

Abstract
The renin‐angiotensin system (RAS) may play a role in vascular aging. The authors hypothesized that blockade of the angiotensin II type 1 receptor with an angiotensin receptor blocker in healthy elderly subjects improves vascular compliance and endothelial function. Thirty‐five healthy elderly subjects were randomized to valsartan or placebo in a double‐blind crossover study after baseline testing for pulse wave velocity, aortic augmentation index, and brachial artery flow‐mediated dilation. Angiotensin II type 1 receptor blockade with valsartan improved vascular compliance but not flowmediated dilation. Changes in pulse wave velocity with valsartan were correlated with change in central systolic blood pressure and pulse pressure and remained associated on multivariate analysis. Change in pulse wave velocity after adjusting for degree of blood pressure change, age, and sex remained correlated with assignment to the angiotensin receptor blocker but not placebo. These data suggest that angiotensin II type 1 receptor blockade improves aging‐related vascular compliance without alterations in flow‐mediated dilation. Mechanisms regulating compliance and endothelial function are complex and may not necessarily converge in aging.
Aging is associated with progressive arterial stiffening and endothelial dysfunction. 1 , 2 , 3 , 4 , 5 These alterations represent not only important surrogates for risk but may potentially mediate some of the excess cardiovascular risk attributed to the aging process. 6 Changes in vasomotor tone and reactivity with aging are believed to result at least in part from increased generation of reactive oxygen species (ROS) and inactivation of nitric oxide (NO). 4 , 5 , 7 , 8 NO is a potent regulator of vascular homeostasis and endothelial function, and modulation in levels of this molecule at the level of the vasculature have been shown to be accompanied by coordinate changes in large‐vessel arterial compliance and endothelial function. Thus, although large‐vessel vascular compliance is partly structurally determined and relates to vascular architecture and composition, a portion is clearly dynamic and subject to regulation. 9
There is increasing evidence to support the concept that the renin‐angiotensin system (RAS) is activated in aging, particularly at the local, vascular level. 10 , 11 Through its effects on the angiotensin type 1 receptor (AT1R), angiotensin II modulates key enzymes involved in generation of ROS that under normal circumstances play an important role in cellular homeostasis and modulation of physiologic processes such as cell growth and mitogenesis. 12 , 13 , 14 The RAS is a key regulator of collagen synthesis, with activation of this system associated with myocardial and vascular fibrosis. 15 , 16 Previous studies have shown that AT1R antagonists significantly reduce arterial stiffness in hypertensive subjects, 17 , 18 while they improve endothelial function in patients with coronary artery disease. 19 In prior studies in the elderly, while nonspecific strategies such as antioxidants that target excess generation of ROS are ineffective, approaches addressing specific pathways that mediate excess ROS generation have yielded more favorable results. 20 , 21 The objective of this study was to evaluate whether an AT1R antagonist, valsartan, could improve large‐vessel arterial vascular compliance and endothelial function through the aforementioned effects in healthy normotensive elderly subjects, thereby suggesting a role of angiotensin II in mediating the vascular dysfunction related solely to the aging process.
METHODS
All studies were performed after informed consent and with prior approval from the University of Michigan Institutional Review Board.
Study Subjects
All subjects in the study were healthy individuals older than 65 years with normal left ventricular function and normal renal function. Exclusion criteria included the following: (1) hypertension (blood pressure [BP] >139/90 mm Hg); (2) hyperlipidemia (total cholesterol >220 mg/dL); (3) current smoking; (4) diabetes (fasting glucose >100 mg/dL); (5) obesity (defined as >85th percentile for body mass index); (6) the use of angiotensin‐converting enzyme (ACE) inhibitors, hormone replacement therapy, statins, or AT1R antagonists; (7) unstable medical conditions or renovascular disease; and (8) initiation of over‐the‐counter vitamins such as vitamin A, C, or E within 6 months of study initiation.
