

Abstract
Human stem cell-derived models provide the promise of accelerating our understanding of brain disorders. But, not knowing whether they possess the ability to mature beyond late mid-fetal stages potentially limits their utility. So, we leveraged a directed differentiation protocol to comprehensively assess maturation in vitro. Based on genome-wide analysis of the epigenetic clock, transcriptomics, as well as RNA-editing, we observe that 3D human cortical organoids reach postnatal stages between 250–300 days, a timeline paralleling in vivo development. We demonstrate the presence of several known developmental milestones, including switches in the histone deacetylase complex and NMDA receptor subunits, which we confirm at the protein and physiological levels. These results suggest that important components of an intrinsic in vivo developmental program persist in vitro. We further map neurodevelopmental and neurodegenerative disease risk genes onto in vitro gene expression trajectories to provide a resource and webtool (GECO) to guide disease modeling.
Introduction
The scarcity and inaccessibility of tissue from many developmental stages in patients with neurodevelopmental and neurodegenerative disorders highlights the need for advanced in vitro models of human brain development and maturation1, 2. Indeed, recent advances make it possible to differentiate human pluripotent stem cells, in vitro, into self-organizing, three-dimensional (3D) cellular ensembles that recapitulate several features of human brain development3–5. One advantage of these organoid models is that they can be maintained for long periods of time5, 6. However, they have not been comprehensively shown to progress beyond stages that are equivalent to mid-fetal cortical development,3, 4, 7, 8 and most organoid cultures have not been maintained for prolonged periods of time in vitro3, 4, 7, 9 .
We previously developed a directed method of differentiation of human pluripotent stem cells in suspension that does not involve embedding into matrices5. This approach generates dorsal forebrain organoids named human cortical spheroid (hCS) with high reliability that can be cultured for more than 20 months progressing from neurogenesis to astrogenesis6, 10. However, no systematic, unbiased functional analysis has been performed to demonstrate maturation matching perinatal or postnatal stages3–5, 7, 8. Reaching these late stages is essential to model neurodevelopmental, neuropsychiatric and childhood neurodegenerative disorders, as well as for modeling neurodegenerative disorders. Moreover, it is not known whether there are intrinsic programs that underlie important physiological transitions during development and maturation, such as NMDA isoform shifts and RNA editing, neither of which have been studied in 3D or 2D human stem cell-based models to date3–5, 7, 8 .
Here, we leverage the ability to maintain human cortical organoids in long-term cultures and perform functional genomic assays to rigorously assess correspondence to in vivo developmental progression and maturation. We demonstrate substantial parallels between in vitro and in vivo development at the epigenetic and transcriptomic levels, as well as preservation of known physiological transitions, including NMDA receptor signaling. By mapping risk genes onto gene expression trajectories across development in this system, we also provide a searchable resource (GECO, Gene Expression in Cortical Organoids) to facilitate the choice of appropriate in vitro time points for modeling specific brain disease-causing genes.
Results
We cultured hCS differentiated from 6 hiPSC lines derived from 5 different individuals for up to 694 days in vitro (summarized in Extended Data Figure 1a and Supplementary Tables 1 and 2) using a protocol that yields highly reliable hCS differentiation across experiments and cell lines5, 11. In total, we collected 62 samples for RNA sequencing (from 4 individuals, 5 hiPSC lines) and 50 samples for DNA methylation (from 5 individuals, 6 hiPSC lines) at 13 time points (Extended Data Figure 1a and Supplementary Tables 1 and 2). Due to the challenging nature of these long-term cultures, samples were collected at various time points up to the following maximum differentiation day for each line: 0307–1, 280 days; 1205–4, 587 days; 2242–1, 652 days; 8858–1, 694 days; 8858–3, 652 days; H2096, 371 days.
Methylation and transcriptional maturation of hCS
To assess whether hCS maturation over a long period of in vitro development paralleled in vivo development, we first analyzed the epigenetic clock12, which predicts chronological age based on the methylation status of CpG islands. We note that the epigenetic clock is unable to predict the precise culture age based on in vivo methylation, as the epigenetic clock is not calibrated for the tissue, or for the newer array type that we used13, 14. Nevertheless, we observed a highly significant, monotonic correlation between the length of differentiation of the hCS in vitro and their predicted methylation age (r = +0.76, p = 1.57e−10; Figure 1a), consistent with continuous and progressive maturation of these cultures over time.
Figure 1. Methylation and transcriptional maturation in long-term hCS.
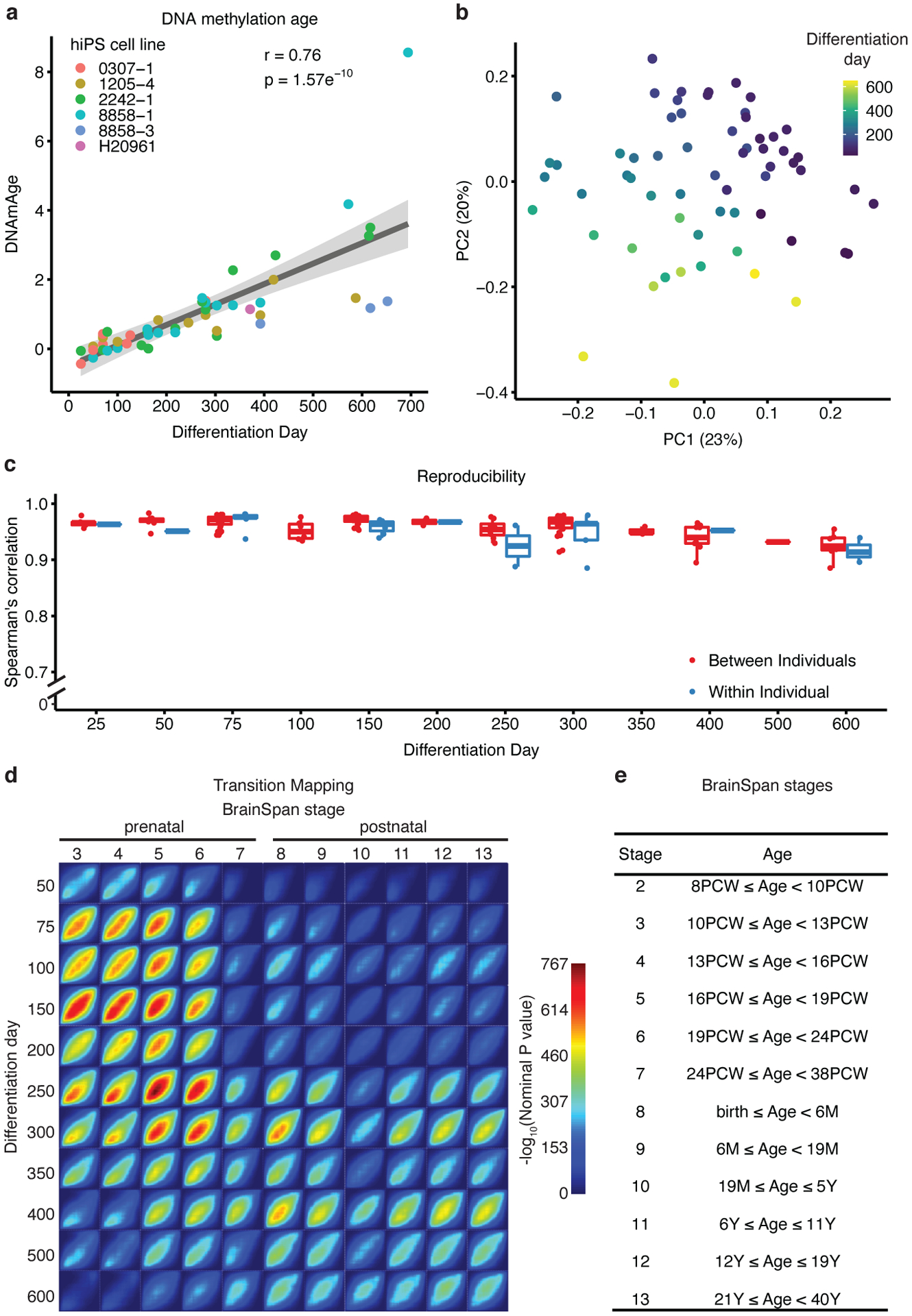
(a) The predicted methylation age (DNAmAge) of hCS is monotonically correlated with the in vitro differentiation day (r= +0.76, p = 1.57e−10, two-sided Pearson correlation test, n = 50 from 6 hiPSC lines derived from 5 individuals). Colors denote individual hiPSC lines. The shaded grey area represents the 95% confidence interval. (b) Scatter plot of the first two principal components (PC) of gene expression data. Color represents differentiation day and shape represents the hiPSC line. Numbers in brackets on axis titles are the percent of variance explained by the PC. (c) Spearman’s correlation of gene expression between samples from the same timepoint which were derived either from different individuals (red) or from the same individual (blue) (n = 62 samples from 6 hiPSC lines derived from 5 individuals).Boxplots show: center – median, lower hinge – 25% quantile, upper hinge – 75% quantile, lower whisker – smallest observation greater than or equal to lower hinge −1.5x interquartile range, upper whisker – largest observation less than or equal to upper hinge +1.5x interquartile range. (d) Transition mapping (TMAP) of gene expression of hCS (compared to differentiation day 25) and human primary tissue from the BrainSpan dataset (compared to stage 2). (e) BrainSpan stages and corresponding age. PCW – post conception weeks, M – months, Y – Years.
To predict culture age more precisely using an independent genome-wide approach, we next applied a validated framework that permits unbiased, quantitative statistical comparison of gene expression during cortical maturation in vitro to the BrainSpan dataset, which represents an in vivo reference for cortical development15–17. To assess the quality of the data, we first sought to verify whether the main driver of variance of gene expression in the system is the time of in vitro differentiation and not other less relevant covariates (e.g. batch, individual or line). Principal component (PC) analysis of gene expression revealed that the top five PCs, which together account for 57.1% of variability, were all associated with differentiation time (Figure 1b and Extended Data Figure 1b). Hierarchical clustering also showed that the samples clustered by differentiation day, not by other covariates (Extended Data Figure 1c). Variance partitioning further demonstrated that the largest driver of variance was time of differentiation (median variance explained 29.2%), while the median value of variance explained for differentiation and cell line was < 0.01% (Extended Data Figure 1d). Reproducibility between samples from the same time point was high both between individuals (mean Spearman correlation 0.96, range 0.88–0.98) and within individuals (mean Spearman correlation 0.95, range 0.88–0.98) similar to what we have previously reported11, 18. We note that, as differentiation progressed, this high correlation between lines decreased slightly from 0.96 at day 25 (range 0.95–0.98) to 0.92 at day 600 (range 0.88–0.95) (Figure 1c).
Cellular stress pathways (i.e. endoplasmic reticulum, glycolysis pathways) have previously been proposed to be upregulated during development of in vitro 3D organoids19, 20. We were able to detect moderate expression levels of genes in these pathways both in vivo in BrainSpan, as well as in vitro (Extended Data Figure 2a–b). We also detected ubiquitous expression of genes in these pathways in a large single cell dataset consisting of 40,000 cells collected from in vivo developing human cerebral cortex, consistent with the BrainSpan results21 (Extended Data Figure 2c). Although we do observe that stress pathway genes are expressed at slightly higher levels in vitro than in vivo, they follow a flat trajectory over time (Extended Data Figure 2a–b). This is more consistent with a homeostatic state, and the absence of progressive stress or dysfunction.
We next compared changes in gene expression during the maturation of hCS to transcriptome changes observed in cortical development in vivo in humans16, 17 using transition mapping, a quantitative method based on the rank-rank hypergeometric test15. At early time points (i.e., prior to 250 days in culture), hCS mapped to prenatal stages (stages 3–7), consistent with the observed developmental progression in vitro5, 11 (Figure 1d–e). By day 250, we started to observe a postnatal signature that became more evident by day 300. Between day 250 to day 300, hCS displayed a similar overlap with both prenatal and postnatal stages, while after day 300, they showed increasing overlap with postnatal stages (stages 8–13) (Figure 1d–e). Based on this analysis, the predicted transition between prenatal and postnatal stages occurs around 250–300 days (~8–10 months) of in vitro differentiation.
Gene expression and gene network correspondence
Seeing this strong overall correspondence between in vitro and in vivo transcriptomes, we next compared known biological processes occurring during the maturation of hCS to those occurring during in vivo cortical development. Gene set enrichment of differentially expressed genes spanning prenatal stages in vitro (between day 25 and day 200; 1,940 downregulated genes, 2,122 upregulated genes at FDR<0.05; Extended Data Figure 3a and Supplementary Table 3) showed that early embryonic proliferation and developmental processes were downregulated, while neuronal and synaptic related genes were upregulated, analogous to what was reported in vivo16 (Extended Data Figure 3b and Supplementary Table 4). In contrast, in vitro stages that were similar to early postnatal stages (between day 200 and day 400; 992 downregulated genes, 1,369 upregulated genes at FDR<0.05; Extended Data Figure 3a and Supplementary Table 3), showed enrichment for processes related to translation and cortical neuronal development, including downregulation of forebrain generation of neurons, whereas terms associated with non-neuronal cell development and synaptic function (e.g., regulation of long term synaptic potentiation, neurotransmitter metabolic process) were upregulated, again corresponding to known in vivo processes16 (Extended Data Figure 3b and Supplementary Table 4).
These parallels of in vivo biological processes being preserved in vitro were also observed when examining the expression trajectories of markers for the major hCS cell types, including progenitors, layer-specific cortical neurons and developing and maturing astrocytes (Figure 2a–c). The expression of these cell markers followed trajectories similar to those found in vivo (Extended Data Figure 3c) with radial glia markers peaking earliest, followed by neuronal markers and subsequently mature astrocyte markers reaching their highest level later, at around 350–400 days, when cultures start to resemble postnatal stages (Figure 2a–c and Extended Data Figure 3d). It is important to note that the down regulation of neuronal markers at late stages of differentiation, in both the hCS and in vivo, may be due to the increase in the proportion of astrocytes, rather than absolute reduction in the expression of these genes.
Figure 2. Biological processes and cell type marker changes in long-term hCS.
