

Abstract
Evidence suggests that renin‐angiotensin‐aldosterone system inhibition ameliorates endothelial dysfunction. The authors examined the effect of amlodipine besylate/benazepril HCl combination treatment compared with amlodipine besylate monotherapy in modulating endothelial dysfunction. This multicenter, double‐blind, 12‐week study randomized 70 hypertensive subjects with at least one other endothelial dysfunction risk factor to amlodipine besylate/benazepril HCl (5/20 mg/d force‐titrated to 5/40 mg/d) or amlodipine besylate monotherapy (5 mg/d force‐titrated to 10 mg/d). Both the combination and monotherapy produced significant median increases from baseline in percentage flow‐mediated vasodilation (2.0% and 1.2%, respectively) and percentage change in percent flow‐mediated vasodilation (25% and 16%, respectively). These improvements were numerically larger with combination treatment, but between‐group differences did not achieve statistical significance. Reductions in systolic and diastolic blood pressure were significantly greater (P=.0452/P=.0297) with combination treatment (−18.6/−12.3 mm Hg) than with monotherapy (−14.8/−9.1 mm Hg). A highly positive correlation between change in systolic blood pressure and change in percent of flow‐mediated vasodilation was demonstrated only for combination treatment.
Endothelial dysfunction (ED) is associated with numerous cardiovascular diseases (eg, atherosclerosis) as well as diseases and processes with cardiovascular components. The presence of additional risk factors, such as hypertension, diabetes, dyslipidemia, and obesity, serves to accelerate development and/or increase the severity of cardiovascular disease. The mechanisms by which ED leads to atherosclerosis are not fully elucidated but are thought to be related to activation of endothelial‐derived agents such as vascular cell adhesion molecules, which have been shown to lead to oxidative stress, lipid peroxidation, inflammation, cellular adhesion, and cytotoxicity. 1 , 2 Preclinical and clinical studies have demonstrated that angiotensin II plays a key role in the development of ED. 3 Thus, the use of agents that interrupt the renin‐angiotensin‐aldosterone system such as angiotensin‐converting enzyme (ACE) inhibitors and angiotensin receptor blockers have been advocated to improve vascular function. Calcium antagonists, particularly lipophilic members including amlodipine, have been shown to have antioxidant and antiatherosclerotic effects. 4 , 5 , 6 , 7 , 8
Endothelial function has been assessed in the coronary and peripheral vascular systems. Forearm circulation and flow‐mediated vasodilation (FMD) of the brachial artery reflect endothelial functional changes similar to those found in the coronary arteries of patients with coronary artery disease. 9 The percentage of FMD (%FMD) can be determined using hyperemic techniques or by the use of vasodilators. Measurement of %FMD has been instrumental in demonstrating the effect of various therapeutic agents on endothelial function as early as 6 weeks after initiating treatment.
Not all classes of antihypertensive agents have been shown to improve endothelial dysfunction. Thiazide diuretics have been shown to have no effect, 10 while β‐blockers have a weak effect on %FMD. 11 Significant effects on %FMD have been demonstrated for angiotensin receptor blockers, 12 hydroxymethylglutaryl coenzyme A reductase inhibitors (statins) 13 and ACE inhibitors. 14 Negligible improvement in %FMD has been shown with a low dose (5 mg) of the calcium channel blocker (CCB) amlodipine. 14
These trials confirm that inhibition of the renin‐angiotensin‐aldosterone system ameliorates ED. However, there is a paucity of data on the effect of combination therapy with amlodipine besylate/benazepril HCl in modulating ED. Furthermore, dose responses on ED with these agents have not been established. The Exploring Lotrel in Hypertensive Patients With Endothelial Dysfunction (EXPLORE) trial was designed to compare the effects of a 12‐week regimen of once‐daily amlodipine besylate/benazepril HCl with those of amlodipine besylate on endothelial function in subjects with hypertension and at least one other risk factor for ED.
