

Abstract
Hypertension and cardiovascular disease are leading causes of morbidity and mortality. Accumulating data demonstrate a relationship between hypertension and several vascular and metabolic abnormalities that are components of the cardiometabolic syndrome. The components of the cardiometabolic syndrome include insulin resistance/hyperinsulinemia, central obesity, dyslipidemia, hypertension, microalbuminuria, increased inflammation, and oxidative stress. There is growing evidence that tissue activation of the renin‐angiotensin‐aldosterone system participates in endothelial dysfunction, microalbuminuria, insulin resistance, and subsequent cardiovascular and chronic kidney disease. The notion that hypertension is a metabolic as well as a vascular disease opens a new paradigm for the treatment of this disorder.
Hypertension (HTN) is a leading cause of morbidity and mortality. In the United States, it is estimated that more than 52 million people—roughly 30% of the total population—have HTN. 1 Until recently, HTN was considered an independent morbidity leading to chronic cardiovascular disease (CVD) complications, in particular coronary heart disease, chronic kidney disease (CKD), and stroke. However, recent advances in the study of the mechanisms that contribute to HTN and its complications have led to a more comprehensive understanding of this disorder. It is well documented that hypertensive patients exhibit more frequent impairment of insulin resistance/hyperinsulinemia, dyslipidemia, microalbuminuria, and obesity than nonhypertensive individuals. 2 , 3
The obesity epidemic experienced by industrialized and nonindustrialized countries has contributed to a rise in the prevalence of HTN and other components of the cardiometabolic syndrome (CMS), and subsequent CVD and CKD. 3 , 4 Recent data indicate a 110% increase in the prevalence of obesity, and at least a 65% prevalence of excess body weight in the US population. 4 The obesity epidemic becomes even more dramatic if we consider the increasing prevalence of overweight American children and adolescents (10%–15%). 5 HTN and obesity cluster with other CVD risk factors that provide a higher probability of CVD and CKD compared with isolated components of CMS. 6 , 7
CARDIOMETABOLIC SYNDROME
Despite the recent impressive progress in the understanding of HTN and CMS, the causal link between these two conditions remains to be better defined. Further under standing of underlying pathophysiologic factors should lead to improved treatment strategies. Characteristics associated with CMS have been described since the 1920s. The disorder has received multiple names, including syndrome X, the deadly quartet, dysmetabolic syndrome, plurimetabolic syndrome, insulin resistance syndrome, and finally, CMS. 2 , 7 , 8 Unfortunately, the epidemiologic trends of CMS were difficult to follow until uniform diagnostic criteria were developed. Since 1998, definitions of CMS (Table I) promulgated by the World Health Organization, and later by the National Cholesterol Education Program (NCEP) Adult Treatment Panel‐III (ATP‐III), 9 have allowed for a better assessment and tracking of the epidemiology and features of CMS.
Table I.