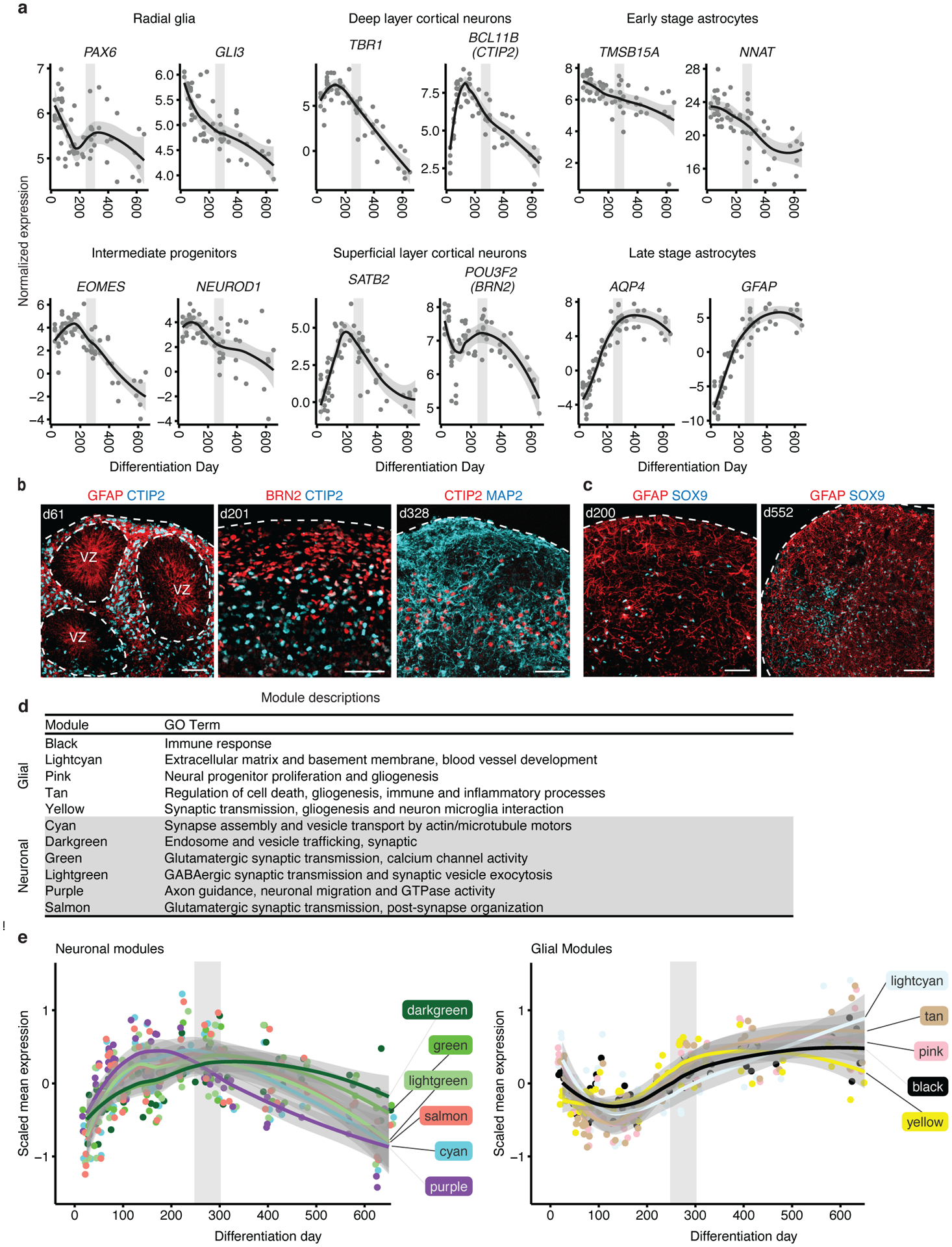
(a) Normalized expression of marker genes for neurons, intermediate progenitors, astrocytes, and radial glia as well as upper and deep layer neurons. Neuronal and intermediate progenitor markers are initially expressed at high levels and decrease after day 250. Astrocyte markers increase in expression with time and peak after day 300. Radial glia markers decrease in expression as hCS advance in differentiation. (b) Immunohistochemistry of progenitors and neuronal markers at day 61 (d61; line 0524–1), day 201 (d201; line 8858–1) and day 328 (d328; line 2242–1) showing expression of GFAP in ventricular zone (VZ)–like regions and the deep and upper layer markers CTIP2 (also known as BCL11B) and BRN2 (also known as POU3F2). (c) Immunohistochemistry for the astrocyte markers GFAP and SOX9 at day 200 (d200; line 2242–1) and day 552 (d552; line 8858–1). Immunohistochemistry experiments were performed twice with similar results (1–3 hCS per line from at least 2 hiPSC lines were included). Scale bars, 50 μm (b-c) (d) Annotation of groups and GO term annotation of in vivo WGCNA modules performed by Stein et al15. (e) Scaled mean expression of neuronal and glial module genes. The neuronal modules peaked at ~day 200; the glial modules decreased in expression until about differentiation day 150 and then increased in expression to peak around day 500. In (a) and (e) the shaded grey area around the trajectory line represents the 95% confidence interval and the vertical grey denotes the shift from prenatal to postnatal gene expression based on matching to in vivo patterns. In (a) and (e), n = 62 samples from 6 hiPSC lines derived from 5 individuals
Notably, in agreement with previous reports showing that oligodendrocyte precursor cells (OPCs), GABAergic neurons and microglia were not present or present in low proportion in hCS11, 22, 23, we found that the expression trajectories of markers for these cells types either did not follow cortical in vivo trajectories (i.e., GAD1 and PLP1) or were not detectable (i.e., AIF1, ITGAM) in hCS (Extended Data Figure 3e). Markers for neuronal activity were only partially preserved in hCS (i.e., NPAS4 and ARC; Extended Data Figure 3f), which is likely due to lack of typical physiological inputs.
To provide a more refined view of the trajectories of specific biological processes, we leveraged co-expression modules defined from in vivo brain development to directly examine the trajectories of in vivo processes in hCS15. These in vivo modules were previously annotated based on enrichment for processes associated with specific cell types15 (Figure 2d). We verified that in vitro modules significantly overlapped with these in vivo modules (Extended Data Figure 4). As seen for single genes, the trajectories of these modules followed the expected in vivo sequence. Namely, the neuronal modules peaked at times matching prenatal stages (100–300 days in vitro; Figure 2e) matching what is seen in vivo15, 16. The glial processes were upregulated at early stages (day 25; Figure 2e) corresponding to the proliferation of radial glia in vivo16 and then, again, at postnatal stages (400–600 days of differentiation; Figure 2e) corresponding to the development and maturation of astrocytes in vivo6. Overall, these analyses demonstrate that the in vivo gene expression trajectories are paralleled during long-term in vitro hCS differentiation11, 22.
Preservation of RNA editing
RNA editing, a major RNA processing mechanism, is dynamically regulated during in vivo brain development24. RNA editing has been shown to display developmental trajectories that vary with maturation in vivo and are dependent on neuronal activity25. Recently, disruption of RNA editing was suggested to play a role in autism spectrum disorder (ASD), further supporting its importance in brain function and dysfunction26. Despite its importance in brain development, global patterns of RNA editing have not been explored in hiPSC-derived brain organoids.
To assess editing in vivo, we identified two modules of coordinated RNA editing in the in vivo BrainSpan data16: BSeditM1 which increased in editing rates during development especially in late prenatal and early childhood (stages 6–9) and BSeditM2 which was slightly decreased in editing rates during development (Figure 3a). Both in vivo modules were moderately preserved in the in vitro data (ZsummaryBSeditM1 = 9.3, ZsummaryBSeditM2 = 5.3) (Figure 3b), showing preservation of in vivo RNA editing processes in hiPSC-derived brain organoids for the first time.
Figure 3. RNA editing in hCS.
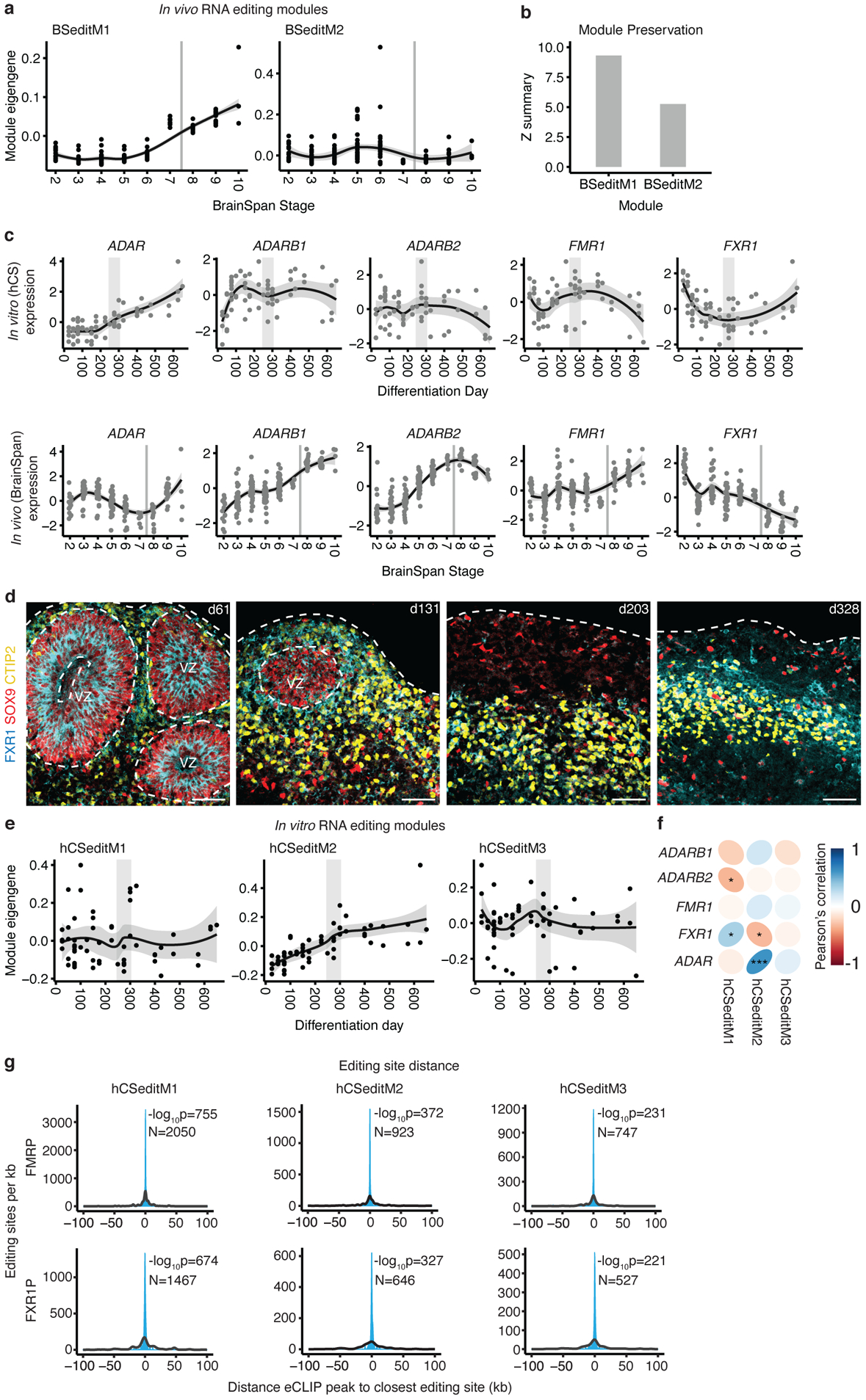
(a) Trajectories of in vivo (BrainSpan) RNA editing modules. (b) Preservation scores (Z summary) of the in vivo RNA editing modules in hCS. (c) Trajectories of RNA editing enzymes in hCS (top) and in vivo from Brain Span (bottom). (d) Immunohistochemistry of the RNA editing regulator FXR1 with the glial and neuronal markers GFAP and CTIP2 (also known as BCL11B) at day 61 (d61; line 0524–1), at day 131 (d131; line 1205–4), at day 200 (d203; line1205–4), and at day 328 of differentiation (d328; line 2242–1). VZ - ventricular zone. Scale bars, 50 μm. Immunohistochemistry experiments were performed once for d61, twice for d131 and d203, and 3 times for d328 (1–3 hCS per line from at least 2 hiPSC lines were included) (e) Trajectories of the three hCS RNA editing modules. (f) Correlation of module eigenvalues with the expression of the major known RNA editing enzymes and regulators. (g) Distributions showing the closest distances between editing sites from hCS editing modules and FMRP or FXR1P eCLIP peaks (blue). The median of 10,000 sets of control sites (black) is depicted for comparison. See methods for details of permutation-based tow sided P-value calculation. N indicates the number of editing sites shown. *FDR, 0.05, ***FDR < 0.005. In (a), (c) and (e), the shaded grey area around the trajectory represents the 95% confidence interval, vertical grey lines represent birth and vertical grey bars denote the shift from prenatal to postnatal gene expression based on matching to in vivo patterns. In (c) top row and (e), n = 62 samples from 6 hiPSC lines derived from 5 individuals. In (a) and (c) bottom row, n = 196 from 24 individuals).
We next examined the expression of the major RNA editing enzymes and regulators. In vivo, ADAR increases postnatally, ADARB1 expression increases prenatally, followed by relatively stable expression postnatally, while FXR1 expression decreases prenatally26; these followed a similar trend in vitro (Figure 3c). Expression levels of FMR1 increased prenatally both in vivo and in hCS. However, the subsequent postnatal increase in expression seen in vivo was not observed in hCS (Figure 3c). Immunohistochemistry confirmed the transcript level analysis and suggested that FXR1 predominantly localized to ventricular zone areas and appeared down-regulated at later stages in neurons (Figure 3d).
To test whether ADAR, ADARB1 and FXR1 potentially drive editing in vitro, we identified editing modules in hCS (Figure 3e) and correlated the module eigenvalues with the expression of the editing enzymes and regulators. We found that one module, hCSeditM2, was significantly correlated with the expression of the editing related genes FXR1 (r = −0.32, FDR = 0.04) and ADAR (r = +0.60, FDR = 5.1e−6) (Figure 3f). Interestingly, FXR1 has previously been shown to inhibit editing by interacting with ADAR26 and this interaction regulates RNA editing sites dysregulated in ASD26. hCSeditM2 also significantly overlapped with the temporally increasing in vivo module BSeditM1 (OR = 1.8, FDR = 2.3e−4; Extended Data Figure 5a). The hCSeditM2 module eigengene increased in expression over stages matching prenatal in vivo development (before 250 days) and plateaued at stages resembling postnatal periods (after 350 days) (Figure 3e), very similar to the trajectory of the increasing in vivo module BSeditM1 (Figure 3a).
To further support their potential to regulate RNA editing, we tested whether FMRP and FXR1P locally bind to the mRNA in close proximity to the editing sites of the genes in the module by integration with enhanced cross-linking and immunoprecipitation (eCLIP) data from the FMRP and FXR1P proteins in the adult human brain26. We observed that the editing sites that we identified in both the hCS and the BrainSpan modules have highly significant proximity to both FMRP and FXR1P binding sites (Figure 3g and Extended Data Figure 5b). These results support the claim that FMRP and FXR1P regulate brain-related editing modules through proximity mediated interactions26. To investigate whether the target editing sites of FMRP or FXR1P were similar between BrainSpan and hCS modules, we measured the overlap of editing sites within 1 kb of a CLIP site of FMRP or FXR1P. We found that FXR1P targeted editing sites significantly overlapped between BSeditM1 and hCSeditM1 (OR = 7.71, FDR = 6.18e−58) and hCSeditM2 (OR = 2.16, FDR = 5.31e−4), which also showed a conserved trajectory, increasing over in vitro differentiation time (Extended Data Figure 5c). Taken together, the methylation, gene expression and RNA editing data, paint a coherent picture, indicating that hCS reach a state of postnatal maturation between 250 and 300 days.
Canonical isoform switching in development
To further validate known transitions that occur with development and maturation, we assessed isoform switches in specific protein complexes related to histone modifying complexes and neurotransmitter signaling that characterize the transition from prenatal to postnatal stages of brain development27–29. One canonical switch is in the histone deacetylation complex (HDAC), in which the more highly expressed isoform– HDAC2, diminishes and HDAC1 and HDAC11 isoforms increase in expression27, 30 (Figure 4a), which plays a role in fate specification and synapse maturation27. Indeed, we observed a switch in the HDACs with an increase in HDAC1 and HDAC11 and a concomitant decrease in HDAC2 in hCS. We note that while HDAC1 is downregulated postnatally in vivo, it remains upregulated in hCS (Figure 4a). We speculate that perhaps cell types not present in hCS, or activity-dependent processes that occur in vivo31 may contribute to changes in HDAC enzyme expression in vitro at later stages. We also assessed whether these enzymes show cell type-specific enrichment, which could contribute to the differences in postnatal expression. Using immunohistochemistry, we observed that HDAC2 was expressed in both progenitors and neurons, although it appeared more highly expressed in progenitors, consistent with its down regulation over time (Figure 4b and Extended Data Figure 6a). Additionally, using a mid-fetal single cell dataset21, we found that HDAC1, HDAC2 and HDAC11 did not show significant cell type-specific enrichment at that timepoint (Extended Data Figure 6b), which is more equivalent to the later time point in vitro (day 131).
