

Abstract
A low-frequency variant of sushi, von Willebrand factor type A, EGF and pentraxin domain containing protein 1 (SVEP1), an extracellular matrix protein, is associated with risk of coronary disease in humans independent of plasma lipids. Despite a robust statistical association, if and how SVEP1 might contribute to atherosclerosis remained unclear. Here, using Mendelian randomization and complementary mouse models, we provide evidence that SVEP1 promotes atherosclerosis in humans and mice and is expressed by vascular smooth muscle cells (VSMCs) within the atherosclerotic plaque. VSMCs also interact with SVEP1, causing proliferation and dysregulation of key differentiation pathways, including integrin and Notch signaling. Fibroblast growth factor receptor transcription increases in VSMCs interacting with SVEP1, and is further increased by the coronary disease-associated SVEP1 variant p.D2702G. These effects ultimately drive inflammation and promote atherosclerosis. Taken together, our results suggest that VSMC-derived SVEP1 is a pro-atherogenic factor, and support the concept that pharmacological inhibition of SVEP1 should protect against atherosclerosis in humans.
One Sentence Summary:
Reducing SVEP1 confers protection from atherosclerosis and may be a therapeutic target for the treatment and prevention of coronary artery disease.
Introduction
Cardiometabolic diseases are leading causes of morbidity and mortality and their prevalence is increasing (1–8). Although approved treatments can help ameliorate these diseases, residual disease risk remains a substantial problem. Statin medications, for example, lower plasma cholesterol concentrations and reduce risk of coronary events by 20–30% (9), highlighting both substantial residual risk and an unmet need for identifying alternative treatment strategies. Human genetics is a powerful approach to uncover potential therapeutic targets and to date more than 160 loci have been robustly associated with coronary artery disease (CAD) (10). At most loci, however, the causal gene is unknown, presenting a major bottleneck and hindering the translation of these findings into new therapies. We previously performed a large-scale exome-wide association study of low-frequency protein altering variation in an attempt to identify genes for CAD and discovered a highly conserved missense polymorphism in SVEP1 (p.D2702G) that associated with an increased risk of disease (Odds Ratio = 1.14 per risk allele) (11). This CAD risk variant (hereafter referred to as SVEP1CADrv) was not associated with an effect on plasma lipids but had a modest positive association with blood pressure and type 2 diabetes (11), suggesting this variant may broadly contribute to the progression of cardiometabolic disease.
SVEP1, also known as polydom, encodes a large extracellular matrix protein with sushi (complement control protein), von Willebrand factor type A, epidermal growth factor-like (EGF), and pentraxin domains (12, 13). This gene was originally discovered in a screen for Notch-interacting proteins, as it contains Notch-like repeat EGF-domains (13). The only protein currently known to directly interact with SVEP1 is integrin α9β1 (14), a provisional matrix-binding integrin that is linked to increased blood pressure in humans (15, 16). Integrin α9β1 binds to the same protein domain that harbors the variant residue in SVEP1CADrv (14) and both proteins also play critical roles in development, including lymphatic patterning (17, 18).
Despite strong statistical evidence linking SVEP1 with CAD, its direct causality and potential disease-associated mechanisms were unclear. Here, we sought to determine if and how SVEP1 may influence the development of atherosclerosis. Given the overlapping disease associations between SVEP1 and integrin α9β1, their shared biological functions, and the proximity of the variant to integrin α9β1’s binding site, we focused our mechanistic studies on cell types that play a prominent role in atherosclerosis and express either SVEP1, Integrin alpha-9 (ITGA9) , or both of these genes.
Results
SVEP1 is expressed by arterial VSMCs under pathological conditions
To begin characterizing the role of SVEP1 in the pathogenesis of atherosclerosis, we searched for disease-relevant tissues and cell types that express SVEP1. Expression data from the Genotype-Tissue Expression (GTEx) project indicated that human arterial tissue, including coronary arteries, express SVEP1 (fig. S1A). To confirm arterial expression, we used in situ hybridization on tissue explants from the aortic wall and left internal mammary artery (LIMA) of patients with established coronary artery disease. SVEP1 expression was detected within cells staining with the vascular smooth muscle cell marker smooth muscle α-actin (SMα-actin) (Fig. 1A). VSMCs are known to increase synthesis of certain extracellular matrix proteins in response to various pathological stimuli (19); therefore, we assessed expression data from relevant disease specimens to determine if this also applies to SVEP1. Indeed, SVEP1 expression was higher within human atherosclerotic tissue from carotid explants relative to patient-paired adjacent and macroscopically intact tissue (20) (fig. S1B). Athero-prone arterial tissue explants from patients with diabetes also expressed more SVEP1 compared to patients without diabetes (19) (fig. S1C).
Fig. 1. SVEP1 is expressed by VSMCs under pathological conditions.
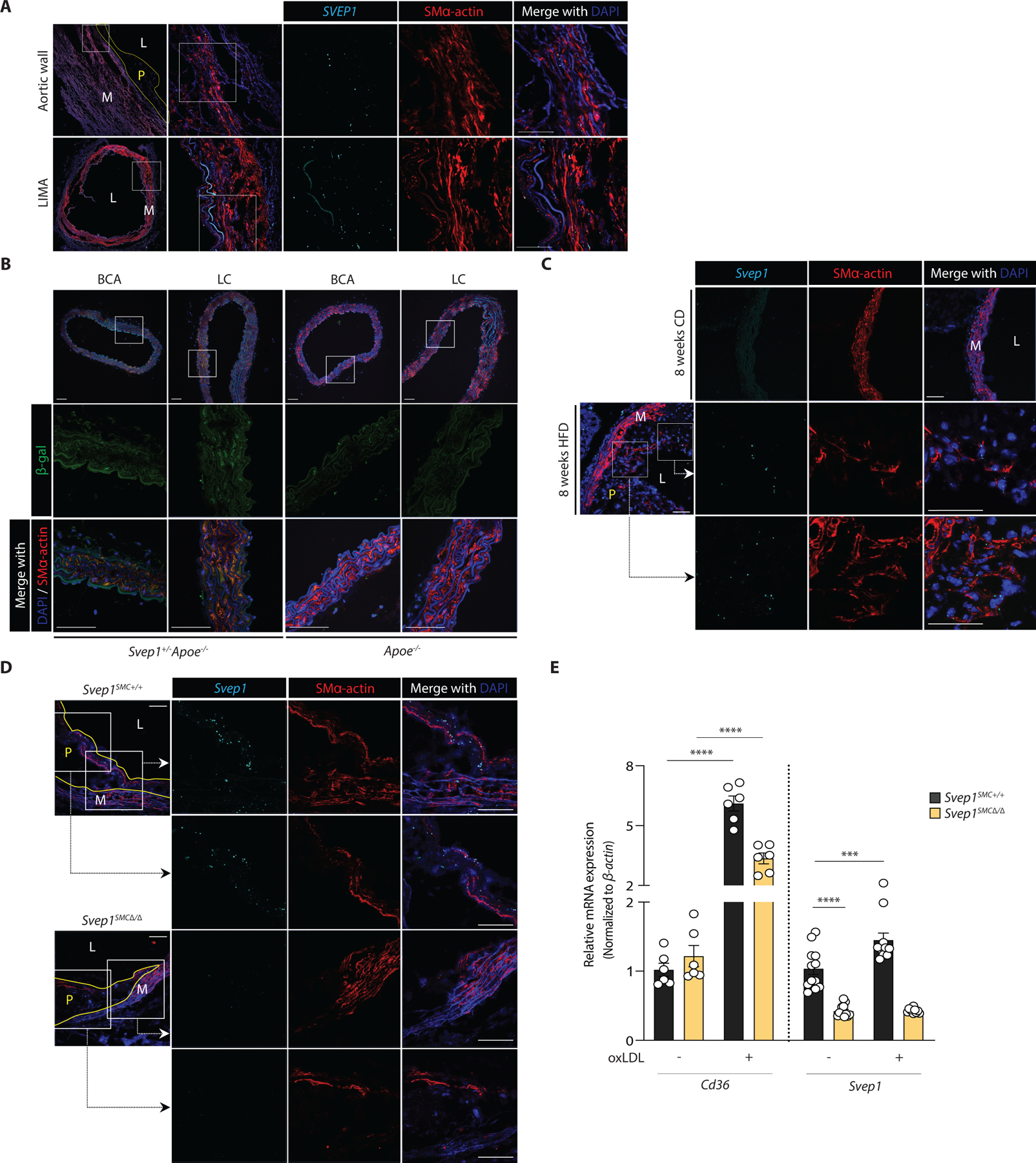
(A) Expression of SVEP1 in human aortic wall and LIMA cross-sections from patients using in situ hybridization (ISH).
(B) β-gal expression in the aortic root, BCA (brachiocephalic artery), LC (lesser curvature) from 8-week-old Svep1+/−Apoe−/− and Apoe−/− mice.
(C) Expression of Svep1 using ISH in the aortic root from young (8-week-old) CD-fed and 8 weeks of HFD-fed Apoe−/− mice.
(D) Expression of Svep1 using ISH in the aortic root from Svep1SMC+/+ and Svep1SMC∆/∆ mice after 8 weeks of HFD feeding. Outlined areas indicate the regions magnified in the next panels. Tissues in (A-D) were co-stained with the VSMC marker, SMα-actin. Scale bars, 50 µm. M, media; L, lumen; P, plaque.
(E) Cd36 and Svep1 expression in primary VSMCs from Svep1SMC+/+ and Svep1SMC∆/∆ mice with or without the addition of oxLDL for 48 hr (n = 6–12/group, N = 2). Data were analyzed with an unpaired nonparametric Mann-Whitney test. The bar graphs depict the mean ± SEM. ***P < 0.001; ****P < 0.0001.
To determine whether murine Svep1 expression recapitulated human SVEP1 expression, and may therefore be a viable animal model to study its effects on disease, we obtained mice expressing a lacZ reporter under the native Svep1 promotor of a single allele because mice with homozygous Svep1 deficiency have developmental defects and die from edema at embryonic day 18.5 (17, 21). Within healthy arterial tissue of young mice, we observed low β-gal expression, mostly colocalizing with VSMCs (Fig. 1B). These data are consistent with published single-cell studies that identify VSMCs within the healthy murine aorta as a minor source of Svep1 expression (22) (fig. S1D). To determine if murine Svep1 expression was increased in the development of atherosclerosis, as in humans, we assayed expression within mouse arterial tissue after inducing experimental atherosclerosis by feeding atheroprone (Apoe−/−) mice a Western, high-fat diet (HFD) for 8 weeks. Apoe−/− mice fed a standard chow diet (CD) served as non-atherogenic controls. After 8 weeks of an atherogenic HFD, we observed a 2-fold increase in Svep1 expression relative to CD fed control mice (Fig. 1C, S1E). This expression was colocalized with neointimal cells that co-stained with SMα-actin, suggesting VSMC expression (Fig. 1C).
Multiple cell types have been demonstrated to gain expression of VSMC markers in the context of atherosclerosis (23). Therefore, to test the hypothesis that VSMC-derived cells within the neointima are the predominate source of SVEP1, we generated Apoe−/− mice with VSMC-specific knockout of Svep1 (Svep1flox/floxMyh11-CreERT2Apoe−/−; hereafter referred to as Svep1SMC∆/∆) and mice with unaltered Svep1 expression (Svep1+/+Myh11-CreERT2Apoe−/−; hereafter referred to as Svep1SMC+/+), which served as controls. Svep1 expression was assessed using in situ hybridization within the neointima of the aortic root of both groups after 8 weeks of HFD feeding. Indeed, while we observed robust Svep1 expression in control mice, neointimal Svep1 expression was nearly undetectable in Svep1SMC∆/∆ mice (Fig. 1D, S1F). These data indicate that VSMC-derived cells are the predominant source of SVEP1 in atherosclerotic plaque.
Given the increased expression of Svep1 under atherosclerotic conditions in mice and humans, we tested the ability of atheroma-associated oxidized low-density lipoprotein (oxLDL) to directly induce Svep1 expression in VSMCs. Exposure to oxLDL increased Svep1 expression by 48% in primary VSMCs from Svep1SMC+/+ mice but not Svep1SMC∆/∆ mice, compared to vehicle-treated control cells (Fig. 1E). Both Svep1SMC+/+ and Svep1SMC∆/∆ cells increased expression of Cd36, indicating they were activated upon binding of oxLDL with its receptor (24). Exposure to oxLDL also modestly induced SVEP1 expression in human primary coronary artery smooth muscle cells (CASMCs, fig. S1G).