Subjects were invited by the University of Michigan and Mount Sinai schools of medicine to participate if they met the inclusion/exclusion criteria. Subjects were solicited through advertisements and telephone calls. A total of 35 subjects met the initial screening criteria and underwent a history and physical examination, blood chemistry with fasting lipids, and an echocardiography screen to ensure normal systolic function. Individuals who met the lipid and chemistry criteria then underwent baseline blood sampling before being randomized to receive either valsartan (160 mg/d) or placebo in a double‐blinded randomized fashion for a week, as outlined in Figure 1. The dose was then gradually doubled to 320 mg/d over a period of 2‐3 weeks, provided the patients could tolerate this dose and there was not a >10% decrease in systolic BP (SBP). At the end of 13 weeks, treatment was discontinued for 4 weeks (washout phase), and patients were switched to the other arm and treatment continued for an additional 13 weeks. Vascular and laboratory studies were then repeated at the end of this period. Two subjects dropped out during the course of the study; the data presented reflect the remaining 33 subjects.
Figure 1.
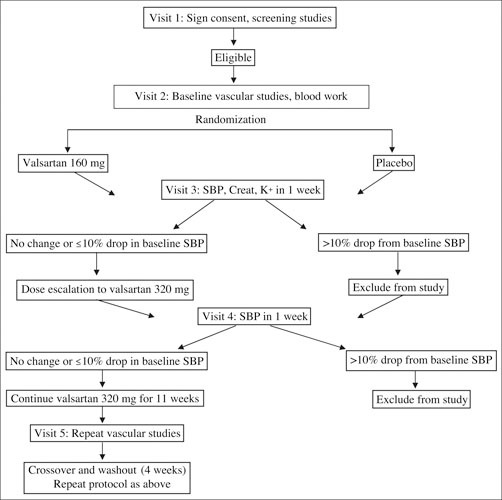
Study flow. SBP indicates systolic blood pressure; Creat, creatinine.
Brachial Artery Vasoreactivity Studies
Flow‐mediated dilation (FMD) of the brachial artery was determined according to established and validated methodology. 22 Images were obtained with a 10‐MHz linear array transducer and an HP Image Point ultrasound system (Hewlett Packard, Andover, MA). Imaging was performed in the morning with the subject in a fasting state and resting quietly for at least 10 minutes in a light‐and temperature‐controlled room. After baseline measurements of brachial artery diameter, a BP cuff was inflated to 200 mm Hg over the proximal portion of the right forearm for 4 minutes. FMD was determined 1 minute after release of the cuff. 23 Brachial artery diameter was then allowed to return to baseline over a period of 15 minutes. Endothelial‐independent responsiveness was then evaluated with 0.4 mg of nitroglycerin administered sublingually with brachial artery images obtained 3 minutes later. Four triggered events (defined as the onset of the R wave on the electrocardiogram [ECG]) for each intervention were recorded and acquired through a frame grabber attached to a computer. Each triggered event consisted of 6 sequential frames. The media‐adventitia interface in a linear portion of the vessel was chosen for analysis. The end point of measurement was the percentage change in diameter in response to reactive hyperemia and to nitroglycerin. For follow‐up studies, the images from the subject's initial study were recalled on a Matrox secondary monitor (Matrox, Quebec, Canada) and the vessel wall segments matched accurately to ensure that measurements were being made in the same segment. An independent reviewer not familiar with the treatment status of the patient reviewed the images.
Arterial Compliance and Central Blood Pressure Determination
Arterial stiffness was assessed by measuring carotid‐femoral pulse wave velocity (PWV) and central augmentation index by radial tonometry. The protocol and the reproducibility/reliability data in our laboratory have been reported previously. 24 , 25 A hand‐held high‐fidelity tonometer (SPC‐301; Millar Instruments, Houston, TX) was placed over the carotid and then the femoral arteries to record pressure waves. Simultaneous ECG tracings and pressure waves were recorded. The length of the descending aorta was approximated by subtracting the manubrium‐carotid artery distance from the manubrium‐femoral artery distance. PWV in m/s was calculated in an automated fashion by proprietary software (AtCor, version 7.0; AtCor Medical, Sydney, Australia). Four PWV measurements were recorded for each subject. A measurement was excluded if the pressure contour was of poor quality or if a significant difference (>15%) in heart rate was found between the carotid and femoral measurements. A subject's PWV was the average of the technically acceptable measurements. Waveforms were also recorded at the level of the radial artery. After 20 sequential waveforms had been acquired, a validated generalized transfer function was used to generate the corresponding central aortic pressure waveform. Aortic augmentation index and central aortic blood pressures were derived from this waveform using the technique of pulse waveform analysis. 26
Sample Size Estimates and Statistics
This study was originally designed with the intent of detecting a ≥2% change in endothelium‐dependent forearm dilation in the subjects receiving the angiotensin receptor blocker (ARB) regimen. Using an effect size of 2.0/4.0 (0.50), a sample size of 34 subjects was estimated as required for a crossover‐type design, assuming an α of .05 and power of 80%. All values were expressed as means ± SD unless specified. Changes in reactive hyperemia, nitroglycerin, and other baseline vascular outcome measures in the various groups were compared using analysis of variance (ANOVA). Statistical significance was established at P≤.05. Univariate Pearson correlation coefficients and significance were performed to assess the relationship among known predictors of endothelial function, arterial compliance as measured by PWV, and radial augmentation index. These variables included age, sex, baseline SBP, pulse pressure, body mass index, lipid panel, and baseline diameter of the brachial artery. Variables showing an association with endothelial function or compliance (P<.05) on univariate analysis were included in a multivariate model. All statistical analyses were performed using SAS software (version 9.1, SAS Institute, Inc, Cary, NC).