Figure 4. Developmental isoform switches in hCS.
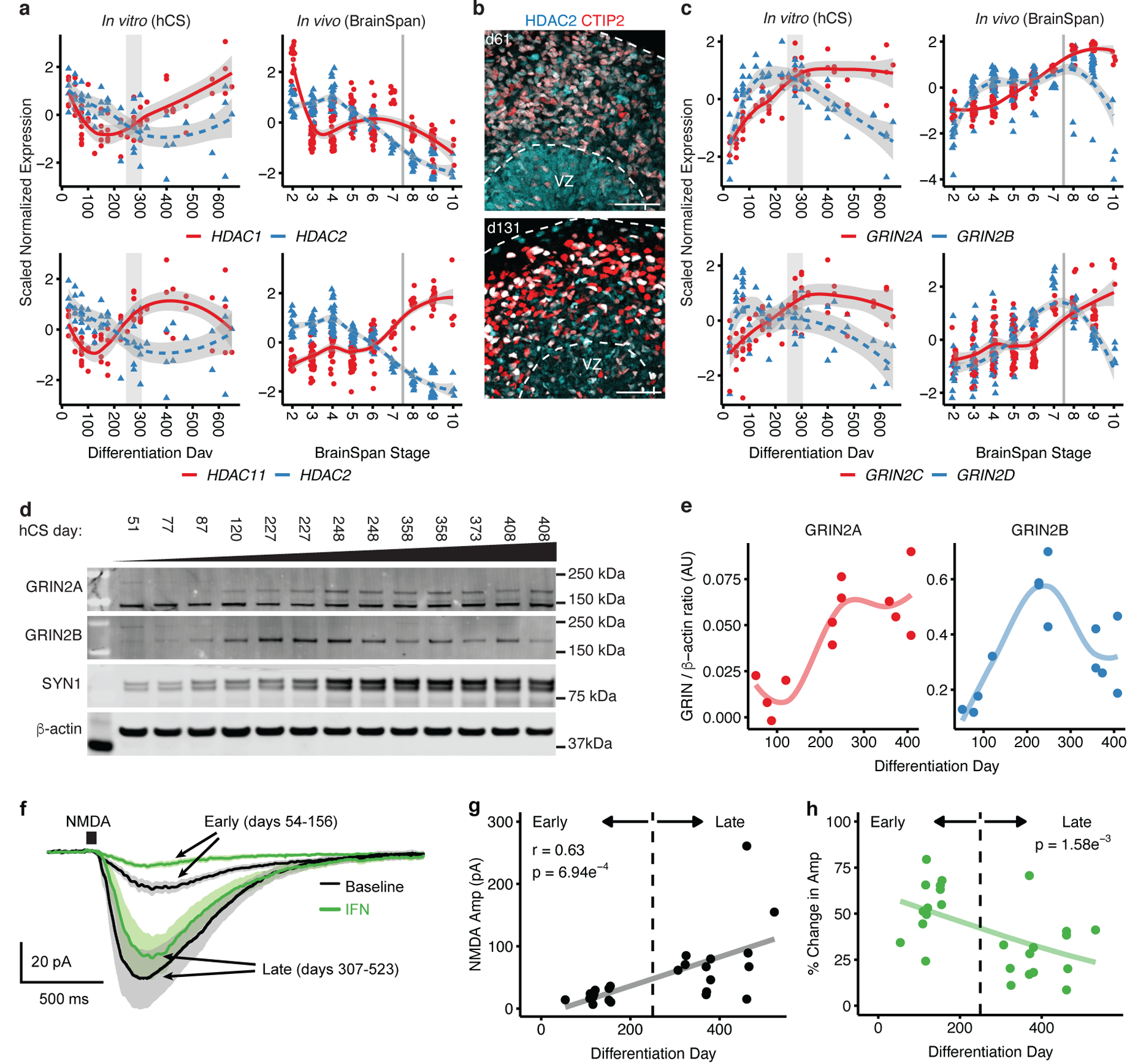
(a) Expression trajectories of histone deacetylase (HDAC) subunits. In vivo (right) and in vitro (left), HDAC2 expression decreases while the expression of both HDAC1 (top) and HDAC11 (bottom) increases. The shaded grey area around the trajectory line represents the 95% confidence interval. (b) Immunohistochemistry for HDAC2 and deep layer marker CTIP2 (also known as BCL11B) at day 61 (d61; line 0524–1) and day 131 (d131; line 1205–4). VZ, ventricular zone. Scale bars, 50 μm. Immunohistochemistry experiments were performed once for d61 or twice for d131 (1–3 hCS per line from at least 2 hiPSC lines were included). (c) Expression trajectories of NMDA receptor subunits. In vivo (right) and in vitro (left), GRIN2A (NR2A) and GRIN2B (NR2B) (top), as well as GRIN2C (NR2C) and GRIN2D (NR2D) (bottom). In (a) and (c), the shaded grey area around the trajectory represents the 95% confidence interval, vertical grey lines represent birth and vertical grey bars denote the shift from prenatal to postnatal gene expression based on matching to in vivo patterns. For in vitro data, n = 62 samples from 6 hiPSC lines derived from 5 individuals; for in vivo data, n = 196 from 24 individuals. (d) Western blots for GRIN2A, GRIN2B, Synpsin-1. β-actin was used as a loading control. The images shown were cropped (uncropped images are included in Source Data 1). Cell lines used are 1205–4 (samples 1, 2, 3, 4, 6, 8, 10, 11 and 13) and 0524–1 (samples 5, 7, 9 and 12). Western blot experiments were run 3 times with similar results. (e) Quantification of GRIN2A and GRIN2B protein levels from (d) (n = 13 from 2 hiPSC lines). (f) Average whole-cell voltage clamp recordings of NMDA responses (10 mM NMDA, 50 ms pulse) at early (red, days 54–156) and late (black, days 307–523) stages of hCS development. Standard error of the mean (SEM) are depicted by the grey and pink lines. Neurons were identified with a fluorescent reporter (Syn1::GFP). (g) Increased maximum NMDA response amplitudes over developmental time (r = 0.63, p = 6.94e−4). Black line represents the linear fit of the data. (h) Percent reduction of maximum NMDA responses by the NR2B-containing NMDA receptor blocker ifenprodil (IFN, 10 μM) is significantly reduced with time. Significance was measured using a beta regression with logit link function, B = −0.003, p = 1.58e−3. One cell was patched per hCS for a total of 25 cells from two hiPSC lines (8858–1, 1205–4).
Another critical neurodevelopmental switch is the change in the NMDA receptor subunits from GRIN2B (also known as NR2B and GluN2B) to GRIN2A (also known as NR2A or GluN2A) (Figure 4c)28 and from GRIN2D to GRIN2C29 (Figure 4c). The progressive change in subunit expression results in a switch around birth28 and has a profound effect on channel function, including how it responds to allosteric modulators32. In vivo, we observed the subunit shift at the transcriptional level occurring soon after birth (BrainSpan stage 8; 0–6 months after birth; Figure 4c). Interestingly, in hCS we observed a switch in NMDA receptor subunits at the time when cultures are predicted to transition from prenatal to postnatal stages based on their transcriptomic patterns (day 250–300 of differentiation; Figure 4c). Using western blotting for GRIN2A and GRIN2B in hCS from day 51 to day 408, we found that the level of GRIN2A appeared to increase with time and the level of GRIN2B seemed to peak at 200–250 days and decreased at later time points (350–400 days; Figure 4d–e and Source Data 1). The protein trajectories mirrored the trajectories seen at the RNA level (Figure 4c), with the switch between the two subunits occurring between 250 and 300 days (Figure 4c–e).
This switch that results in a greater contribution of the GRIN2A subunit to the NMDA complex is associated with changes in the functional properties of the channel32. To verify that this results in physiological changes in hCS neurons, we measured the magnitude of NMDA receptor activity at early (days 54–156 of differentiation; GRIN2B predominant) and late (days 307–523 days of differentiation; NR2A predominant) developmental time points. We performed voltage-clamp recording of neurons in hCS slices while applying pulses of NMDA to measure the summation of the NMDA response throughout the cell independent of subcellular receptor localization (Figure 4f). We found that the magnitude of NMDA responses increased over in vitro time (r = +0.63, p = 6.94e−4; Figure 4g). Importantly, there was a significant negative association between the reduction in total NMDA response after application of the GRIN2B-specific antagonist ifenprodil (IFN) and the total time of differentiation mirroring the reduction seen in the GRIN2B subunit (logit regression B = −0.003, p = 1.58e−3) (Figure 4h). This demonstrates that changes observed in hCS at the RNA and protein level result in physiological changes that are consistent with the presence of a greater proportion of GRIN2B-enriched NMDA receptors at early compared to later developmental time points.
Disease risk gene trajectories
One of the goals of these model systems is to guide a biological understanding of brain disorders. We therefore mapped genes associated with autism spectrum disorder (ASD; Figure 5a), intellectual disability (ID; Figure 5b) and schizophrenia (SCZ; Figure 5c) onto these gene expression data to see if there were specific expression patterns associated with subsets of risk genes and whether they were shared between disorders.
Figure 5. Mapping neurodevelopmental and neuropsychiatric disorder genes onto hCS differentiation.
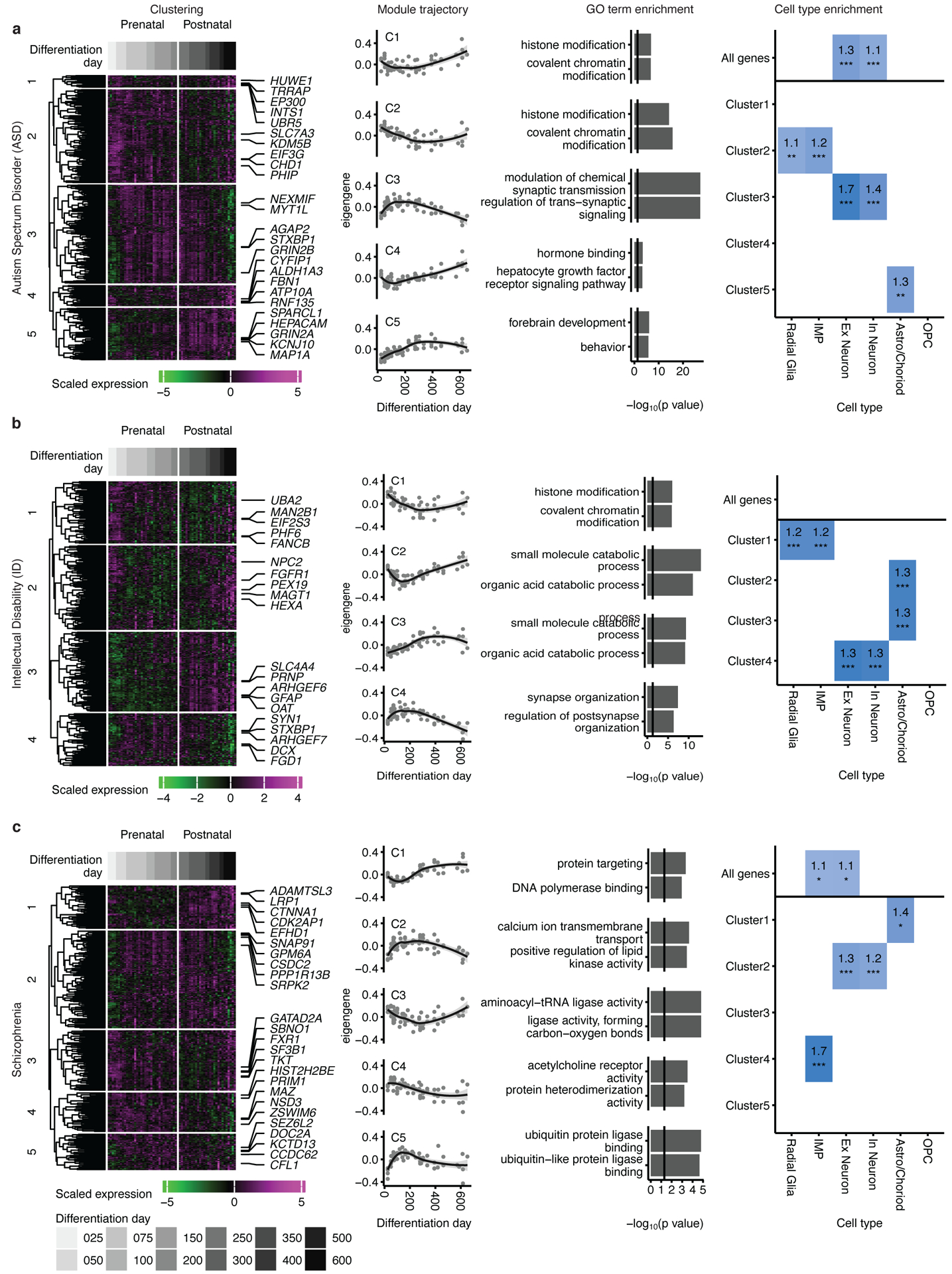
Mapping of genes associated with autism spectrum disorder (a), intellectual disability (b) and schizophrenia (c) onto hCS differentiation trajectories. The first column shows clustering of scaled normalized expression of genes associated with a disorder. Genes (in rows) are clustered using hierarchical clustering on the Euclidean distance between genes. Samples (columns) are ordered by differentiation day (represented by gray bars) with the earliest days on the left and latest time points on the right. The 5 most representative genes (highest correlation with the cluster eigengene) are shown. The second column shows the cluster eigengenes (first principal component) for the identified gene clusters. The shaded grey area around the trajectory line represents the 95% confidence interval. The third column shows the top GO-terms enriched in the identified clusters. The fourth column shows cell types over expressed in either all the genes associated with a disorder (above line) or in the genes from the identified clusters. Number and color represent the fold change. Significance was tested using a one-sided permutation test with 100,000 permutations. P values were corrected for multiple testing using the Benjamini-Hochberg method. * FDR < 0.05, ** FDR < 0.01, *** FDR < 0.005, n = 62 samples from 6 hiPSC lines derived from 5 individuals.
We performed unsupervised hierarchical clustering of disorder associated genes based on the temporal expression patterns in hCS, which identified clusters representing distinct temporal trajectories in each disorder (Figure 5 and Supplementary Table 5). Annotation with gene ontology (GO) and a cell type enrichment analyses (Methods) revealed that each of these clusters represent different developmental trajectories, as well as specific biological processes and cell types (Figure 5).
ASD and SCZ risk genes clustered into 5 developmental trajectories each, whereas ID genes formed 4 clusters. We found three trajectory patterns shared across disorders. One trajectory seen in ASD-C3, ID-C4 and SCZ-C2 peaked at around 150 days of differentiation (Figure 5). These clusters were all enriched for both excitatory and inhibitory neuronal genes as well as for synaptic related GO terms, such as modulation of chemical synaptic transmission (ASD-C3), synapse organization (ID-C4) and calcium ion transmembrane transport (SCZ-C2) (Figure 5). Another shared trajectory was seen in ASD-C2 and ID-C1, which were highly expressed at the peak of neurogenesis (< 150 days) and gliogenesis (> 300 days) (Figure 5a–b). These clusters were enriched for progenitor cell type genes (radial glia and intermediate progenitors) and for histone modification and covalent chromatin modification GO terms, and they were not conserved in SCZ. Although SCZ-C3 showed a similar trajectory to ASD-C2 and ID-C1, it was not enriched for any cell type, but was enriched for protein translation-related GO terms, such as aminoacyl-tRNA ligase activity (Figure 5c). The third shared trajectory peaked in expression at later stages of differentiation (> 250 days) and was found in ASD-C5, ID-C3, and SCZ-C1. These clusters were all enriched for astrocyte genes; however, they did not share common biological processes across disorders, indicating that different pathways may be impacted (Figure 5). These three trajectories were also seen in genes associated with epilepsy (Extended Data Figure 7a). Most of the epilepsy genes (62%) were found in the cluster epilepsy-C3 that peaked at day 150. This cluster was enriched for GO terms related to ion channel activity and for excitatory and inhibitory neuronal genes. However, a substantial proportion of genes (20%; epilepsy-C2) peaked at much later stages of differentiation (> 250 days) and was not enriched for any cell type (Extended Data Figure 7a).