Taken together, these data demonstrate that SVEP1 is produced locally by VSMCs in atherosclerotic disease and are consistent with prior studies which concluded that SVEP1 is produced by cells of mesenchymal origin (17, 21). Further, these data suggest that SVEP1 may play a direct role in the pathogenesis of atherosclerosis and that mouse models are an appropriate means to interrogate this question.
SVEP1 drives atherosclerotic plaque development
To study the effect of Svep1 on atherosclerosis, we fed Apoe−/− and Svep1+/−Apoe−/− mice a HFD for 8 weeks and analyzed the resulting atherosclerotic plaque burden. There were no observed differences between genotypes in body weight, plasma total cholesterol, triglycerides, and glucose (Fig. 2A, B). Relative to controls, however, Svep1+/−Apoe−/− mice had a reduction in plaque burden (as characterized by the percentage of surface area staining positive with Oil Red O) in the aortic arch and whole aorta by en face preparations, as well as in sectioned aortic roots (Fig. 2C, D). The effect of Svep1 haploinsufficiency on reducing the development of atherosclerotic plaque in the aortic arch and en face aorta was notably greater than in the aortic root, perhaps reflecting differences in VSMC embryonic origin and biology (25). Svep1 haploinsufficiency also resulted in reduced macrophage staining within the aortic root neointima, as determined by the percentage of area staining positive for Mac3 (Fig. 2E). We did not appreciate marked differences in measures of plaque stability, such as area staining positive for VSMC markers or necrotic core size, although collagen content was modestly higher in atheromas from control mice compared to Svep1+/−Apoe−/− mice (fig. S2).
Fig. 2. Svep1 haploinsufficiency abrogates atherosclerosis.
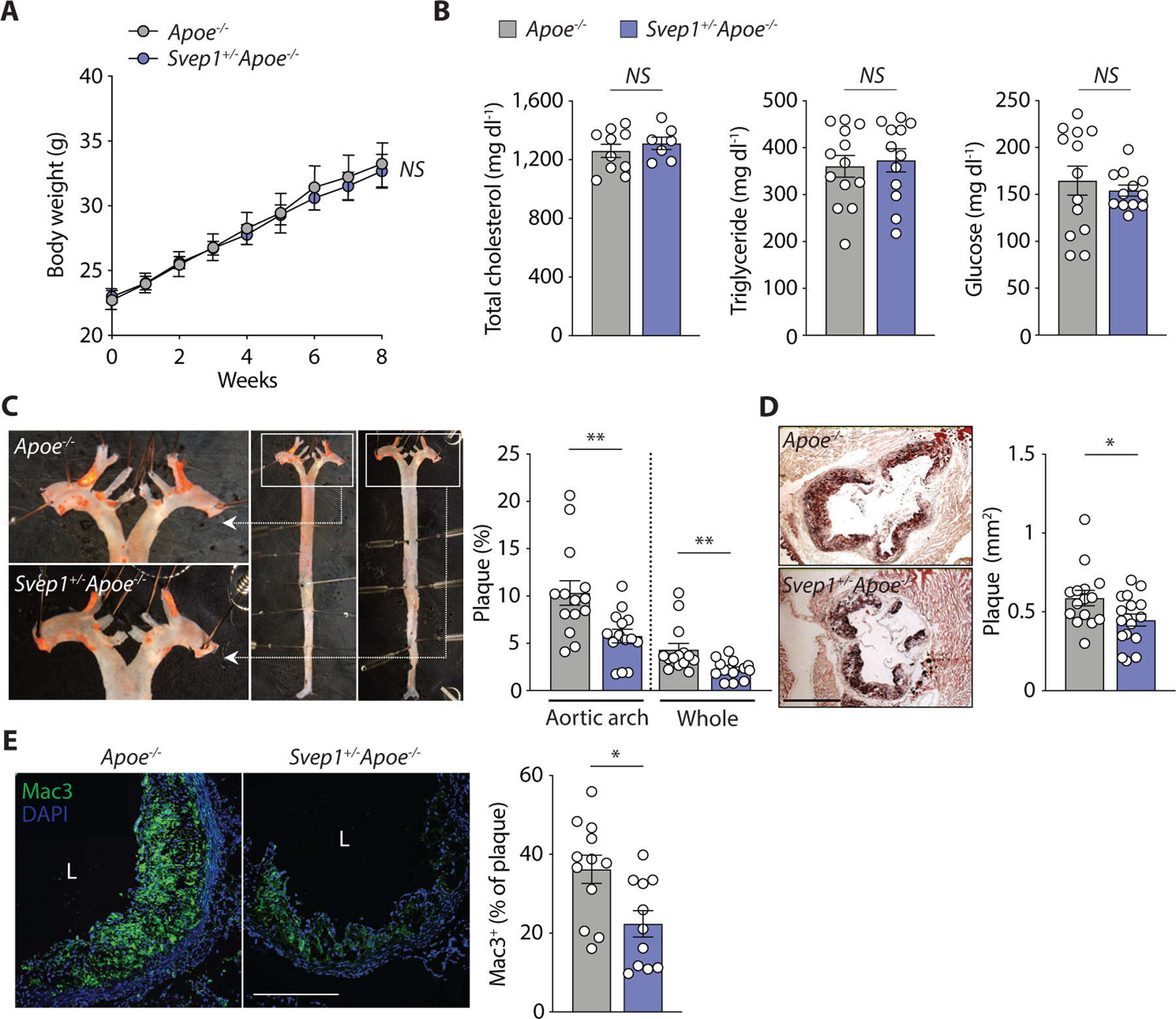
(A) Body weight of Apoe−/− and Svep1+/−Apoe−/− mice during HFD feeding.
(B) Plasma total cholesterol, triglycerides, and glucose of mice after HFD feeding.
(C) En face Oil Red O-stained murine aortas. Outlined areas indicate the aortic arch regions magnified in left panels. Quantification of Oil Red O-stained area in each aortic arch and whole artery.
(D) Oil Red O-stained murine aortic root cross-sections. Quantification of Oil Red O-stained area. Scale bar, 500 µm.
(E) Mac3 staining in murine aortic root sections. Quantification of Mac3 as a percentage of plaque area. Scale bar, 200 µm. M, media; L, lumen; P, plaque. n = 7–17/group (A-E). Data were analyzed with a one-way ANOVA (A) or unpaired nonparametric Mann-Whitney test (B-E). The bar graphs depict the mean ± SEM. *P < 0.05; **P < 0.01; NS, not significant.
We then tested the hypothesis that the atherogenic effects of SVEP1 could be attributed to its synthesis by VSMCs using Svep1SMC∆/∆ and Svep1SMC+/+ mice, as previously described. As with Svep1 haploinsufficiency, loss of Svep1 in VSMCs did not significantly alter body weight, plasma cholesterol, triglycerides and glucose concentrations (Fig. 3A, B) following 8 weeks of HFD feeding. Also consistent with our Svep1 haploinsufficiency model, Svep1SMC∆/∆ mice had decreased plaque burden plaque in the aortic arch, whole aorta, and aortic root (Fig. 3C, D), as compared to Svep1SMC+/+ control mice. Additionally, atheromas from Svep1SMC∆/∆ mice contained less macrophage staining and necrotic core area, indicators of plaque instability, and unaltered collagen content (fig. S3A-C).
Fig. 3. VSMC-specific Svep1 deficiency reduces atherosclerosis and plaque complexity.
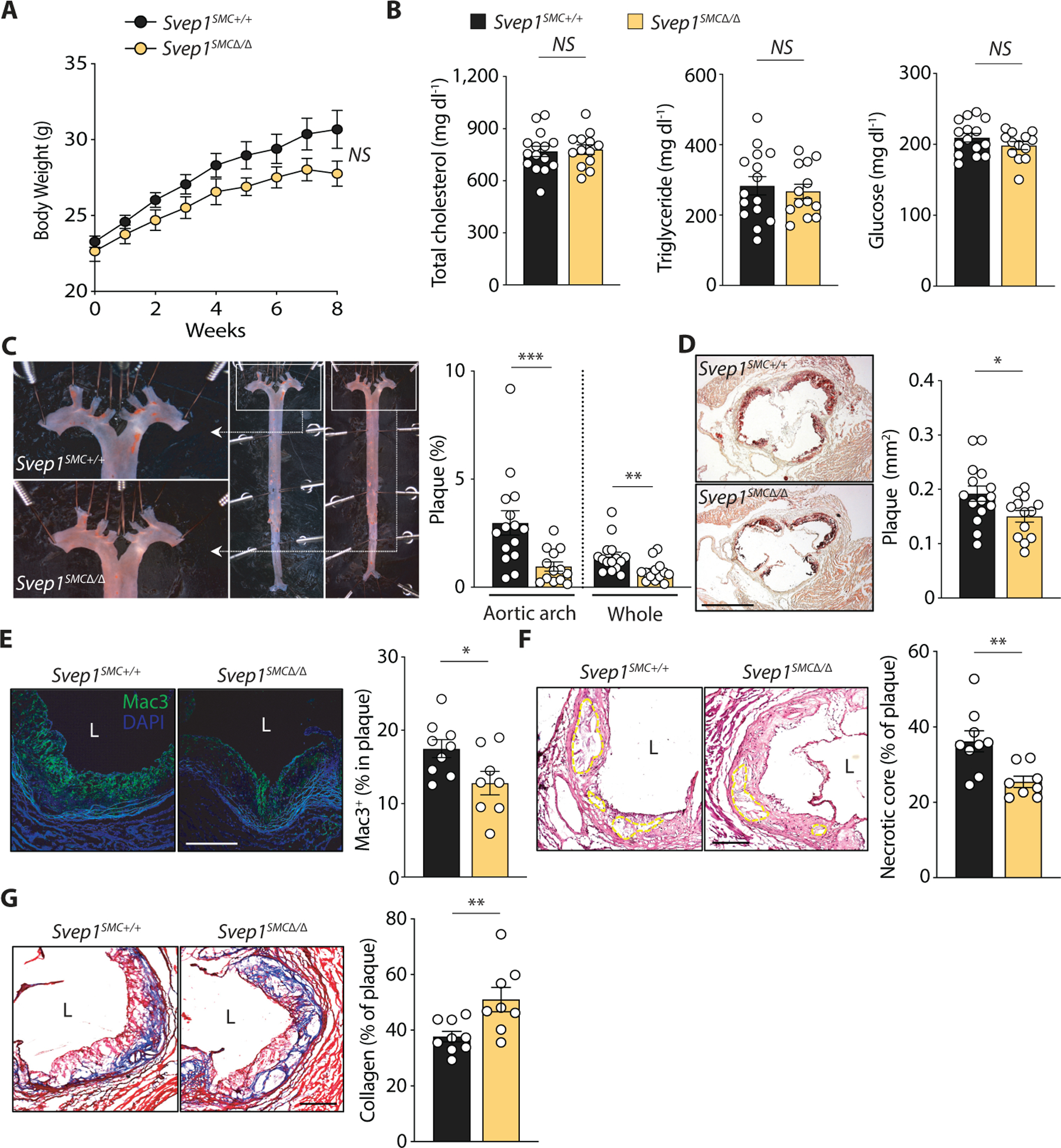
(A) Body weight of Svep1SMC+/+ and Svep1SMC∆/∆ mice during HFD feeding.
(B) Total plasma cholesterol, triglycerides, and glucose in mice.
(C) En face Oil Red O-stained murine aortas. Outlined areas indicate the aortic arch regions magnified in left panels. Quantification of Oil Red O-stained area in each aortic arch and whole artery.
(D) Oil Red O-stained murine aortic root cross-sections. Quantification of Oil Red O-stained area. Scale bar, 500 µm.
(E) Mac3 staining of murine aortic roots. Quantification of Mac3 as a percentage of plaque area.
(F) Necrotic core of murine aortic roots outlined on H&E-stained sections. Quantification of necrotic core as a percentage of plaque area.
(G) Collagen staining of murine aortic roots using Masson’s trichrome stain. Quantification of collagen as a percentage of plaque area. Scale bars, 200 µm. M, media; L, lumen; P, plaque. n = 13–15/group (A- D) or 8–9/group (E-G). Data were analyzed with a one-way ANOVA (A) or unpaired nonparametric Mann-Whitney test (B-G). The bar graphs depict the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant.
