

SUMMARY
During development, two cell types born from closely-related progenitor pools often express identical transcriptional regulators despite their completely distinct characteristics. This phenomenon implies the need for a mechanism that operates to segregate the identities of the two cell types throughout differentiation after initial fate commitment. To understand this mechanism, we investigated the fate specification of spinal V2a interneurons, which share important developmental genes with motor neurons (MNs). We demonstrate that the paired homeodomain factor Chx10 functions as a critical determinant for V2a fate and is required to consolidate V2a identity in postmitotic neurons. Chx10 actively promotes V2a fate, downstream of the LIM-homeodomain factor Lhx3, while concomitantly suppressing the MN developmental program by preventing the MN-specific transcription complex from binding and activating MN genes. This dual activity enables Chx10 to effectively separate the V2a and MN pathways. Our study uncovers a widely-applicable gene regulatory principle for segregating related cell fates.
Keywords: Chx10, Vsx2, Lhx3, Sox14, V2a interneurons, motor neurons, transcription factor, spinal cord, development
Graphical Abstract
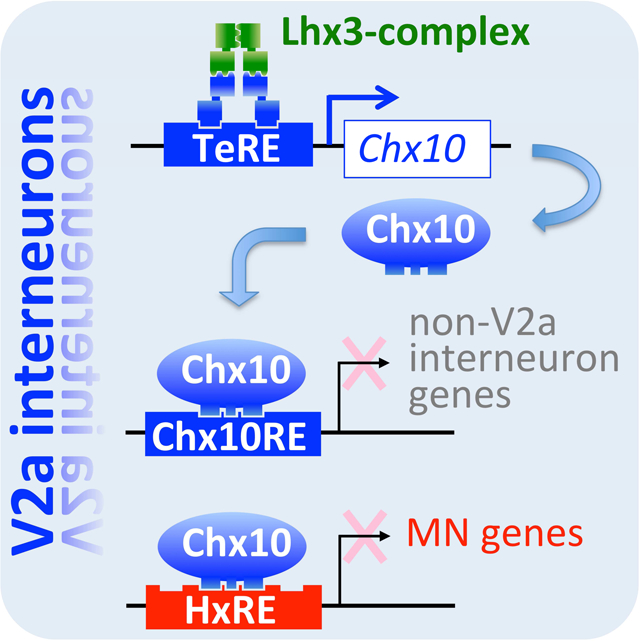
eTOC
Clovis et al. describe the mechanism through which spinal V2a interneurons are specified. They find that the best-known marker for V2a interneurons, Chx10, is the major determinant of V2a fate specification. Chx10 upregulates the expression of V2a interneuron genes while suppressing the expression of non-V2a interneuron and motor neuron genes.
INTRODUCTION
During embryonic organogenesis, many cell types are born in a spatially and temporally controlled manner to form a functional tissue. One of the most fundamental questions in developmental biology is how closely related cell types are produced from similar progenitors and yet acquire and maintain completely distinct cell identities during later stages of organogenesis. This relatively poorly understood process involves intricate gene regulatory networks that operate during sequential steps of cell fate commitment, specification and differentiation.
The gene regulatory networks for motor neurons (MNs) and V2a interneurons (V2aINs) provide an ideal platform to address this topic. The morphogen Sonic hedgehog (shh) is secreted from the notochord and floor plate, and patterns neuroepithelial cells along the dorso-ventral axis, leading to the formation of the two neighboring progenitor domains, the pMN and p2 domains (Fig. 1A) (Catela et al., 2015; Lee and Pfaff, 2001). Progenitor cells in the pMN and p2 domains produce MNs and V2aINs, respectively. While pMN cells upregulate the LIM-homeodomain (HD) transcription factors Isl1 and Lhx3 right before differentiation to MNs, p2 cells upregulate Lhx3, but not Isl1, shortly before cell cycle exit (Fig. 1A) (Ericson et al., 1992; Pfaff et al., 1996; Sharma et al., 1998; Tsuchida et al., 1994). Isl1 and Lhx3 form a hexameric complex with a self-dimerizing cofactor NLI, herein referred to as Isl1-Lhx3-complex (aka, the MN-hexamer; Fig. 1A) (Lee et al., 2008; Lee and Pfaff, 2003; Seo et al., 2015; Thaler et al., 2002). The Isl1-Lhx3-complex directly controls a wide range of MN genes and plays crucial roles in the fate specification of MNs (Cho et al., 2014; Lee et al., 2008; Lee et al., 2012; 2013; Lee and Pfaff, 2003; Lee et al., 2004; Mazzoni et al., 2013; Thaler et al., 2002; Thiebes et al., 2015). The misexpression of Lhx3 alone drives formation of ectopic V2aINs marked by Chx10 in the developing spinal cord (Tanabe et al., 1998; Thaler et al., 2002). Lhx3 also binds to NLI and forms a tetrameric complex herein referred to as Lhx3-complex (aka, the V2-tetramer; Fig. 1A) (Joshi et al., 2009; Thaler et al., 2002). It has remained unclear whether the Lhx3-complex directly induces the expression of an array of V2aIN genes similarly to the Isl1-Lhx3-complex, or it indirectly triggers V2aIN fate by upregulating other transcription factors that serve as its downstream effectors to induce V2a-identity. The segregation of pMN and p2 domains is initiated by the cross-repressive actions of two transcription factors, Olig2 and Irx3, in the progenitor cells (Lee and Pfaff, 2001; Lu et al., 2002; Novitch et al., 2001; Zhou and Anderson, 2002). Interestingly, pMN cells and newborn MNs maintain cell fate plasticity and can switch their fate into V2aINs when the MN pathway is dysregulated (Arber et al., 1999; Lee et al., 2008; Lu et al., 2002; Song et al., 2009; Thaler et al., 1999; Zhou and Anderson, 2002). These results strongly suggest that the mechanisms that separate MN and V2aIN identities continue to operate even after the progenitor cells are committed to MN or V2aIN fate. However, the precise regulatory mechanisms that consolidate V2aIN fate after cell cycle exit have yet to be clarified.
Figure 1. Chx10 is a critical downstream effector of Lhx3 in promoting V2aIN fate.
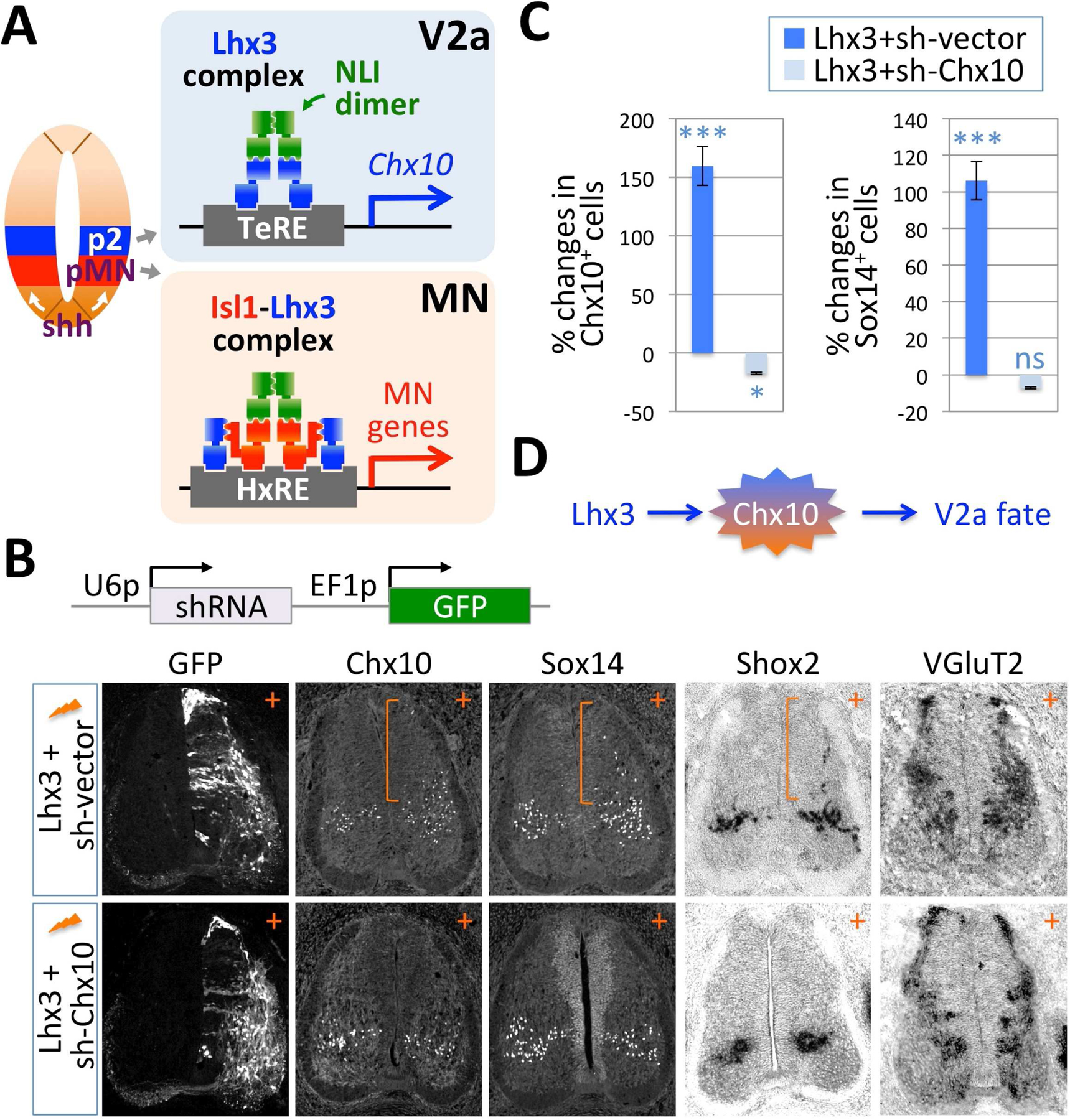
(A) Illustration of the developing spinal cord showing the specification of V2aINs and MNs from the p2 and pMN progenitor domains respectively, in response to graded Shh signaling from the floor plate. The Lhx3-complex is needed to drive V2aIN fate, whereas the Isl1-Lhx3-complex triggers the expression of MN genes. HxRE, hexamer response element; TeRE, tetramer response element.