We next mapped genes associated with two neurodegenerative disorders: Alzheimer’s disease (AD; Figure 6a) and Parkinson’s disease (PD; Figure 6b). We found that the majority of clusters (3/4 in AD and 3/4 in PD) showed peak levels of expression at late time points (>250 days), corresponding to predicted postnatal stages. One of these late peeking clusters in AD (AD-C1) contained the familial Alzheimer’s associated genes APOE, APP and PSEN2, and was associated with amyloid-beta formation, a hallmark of AD (Figure 6a). In PD, genes associated with mendelian forms of the disease, such as PRKN, UCHL1, SNCA, PARK7, PINK1 and LRRK2, were all found in clusters that peaked later in differentiation (>250 days; PD-C1and PD-C2; Figure 6b). Genes associated with two other related neurodegenerative diseases–progressive supranuclear palsy (PSP) and frontotemporal dementia (FTD), formed two clusters that also peaked late in differentiation (Extended Data Figure 7b).
Figure 6. Mapping neurodegenerative disorder genes onto hCS differentiation.
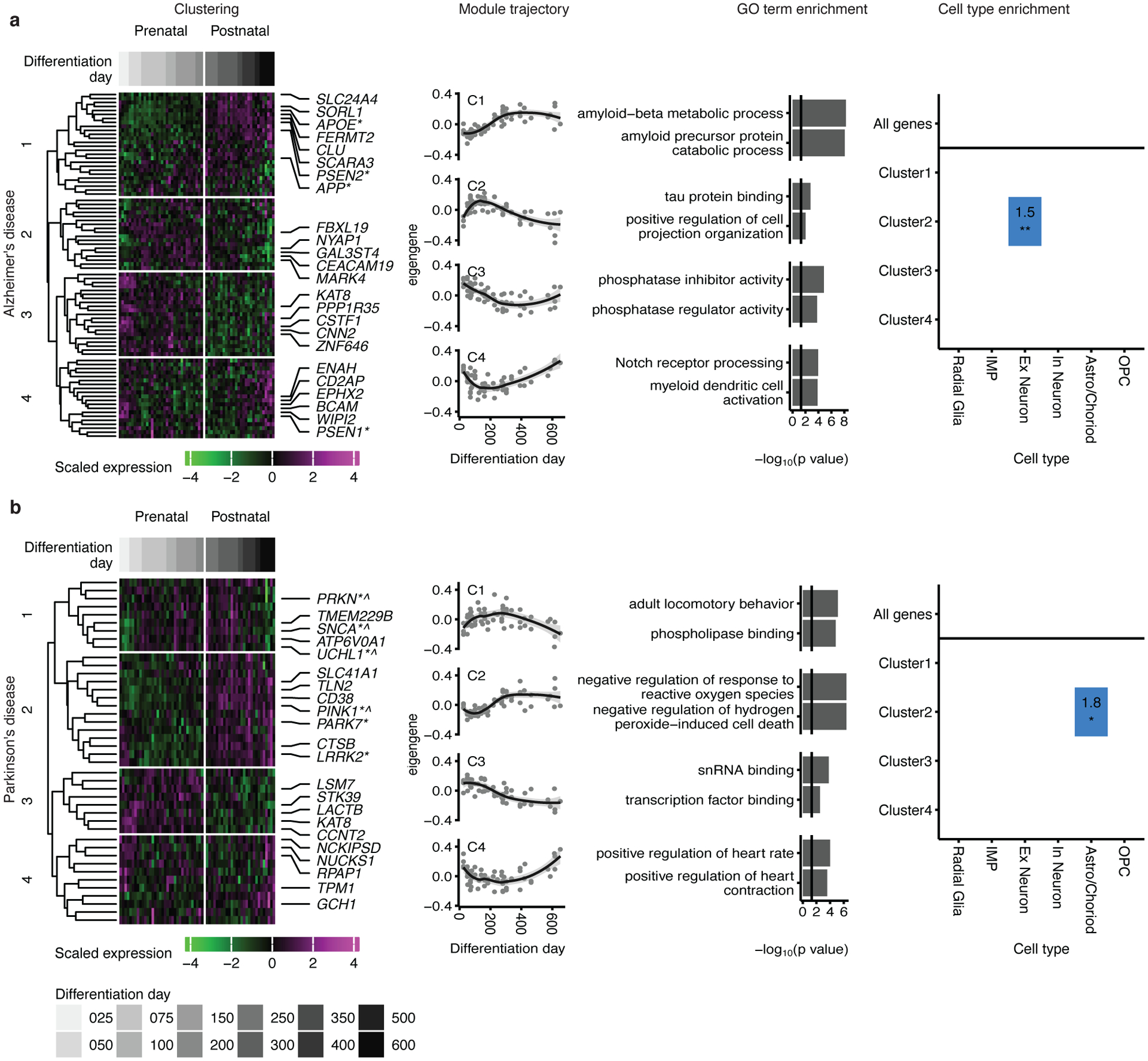
Mapping of genes associated with Alzheimer’s disease (a) and Parkinson’s disease (b) onto hCS differentiation. The first column shows clustering of scaled normalized expression of genes associated with a disorder. Genes (in rows) are clustered using hierarchical clustering on the Euclidean distance between genes. Samples (columns) are ordered by differentiation day (represented by gray bars) with the earliest days on the left and latest time points on the right. The 5 most representative genes (highest correlation with the cluster eigengene) and genes associated with familial forms of the disease are shown. *, Familial gene; ^, Familial gene that is also a hub gene. The second column is the cluster eigengenes (first principal component) for the identified gene clusters. The shaded grey area around the trajectory line represents the 95% confidence interval. The third column is the top GO-terms enriched in the identified clusters. The fourth column is cell types over expressed in either all the genes associated with a disorder (above line) or in the genes from the identified clusters. Number and color represent the fold change. Significance was tested using a one-sided permutation test with 100,000 permutations. P values were corrected for multiple testing using the Benjamini-Hochberg method. * FDR < 0.05, ** FDR < 0.01, *** FDR < 0.005. n = 62 samples from 6 hiPSC lines derived from 5 individuals.
At least one cluster in each disorder peaked in expression at later stages of neural differentiation (>250 days), which emphasizes the need for long term maturation to study the role of these disease-related genes. These clusters can guide the selection of appropriate time points and cell types to model specific disorders with hiPSC-derived neural cultures. Genes in some of the clusters, such as ASD-C3, ID-C4, SCZ-C2, epilepsy-C3, AD-C2, and PD-C3, could be used in early stage hCS models, whereas genes in clusters ASD-C5, ID-C3, SCZ-C1 and AD-C1, epilepsy-C2, AD-C1, PD-C2 and FTD/PSP-C1, may require cultures beyond 250 days in vitro. To allow detailed comparison between in vivo and hCS gene trajectories, we provide a webtool named GECO (Gene Expression in Cortical Organoids), which allows the concomitant examination of gene trajectories in hCS and in BrainSpan (https://labs.dgsom.ucla.edu/geschwind/files/view/html/GECO.html).
Discussion
We performed multiple independent analyses of functional genomic features to comprehensively assess to what extent can in vitro hCS recapitulate in vivo cortical maturation. We observe substantial convergence across these different data types, which indicates attainment of early postnatal maturation between 250–300 days in vitro. To our knowledge, this is the first multi-level demonstration that an in vitro model of human neural development has matured sufficiently to acquire some postnatal features. This includes several important features of RNA editing and a major physiological transition involving the switch in the ratio of NMDA receptor subunits that occurs postnatally in mammals28. Our results suggest that hCS mature both at the level of individual cells, as evident by the NMDA receptor isoform switch, as well as some aspects of cellular composition, as shown by the emergence of upper layer neurons and astrocytes at later stages. It is important to note that some changes in gene expression are likely due to changes in cell proportions.
We also show that genes associated with neurodevelopmental and neurodegenerative disorders are not monolithic in their expression, but fall into distinct development trajectories. These trajectories, include those peaking both before 100 days and after 250 days (e.g., ASD-C2 and ID-C1), which represent histone modification in progenitor cells; those peaking between 100–150 days (e.g., ASD-C3, ID-C4 and SCZ-C2), which represent synaptic structure and function in neuronal cell types; and those with late expression trajectories, which are related to astrocyte biology (e.g., ASD-C5, ID-C3, and SCZ-C1). This timing should be considered when establishing in vitro models of disease. For example, mutations in the astrocyte-related gene HEPACAM, which is part of ASD-C5 cluster (Figure 5a), should be studied at later stages of differentiation (> 250 days), while the consequences of mutations in the neuronal transcription factor MYT1L, which is part of ASD-C3 (Figure 5a), can likely be probed in early stages of neural differentiation.
The majority of these cluster trajectories follow in vivo trajectories. One exception is the neuronal activity dependent clusters (i.e., ASD-C3, SCZ-C2 and epilepsy-C3), which do not show strong fidelity at late stages of differentiation (>400 days) likely due to the lack of extensive network activity in hCS. We provide the GECO tool to allow detailed comparison between in vivo and hCS trajectories. We note that the gene trajectories presented in this web tool were derived from a directed hCS differentiation protocol and other differentiation protocols and cell lineages from other brain regions may exhibit a different timeline of maturation.
One caveat of this study is that culturing hCS for long periods of time is not trivial, leading us to collect samples as they became available. This is the reason three lines do not have samples from the entire time course. It is also important to note that, after 400 days of differentiation, we observed an increase in the variability of the hCS differentiation (Figure 1c). Reducing this variability will be essential for modeling disease at these very late stages of differentiation, as will development of methods to accelerate this process.
Previous studies in brain organoids have suggested that glycolysis and ER stress are highly upregulated in these systems, reflecting a state of cell stress19, 20. If this were the case, we would expect to see these pathways increasing over time, reflective of progressive cellular stress in vitro, since cellular stress is not a homeostatic state33. However, our analysis shows that the trajectories of these pathways remain flat during differentiation up to 21 months in vitro. Moreover, we were able to detect robust levels of many ER stress and glycolysis genes in vivo in the BrainSpan dataset. Our ability to detect more robust in vivo expression of genes involved in these stress pathways may be due to higher levels of gene detection in bulk RNA sequencing compared to the single cell RNA sequencing data used previously19, 34. However, we note that we were also able to detect them at similar levels in another in vivo single cell data set from fetal cortex21. Thus, our interpretation of these data is that the slightly higher, but relatively constant elevation of these genes involved in glycolysis in vitro likely reflects the different, but homeostatic, metabolic state with respect to glucose utilization that has been observed across in vitro cell culture systems35, 36.
Our findings also support the interpretation that key features of human corticogenesis are be guided by an internal differentiation clock. This is consistent with findings in mouse showing that major features of the progression of cortical neurogenesis are governed by intrinsic factors and do not require extrinsic signals from other brain regions, observations that warrant further study and refinement37, 38. Although reaching later stages of development in vitro is currently time consuming (> 250 days), it nevertheless extends the value of hiPSC-derived in vitro 3D cultures by providing a platform to study the processes occurring during late fetal and early post-natal stages of brain development. The presence of continual time-dependent aging in hCS also indicates that these cellular models could be used to model epigenetic aspects of aging, which has been shown to capture key biological features associated with aging biology39.
It is important to note that while our model system was able to capture key features of in vivo human corticogenesis, some important aspects still require further investigation. Alternative splicing plays an important role in neural development40 however detecting significant alternative splicing will require a more deeply sequenced and extensive set of in vivo and in vitro samples. Another aspect requiring further investigation is on the role of cells not born in the dorsal forebrain on the maturation of hCS. For example, ventral forebrain-derived GABAergic neurons promote synaptic maturation and network maturation31. These effects could be studied in forebrain assembloids, in which we have previously shown that GABAergic interneurons migrate and functionally integrate into the cortical network22. It will be important to assess how network dynamics mature in the presence of GABAergic neurons to establish the excitation-inhibition (E/I) balance, which has been linked to neurodevelopmental disorders41. Microglia, which are mesoderm derived, also play an important role in the developmental maturation of the cerebral cortex42,, and previous studies have shown successful integration of microglia-like cells into human brain organoids43. Lastly, strategies to derive oligodendrocytes23 or vascular-forming endothelial cells44 in assembloids will also be useful to investigate how they modulate developmental trajectories in long term hCS cultures.
Additionally, while we identified preservation of some aspects of RNA editing in hCS this process was not complete. For instance, ADRAB2 had a far less dynamic pattern in hCS than in vivo. Moreover, while the prenatal expression trajectories of FXR1 and FMR1 in hCS were similar to those seen in vivo, hCS at later stages did not track the in vivo trajectories. We speculate that this could be related to activity-dependent processes25, 45. For example, RNA editing of the AMPA receptor GluA2 (GRIA2 gene) is highly dependent on neuronal activity45. Our results suggest that alterations in the balance of the RNA editing process may be used as a measure for optimizing and enhancing the functional similarities between these 3D in vitro models and in vivo brain development. Another important challenge remains to find ways to enhance this maturation speed to further facilitate more efficient in vitro modeling, including features that may be dependent on certain forms of neuronal activity and aging. This is especially true for modeling neurodegenerative disorders. Our approach provides a framework for comprehensive analysis of such features.
Methods
Culture of hiPSC and differentiation into hCS.
All hiPSC lines used in this study were validated using previously described standardized methods5, 22, 46. Cultures were maintained mycoplasma free and were periodically tested for mycoplasma contamination. A total of 6 hiPSC lines were collected from 5 healthy subjects (4 male and 1 female). The hiPSC H20961 line was derived by the Gilad Laboratory (University of Chicago). Approval for this study was obtained from the Stanford IRB panel, and informed consent was obtained from all subjects. hiPSC were cultured on inactivated mouse embryonic fibroblast feeders (EmbryoMax PMEF; Millipore) in DMEM/F12 (1:1, Life Technologies, 11330) containing 20% knockout serum (Life Technologies, 10828), 1 mM non-essential amino acids (Life Technologies, 11140), 1:200 GlutaMax (Life Technologies, 35050), 0.1 mM β-mercaptoethanol (Sigma-Aldrich, M3148), and 10 ng/ml FGF2 (R&D Systems, 233-FB) diluted at 0.1% BSA in DPBS (Life Technologies, 14190).
hCS were generated as previously described5. Intact hiPSC cells colonies were lifted using 0.7 mg/ml dispase and transferred to ultra-low-attachment plastic dishes (Corning) in the same hiPSC medium without FGF2 but supplemented with 5 μM dorsomorphin (Sigma-Aldrich), 10 μM SB-431542 (Tocris) both of which are SMAD inhibitors, and 10 μM Y-27632 (EMD Chemicals) which is a ROCK inhibitor. From day 2 (48 hours of differentiation), the medium supplemented with dorsomorphin and SB-431542 was changed daily. From day six until day 24, neural spheroids were grown in neurobasal-A (Life Technologies, 10888) neural medium supplemented with B-27 supplement without vitamin A (Life Technologies, 12587), 1:100 GlutaMax (Life Technologies), 1:100 penicillin and streptomycin (Life Technologies, 15070) and with 20 ng/ml EGF (R&D Systems, 236-EG) and 20 ng/ml FGF2 (R&D Systems, 233-FB). From day 25 to 42, the neural medium was supplemented with 20 ng/ml BDNF (Peprotech, 450–02) and 20 ng/ml NT3 (Peprotech, 450–03) and medium was changed every other day. From day 43 onwards, hCS were maintained in unsupplemented neural medium with medium changes every four days. hCS of similar diameter were randomly selected for experiments.