METHODS
This multicenter, randomized, double‐blind study was conducted at 7 hospital‐based centers in Ohio, Colorado, and the eastern United States. The primary objective was to compare the effect of amlodipine besylate/benazepril HCl (5/20 mg force‐titrated to 5/40 mg) with that of amlodipine besylate monotherapy (5 mg force‐titrated to 10 mg) on endothelium‐dependent FMD, via brachial artery reactivity testing (BART), after 12 weeks of daily treatment in hypertensive subjects at risk for ED. Other objectives were to examine the effects on blood pressure (BP), serum markers of ED and vascular inflammation, and pedal (ankle) edema. This small study was intended to identify trends in the data, and its size was not based on statistical power considerations.
Subjects were recruited from investigators' private practices and through advertising from December 17,2002 through March 12, 2004. Men and women aged 21–75 years, inclusive, with systolic BP (SBP) ≥140 mm Hg and ≤180 mm Hg and at least one additional risk factor for ED were eligible for enrollment in the study. These risk factors included fasting plasma glucose 110–125 mg/dL, waist circumference ≥40 inches (102 cm) in men and ≥35 inches (89 cm) in women, body mass index >25 kg/m2, triglycerides >150 mg/dL, high‐density lipoprotein cholesterol <40 mg/dL in men and <45 mg/dL in women, family history of premature coronary artery disease, or a first‐degree relative with type 2 diabetes. Excluded were subjects with liver, kidney, overt coronary artery, or pulmonary disease or dysfunction; diabetes; and women of child‐bearing potential. Subjects who required concomitant antioxidant agents or vitamins, hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), long‐acting nitrates, or any medication that altered the renin‐angiotensin system were also excluded. The study protocol was approved by the respective trial site institutional review boards and was carried out according to Good Clinical Practice guidelines and the Declaration of Helsinki. All subjects provided written informed consent before enrollment into the study.
Study Protocol
Subjects entered a 3‐week placebo run‐in phase, during which a baseline FMD measurement via BART was performed. After the placebo run‐in, eligible subjects underwent baseline evaluations of serum markers of endothelial function and vascular inflammation and were randomly assigned to receive amlodipine besylate/benazepril HCl (5/20 mg once daily) or amlodipine besylate (5 mg once daily). After 6 weeks of active treatment, the dose of study medication was force‐titrated to amlodipine besylate/benazepril 5/40 mg once daily for subjects previously randomized to combination therapy or to amlodipine besylate 10 mg once daily for subjects previously randomized to monotherapy for the remaining 6 weeks of the study. The %FMD via BART, serum markers of ED, and vascular inflammation were evaluated at week 6 before force‐titration and again at week 12. Unacceptable ultrasounds were repeated within 72 hours of the original test at baseline and at week 12. Vital signs and adverse events were monitored at weeks 2, 6, 8, and 12.
Randomization numbers were generated by the sponsor using a validated system that automated the random assignment of treatment groups to randomization numbers. The active study drug was identified by a random number, associated with a specific treatment regimen. At each site, randomization numbers were assigned sequentially in the order in which subjects were randomized. Both study participants and investigators were unaware of the treatment assignments. Blinding of the investigational drug was maintained by use of matching capsules.
Evaluations
The primary efficacy variable was the change from baseline in %FMD via the BART procedure at week 12. The change from baseline in SBP and diastolic BP (DBP) was also evaluated. Secondary efficacy variables were the change from baseline to week 12 in circulating levels of interleukin (IL) 6, intracellular adhesion molecule‐1, vascular cell adhesion molecule, oxidized low‐density lipoprotein (LDL) cholesterol autoantibody, tumor necrosis factor α, and plasminogen activator inhibitor‐1. Comparisons between the groups for the change from baseline to week 6 and the change from week 6 to week 12 on endothelium‐dependent %FMD, via BART, were performed. Correlation between the change in %FMD and relevant variables was explored using the Spearman correlation procedure.