(B) Illustration of the shRNA expression plasmid, which encodes both shRNA and GFP expression cassettes. The expression of the shRNA sequence designed against Chx10 3’UTR (sh-Chx10) is driven by the U6 promoter (U6p), while the expression of GFP is driven by the EF1 promoter (EF1p). Transverse sections of chick spinal cord 3 days after co-electroporation with Lhx3 together with sh-vector or sh-Chx10. GFP marks electroporated cells. Immunostaining with Chx10 and Sox14 antibodies and in situ hybridizations for Shox2 and VGluT2 mRNAs reveal that Lhx3 drives ectopic expression of V2aINs markers in the dorsal spinal cord (brackets). Co-electroporation of sh-Chx10 with Lhx3 prevents Lhx3 from inducing ectopic expression of V2aINs markers. +, electroporated side of the spinal cord.
(C) Quantification of the changes in the number of Chx10+ and Sox14+ cells in chick spinal cords following the electroporation of Lhx3+sh-vector (dark blue) or Lhx3+sh-Chx10 (light blue). Percent changes of total Chx10+ cells (left graph) or Sox14+ cells (right graph) in the electroporated (+) side over control side are presented. The zero means the equal number of Chx10+ or Sox14+ cells between the electroporated and control sides. The error bars represent the standard error of the mean. The statistics represent the comparison to unelectroporated control sides. ***, p<0.005; *, p<0.05; ns, non-significant; n≥6 embryos.
(D) Model of the gene regulatory pathway of V2aIN development involving Lhx3 and Chx10. Upregulation of Chx10 is necessary for Lhx3 to induce V2aIN fate.
While Lhx3 triggers the V2aIN fate specification, it is also expressed in MNs (Sharma et al., 1998; Tsuchida et al., 1994), thus warranting additional mechanisms to block erroneous activation of MN genes in differentiating V2aINs. It is possible that Lhx3 induces the expression of other effector transcription factors only in V2aINs, rather than directly upregulating the expression of V2aIN genes. Those V2a-specific transcription factors may then activate the expression of V2aIN genes. One such candidate is Chx10 (Vsx2), a paired HD transcription factor with the CVC domain. Chx10 expression, restricted to V2aINs within the developing spinal cord (Ericson et al., 1997), is induced ectopically by the misexpression of Lhx3 in the neural tube (Tanabe et al., 1998; Thaler et al., 2002). Chx10 is the best-known marker of V2aINs. However, the role of Chx10 in V2aIN specification and differentiation has not been explored.
In this report, we show that Chx10 promotes V2aIN fate by actively promoting V2a identity, while simultaneously suppressing non-V2a interneuron and MN identities. We further show that Chx10 employs two distinct modes of action in promoting V2a fate and suppressing MN identity. While Chx10 functions as a transcriptional repressor to activate V2aIN program, it acts as a DNA-binding competitor of the Isl1-Lhx3-complex in suppressing the expression of MN genes. Our findings represent a generalizable phenomenon likely occurring during the organogenesis of many tissues that require segregation of cell fates derived from related progenitor pools during development.
RESULTS
Chx10 mediates the activity of Lhx3 in inducing V2aIN fate
To test whether Chx10 contributes to Lhx3-directed induction of V2aIN identity, we devised a short-hairpin RNA (shRNA) construct targeting the 3’UTR region of the chick Chx10 transcript (Fig. 1B). We misexpressed Lhx3 with shRNA-Chx10 or shRNA-vector control in neural progenitors of the developing spinal cord using in ovo electroporation and monitored the formation of ectopic V2aINs three days post-electroporation. Co-electroporation of Lhx3 with shRNA-Chx10 led to a 17.5% reduction of Chx10+ cells in the electroporated side compared to the control side (Fig. 1B,C), confirming effective knock-down efficiency of shRNA-Chx10. The V2aIN marker Sox14 expression was induced as soon as V2aINs were born, and remained co-expressed with Chx10 in most V2aINs (Fig. S1A). The V2aIN marker Shox2 has been reported to be expressed in ~60% of V2aINs at P0 (Dougherty et al., 2013; Joshi et al., 2009; Thaler et al., 2002). We found that Lhx3 triggers the ectopic expression of Chx10, Sox14 and Shox2 (Fig. 1B). Quantification revealed an increase of 160% and 106% of Chx10+ and Sox14+ cells, respectively, in the Lhx3-electroporated spinal cord relative to the unelectroporated control side (Fig. 1C). Lhx3 also induced the expression of vesicular gluatamate transporter 2 (VGluT2), a marker of glutamatergic neurons (Fig. 1B). Thus, Lhx3 is sufficient to induce the formation of V2aINs in spinal neural progenitors. With shRNA-Chx10, however, Lhx3 failed to trigger the expression of other V2a marker genes, such as Sox14, Shox2 and VGluT2 (Fig. 1B,C). Lhx3 did not trigger ectopic MN formation with shRNA-Chx10 or shRNA-vector control (Fig. S1B). These data suggest that Chx10 is a crucial mediator of the activity of Lhx3 to induce V2aIN fate (Fig. 1D).
Development of V2aINs and MNs is perturbed in Chx10-null mice
To elucidate the role of Chx10 in the developing spinal cord, we analyzed Chx10orJ/orJ mice (Burmeister et al., 1996). Immunohistochemical analyses with a Chx10 antibody, which detects the N-terminal region of the mouse Chx10 protein, confirmed that the expression of Chx10 protein is reduced to undetectable levels in Chx10orJ/orJ spinal cords (Fig. 2A,S2A).
Figure 2. Specification of V2aIN fate requires Chx10.
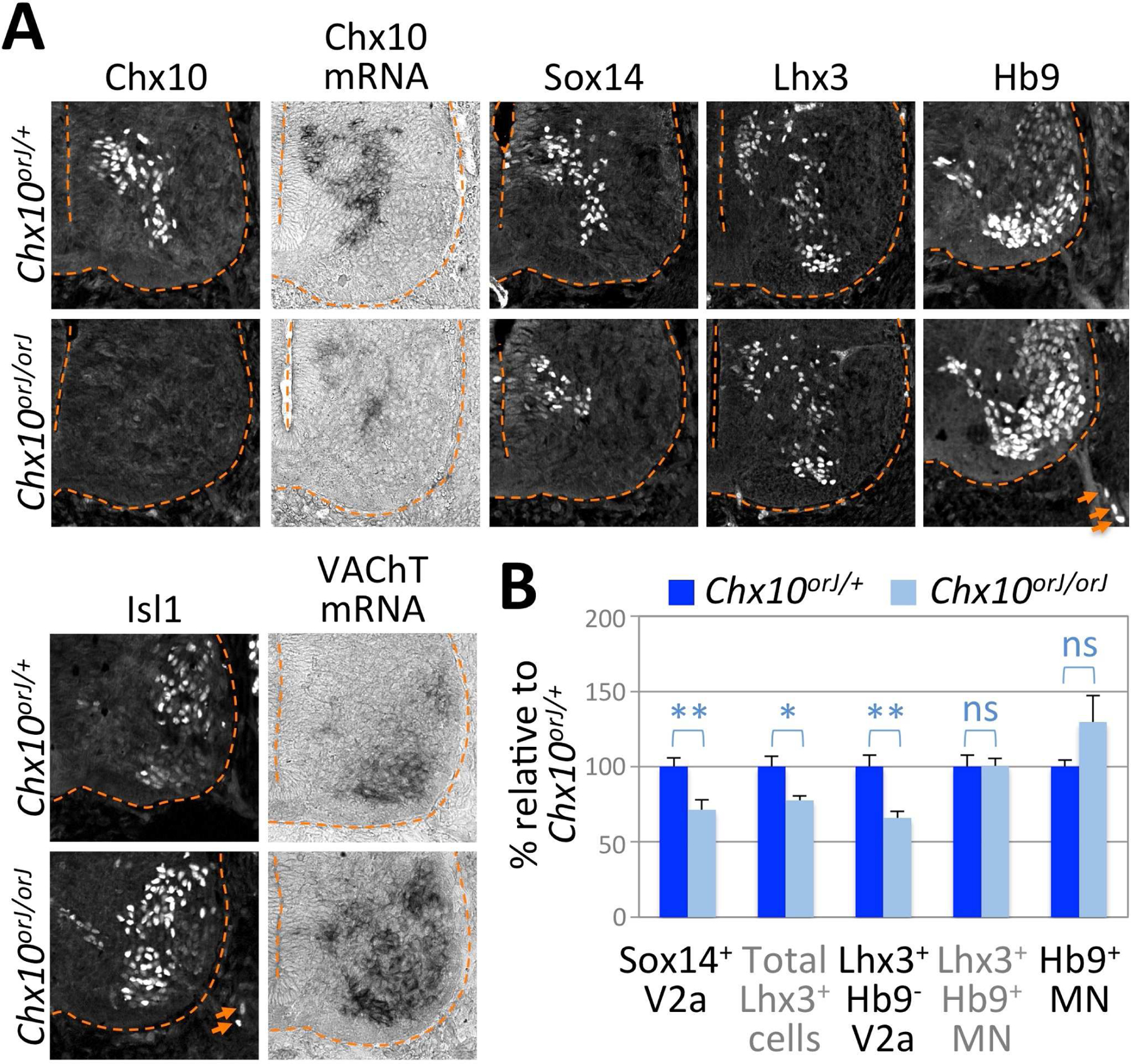
(A) Immunohistochemical and in situ hybridization analyses of E12.5 Chx10orJ/+ and Chx10orJ/orJ littermates. The ventral spinal cords are shown. Sox14/Lhx3-expressing V2aINs are reduced in Chx10orJ/orJ spinal cords. While the number of Hb9+/Isl1+ MNs did not significantly change, Hb9+/Isl1+ MNs emigrate abnormally from the spinal cord into the motor axonal track outside of the spinal cord (arrows) in Chx10orJ/orJ mice. The expression of the cholinergic gene VAChT is also increased in the ventral spinal cords of Chx10orJ/orJ mice.
(B) Quantification of the number of V2aINs and MNs in E12.5 mice. The change of the number of V2aINs and MNs in Chx10orJ/orJ spinal cords is relative to their control littermates. The error bars represent the standard error of the mean. **, p<0.01; *, p<0.05; ns, non-significant; n≥5 embryos.