Given the observations that loss of Svep1 in VSMCs resulted in a dramatic reduction in plaque size in the setting of 8 weeks of HFD feeding, we extended the length of plaque development to investigate the effect of SVEP1 on advanced plaque lesions. After treatment with tamoxifen, Svep1SMC+/+and Svep1SMC∆/∆ mice were fed HFD for 16 weeks. Again, no differences were observed in body weight (fig. S3D), plasma cholesterol, and glucose concentrations (fig. S3E) between groups. Triglycerides were higher in the Svep1SMC∆/∆ mice with nominal significance (P = 0.046), but this was not observed at other timepoints or in the haploinsufficiency model. Although we did not detect a statistically significant effect of VSMC-specific Svep1 deletion on atherosclerotic plaque burden (fig. S3F, G), plaques from Svep1SMC∆/∆ mice tended to be smaller and were both less complex and more stable than controls. These indicators of an altered plaque phenotype include decreased neointimal macrophage staining (Fig. 3E) and necrotic core size (Fig. 3F), in addition to greater collagen content (Fig. 3G). Taken together, these experimental atherosclerosis data suggest that SVEP1 drives atherosclerosis and increases plaque complexity in mice.
SVEP1 is causally related to cardiometabolic disease in humans
Due to the relationship we discovered between SVEP1 depletion and reduced atherosclerosis across our mouse models, we wondered if the human SVEP1 CAD-associated D2072G missense polymorphism was associated with altered SVEP1 expression in humans. We did not find that this allele (or other alleles in linkage disequilibrium) associated with changes in SVEP1 mRNA expression in GTEx (fig. S4A), however we did find that the 2702G risk variant (SVEP1CADrv) was associated with a significant increase in circulating plasma SVEP1 protein concentration (P = 8 × 10−14; Fig. 4A) as measured by two independent aptamers (fig. S4B) from participants in the INTERVAL study (26), suggesting that increased SVEP1 protein concentrations were associated with increased risk of CAD. We then tested if this was true for other genetic variants influencing SVEP1 plasma protein concentrations. Using published data from the INTERVAL study (26), we cataloged cis-acting variants that associated with SVEP1 protein concentration at a genome-wide (P < 5 × 10−8) level of statistical significance (Fig. 4B). We performed Mendelian randomization (27) using a subset of these variants in linkage equilibrium (r2 < 0.3) and found that increased SVEP1 protein was causally related to increased CAD risk (P = 7 × 10−11; Fig. 4C, D). We also asked if SVEP1 protein concentration was causally related to increased risk for hypertension and type 2 diabetes due to the prior associations we observed for the SVEP1CADrv allele with these risk factors. Indeed, we found that increased SVEP1 protein was causally related to both hypertension (P = 2 × 10−15; fig. S4C) and type 2 diabetes (P = 0.0004; fig. S4D).
Fig. 4. Plasma SVEP1 is causally related to CAD in humans.
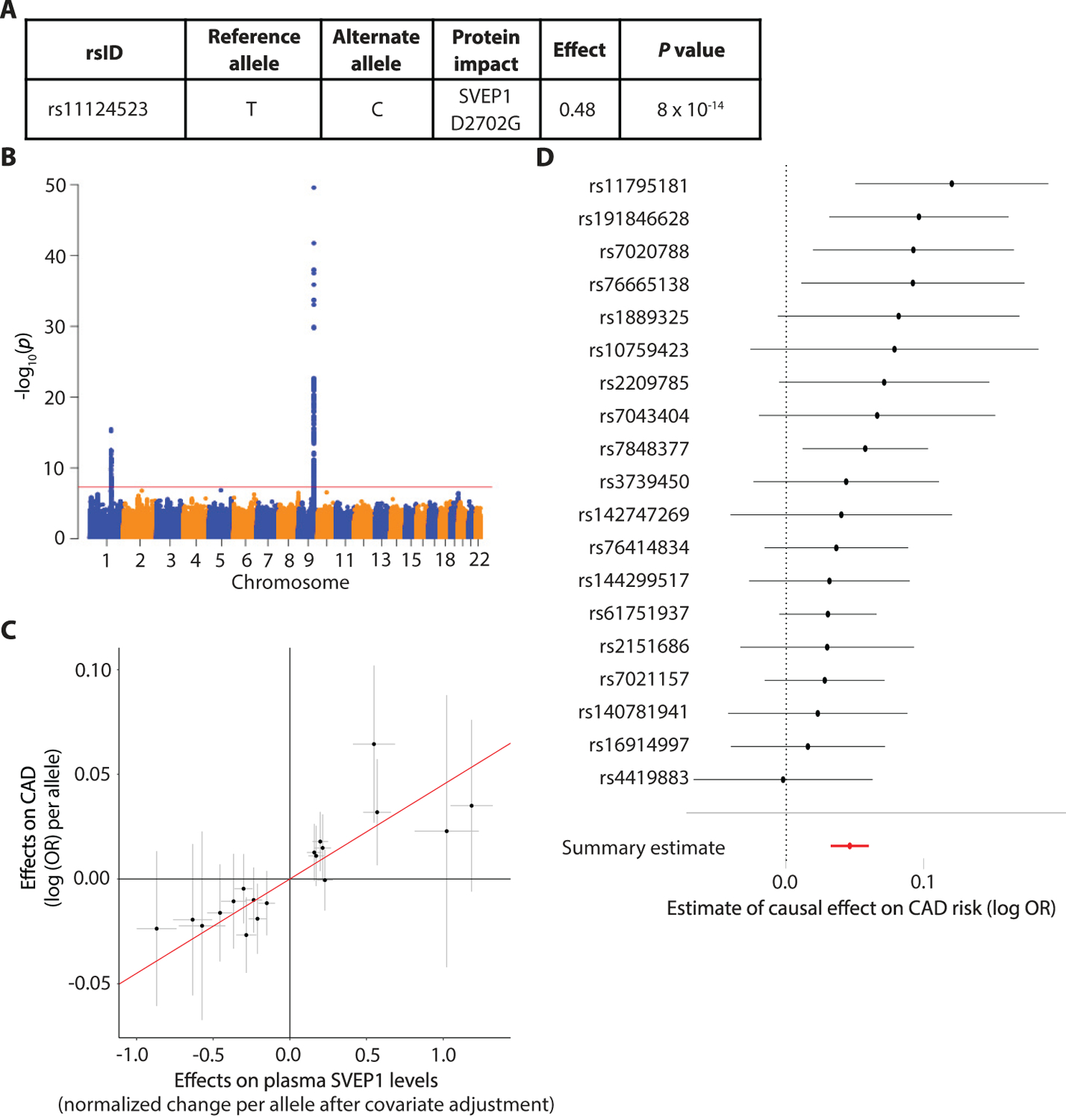
(A) The effect of the CAD-associated SVEP1 p.D2702G allele on plasma SVEP1 expression in humans. Effect refers to the change per alternative allele (2702G) in units of normalized protein concentration after adjusting for covariates as previously described (26).
(B) Genome-wide Manhattan plot for variants associated with plasma SVEP1 in humans. The –log10(P) of the association with SVEP1 concentration is plotted for each variant across the genome according to chromosomal position (X-axis). The red line indicates genome-wide significance (P < 5 × 10−8). The association peak on chromosome 9 overlies the SVEP1 locus.
(C) Estimated effect (with 95% confidence intervals) of each variant included in the Mendelian randomization analysis on plasma SVEP1 expression and CAD risk. The red line indicates the causal effect estimate (P = 7 × 10−11).
(D) The estimated causal effect (with 95% confidence intervals) of each SNP included in the Mendelian randomization analysis for a one unit increase in SVEP1 concentration is plotted along with the overall summary estimate from the causal analysis.
To investigate how the human SVEP1CADrv missense polymorphism might impact CAD risk, we generated homozygous mice harboring the human SVEP1CADrv at the orthologous murine position (Svep12699G/2699G; hereafter referred to as Svep1G/G). These mice were bred with Apoe−/− mice to generate Svep1G/GApoe−/− mice. We were not able to detect differences in body weight, serum total cholesterol, triglycerides, and glucose (fig. S4E-H) between groups after feeding HFD. We also did not appreciate a significant difference between groups in the development of atherosclerotic plaque at either 8 or 16 weeks of HFD feeding (fig. S4I, J). Although our prior human genetic study revealed a robust association with an increased risk of CAD, the effect of the SVEP1CADrv in humans was modest, in which each copy of the G allele was associated with a 14% increased risk of disease. If an effect size in mice is similarly modest, further investigation would require a very large number of animals, presenting both pragmatic and ethical barriers. To circumvent these concerns, subsequent functional interrogation of the SVEP1CADrv was performed in vitro.
SVEP1 induces proliferation and integrin signaling in VSMCs
To begin characterizing the mechanism by which SVEP1 drives atherosclerosis, we sought to identify receptors and associated cell types that interact with SVEP1 in the extracellular space. Integrin α9β1 is the only protein known to interact with SVEP1 and the two proteins colocalize in vivo (14). Integrins are transmembrane, heterodimeric receptors that respond to the extracellular environment and influence numerous aspects of atherosclerosis (28, 29). Therefore, we hypothesized that integrin α9β1 (and associated cell-types) may be involved in SVEP1-mediated atherosclerosis. The α9 subunit (ITGA9) is known to exclusively heterodimerize with β1 (ITGB1), therefore assessing ITGA9 expression is a reliable proxy for integrin α9β1 expression. Integrin α9β1 expression has been documented in airway epithelium, smooth muscle, skeletal muscle, hepatocytes, and epithelial cells (14, 30–36), yet arterial tissue expresses the most ITGA9 of all GTEx tissues (fig. S5A). In situ hybridization confirmed that ITGA9 is broadly expressed in the human aortic wall and LIMA, predominately colocalizing with VSMCs (Fig. 5A). Likewise, VSMCs of the murine aorta expressed Itgα9 (Fig. 5B). Consistent with these data, single cell studies of the murine aorta indicated that VSMCs express Itgα9 (fig. S5B) (22). Given the established role of VSMCs in CAD (23), their expression of integrin α9β1, and the local expression patterns of SVEP1 in disease, we tested the hypothesis that VSMCs respond to SVEP1 in a cell-autonomous manner to promote atherosclerosis.
Fig. 5. SVEP1 induces Itgα9-dependent proliferation in VSMCs.
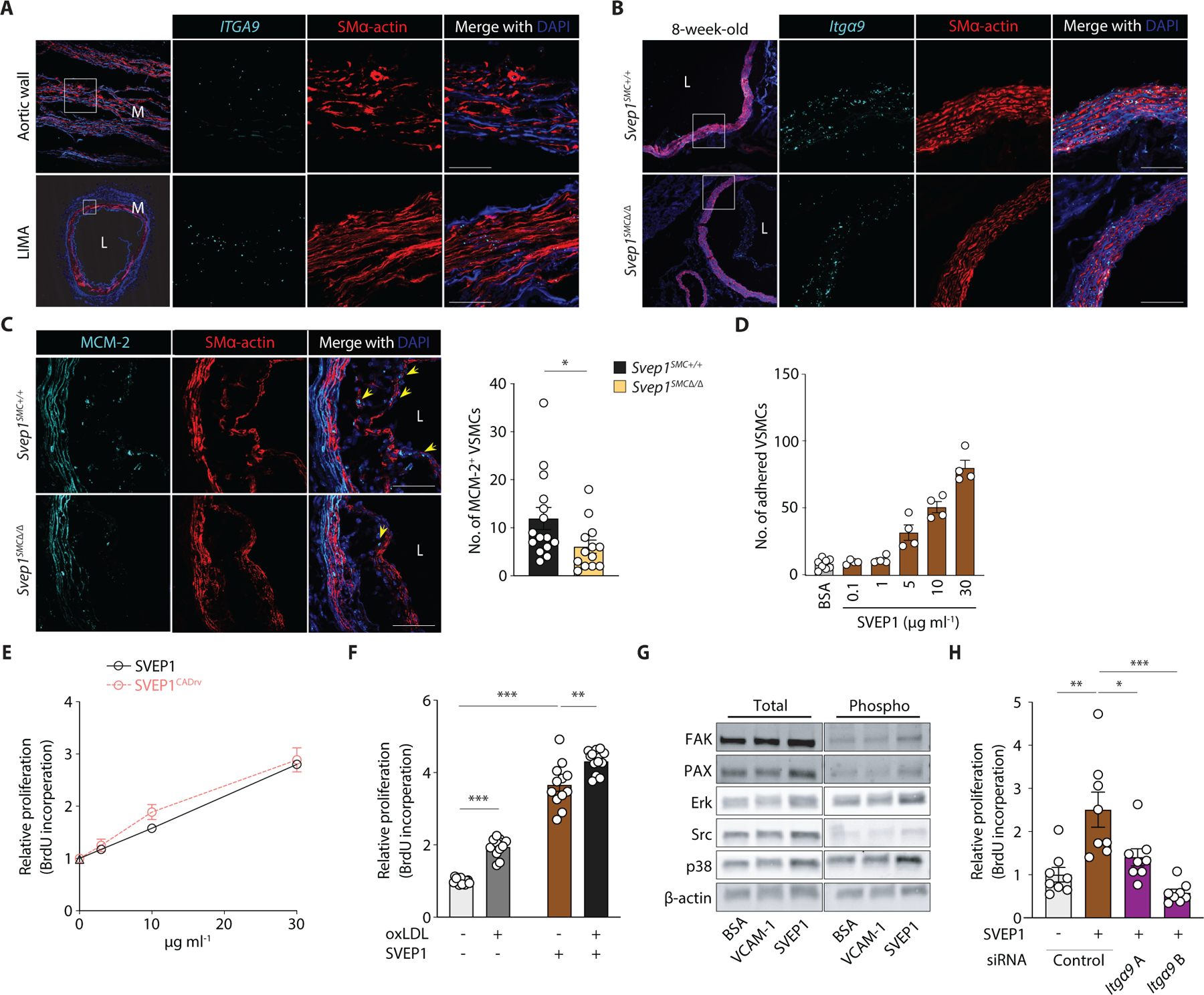
(A) ITGA9 expression in human aortic wall and LIMA cross-sections from patients using ISH. M, media; L, lumen.