Brachial Arterial Reactivity Test
BART was performed as per a previously published method. 15 In brief, the brachial artery was imaged in the longitudinal view using a high‐frequency transducer, and the study was recorded on a super‐VHS videotape. The same location of the brachial artery was imaged to allow for comparison between baseline and flow‐mediated images. The brachial artery vasomotor diameter response to high‐flow stimulus (hyperemia) was accomplished with a 5‐minute upper arm occlusion. The Doppler velocity of blood flow was obtained immediately after cuff release. Ultrasound images of the brachial artery were obtained up to 1.5–2 minutes post‐cuff release. The 1‐minute post‐cuff release amount of vasodilatation was measured by a core laboratory at the University of Maryland. The core personnel were blinded to the study medication.
Laboratory Measurements
The laboratory assays, all commercially available, included evaluation of the complete blood cell count, chemistry studies, lipid profile, urinalysis, and markers of inflammation. The inflammatory markers included IL‐6, intracellular adhesion molecule‐1, vascular cell adhesion molecule, plasminogen activator inhibitor‐1, tumor necrosis factor α, and oxidized LDL autoantibodies.
Statistical Analyses
This small study was designed to identify trends in the data, and the sample size was not determined based on statistical power considerations. It was determined that 60 randomized subjects who completed the assessments would be sufficient to test the hypothesis of no mean change from baseline within each treatment group. Any statistical tests performed to explore the data were used only to highlight any interesting comparisons that may warrant further consideration.
The safety analysis included all randomized subjects who received at least one dose of study medication. The completers population, defined as all randomized subjects who had both baseline and week 12 assessments of the primary efficacy variable, was used for efficacy analyses. Additional efficacy analyses examining the change from baseline to week 6 and the change from week 6 to week 12 used observed cases (subjects who had valued assessments at both time points).
Baseline clinical and demographic characteristics and safety data were summarized with appropriate descriptive statistics (mean, SD, median, and range for continuous variables and number and percentage for categoric variables). Homogeneity between treatment groups was tested using the chi‐square test for categoric variables and 1‐way analysis of variance for continuous variables. For the primary efficacy variable, medians of %FMD at baseline and postbaseline were computed, and the significance of any differences in distributions between the treatment groups was assessed using the Wilcoxon rank sum test. Within‐group comparisons were made using the Wilcoxon signed rank test. Secondary efficacy variables were analyzed using similar methods. Changes in BP were compared using an analysis of covariance model with treatment and center as main effects and baseline %FMD level as the covariate in the model.
Comparisons between the groups for the change from baseline to week 6 and the change from week 6 to week 12 were made using observed cases (subjects who had valued assessments at both time points). An exploratory analysis to examine the correlation between the change in %FMD and relevant variables was performed using the Spearman rank correlation test. As appropriate, multiple linear regression models were used to assess the relationship between the change in %FMD and other relevant variables.
RESULTS
Of the 70 subjects randomly assigned to treatment, 61 completed the trial and 61 (29 amlodipine besylate/benazepril HCl and 32 amlodipine besylate) comprised the completers population (randomized subjects with both baseline and week 12 assessments of the primary efficacy variable; Figure 1). The mean age of subjects in the safety population was 60.5±9.8 years; most (60%) were younger than 65 years, and a slight majority were men (54.3%) (Table I). The mean baseline sitting SBP and DBP were 145.6±11.5 mm Hg and 87.3±12.4 mm Hg for the combination treatment group and 147.1±12.7 mm Hg and 88.4±11.4 mm Hg for the amlodipine alone group.
Figure 1.
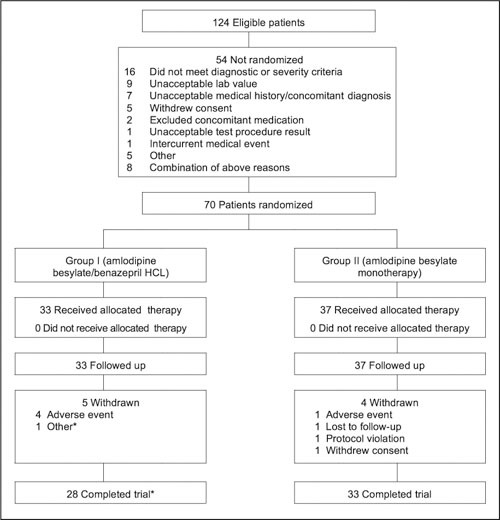
Patient disposition chart showing the number of eligible subjects, randomized subjects, and completers. *One subject in the combination arm completed all study evaluations but did not return for the final study visit. This subject technically did not complete the study, but was included in the completers population.