As Chx10 is the best-known marker of V2aINs and the production of Chx10 transcripts should not be affected by the point mutation in the Chx10orJ allele, we first investigated V2aIN development in Chx10orJ/orJ embryos by analyzing the expression level and pattern of Chx10 transcripts. Chx10 mRNA was markedly reduced in Chx10orJ/orJ embryos compared to littermate control Chx10orJ/+ embryos at E12.5 (Fig. 2A), suggesting that V2aIN identity is compromised without Chx10 protein. Consistently, the number of Sox14+ and Lhx3+ neurons was reduced by 29% and 38%, respectively, in Chx10orJ/orJ embryos compared to their control littermates (Fig. 2A,B,S2B). At E12.5, Lhx3 labels both V2aINs and MMCm-type MNs (Sharma et al., 1998). Double labeling of Lhx3 and the MN marker Hb9 revealed that Lhx3+Hb9− V2aINs was substantially reduced in Chx10orJ/orJ embryos, whereas the number of Lhx3+Hb9+ MNs did not change (Fig. 2B,S2B). Immunostaining with activated Caspase 3 antibody showed no substantial change in cell death in Chx10orJ/orJ spinal cords (Fig. S2C). The number of cells expressing Gata3, a V2b interneuron marker, or Evx1, a V0 interneuron marker, did not change (Fig. S2D), suggesting that the loss of Chx10 did not lead to a fate conversion to V2b or V0 interneurons. Sox14+ V2aINs remained to be reduced in E13.5 and E15.5 Chx10orJ/orJ embryos (Fig. S2G,H).
In Chx10-deficient mice, several MNs were found in the motor axonal track outside of the spinal cord (Fig. 2A,S2E), indicating that some MN soma abnormally escaped the spinal cord via the motor exit point in the absence of Chx10. The MN marker vesicular acetylcholine transporter (VAChT) was expressed more strongly in a wider area in Chx10-null mice than in control littermates (Fig. 2A,S2F), suggesting that the VAChT gene becomes aberrantly upregulated in Chx10-null neurons.
Overall, our data demonstrate that Chx10 is critical for the development of V2aINs, and that without Chx10, MN gene expression and development are also perturbed.
Chx10 triggers V2a fate, while suppressing MN fate, through its DNA-binding activity
Electroporation of Chx10 alone triggered the formation of ectopic Sox14+ V2aINs in the developing chick spinal cord (Fig. 3A). While Lhx3 failed to induce V2aINs in MN area, the expression of Chx10 led to effective generation of V2aINs in MN area (Fig. 1B,3A). Chx10 also induced the expression of Shox2 and VGlut2 in MN area (Fig. 3A). These results suggest that Chx10 alone is sufficient to induce V2aIN fate.
Figure 3. Chx10 requires its DNA-binding to promotes V2aIN fate and suppress MN fate.
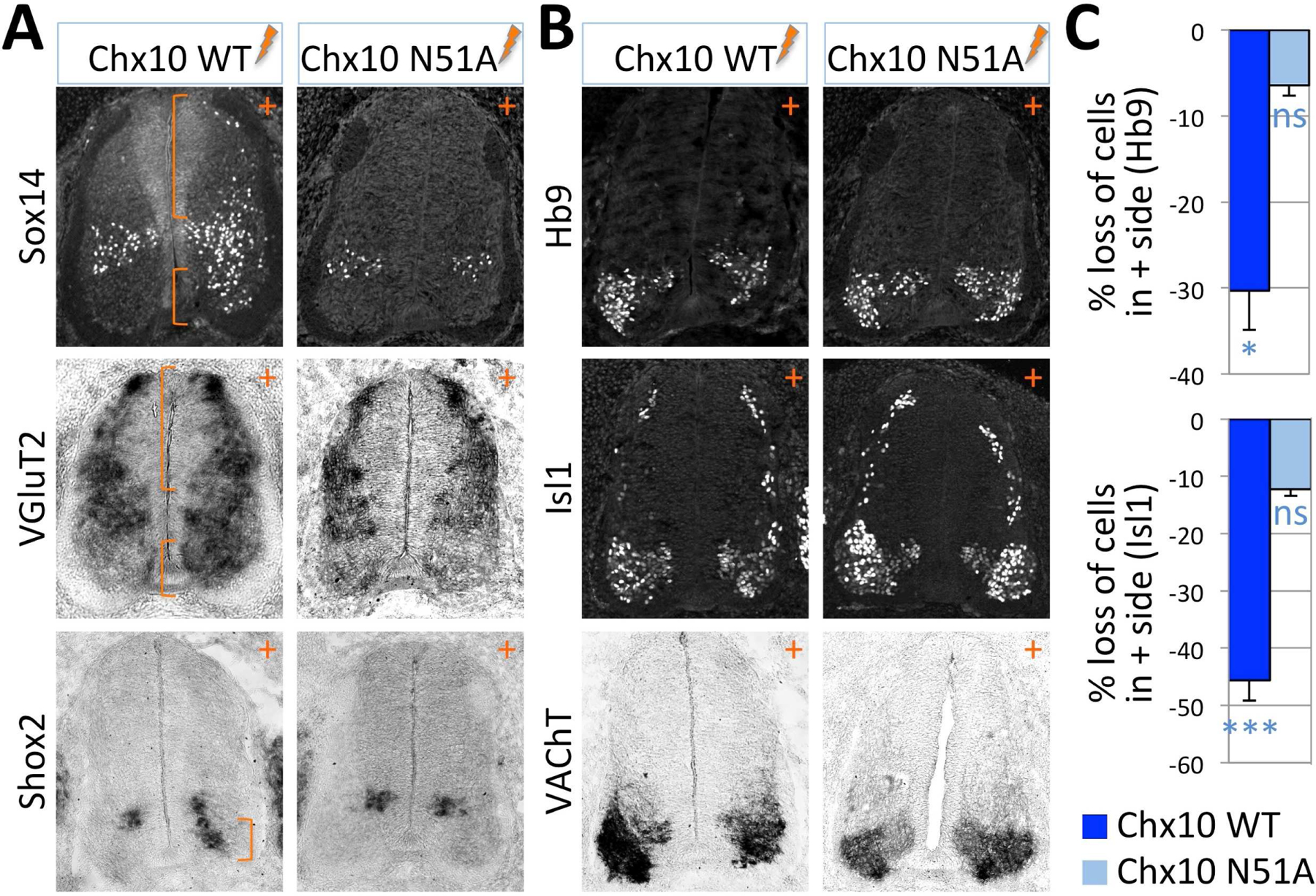
Immunohistochemical and in situ hybridization analyses in chick spinal cords electroporated (+ side) with Chx10 WT or with a DNA-binding defective mutant version of Chx10 (Chx10-N51A) using a panel of V2aIN markers (A) or MN markers (B).
(A) Chx10 WT, but not Chx10-N51A, promotes the expression of V2aIN markers in both dorsal spinal cord and the MN area (brackets).
(B) Chx10 WT, but not Chx10-N51A, inhibits specification of MNs.
(C) Quantification of the repression of MN generation in chick spinal cords electroporated with Chx10 WT (dark blue bars) or Chx10-N51A (light blue bars), compared to LacZ-electroporated control spinal cords. The data is presented as the percent loss of Hb9+ cells (upper panel) or Isl1+ cells (lower panel). The error bars represent the standard error of the mean. The statistics represent the comparison to LacZ-electroporated spinal cords. ***, p>0.005; *, p<0.05; ns, non-significant; n≥6 embryos.
To test whether Chx10 converts presumptive MNs to V2aINs, we examined the generation of MNs in the neural tube electroporated with Chx10. Upon the expression of Chx10, the number of Hb9+ MNs and Isl1+ MNs decreased by 30% and 46%, respectively (Fig. 3B,C,S3). Likewise, the expression of VAChT, a cholinergic gene, was reduced by Chx10 (Fig. 3B). Thus, Chx10 triggers the generation of V2aINs at the expense of MNs in the ventral spinal cord.
To test whether the DNA-binding activity of Chx10 is required to activate V2aIN differentiation and inhibit MN fate, we employed the DNA-binding defective Chx10-N51A mutant (Dorval et al., 2005). Despite a high level of expression of Chx10-N51A comparable to that of Chx10 wild-type, Chx10-N51A neither induced ectopic V2aINs nor inhibited differentiation of MNs (Fig. 3). These results suggest that binding to Chx10-response elements is required for Chx10 to drive V2aIN fate acquisition and suppress MN differentiation.
Taken together, our data demonstrate that Chx10 is a critical and active regulator of V2aIN identity.
Chx10 effectively triggers V2a fate and suppresses MN fate in differentiating ESCs
To investigate whether Chx10 can actively promote V2aIN fate while concomitantly repressing MN identity, we utilized a well-established differentiation paradigm of mouse embryonic stem cells (ESCs), which directs ESCs to acquire MN identity (Wichterle et al., 2002). To monitor the activity of Chx10 in this context, we generated doxycycline (Dox)-dependent Chx10-inducible ESCs (Chx10-ESCs), in which Chx10 coding sequences were inserted downstream of the tetracycline response element (TRE) and the reverse tetracycline transactivator (rtTA) was integrated into the constitutively active Rosa26 locus (Fig. 4A). In Chx10-ESCs, the expression of Flag epitope-tagged Chx10 was robustly induced by Dox treatment (Fig. 4B). We differentiated Chx10-ESCs to MNs following the protocol (Wichterle et al., 2002), in the presence of vehicle or Dox (i.e. Chx10 expression) (Fig. 4A,C). Chx10-ESCs differentiated to MNs when incubated with vehicle, as revealed by strong Hb9-expression (Fig. 4C). However, they failed to differentiate into Hb9+ MNs when Chx10 expression was induced by Dox (Fig. 4C). Several remaining Hb9+ MNs in Dox-treated condition expressed low levels of Chx10, exhibiting complementary pattern of expression between Chx10 and Hb9 (Fig. 4C). These results indicate that Chx10 inhibits differentiation of ESCs to MNs.
Figure 4. Chx10 actively promotes V2aIN fate and suppresses MN development in differentiating ESCs.
