

Abstract
Hypertension is a major risk factor for cardiovascular disease, and reduction of elevated blood pressure significantly reduces the risk of cardiovascular events. Endothelial dysfunction, which is characterized by impairment of nitric oxide (NO) bioavailability, is an important risk factor for both hypertension and cardiovascular disease and may represent a major link between the conditions. Evidence suggests that NO plays a major role in regulating blood pressure and that impaired NO bioactivity is an important component of hypertension. Mice with disruption of the gene for endothelial NO synthase have elevated blood pressure levels compared with control animals, suggesting a genetic component to the link between impaired NO bioactivity and hypertension. Clinical studies have shown that patients with hypertension have a blunted arterial vasodilatory response to infusion of endothelium‐dependent vasodilators and that inhibition of NO raises blood pressure. Impaired NO bioactivity is also implicated in arterial stiffness, a major mechanism of systolic hypertension. Clarification of the mechanisms of impaired NO bioactivity in hypertension could have important implications for the treatment of hypertension.
Recent data demonstrate that the global burden of hypertension is an important and increasing health problem worldwide and that awareness and control of hypertension vary considerably. 1 Within the next 20 years, the percentage of the adult population that will be affected by hypertension is predicted to increase by 60% to a total of more than 1.5 billion individuals. 1 In economically developed countries, the level of adequate blood pressure (BP) control (<140/90 mm Hg) among patients receiving antihypertensive treatment ranges from approximately 30%–50%. 2 , 3 , 4
Endothelial function is the first step in the development of atherosclerotic disease. It is present in the early course of all known cardiovascular (CV) risk factors and is characterized by impaired bioavailability of nitric oxide (NO). Data from large observational studies beginning in the 1990s demonstrate that diastolic and systolic hypertension are associated with increased risk for the development of CV disease and stroke. 5 The relationship between BP increase and stroke, however, is higher than for CV disease. The role of pulse pressure, which is the difference between systolic and diastolic BP, as a predictor of CV events is controversial, although it may help to identify subjects with isolated systolic hypertension with an increased risk. Treatment of hypertension reduces the risk for CV events and stroke by 20% and up to 40%, respectively. 6 , 7 , 8 There are some differences between antihypertensive drug classes and some within‐class differences, in particular with β‐blockers. 9 Thus, drugs that possess pleiotropic effects beyond BP lowering, such as improvement of endothelial function via increased bioavailability of NO, may be of particular interest.
Endothelial dysfunction is associated with hypertension and other conventional CV risk factors, including hypercholesterolemia, diabetes, smoking, and aging, 10 and is a significant independent risk factor for CV events in hypertensive patients. 11 Impaired NO bioactivity plays a major role in endothelial dysfunction. 12 Understanding the role of NO in regulating BP may have implications for improving hypertension treatment and reducing the risk of CV morbidity and mortality. This article reviews the association between impaired NO bioactivity and hypertension and the potential for therapeutic modulation of NO to reduce BP.
No, Endothelial Function, and BP
NO is a simple but pluripotent molecule that is predominantly synthesized in the vascular endothelium. NO is generated from l‐arginine by endothelial NO synthase (eNOS), which metabolizes l‐arginine to NO. NO stimulates guanylyl cyclase to form 3′,5′‐cyclic guanosine monophosphate, which results in vasodilatation of vascular smooth muscle cells, prevention of platelet adhesion and aggregation, and exertion of anti‐inflammatory, antiproliferative, and antimigratory effects on leukocytes, endothelial cells, and vascular smooth muscle cells, thus providing protection from atherosclerosis (Figure 1).
Figure 1.

