, Okuno M, Hiyoshi M, Kitayama J, Akiyama T. The miR-363-GATA6-Lgr5 pathway is critical for colorectal tumourigenesis. Nat Commun. 2014; 5:3150. 10.1038/ncomms4150 [DOI] [PubMed] [Google Scholar]
- 27.Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009; 324:1710–13. 10.1126/science.1174381 [DOI] [PubMed] [Google Scholar]
- 28.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008; 132:875–86. 10.1016/j.cell.2008.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Q, Zhang J, Cao W, Wang X, Xu Q, Yan M, Wu X, Chen W. Dysregulated miR-363 affects head and neck cancer invasion and metastasis by targeting podoplanin. Int J Biochem Cell Biol. 2013; 45:513–20. 10.1016/j.biocel.2012.12.004 [DOI] [PubMed] [Google Scholar]
- 30.Ou Y, Zhai D, Wu N, Li X. Downregulation of miR-363 increases drug resistance in cisplatin-treated HepG2 by dysregulating Mcl-1. Gene. 2015; 572:116–22. 10.1016/j.gene.2015.07.002 [DOI] [PubMed] [Google Scholar]
- 31.Hu F, Min J, Cao X, Liu L, Ge Z, Hu J, Li X. MiR-363-3p inhibits the epithelial-to-mesenchymal transition and suppresses metastasis in colorectal cancer by targeting Sox4. Biochem Biophys Res Commun. 2016; 474:35–42. 10.1016/j.bbrc.2016.04.055 [DOI] [PubMed] [Google Scholar]
- 32.Zhu S, Zhang J, Lv Y. Glaucocalyxin a inhibits hydrogen peroxide-induced oxidative stress and inflammatory response in coronary artery smooth muscle cells. Clin Exp Pharmacol Physiol. 2020; 47:765–70. 10.1111/1440-1681.13253 [DOI] [PubMed] [Google Scholar]
- 33.Zeng X, Li X, Chen Z, Yao Q. DPP-4 inhibitor saxagliptin ameliorates oxygen deprivation/reoxygenation-induced brain endothelial injury. Am J Transl Res. 2019; 11:6316–25. [PMC free article] [PubMed] [Google Scholar]
- 34.Goettsch C, Goettsch W, Muller G, Seebach J, Schnittler HJ, Morawietz H. Nox4 overexpression activates reactive oxygen species and p38 MAPK in human endothelial cells. Biochem Biophys Res Commun. 2009; 380:355–60. 10.1016/j.bbrc.2009.01.107 [DOI] [PubMed] [Google Scholar]
- 35.Ivey KN, Srivastava D. microRNAs as developmental regulators. Cold Spring Harb Perspect Biol. 2015; 7:a008144. 10.1101/cshperspect.a008144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Li Q, Li R, Ren P, Dong S. MicroRNA-363-3p inhibits papillary thyroid carcinoma progression by targeting PIK3CA. Am J Cancer Res. 2017; 7:148–58. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Singh U, Jialal I. Oxidative stress and atherosclerosis. Pathophysiology. 2006; 13:129–42. 10.1016/j.pathophys.2006.05.002 [DOI] [PubMed] [Google Scholar]
- 38.He H, Xiao S, Xu G, Wang B, Zou Z, Qin X, Yu C, Zhang J. The NADPH oxidase 4 protects vascular endothelial cells from copper oxide nanoparticles-induced oxidative stress and cell death. Life Sci. 2020; 252:117571. 10.1016/j.lfs.2020.117571 [DOI] [PubMed] [Google Scholar]
- 39.Tan YC, Abdul Sattar M, Ahmeda AF, Abdul Karim Khan N, Murugaiyah V, Ahmad A, Hassan Z, Kaur G, Abdulla MH, Johns EJ. Apocynin and catalase prevent hypertension and kidney injury in cyclosporine A-induced nephrotoxicity in rats. PLoS One. 2020; 15:e0231472. 10.1371/journal.pone.0231472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiménez-Altayó F, Meirelles T, Crosas-Molist E, Sorolla MA, Del Blanco DG, López-Luque J, Mas-Stachurska A, Siegert AM, Bonorino F, Barberà L, García C, Condom E, Sitges M, et al. Redox stress in marfan syndrome: dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic Biol Med. 2018; 118:44–58. 10.1016/j.freeradbiomed.2018.02.023 [DOI] [PubMed] [Google Scholar]