(B) Expression of Itgα9 in the aortic root from 8-week-old Svep1SMC+/+ and Svep1SMC∆/∆ mice using ISH. Outlined areas indicate the regions magnified in the next panels. Scale bar, 50 µm.
(C) MCM-2 immunofluorescent staining of aortic root regions from Svep1SMC+/+ and Svep1SMC∆/∆ mice after 8 weeks of HFD feeding. Yellow arrows indicate MCM-2+/SMα-actin+ cells within plaque. Quantification of MCM-2+/SMα-actin+ cells (n = 13–15/group). Scale bars = 50 µm. Tissues in (A-C) were co-stained with the VSMC marker, SMα-actin.
(D) Adhesion of murine VSMCs to increasing concentrations of immobilized SVEP1. Adhered cells were counted manually and normalized to wells lacking SVEP1.
(E) Proliferation of murine VSMCs in response to increasing concentrations of immobilized SVEP1 and SVEP1CADrv using a BrdU incorporation assay.
(F) Svep1SMC∆/∆ murine VSMCs were incubated in wells precoated with 30 µg ml−1 SVEP1 protein or BSA (as vehicle control) and treated with or without 50 µg ml−1 oxLDL in the culture media for 36 hr. Proliferation was determined by BrdU incorporation.
(G) Immunoblots of integrin signaling kinases and downstream kinases of murine VSMCs adhered to control, VCAM-1, or SVEP1-treated plates. β-actin was used as loading control.
(H) Murine VSMCs were transfected with control or Itga9-targetted siRNAs and grown on immobilized SVEP1 or BSA. Proliferation was determined by BrdU incorporation. n = 4–12/group; N = 2–3 for D-H. Data were analyzed using an unpaired nonparametric Mann-Whitney test (C) or a two-tailed t-test (F and H). The bar graphs depict the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
The extracellular matrix (ECM) plays a critical role in orchestrating cellular responses to tissue injury, including promoting cell proliferation and differentiation (23, 37). We therefore assessed the proliferation of neointimal Svep1SMC∆/∆ and Svep1SMC +/+ VSMCs using immunofluorescent staining of the proliferation marker mini-chromosome maintenance protein-2 (MCM-2). Among cells expressing smooth muscle actin, fewer stained positive for MCM-2 in Svep1SMC∆/∆ mice as compared to Svep1SMC +/+ controls after HFD feeding for 8 weeks (Fig. 5C), suggesting SVEP1 induces VSMC proliferation.
To further explore the effects of SVEP1 on VSMCs, we generated and purified recombinant SVEP1 and its orthologous CAD risk variant (SVEP1CADrv) using a mammalian expression system. We tested the response of primary VSMCs to SVEP1 that was immobilized on culture plates, reflecting an overexpression-like assay while maintaining its physiologic context as an extracellular matrix protein (in contrast to genetic overexpression). VSMCs adhered to SVEP1 in a dose dependent manner (Fig. 5D). Exposure to both SVEP1 variants induced dose-dependent VSMC proliferation, based on bromodeoxyuridine (BrdU) incorporation (Fig. 5E). As a point of reference, we used oxLDL, a proliferative stimulus relevant to atherosclerosis, in addition to SVEP1 to test VSMC proliferation; strikingly, SVEP1 induced more VSMC proliferation than oxLDL (Fig. 5F). Exposure to a combination of oxLDL and SVEP1, as exists within the atheromatous environment, caused the greatest amount of VSMC proliferation (Fig. 5F). Human coronary artery smooth muscle cells (CASMCs) also proliferated in response to SVEP1 (fig. S5C). Murine macrophages exposed to SVEP1 did not proliferate in the absence or presence of oxLDL (fig. S5D), suggesting that SVEP1 is not a proliferative stimulus for all cell types.
Integrin α9β1 is expressed by VSMCs, binds to SVEP1, and drives proliferation in some cell types (30). Therefore, to begin to interrogate the molecular mechanisms by which SVEP1 influences VSMCs, we tested whether SVEP1 exposure could induce integrin signaling in VSMCs. We seeded cells to wells coated with bovine serum albumin (as an inert protein control), Vascular cell adhesion molecule 1 (VCAM-1, a low affinity integrin α9β1 ligand), or SVEP1 (a high affinity integrin α9β1 ligand). Cells adherent to SVEP1 had increased phosphorylation of canonical integrin signaling kinases, such as focal adhesion kinase (FAK), Paxillin (Pax), and Src, as well as downstream MAPK kinases, ERK and p38 (Fig. 5G), relative to an inert protein control. SVEP1CADrv had similar effects as SVEP1 on integrin signaling in VSMCs (fig. S5E). We then tested if SVEP1-induced proliferation was dependent on integrin α9β1. Since Itgα9 exclusively heterodimerizes with Itgβ1, we used siRNA knockdown of Itgα9 to disrupt integrin α9β1. The proliferative effect of SVEP1 was completely inhibited by knockdown of Itgα9 using two different siRNA constructs (Fig. 5H), suggesting that integrin α9β1 is necessary for SVEP1-induced VSMC proliferation.
SVEP1 and SVEP1CADrv regulate key VSMC differentiation pathways
We sought to characterize the response of primary VSMCs to the wildtype SVEP1 and SVEP1CADrv proteins using an unbiased methodology. Cells were collected after 20 hours of growth on the indicated substrate and transcriptomic analysis was performed using RNA-sequencing. Pathway and gene ontology analysis was used to determine the shared and unique transcriptional response to the SVEP1 variants. Consistent with previous findings, cell adhesion and proliferation-related pathways and terms were enriched in the shared transcripts of cells exposed to either SVEP1 variant. These include ECM-receptor interaction, focal adhesion, integrin-mediated signaling, positive regulation of cell proliferation, and proliferative and mitogenic pathways (Fig. 6A, B, table S1). A striking number of differentiation and development-related pathways and terms were also enriched in cells exposed to the SVEP1 variants. These include angiogenesis, cell differentiation, and wound healing, among others (Fig. 6A, B, table S1).
Fig. 6. SVEP1 modulates key VSMC-developmental pathways.
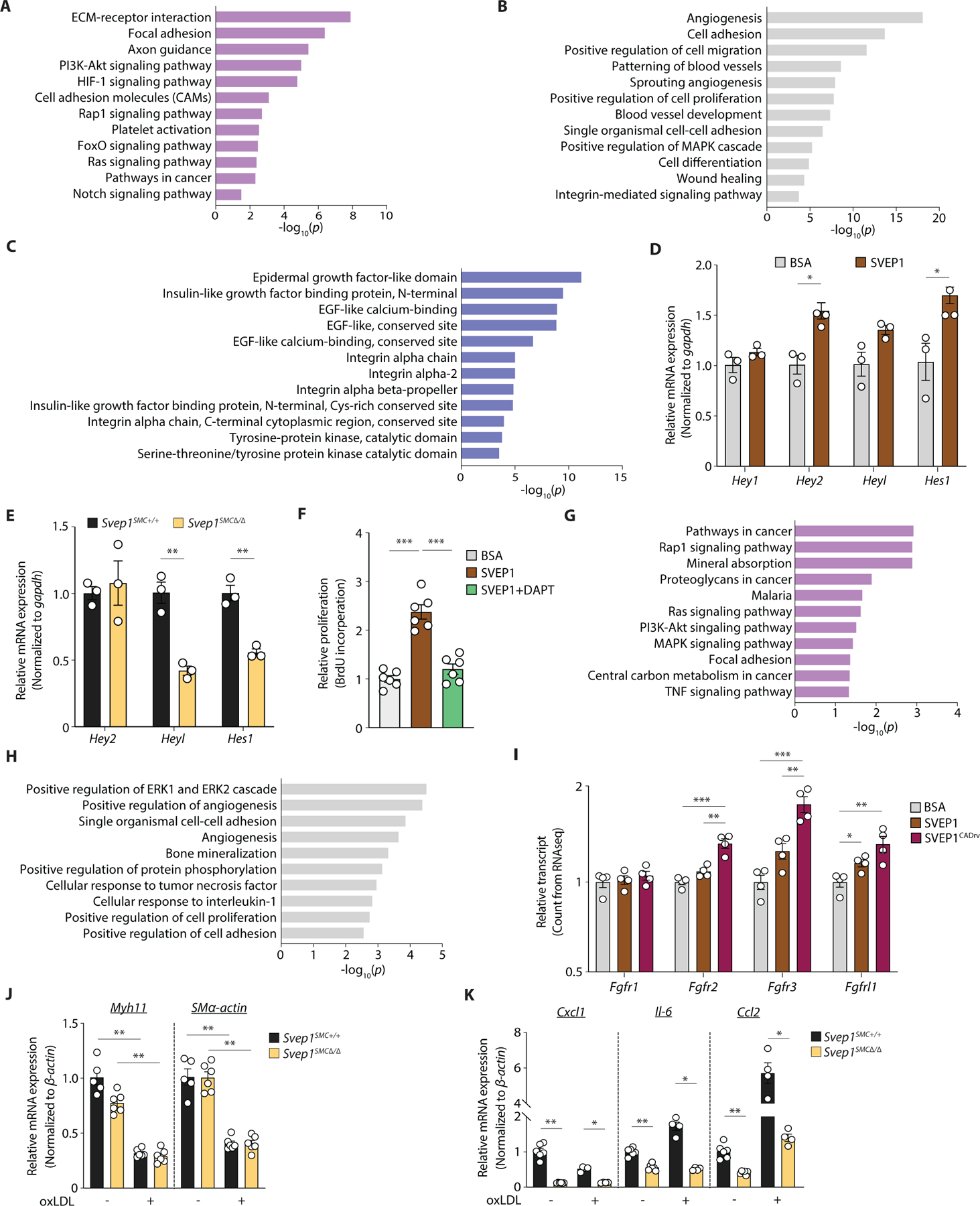
(A-C) Common transcriptional response of murine VSMCs to SVEP1 and SVEP1CADrv proteins. Dysregulated (A) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, (B) Gene Ontoloty (GO) term molecular functions, and (C) InterPro domains. Top 5 dysregulated categories plus additional, select categories are included. Full results are available in table S1. Bars represent -log10 of P values (n = 4/group).
(D) Transcription of canonical Notch target genes in murine VSMCs after 4 hours of adhesion to SVEP1, relative to BSA.
(E) Basal transcription of Notch target genes in Svep1SMC+/+ and Svep1SMC∆/∆ murine VSMCs.
(F) Proliferation of murine VSMCs in response to immobilized SVEP1. Cells were treated with DMSO (carrier) or 25 µM DAPT. Proliferation was determined by BrdU incorporation. n = 3–6/group; N = 2–3 for D-F.
(G-H) Differential transcriptional response of murine VSMCs to SVEP1 and SVEP1CADrv proteins. Dysregulated (G) KEGG pathways, and (H) GO term molecular functions. Top 5 dysregulated categories plus additional, select categories are included. Full results are available in table S1. Bars represent -log10 of P values.
(I) Bar graph of Fgfr transcript counts from murine VSMC RNAseq. Each transcript is normalized to the BSA control group. n = 4/group for G-I.