Endothelium‐derived vasoactive substances. Nitric oxide (NO) is released from endothelial cells in response to shear stress and activation of a variety of receptors. NO exerts vasodilating and antiproliferative effects on smooth muscle cells and inhibits thrombocyte aggregation and leukocyte adhesion. Endothelin‐1 (ET‐1) exerts its major vascular effects—vasoconstriction and cell proliferation—through activation of specific endothelin‐A (ETA) receptors on vascular smooth muscle cells. In contrast, endothelin B (ETB) receptors mediate vasodilation via release of NO and prostacyclin. In addition, ETB receptors in the lung were shown to be a major pathway for the clearance of ET‐1 from plasma. AI indiBcates angiotensin I; AII, angiotensin II; Thr, thrombine; TGFBβ1, transforming growth factor β; AcCh, acetylcholine; 5‐HT, 5‐hydroxytryptamine (serotonin); ADP, adenosine diphosphate; BK, bradykinin; ACE, angiotensin‐converting enzyme; AT1, angiotensin II type 1 receptor; T, thromboxane receptor; bET‐1, big ET‐1; ECE, endothelin‐converting enzyme; M, muscarinergic receptor; S1, serotoninergic receptor; B2, bradykinin receptor; NOS, NO synthase; L‐Arg, L‐arginine; EDHF, endothelium‐derived hyperpolarizing factor; TXA2, thromboxane; PGH2, prostaglandin H2; PGI2, prostacyclin; cAMP, cyclic adenosine monophosphate; and cGMP, cyclic 3′,5′‐guanosine monophosphate. Adapted from Lülscher et al. 107
Attenuated NO bioavailability, the main characteristic of endothelial dysfunction, is present in arterial hypertension. 10 , 13 , 14 Hypertensive subjects have increased generation of reactive oxygen species (ROS), which scavenge NO, thereby reducing NO bioavailability. A decade ago it was shown in animal models that deletion of the eNOS gene (eNOS‐/‐) as well as chronic inhibition of NO synthesis (NOS) with Nω‐nitro‐l‐arginine methyl ester (l‐NAME) leads to the development of arterial hypertension. 15 , 16 l‐NAME infusion also induces endothelial dysfunction in humans, 17 as does the NOS inhibitor Ng‐monomethyl‐l‐arginine (l‐NMMA) (Figure 2). 18
Figure 2.
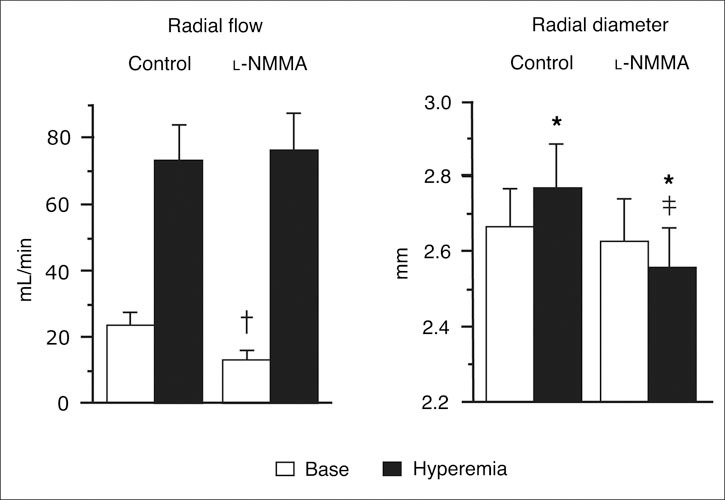
Bar graphs showing radial artery flow (mL/min) and radial artery diameter (mm) measured at baseline (Base) and during reactive hyperemia before and after infusion of Ng‐monomethyl‐l‐arginine (l‐NMMA). All results are expressed as mean ± SEM of 8 subjects. *P<.01 vs base. †P<.05. ‡P<.01 vs corresponding control value. Reproduced with permission from Joannides et al. 108
NO bioavailability can be improved with pharmacologic and nonpharmacologic approaches. Regular physical exercise improves endothelial function in hypertensive subjects, 19 and beneficial effects of restored NOS by administration of NOS cofactors such as tetrahydrobiopterin (BH4) or l‐arginine have been demonstrated in several animal models as well as in patients. BP increases are prevented 20 , 21 and endothelial dysfunction is improved after administration of BH4 in insulin‐resistant rats, 22 in healthy subjects with glucose‐induced impairment of endothelial function, 23 and in patients with established coronary artery disease 24 and chronic heart failure. 25