RESULTS
Figure 1 displays the study flow. Table I summarizes the characteristics of the 33 individuals enrolled in the study. All subjects were nonobese (mean body mass index, 27±4 kg/m2) healthy individuals with no known cardiovascular disease. None of the patients had a family history of premature coronary or vascular disease. The mean age was 71±6 years, and 60% were men. Mean glomerular filtration rate by the modified diabetes and renal disease equation 27 was 65±10 mL/min. Mean total cholesterol, low‐density lipoprotein cholesterol, and tri‐glyceride levels did not change during the course of the trial (data not shown). Table II depicts hemodynamic changes in the trial. Valsartan (320 mg/d) resulted in a 9‐mm Hg decrease in central aortic SBP compared with a 5‐mm Hg decrease in the placebo group at the end of 13 weeks of therapy (net decrease of 4 mm Hg), with no discernible effect on central aortic diastolic pressure. Central aortic pulse pressure decreased from 46.8±12 to 43.3±10 mm Hg in the placebo group (P=.03) and from 44.0±10 to 38.11±10 mm Hg (P=.0002) in the valsartan group. Figure 2 depicts a box plot of mean FMD values in the placebo group compared with the ARB group. Mean FMD changed from 3.4%±4.6% to 3.0%±4.4% in the placebo arm compared with 2.9%±5.0% to 3.7%±4.7% in the valsartan arm (P=nonsignificant for both groups compared with baseline values and between groups). Mean nitroglycerin‐mediated dilation changed from 14.8%±5.2% to 15.4%±5.6% following placebo vs 16.2%±6.3% to 17.1%±7.1% following valsartan (P=.81 and P=.55, respectively, by ANOVA) (Figure 2).
Figure 2.
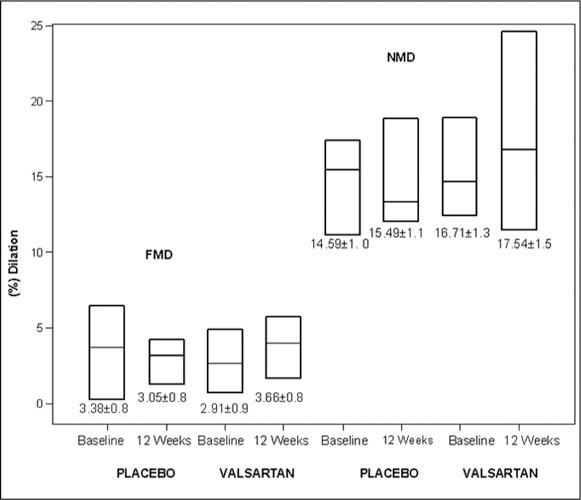
Flow‐mediated dilation (FMD) and nitroglycerin‐mediated dilation (NMD) in response to placebo and valsartan treatment. Changes between groups were not significant. Figure depicts median and 25th–75th percentile as a box plot. Mean ± SEM values are depicted below each box.