(J, K) qPCR of (J) VSMC markers, and (K) inflammatory markers of murine VSMC cultured with or without 50 µg ml−1 oxLDL for 24 hr (n = 4–6/group; N = 2). Data were analyzed using a two-tailed t-test (D-F and I-K). The bar graphs depict the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
SVEP1 contains different and repeating domains that are known to play critical developmental roles and may therefore be governing the effects of SVEP1 on VSMCs. Further, although Svep1−/− and Itgα9−/− mice have similar phenotypes of edema and lymphatic defects (17, 21), the phenotype of Svep1−/− mice is markedly more severe [death by embryonic day 18.5 vs postnatal day 12 (38)], suggesting that ITGA9 may have partial redundancy with an additional receptor(s) for SVEP1. To search for evidence of additional domain interactions, we cross-referenced the transcriptional profile of VSMCs to the SVEP1 variants with InterPro (39), a database of protein domains. In addition to integrin-related domains, transcripts that code for EGF-like domain-containing proteins were highly differentially expressed in cells exposed to SVEP1 (Fig. 6C). Repeat EGF-like domains often interact, as occurs in Notch signaling, suggesting SVEP1’s repeat EGF-like domains may be playing an important, but as of yet undescribed role in the biological function of SVEP1 (39). Indeed, transcripts related to Notch signaling were dysregulated in cells exposed to SVEP1 (Fig. 6A).
As an orthogonal approach to interrogating SVEP1’s mechanisms and potential binding partners, we sought to identify homologues in distantly related species. The Drosophila protein, uninflatable, is a potential orthologue of SVEP1 (40) and contains a region defined by three ephrin-receptor like domains, followed by tandem EGF-repeats and a Laminin-G domain (41), mirroring a region of SVEP1 that contains a highly similar sequence of domains. Inhibition of uninflatable in Drosophila larvae results in defective tracheal development, analogous to the vascular defects observed in zebrafish Svep1 mutants (42, 43). Uninflatable has been shown to bind and modulate Notch signaling in Drosophila (42, 44, 45). These findings, in addition to the RNAseq analysis, led us to hypothesize that SVEP1 may also modulate Notch signaling. VSMCs express multiple Notch receptors (46), thus, we tested the impact of SVEP1 on Notch signaling in VSMCs. This was assessed by seeding VSMCs on tissue culture plates treated with SVEP1 or BSA (as an inert control protein) for 4 hours, since Notch signaling is highly temporally regulated (47). Cells grown on SVEP1 had increased expression of canonical Notch targets Hey2 and Hes1 even without overexpression of a Notch receptor (Fig. 6D). Conversely, primary VSMCs collected from Svep1SMC∆/∆ mice had decreased transcription of Notch target genes (Fig. 6E), supporting the regulation of Notch signaling by SVEP1. SVEP1-induced proliferation was also completely abrogated upon Notch inhibition by the γ-secretase inhibitor DAPT (Fig. 6F). It is possible that Notch and integrin receptors may cooperatively regulate the effects of SVEP1, similar to that reported on non-canonical ECM Notch regulators Microfibrillar-associated protein 5 (MFAP5) and Epidermal growth factor-like protein 7 (EGFL7) (48).
Our experimental atherosclerosis models and Mendelian randomization analysis indicate that both SVEP1 variants are atherogenic, with SVEP1CADrv having the greater atherogenicity of the two. We therefore investigated the differential transcriptional responses of VSMCs to the SVEP1 variants. This analysis revealed that many proliferation-related pathways were disproportionately regulated by the variants (Fig. 6G, H, table S1). Further exploration identified differential expression of the fibroblast growth factor (FGF) receptor family between the variants. The FGFR family is also sub-categorized within several of the most differentially regulated pathways and terms. FGF signaling is proatherogenic in VSMCs (49), so we assessed the effect of each variant on the direction and magnitude of transcription of each FGF receptor expressed by VSMCs. Consistent with their relative atherogenicities, SVEP1 increased expression of FGF receptors but exposure to SVEP1CADrv resulted in higher expression of FGF receptors (Fig. 6I). These data suggest that increased FGF signaling may contribute to the increased CAD risk associated with SVEP1CADrv.
Given the fundamental role of integrin, Notch, and FGFR signaling in regulating VSMC phenotype, we assessed the effects of SVEP1 in response to oxLDL, an inflammatory stimulus relevant to atherosclerosis. Upon oxLDL stimulation, both Svep1SMC+/+ and Svep1SMC∆/∆ VSMCs decreased the expression of contractile markers Myh11 and SMα-actin (Fig. 6J), and increased expression of the inflammatory marker C-C motif chemokine ligand 2 (Ccl2) (Fig. 6K), confirming an inflammatory response to oxLDL. C-X-C motif chemokine ligand 1 (Cxcl1), interleukin-6 (Il-6), and Ccl2 expression was lower in Svep1SMC∆/∆ VSMCs than Svep1SMC+/+ controls, suggesting that SVEP1 may be a pro-inflammatory stimulus in VSMCs under atherosclerotic conditions.
SVEP1 promotes inflammation in atherosclerosis
To investigate how the loss of Svep1 influences pathways involved in the development of atherosclerosis at the tissue level, we performed RNA-seq analyses on mRNA extracted from aortic arches of Svep1SMC+/+and Svep1SMC∆/∆ mice after 8 weeks of HFD. Loss of Svep1 in VSMCs altered inflammatory pathways upon induction of atherosclerosis, including cytokine-cytokine receptor interaction, chemokine signaling, and NF-kappa B signaling pathways (Fig. 7A). Both cell adhesion molecules (CAMs) and ECM-receptor interaction were also dysregulated in the atherosclerotic aortic arches from Svep1SMC∆/∆ (Fig. 7A, B and table S2). Quantitative PCR using cDNA from the aortic arches of the same mice was used to validate the RNA-seq results. Specifically, Ccl2, Spp1 (secreted phosphoprotein 1, also known as osteopontin), and Cxcl5 (C-X-C motif chemokine ligand 5) were decreased in Svep1SMC∆/∆ mice, as compared to Svep1SMC+/+ mice (fig. S6A). Despite these differences, we did not find a significant alteration in circulating inflammatory mediators in these mice, suggesting SVEP1 influences local tissue inflammation but not systemic inflammation (fig. S6B). These data are also consistent with our observations that Svep1 depletion decreases neointimal macrophage staining in atherosclerotic plaque.
Fig. 7. SVEP1 promotes inflammation in atherosclerosis.
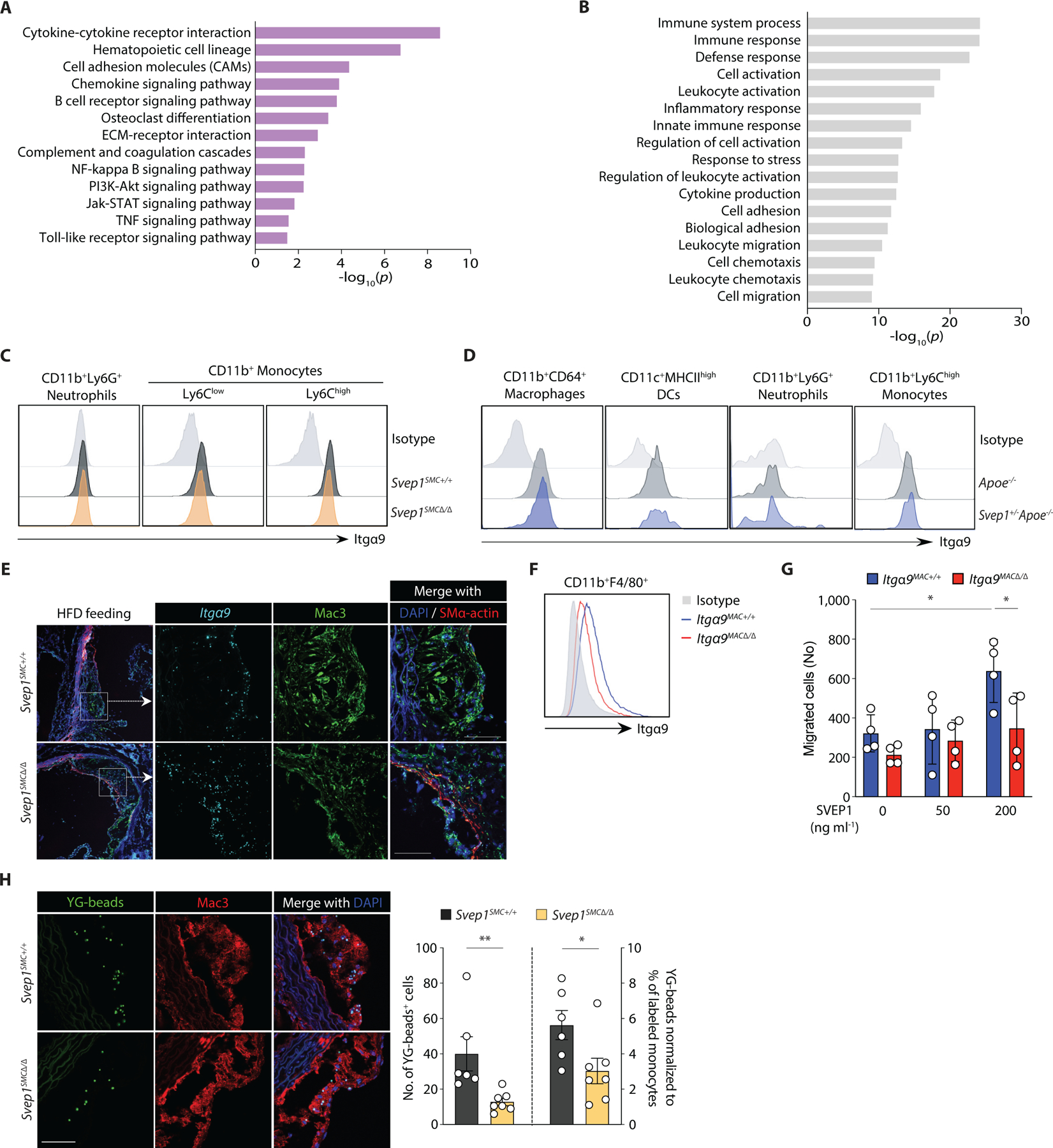
(A-B) Differential transcriptional profile of atherosclerotic aortic arches from Svep1SMC+/+ and Svep1SMC∆/∆ mice. Dysregulated (A) KEGG pathways, and (B) GO term molecular functions. Top 5 dysregulated categories plus additional, select categories are included. Full results are available in table S2. Bars represent -log10 of P values (n = 3–4/group).
(C) Histogram for Itgα9β1 expression in mouse blood neutrophils (CD11b+Ly6G+), Ly6Clow (CD11b+Ly6Clow), and Ly6Chigh (CD11b+Ly6Chigh) monocytes from Svep1SMC+/+ and Svep1SMC∆/∆ mice after 8 weeks of HFD.
(D) Histogram of Itgα9β1 expression in the subpopulations of aortic leukocytes. Macrophages (CD64+CD11b+), DCs (CD11c+MHCIIhigh), neutrophils (CD11b+Ly6G+), and Ly6Chigh (CD11b+Ly6Chigh) monocytes from Apoe−/− and Svep1+/−Apoe−/− mice after 8 weeks of HFD (n = 3–4/group; N = 3).
(E) Expression of Itga9 in the aortic roots from Svep1SMC+/+ and Svep1SMC∆/∆ mice using ISH after 8 weeks of HFD. Tissues were co-stained for Mac3 and SMα-actin. Scale bars, 50 µm.
(F) Expression of Integrin alpha-9 in BMDM from Itgα9MAC+/+ and Itgα9MAC∆/∆ mice (n = 3/group; N = 3).
(G) Migratory response of thioglycolate-elicited murine macrophages from Itgα9MAC+/+ and Itgα9MAC∆/∆ were determined using a chemotaxis chamber incubated with 0, 50, and 200 ng ml−1 of SVEP1 protein. Migrated cells were counted by an automated microscope and expressed as cells per field of view (n = 4/group; N = 2).
(H) In vivo monocyte recruitment assay. YG-bead uptake within plaque lesion in the aortic root regions from Svep1SMC+/+ and Svep1SMC∆/∆ mice. Quantification of YG-bead uptake showing the total number of YG-beads per section (left Y axis), and the number of YG-beads normalized to the percentage of labeled Ly6Clow monocytes (right Y axis) (n = 6–7/group). Scale bar, 50 µm. Data were analyzed using two-tailed t-test (G) or unpaired nonparametric Mann-Whitney test (H). The bar graphs depict the mean ± SEM. *P ≤ 0.05; **P < 0.01.