RNA-sequencing.
RNA-sequencing was performed as previously described11. Briefly, libraries were prepared using Truseq stranded RNA RiboZero Gold (Illumina) and were sequenced using 100-bp paired end reads on an Illumina HiSeq 4000. Reads were then mapped to hg38 with Gencode v25 annotations using STAR (v2.5.2b)47. Gene expression levels were quantified using RSEM (v1.3.0)48. Genes with low levels of expression (less than 10 reads in more than 20% of the samples) were removed from the analysis. Outliers were then removed using standardized sample network connectivity (Z scores smaller than −3)49. This method identified two samples as outliers, both of which were > 600 days. To quantify the technical variation in the RNA sequencing we calculated the first five principal components of the Picard sequencing metrics (http://broadinstitute.github.io/picard/; v2.5.0). These principal components referred to as seqPC1-seqPC5 were then included in the linear model.
To help control for variability between the individuals racial background we used the GATK (v3.3) haplotype caller to call SNPs from the aligned reads50. We filtered for sites with missing genotypes (> 5%), rare minor allele frequency (< 0.05) and out of Hardy–Weinberg equilibrium (< 1e−6)51. Genetic ancestry was inferred by running multi-dimensional scaling (MDS) on these high-quality SNPs together with HapMap3.3 (hg38). The first two MDS values referred to as ancestryPC1/2 were then included in our linear model. For principal component analysis (PCA), as well as to visualize single gene trajectories, gene expression was normalized using CQN (without quantile normalization, sqn=FALSE) (v1.28.0) and ancestryPC1-2 and SeqPC1-5 were regressed out prior to batch correction using Combat52 from the sva package (v3.30.0) in R. Single gene trajectories trends lines were fitted using the loess method53 from the ggplot2 package54 in R. PCA was calculated using the prcomp function in R on scaled normalized and batch corrected counts.
BrainSpan RNA sequencing data analysis.
The BrainSpan RNA sequencing data16 was used as an in vivo refence for the analysis. To quantify the gene expression at each developmental stage the cortical samples were aligned to hg38 using Gencode v25 annotations via STAR47. Gene expression we then quantified using the union exon model in featureCounts55. We removed low quality samples in which the RIN was lower than 8, there were less than 25% coding bases or ribosomal bases made up more than 25% of total bases (as called by Picard tools). Genes with low levels of expression (less than 10 mapped reads in more than 80% of the samples) in a given developmental stage were removed. We retained 196 samples from 24 individuals (9 female and 15 male).
Transition mapping.
To compare the in vivo and in vitro changes in gene expression during maturation we used transition mapping15 which utilizes a rank-rank hyper geometric test56. To this end both in vivo and in vitro gene expression levels were normalized using the trimmed mean of M-values (TMM) method from the edgeR package57 (v3.24.0). In vitro samples were grouped to the closest day 25th day in the first 100 days, closest 50th day in between days 100 and 400, closest 100 day until day 600 and timepoints above day 600 were included in the 600 day group resulting in the following groups 25, 50, 75, 100, 150, 200, 250, 300, 350, 400, 450, 500, 600. Fold change was calculated for each differentiation day (in vitro) or developmental stage (in vivo) by comparing it to the baseline values of the earliest day in vitro (day 25) or earliest stage in vivo (stage 2; 8–10 post conception weeks, PCW16) using the limma-voom method58 from the limma package (v3.38.2) in R. To account for multiple samples coming from the same individual we used brainID (for the BrainSpan data) or IndividualID (for the hCS data) as blocking factors in the model. The linear model used was ~ 0 + Differentiation day + batch + PC1 + racePC2 + SeqPC1 + SeqPC2 + SeqPC3 + SeqPC4 + SeqPC5 for the hCS data and ~ 0 + Period + Ethnicity + PMI + SeqPC1 + SeqPC2 for the brain span data. Gene were then ranked by logFC and the rank-rank hypergeometric test56 was used to calculate the significance of the overlap of the gene list using a step size of 200 genes15
DNA methylation age.
To calculate the DNA methylation (DNAm) age of the samples12 DNA was extracted using the AllPrep DNA/RNA/miRNA Universal Kit (Qigen, 80224). Methylation levels were measured using the Infinium MethylationEPIC BeadChip Kit (Illumina), normalized using the Noob method59 and were then used to calculate DNAmage12. DNAmage was averaged over technical replications. We only evaluated clocks that were designed for non-blood tissues, i.e. the pan tissue clock12 and the in vitro clock14. We were unable to predict the culture age using this methylation age, as these epigenetic clocks are not calibrated for the array type13 and those using these arrays do not perform well in brain tissue14. Results were similar between the two methods and we present the results of the pan tissue clock.
Gene set enrichment analysis (GSEA).
Gene set enrichment analysis was performed using the fgsea package (v1.8.0)60 on all genes ranked by log fold-change (using limma-voom as described above) at different time points. GO gene sets (v7.0) were downloaded from http://software.broadinstitute.org/gsea/msigdb/. Sets with less than 30 or more than 500 genes were omitted. p-values were calculated using 1,000,000 permutations and were corrected using the BH method. Gene sets with FDR < 0.05 were considered to be significant and the top 3 up and down regulated sets were plotted.
Weighted Gene Network Analysis.
To compare trajectories of genes networks between in vivo an in vitro data sets, previously described in vivo network modules were used15. To assure that the same networks were present in vivo weighted gene network analysis (WGCNA) was performed on the in vitro data using a soft power of 12, minimal module size = 100, deep split = 2, cut height for creation of modules = 0.9999 and cut height for merging modules of 0.1. The modules were then tested for overlap with the in vivo modules using Fisher’s exact test61. To visualize the trajectories of the different modules, which overlapped with the in vitro module, the normalized average expression was calculated using the moduleEigengenes function from the WGCNA package62 (v1.68) in R. The trend line was fitted using the loess method53 from the ggplot2 package54 in R.
Human tissue.
Human brain tissue was obtained under a protocol approved by the Research Compliance Office at Stanford University. PCW21 forebrain tissue was fixed immediately upon arrival.
Immunohistochemistry.
Immunohistochemistry was performed as described22, 63. Briefly, hCS were fixed in 4% paraformaldehyde (PFA) for 2 hours at 4°C. Samples were then washed with PBS three times, transferred to a 30% sucrose solution and 48–72 hours later, embedded and snap frozen in a 30% sucrose and OCT solution (1:1 ratio; Tissue-Tek OCT Compound, 4583, Sakura Finetek). 16μm cryosections were obtained using a cryostat (Leica). Human cortical tissue was fixed overnight in 4% paraformaldehyde and 30 μm cryosections were made. All sections were incubated for 1 hour at room temperature with blocking solution (10% normal donkey serum and 0.3% Triton-X in PBS), and then overnight with primary antibodies. The following primary antibodies were used: anti-BRN2 (Mouse, 1:500, Millipore, MABD51), anti-CTIP2 (Rat, 1:300, Abcam, ab18465), anti-FXR1 (Mouse, 1:50; Santa Cruz, sc-374148), anti-GFAP (Rabbit, 1:1,000, Dako, Z0334), anti-GFAP (Rat, 1:1,000, Thermo Fisher Scientific, 13–0300), anti-HDAC2 (Mouse, 1:50, Santa Cruz, sc-9959), anti-MAP2 (1:5,000, Synaptic Systems, 188004), and anti-SOX9 (Goat, 1:500, R&D Systems, AF3075). After three PBS washes, sections were incubated with Alexa Fluor secondary antibodies (1:1,000, Life Technologies) for 1 hour at room temperature. Nuclei were visualized with Hoechst 33258 (Thermo Fisher Scientific, H3569). Glass coverslips were mounted on microscopy slides using Aquamount (Thermo Scientific). Images were taken using a SP8 confocal microscope and processed using ImageJ (Fiji).
Western blotting.
hCS protein lysates were prepared using RIPA buffer system (Santa Cruz, sc-24948) and protein concentrations were quantified using the Bicinchoninic Acid (BCA) assay (Pierce, ThermoFisher 23225). 8 μg of protein per sample per lane were loaded and run on a 4–12% Bis-Tris PAGE gel (NuPAGE 4–12% Bis-Tris Protein Gel, Invitrogen) and transferred onto a PVDF membrane (Immobulin-FL, EMD Millipore). Membranes were blocked with 5% milk in PBST for 1 hour at room temperature (RT) and incubated with primary antibodies against β-actin (mouse, 1:50,000, Sigma, A5316), synapsin-1 (rabbit, 1:1000, Cell Signaling, 5297S) overnight at 4°C and antibodies against the NMDA receptor 2A (GRIN2A, rabbit, 1:1000, Cell Signaling, 4205S), NMDA receptor 2B (GRIN2B, rabbit, 1:1000, Cell Signaling, 4207S)) for 72 hours at 4°C. Membranes were washed 3 times with PBST and then incubated with near-infrared fluorophore-conjugated species-specific secondary antibodies: Goat Anti-Mouse IgG Polyclonal Antibody (IRDye 680RD, 1:10,000, LI-COR Biosciences, 926–68070) or Goat Anti-Rabbit IgG Polyclonal Antibody (IRDye 800CW, 1:10,000, LI-COR Biosciences, 926–32211) for 1 hour at RT. Following secondary antibody application, membranes were washed 3 times with PBST, once with PBS, and then imaged using a LI-COR Odyssey CLx imaging system (LI-COR). Protein band intensities were quantified using Image Studio Lite (LI-COR) with built-in background correction and normalization to β-actin controls. One sample showed unexpectedly low levels of synapsin-1 and was not included in the analyses (Source Data 1).
Measurements of NMDA currents.
hCS were prepared for whole cell recordings as previously described5. Briefly, hCS were infected with AAV-hSyn1::GFP two weeks before recording. Slices (200 μm thick) were prepared using a Leica VT1200 microtome (Leica) and allowed to recover for one hour before recordings at 32°C in bicarbonate buffered artificial cerebrospinal fluid (aCSF) containing 126 mM NaCl (Sigma, 59222C), 2.5 mM KCl (Sigma, P3911), 1.25 mM NaH2PO4 (Fisher Scientific/Acros Organics, AC424390025), 2 mM MgCl2 (Sigma, M8266), 2 mM CaCl2 (Sigma, C4901), 26 mM NaHCO3 (Sigma, S5761) and 10 mM glucose (Fisher Scientific/Acros Organics, AC410950010). Whole cell patch clamp recordings were performed at room temperature (~22–25°C). Slices were superfused with aCSF at a rate of 3 ml/min. Whole cell patch clamp recordings were collected using a MultiClamp 700A amplifier (Molecular Devices), Axon Digidata 1550B digitizer (Molecular Devices), and Clampex 11.0 software (Molecular Devices). Borosilicate glass pipettes (3–5 MΩ) were used to obtain intracellular recordings. Fluorescently labeled neurons were randomly selected for patching. Pipettes were filled with internal solution containing 120 mM potassium gluconate, 11 mM KCl, 1 mM MgCl2, 1mM CaCl2, 10 mM HEPES (Sigma, H4034), 1 mM EGTA (Sigma, E4378), and the pH was adjusted to 7.4. NMDA (Sigma, M3262) was applied to the cell using a 2–3 MΩ borosilicate glass pipette positioned 25 μm away from the cell body. Pulses (50 ms) of NMDA were produced using a Picospritzer II (General Valve Corporation). Cell recording quality was monitored by measuring access resistance and cells which deviated by more than 15% during the course of the recording were discarded. NR2B containing NMDA receptors were blocked by adding ifenprodil tartrate salt (IFN; 10 μM; Sigma, I2892) to aCSF. hCS derived from two cell lines (8858–3 and 1205–4) were used for these recordings. One cell was patched per hCS, for a total of 25 neurons from 25 hCS (Supplementary Table 1). Association between the proportion of change in amplitude after adding IFN was tested using beta regression64 with a logit link function using the betareg package (v3.1–3) in R.
RNA editing identification.
RNA-seq reads were mapped to GRCh37 genome and transcriptome using HISAT265 (v2.1.0) with parameters accounting for the respective strand specificities of the BrainSpan and cortical spheroid datasets. Uniquely mapped reads were retained for further analysis. We then used previously developed procedures to identify RNA editing sites26, 66, 67. In brief, first we used unmapped reads to find editing sites in hyper-edited regions68. Adenosines in unmapped reads were converted into guanosines and aligned with HISAT2 to a modified hg19 genome where adenosines were also substituted with guanosines26. These hyper-edited reads were then combined with the original uniquely mapped reads. Next, candidate editing sites were identified as mismatches between reads and the reference genome. A log-likelihood test and posterior filters were then applied to eliminate editing sites likely caused by sequencing errors and other technical artifacts67. To eliminate rare genomic variants from identified RNA editing sites, we filtered for sites found across multiple individuals. Specifically, editing sites were required to be found with at least 5 total reads and 2 reads edited across 5 unique individuals. In our cortical spheroid dataset, we reduced the requirement to 4 unique individuals, due to sample size. A total of 109,487 and 19,046 editing sites were identified in the BrainSpan and cortical spheroid datasets respectively.
Weighted co-editing network analysis for RNA editing sites.
Modules of RNA editing sites were found using the WGCNA (weighted gene co-expression network analysis) package62. To obtain accurate representation of topological overlap, we first filtered for editing sites with ≥5 total reads in at least 80% of samples, zero variance or too much missing data using the goodSamplesGenes function in the WGCNA package. To facilitate finding modules corresponding to developmental time in the BrainSpan dataset we required nonzero editing in at least 51% of samples from at least one period. Given the limited sample size of our cortical spheroid dataset, this filter was only applied to the BrainSpan samples.
For the cortical spheroids we adjusted RNA editing levels to avoid individual modules driven by single individuals. For each editing site a linear model was constructed between editing level against differential day, individual, and batch. Editing levels were adjusted by subtracting out the maximum likelihood beta estimates for individuals and batch. A soft threshold power of 10 was used to fit scale-free topology. To preclude modules driven by outlier samples, we followed our previous bootstrapping strategy26, 69, 70, where modules were obtained using consensus topological overlap from 100 bootstraps. In brief, for each bootstrap, samples up to the original sample size were randomly re-sampled with replacement. Signed topological overlap matrices were obtained from the corresponding matrix of editing sites using TOMsimilarity (adjacency(corFnc = “cor”, type = “signed”, power = 10, corOptions = list(method = ‘spearman’, use = “pairwise.complete.obs”), TOMType = “signed”). The boot-strapped matrices were then recalibrated from 20,000 random matrix entries. The consensus topological overlap matrix was taken as the median across all recalibrated boot-strapped matrices.
To find associations of modules with various biological processes, we defined the eigenvalue as the first principal component of each module. The time course trajectory of each module was determined by plotting each eigenvalue against differentiation day (for cortical spheroids) and period (for BrainSpan). Associations between the modules and RNA editing enzymes were evaluated by correlating the eigenvalue against adjusted gene expression values for established RNA editing enzymes encoded by ADAR1, ADAR2, and ADAR371, and against FMR1 and FXR1, that encode for proteins which were recently shown to also exhibit RNA editing regulation26. Modules found in BrainSpan samples and cortical spheroids were tested for significant overlap of member editing sites using Fisher’s Exact test. Only editing sites found in both BrainSpan and cortical spheroids were considered for this test.