Definitions of the Cardiometabolic Syndrome
| Source | ||
|---|---|---|
| Parameter | National Cholesterol Education Program* | World Health Organization** |
| Insulinemia | — | ≥75% of normal or insulin resistance (clamp technique) |
| Fasting glycemia (mg/dL) | ≥110 | ≥110 or OGTT (2hr) ≥140 |
| Body shape | ||
| Men | Waist ≥ 102 cm | Waist‐to‐hip ratio ≥0.90 or BMI ≥30 kg/m2 |
| Women | Waist ≥88 cm | Waist‐to‐hip ratio ≥0.88 or BMI ≥30 kg/m2 |
| Triglycerides (mg/dL) | ≥150 | ≥150 |
| High‐density lipoprotein cholesterol (mg/dL) | ||
| Men | <40 | <35 |
| Women | <50 | <39 |
| Blood pressure (mm Hg) | ≥130/80 | ≥140/90 |
| Renal function | — | Microalbuminuria ≥20 mg/min or urine albumin—creatine ratio ≥30 mg/g |
| OGTT=oral glucose tolerance test; BMI=body mass index; *requires at least three criteria; **requires fasting insulin ≥75% of normal or fasting glycemia ≥100 mg/dL plus at least two other criteria | ||
The World Health Organization criteria lend more importance to the presence of insulin resistance, while the NCEP definition is more selective for the presence of obesity. Of the two, the NCEP definition is the more widely used. According to data from the Third National Health and Nutrition Examination Survey (NHANES III), the overall prevalence of CMS in the US population is 22.8% in men and 22.6% in women, 10 depending on age, gender, and ethnicity. The frequency of CMS increases dramatically in both sexes between the third and the sixth decades of life and appears to plateau thereafter. In women, the peak incidence of both CMS and CVD is between ages 60 and 80 years—the most rapidly growing segment of our population. 3 , 9 , 10
Along with HTN, insulin resistance and hyperinsulinemia are key pathophysiologic elements of CMS (Table II). Available studies indicate approximately 50% prevalence of insulin resistance in patients with HTN. 7 Despite experimental data linking HTN to hyperinsulinemia/insulin resistance, epidemiologic evidence has been difficult to establish. Retrospective data have provided some evidence of the association. 11 In one study, insulin resistance was directly measured in 1146 white, normotensive, normoglycemic European participants by means of the euglycemic hyperinsulinemic clamp technique, as well as by measurement of serum insulin levels. 11 In a homogeneous sub‐population of 333 subjects in which appropriate measurements were available, there was a significant association between HTN and insulin resistance/hyperinsulinemia, which was independent of age, gender, and presence or absence of obesity. According to this analysis, a reduction of 30% in insulin sensitivity predicted an increase in diastolic blood pressure (BP) of 2 mm Hg. This increase, although small in absolute numbers, may help to explain the increasing trends of HTN in association with the growing epidemic of CMS. Also in this analysis, the influence of insulin resistance proved to be stronger than 10 years of aging or an increase of three units of body mass index. This suggests that in people with normal or moderately high fasting insulinemia, the influence of insulin resistance on BP was stronger than the effect of increasing age or obesity.
Table II.
Cardiovascular Disease Risks That Cluster With Hypertension
| Central obesity* |
| Insulin resistance* |
| Low high‐density lipoprotein cholesterol level* |
| High triglyceride level* |
| Small, dense low‐density lipoprotein particles* |
| Microalbuminuria* |
| Chronic kidney disease |
| Elevated inflammatory markers |
| Impaired endothelial function |
| Increased cardiovascular oxidative stress (ROS) |
| Abnormal coagulation/fibrinolytic profiles |
| Hyperlipidemia |
| ROS=reactive oxygen species; *components of the cardiometabolic syndrome |
PATHOGENESIS OF HTN IN CMS
A causal relationship implicating insulin resistance/hyperinsulinemia as a cause of HTN is difficult to prove; however, population‐based studies suggest that serum insulin levels can predict BP and future development of HTN in different healthy populations, such as women, children, and adolescents. 12 , 13 , 14 Rodent models that exhibit insulin resistance such as the Dahl hypertensive, Zucker obese, and spontaneously hypertensive rats exhibit HTN. 15 In humans, insulin resistance as determined by the oral glucose tolerance test was established in a nonobese Japanese population with high‐normal BPs, and has also been confirmed in first‐degree normotensive relatives of hypertensive patients. 16 , 17 Treatment of HTN reduces rates of stroke as well as other CVD events to a greater extent than treatment of other CVD risk factors. 18
Early experimental studies related hyperinsulinemia per se to the pathogenesis of HTN via increased renal reabsorption and diminished excretion of sodium, leading to vascular volume expansion. 19 , 20 Insulin infusion at high doses can also acutely induce activation of the sympathetic nervous system in humans; this is probably related to an increase in the circulating levels of catecholamines. 21 In normotensive Sprague‐Dawley rats, chronic hyperinsulinemia raises BP, an effect that can be prevented by renal denervation—a finding that underscores the importance of renal‐neural autonomic integrity. 22 Thus, the combined effects of hyperinsulinemia and insulin resistance could act synergistically to enhance and perpetuate HTN and a subsequent increase in CVD.