Integrins play a critical role in the immune response, we therefore asked whether immune cells may also express integrin α9β1 and interact with SVEP1 in atherosclerosis. In human peripheral blood cells, moderate integrin α9β1 expression was detected by neutrophils and low expression was detected by CD14lowCD16+ non-classical, CD14highCD16+ intermediate, and CD14+CD16- classical monocytes (fig. S7A) as previously reported (50). Given that monocytes alter their expression profiles upon tissue entry and differentiation into macrophages (51), we sought to test if macrophages in atherosclerotic plaque express ITGA9. Indeed, ITGA9 expression was detected in CD68+ macrophages within human atherosclerotic plaque by in situ hybridization (fig. S7B).
We then sought to further assess the expression of integrin α9β1 expression in circulating murine leukocyte subsets. High expression of integrin α9β1 was detected in both Ly6Chi and Ly6Clow monocytes and we could detect low expression in neutrophils (Fig. 7C). These expression patterns were unaltered in heterozygous Svep1 deficiency (fig. S7C) and we did not observe an induction of integrin α9β1 expression upon oxLDL treatment in any cell type tested (fig. S7D, E). Considering that integrin α9β1 is expressed by monocyte subsets in peripheral mouse blood, we further analyzed its expression in myeloid cells from the aortas of Apoe−/− and Svep1+/−Apoe−/− mice following 8 weeks of HFD feeding. We discovered that integrin α9β1 was expressed in both macrophages and Ly6Chi monocytes of these mice (Fig. 7D), consistent with human expression data. We similarly detected robust expression of Itgα9 by neointimal macrophages using in situ hybridization (Fig. 7E).
Since integrin α9β1 is expressed on monocytes/macrophages, we sought to better understand whether SVEP1 could be directly interacting with integrin α9β1 on these cells. To test this, we generated mice with myeloid cell lineage-specific knockout of Itgα9 using LysM-Cre (Itgα9flox/floxLysM-Cre, hereafter referred to as Itga9MAC∆/∆). Itga9+/+LysM-Cre mice (referred to as Itga9MAC+/+) served as controls. First, we confirmed that bone marrow-derived macrophages from Itga9MAC∆/∆ animals had a reduction in the amount of integrin α9β1 that was present on the cell surface (Fig. 7F). We then tested the ability of peritoneal macrophages from these animals to migrate in response to SVEP1 using a trans-well migration assay. SVEP1 exposure induced a dose-dependent trans-well migration of macrophages from Itga9MAC+/+ control animals but not from Itga9MAC∆/∆ mice (Fig. 7G). This suggests that SVEP1 and integrin α9β1 may directly interact to augment myeloid cell homing or migration. Consistent with this, THP-1 cells, a human monocytic cell line, adhered to SVEP1 in a dose-dependent manner (fig. S7F). Integrin signaling was also activated in THP-1 cells upon exposure to SVEP1 or SVEP1CADrv and no differences were observed between the variants (fig. S7G).
To test if SVEP1 had similar effects on leukocytes in vivo, we performed an in vivo monocyte recruitment assay in Svep1SMC+/+ and Svep1SMC∆/∆ mice. After 8 weeks of HFD feeding, we injected yellow-green (YG) latex beads intravenously to label circulating Ly6Clow monocytes. Flow cytometry was performed three days after intravenous bead injection (to confirm labeling) and the aortic tissues were isolated for histology on the fourth day following bead injection (to assess recruitment). We confirmed that YG beads were preferentially labeled on Ly6Clow monocytes and not on Ly6Chigh monocytes, indicating efficient bead labeling of circulating monocytes (fig. S7H). We did not observe a difference between groups in the efficiency of bead labeling for monocyte subsets (fig. S7I). Next, we quantified the number of labeled monocytes recruited into atherosclerotic plaques of aortic roots using fluorescent microscopy. Svep1SMC∆/∆ mice had fewer YG beads per atheroma, with or without normalization to the percentage of labeled monocytes, relative to Svep1SMC+/+ mice (Fig. 7H). Taken together, these data support SVEP1’s role in promoting inflammation in atherosclerosis, either indirectly by promoting an inflammatory VSMC phenotype, directly by interacting with integrin α9β1 on circulating or tissue leukocytes, or a combination of these processes.
Discussion
Human genomic studies hold great promise in identifying therapeutic targets for disease (52), but a substantial limitation in translating their findings is the identification of specific causal genes that underlie the observed statistical associations. In a previous study, we identified a low-frequency polymorphism in SVEP1 that robustly associated with coronary artery disease risk in humans (11), but it was not clear if SVEP1 was the causal gene in the locus. Here, we present evidence that SVEP1 is causal in coronary artery disease using experimental mouse models and Mendelian randomization.
Atherosclerosis is a complex, multifactorial disease process with numerous cell types playing a role in its pathogenesis. This presents an arduous challenge when validating genomic risk loci and testing their mechanisms. The SVEP1CADrv does not associate with changes in plasma lipid concentrations (11), prompting us to explore how SVEP1 might influence other aspects of disease pathogenesis. We used human and mouse expression data at the cell and tissue level to develop mechanistic hypotheses, which we then tested using in vivo and in vitro approaches. Specifically, high basal arterial expression of both SVEP1 and ITGA9, and increased SVEP1 expression under pathological conditions, led us to hypothesize that these proteins may influence local disease processes. Upon exposure to various pathologic stimuli, VSMCs can undergo a “phenotype shift”, in which they lose their quiescent, contractile properties and become migratory, proliferative, inflammatory, and synthetic (23, 53). VSMCs gain properties of matrix-synthesizing fibroblasts during atherosclerosis (54), making VSMCs our primary candidates for the source of SVEP1 within atherosclerotic plaque. In fact, consistent with prior reports which concluded that SVEP1 is not produced by endothelial or immune cells (21) ,we observed negligible Svep1 expression in the plaques of Svep1SMC∆/∆ animals. Thus, our results provide strong evidence that atherogenic SVEP1 is indeed synthesized by VSMC-derived cells within the atherosclerotic plaque.
Using two independent mouse models in which Svep1 was depleted either partially in all cells (Svep1SMC+/−) or fully in only VSMCs (Svep1SMC∆/∆), we demonstrated that depleting SVEP1 resulted in a significant reduction in the development of atherosclerotic plaque with a magnitude of effect similar to murine models of other CAD risk loci (54, 55). We then used expression of ITGA9 to identify disease-relevant cell types that may respond to SVEP1. This led to the hypothesis that SVEP1 may be interacting with VSMCs by an autocrine mechanism or monocytes by a paracrine mechanism to promote atherosclerosis. VSMCs play a particularly complex and intriguing role in atherosclerosis and warrant further discussion. Recent lineage tracing studies have challenged the notion that VSMCs play a protective role in atherosclerosis (23) by demonstrating that a large, heterogenous population of cells within plaque are derived from VSMCs (23, 53, 56). Furthermore, numerous CAD risk loci have now been linked to VSMCs (57). This study demonstrates that SVEP1 influences the behavior of VSMCs by regulating pathways with vital roles in VSMC biology. These pathways include integrin, Notch, and FGFR signaling, each of which has been shown to contribute to atherosclerosis (29, 49, 58, 59). Recent studies have provided novel insights into the regulation of VSMC phenotype in atherosclerosis by various transcription factors (54, 56, 60). The ECM also plays a fundamental role in regulating VSMC phenotype and is amenable to pharmacologic intervention. Current strategies for the treatment and prevention of CAD consist of lowering risk factors, such as plasma lipids, yet substantial residual risk remains despite effective treatment. Intervening on VSMCs may be a powerful complimentary approach to these traditional therapies.
In addition to its association with CAD, our Mendelian randomization results suggest that circulating SVEP1 causally underlies risk of hypertension and type 2 diabetes. Although the source of SVEP1 in human plasma is unknown, other ECM proteins have been detected in the circulation of patients with atherosclerosis, suggesting that plasma concentrations of these proteins may reflect tissue concentrations and atherosclerotic remodeling (61, 62). The mechanisms by which the genetic variants used in the Mendelian randomization affect plasma SVEP1 are unclear. Two reasonable hypotheses include modification of protein secretion or degradation. Alternatively, it is possible that protein-modifying polymorphisms could alter the affinity of aptamer binding (and thus impact the estimated plasma protein concentrations). It is unlikely that altered aptamer binding is a major contributor, however, because differential plasma SVEP1 concentrations were observed using two independent aptamers and because the majority of genetic variants linked to altered plasma concentrations were not located in protein-coding DNA segments. Further studies will be required to determine the precise mechanisms by which these variants affect plasma protein concentrations. Regardless, the power of the two sample Mendelian randomization framework is that these alleles are allocated randomly at birth and are associated with plasma SVEP1 concentrations in the absence of disease, suggesting that the presence of disease is not driving altered SVEP1 concentrations, but rather that altered SVEP1 expression is causally related to disease. This further suggests that circulating SVEP1 may be useful as a predictive biomarker.
Additional human genetic data support a broader role of SVEP1 in cardiometabolic disease. The alpha subunit of integrin α9β1, which binds to SVEP1 (14) with an affinity that far exceeds its other known ligands (63–66), is also associated with blood pressure in multiple studies (15, 16). Overexpression of disintegrin and metalloproteinase with thrombospondin motifs-7 (ADAMTS-7), another CAD risk locus, in primary rat VSMCs alters the molecular mass of SVEP1 (67). The overlapping disease associations and molecular interactions between these three risk loci converge on SVEP1 and point to a regulatory circuit with a prominent, yet unexplored role in cardiometabolic disease. Further studies will be required to validate their interactions and mechanisms in vivo, and to explore the potential of targeting this pathway for the treatment of cardiometabolic disease.
Our in vivo data were limited to the Apoe−/− mouse model and our functional studies were limited to VSMCs and myeloid cells. Additional confirmatory studies in other model systems and cell types are needed to further evaluate the therapeutic potential of targeting SVEP1. In addition, as SVEP1 is critical for development, we chose to deplete SVEP1 in our VSMC-specific model starting at 6 weeks of age and we do not know how long the protein is retained in the ECM. Finally, although VSMC-specific depletion of SVEP1 did not result in any obvious deleterious impact, we did not rigorously evaluate those animals for more subtle adverse effects.
Our complementary mouse models demonstrate that Svep1 haploinsufficiency and VSMC-specific Svep1 deficiency significantly abrogate the development of atherosclerosis. Each intervention appeared to be well tolerated by mice, as we did not observe any adverse response to SVEP1 depletion. Similarly, our Mendelian randomization analyses suggest there may be a therapeutic window to safely target SVEP1. These findings suggest that targeting SVEP1 or selectively modulating its interactions may be a viable strategy for the treatment and prevention of coronary artery disease.
Materials and Methods
Study design
We designed this study to experimentally determine if SVEP1 causally contributes to risk for atherosclerosis. We used primary tissues harvested from humans and mice to determine if SVEP1 was produced in tissues and cell types of relevance to atherosclerosis. We utilized Mendelian randomization to determine if SVEP1 is causally related to cardiovascular disease in humans. To study the mechanism by which SVEP1 may promote atherosclerosis, we performed animal studies in HFD fed Apoe−/− mice. The number of animals used in each study group are specified in the figure legends. The in vitro experimental data included in this manuscript are representative of multiple experimental outcomes. Experiments were not performed in a blinded fashion. All animal studies were performed according to procedures and protocols approved by the Animal Studies and Institutional Animal Care and Use Committees of the Washington University School of Medicine. All human research participants provided written informed consent for the studies described below which were conducted according to procedures and protocols approved by the Human Research Protection Office and Institutional Review Board of the Washington University School of Medicine.