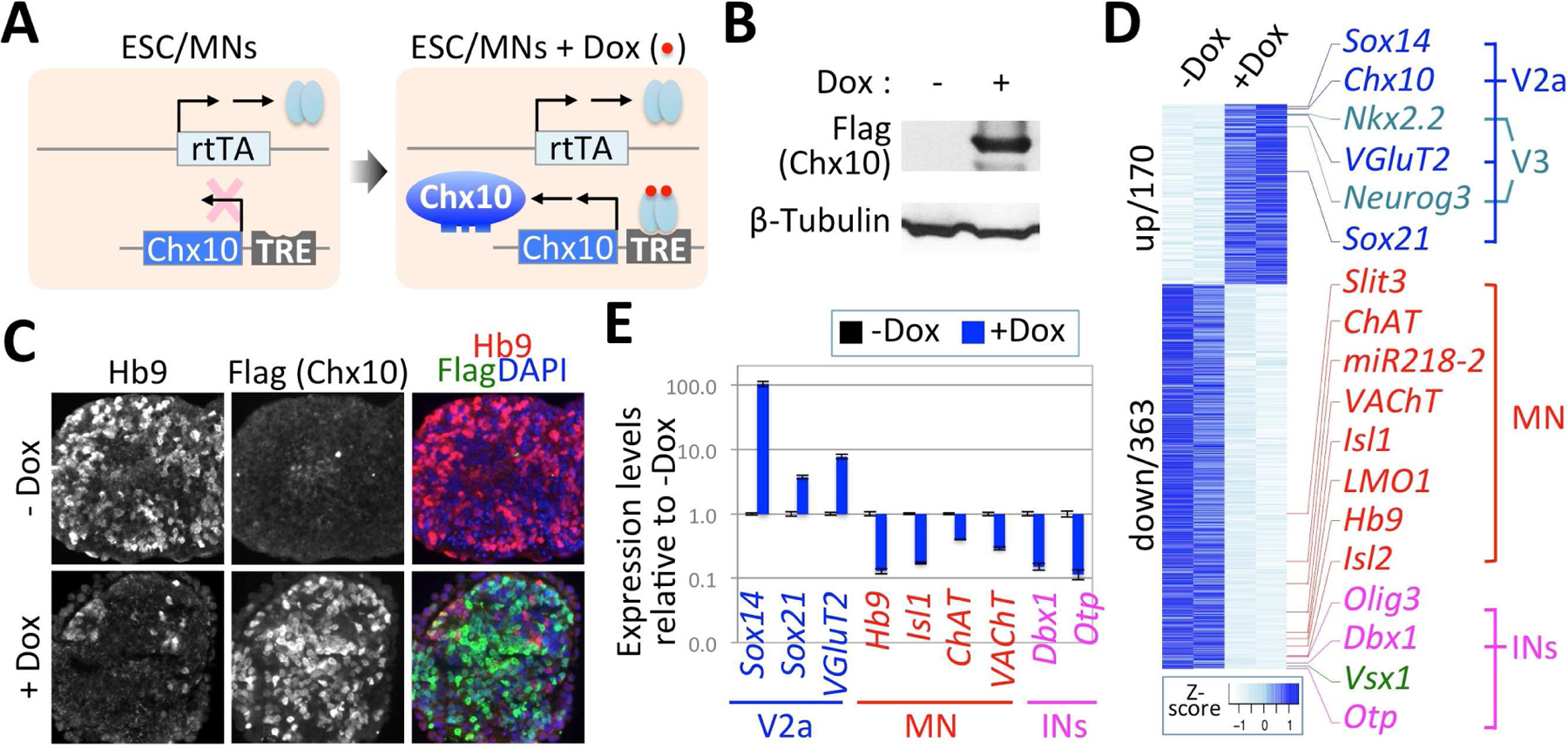
(A) Schematic representation of Chx10-inducible ESCs directed to differentiate into MNs (ESC/MNs) before and after treatment with Doxycycline (Dox). TRE, tetracycline response element; rtTA, reverse tetracycline transactivator.
(B) Western analyses show induction of Chx10 after treatment with Dox.
(C) Cell differentiation analyses in Chx10-ESC, directed to differentiate into MNs, with or without Dox. Upregulation of Chx10 by Dox is correlated with a strong downregulation of the MN marker Hb9.
(D) RNAseq analyses reveal Chx10-mediated transcriptome changes in ESC-derived MNs. The 533 genes that show a significant change (fold change 1.5, p≤0.01) are sorted in a heat map by fold change and biological replicates. Selected genes expressed in MNs and interneurons are highlighted.
(E) The expression levels of selected genes are significantly altered by Chx10 induction in ESC-derived MNs, as obtained with qRT-PCR analyses. Y-axis shows log10 values of gene expression levels in Dox-treated MNs relative to vehicle-treated control MNs (-Dox). Error bars represent the standard deviation of the mean.
To systematically investigate the effect of Chx10 on MN differentiation, we performed RNAseq analyses in ESC-derived MNs, and analyzed the transcriptome changes triggered by Chx10 expression (i.e. Dox treatment) (Fig. 4D, Table S1). Chx10 led to a significant change in the level of 533 genes (fold change 1.5, p≤0.01). Among these genes, 68% (363 genes) were downregulated by Chx10, while 32% (170 genes) were upregulated. Many MN genes, such as Hb9, Isl1, Isl2, choline acetyltransferase (ChAT) and VAChT, were suppressed by Chx10. The expression of interneuron genes, such as Dbx1 and Olig3 that direct V0 and dorsal interneuron dI1–3 fates, respectively, was repressed. Chx10 strongly induced Sox14, a V2aIN gene, by ~149 folds. Other known V2aIN genes, Sox21 and VGluT2, were also upregulated by Chx10. Chx10-directed induction of V2aIN genes and repression of MN and non-V2a interneuron genes were confirmed by independent quantitative RT-PCR analyses (Fig. 4E). These genome-wide transcriptome analyses establish that Chx10 can actively promote V2aIN fate, while suppressing MN development, in the cells that are directed to acquire MN fate.
Dual regulatory modes of Chx10 to control V2aIN and MN fates
Chx10 acts as both transcriptional repressor and activator in chick neuronal cultures (Dorval et al., 2005). To test whether Chx10 acts as a transcriptional repressor or activator to drive V2aIN fate and suppress MN identity, we generated constitutive activator and repressor forms of Chx10 by fusing the C-terminal half of Chx10, which contains the DNA-binding HD, to VP16 transcriptional activation domain or engrailed (EnR) transcriptional repression domain (Fig. 5A). Similarly to Chx10 wild-type, EnR-Chx10 triggered the formation of ectopic V2aINs in both dorsal neural tube and MN area, as indicated by ectopic upregulation of Sox14, Shox2 and VGluT2 (Fig. 5B). However, expression of VP16-Chx10 resulted in a drastic reduction of Sox14+ V2aINs (Fig. 5B). Consistently, expression of VGluT2 and Sox14 was repressed by VP16-Chx10 (Fig. 5B), suggesting that VP16-Chx10 functions as a dominant negative in the V2aIN differentiation pathway. Therefore, Chx10 functions as a transcriptional repressor to drive V2a fate specification.
Figure 5. Chx10 specifies V2a fate through its transcriptional repressor activity and inhibits MN development independently of its transcriptional activity.
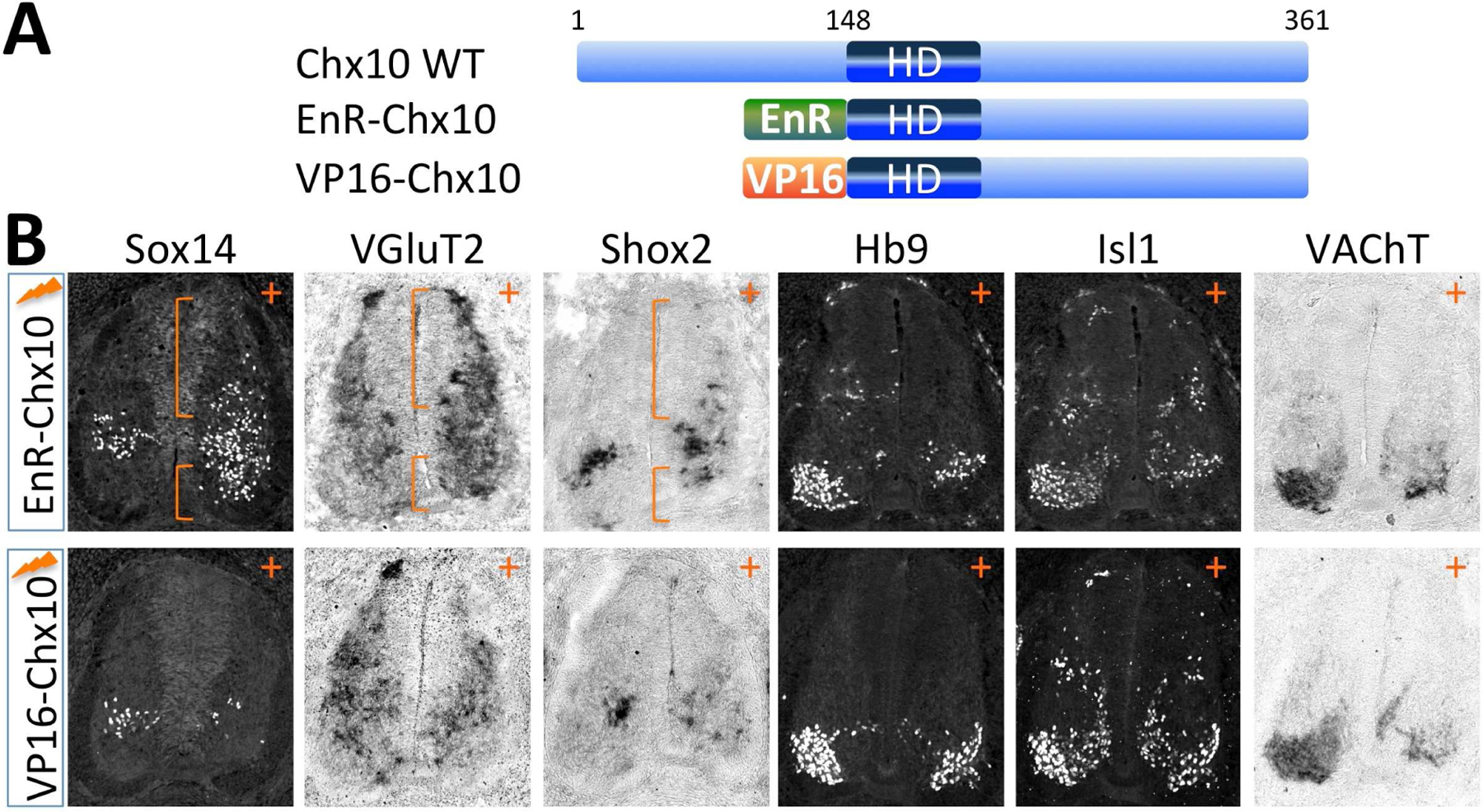
(A) Schematic representation of Chx10 fusions. EnR, Engrailed repressor domain; VP16, VP16 activator domain; HD, homeodomain.
(B) Immunohistochemical and in situ hybridization analyses in chick spinal cords electroporated (+ side) with EnR-Chx10 or with VP16-Chx10. EnR-Chx10 promotes the expression of V2aIN markers and inhibits MN specification, whereas VP16-Chx10 inhibits generation of both V2aINs and MNs. Ectopic V2aINs in the dorsal spinal cord and the MN area are marked with brackets.