Figure 3 depicts change in PWV in the trial. Mean PWV decreased in the ARB group from 7.87±1.2 m/s to 7.30±1.35 m/s (Δ=0.4 m/s; P<.01 by ANOVA) in comparison with the mean PWV in the placebo group, which changed from 7.89±1.5 m/s to 7.86±1.4 m/s (Δ=0.03; P=nonsignificant by ANOVA). Mean augmentation index decreased in both the placebo and valsartan groups (25.5%±7.6% to 24.4%±8.6% and 22.9%±9.7% to 21.8%±10%, respectively) with no differences between groups or between initial and final values. A secondary responder analysis demonstrated that 45% of subjects decreased their PWV after valsartan compared with 20% of the placebo (P<.05 for the comparison). Linear regression analysis was performed to determine the relationship between baseline variables and response to therapy with valsartan. The univariate predictors of change in PWV included change in central SBP (Figure 4) (r 2=0.249; P=.006) and change in central aortic pulse pressure (r 2=0.138; P=.047). On multivariate analysis, changes in central aortic SBP (P=.0086) and central aortic pulse pressure (P=.0492) with the ARB remained significant after adjustment for age and sex. In contrast, change in PWV with the placebo group did not correlate with change in central aortic SBP or pulse pressure. To discern whether the change in PWV seen with treatment with valsartan was dependent on treatment status rather than reduction in BP, we assessed the relationship between change in PWV according to treatment status, after adjusting for age, sex, and degree of BP lowering. Changes in PWV remained correlated with assignment to valsartan, but not placebo, after adjusting for reduction in SBP.
Figure 3.
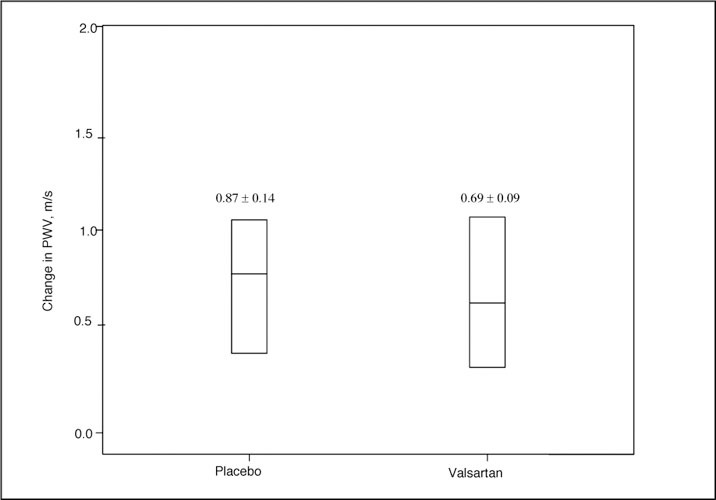
Change in pulse wave velocity (PWV) in response to placebo and valsartan treatment. Values above the box plots represent mean ± SEM.
Figure 4.
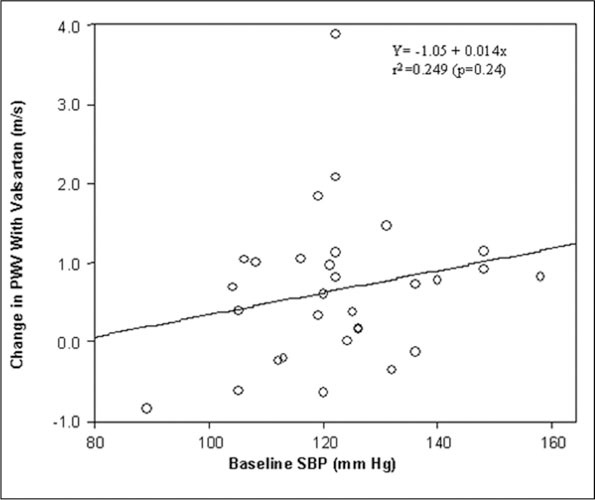
Correlation plot of changes in systolic blood pressure (SBP) and pulse wave velocity (PWV) after valsartan treatment. The solid line indicates the regression line.
DISCUSSION
We have demonstrated in this study that AT1R blockade improves vascular compliance in healthy normotensive aged subjects without prior cardiovascular disease or other traditional risk factors for vascular dysfunction other than aging. Alteration in large‐vessel arterial compliance was not, however, accompanied by changes in FMD in the brachial artery. These changes, although predicted by the reduction in BP, remained significant after accounting for the degree of BP reduction, suggesting an effect of AT1R blockade per se. These findings suggest that blockade of the RAS can improve the aging‐related impairment in vascular compliance and that this may occur without concomitant improvement in endothelial dysfunction.