Mice
Svep1+/− mice were made by KOMP (knockout mouse project), and these mice were then crossed with mice expressing the flippase FLP recombinase under the control of the promoter of the human actin beta gene (hATCB) to generate Svep1flox/flox (Svep1∆/∆) mice. CRISPR/Cas9 genome editing technology was used in collaboration with the Washington University School of Medicine Genome Engineering and Transgenic Micro-Injection Cores to generate Svep1G/G mice on a C57BL/6 background harboring the SVEP1 mutation at the homologous murine position (p.D2699G). Svep1+/− and Svep1G/G mice were crossed with Apoe−/− mice (#002052, Jackson Laboratory) to get Svep1+/−Apoe−/− and Svep1G/+Apoe−/− mice, which we maintained as breeders to generate experimental and control mice. We crossed Svep1∆/∆ mice with Myh11-CreERT2 (#019079, Jackson Laboratory) mice to generate Svep1∆/+Myh11-CreERT2 mice. Svep1∆/+Myh11-CreERT2 males were then crossed with Svep1∆/+ females to generate experimental Svep1∆/∆Myh11-CreERT2 and control Svep1+/+Myh11-CreERT2 male littermate mice. Finally, Svep1∆/∆Myh11-CreERT2 males were crossed with Apoe−/− females. We maintained Svep1∆/+Myh11-CreERT2Apoe−/− males and Svep1∆/+Apoe−/− females as breeders to generate experimental Svep1∆/∆Myh11-CreERT2Apoe−/− (Svep1SMC∆/∆) and control Myh11-CreERT2Apoe−/− (Svep1SMC+/+) mice. To activate Cre-recombinase, mice were injected intraperitoneally with 1 mg of tamoxifen (#T5648, Sigma-Aldrich) in 0.1 mL peanut oil (#P2144, Sigma-Aldrich) for 10 consecutive days starting at 6 weeks of age. Tamoxifen treatment was performed with all experimental and control mice in an identical manner. Itgα9flox/flox (Itgα9∆/∆) mice were gifts from Drs. Dean Sheppard and Livingston Van De Water (Albany Medical College, New York), and LysM-Cre mice were provided from Dr. Babak Razani (Washington University School of Medicine, Saint Louis). We crossed Itgα9fl/fl mice with LysM-Cre mice, and maintained Itgα9fl/+LysM-Cre mice as breeders to generate Itgα9fl/flLysM-Cre (Itgα9MAC∆/∆) and control Itgα9+/+LysM-Cre (Itgα9MAC+/+) mice. All mice were housed in a pathogen-free environment at the Washington University School of Medicine animal facility and maintained on a 12 hr light/12 hr dark cycle with a room temperature of 22 ± 1°C. Table S3 lists primer information.
Statistical analysis
For animal model data, a two-group independent t-test, one-way analysis of variance (ANOVA), or two-way ANOVA were used, provided the data satisfied the Shapiro-Wilk normality test. Otherwise, the Mann-Whitney U test, Kruskal-Wallis one-way ANOVA test, and Friedman two-way ANOVA test were used. Bonferroni correction was used for post-hoc multiple comparison in ANOVA. Unless otherwise stated, cellular assays were analyzed by an unpaired, two-tailed, heteroscedastic t-test. Statistical analyses were performed with GraphPad Prism. Individual data points are reported in data file S1.
Supplementary Material
Fig. S1. SVEP1 expression in health and disease.
Fig. S2. Svep1 haploinsufficiency does not significantly alter plaque composition after 8 weeks of HFD feeding.
Fig. S3. VSMC-specific Svep1 deficiency reduces atherogenesis and plaque complexity, continued.
Fig. S4. The effect of the CAD-associated SVEP1 D2702G missense polymorphism and SVEP1 protein concentration in humans and mice.
Fig. S5. Itgα9 is expressed in both VSMCs and macrophages in mouse arteries.
Fig. S6. SVEP1 promotes local inflammation in atherosclerosis.
Fig. S7. SVEP1 promotes local inflammation in atherosclerosis, continued.
Table S3. Primer table.
Table S1. (Microsoft Excel format). VSMC transcriptional response to SVEP1 variants.
Table S2. (Microsoft Excel format). Transcriptional profile of atherosclerotic aortic arches.
Data file S1. (Microsoft Excel format). Individual data points.
Acknowledgements
We thank all the members of the Stitziel Lab for helpful discussion and Upasana Pudupakkam for technical assistance. We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. The Center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant# UL1TR002345 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health, and NIH Roadmap for Medical Research. We thank the Bursky Center for Human Immunology and Immunotherapy Programs at Washington University, Immunomonitoring Laboratory for help with analysis of mouse plasma. This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH. The Svep1 mouse strain used for this research was created from ES cell clone HEPD0747_6_B06, generated by the European Conditional Mouse Mutagenesis Program which was then made into mice and provided to the KOMP Repository (www.komp.org) by the Jackson Laboratory as part of the KOMP2 Project. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. Data used for the analyses described in this manuscript were obtained from the GTEx Portal on 04/30/2020.
Funding: This work was supported in part by grants from the National Institutes of Health (NIH) T32GM007200 and T32HL134635 to JSE, NIH T32HL007081 to EPY, NIH R01HL53325 to RPM, along with NIH grants R01HL131961, UM1HG008853, UL1TR002345, an investigator-initiated research grant from Regeneron Pharmaceuticals, a career award from the National Lipid Association, and by the Foundation for Barnes-Jewish Hospital to NOS.
Footnotes
This manuscript has been accepted for publication in Science Translational Medicine. This version has not undergone final editing. The definitive version was published in Science Translational Medicine on March 24 2021 in Volume 13, Issue 586, DOI: http://10.1126/scitranslmed.abe0357. Please refer to the complete version of record at www.sciencetranslationalmedicine.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior written permission of AAAS.
Competing interests: IHJ, JSE, AA, and NOS are inventors on U.S. Patent Application 62/962,736 submitted by Washington University that covers compositions and methods of treatment targeted to SVEP1-mediated disorders. NOS has received investigator-initiated research funds from Regeneron Pharmaceuticals unrelated to the content of this study. The other authors have no conflicts.
Data and materials availability: All data associated with this study are included in the manuscript or Supplementary Materials. Biological materials are available from the Stitziel Laboratory under a material transfer agreement with Washington University.
References
- 1.Weber C, Noels H, Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 17, 1410–1422 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Dzau VJ, Braun-Dullaeus RC, Sedding DG, Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med 8, 1249–1256 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Liu X, Ntambi JM, Atherosclerosis: keep your macrophages in shape. Nat Med 15, 1357–1358 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Hansson GK, Klareskog L, Pulling down the plug on atherosclerosis: cooling down the inflammasome. Nat Med 17, 790–791 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Ross R, Genetically modified mice as models of transplant atherosclerosis. Nat Med 2, 527–528 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Randolph GJ, Proliferating macrophages prevail in atherosclerosis. Nat Med 19, 1094–1095 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Rader DJ, FitzGerald GA, State of the art: atherosclerosis in a limited edition. Nat Med 4, 899–900 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Virella G, Lopes-Virella MF, Humoral immunity and atherosclerosis. Nat Med 9, 243–244; author reply 244–245 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Baigent AKC, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R, Cholesterol Treatment Trialists’ (CTT) Collaborators, Efficacy and Safety of Cholesterol-Lowering Treatment: Prospective Meta-Analysis of Data From 90,056 Participants in 14 Randomised Trials of Statins. Lancet 366, 1267–1278 (2005). [DOI] [PubMed] [Google Scholar]
- 10.van der Harst P, Verweij N, Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circulation research 122, 433–443 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stitziel NO, Stirrups KE, Masca NG, Erdmann J, Ferrario PG, Konig IR, Weeke PE, Webb TR, Auer PL, Schick UM, Lu Y, Zhang H, Dube MP, Goel A, Farrall M, Peloso GM, Won HH, Do R, van Iperen E, Kanoni S, Kruppa J, Mahajan A, Scott RA, Willenberg C, Braund PS, van Capelleveen JC, Doney AS, Donnelly LA, Asselta R, Merlini PA, Duga S, Marziliano N, Denny JC, Shaffer CM, El-Mokhtari NE, Franke A, Gottesman O, Heilmann S, Hengstenberg C, Hoffman P, Holmen OL, Hveem K, Jansson JH, Jockel KH, Kessler T, Kriebel J, Laugwitz KL, Marouli E, Martinelli N, McCarthy MI, Van Zuydam NR, Meisinger C, Esko T, Mihailov E, Escher SA, Alvar M, Moebus S, Morris AD, Muller-Nurasyid M, Nikpay M, Olivieri O, Lemieux Perreault LP, AlQarawi A, Robertson NR, Akinsanya KO, Reilly DF, Vogt TF, Yin W, Asselbergs FW, Kooperberg C, Jackson RD, Stahl E, Strauch K, Varga TV, Waldenberger M, Zeng L, Kraja AT, Liu C, Ehret GB, Newton-Cheh C, Chasman DI, Chowdhury R, Ferrario M, Ford I, Jukema JW, Kee F, Kuulasmaa K, Nordestgaard BG, Perola M, Saleheen D, Sattar N, Surendran P, Tregouet D, Young R, Howson JM, Butterworth AS, Danesh J, Ardissino D, Bottinger EP, Erbel R, Franks PW, Girelli D, Hall AS, Hovingh GK, Kastrati A, Lieb W, Meitinger T, Kraus WE, Shah SH, McPherson R, Orho-Melander M, Melander O, Metspalu A, Palmer CN, Peters A, Rader D, Reilly MP, Loos RJ, Reiner AP, Roden DM, Tardif JC, Thompson JR, Wareham NJ, Watkins H, Willer CJ, Kathiresan S, Deloukas P, Samani NJ, Schunkert H, Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med 374, 1134–1144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilges D, Vinit MA, Callebaut I, Coulombel L, Cacheux V, Romeo PH, Vigon I, Polydom: a secreted protein with pentraxin, complement control protein, epidermal growth factor and von Willebrand factor A domains. Biochem J 352 Pt 1, 49–59 (2000). [PMC free article] [PubMed] [Google Scholar]
- 13.Shur I, Socher R, Hameiri M, Fried A, Benayahu D, Molecular and cellular characterization of SEL-OB/SVEP1 in osteogenic cells in vivo and in vitro. J Cell Physiol 206, 420–427 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Sato-Nishiuchi R, Nakano I, Ozawa A, Sato Y, Takeichi M, Kiyozumi D, Yamazaki K, Yasunaga T, Futaki S, Sekiguchi K, Polydom/SVEP1 is a ligand for integrin alpha9beta1. J Biol Chem 287, 25615–25630 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T, Aulchenko Y, Lumley T, Kottgen A, Vasan RS, Rivadeneira F, Eiriksdottir G, Guo X, Arking DE, Mitchell GF, Mattace-Raso FU, Smith AV, Taylor K, Scharpf RB, Hwang SJ, Sijbrands EJ, Bis J, Harris TB, Ganesh SK, O’Donnell CJ, Hofman A, Rotter JI, Coresh J, Benjamin EJ, Uitterlinden AG, Heiss G, Fox CS, Witteman JC, Boerwinkle E, Wang TJ, Gudnason V, Larson MG, Chakravarti A, Psaty BM, van Duijn CM, Genome-wide association study of blood pressure and hypertension. Nat Genet 41, 677–687 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeuchi F, Isono M, Katsuya T, Yamamoto K, Yokota M, Sugiyama T, Nabika T, Fujioka A, Ohnaka K, Asano H, Yamori Y, Yamaguchi S, Kobayashi S, Takayanagi R, Ogihara T, Kato N, Blood pressure and hypertension are associated with 7 loci in the Japanese population. Circulation 121, 2302–2309 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Karpanen T, Padberg Y, van de Pavert SA, Dierkes C, Morooka N, Peterson-Maduro J, van de Hoek G, Adrian M, Mochizuki N, Sekiguchi K, Kiefer F, Schulte D, Schulte-Merker S, An Evolutionarily Conserved Role for Polydom/Svep1 During Lymphatic Vessel Formation. Circulation research 120, 1263–1275 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samuelov L, Li Q, Bochner R, Najor NA, Albrecht L, Malchin N, Goldsmith T, Grafi-Cohen M, Vodo D, Fainberg G, Meilik B, Goldberg I, Warshauer E, Rogers T, Edie S, Ishida-Yamamoto A, Burzenski L, Erez N, Murray SA, Irvine AD, Shultz L, Green KJ, Uitto J, Sprecher E, Sarig O, SVEP1 plays a crucial role in epidermal differentiation. Exp Dermatol 26, 423–430 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cangemi C, Skov V, Poulsen MK, Funder J, Twal WO, Gall MA, Hjortdal V, Jespersen ML, Kruse TA, Aagard J, Parving HH, Knudsen S, Hoilund-Carlsen PF, Rossing P, Henriksen JE, Argraves WS, Rasmussen LM, Fibulin-1 is a marker for arterial extracellular matrix alterations in type 2 diabetes. Clin Chem 57, 1556–1565 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Ayari H, Bricca G, Identification of two genes potentially associated in iron-heme homeostasis in human carotid plaque using microarray analysis. J Biosci 38, 311–315 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Morooka N, Futaki S, Sato-Nishiuchi R, Nishino M, Totani Y, Shimono C, Nakano I, Nakajima H, Mochizuki N, Sekiguchi K, Polydom Is an Extracellular Matrix Protein Involved in Lymphatic Vessel Remodeling. Circulation research 120, 1276–1288 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Kalluri AS, Vellarikkal SK, Edelman ER, Nguyen L, Subramanian A, Ellinor PT, Regev A, Kathiresan S, Gupta RM, Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation 140, 147–163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett MR, Sinha S, Owens GK, Vascular Smooth Muscle Cells in