GENETICS AND ENVIRONMENT
A genetic basis for the relationship between HTN and insulin resistance has been documented in animal models. Both HTN and insulin resistance are found in rodent models such as the Zucker obese and Goto‐Kakizaki rats. Both models have regions in chromosome 2, 3, and 19 that are related to components of CMS. 23 The recent dramatic increase in CMS and CVD suggests factors beyond a genetic predisposition. Obesity in industrialized countries has been related to sedentary lifestyle and the high‐calorie, high‐fat “Western diet.” It has been demonstrated in experimental models that glucose and lipid loads, and mixed fast food (which contains large amounts of saturated fats and carbohydrates) are able to trigger inflammatory responses linked to insulin resistance and oxidative stress. 24 The mechanisms involved include activation of inflammatory pathways as well as enhancement of the expression of certain oxidase constituents, which leads to the generation of superoxide anions. 15 , 25 , 26 In men, glucose infusion acutely induces activation of numerous factors that are involved in pathways that lead to endothelial dysfunction. 27 Interestingly, obese humans under basal conditions display activation of the same genes that are triggered by acute administration of glucose and lipids. The composition of the diet seems more important than the total amount of calories ingested, since isocaloric diets with a greater relative composition of fruits, vegetables, and fiber instead of sugar and fat do not induce proinflammatory changes or reactive oxygen species (ROS) production. Proinflammatory and oxidative changes can be reversed by fasting, as well as by reduction in the number of calories consumed. 28
MECHANISMS OF ENDOTHELIAL DYSFUNCTION ASSOCIATED WITH INSULIN RESISTANCE
Insulin resistance/hyperinsulinemia is central to the development of CMS, HTN, and dyslipidemia. 23 Insulin resistance describes an impaired response of different tissues to the metabolic actions of insulin. Insulin resistance is not universal, and some tissues retain insulin sensitivity while others do not. Traditionally, insulin resistance has been described in muscle, liver, and adipose tissue; however, there is mounting evidence that cardiovascular tissue is also influenced by the presence of insulin and its homologous peptide insulin‐like growth factor 1 (IGF‐1). 29 Binding of insulin and IGF‐1 triggers intracellular phenomena which enhance endothelial NO synthase and NO production. 30 NO is an endothelial vasodilator that has vascular protective actions including regulation of the coagulation cascade and fibrinolysis, promotion of vascular smooth muscle relaxation, and control of vascular growth and remodeling. 31 Insulin resistance contributes to both reduced NO production and increased oxidative destruction of NO. 15
Insulin displays intrinsic anti‐inflammatory activity 28 and causes vasorelaxation, in part through increases in endothelial NO production, which induces a series of enzyme changes that result in vascular smooth muscle cell relaxation. 15 In addition, insulin stimulates the activity of the Na+‐K+ adenosinetriphosphatase pump, leading to a subsequent reduction in intracellular Ca++ concentration. 15 Thus, insulin/IGF‐1 acts in conjunction with angiotensin II (AII) to promote left ventricular hypertrophy, vascular remodeling, and glomerular mesangial expansion. 29 This enhancement of cardiac hypertrophy is mediated through increased signaling via several growth pathways. 15
ROLE OF THE RENIN‐ANGIOTENSIN‐ALDOSTERONE SYSTEM (RAAS) AND OXIDATIVE STRESS