Previous studies have demonstrated that aging is consistently associated with a progressive decline in the vasodilatory capacity of conduit and resistance vessels. 2 , 28 While a number of etiologies to explain this hallmark of aging have been set forth, a prominent aspect of the reduced capacity in response to stimuli such as shear stress or agonists appears to be related to the excess generation of ROS in the vessel wall. 4 , 5 , 29 Excess generation of ROS degrades NO, resulting in reductions in this key molecule and disruption of cellular homeostatic pathways. 4 While the precise mechanisms underlying the excess generation of ROS in aging are controversial, they very likely involve a multiplicity of sources. 4 , 21 , 30 , 31 An important generator of ROS, at least in the arterial wall endothelium and smooth muscle, is the vascular nicotinamide adenine dinucleotide phosphate (reduced) oxidase family of enzymes. 30 The latter oxidase is prominently regulated by angiotensin II, as evidenced by increased generation of ROS with the latter. 32 Conversely, AT1R antagonism reduces ROS generation and restores and/or improves endothelium‐dependent vasodilation in patients with coronary artery disease and risk factors for atherosclerosis such as hypertension, hypercholesterolemia, and diabetes. 19 , 33 Consistent with a potential role for angiotensin II in the vasculopathy of aging, expression of angiotensin II, ACE, and the AT1R have been reported to be increased in aging. 34 Thus there is significant experimental rationale to suspect that the local vascular RAS may mediate some of the vascular alterations seen in conditions such as aging. One could therefore hypothesize that antagonizing the RAS could potentially improve age‐related arterial dysfunction. In a previous small study, however, we failed to show improvement in FMD with losartan, a noncompetitive AT1R antagonist, in healthy older individuals. 35 In the previous study, we hypothesized that the lack of effect of AT1R antagonism in this study was probably due to inadequate blockade of the receptor due to the use of a low dose of the drug (50 mg of losartan) as part of a once‐daily regimen. According to pharmacokinetic data, this regimen does not produce a sustained 24‐hour inhibition of the RAS, due to the 6‐hour half‐life of losartan. 36 We therefore tested the effects of a regimen that ensures complete blockade of the AT1R, employing higher doses of a competitive AT1R antagonist valsartan.
The design of the present study was intended to circumvent other potential risk factors that may independently alter endothelial function. As a result, the subjects studied were healthy, with no risk factors for vascular dysfunction other than aging. Additionally, a specific design aspect related to the fact that we wished to avoid excess BP lowering with valsartan to decrease the likelihood of BP lowering being a significant driver of changes in the vasculature. Individuals with excessive decreases in systolic BP were excluded during the run‐in phase of the study. We hypothesized that if NO is a key regulator of conduit vessel compliance and endothelial function, alterations in FMD should result in parallel improvements in compliance as determined by measurements of aortic PWV and radial artery augmentation index.
Our data indicate that changes in compliance may occur independently of changes in FMD. Based on our results, several different scenarios are possible. One possibility is that mechanisms that regulate large‐vessel compliance may be distinct from those that regulate endothelial function. How may AT1R antagonism alter arterial compliance? Alterations in BP (pulse pressure), interstitial matrix composition, and reduction in vasoconstriction may all have contributed to improvements in compliance. In this study, changes in aortic compliance in response to valsartan were strongly correlated with alterations in central SBP and aortic pulse pressure. It is possible that AT1R antagonism through favorable effects on interstitial collagen(s) may have mediated these effects, even within the short duration of this trial 37 ; however, we did not measure these parameters in this trial. Our results are consistent with prior studies that have demonstrated an impact of AT1R antagonism on compliance in younger hypertensive patients and underscores the importance of targeting the RAS as a therapeutic modality to improve arterial function. 38 , 39
The lack of effect of valsartan on FMD, despite significant reductions in SBP, suggests a mechanism unique to aging that may not easily respond to modulation of traditional predictors of endothelial function, at least in the short term. While it is possible that endothelial function may have improved with more prolonged inhibition of the AT1R, it is also possible that it may be necessary to simultaneously target multiple maladaptive pathways in aging. These may include modulation of abnormal levels of tetrahydrobiopterin (an essential cofactor required for NO synthase function) or the substrate L‐arginine, which may result from either deficiency or up‐regulation of the arginase pathway. 31 , 40 , 41
CONCLUSIONS
Short‐term AT1R blockade improves aging‐related vascular compliance without restoring the associated peripheral conduit artery endothelial dysfunction. These findings may have important implications for the treatment of aging‐related alteration in vessel wall function and for the design of clinical trials targeting the vasculature in the elderly.