Expression of EnR-Chx10 or VP16-Chx10 led to a decrease of Hb9+/Isl1+ MNs in chick neural tube (Fig. 5B). Similarly, both fusions reduced VAChT expression (Fig. 5B). Therefore, Chx10 inhibits MN development independently of its transcriptional activity, which is distinct from the requirement of its transcriptional repression activity in promoting V2aIN fate.
Identification of Chx10-bound genomic elements
Our findings suggest that Chx10 suppresses MN genes by competitively binding to the response elements bound and activated by MN fate-determining transcription factors such as the Isl1-Lhx3-complex. Furthermore, the Isl1-Lhx3-binding motif is similar to the optimal binding site of Chx10, identified in vitro (Lee et al., 2008; Lee et al., 2013; Wilson et al., 1993). Thus, Chx10 may compete with the Isl1-Lhx3-complex to bind to an important set of MN genes, thereby inhibiting their activation by Isl1-Lhx3. To test this hypothesis in an unbiased genome-wide manner, we performed ChIPseq analyses in Chx10-ESCs and mapped Chx10-binding genomic loci (Table S2). Among the 363 genes that were downregulated by Chx10, 232 genes were associated with Chx10-bound ChIPseq peaks (Table S3), suggesting that this set of genes are likely direct target genes of Chx10. In support of our hypothesis, these genes included MN genes, including Hb9, Isl1, ChAT and VAChT (Table S3). Non-V2a interneuron genes, such as Dbx1, Olig3 and Otp, also recruited Chx10 (Fig. S4, Table S3) and became downregulated by Chx10 (Fig. 4D,E), suggesting that Chx10 represses a subset of non-V2a interneuron genes via its repressor activity.
Chx10 also bound the promoter of the Vsx1 gene (Fig. S5A). Chx10 and Vsx1 are the only Paired-like HD genes with the CVC domain in the mouse genome (Chow et al., 2001). Vsx1 is expressed in differentiating V2 interneurons in zebrafish (Kimura et al., 2008). Vsx1 displayed similar expression pattern in the mouse spinal cord but appeared to be downregulated in more laterally located, mature Chx10+ V2aINs (Fig. S5C). Combined with our RNAseq and RT-PCR results that Chx10 represses the expression of Vsx1 (Fig. 4D,S5B), these results suggest that Chx10 negatively controls Vsx1 transcription in the developing spinal cord. Interestingly, misexpression of Vsx1 in chick neural tube results in generation of ectopic Sox14+ cells and inhibition of Hb9+ MN formation, resembling the activity of Chx10, although the activity of Vsx1 in inducing ectopic Sox14+ cells was weaker than that of Chx10 in the dorsal spinal cord (Fig. S5D,3A). These results indicate the functional redundancy between Chx10 and Vsx1 in directing V2a specification and inhibiting MN differentiation. The redundant action and regulatory feedback between Chx10 and Vsx1 likely contributes to the gene network in V2a-MN development.
Next, we compared Chx10-bound ChIPseq peaks to Isl1-Lhx3-bound ChIPseq peaks (Lee et al., 2013), and identified 927 genomic regions that recruit both Chx10 and Isl1-Lhx3 (Fig. 6A, Table S4). To identify DNA motifs that are significantly enriched in this set of ChIPseq peaks, we analyzed the sequences of the 927 genomic regions using two complementary motif discovery algorithms, MEME and DREME (Bailey et al., 2009; Machanick and Bailey, 2011). Both algorithms discovered an almost identical motif as the most significantly enriched DNA sequences (Fig. 6A). This motif resembles the previously identified binding sites of Isl1-Lhx3 and Chx10 (Fig. 6A) (Lee et al., 2008; Lee et al., 2013; Wilson et al., 1993) and is enriched in the center of the common peaks for Chx10 and Isl1-Lhx3 (Fig. 6B). Thus, this motif likely serves as a direct binding element for both Chx10 and Isl1-Lhx3.
Figure 6. Chx10 and the Isl1-Lhx3-complex bind common genomic regions linked MN genes.
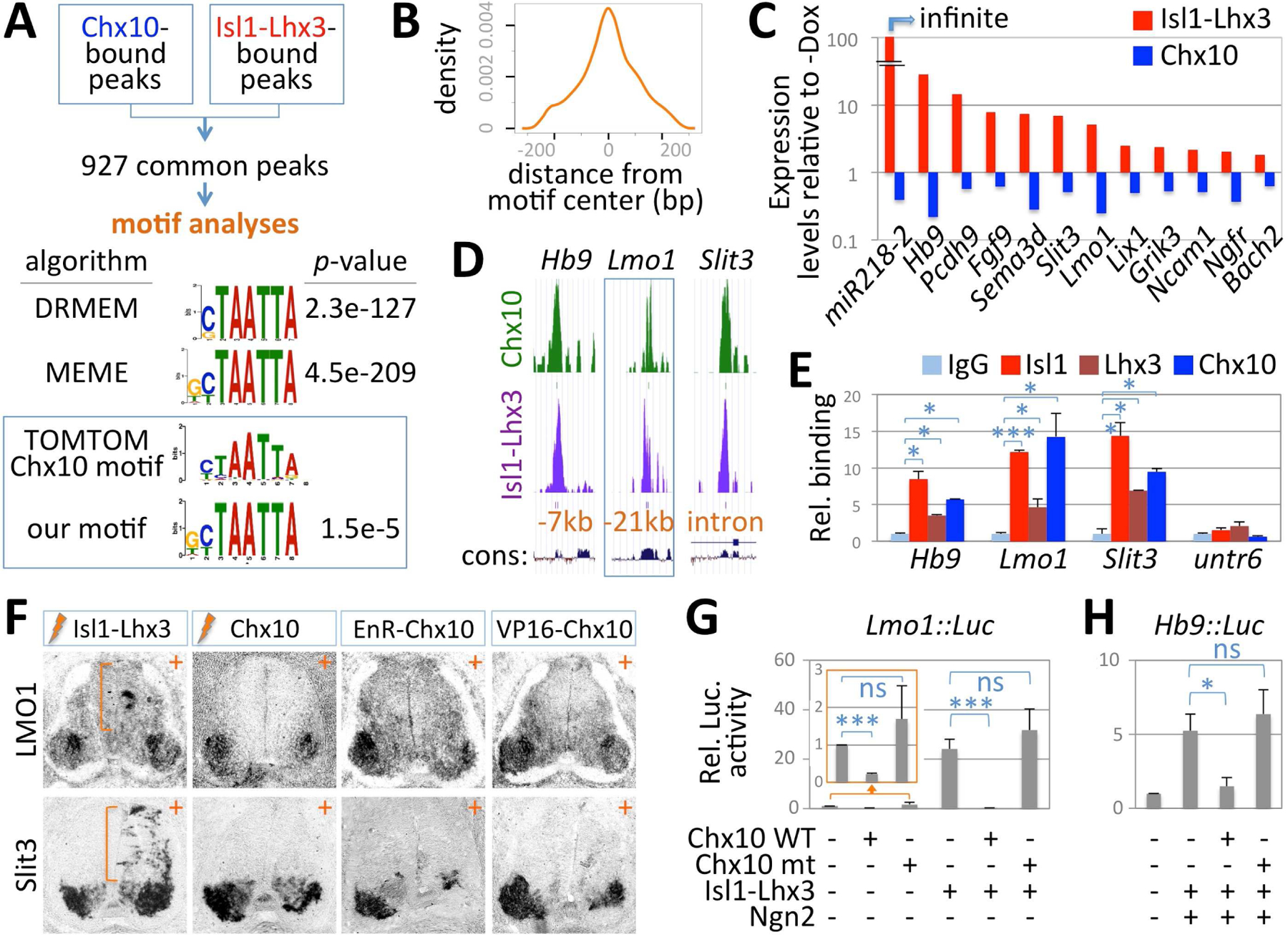
(A) Strategy to analyze ChIPseq datasets for Chx10 and Isl1-Lhx3. DREME and MEME algorithms identified a nearly identical DNA motif that is enriched in the 927 common genomic regions recruiting both Chx10 and Isl1-Lhx3. TOMTOM algorithm demonstrated that this motif resembles Chx10-binding motif in the database.
(B) Graphical representation of the frequency of the motif from the 927 common ChIPseq peaks in relation to the central position (0 in X-axis) of the peaks.
(C) Fold change of expression for 12 genes associated with Chx10/Isl1-Lhx3 common ChIPseq peaks, which are downregulated by Chx10 (blue) in Chx10-ESCs while being upregulated by Isl1-Lhx3 (red) in Isl1-Lhx3-ESCs, as obtained with RNAseq analyses. Y-axis shows log10 values of gene expression levels in Dox-treated ESCs relative to vehicle treated controls (-Dox).
(D) Overlapping peaks for Isl1-Lhx3 (purple) and Chx10 (green) from ChIPseq datasets, associated with MN genes Hb9, Lmo1 and Slit3. The peaks are located upstream of Hb9 and Lmo1 coding regions and intron of the Slit3 gene.
(E) ChIP-qPCR analyses in E12.5 mouse spinal cords show that Isl1, Lhx3 and Chx10 are recruited to the ChIPseq peak regions associated with Hb9, Lmo1 and Slit3, but not to the Untr6 gene, a negative control genomic region. The error bars represent the standard deviation. ***, p<0.005; *, p<0.05, compared to IgG.
(F) In situ hybridization analyses in chick spinal cords electroporated (+ side) with Isl1-Lhx3, Chx10, EnR-Chx10 or VP16-Chx10. Isl1-Lhx3 promotes the expression of Lmo1 and Slit3 in the dorsal spinal cord (bracket), whereas Chx10, EnR-Chx10 and VP16-Chx10 inhibit the expression of LMO1 and Slit3 in MNs.
(G,H) Luciferase assays in P19 cells using luciferase reporters linked to a Chx10/Isl1-Lhx3-bound genomic element upstream of the Lmo1 (G) or Hb9 (H) genes. Each reporter was co-transfected with constructs shown below the graph. Chx10 WT, but not Chx10 DNA-binding mutant (Chx10 mt, Chx10-N51A), represses the transcriptional activation of the reporter gene. The error bars represent the standard deviation. ***: p<0.005, * p<0.05, ns: non-significant.
Overall, we identified a set of direct target genes for Chx10, which include many known MN genes. Furthermore, these analyses demonstrate that Chx10 share many genomic target elements with Isl1-Lhx3.