Oxidative stress, which is promoted by the RAAS, is a condition of increased oxidant production in cells characterized by the release of free radicals and resulting in cellular degeneration. 15 All components of the RAAS system are expressed in adipose tissue, especially in visceral adipose tissue. 29 , 32 The RAAS modulates adipocyte growth and differentiation and adipose tissue adipokine production. 32 Beyond stimulation of aldosterone secretion and promotion of renal tubular reabsorption of sodium, AII also antagonizes insulin and IGF‐1 actions (Figure). Through binding to the AII type 1 receptor, AII interferes with the delicate balance between antioxidants and increased reactive oxygen species (ROS). AII and aldosterone both promote the production of ROS in adipose tissue, skeletal muscle, and cardiovascular tissue. 15 , 33 , 34 Oxidative stress also induces modifications in DNA, proteins, lipid peroxidation, and gene expression patterns, and causes a shift toward proinflammatory and proatherogenic patterns. 33 ROS react with NO to form peroxynitrite, a potent oxidant that enhances oxidation and inflammation in various tissues and disrupts the integrity of cellular membranes. Enhanced production of ROS depletes NO reserves, impairs endothelium‐dependent vasodilatation, promotes vascular inflammation, and creates a prothrombotic environment in the vasculature. 34 Oxidative stress in CMS is enhanced by increased free fatty acids and plasma triglycerides that also have a proinflammatory effect. 35 Oxidative stress creates altered production of adipokines that in turn promotes low‐grade inflammation, endothelial dysfunction, and a state of atherogenesis. 32 , 36 , 37 , 38 Recent work in our laboratory demonstrates that blockade of AII type 1 receptorabrogates exaggerated production of ROS and corrects insulin resistance in a rodent hypertensive model. 38
Figure.
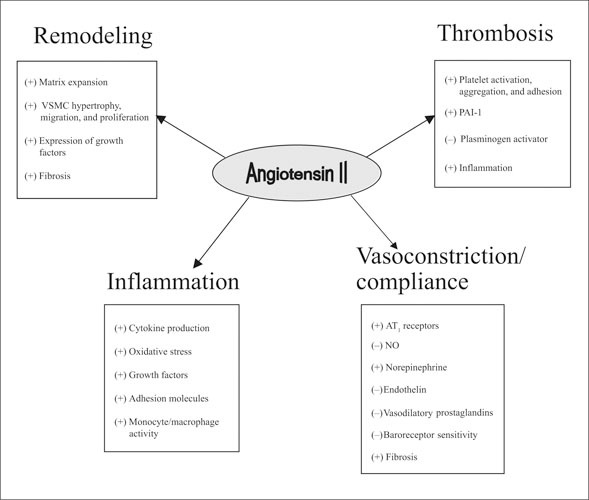
Systemic effects of angiotensin II. +=increased; −=decreased; VSMC=vascular smooth muscle cell; PAI‐1=plasminogen activator inhibitor type 1; AT1=angiotensin II type 1
ADIPOSITY
The concept of body fat as a simple deposit of energy without any metabolic role has been abandoned. Adipose tissue—which includes adipocytes, preadipocytes, and stromal tissue—is a metabolically active organ and plays a key role in energy homeostasis. 15 , 32 , 37 Visceral adipose tissue of patients with insulin resistance and type 2 diabetes is dysfunctional and is a source of chronic low‐grade inflammation. 37 Proinflammatory cytokines and adipokines that have been related to the development of insulin resistance and CMS, including interleukin‐6 and tumor necrosis factor‐α, are produced in adipose tissue. 32 There is accumulating evidence that enhanced production of these cytokines plays a key role in promoting thrombosis, vascular inflammation, and adhesiveness as well as promoting the formation of vulnerable plaque. 32 In states of insulin resistance, there is also diminished production of adiponectin, an adipokine which improves the action of insulin in diminishing vascular inflammation. 37
THERAPEUTIC IMPORTANCE OF RAAS BLOCKADE
There is a growing body of evidence regarding the role of the RAAS in promoting endothelial dysfunction and microalbuminuria in CMS. 15 , 34 RAAS inhibition with angiotensin‐converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) is associated with a decreased incidence of new‐onset type 2 diabetes mellitus and improvement of CVD outcomes. The beneficial effects of RAAS inhibition on CVD in patients with insulin resistance or overt type 2 diabetes mellitus may best be explained by the blockade of the different pathologic phenomena induced by the activation of the RAAS: increased oxidative stress; increased vasoconstriction; promotion of a proinflammatory, procoagulatory, and proliferative environment; and the disruption of insulin signaling pathways. Clinically, these beneficial implications result in reduced CVD.