- 41.Dong Y, Li P, Ni Y, Zhao J, Liu Z. Decreased microRNA-125a-3p contributes to upregulation of p38 MAPK in rat trigeminal ganglions with orofacial inflammatory pain. PLoS One. 2014; 9:e111594. 10.1371/journal.pone.0111594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo Y, Zhang C, Tang F, Zhao J, Shen C, Wang C, Yu P, Wang M, Li Y, Di JI, Chen R, Rili G. Bioinformatics identification of potentially involved microRNAs in Tibetan with gastric cancer based on microRNA profiling. Cancer Cell Int. 2015; 15:115. 10.1186/s12935-015-0266-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anne SL, Govek EE, Ayrault O, Kim JH, Zhu X, Murphy DA, Van Aelst L, Roussel MF, Hatten ME. Wnt3 inhibits cerebellar granule neuron progenitor proliferation and medulloblastoma formation via MAPK activation. PLoS One. 2013; 8:e81769. 10.1371/journal.pone.0081769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia F, Wang C, Jin Y, Liu Q, Meng Q, Liu K, Sun H. Luteolin protects HUVECs from TNF-α-induced oxidative stress and inflammation via its effects on the Nox4/ROS-NF-κB and MAPK pathways. J Atheroscler Thromb. 2014; 21:768–83. 10.5551/jat.23697 [DOI] [PubMed] [Google Scholar]
- 45.Ji JF, Jiao WZ, Cheng Y, Yan H, Su F, Chi LL. ShenFu preparation protects AML12 cells against palmitic acid-induced injury through inhibition of both JNK/Nox4 and JNK/NFκB pathways. Cell Physiol Biochem. 2018; 45:1617–30. 10.1159/000487728 [DOI] [PubMed] [Google Scholar]
- 46.Touati S, Montezano AC, Meziri F, Riva C, Touyz RM, Laurant P. Exercise training protects against atherosclerotic risk factors through vascular NADPH oxidase, extracellular signal-regulated kinase 1/2 and stress-activated protein kinase/c-Jun N-terminal kinase downregulation in obese rats. Clin Exp Pharmacol Physiol. 2015; 42:179–85. 10.1111/1440-1681.12338 [DOI] [PubMed] [Google Scholar]
- 47.Nishiyama A, Yao L, Nagai Y, Miyata K, Yoshizumi M, Kagami S, Kondo S, Kiyomoto H, Shokoji T, Kimura S, Kohno M, Abe Y. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension. 2004; 43:841–48. 10.1161/01.HYP.0000118519.66430.22 [DOI] [PubMed] [Google Scholar]
- 48.Zhou P, Huang G, Zhao Y, Zhong D, Xu Z, Zeng Y, Zhang Y, Li S, He F. MicroRNA-363-mediated downregulation of S1PR1 suppresses the proliferation of hepatocellular carcinoma cells. Cell Signal. 2014; 26:1347–54. 10.1016/j.cellsig.2014.02.020 [DOI] [PubMed] [Google Scholar]
- 49.Ren B, She Q. Study on the association between IL-1β, IL-8 and IL-10 gene polymorphisms and risk of coronary artery disease. Int J Clin Exp Med. 2015; 8:7937–43. [PMC free article] [PubMed] [Google Scholar]
- 50.Tsitsiou E, Lindsay MA. microRNAs and the immune response. Curr Opin Pharmacol. 2009; 9:514–20. 10.1016/j.coph.2009.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu WJ, Lei T, Yin Z, Pan JH, Chai YS, Xu XY, Yan YX, Wang ZH, Ke J, Wu G, Xu RH, Paranjpe M, Qu L, Nie H. Anti-atherosclerosis and cardio-protective effects of the angong niuhuang pill on a high fat and vitamin D3 induced rodent model of atherosclerosis. J Ethnopharmacol. 2017; 195:118–26. 10.1016/j.jep.2016.11.015 [DOI] [PubMed] [Google Scholar]
- 52.Yang Q, Gong ZJ, Zhou Y, Yuan JQ, Cheng J, Tian L, Li S, Lin XD, Xu R, Zhu ZR, Mao C. Role of drosophila alkaline ceramidase (dacer) in drosophila development and longevity. Cell Mol Life Sci. 2010; 67:1477–90. 10.1007/s00018-010-0260-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang L, Zheng XL, Sun H, Zhong YJ, Wang Q, He HN, Shi XW, Zhou B, Li JK, Lin Y, Zhang L, Wang X. Catalase suppression-mediated H(2)O(2) accumulation in cancer cells by wogonin effectively blocks tumor necrosis factor-induced NF-κB activation and sensitizes apoptosis. Cancer Sci. 2011; 102:870–76. 10.1111/j.1349-7006.2011.01874.x [DOI] [PubMed] [Google Scholar]
- 54.Diaz D, Prieto A, Reyes E, Barcenilla H, Monserrat J, Alvarez-Mon M. Flow cytometry enumeration of apoptotic cancer cells by apoptotic rate. Methods Mol Biol. 2015; 1219:11–20. 10.1007/978-1-4939-1661-0_2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.