Chx10 and Isl1-Lhx3 bind the same genomic regions linked to MN genes
Our studies suggest a model that, during V2aIN differentiation, Chx10 represses important MN genes by competitively binding to the response elements that are occupied and activated by Isl1-Lhx3 in MNs (Fig. 7E). To identify the genes that are co-regulated by Chx10 and Isl1-Lhx3 via the same regulatory elements, we compared Chx10/Isl1-Lhx3 common ChIPseq peaks with two RNAseq datasets, which identified transcriptome changes upon expression of Chx10 or Isl1-Lhx3 (Table S1) (Lee et al., 2012). These analyses revealed 12 genes, which are associated with Chx10/Isl1-Lhx3-common ChIPseq peaks and are downregulated by Chx10, while being upregulated by Isl1-Lhx3 (Fig. 6C,D,S6A). These 12 genes have been implicated in MN development or neuronal functions (see discussion).
Figure 7. Chx10 is necessary for the aberrant acquisition of V2a identity in Hb9-deficient MNs.
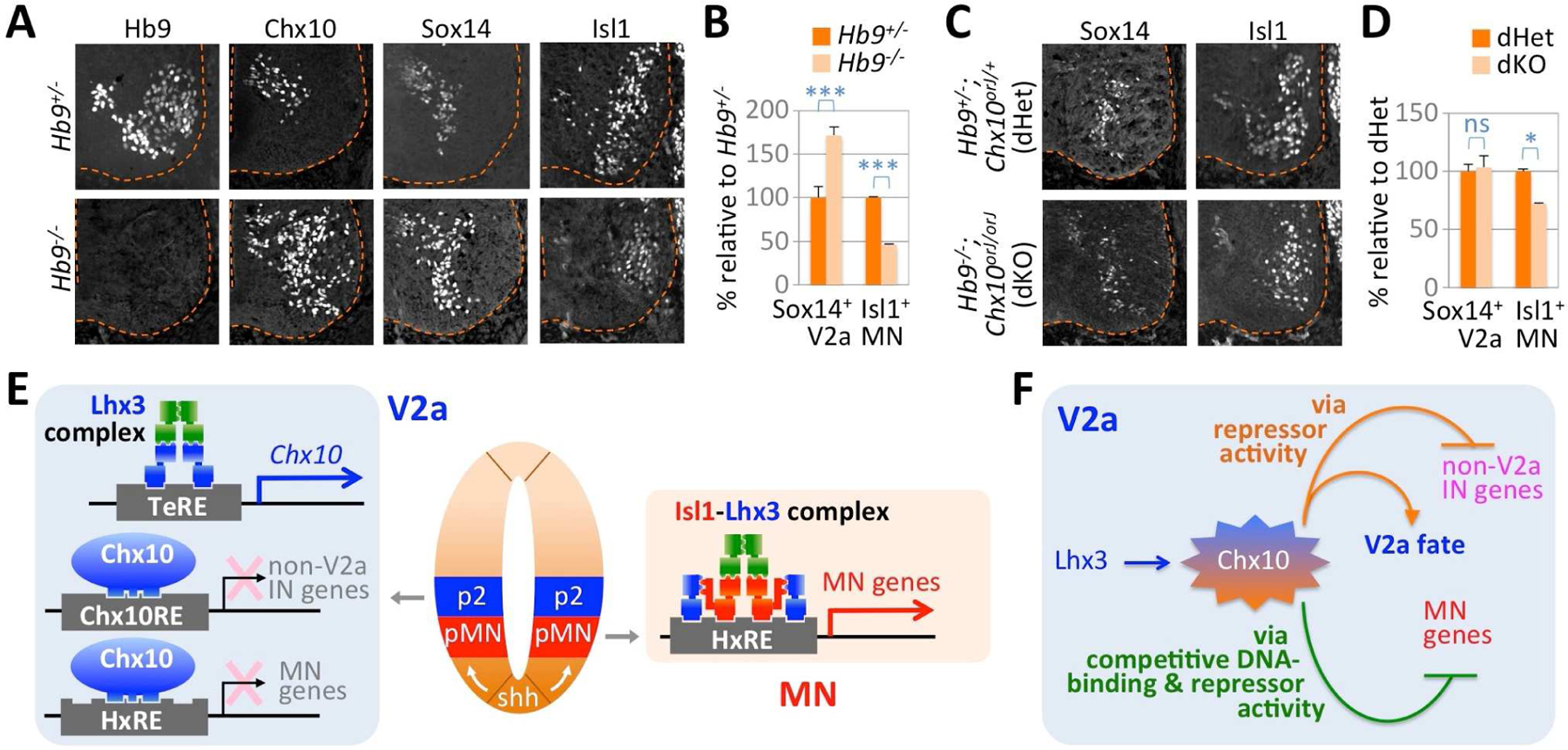
(A) Immunohistochemical analyses of E12.5 Hb9+/− and Hb9−/− littermates. The ventral spinal cords are shown. In Hb9-null spinal cord, Chx10 and Sox14 are ectopically upregulated in the MN area while Isl1 expression is markedly reduced.
(B) Quantification of Sox14+ V2aINs and Isl1+ MNs in Hb9-null spinal cords relative to Hb9+/− control littermates at E12.5. The error bars represent the standard error of the mean. ***, p<0.005; n> 3 embryos.
(C) Immunohistochemical analyses of E12.5 Hb9+/−;Chx10orJ/+ (dHet) and Hb9−/−;Chx10orJ/orJ (dKO) littermates. The ventral spinal cords are shown.
(D) Quantification of Sox14+ V2aINs and Isl1+ MNs in dKO spinal cords relative to dHet control littermates at E12.5. The error bars represent the standard error of the mean. *, p<0.05; ns, non-significant; n> 3 embryos.
(E) In V2aINs, the Lhx3-complex upregulates Chx10 by directly binding the TeRE in the Chx10 gene. Chx10 binds the HxRE associated with MN genes and represses MN genes. Chx10 also binds and inhibits non-V2a interneuron genes. In MNs, the Isl1-Lhx3-complex binds and upregulates MN genes.
(F) Gene regulatory mechanisms to precisely establish V2aIN identity, separated from MN or non-V2a interneuron fate. Lhx3 induces the expression of Chx10. In turn, Chx10 functions as a transcriptional repressor to activate V2aIN program and to inhibit non-V2a interneuron genes. Chx10 also functions as a DNA-binding competitor of the Isl1-Lhx3-complex to suppress MN genes.
To further test our model (Fig. 7E), we investigated the in vivo recruitment of Isl1-Lhx3 and Chx10 to their common binding elements in Hb9, LMO1 and Slit3/miR218-2 loci. The endogenous Isl1, Lhx3 and Chx10 proteins were recruited to each of the common binding sites in the developing spinal cord, as shown by ChIP in E12.5 mouse spinal cord (Fig. 6E). Next, we monitored the binding of Isl1-Lhx3 to Hb9, LMO1 and Slit3/miR218-2 loci in the presence of Chx10 wild-type or Chx10-N51A in P19 cells. Chx10 wild-type, but not Chx10-N51A, inhibited the recruitment of Isl1-Lhx3 to their common binding loci (Fig. S6B). While Hb9 and miR-218 are expressed in MNs and induced by Isl1-Lhx3 (Tanabe et al., 1998; Thaler et al., 2002; Thiebes et al., 2015), the regulation of Lmo1 and Slit3 genes in the neural tube remains unknown. In the developing spinal cord, the expression of LMO1 and Slit3 were highly and specifically induced as MNs emerged from the progenitor zone (see unelectroporated control sides in Fig. 6F). Interestingly, Isl1-Lhx3 ectopically upregulated LMO1 and Slit3 in the dorsal spinal cord, whereas Chx10 inhibited their expression in MNs (Fig. 6F). As expected, both EnR-Chx10 and VP16-Chx10 suppressed LMO1 and Slit3 expression in MNs (Fig. 6F). These results support our model that Chx10 effectively represses MN genes, at least in part, by competing with Isl1-Lhx3 for the same response elements (Fig. 7E).
To investigate how Chx10 and Isl1-Lhx3 influence the transcriptional activity of their common target genomic elements, we generated a luciferase reporter linked to a Chx10/Isl1-Lhx3-bound genomic element upstream of the Lmo1 gene (Fig. 6D). We monitored how the expression of Isl1-Lhx3, Chx10 or both affects the transcriptional activity of the Lmo1 genomic element using the Lmo1::Luc reporter in P19 mouse embryonic cells (Fig. 6G). While Isl1-Lhx3 potently activated the Lmo1 element, Chx10 repressed its activity. Furthermore, Chx10 inhibited Isl1-Lhx3-dependent activation of the reporter in a DNA-binding activity-dependent manner. VP16-Chx10 activated the Lmo1::Luc, while EnR-Chx10 suppressed it (Fig. S6C), suggesting that VP16-Chx10 is capable of DNA-binding and likely functions as DNA-binding competitor of Isl1-Lhx3 in MN inhibition (Fig. 5,6F). Chx10 acted similarly on the Hb9::Luc reporter (Fig. 6H).
Our data establish that, during V2aIN specification, Chx10 represses a subset of MN genes, which are activated by Isl1-Lhx3 in MNs via shared response elements.
The loss of Chx10 partially restores the V2aIN-MN gene network disrupted in Hb9-null mice
To further investigate the role of Chx10 in the V2aIN-MN gene network, we took advantage of the Hb9-null mouse model, in which MNs are drastically reduced while Chx10 is aberrantly upregulated in the MN area (Fig. 7A) (Arber et al., 1999; Thaler et al., 1999). We hypothesized that the aberrant upregulation of Chx10 contributes to the reduction of MNs and it also simultaneously promotes V2aIN genes in Hb9-deficient MNs. To test this hypothesis, we generated Hb9;Chx10 double knockout (dKO) mice and monitored whether the deletion of Chx10 rescues MN developmental deficits in Hb9-null spinal cords. While Hb9-null mice showed 54% loss of Isl1+ MNs, Hb9;Chx10 dKO displayed 28% reduction of Isl1+ MNs compared to their respective control littermates (Fig. 7A–D). The Isl1+ MNs in Hb9;Chx10 dKO expressed other MN markers, such as Lhx3 and FoxP1 (Fig. S7). Thus, preventing the expression of Chx10 in Hb9-depleted cells partially restored the compromised MN development, indicating that misexpressed Chx10 in Hb9-deficient MNs is in part responsible for the MN deficits in Hb9-null mice.
To test whether misexpressed Chx10 promotes V2aIN identity in Hb9-depleted MNs, we performed immunostaining with Sox14 antibodies. Ectopic Sox14+ cells were found in the ventral spinal cord of Hb9-null mice, as revealed by ~53% increase of Sox14+ cells (Fig. 7A,B). Interestingly, however, Hb9;Chx10 dKO mice showed no significant increase of Sox14+ cells compared to control littermates (Fig. 7C,D). These results indicate that the ectopic induction of Chx10 drives the aberrant acquisition of V2a identity in Hb9-null MNs.