Disclosure: This study was funded by a research grant from Novartis Pharmaceuticals Corporation, East Hanover, NJ.
Reference
- 1. Luscher TF, Dohi Y, Tschudi M. Endothelium‐dependent regulation of resistance arteries: alterations with aging and hypertension. J Cardiovasc Pharmacol. 1992;19:S34–S42. [PubMed] [Google Scholar]
- 2. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–146. [DOI] [PubMed] [Google Scholar]
- 3. Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490–497. [DOI] [PubMed] [Google Scholar]
- 4. Tschudi MR, Barton M, Bersinger NA, et al. Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. J Clin Invest. 1996;98:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Der Loo B, Labugger R, Skepper JN, et al. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sutton‐Tyrrell K, Najjar SS, Boudreau RM, et al. Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well‐functioning older adults. Circulation. 2005;111:3384–3390. [DOI] [PubMed] [Google Scholar]
- 7. Payne JA, Reckelhoff JF, Khalil RA. Role of oxidative stress in age‐related reduction of NO‐cGMP‐mediated vascular relaxation in SHR. Am J Physiol Regul Integr Comp Physiol. 2003;285:R542–R551. [DOI] [PubMed] [Google Scholar]
- 8. Blackwell KA, Sorenson JP, Richardson DM, et al. Mechanisms of aging‐induced impairment of endothelium‐dependent relaxation: role of tetrahydrobiopterin. Am J Physiol Heart Circ Physiol. 2004;287:H2448–H2453. [DOI] [PubMed] [Google Scholar]
- 9. Kinlay S, Creager MA, Fukumoto M, et al. Endothelium‐derived nitric oxide regulates arterial elasticity in human arteries in vivo. Hypertension. 2001;38:1049–1053. [DOI] [PubMed] [Google Scholar]
- 10. Anderson S. Ageing and the renin‐angiotensin system [editorial]. Nephrol Dial Transplant. 1997;12:1093–1094. [DOI] [PubMed] [Google Scholar]
- 11. Wang M, Takagi G, Asai K, et al. Aging increases aortic MMP‐2 activity and angiotensin II in nonhuman primates. Hypertension. 2003;41:1308–1316. [DOI] [PubMed] [Google Scholar]
- 12. Rajagopalan S, Kurz S, Munzel T, et al. Angiotensin II‐mediated hypertension in the rat increases vascular super‐oxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension. 2003;42:1075–1081. [DOI] [PubMed] [Google Scholar]
- 14. Sorescu D, Szocs K, Griendling KK. NAD(P)H oxidases and their relevance to atherosclerosis. Trends Cardiovasc Med. 2001;11:124–131. [DOI] [PubMed] [Google Scholar]
- 15. Brilla CG, Zhou G, Rupp H, et al. Role of angiotensin II and prostaglandin E2 in regulating cardiac fibroblast collagen turnover. Am J Cardiol. 1995;76:8D–13D. [DOI] [PubMed] [Google Scholar]
- 16. Zhou G, Kandala JC, Tyagi SC, et al. Effects of angiotensin II and aldosterone on collagen gene expression and protein turnover in cardiac fibroblasts. Mol Cell Biochem. 1996;154:171–178. [DOI] [PubMed] [Google Scholar]
- 17. Mahmud A, Feely J. Effect of angiotensin II receptor blockade on arterial stiffness: beyond blood pressure reduction. Am J Hypertens. 2002;15:1092–1095. [DOI] [PubMed] [Google Scholar]
- 18. Klingbeil AU, John S, Schneider MP, et al. AT1‐receptor blockade improves augmentation index: a double‐blind, randomized, controlled study. J Hypertens. 2002;20:2423–2428. [DOI] [PubMed] [Google Scholar]
- 19. Hornig B, Landmesser U, Kohler C, et al. Comparative effect of ACE inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease: role of superoxide dismutase. Circulation. 2001;103:799–805. [DOI] [PubMed] [Google Scholar]
- 20. Carlsson CM, Pharo LM, Aeschlimann SE, et al. Effects of multivitamins and low‐dose folic acid supplements on flow‐mediated vasodilation and plasma homocysteine levels in older adults. Am Heart J. 2004;148:E11. [DOI] [PubMed] [Google Scholar]