Enhanced crosslinking and immunoprecipitation (eCLIP) analysis of RNA editing sites
To investigate whether RNA editing in modules are targetable by FMRP and FXR1P proteins, we obtained eCLIP datasets of FMRP and FXR1P binding sites in postmortem human frontal cortex26. Comparison of distances between eCLIP peaks and module editing sites was also performed using published methods26. Briefly, for member editing sites within each BrainSpan or hCS module, the closest distances from eCLIP peaks compared to the null background consisting of distances between peaks and gene-matched random adenosines were compared over 10,000 sets of controls. P-values were obtained by calculating the area under curve (AUC) of the cumulative distribution of distances of editing sites to CLIP peaks in the interval 0–100,00kb. AUC values of the 10,000 sets of controls were modeled by a Gaussian distribution, which was then used to calculate a one-sided P-value for the AUC of the module editing sites. To test overlap of FMRP and FXR1P targeted editing sites between hCS and BS modules, an editing was labelled “targetable” if within 1000 bp of the nearest FMRP or FXR1P CLIP peak. Editing sites residing in genes expressed with RPKM < 5 in adult frontal cortex were not coverable by CLIP analysis and excluded from analysis. The significance of overlap of targetable editing sites between hCS and BS modules was evaluated using Fisher’s Exact Test. Only editing sites found in both BrainSpan and hCS were considered for this test.
Mapping of disease genes.
Genes associated with autism spectrum disorder (ASD; https://gene.sfari.org/database/gene-scoring/), intellectual disability (ID)72, epilepsy21, schizophrenia (SCZ)73, Alzheimer’s disease (AD)74, 75, Parkinson’s disease76, 77 (PD) and Progressive Supranuclear Palsy (PSP)78 and Frontotemporal Dementia (FTD)79 were analyzed. For the ASD genes only high confidence genes (gene score < 2 or syndromic genes) were analyzed. For AD and PD, we combined common variants from the genome wide association studies (GWAS) with genes associated with rare, familial forms of these diseases. For AD these genes are: APOE, APP, PSEN1 and PSEN275. For PD these genes are PINK1, SNCA, LRRK2, PRKN, UCHL1, and PARK777. As there are only a small number of genes associated with the neurodegenerative diseases PSP (9 genes) and FTD (13 genes) and they are considered part of a frontal lobar degeneration spectrum, these were combined. Genes were clustered by their expression in the hCS using hierarchal clustering on the Euclidean distance between the genes. Cluster eigengenes were calculated using the moduleEigengenes function from the WGCNA package. The gene in each cluster were correlated to the cluster module eigengene and the top 5 genes were annotated on the heatmap. GO terms enrichment was performed using the enrichGO function from the clusterProfiler package80 (v3.12.0). Enrichment was performed on biological process and molecular function GO terms. All genes expressed in the hCS were used as background. Cell type enrichment was performed using the bootstrap.enrichment.test from the EWCE package81 (v0.99.2) on hCS single cell data11 with 100,000 permutations. All genes expressed in both the current dataset and the single cell dataset were used as background. This method tests whether a list of genes has a higher level of expression in a specific cell type that would be expected by chance.
Statistics
Statistical analyses, including Fisher’s exact test, beta regression, Spearman correlation rank-rank hyper-geometric tests, were performed as detailed in legends and methods. As these tests do not depend on the distribution being normal, no test for normality was performed. No statistical methods were used to pre-determine sample sizes, but our sample sizes per time point are similar to those reported in previous publications5, 6, 10. Due to the nature of these long-term cultures, collection of samples was not performed blind to the differentiation stage.
Reporting summary
Further information on research design is available in the Life Science Reporting Summary linked to this article.
Data availability.
Gene expression data and methylation data are available in the Gene Expression Omnibus (GEO) under accession numbers GSE150122 and GSE150123. The accompanying GECO webtool can be accessed at https://labs.dgsom.ucla.edu/geschwind/files/view/html/GECO.html. The BrainSpan data are available in the database of Genotypes and Phenotypes (dbGaP) under Study Accession phs000755.v2.p1. Single cell data from human fetal cerebral cortex can be found at http://geschwindlab.dgsom.ucla.edu/pages/codexviewer and at dbGaP under Study accession phs001836. eCLIP data for FXR1 and FMR1are available in GEO with accession number GSE107895. Human cortical organoid single cell sequencing data are available in GEO with accession number GSE107771.
Code availability
The code and data used in this manuscript can be found at https://github.com/dhglab/human_cortical_organoid_maturation.
Extended Data
Extended Data Fig. 1. Data description and quality.
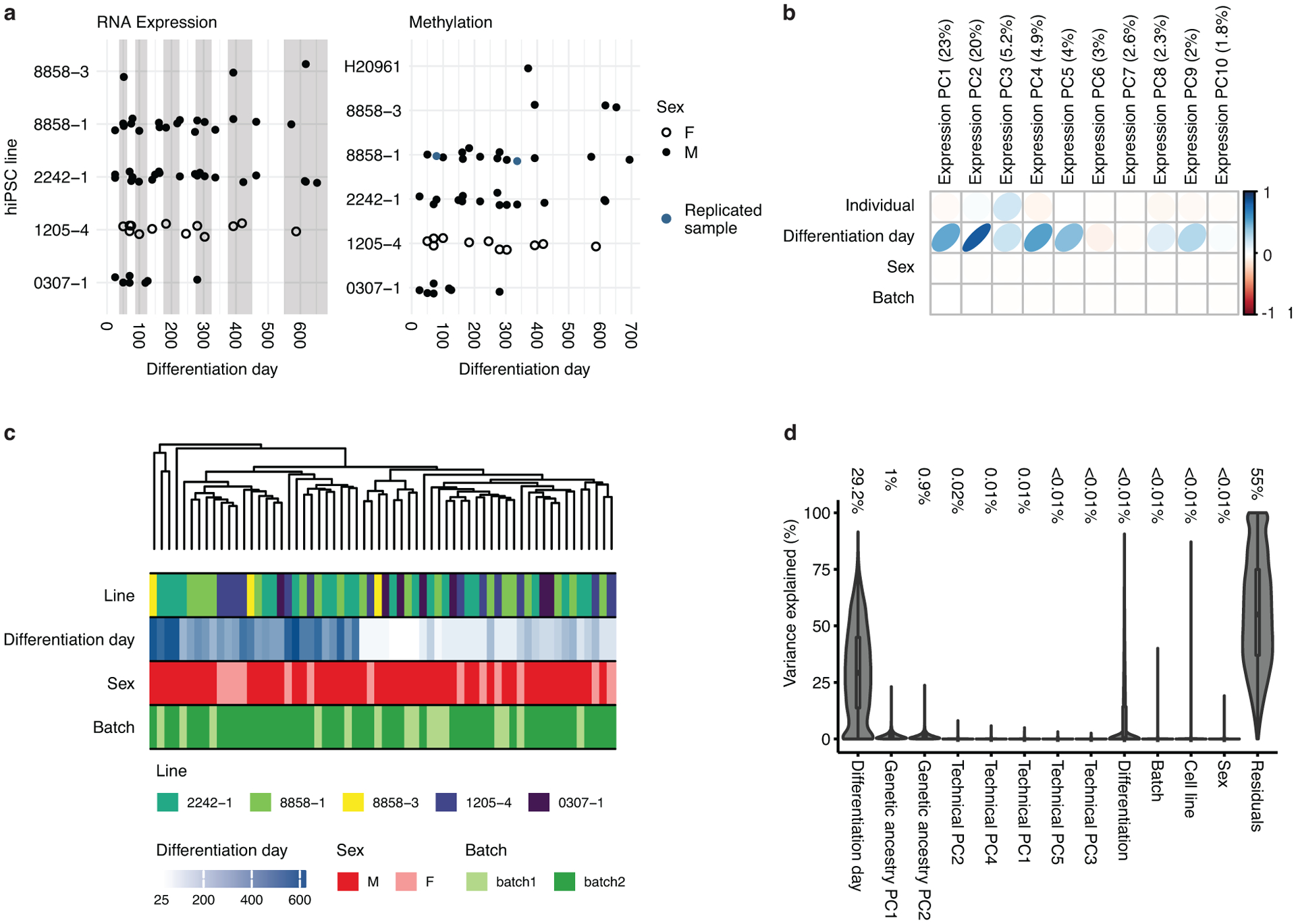
(a) Time points and hiPSC line information for the 62 samples used for RNA sequencing (left). Samples were differentiated from 5 cell lines derived from 4 individuals. Time points and hiPSC information for the 50 samples used for DNA methylation (right). Samples were differentiated from 6 cell lines derived from 5 individuals (see Supplementary Tables 1 and 2). Two samples (blue) were hybridized in replicate for quality control purposes and their values were averaged. Each point represents one sample from a specific cell line (y-axis) and differentiation day (x-axis). Full circles represent sample coming from males and rings represent samples coming from females. Grey and white background shading show aggregation of differentiation days into stages. (b) Principal component analysis (PCA) of the expression data. The values represent the adjusted r squared of the PC with the covariates indicated. The numbers in brackets on axis titles are the percent of variance explained by the PC. The first 5 PCs, which explain 57.1% of the total variance, show high association with differentiation day. (c) Dendrogram of hierarchical clustering of samples demonstrating that differentiation day but no other covariates (individual, Sex, batch) is driving the clustering of samples. (d) Violin plots of the variance explained by each of the covariates for each gene. Outlines represent the density of the percent of variance explained. The numbers are the median value of percent of explained variance for each variable. Boxplots in d show: center – median, lower hinge – 25% quantile, upper hinge – 75% quantile, lower whisker – smallest observation greater than or equal to lower hinge −1.5x interquartile range, upper whisker – largest observation less than or equal to upper hinge +1.5x interquartile range. n = 62 samples from 6 hiPSC lines derived from 5 individuals.
Extended Data Fig. 2. Cell stress in hCS.
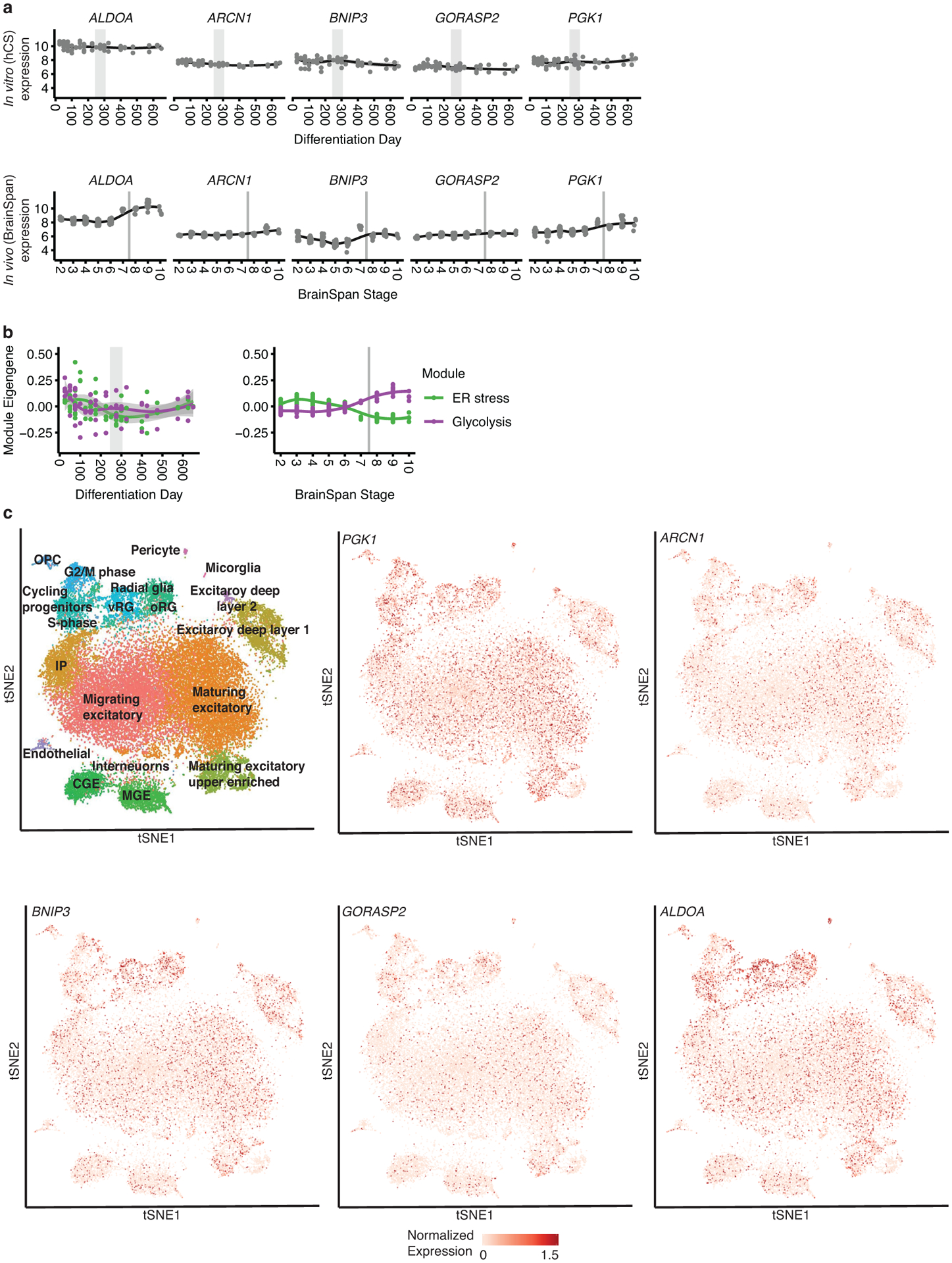
(a) Trajectories of metabolic cell stress genes20 hCS (left) and in vivo (right). (b) In vitro and in vivo module eigen genes of glycolysis (organoid.Sloan.human.ME.paleturquoise) and ER stress (organoid.human.ME.darkred) previously suggested to be upregulated in vitro20. Grey areas denote time of shift from prenatal to postnatal gene expression. In (a) and (b) shaded grey area around the trajectory represents the 95% confidence interval, vertical grey lines represent birth and vertical grey bars denote the shift from prenatal to postnatal gene expression based on matching to in vivo patterns. For in vitro data n = 62 samples from 6 hiPSC lines derived from 5 individuals and for in vivo data n = 196 from 24 individuals. (c) Scatterplot visualization of cells in in developing fetal cortex colored by major cell types22. vRG, ventral radial glia; oRG, outer radial glia; CGE, caudal ganglionic eminence; MGE, medial ganglionic eminence; OPC, oligodendrocyte precursor cell; IP, intermediate progenitors.
Extended Data Fig. 3. Changes in biological processes between early and later stages of differentiation.