Data concerning the effect of antihypertensive treatment with β‐blockers on endothelial function depend on the specific β‐blocker; the beneficial effect of an NO‐releasing β‐blocker, nebivolol, on endothelial function results from the increase in NO and not from the β‐blocking effects of the drug. 26 Angiotensin‐converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) improve endothelial function partly independent of arterial pressure reduction, 27 , 28 and calcium antagonists (dihydropyridine‐like agents in particular) improve endothelial dysfunction, accompanied by a simultaneous improvement in several markers of oxidative stress. 29 , 30 , 31 These antioxidant actions are particularly important because oxidative stress, and the resulting scavenging of NO by excessive ROS, is believed to be a major cause of impaired NO bioactivity. 32 The term oxidative stress refers to conditions under which excessive production of ROS, possibly triggered by CV risk factors such as hypertension, smoking, obesity and dyslipidemia, overcomes antioxidant defense mechanisms, such as NO bioactivity, leading to oxidation of biologic macromolecules including lipids, DNA, protein and carbohydrates, vascular inflammation, and the development of atherosclerosis and CV disease. 32
While research data show that NO contributes to regulation of BP and that impaired NO bioactivity is associated with hypertension, the etiology and mechanisms of these relationships, particularly the question of whether impaired NO precedes or follows hypertension, remain unclear. 33 Impaired NO‐dependent vasodilation has been shown to precede hypertension in black patients with normotension and in small studies involving normotensive offspring of hypertensive parents (Figure 3). 34 A large trial of the Framingham Heart Study 35 population (N=2883), however, found that the estimated heritability of endothelial function, measured as flow‐mediated dilation of the brachial artery, was modest (0.14); this analysis generally could not determine whether endothelial dysfunction was a cause or consequence of hypertension. Potential mechanisms for the pathogenic link between impaired NO and hypertension include defects in the l‐arginine/NO pathway, leading to decreased NO production; genetic polymorphisms in eNOS; reduced availability of cofactors essential to NO formation; increased levels of circulating NO inhibitors; and destruction of NO by ROS. 32 , 34 , 36 , 37 , 38 , 39 , 40
Figure 3.
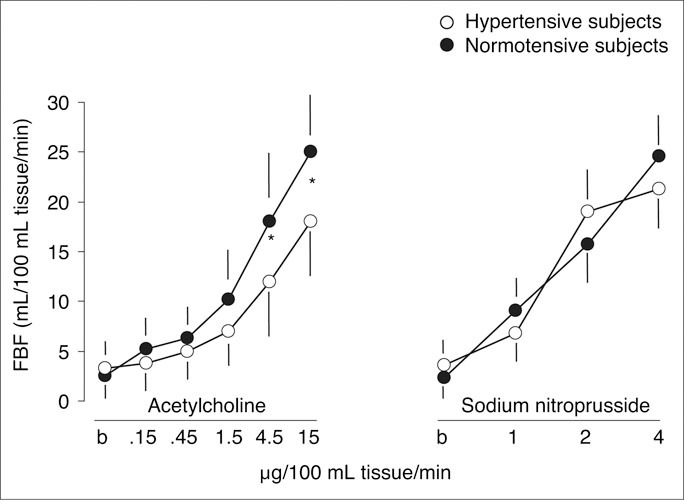
In 34 normotensive subjects (mean age, 23 years) with a family history of essential hypertension, brachial artery vasodilation in response to acetylcholine, as represented by forearm blood flow (FBF) measured with strain‐gauge plethysmography, was significantly blunted compared with normotensive subjects with no family history of hypertension. FBF increased from 3.9 mL/min to a maximum of 18.9 mL/min with the highest dose of acetylcholine in hypertensive subjects, compared with an increase of 3.8 mL/min to 26.2 mL/min with the highest dose in normotensive subjects (P<.01). The FBF response to sodium nitroprusside, a nonendothelium‐dependent vasodilator, was similar between the 2 groups. *P<.01 hypertensive vs normotensive subjects. Reproduced with permission from Taddei et al. 34
Data on a possible genetic mechanism of NO impairment in hypertension have been inconsistent. Several studies have identified specific eNOS gene mutations as being significantly more prevalent in patients with hypertension than in normotensive subjects, 41 while other studies found no association between these genotypes and hypertension. 42 , 43 These polymorphisms probably have only a small effect on NO production, however, and may only become significant in the presence of other risk factors or mechanisms.