DISCUSSION
While the gene regulatory pathway to specify MN fate has been well characterized (Lee et al., 2008; Lee and Pfaff, 2001), much less has been known for the transcriptional mechanisms for specifying V2aINs, whose gene expression profile is related to that of MNs at progenitor and early differentiation stages. Despite being the best marker of V2aINs, the actual role of Chx10 in V2aINs remains unknown. Our analyses of the action of Chx10 demonstrate that Chx10 plays a crucial role in V2aIN fate specification and differentiation in the developing spinal cord. Our comparative genomics analyses, coupled with embryonic studies, uncovered how the Chx10-directed V2aIN pathway interacts with the Isl1-Lhx3-directed MN pathway in the gene regulatory network of spinal cord development (Fig. 7E). This gene network for V2aINs and MNs provides important insights into a fundamental question in developmental biology; how two closely related cell fates are specified and maintain disparate cell identities despite the expression of common transcriptional regulators during organogenesis. Many transcriptional repressors contribute to patterning, cell fate specification, differentiation and organogenesis in various developmental contexts. Thus, the mechanisms by which Chx10 promotes and consolidates V2aIN fate are applicable to many other developmental contexts that require the action of transcriptional repressors.
Our study revealed that the mechanism by which Lhx3 directs the V2aIN fate is distinct from the mechanism by which Isl1-Lhx3 triggers the MN fate. The Isl1-Lhx3-complex drives MN differentiation by directly binding and inducing a wide range of MN genes, whereas the potential of Lhx3 to trigger V2aIN differentiation is mediated largely by its downstream target Chx10, as demonstrated by our results showing that Lhx3 fails to trigger V2aIN generation without Chx10 (Fig. 1B,7E). We also found that Chx10 triggers the formation of ectopic V2aINs without upregulating Lhx3 in the neural tube (Fig. 3A, data not shown), suggesting that Chx10 does not need Lhx3 to trigger V2aIN fate. Furthermore, Chx10 is much more effective in inducing V2aIN fate than Lhx3 in the MN area. Lhx3 is unable to induce V2aIN genes in MN domain likely due to the presence of Isl1 and LMO4, which block the formation of the Lhx3-complex that upregulates Chx10 (Fig. 1A) (Lee et al., 2008; Thaler et al., 2002). However, Chx10 effectively drives V2aIN differentiation in the MN area (Fig. 3A). The ability of Chx10 to suppress the activity of Isl1-Lhx3 likely enables Chx10 to effectively convert presumptive MNs into V2aINs. These findings demonstrate that MNs are inherently capable of activating V2aIN genes, and this property is unveiled when the MN program is suppressed by Chx10. Consistently, V2aIN program is aberrantly activated in MNs of Hb9-null mice (Fig. 7A,B). Our analysis of Hb9;Chx10 dKO mice shows that ectopic induction of V2aIN program in Hb9-null mice is substantially corrected by eliminating Chx10 (Fig. 7C,D). This data reinforces the idea that Chx10 is an active driver of V2aIN fate. It also indicates that the main function of Hb9 with regard to inhibition of V2aIN fate is to suppress the Chx10 gene. Indeed, we have previously found that Hb9 directly binds and represses the Chx10 gene (Lee et al., 2008).
The action of Chx10 in segregating MN and V2aIN fates is expected to be particularly important in V2aINs derived from the progenitor cells located in the boundary between pMN and p2 domains (Fig. 7E). The progenitors at the borderline have to interpret slightly different concentration of Shh and take on either pMN or p2 identities. Without Chx10, presumptive V2aINs may upregulate MN genes but fail to observe MN-periphery boundaries and get lost via the motor exit point, contributing to the reduction of V2aINs.
Our results revealed that Chx10 primarily functions as a transcriptional repressor to induce V2aIN fate, whereas Chx10 inhibits MN fate regardless of its inherent transcriptional activity (Fig. 5,6F,7F). Chx10 competes with Isl1-Lhx3 by binding the shared response elements linked to several MN genes. Consistently, our comparative ChIPseq analyses identified common binding sites for Isl1-Lhx3 and Chx10. Many of these sites control genes that play a role in MN fate specification, differentiation and function. Hb9, LMO and miR-218 are important to establish MN identity and MN-specific gene expression pattern (Amin et al., 2015; Arber et al., 1999; Lee et al., 2008; Thaler et al., 1999; Thiebes et al., 2015). Slit-Robo and Semaphorin-Neuropilin signaling pathways are important for MN cell body positioning and motor axon guidance (Bai et al., 2011; Bron et al., 2007; Hojae Lee et al., 2015). FGF, NCAM and NGFR signaling pathways play a role in MN survival (Garces et al., 2000; Nishimune et al., 2005; Xie et al., 2003). The Lix1 gene is deleted in autosomal recessive spinal muscular atrophy, a MN disease (Fyfe, 2006). These findings strongly support our model that Isl1-Lhx3 and Chx10 commonly target a range of MN genes and regulate their transcription oppositely (Fig. 7E). Given that we used stringent cut-off in comparative analyses of two ChIPseq and two RNAseq datasets, the common target MN genes of Isl1-Lhx3 and Chx10 are likely to be more than the 12 genes identified in this study.
Chx10 is likely to mobilize transcriptional activators to upregulate V2aIN genes, given that it acts as a transcriptional repressor in directing V2aIN fate. V2aIN-specific transcription factors downstream of Chx10, such as Sox14, may function as activators to upregulate V2aIN genes, forming a positive feedback loop to reinforce V2aIN fate choice. Alternatively, a relatively widely expressed neuronal transcription factor may be responsible for activating V2aIN genes, and Chx10 may be important to suppress the inhibitor of this transcriptional activator.
Importantly, our findings represent a generalizable phenomenon likely occurring during the organogenesis of many different tissues that require segregation of cell fates derived from closely related progenitor pools during development.
EXPERIMENTAL PROCEDURES
Mouse lines
The Chx10orJ/orJ and Hb9−/− mutant mice have been previously described (Burmeister et al., 1996; Thaler et al., 1999). Chx10orJ/orJ mice were obtained from the Jackson Laboratories (Bar Harbor, ME). Animals were housed and cared for according to IACUC guidelines.
Chick in ovo electroporation, Immunohistochemistry and in situ hybridization assay
In ovo electroporation, immunohistochemistry and in situ hybridization assays was performed as previously described on chick or mouse embryos cryosectioned at 13–18 μm (Lee et al., 2004). The following primary antibodies were used: Mouse anti-Hb9/MNR2 (on chick tissue, DSHB, 5C10), rabbit anti-Hb9 (on mouse tissue) (Thaler et al., 1999), rabbit anti-Isl1 (K5) (Tsuchida et al., 1994), guinea pig anti-Lhx3 (Sharma et al., 1998), guinea pig anti-Chx10 (Lee et al., 2008; Thaler et al., 1999), rabbit anti-Chx10 (Homemade), guinea pig anti-Sox14 (Homemade), rat anti-Gata3 (Absea Biotechnology Ltd., Beijing, China), rabbit anti-Evx1 (Moran-Rivard et al., 2001), mouse anti-Flag (Sigma) and rabbit anti-activated Caspase 3 (Cell Signaling Technology, #9661, Danvers, MA, USA). We generated the antibodies against Sox14 and Chx10, using the proteins corresponding to amino acids 1–154 of mouse Chx10 protein or 67–243 of chick Sox14 (NM_204761).
Culture, generation and differentiation of Chx10-inducible ESCs
Chx10-ESCs were generated from the A172LoxP ESC line (Iacovino et al., 2011). For MN differentiation assays, Chx10-ESC aggregates (embryoid bodies, EBs) were treated with all trans retinoic acid (0.5 μM) and a Shh agonist purmorphamine (1 μM; Calbiochem) for two days, and then cultured with either doxycycline (2 μg/mL) or vehicle for additional two to three days before the analyses.
RT-PCR, RNAseq and ChIPseq assays
Total RNA was extracted using RNeasy Mini kit (Qiagen) and reverse transcription was performed using SuperScript III (Invitrogen). RNA-seq libraries were prepared according to the Illumina TruSeq protocol, validated using the bioanalyzer and real-time PCR, and then sequenced on the Illumina HiSeq 2000. For ChIPseq, ChIP DNA samples from Chx10-ESC-derived MNs were prepared for sequencing according to the Illumina protocol, and sequenced on the Illumina HiSeq 2000. The peak calling was conducted with MACS software (Zhang et al., 2008). MEME-ChIP Suite (Bailey et al., 2009; Machanick and Bailey, 2011) and TOMTOM algorithm (Gupta et al., 2007) was used for motif analysis.
Statistical analysis
Statistical differences were determined by two-tailed Student t test. Statistical significance is displayed as * (P < 0.05), ** (P < 0.01), *** (P < 0.005), ns (non-significant).
Supplementary Material
Highlights.
Chx10 is required for Lhx3 to specify V2a interneurons
Chx10 upregulates the expression of V2a interneuron genes
Chx10 suppresses the expression of non-V2a interneuron and motor neuron genes
These activities enable Chx10 to effectively consolidate the V2a pathway
ACKNOWLEDGEMENTS
We are grateful to Younjung Park for the excellent technical support to raise antibodies and manage mouse colonies; to Dr. Samuel P. Pfaff for Hb9 mutant mice; to Dr. Anthony P. Barnes for critically reading our manuscript. This research was supported by grants from NIH/NINDS (R01 NS054941), the American Heart Association Award (to S.-K.Lee), NIH/NIDDK (R01 DK064678) (to J.W.Lee), NIH/NIDDK (R01 DK103661) (to J.W.Lee and S.-K.Lee), and Basic Science Research Program (NRF-2015R1A2A1A15055611) and Bio & Medical Technology Development Program (NRF-2012M3A9C6050508) and the Global Core Research Center (GCRC) funded by the Korean government (MSIP) (2011-0030001) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and future Planning.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes 3 figures, 4 tables, and supplemental experimental procedures.
ACCESSION NUMBERS
ChIP-seq and RNA-seq data are deposited under accession number GSE83874.