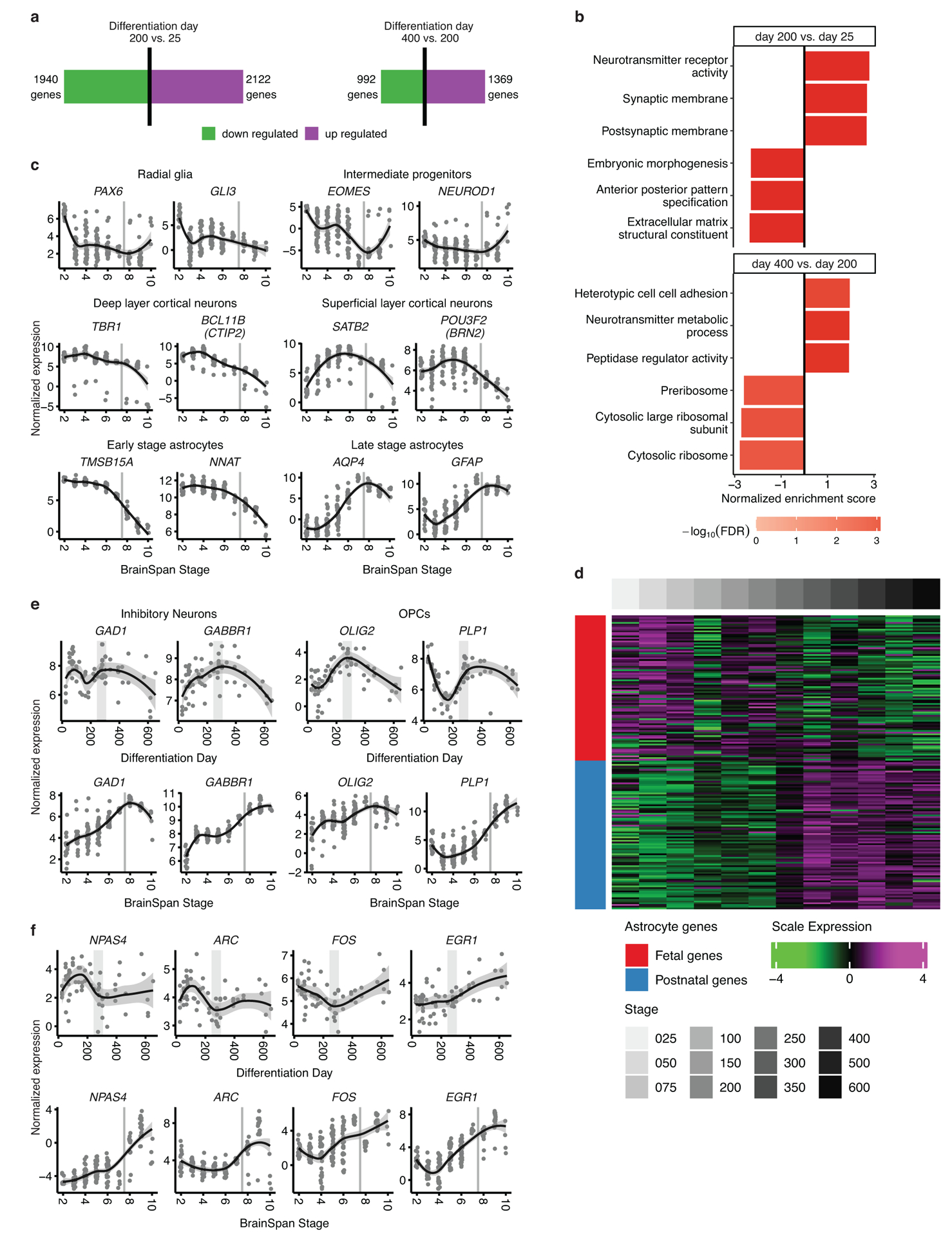
(a) Number of differentially expressed genes when comparing differentiation day 200 to differentiation day 25 (left) and differentiation day 400 to differentiation day 200 (right). Red bar represents up-regulated genes and the green bar represents down-regulated genes. (b) Top 3 up- and downregulated GO terms enriched in genes ranked by logFC using gene set enrichment analysis, (GSEA; FDR < 0.05). (c) Normalized expression of marker genes in vivo for neurons, intermediate progenitors, astrocytes, and radial glia as well as upper and deep layer cortical neurons. (d) Scaled expression of fetal and mature astroglial genes7 during differentiation. A shift between fetal and mature gene sets occurs at ~250 days of hCS differentiation. (e) Normalized expression of marker genes for inhibitory neurons and oligodendrocyte precursor cells (OPCs) that are not preserved in hCS. (f) Normalized expression of activity dependent genes that are not preserved in hCS. In (c), (e) and (f) shaded grey area around the trajectory represents the 95% confidence interval, vertical grey lines represent birth and vertical grey bars denote the shift from prenatal to postnatal gene expression based on matching to in vivo patterns. For in vitro data n = 62 samples from 6 hiPSC lines derived from 5 individuals and for in vivo data n = 196 from 24 individuals.
Extended Data Fig. 4. Overlap between hCS and in vivo WGCNA modules.
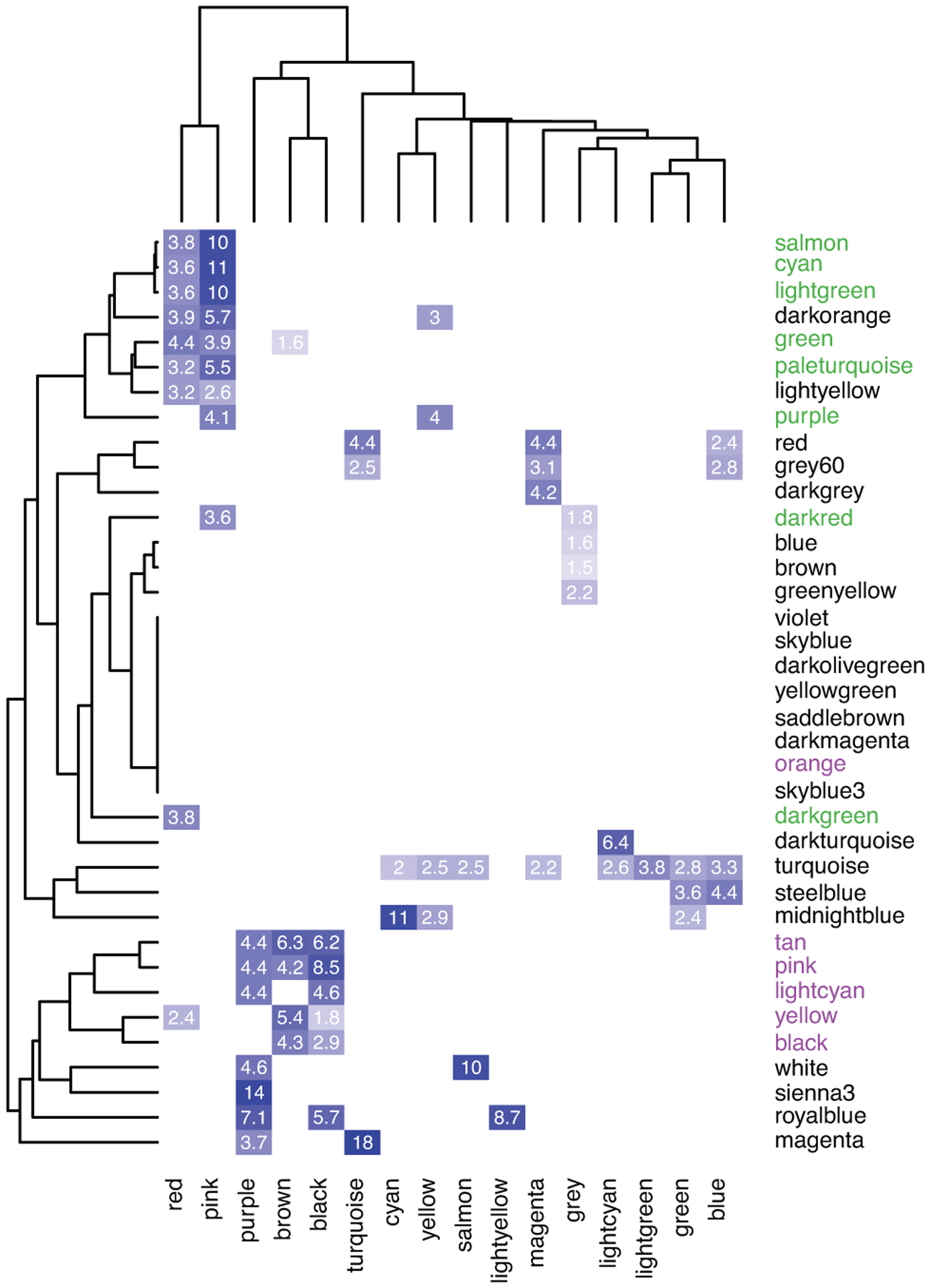
Overlap of genes in hCS and the BrainSpan in vivo modules. Significant ORs are presented. Modules were clustered using complete-linkage hierarchal clustering. Color represents the OR of each overlap. In vivo neuronal modules (green) and glial modules (purple) are marked.
Extended Data Fig. 5. Overlap between hCS and in vivo editing modules.
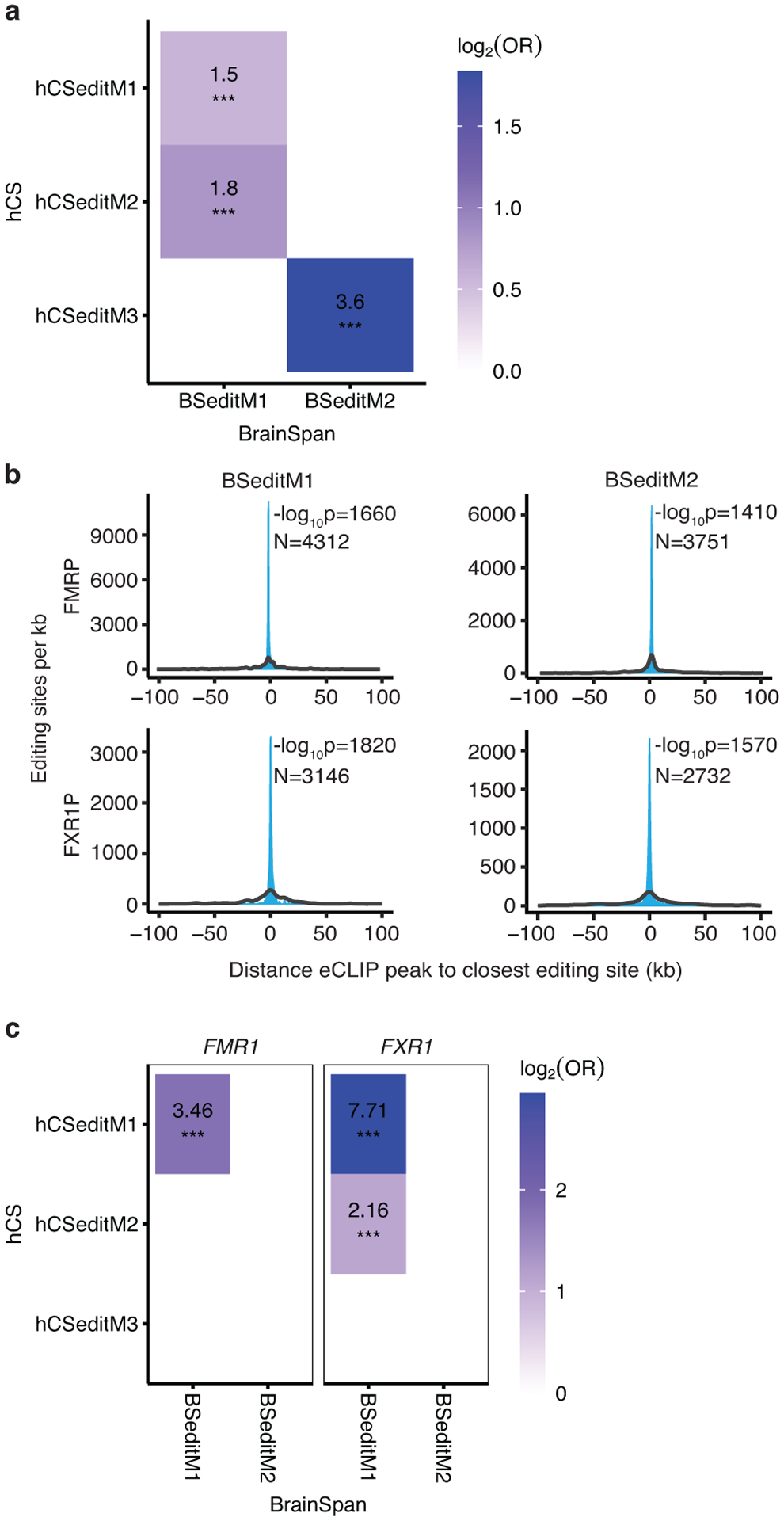
(a) Overlap of editing sites in hCS and BrainSpan in vivo modules. Significant ORs are presented. (b) Distributions showing the closest distances between editing sites from BrainSpan editing modules and FMRP or FXR1P eCLIP peaks (blue). The median of 10,000 sets of control sites (black) is depicted for comparison. See Methods for details of P-value calculation. N, number of editing sites shown. (c) Overlap of editing sites within 1000bp of a CLIP site in hCS and BrainSpan in vivo modules. Significant ORs are presented. *** FDR < 0.005
Extended Data Fig. 6. Expression of select genes in the in-vivo fetal cortex.
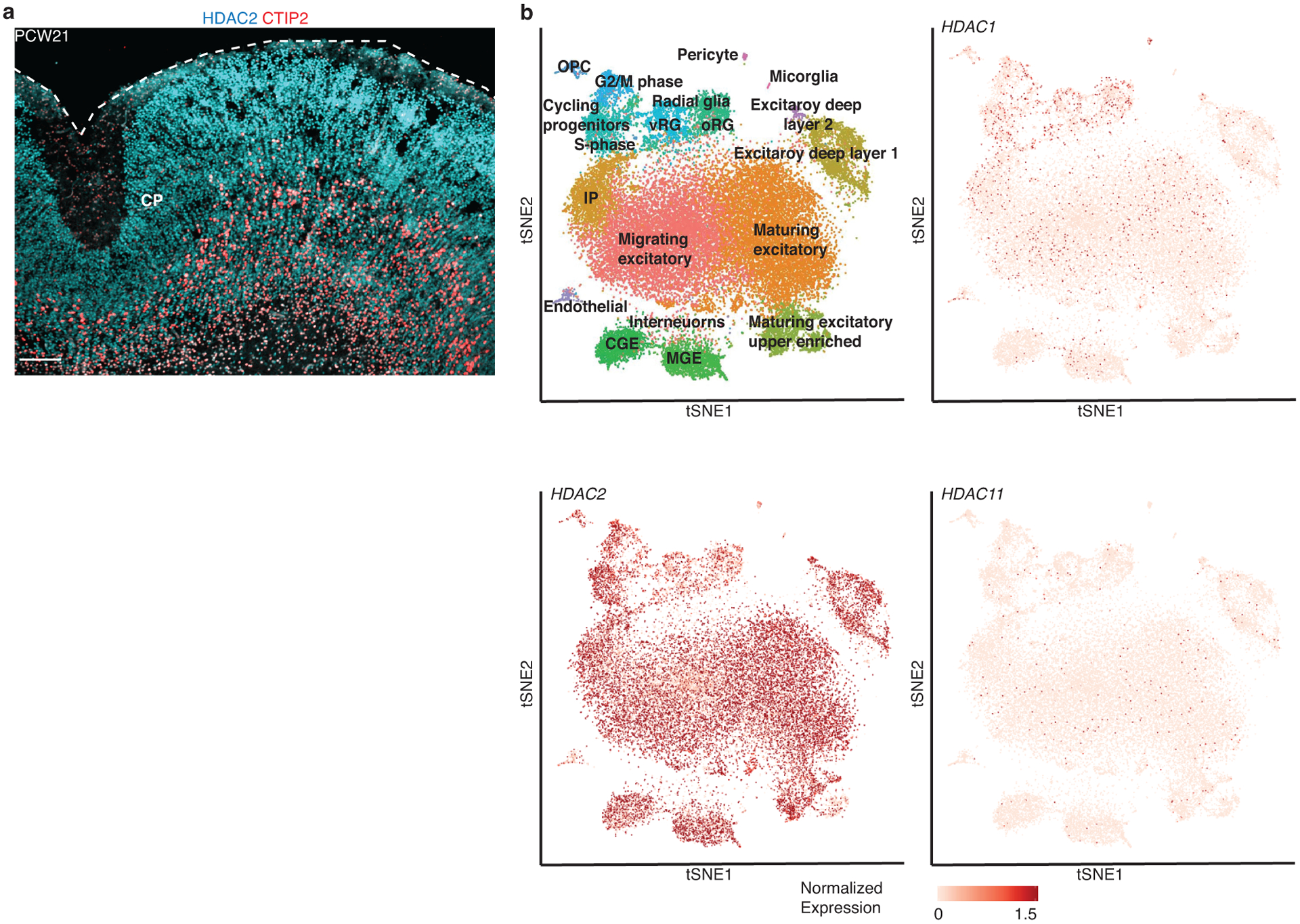
(a) Immunohistochemistry of HDAC2 and the deep layer marker CTIP2 (BCL11B) at post conception week 21 (PCW21). CP, cortical plate. Scale bars, 100 μm. The Immunohistochemistry experiment was performed once. (b) Scatterplot visualization of cells in developing fetal human cerebral cortex colored by major cell types22. vRG, ventral radial glia; oRG, outer radial glia; CGE, caudal ganglionic eminence; MGE, medial ganglionic eminence; OPC, oligodendrocyte precursor cell, IP, Intermediate progenitors.