Table I.
Demographics of Study Population
| Variable | Amlodipine Besylate/Benazepril HCl (n=33) | Amlodipine Besylate Monotherapy (n=37) | All Subjects (N=70) |
|---|---|---|---|
| Age, y | 61.0±10.7 | 60.0±9.1 | 60.5±9.8 |
| <65 | 19 (57.6) | 23 (62.2) | 42 (60.0) |
| ≥65 | 14 (42.4) | 14 (37.8) | 28 (40.0) |
| Sex | |||
| Male | 17(51.5) | 21 (56.8) | 38 (54.3) |
| Female | 16 (48.5) | 16 (43.2) | 32 (45.7) |
| Race | |||
| Caucasian | 22 (66.7) | 22 (59.5) | 44 (62.9) |
| Black | 8 (24.2) | 13(35.1) | 21 (30.0) |
| Asian | 2 (6.1) | 0 (0.0) | 2 (2.9) |
| Other | 1 (3.0) | 2 (5.4) | 3 (4.3) |
| Blood pressure, mm Hg | |||
| Systolic | 145.6±11.5 | 147.1±12.7 | 146.4±12.1 |
| Diastolic | 87.3±12.4 | 88.4±11.4 | 87.9±11.8 |
| Risk factors | |||
| Height, cm | 169.8±10.96 | 170.3±10.29 | 170.1±10.53 |
| Weight, kg | 88.1±21.17 | 91.9±20.61 | 90.1±20.81 |
| Waist circumference, cm | 100.7±15.82 | 103.4±18.42 | 102.1±17.17 |
| Total cholesterol, mg/dL | 212.3±41.54 | 207.0±38.32 | |
| LDL cholesterol, mg/dL | 125.6±40.18 | 128.6±29.91 | |
| HDL cholesterol, mg/dL | 54.8±15.51 | 49.5±12.45 | |
| Triglycerides, mg/dL | 160.7±100.75 | 156.5±108.25 | |
| Fasting glucose, mg/dL | 94.9±11.87 | 97.8±12.59 | |
| Demographic and baseline characteristics based on the safety population. Data are presented as mean ± SD or number (percentage). LDL indicates low‐density lipoprotein; HDL, high‐density lipoprotein. | |||
Efficacy
Both groups had similar baseline FMD and statistically significant increases from baseline in median %FMD and in median percentage change from baseline in %FMD (Table II). These improvements were numerically larger with combination treatment than with monotherapy, but the between‐group differences did not achieve statistical significance. Both treatments produced statistically significant reductions from baseline to end of study in SBP and DBP (Figure 2). These reductions were significantly greater in the combination group compared with monotherapy for both SBP (−18.6 mm Hg and −14.8 mm Hg, respectively; P=.0452) and DBP (−12.3 mm Hg and −9.1 mm Hg, respectively; P=.0297) and occurred earlier in the course of therapy.
Table II.
Change and Percentage Change in %FMD at Week 12
| %FMD | Amlodipine Besylate/Benazepril HCl (n=29) | Amlodipine Besylate (n=32) |
|---|---|---|
| Baseline (median) | 8.10 | 7.05 |
| Week 12 (median) | 10.30 | 8.65 |
| Change at week 12 (median) | 2.00 | 1.20 |
| P value* | .0008 | .0079 |
| P value vs combination† | .1729 | |
| Change in %FMD, % | ||
| Median | 24.7 | 16.4 |
| P value* | .0016 | .0009 |
| P value vs combination† | .4529 | |
| %FMD indicates percentage of flow‐mediated vasodilation. *From Wilcoxon signed rank test. †From Wilcoxon rank sum test. | ||
For secondary efficacy variables, there were no statistically significant differences between the treatments in the change from baseline in IL‐6, intracellular adhesion molecule‐1, vascular cell adhesion molecule, tumor necrosis factor a, or plasminogen activator inhibitor 1 (data not shown). For oxidized LDL cholesterol autoantibody, there was a nonsignificant decrease from baseline in the combination treatment group (−34.4 mU/mL) and a significant increase in the monotherapy group (262.4 mU/mL; P=.044). The between‐group difference for oxidized LDL cholesterol autoantibody was statistically significant (P=.0300); however, because of the small sample size, caution should be used in interpreting this result.