Drugs targeted to inhibit the RAAS appear to lessen the development of type 2 diabetes in persons with HTN. 15 An evaluation of the effect of captopril compared with conventional therapy (β blockers and diuretics) showed a significant reduction of 30% in the incidence of type 2 diabetes. In the subgroup of patients who were diabetic before the beginning of the study, a significant reduction in fatal and nonfatal CVD was demonstrated. 39 Similar findings have been noted with other RAAS blockers in the prevention of new‐onset diabetes. 40
ARBs have also shown beneficial effects. Trials with losartan and valsartan have reported a reduction of more than 20% in new‐onset diabetes compared with an atenolol‐ or amlodipine‐based treatment regimen. 41 , 42 A combination of ACEIs and ARBs also reduces the incidence of new‐onset diabetes. 43
Collectively, these studies, as well as other already published and ongoing clinical trials, 44 support not only the concept of RAAS inhibition as a means of controlling HTN, but also as a strategy to reduce endothelial dysfunction, micro‐albuminuria, and the progression of patients with insulin resistance to clinical diabetes.
CONCLUSIONS
The relationship between HTN and CMS is complex and encompasses many interactive dysfunctional regulatory systems that all contribute to increased CVD. It involves not only vascular and hemodynamic changes that accompany HTN, but also a myriad of complex metabolic abnormalities that collectively constitute CMS. Insulin resistance/hyperinsulinemia and obesity promote a chronic low‐grade inflammatory state that characterizes dysfunctional adipose tissue, activation of the RAAS system, and oxidative stress, resulting in endothelial dysfunction, microalbuminuria, and increased CVD. From a clinical standpoint, development of uniform criteria to diagnose CMS allows us to quantify and identify at‐risk populations in whom intensive intervention should significantly prevent CVD. Pharmacologic treatment strategies, most importantly RAAS inhibition, as well as nonpharmacologic approaches, specifically diet and physical activity, address more comprehensively the treatment of HTN and CMS and the prevention of CVD and CKD.
References
- 1. Hajjar I, Kotchen TA. Trends in prevalence, awareness, treatment and control of hypertension in the United States, 1988–2000. JAMA. 2003;290:199–206. [DOI] [PubMed] [Google Scholar]
- 2. Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. [DOI] [PubMed] [Google Scholar]
- 3. Sowers JR, Haffner S. Treatment of cardiovascular and renal risk factors in the diabetic hypertensive. Hypertension. 2002;40:781–788. [DOI] [PubMed] [Google Scholar]
- 4. Stein CJ, Colditz GA. The epidemic of obesity. J Clin Endocrinol Metab. 2004;89:2522–2525. [DOI] [PubMed] [Google Scholar]
- 5. Flegal KM, Carroll MD, Ogden CL, et al. Prevalence and trends in obesity among US adults, 1999–2000. JAMA. 2002;288:1723–1727. [DOI] [PubMed] [Google Scholar]
- 6. DeFronzo RA, Ferrannini E. Insulin resistance: a multi‐faceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173–194. [DOI] [PubMed] [Google Scholar]
- 7. Sowers JR. Obesity as a cardiovascular risk factor. Am J Med. 2003;115(suppl 8A):37S–41S. [DOI] [PubMed] [Google Scholar]
- 8. McFarlane SI, Banerji M, Sowers JR. Insulin resistance and cardiovascular disease. J Clin Endocrinol Metab. 2001;86:713–718. [DOI] [PubMed] [Google Scholar]