- 21. Higashi Y, Sasaki S, Nakagawa K, et al. Tetrahydrobiopterin improves aging‐related impairment of endothelium‐dependent vasodilation through increase in nitric oxide production. Atherosclerosis. 2005;186:390–395. [DOI] [PubMed] [Google Scholar]
- 22. Celermajer DS, Sorensen KE, Gooch VM, et al. Non‐invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340:1111–1115. [DOI] [PubMed] [Google Scholar]
- 23. Anderson TJ, Uehata A, Gerhard MD, et al. Close relation of endothelial function in the human coronary and peripheral circulations. J Am Coll Cardiol. 1995;26:1235–1241. [DOI] [PubMed] [Google Scholar]
- 24. Sengstock D, Vaitkevicius PV, Supiano MA. Does increased arterial stiffness increase the risk for postural hypotension? Am J Geriatr Cardiol. 2005;14:224–229. [DOI] [PubMed] [Google Scholar]
- 25. Sengstock DM, Vaitkevicius PV, Supiano MA. Arterial stiffness is related to insulin resistance in nondiabetic hypertensive older adults. J Clin Endocrinol Metab. 2005;90:2823–2827. [DOI] [PubMed] [Google Scholar]
- 26. O'Rourke MF, Gallagher DE. Pulse wave analysis. J Hypertens Suppl. 1996;14:S147–S157. [PubMed] [Google Scholar]
- 27. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis. 2002;39:S1‐266. [PubMed] [Google Scholar]
- 28. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346–354. [DOI] [PubMed] [Google Scholar]
- 29. Muscari C, Giaccari A, Giordano E, et al. Role of reactive oxygen species in cardiovascular aging. Mol Cell Biochem. 1996;160–161:159–166. [DOI] [PubMed] [Google Scholar]
- 30. Hamilton CA, Brosnan MJ, Mclntyre M, et al. Superoxide excess in hypertension and aging: a common cause of endothelial dysfunction. Hypertension. 2001;37:529–534. [DOI] [PubMed] [Google Scholar]
- 31. Eskurza I, Myerburgh LA, Kahn ZD, et al. Tetrahydrobiopterin augments endothelium‐dependent dilatation in sedentary but not in habitually exercising older adults. J Physiol. 2005;568:1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rajagopalan S, Kurz S, Munzel T, et al. Angiotensin II‐mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schiffrin EL, Touyz RM Multiple actions of angiotensin II in hypertension: benefits of AT1 receptor blockade. J Am Coll Cardiol. 2003;42:911–913. [DOI] [PubMed] [Google Scholar]
- 34. Duggan J, Kilfeather S, O'Brien E, et al. Effects of aging and hypertension on plasma angiotensin II and platelet angiotensin II receptor density. Am J Hypertens. 1992;5:687–693. [DOI] [PubMed] [Google Scholar]
- 35. Rajagopalan S, Brook R, Mehta RH, et al. Effect of losartan in aging‐related endothelial impairment. Am J Cardiol. 2002;89:562–566. [DOI] [PubMed] [Google Scholar]
- 36. Oparil S. Newly emerging pharmacologic differences in angiotensin II receptor blockers. Am J Hypertens. 2000;13:18S–24S. [DOI] [PubMed] [Google Scholar]
- 37. Weber KT. From inflammation to fibrosis: a stiff stretch of highway. Hypertension. 2004;43:716–719. [DOI] [PubMed] [Google Scholar]
- 38. Shargorodsky M, Leibovitz E, Lubimov L, et al. Prolonged treatment with the AT1 receptor blocker, valsartan, increases small and large artery compliance in uncomplicated essential hypertension. Am J Hypertens. 2002;15:1087–1091. [DOI] [PubMed] [Google Scholar]
- 39. Munakata M, Nagasaki A, Nunokawa T, et al. Effects of valsartan and nifedipine coat‐core on systemic arterial stiffness in hypertensive patients. Am J Hypertens. 2004;17:1050–1055. [DOI] [PubMed] [Google Scholar]
- 40. White AR, Ryoo S, Li D, et al. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension. 2006;47:245–251. [DOI] [PubMed] [Google Scholar]
- 41. Berkowitz DE, White R, Li D, et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108:2000–2006. [DOI] [PubMed] [Google Scholar]