Additional analyses examined the change from baseline to week 6 and the change from week 6 to week 12 in efficacy variables. There were no statistically significant changes from baseline to week 6 or from week 6 to week 12 in %FMD, percentage change in %FMD, or in any marker of ED of vascular inflammation.
Correlation coefficients were estimated to see whether there was any association between variables (%FMD, SBP, DBP, fasting plasma glucose, age, gender, and body mass index) and whether there was any association between the changes from baseline in those variables. The change from baseline in SBP and the change in %FMD were highly correlated (r 2=0.55) for the combination therapy group (completers population), but not the monotherapy group. As expected, baseline SBP and baseline DBP were highly correlated, both in the combination group (r 2=0.69) and when both treatment groups were combined (r 2=0.56). Similarly, the change from baseline in SBP and the change from baseline in DBP were highly correlated in the combination group (r 2=0.61), the monotherapy group (r 2=0.62), and when both treatment groups were combined (r 2=0.59).
Adverse Events
The majority of adverse events reported were mild to moderate in severity. The incidence of adverse events was 73% and 62% with combination therapy and monotherapy, respectively. The most common adverse events, occurring in 3 or more subjects, are presented in Table III. Cough was more commonly reported with combination therapy than with monotherapy (21% vs 11%, respectively), as was nausea (18% vs 0%, respectively). The monotherapy group reported higher incidences of peripheral edema (19% vs 0%) and joint swelling (14% vs 3%) than did the combination group.
Table III.
Most Frequently Reported Adverse Events*
| Amlodipine Besylate/Benazepril HCl, No. (%) | Amlodipine Besylate Monotherapy, No. (%) | |
|---|---|---|
| Subjects studied | ||
| Total | 33 | 37 |
| Total with adverse events | 24 (72.7) | 23 (62.2) |
| Total withdrawn for an adverse event | 4(12.1) | 1 (2.7) |
| Adverse event | ||
| Cough | 7 (21.2) | 4 (10.8) |
| Peripheral edema | 0(0) | 7 (18.9) |
| Nausea | 6 (18.2) | 0(0) |
| Headache | 3 (9.1) | 5 (13.5) |
| Joint swelling | 1 (3.0) | 5 (13.5) |
| Rhinitis | 0(0) | 3 (8.1) |
| *Adverse events reported by 3 or more subjects. | ||
Five subjects (4 in the combination group; 1 on monotherapy) discontinued the study for adverse events, all of which were suspected by the investigator to be related to study medication. Subjects taking combination therapy discontinued the study for dizziness (one subject), cough (2 subjects), and cough with nausea (1 subject). One subject on monotherapy withdrew because of ankle swelling.
There were no clinically significant changes from baseline in laboratory test results or vital signs (excluding reductions in BP) in either treatment group.
No deaths occurred during the study. One subject in each treatment group experienced an adverse event of severe intensity. One subject in the combination therapy group had severe worsening cough that was suspected to be related to study treatment, and one amlodipine subject had severe sinusitis that was not suspected to be treatment‐related.
DISCUSSION
In the present study, there were no statistically significant changes in absolute %FMD or in percent change in %FMD with amlodipine besylate/benazepril HCl compared with amlodipine besylate monotherapy. A positive trend suggests but does not prove that the combination of an ACE inhibitor and a CCB may be more beneficial to endothelial function than a CCB alone. This, however, may be the result of a greater BP reduction with combination therapy than with monotherapy. It was difficult to interpret the changes in circulating markers of ED/vascular inflammation due to insufficient sample size. The adverse events reported were consistent with product labeling for the study medications.