- 9. Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults . Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA. 2001;285:2486–2496. [DOI] [PubMed] [Google Scholar]
- 10. Park YW, Zhu S, Palaniappan L, et al. The metabolic syndrome: prevalence and associated risk factor findings in the US population from the Third National Health and Nutrition Examination Survey, 1988–1994. Arch Intern Med. 2003;163:427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ferrannini E, Natali A, Capaldo B, et al. Insulin resistance, hyperinsulinemia and blood pressure: role of age and obesity. European Group for the Study of Insulin Resistance (EGIR) . Hypertension. 1997;30:1144–1149. [DOI] [PubMed] [Google Scholar]
- 12. Lissner L, Bengtsson C, Lapidus L, et al. Fasting insulin in relation to subsequent blood pressure changes and hypertension in women. Hypertension. 1992;20:797–801. [DOI] [PubMed] [Google Scholar]
- 13. Taittonen L, Uhari M, Nuutinen M, et al. Insulin and blood pressure among healthy children. Cardiovascular risk in young Finns. Am J Hypertens. 1996;9:194–199. [DOI] [PubMed] [Google Scholar]
- 14. Raitakari OT, Porkka KVK, Ronnemaa T, et al. The role of insulin in clustering of serum lipids and blood pressure in children and adolescents. Diabetologia. 1995;38:1042–1050. [DOI] [PubMed] [Google Scholar]
- 15. Sowers JR. Insulin resistance and hypertension. Am J Physiol Heart Circ Physiol. 2004;286:H1597–H1602. [DOI] [PubMed] [Google Scholar]
- 16. Kanauchi M, Yamano S, Kanauchi K, et al. Homeostasis model assessment of insulin resistance, quantitative insulin sensitivity check index, and oral glucose insulin sensitivity index in nonobese, nondiabetic subjects with high‐normal blood pressure. J Clin Endocrinol Metab. 2003;88:3444–3446. [DOI] [PubMed] [Google Scholar]
- 17. Vlasakova Z, Pelikanova T, Karasova L, et al. Insulin secretion, sensitivity, and metabolic profile of young healthy offspring of hypertensive parents. Metabolism. 2004;53:469–475. [DOI] [PubMed] [Google Scholar]
- 18. Collins R, Peto R, MacMahon S, et al. Blood pressure, stroke and coronary heart disease, II: short‐term reductions in blood pressure: overview of randomized drug trials in their epidemiological context. Lancet. 1990;335:827–838. [DOI] [PubMed] [Google Scholar]
- 19. DeFronzo RA, Cooke CR, Andres R, et al. The effect of insulin in renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest. 1975;55:845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baum M. Insulin stimulates volume absorption in the rabbit proximal convoluted tubule. J Clin Invest. 1987;79:1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kern W, Peters A, Born J, et al. Changes in blood pressure and plasma catecholamine levels during prolonged hyperinsulinemia. Metabolism. 2005;54:391–396. [DOI] [PubMed] [Google Scholar]
- 22. Huang WC, Fang TC, Cheng JT. Renal denervation prevents and reverses hyperinsulinemia‐induced hypertension in rats. Hypertension. 1998;32:249–254. [DOI] [PubMed] [Google Scholar]
- 23. Natali A, Ferrannini E. Hypertension, insulin resistance, and the metabolic syndrome. Endocrinol Metab Clin North Am. 2004;33:417–429. [DOI] [PubMed] [Google Scholar]
- 24. Pereira MA, Kartashov AI, Ebbeling CB, et al. Fast‐food habits, weight gain, and insulin resistance (the CARDIA study): 15‐year prospective analysis. Lancet. 2005;365:36–42. [DOI] [PubMed] [Google Scholar]
- 25. Mohanty P, Hamouda W, Garg R, et al. Glucose challenge stimulates reactive species (ROS) generation by leucocytes. J Clin Endocrinol Metab. 2000;85:2970–2973. [DOI] [PubMed] [Google Scholar]