In addition, the reduction of NO cofactors and the increase of NO inhibitors may be correlated with impaired NO bioactivity; these mechanisms, however, are only 2 of many factors in oxidative stress that may originate with preexisting systemic vascular disease states, such as hypertension and dyslipidemia. 44
Hypertension, Inflammation, and Oxidative Stress
An increasing body of evidence suggests that low‐grade inflammation and oxidative stress account in part for hypertension‐induced endothelial dysfunction and that C‐reactive protein (CRP) levels may be associated with the future development of hypertension. 32 , 45 , 46 Women with baseline CRP levels >3.5 mg/L have higher rates of hypertension and self‐reported systolic BPs of at least 140 mm Hg or diastolic BPs of at least 90 mm Hg. 46 In addition, there is a linear relationship between increasing BP and increasing CRP levels in women. 45 During 8 years of follow‐up, both parameters were strong predictors for CV events, and the predictive values of CRP and elevated BP in combination are additive. In untreated human hypertension, CRP levels have recently been found to be increased dependent on systolic BP levels. 47 , 48 Most importantly, an increase in CRP level is independently associated with elevated systolic BP and pulse pressure (but not diastolic BP), as well as with other classical CV risk factors. 47 CRP is also a strong risk factor for ischemic stroke, independent of the severity of the underlying atherosclerotic disease. 49
Although an active role of CRP in the development of CV disease has been questioned, 50 systemic inflammation has been accepted as a CV risk factor. 51 Engström and colleagues 52 demonstrated that increased levels of inflammation‐sensitive plasma proteins are associated with an increased incidence of hypertension. 52 Chronic inflammation presents with activation of the cyclooxygenase system, increased production of ROS, and increased synthesis of CRP and proinflammatory cytokines, which have been shown to correlate with arterial stiffness in untreated hypertensive patients. 53 Data suggest a pivotal role for inflammation in the development of vascular disease and, in particular, hypertension. A causal link between CRP and hypertension, however, remains elusive. Recent data suggest that elevated CRP levels may not lead to elevated BP. 54
Increased ROS and an altered balance between NO and ROS lead to impaired bioavailability of NO, resulting in decreased endothelium‐dependent vasodilation, which, in turn, causes or exacerbates hypertension. 44 , 55 Oxidation‐induced impairment of NO also results in reduced opposition to the vasoconstrictive and hypertensive effects of angiotensin II. Angiotensin II decreases NO bioavailability by promoting oxidative stress. ACE inhibitors have been shown to reduce CV events in several large‐scale randomized trials. 56 , 57 ARBs selectively block the angiotensin II type 1 receptor (Figure 4), thus inhibiting most of the deleterious effects of angiotensin II; they have also been shown to provide CV benefit, 58 , 59 , 60 as have other antihypertensive agents including diuretics, calcium channel blockers, and β‐blockers. The integral role of oxidative stress in NO impairment in hypertension has been demonstrated by in vivo and in vitro studies showing that substances that protect against superoxide‐induced damage, such as superoxide dismutase and the antioxidant vitamin C, restore endothelium‐dependent vasodilation in hypertensive animals and humans. 61 , 62
Figure 4.

Cardiovascular regulation occurs with the interaction of the sympathetic nervous system (SNS) and the renin‐angiotensin system (RAS) with the vascular endothelium. AT indicates angiotensin II type 1 receptor; ET, endothelin; AT II, angiotensin II; ACEI, angiotensin‐converting enzyme inhibitor; Ach, acetylcholine; M, muscarinergic receptor; ETB, endothelin B receptor; ARB, angiotensin receptor blocker; NO, nitric oxide; PGI2, prostacyclin; ETA, endothelin A receptor; and Bβ2, bradykinin receptor. Reproduced with permission from Wenzel et al. 105
Other important mechanisms of hypertension in which NO impairment plays a major role, with possible implications for antihypertensive treatment, include arterial stiffness and chronic sympathetic nervous system (SNS) activation (Figure 4).
Arterial Stiffness
Recently, the significance of arterial stiffness, or rigidity, as a determinant of future CV events has been demonstrated in retrospective and prospective trials. 63 Although aging is the major factor for vascular stiffness, several CV risk factors, such as smoking, hypertension, and diabetes mellitus, alter the structure and function of the vascular wall and endothelial components. Increased vascular stiffness leads to greater afterload stress on the heart. Increased aortic wall stiffness raises resistance to stroke volume, necessitating a higher systolic pulse and increased systolic BP; this causes higher systolic pulse wave velocity (PWV) and premature wave reflection, arriving in late systole rather than early diastole. 63 , 64 This effect further increases systolic BP and decreases diastolic BP, resulting in increased pulse pressure. PWV has been used as an index for vascular stiffness and as a surrogate marker for atherosclerosis in patients with hypertension, diabetes, and renal failure. 65 , 66 , 67 The development of atherosclerosis contributes to arterial stiffness. Therefore, mechanisms involved in the development and progression of atherosclerosis, such as endothelial dysfunction characterized by reduced NO bioavailability, directly impact arterial stiffness.