ED comprises abnormal vasodilation, expression of inflammatory mediators, and a tendency toward platelet aggregation. 16 The %FMD of the brachial artery is indicative of ED and a possible indicator of adverse cardiovascular events. 17 , 18 The molecules involved in vascular relaxation include nitric oxide, prostacyclin, and hyperpolarizing factor. 19 Components of the renin‐angiotensin system are thought to have proatherogenic effects. 20 Nitric oxide down‐regulates endothelial synthesis of ACE and angiotensin II type I receptors and thus reduces angiotensin II activity, which likely provides protection against atherosclerosis. 21 As a class, CCBs work independently of the endothelium by reducing calcium inflow in the voltage‐dependent channels of underlying vascular smooth muscle cells, thereby resulting in dilation of large conduit and resistance arteries. However, in the vascular wall, dihydropyridine CCBs inhibit the effects of angiotensin I and endothelin‐1 in the vascular smooth muscle and allow for the vasodilatory effects of nitric oxide. 22 There are experimental studies indicating that a combination of ACE inhibitor and CCB (dihydropyridine) may produce additive or synergistic benefits for endothelial function. One study indicates that the mode of action whereby amlodipine enhances nitric oxide production induced by an ACE inhibitor is via a kininmediated mechanism in coronary microvessels. 23 The trend toward reduction in oxidized LDL with combination therapy may also be consistent with antioxidant effects of these two agents. Thus, it is possible that the combination of ACE/CCB acts synergistically to improve endothelial function.
The present study is limited since the combination drug intervention group achieved a significantly lower BP than single drug treatment and it is therefore not possible to determine whether the trend in endothelial improvement was due to BP lowering alone or an actual pleiotropic effect from the combination of the ACE inhibitor and CCB.
Our study is the first that we know of to date investigating the combination of an ACE inhibitor and a CCB on %FMD of the brachial artery. The trend in improvement with combination therapy compared with treatment with a dihydropyridine CCB alone is consistent but clearly not confirmatory of the presumed synergistic effect of these two agents on vascular health. The observation that both treatment groups had a significant improvement in %FMD compared with baseline suggests that both classes of drugs favorably affect endothelial function.
In conclusion, there was no significant improvement in ED with combination treatment, but there were some positive changes in %FMD and a significant and rapid decrease in both SBP and DBP compared with monotherapy. This suggests that the combination of ACE/CCB may have more favorable effects on vascular health than a CCB alone. Future studies are needed to assess the impact of endothelial modulation in the prevention of cardiovascular events.
Acknowledgment: We thank Barbara Liptak for her assistance with this manuscript.
References
- 1. Ross R. Atherosclerosis‐an inflammatory disease. N Engl J Med. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 2. Joannides R, Haefeli WE, Linder L, et al. Nitric oxide is responsible for flow‐dependent dilatation of human peripheral conduit arteries in vivo. Circulation. 1995;91:1314–1319. [DOI] [PubMed] [Google Scholar]
- 3. Harrison DG, Cai H, Landmesser U, et al. Interactions of angiotensin II with NAD (P)H oxidase, oxidant stress and cardiovascular disease. J Renin Angiotensin Aldosterone Syst. 2003;4:51–61. [DOI] [PubMed] [Google Scholar]
- 4. Chen L, Haught WH, Yang B, et al. Preservation of endogenous antioxidant activity and inhibition of lipid peroxidation as common mechanisms of antiatherosclerotic effects of vitamin E, lovastatin and amlodipine. J Am Coll Cardiol. 1997;30:569–575. [DOI] [PubMed] [Google Scholar]