REFERENCES
- Amin ND, Bai G, Klug JR, Bonanomi D, Pankratz MT, Gifford WD, Hinckley CA, Sternfeld MJ, Driscoll SP, Dominguez B, Lee K-F, Jin X, Pfaff SL, 2015. Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science 350, 1525–1529. doi: 10.1126/science.aad2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S, Han B, Mendelsohn M, Smith M, Jessell TM, Sockanathan S, 1999. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. NEURON 23, 659–674. [DOI] [PubMed] [Google Scholar]
- Bai G, Chivatakarn O, Bonanomi D, Lettieri K, Franco L, Xia C, Stein E, Ma L, Lewcock JW, Pfaff SL, 2011. Presenilin-dependent receptor processing is required for axon guidance. Cell 144, 106–118. doi: 10.1016/j.cell.2010.11.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS, 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–8. doi: 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron R, Vermeren M, Kokot N, Andrews W, Little GE, Mitchell KJ, Cohen J, 2007. Boundary cap cells constrain spinal motor neuron somal migration at motor exit points by a semaphorin-plexin mechanism. Neural Dev 2, 21. doi: 10.1186/1749-8104-2-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister M, Novak J, Liang MY, Basu S, Ploder L, Hawes NL, Vidgen D, Hoover F, Goldman D, Kalnins VI, Roderick TH, Taylor BA, Hankin MH, McInnes RR, 1996. Ocular retardation mouse caused by Chx10 homeobox null allele: impaired retinal progenitor proliferation and bipolar cell differentiation. Nat Genet 12, 376–384. doi: 10.1038/ng0496-376 [DOI] [PubMed] [Google Scholar]
- Catela C, Shin MM, Dasen JS, 2015. Assembly and function of spinal circuits for motor control. Annu Rev Cell Dev Biol 31, 669–698. doi: 10.1146/annurev-cellbio-100814-125155 [DOI] [PubMed] [Google Scholar]
- Cho H-H, Cargnin F, Kim Y, Lee B, Kwon R-J, Nam H, Shen R, Barnes AP, Lee JW, Lee S, Lee S-K, 2014. Isl1 Directly Controls a Cholinergic Neuronal Identity in the Developing Forebrain and Spinal Cord by Forming Cell Type-Specific Complexes. PLoS Genet 10, e1004280. doi: 10.1371/journal.pgen.1004280.s007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow RL, Snow B, Novak J, Looser J, Freund C, Vidgen D, Ploder L, McInnes RR, 2001. Vsx1, a rapidly evolving paired-like homeobox gene expressed in cone bipolar cells. Mech Dev 109, 315–322. [DOI] [PubMed] [Google Scholar]
- Dorval KM, Bobechko BP, Ahmad KF, Bremner R, 2005. Transcriptional activity of the paired-like homeodomain proteins CHX10 and VSX1. J Biol Chem 280, 10100–10108. doi: 10.1074/jbc.M412676200 [DOI] [PubMed] [Google Scholar]
- Dougherty KJ, Zagoraiou L, Satoh D, Rozani I, Doobar S, Arber S, Jessell TM, Kiehn O, 2013. Locomotor rhythm generation linked to the output of spinal shox2 excitatory interneurons. NEURON 80, 920–933. doi: 10.1016/j.neuron.2013.08.015 [DOI] [PubMed] [Google Scholar]
- Ericson J, Rashbass P, Schedl A, Brenner-Morton S, Kawakami A, van Heyningen V, Jessell TM, Briscoe J, 1997. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell 90, 169–180. [DOI] [PubMed] [Google Scholar]
- Ericson J, Thor S, Edlund T, Jessell TM, Yamada T, 1992. Early stages of motor neuron differentiation revealed by expression of homeobox gene Islet-1. Science 256, 1555–1560. [DOI] [PubMed] [Google Scholar]
- Fyfe JC, 2006. An 140-kb deletion associated with feline spinal muscular atrophy implies an essential LIX1 function for motor neuron survival. Genome Res 16, 1084–1090. doi: 10.1101/gr.5268806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garces A, Nishimune H, Philippe J, Pettmann B, deLapeyrière O, 2000. FGF9: a motoneuron survival factor expressed by medial thoracic and sacral motoneurons. J Neurosci Res 60, 1–9. [DOI] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS, 2007. Quantifying similarity between motifs. Genome Biol 8, R24. doi: 10.1186/gb-2007-8-2-r24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovino M, Bosnakovski D, Fey H, Rux D, Bajwa G, Mahen E, Mitanoska A, Xu Z, Kyba M, 2011. Inducible cassette exchange: a rapid and efficient system enabling conditional gene expression in embryonic stem and primary cells. Stem Cells 29, 1580–1588. doi: 10.1002/stem.715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi K, Lee S, Lee B, Lee J, Lee S, 2009. LMO4 controls the balance between excitatory and inhibitory spinal V2 interneurons. NEURON 61, 839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Satou C, Higashijima S-I, 2008. V2a and V2b neurons are generated by the final divisions of pair-producing progenitors in the zebrafish spinal cord. Development 135, 3001–3005. doi: 10.1242/dev.024802 [DOI] [PubMed] [Google Scholar]
- Lee Hojae, Kim M, Kim N, Macfarlan T, Pfaff SL, Mastick GS, Song M-R, 2015. Slit and Semaphorin signaling governed by Islet transcription factors positions motor neuron somata within the neural tube. Exp Neurol 269, 17–27. doi: 10.1016/j.expneurol.2015.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee B, Joshi K, Pfaff S, Lee J, Lee S, 2008. A regulatory network to segregate the identity of neuronal subtypes. Dev Cell 14, 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Seunghee, Cuvillier JM, Lee B, Shen R, Lee JW, Lee S-K, 2012. Fusion protein Isl1-Lhx3 specifies motor neuron fate by inducing motor neuron genes and concomitantly suppressing the interneuron programs. Proceedings of the National Academy of Sciences 109, 3383–3388. doi: 10.1073/pnas.1114515109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Seunghee, Shen R, Cho H-H, Kwon R-J, Seo SY, Lee JW, Lee S-K, 2013. STAT3 promotes motor neuron differentiation by collaborating with motor neuron-specific LIM complex. Proceedings of the National Academy of Sciences 110, 11445–11450. doi: 10.1073/pnas.1302676110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Pfaff S, 2001. Transcriptional networks regulating neuronal identity in the developing spinal cord. Nat Neurosci 4, 1183–1191. [DOI] [PubMed] [Google Scholar]
- Lee Soo-Kyung, Pfaff SL, 2003. Synchronization of neurogenesis and motor neuron specification by direct coupling of bHLH and homeodomain transcription factors. NEURON 38, 731–745. [DOI] [PubMed] [Google Scholar]
- Lee Soo-Kyung, Jurata LW, Funahashi J, Ruiz EC, Pfaff SL, 2004. Analysis of embryonic motoneuron gene regulation: derepression of general activators function in concert with enhancer factors. Development 131, 3295–3306. doi: 10.1242/dev.01179 [DOI] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH, 2002. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 109, 75–86. [DOI] [PubMed] [Google Scholar]
- Machanick P, Bailey TL, 2011. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. doi: 10.1093/bioinformatics/btr189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Closser M, Morrison CA, Nedelec S, Williams DJ, An D, Gifford DK, Wichterle H, 2013. Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nat Neurosci 16, 1219–1227. doi: 10.1038/nn.3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Rivard L, Kagawa T, Saueressig H, Gross MK, Burrill J, Goulding M, 2001. Evx1 is a postmitotic determinant of v0 interneuron identity in the spinal cord. NEURON 29, 385–399. [DOI] [PubMed] [Google Scholar]
- Nishimune H, Bernreuther C, Carroll P, Chen S, Schachner M, Henderson CE, 2005. Neural adhesion molecules L1 and CHL1 are survival factors for motoneurons. J Neurosci Res 80, 593–599. doi: 10.1002/jnr.20517 [DOI] [PubMed] [Google Scholar]
- Novitch B, Chen A, Jessell T, 2001. Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. NEURON 31, 773–789. [DOI] [PubMed] [Google Scholar]
- Pfaff S, Mendelsohn M, Stewart C, Edlund T, 1996. Requirement for LIM Homeobox Gene Isl1 in Motor Neuron Generation Reveals a Motor Neuron–Dependent Step in Interneuron Differentiation. Cell. [DOI] [PubMed] [Google Scholar]
- Seo SY, Lee B, Lee S, 2015. Critical Roles of the LIM Domains of Lhx3 in Recruiting Coactivators to the Motor Neuron-Specifying Isl1-Lhx3 Complex. Mol Cell Biol 35, 3579–3589. doi: 10.1128/MCB.00335-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Sheng H, Lettieri K, Li H, Karavanov A, Potter S, Westphal H, Pfaff S, 1998. LIM homeodomain factors Lhx3 and Lhx4 assign subtype identities for motor neurons. Cell 95, 817–828. [DOI] [PubMed] [Google Scholar]
- Song M-R, Sun Y, Bryson A, Gill GN, Evans SM, Pfaff SL, 2009. Islet-to-LMO stoichiometries control the function of transcription complexes that specify motor neuron and V2a interneuron identity. Development 136, 2923–2932. doi: 10.1242/dev.037986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y, William C, Jessell TM, 1998. Specification of motor neuron identity by the MNR2 homeodomain protein. Cell 95, 67–80. [DOI] [PubMed] [Google Scholar]
- Thaler J, Harrison K, Sharma K, Lettieri K, Kehrl J, Pfaff SL, 1999. Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. NEURON 23, 675–687. [DOI] [PubMed] [Google Scholar]
- Thaler JP, Lee S-K, Jurata LW, Gill GN, Pfaff SL, 2002. LIM factor Lhx3 contributes to the specification of motor neuron and interneuron identity through cell-type-specific protein-protein interactions. Cell 110, 237–249. [DOI] [PubMed] [Google Scholar]
- Thiebes KP, Nam H, Cambronne XA, Shen R, Glasgow SM, Cho H-H, Kwon J-S, Goodman RH, Lee JW, Lee S, Lee S-K, 2015. miR-218 is essential to establish motor neuron fate as a downstream effector of Isl1–Lhx3. Nat Commun 6, 1–14. doi: 10.1038/ncomms8718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida T, Ensini M, Morton SB, Baldassare M, Edlund T, Jessell TM, Pfaff SL, 1994. Topographic organization of embryonic motor neurons defined by expression of LIM homeobox genes. Cell 79, 957–970. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter J, Jessell T, 2002. Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397. [DOI] [PubMed] [Google Scholar]
- Wilson D, Sheng G, Lecuit T, Dostatni N, Desplan C, 1993. Cooperative dimerization of paired class homeo domains on DNA. Genes Dev 7, 2120–2134. [DOI] [PubMed] [Google Scholar]
- Xie Y, Yao Z, Chai H, Wong W-M, Wu W, 2003. Expression and role of low-affinity nerve growth factor receptor (p75) in spinal motor neurons of aged rats following axonal injury. Dev Neurosci 25, 65–71. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. doi: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Anderson D, 2002. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell 109, 61–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.