The assumption that the peripheral systemic pressure is an accurate reflection of central arterial BP may be an oversimplification. The relationship between peripheral BP and central arterial BP is dependent on the hemodynamic performance of the vasculature, and this relationship can be profoundly disturbed if there is evidence of vascular disease, particularly arteriosclerosis (arterial aging, arterial stiffening, and loss of compliance). 68 Recent data suggest that pulse pressure in the central arteries is a better predictor of left ventricular mass and carotid intima thickness than peripheral pulse pressure. Moreover, in patients 70 years and older and in those with end‐stage renal failure, central but not peripheral pulse pressure is a powerful, independent predictor of CV and total mortality. 67 , 69 Aortic PWV provides a more direct measure of large artery stiffness and can now be measured reliably using simple, noninvasive equipment. Using such techniques, aortic PWV has been shown to predict CV risk in patients with end‐stage renal disease, among hypertensive individuals, in those with diabetes mellitus, and among older individuals.
In the Anglo‐Scandinavian Cardiac Outcomes Trial (ASCOT), 70 amlodipine‐based treatment did not benefit the primary outcome of coronary heart disease events but was associated with significant reductions in major CV and renal outcomes and death, compared with an atenolol‐based treatment program. In a substudy of ASCOT, the Conduit Artery Function Evaluation, 71 researchers found that although arm BP differed little between treatment groups, there were substantial reductions in trial‐averaged values for central aortic pressures and hemodynamic indices in favor of the amlodipine‐based treatment. Central aortic systolic BP was lower by 4.3 mm Hg and the central aortic pulse pressure was lower by 3.0 mm Hg in the amlodipine group. Thus, central pulse pressure appeared to be a significant determinant of total CV and renal events in this trial.
No and Arterial Stiffness
Arterial stiffness is an important determinant of CV risk in hypertensive patients and can be assessed by intravascular measurements of PWV. It has been shown that endothelium‐derived NO interacts with arterial elasticity in animal models. 72 Data from studies in humans, however, are somewhat inconsistent. While infusion of l‐NMMA increased PWV in healthy volunteers 73 and augmentation index in sheep, 74 other studies observed no effect on PWV in healthy volunteers. 75 , 76 Wilkinson and associates 74 found a significant increase in central pulse pressure (P<.001) but not in peripheral pulse pressure in sheep, suggesting that arterial stiffness is most closely correlated with central BP. Hypertension associated with impaired NO bio‐availability/endothelial dysfunction, however, also causes pathologic structural changes and reduced compliance in small arteries, which may lead to reduced large artery compliance. 64
The impact of antihypertensive drugs on arterial stiffness is controversial, and drugs that reduce BP as well as vascular stiffness may be more effective in reducing CV events. Interestingly, the NO‐stimulating β‐blocker nebivolol, but not atenolol, increases arterial distensibility in vivo. 26 The effect of nebivolol on PWV is significantly attenuated during coinfusion of l‐NAME, demonstrating that nebivolol increases arterial distensibility via NO release. 26 Thus, nebivolol may be of benefit in conditions of increased large artery stiffness, such as isolated systolic hypertension. Other antihypertensive drugs, such as ACE inhibitors and ARBs, also improve arterial stiffness. 77 , 78 This effect is at least in part NO‐dependent because ACE inhibition with perindopril improves NO‐dependent endothelial function in conduit arteries. 79 Other antihypertensive drugs, such as calcium channel blockers, also improve arterial distensibility and endothelial function in hypertensive patients. 80 , 81
No and SNS Activation
There is growing evidence that NO not only has a direct effect on vascular tone but, in addition, impacts vascular tone via interaction with the autonomic nervous system (central and at their peripheral effector sites), resulting in sympathoinhibitory effects in animals 82 and humans. 83 This may also play an important role in the pathogenesis of arterial hypertension. 84
The SNS represents a major regulatory mechanism for the short‐ and long‐term adjustments of the CV system and is an important mediator of vascular tone in humans. Its activity is stimulated by mental stress, pain, cold, exercise, and certain disease states. 85 , 86 Indeed, borderline and essential hypertension as well as accelerated hypertension are associated with sympathetic activation. 87 , 88 , 89 Because normotensive offspring of hypertensive parents exhibit exaggerated response of muscle sympathetic nerve activity (MSNA) to mental stress, the SNS may be involved in the development of hypertension. 85 The exact mechanisms and the interactions with NO, however, are not yet fully understood and may differ in acute and chronic settings, and in central and peripheral systems.