- 5. Digiesi V, Fiorillo C, Cosmi L, et al. Reactive oxygen species and antioxidant status in essential arterial hypertension during therapy with dihydropyridine calcium channel antagonists. Clin Ter. 2000;151:15–18. [PubMed] [Google Scholar]
- 6. Zhou MS, Jaimes EA, Raij L. Inhibition of oxidative stress and improvement of endothelial function by amlodipine in angiotensin II‐infused rats. Am J Hypertens. 2004;17:167–171. [DOI] [PubMed] [Google Scholar]
- 7. Pitt B, Byington RP, Furberg CD, et al. Effect of amlodipine on the progression of atherosclerosis and the occurrence of clinical events. PREVENT Investigators . Circulation. 2000;102:1503–1510. [DOI] [PubMed] [Google Scholar]
- 8. Loaldi A, Polese A, Montorsi P, et al. Comparison of nifedipine, propranolol and isosorbide dinitrate on angiographic progression and regression of coronary arterial narrowings in angina pectoris. Am J Cardiol. 1989;64:433–439. [DOI] [PubMed] [Google Scholar]
- 9. Corretti MC, Anderson TJ, Benjamin EJ, et al. Guidelines for the ultrasound assessment of endothelial‐dependent flow‐mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39:257–265. [DOI] [PubMed] [Google Scholar]
- 10. Verma S, Anderson TJ. Fundamentals of endothelial function for the clinical cardiologist. Circulation. 2002;105:546–549. [DOI] [PubMed] [Google Scholar]
- 11. Muhlen BV, Millgard J, Lind L. Effects of digoxin, furosemide, enalaprilat and metoprolol on endothelial function in young normotensive subjects. Clin Exp Pharmacol Physiol. 2001;28:381–385. [DOI] [PubMed] [Google Scholar]
- 12. Wilmink HW, Banga JD, Hijmering M, et al. Effect of angiotensin‐converting enzyme inhibition and angiotensin II type 1 receptor antagonism on postprandial endothelial function. J Am Coll Cardiol. 1999;34:140–145. [DOI] [PubMed] [Google Scholar]
- 13. Herrington DM, Werbel BL, Riley WA, et al. Individual and combined effects of estrogen/progestin therapy and lovastatin on lipids and flow‐mediated vasodilation in postmenopausal women with coronary artery disease. J Am Coll Cardiol. 1999;33:2030–2037. [DOI] [PubMed] [Google Scholar]
- 14. Anderson TJ, Elstein E, Haber H, et al. Comparative study of ACE‐inhibition, angiotensin II antagonism, and calcium channel blockade on flow‐mediated vasodilation in patients with coronary disease (BANFF study). J Am Coll Cardiol. 2000;35:60–66. [DOI] [PubMed] [Google Scholar]
- 15. Corretti MC, Plotnick GD, Vogel RA. Technical aspects of evaluating brachial artery vasodilatation using high‐frequency ultrasound. Am J Physiol. 1995;268:H1397–H1404. [DOI] [PubMed] [Google Scholar]
- 16. Kinlay S, Libby P, Ganz P. Endothelial function and coronary artery disease. Curr Opin Lipidol. 2001;12:383–389. [DOI] [PubMed] [Google Scholar]
- 17. Suwaidi JA, Hamasaki S, Higano ST, et al. Long‐term follow‐up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. 2000;101:948–954. [DOI] [PubMed] [Google Scholar]
- 18. Schachinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long‐term outcome of coronary heart disease. Circulation. 2000;101:1899–1906. [DOI] [PubMed] [Google Scholar]
- 19. Harrison DG, Cai H. Endothelial control of vasomotion and nitric oxide production. Cardiol Clin. 2003;21:289–302. [DOI] [PubMed] [Google Scholar]
- 20. Luscher TF, Barton M. Biology of the endothelium. Clin Cardiol. 1997;20(11 suppl 2):II‐3–II‐10. [PubMed] [Google Scholar]
- 21. Ichiki T, Usui M, Kato M, et al. Downregulation of angiotensin II type 1 receptor gene transcription by nitric oxide. Hypertension. 1998;31(1 pt 2):342–348. [DOI] [PubMed] [Google Scholar]
- 22. Ruschitzka FT, Noll G, Luscher TF. Combination of ACE inhibitors and calcium antagonists: a logical approach. J Cardiovasc Pharmacol. 1998;31(suppl 2):S5–S16. [DOI] [PubMed] [Google Scholar]
- 23. Zhang X, Xu X, Nasjletti A, et al. Amlodipine enhances NO production induced by an ACE inhibitor through a kininmediated mechanism in canine coronary microvessels. J Cardiovasc Pharmacol. 2000;35:195–202. [DOI] [PubMed] [Google Scholar]