- 26. Aljada A, Mohanty P, Abdo Tripathy D, et al. Increase in intranuclear nuclear factor kappa B and decrease in inhibitor kappa B in mononuclear cells after a mixed meal: evidence for a proinflammatory effect. Am J Clin Nutr. 2004;79:682–690. [DOI] [PubMed] [Google Scholar]
- 27. Aljada A, Ghanim H, Mohanty P, et al. Glucose intake induces an increase in activator protein 1 and early growth response 1 binding activities, in the expression of tissue factor and matrix metalloproteinase in mononuclear cells, and in plasma tissue factor and matrix metalloproteinase concentrations. Am J Clin Nutr. 2004;80:51–57. [DOI] [PubMed] [Google Scholar]
- 28. Dandona P, Aljada A, Chaudhuri A, et al. Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation. 2005;111:1448–1454. [DOI] [PubMed] [Google Scholar]
- 29. Aneja A, El‐Atat A, McFarlane SI, et al. Hypertension and obesity. Recent Prog Horm Res. 2004;59:169–205. [DOI] [PubMed] [Google Scholar]
- 30. Zeng G, Nystrom FH, Ravichardran LV, et al. Roles for insulin receptor, PI3‐kinase, and Akt in insulin‐signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation. 2000;101:1539–1545. [DOI] [PubMed] [Google Scholar]
- 31. Celermajer D. Endothelial dysfunction: does it matter? Is it reversible? J Am Coll Cardiol. 1997;30:325–333. [DOI] [PubMed] [Google Scholar]
- 32. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. [DOI] [PubMed] [Google Scholar]
- 33. Nickenig G, Harrison DG. The AT1‐type angiotensin receptor in oxidative stress and atherogenesis, I: oxidative stress and atherogenesis. Circulation. 2002;105:393–396. [DOI] [PubMed] [Google Scholar]
- 34. Sowers JR. Hypertension, angiotensin II and oxidative stress. N Engl J Med. 2002;346:1999–2001. [DOI] [PubMed] [Google Scholar]
- 35. Tripathy D, Mohanty P, Dhindsa S, et al. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes. 2003;52:2882–2887. [DOI] [PubMed] [Google Scholar]
- 36. Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27:813–823. [DOI] [PubMed] [Google Scholar]
- 38. Blendea MC, Jacobs D, Stump CS. Abrogation of oxidative stress improves insulin sensitivity in Ren‐2 rat model of tissue angiotensin II overexpression. Am J Physiol Endocrinol Metab. 2005;288:E353–E359. [DOI] [PubMed] [Google Scholar]
- 39. Hansson L, Lindholm LH, Niskanen L, et al. Effect of angiotensin‐converting‐enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPPP) randomized trial. Lancet. 1999;353:611–616. [DOI] [PubMed] [Google Scholar]
- 40. Heart Outcomes Prevention Evaluation Study Investigators . Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICROHOPE substudy. Lancet. 2000;355:253–259. [PubMed] [Google Scholar]
- 41. Julius S, Kjeldsen SV, Weber M, et al. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomized trial. Lancet. 2004;363:2022–2031. [DOI] [PubMed] [Google Scholar]
- 42. Dahlof B, Devereux RB, Kjeldsen SE, et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet. 2002;359:995–1003. [DOI] [PubMed] [Google Scholar]
- 43. McMurray JJ, Ostergren J, Swedberg K, et al. Effects of candesartan in patients with chronic heart failure and reduced left‐ventricular systolic function taking angiotensin‐converting enzyme inhibitors: the CHARM‐Added trial. Lancet. 2003;362:767–771. [DOI] [PubMed] [Google Scholar]
- 44. Buse JB, Rosenstock J. Prevention of cardiovascular outcomes in type 2 diabetes mellitus: trials on the horizon. Endocrinol Metab Clin North Am. 2005;34:221–235. [DOI] [PubMed] [Google Scholar]