In animal models, it has been shown that NO regulates vasomotor tone and arterial pressure, at least in part by modulation of central sympathetic neural outflow by neuronal NO in the brain stem. 84 Intravenous infusion of NOS inhibitors reduces NO bioavailability and increases BP, 90 an effect that is accompanied by a decrease in sympathetic nerve activity. 91 This suggests that an acute BP increase might not initially be mediated by the SNS. During chronic NOS blockade in hypertension, however, the SNS seems to play an important role, because its activity is amplified. 92 This sympathoexcitatory effect is more marked after sinoaortic denervation, indicating that sympathetic activation after NOS blockade is buffered by arterial baroreflexes. 93 The sympathetic activation and pressor effects appear to be mediated by central neural action. In rats, the intracerebroventricular injection of l‐NMMA (at doses that have no effect when administrated systemically) induces sympathetic activation and hypertension, 92 , 94 while the application of NO donors to the nucleus tractus solitarius or the hypothalamus of rats produces hypotension. 82 , 95
Experimental data in humans are limited. While most data are consistent with the results obtained in animal models, others have found no evidence for a role of NO in the regulation of sympathetic outflow. In one study, the BP increase observed with NO inhibition via l‐NMMA infusion in healthy human subjects was accompanied by significant decreases in heart rate and MSNA, which were similar to the effects of infusion of phenylephrine (anNO‐independent vasoconstrictor), suggesting no apparent effect of NO on central sympathetic outflow. 96 Another study found that while l‐NMMA infusion raised BP in 8 healthy subjects, the exercise‐induced increase in MSNA during head‐up tilt was similar with and without l‐NMMA infusion. 97 In contrast, a study in 15 healthy male volunteers found that infusion of l‐NMMA at rest increased BP significantly but had no effect on MSNA, while phenylephrine infusion resulted in a similar BP increase; a 50% decrease in MSNA was also documented, probably evoked by the baroreflex response to increased BP. Hence, NO blockade appeared to counteract this sympathetic response (Figure 5). 98 When sodium nitroprusside was coinfused with l‐NMMA, the increase in BP was abolished, but MSNA increased significantly. These findings suggest that inhibition of NO had sympathoexcitatory effects that were masked by the inhibitory response of the baroreflexes, providing the first indication that NO regulates central SNS outflow in humans and that neuronal NO as well as endothelial NO mediate vasomotor tone. 98
Figure 5.
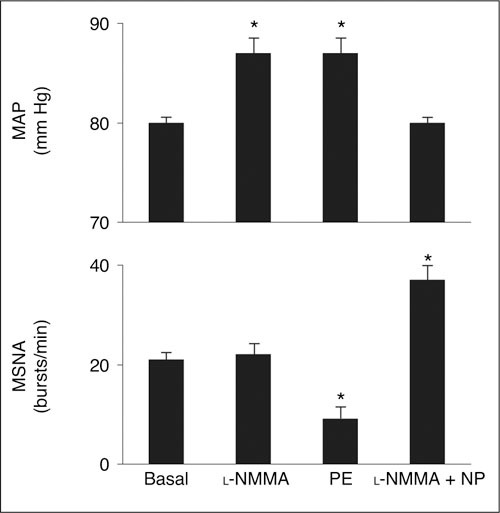
Under resting conditions, infusion of Ng‐monomethyl‐l‐arginine (l‐NMMA) in 5 normotensive male volunteers increased mean arterial pressure (MAP) significantly by about 10% but had no effect on muscle sympathetic nerve activity (MSNA), measured by microelectrodes inserted selectively into muscle nerve fasciculi of the peroneal nerve posterior to the fibular head. By contrast, infusion of phenylephrine (PE), a nitric oxide‐independent vasoconstrictor, increased MAP to a similar extent but also decreased MSNA by about 50%. Coinfusion of nitroprusside (NP) with l‐NMMA resulted in reversal of the increase in MAP observed with l‐NMMA alone, but also resulted in a significant increase in MSNA. *P<.05 vs baseline. Reproduced with permission from Owlya et al. 98
A subsequent clinical study found that while NO inhibition with l‐NAME induced significant BP increases in healthy normotensive subjects (P<.001), α‐adrenergic blockade with phentolamine attenuated the l‐NAME‐induced BP increase by 40% (P<.05), indicating a sympathetic component in NO regulation of BP. 99 The mechanisms of NO regulation of SNS outflow are complex and not entirely understood, however, and may involve multiple modulating factors, including background angiotensin II levels, the cholinergic nervous system, and baroreceptor input. 84 , 100 If the role of neuronal NO in regulation of SNS outflow and BP can be clearly established, then antihypertensive therapies that target this mechanism may be particularly effective in some patients.