Extended Data Fig. 7. Mapping neurodegenerative and epilepsy disorder genes onto hCS differentiation.
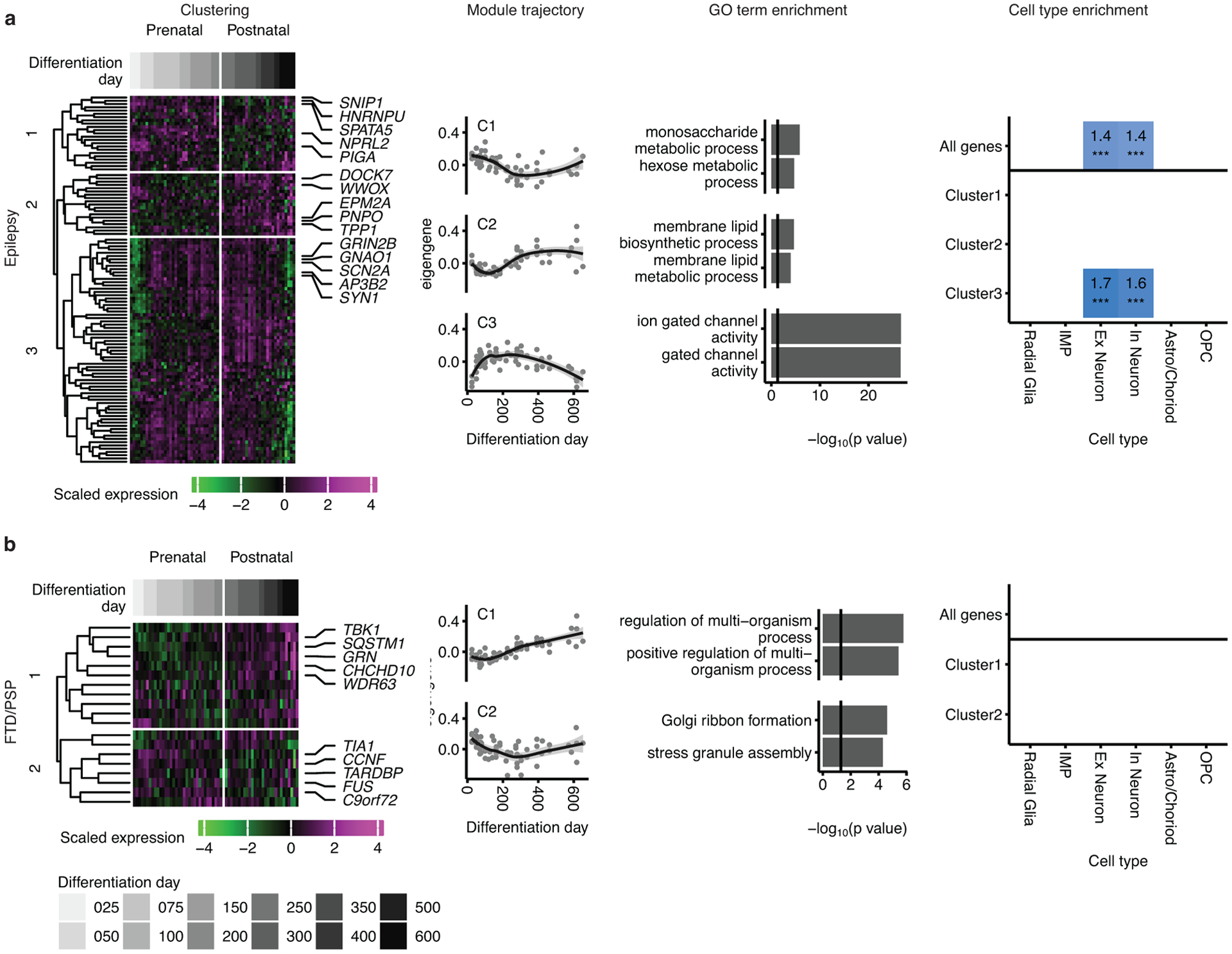
Mapping of genes associated with progressive supranuclear palsy (PSP) and frontotemporal dementia (FTD) (a), and epilepsy (b) onto hCS differentiation trajectories. The first column shows clustering of scaled normalized expression of genes associated with a disorder. Genes (in rows) are clustered using hierarchical clustering on the Euclidean distance between genes. Samples (columns) are ordered by differentiation day (represented by gray bars) with the earliest days on the left and latest time points on the right. The 5 most representative genes (highest correlation with the cluster eigengene) are shown. The second column shows the cluster eigengenes (first principal component) for the identified gene clusters. Shaded grey area around the trajectory line represents the 95% confidence interval. The third column shows the top GO terms enriched in the identified clusters. The fourth column shows cell types over expressed in either all the genes associated with a disorder (above line) or in the genes from the identified clusters. Number and color represent the fold change. Significance was tested using a one-sided permutation test with 100,000 permutations. P values were corrected for multiple testing using the Benjamini-Hochberg method. * FDR < 0.05, ** FDR < 0.01, *** FDR < 0.005. n = 62 samples from 6 hiPSC lines derived from 5 individuals.
Supplementary Material
Acknowledgements
We would like to acknowledge experimental support from members of the Pasca Lab at Stanford University: A.M. Pasca, N. Huber, T. Khan, F. Birey, A. Puno, L. Li, and T. Li., and members of the Pasca lab and Geschwind lab for helpful discussions and support, including Andrew Elkins for setting up the GECO web browser accompanying the manuscript. This work was supported by California Institute of Regenerative Medicine (CIRM) and the National Institute of Mental Health Convergent Neuroscience Consortium (U01 MH115745) (to D.H.G. and S.P.P), the Stanford Human Brain Organogenesis Program in the Wu Tsai Neuroscience Institute (to S.P.P), Bio-X (to S.P.P.), the Wu Tsai Big Idea Grant (to S.P.P.), the Kwan Funds (to S.P.P), the Senkut Research Fund (to S.P. P.), the Autism Science Foundation (ASF) and the Brain and Behavior Research Foundation Young Investigator award (Brain & Behavior Research Foundation) (to A.G.). S.P.P. is a New York Stem Cell Foundation (NYSCF) Robertson Stem Cell Investigator and a Chan Zuckerberg Initiative (CZI) Ben Barres Investigator.
Footnotes
Accession Codes.
Data generated by this study are available at the Gene Expression Omnibus (GEO) with accession codes GSE150122 and GSE150123.
Competing Interests Statement
S.P.P. is listed on a patent held by Stanford University that covers the generation of region-specific brain organoids (U.S. patent 62/477,858). All other authors declare no competing interests.
References
- 1.Pasca SP The rise of three-dimensional human brain cultures. Nature 553, 437–445 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Mertens J, Marchetto MC, Bardy C & Gage FH Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat Rev Neurosci 17, 424–437 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lancaster MA, et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qian X, et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 165, 1238–1254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasca AM, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods 12, 671–678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sloan SA, et al. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 95, 779–790 e776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camp JG, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci U S A 112, 15672–15677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amiri A, et al. Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science 362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quadrato G, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545, 48–53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trevino AE, et al. Chromatin accessibility dynamics in a model of human forebrain development. Science 367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoon SJ, et al. Reliability of human cortical organoid generation. Nat Methods 16, 75–78 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horvath S DNA methylation age of human tissues and cell types. Genome Biol 14, R115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McEwen LM, et al. Systematic evaluation of DNA methylation age estimation with common preprocessing methods and the Infinium MethylationEPIC BeadChip array. Clin Epigenetics 10, 123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horvath S, et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging (Albany NY) 10, 1758–1775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stein JL, et al. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron 83, 69–86 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M, et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang HJ, et al. Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan TA, et al. Neuronal defects in a human cellular model of 22q11.2 deletion syndrome. Nat Med (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pollen AA, et al. Establishing Cerebral Organoids as Models of Human-Specific Brain Evolution. Cell 176, 743–756.e717 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhaduri A, et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polioudakis D, et al. A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron 103, 785–801 e788 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birey F, et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marton RM, et al. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat Neurosci (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hwang T, et al. Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci 19, 1093–1099 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Sanjana NE, Levanon EY, Hueske EA, Ambrose JM & Li JB Activity-dependent A-to-I RNA editing in rat cortical neurons. Genetics 192, 281–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran SS, et al. Widespread RNA editing dysregulation in brains from autistic individuals. Nat Neurosci 22, 25–36 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris MJ, Karra AS & Monteggia LM Histone deacetylases govern cellular mechanisms underlying behavioral and synaptic plasticity in the developing and adult brain. Behav Pharmacol 21, 409–419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheng M, Cummings J, Roldan LA, Jan YN & Jan LY Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147 (1994). [DOI] [PubMed] [Google Scholar]
- 29.Watanabe M, Inoue Y, Sakimura K & Mishina M Developmental changes in distribution of NMDA receptor channel subunit mRNAs. Neuroreport 3, 1138–1140 (1992). [DOI] [PubMed] [Google Scholar]
- 30.Liu H, Hu Q, Kaufman A, D’Ercole AJ & Ye P Developmental expression of histone deacetylase 11 in the murine brain. J Neurosci Res 86, 537–543 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Magueresse C & Monyer H GABAergic interneurons shape the functional maturation of the cortex. Neuron 77, 388–405 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Wyllie DJ, Livesey MR & Hardingham GE Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 74, 4–17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galluzzi L, Yamazaki T & Kroemer G Linking cellular stress responses to systemic homeostasis. Nat Rev Mol Cell Biol 19, 731–745 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Nowakowski TJ, et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sunwoldt J, Bosche B, Meisel A & Mergenthaler P Neuronal Culture Microenvironments Determine Preferences in Bioenergetic Pathway Use. Front Mol Neurosci 10, 305 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma EH, et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8(+) T Cells. Immunity 51, 856–870 e855 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Gaspard N, et al. An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature 455, 351–357 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Shen Q, et al. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat Neurosci 9, 743–751 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Zhang W, Qu J, Liu GH & Belmonte JCI The ageing epigenome and its rejuvenation. Nat Rev Mol Cell Biol 21, 137–150 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Raj B & Blencowe BJ Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 87, 14–27 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Sohal VS & Rubenstein JLR Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry 24, 1248–1257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mosser CA, Baptista S, Arnoux I & Audinat E Microglia in CNS development: Shaping the brain for the future. Prog Neurobiol 149–150, 1–20 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Lin YT, et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 98, 1294 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cakir B, et al. Engineering of human brain organoids with a functional vascular-like system. Nat Methods 16, 1169–1175 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balik A, Penn AC, Nemoda Z & Greger IH Activity-regulated RNA editing in select neuronal subfields in hippocampus. Nucleic Acids Res 41, 1124–1134 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paşca SP, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nature Medicine 17, 1657–1662 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oldham MC, Langfelder P & Horvath S Network methods for describing sample relationships in genomic datasets: application to Huntington’s disease. BMC Syst Biol 6, 63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leek JT, Johnson WE, Parker HS, Jaffe AE & Storey JD The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Devlin SJ Locally Weighted Regression: An Approach to Regression Analysis by Local Fitting AU - Cleveland, William S. Journal of the American Statistical Association 83, 596–610 (1988). [Google Scholar]
- 54.Hadley W Ggplot2 : elegrant graphics for data analysis. in Use R! 1 online resource (Springer,, Switzerland, 2016). [Google Scholar]
- 55.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Plaisier SB, Taschereau R, Wong JA & Graeber TG Rank-rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res 38, e169 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Triche TJ Jr., Weisenberger DJ, Van Den Berg D, Laird PW & Siegmund KD Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res 41, e90 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korotkevich G, Sukhov V & Sergushichev A Fast gene set enrichment analysis. bioRxiv, 060012 (2019). [Google Scholar]
- 61.Langfelder P, Luo R, Oldham MC & Horvath S Is my network module preserved and reproducible? PLoS Comput Biol 7, e1001057 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Langfelder P & Horvath S WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sloan SA, Andersen J, Pasca AM, Birey F & Pasca SP Generation and assembly of human brain region-specific three-dimensional cultures. Nat Protoc 13, 2062–2085 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cribari-Neto F & Zeileis A Beta Regression in R. Journal of Statistical Software; Vol 1, Issue 2 (2010). [Google Scholar]
- 65.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bahn JH, et al. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res 22, 142–150 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee JH, Ang JK & Xiao X Analysis and design of RNA sequencing experiments for identifying RNA editing and other single-nucleotide variants. RNA 19, 725–732 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Porath HT, Carmi S & Levanon EY A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat Commun 5, 4726 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parikshak NN, et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu YE, Parikshak NN, Belgard TG & Geschwind DH Genome-wide, integrative analysis implicates microRNA dysregulation in autism spectrum disorder. Nat Neurosci 19, 1463–1476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nishikura K A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17, 83–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parikshak NN, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang D, et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jansen IE, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51, 404–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cacace R, Sleegers K & Van Broeckhoven C Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement 12, 733–748 (2016). [DOI] [PubMed] [Google Scholar]
- 76.Chang D, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49, 1511–1516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Farrer MJ Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 7, 306–318 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Chen JA, et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol Neurodegener 13, 41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Greaves CV & Rohrer JD An update on genetic frontotemporal dementia. J Neurol 266, 2075–2086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu G, Wang LG, Han Y & He QY clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Skene NG & Grant SG Identification of Vulnerable Cell Types in Major Brain Disorders Using Single Cell Transcriptomes and Expression Weighted Cell Type Enrichment. Front Neurosci 10, 16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Gene expression data and methylation data are available in the Gene Expression Omnibus (GEO) under accession numbers GSE150122 and GSE150123. The accompanying GECO webtool can be accessed at https://labs.dgsom.ucla.edu/geschwind/files/view/html/GECO.html. The BrainSpan data are available in the database of Genotypes and Phenotypes (dbGaP) under Study Accession phs000755.v2.p1. Single cell data from human fetal cerebral cortex can be found at http://geschwindlab.dgsom.ucla.edu/pages/codexviewer and at dbGaP under Study accession phs001836. eCLIP data for FXR1 and FMR1are available in GEO with accession number GSE107895. Human cortical organoid single cell sequencing data are available in GEO with accession number GSE107771.