Atherosclerosis. Circulation research 118, 692–702 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li M. F. Wei, Reddy Sekhar P, Yu Dae-Yeul, Yamamoto Masayuki, Silverstein Roy L, CD36 Participates in a Signaling Pathway That Regulates ROS Formation in Murine VSMCs. J Clin Invest 120, 3996–4006 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majesky MW, Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol 27, 1248–1258 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran P, Oliver-Williams C, Kamat MA, Prins BP, Wilcox SK, Zimmerman ES, Chi A, Bansal N, Spain SL, Wood AM, Morrell NW, Bradley JR, Janjic N, Roberts DJ, Ouwehand WH, Todd JA, Soranzo N, Suhre K, Paul DS, Fox CS, Plenge RM, Danesh J, Runz H, Butterworth AS, Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG, Consortium E-I, Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. European journal of epidemiology 30, 543–552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weng S, Zemany L, Standley KN, Novack DV, La Regina M, Bernal-Mizrachi C, Coleman T, Semenkovich CF, Beta3 integrin deficiency promotes atherosclerosis and pulmonary inflammation in high-fat-fed, hyperlipidemic mice. Proc Natl Acad Sci U S A 100, 6730–6735 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misra A, Feng Z, Chandran RR, Kabir I, Rotllan N, Aryal B, Sheikh AQ, Ding L, Qin L, Fernandez-Hernando C, Tellides G, Greif DM, Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nature communications 9, 2073 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schreiber TD, Steinl C, Essl M, Abele H, Geiger K, Muller CA, Aicher WK, Klein G, The integrin alpha9beta1 on hematopoietic stem and progenitor cells: involvement in cell adhesion, proliferation and differentiation. Haematologica 94, 1493–1501 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen C, Kudo M, Rutaganira F, Takano H, Lee C, Atakilit A, Robinett KS, Uede T, Wolters PJ, Shokat KM, Huang X, Sheppard D, Integrin alpha9beta1 in airway smooth muscle suppresses exaggerated airway narrowing. J Clin Invest 122, 2916–2927 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Danussi C, Petrucco A, Wassermann B, Pivetta E, Modica TM, Del Bel Belluz L, Colombatti A, Spessotto P, EMILIN1-alpha4/alpha9 integrin interaction inhibits dermal fibroblast and keratinocyte proliferation. The Journal of cell biology 195, 131–145 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta SK, Vlahakis NE, Integrin alpha9beta1: Unique signaling pathways reveal diverse biological roles. Cell Adh Migr 4, 194–198 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanayama M, Kurotaki D, Morimoto J, Asano T, Matsui Y, Nakayama Y, Saito Y, Ito K, Kimura C, Iwasaki N, Suzuki K, Harada T, Li HM, Uehara J, Miyazaki T, Minami A, Kon S, Uede T, Alpha9 integrin and its ligands constitute critical joint microenvironments for development of autoimmune arthritis. J Immunol 182, 8015–8025 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Mostovich LA, Prudnikova TY, Kondratov AG, Loginova D, Vavilov PV, Rykova VI, Sidorov SV, Pavlova TV, Kashuba VI, Zabarovsky ER, Grigorieva EV, Integrin alpha9 (ITGA9) expression and epigenetic silencing in human breast tumors. Cell Adh Migr 5, 395–401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roy S, Bingle L, Marshall JF, Bass R, Ellis V, Speight PM, Whawell SA, The role of alpha9beta1 integrin in modulating epithelial cell behaviour. J Oral Pathol Med 40, 755–761 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Johnson JL, Emerging regulators of vascular smooth muscle cell function in the development and progression of atherosclerosis. Cardiovascular research 103, 452–460 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Huang XZ, Wu JF, Ferrando R, Lee JH, Wang YL, Farese RV Jr., Sheppard D, Fatal bilateral chylothorax in mice lacking the integrin alpha9beta1. Mol Cell Biol 20, 5208–5215 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitchell AL, Attwood TK, Babbitt PC, Blum M, Bork P, Bridge A, Brown SD, Chang HY, El-Gebali S, Fraser MI, Gough J, Haft DR, Huang H, Letunic I, Lopez R, Luciani A, Madeira F, Marchler-Bauer A, Mi H, Natale DA, Necci M, Nuka G, Orengo C, Pandurangan AP, Paysan-Lafosse T, Pesseat S, Potter SC, Qureshi MA, Rawlings ND, Redaschi N, Richardson LJ, Rivoire C, Salazar GA, Sangrador-Vegas A, Sigrist CJA, Sillitoe I, Sutton GG, Thanki N, Thomas PD, Tosatto SCE, Yong SY, Finn RD, InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res 47, D351–D360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sonnhammer EL, Ostlund G, InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res 43, D234–239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH, CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res 39, D225–229 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Ward R. E. t., uninflatable encodes a novel ectodermal apical surface protein required for tracheal inflation in Drosophila. Dev Biol 336, 201–212 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghabrial AS, Krasnow MA, Social interactions among epithelial cells during tracheal branching morphogenesis. Nature 441, 746–749 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Loubery S, Seum C, Moraleda A, Daeden A, Furthauer M, Gonzalez-Gaitan M, Uninflatable and Notch control the targeting of Sara endosomes during asymmetric division. Curr Biol 24, 2142–2148 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Xie G, Zhang H, Du G, Huang Q, Liang X, Ma J, Jiao R, Uif, a large transmembrane protein with EGF-like repeats, can antagonize Notch signaling in Drosophila. PLoS One 7, e36362 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis-Knowlton J, Turner JE, Turner A, Damian-Loring S, Hagler N, Henderson T, Emery IF, Bond K, Duarte CW, Vary CPH, Eldrup-Jorgensen J, Liaw L, Characterization of smooth muscle cells from human atherosclerotic lesions and their responses to Notch signaling. Lab Invest 99, 290–304 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schweisguth F, Regulation of notch signaling activity. Curr Biol 14, R129–138 (2004). [PubMed] [Google Scholar]
- 48.Deford P, Brown K, Richards RL, King A, Newburn K, Westover K, Albig AR, MAGP2 controls Notch via interactions with RGD binding integrins: Identification of a novel ECM-integrin-Notch signaling axis. Exp Cell Res 341, 84–91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen PY, Qin L, Li G, Tellides G, Simons M, Smooth muscle FGF/TGFbeta cross talk regulates atherosclerosis progression. EMBO molecular medicine 8, 712–728 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shang T, Yednock T, Issekutz AC, alpha9beta1 integrin is expressed on human neutrophils and contributes to neutrophil migration through human lung and synovial fibroblast barriers. J Leukoc Biol 66, 809–816 (1999). [DOI] [PubMed] [Google Scholar]
- 51.Chistiakov DA, Bobryshev YV, Orekhov AN, Changes in transcriptome of macrophages in atherosclerosis. J Cell Mol Med 19, 1163–1173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Young EP, Stitziel NO, Capitalizing on Insights from Human Genetics to Identify Novel Therapeutic Targets for Coronary Artery Disease. Annu Rev Med 70, 19–32 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z, Vascular smooth muscle cells in atherosclerosis. Nature reviews. Cardiology 16, 727–744 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, Kundu R, Nagao M, Coller J, Koyano TK, Fong R, Woo YJ, Liu B, Montgomery SB, Wu JC, Zhu K, Chang R, Alamprese M, Tallquist MD, Kim JB, Quertermous T, Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med 25, 1280–1289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bauer RC, Tohyama J, Cui J, Cheng L, Yang J, Zhang X, Ou K, Paschos GK, Zheng XL, Parmacek MS, Rader DJ, Reilly MP, Knockout of Adamts7, a novel coronary artery disease locus in humans, reduces atherosclerosis in mice. Circulation 131, 1202–1213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK, KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 21, 628–637 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu B, Pjanic M, Wang T, Nguyen T, Gloudemans M, Rao A, Castano VG, Nurnberg S, Rader DJ, Elwyn S, Ingelsson E, Montgomery SB, Miller CL, Quertermous T, Genetic Regulatory Mechanisms of Smooth Muscle Cells Map to Coronary Artery Disease Risk Loci. Am J Hum Genet 103, 377–388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fukuda D, Aikawa E, Swirski FK, Novobrantseva TI, Kotelianski V, Gorgun CZ, Chudnovskiy A, Yamazaki H, Croce K, Weissleder R, Aster JC, Hotamisligil GS, Yagita H, Aikawa M, Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc Natl Acad Sci U S A 109, E1868–1877 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boucher J, Gridley T, Liaw L, Molecular pathways of notch signaling in vascular smooth muscle cells. Front Physiol 3, 81 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Williams J, Sarmento OF, Alencar GF, Hess DL, Bevard MH, Greene ES, Murgai M, Turner SD, Geng YJ, Bekiranov S, Connelly JJ, Tomilin A, Owens GK, Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat Med 22, 657–665 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sundstrom J, Vasan RS, Circulating biomarkers of extracellular matrix remodeling and risk of atherosclerotic events. Curr Opin Lipidol 17, 45–53 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Langley SR, Willeit K, Didangelos A, Matic LP, Skroblin P, Barallobre-Barreiro J, Lengquist M, Rungger G, Kapustin A, Kedenko L, Molenaar C, Lu R, Barwari T, Suna G, Yin X, Iglseder B, Paulweber B, Willeit P, Shalhoub J, Pasterkamp G, Davies AH, Monaco C, Hedin U, Shanahan CM, Willeit J, Kiechl S, Mayr M, Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J Clin Invest 127, 1546–1560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andrews MR, Czvitkovich S, Dassie E, Vogelaar CF, Faissner A, Blits B, Gage FH, ffrench-Constant C, Fawcett JW, Alpha9 integrin promotes neurite outgrowth on tenascin-C and enhances sensory axon regeneration. J Neurosci 29, 5546–5557 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hakkinen L, Hildebrand HC, Berndt A, Kosmehl H, Larjava H, Immunolocalization of tenascin-C, alpha9 integrin subunit, and alphavbeta6 integrin during wound healing in human oral mucosa. J Histochem Cytochem 48, 985–998 (2000). [DOI] [PubMed] [Google Scholar]
- 65.Smith LL, Cheung HK, Ling LE, Chen J, Sheppard D, Pytela R, Giachelli CM, Osteopontin N-terminal domain contains a cryptic adhesive sequence recognized by alpha9beta1 integrin. J Biol Chem 271, 28485–28491 (1996). [PubMed] [Google Scholar]
- 66.Nishimichi N, Higashikawa F, Kinoh HH, Tateishi Y, Matsuda H, Yokosaki Y, Polymeric osteopontin employs integrin alpha9beta1 as a receptor and attracts neutrophils by presenting a de novo binding site. J Biol Chem 284, 14769–14776 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kessler T ZL, Liu Z, Yin X, Huang Y, Wang Y, Fu Y, Mayr M, Ge Q, Xu Q, Zhu Y, Wang X, Schmidt K, de Wit C, Erdmann J, Schunkert H, Aherrahrou Z, Kong W., ADAMTS-7 inhibits re-endothelialization of injured arteries and promotes vascular remodeling through cleavage of thrombospondin-1. Circulation 131, 1191–1201 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liao Y, Smyth GK, Shi W, featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C, Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang L, Wang S, Li W, RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, Blewitt ME, Asselin-Labat ML, Smyth GK, Ritchie ME, Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res 43, e97 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang da W, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 76.Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ, GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao S, Guo Y, Sheng Q, Shyr Y, Advanced heat map and clustering analysis using heatmap3. Biomed Res Int 2014, 986048 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luo W, Brouwer C, Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 29, 1830–1831 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, Tan VY, Yarmolinsky J, Shihab HA, Timpson NJ, Evans DM, Relton C, Martin RM, Davey Smith G, Gaunt TR, Haycock PC, The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hemani G, Tilling K, Davey Smith G, Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet 13, e1007081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. SVEP1 expression in health and disease.
Fig. S2. Svep1 haploinsufficiency does not significantly alter plaque composition after 8 weeks of HFD feeding.
Fig. S3. VSMC-specific Svep1 deficiency reduces atherogenesis and plaque complexity, continued.
Fig. S4. The effect of the CAD-associated SVEP1 D2702G missense polymorphism and SVEP1 protein concentration in humans and mice.
Fig. S5. Itgα9 is expressed in both VSMCs and macrophages in mouse arteries.
Fig. S6. SVEP1 promotes local inflammation in atherosclerosis.
Fig. S7. SVEP1 promotes local inflammation in atherosclerosis, continued.
Table S3. Primer table.
Table S1. (Microsoft Excel format). VSMC transcriptional response to SVEP1 variants.
Table S2. (Microsoft Excel format). Transcriptional profile of atherosclerotic aortic arches.
Data file S1. (Microsoft Excel format). Individual data points.