NO has also been shown to modulate sympathetic tone peripherally. 101 , 102 Therefore, important interactions in the vasculature, between the SNS and the peripheral l‐arginine‐NO system in the regulation of the vascular tone in humans, must be postulated.
SNS activity is affected by antihypertensive treatment. Indeed, in patients with renovascular hypertension characterized by a high plasma level of angiotensin II, SNS activity is increased. 102 Different responses occurred in patients given treatment with dihydralazine or enalapril: after lowering BP with the vasodilator dihydralazine, an increase in sympathetic nerve activity, heart rate, and plasma angiotensin II concentrations occurred, while no increase in sympathetic nerve activity was observed with enalapril (with the same arterial pressure‐lowering effect). In hypertensive patients, the ARB losartan reduced sympathetic nerve activity as well as improved cardiac baroreceptor sensitivity (Figure 6). 103 In contrast to inhibitors of the renin‐angiotensin system, calcium blockade has no effect on SNS activity in hypertension. 104 Therefore, antihypertensive drugs seem to act differently with respect to SNS activity, despite similar BP‐reducing effects (Figure 7). 105 This effect of renin‐angiotensin system inhibitors on SNS activity could therefore explain the favorable effects of these drugs on the prognosis of patients with heart failure in whom activation of the SNS is an important prognostic factor. Generally, they may be particularly efficacious in blunting or inhibiting the unwanted effects of the SNS in patients with CV disease. 106
Figure 6.
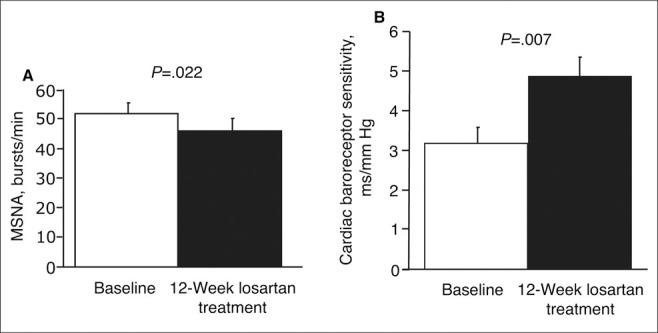
Muscle sympathetic nerve activity (MSNA) as assessed by microneurography was significantly decreased in hypertensive patients after 12 weeks of treatment with losartan (from 52±3.5 to 46±4.2 bursts/min; P=.022) (A). Baroreceptor sensitivity was significantly enhanced after 12 weeks of treatment with losartan (from 3.2±1.3 to 4.9±1.8 ms/mm Hg; P=.007) (B).
Figure 7.

Interaction of the sympathetic nervous system (SNS) with the renin‐angiotensin system (RAS). ACE indicates angiotensin‐converting enzyme; AII, angiotensin II; NO, nitric oxide; and ET‐1, endothelin‐1. Adapted from Wenzel et al. 105
Conclusions
Hypertension is increasingly understood to be a complex disorder that is strongly associated with other risk factors for CV disease. Clinical studies have demonstrated that patients with hypertension have a reduced vasodilatory response to endothelium‐dependent vasodilators, such as acetylcholine, and that blockade of NOS also blunts endothelium‐dependent vasodilation. In addition, there is evidence for a role of eNOS polymorphisms, and experimental studies suggest a possible genetic component to impaired NO bioavailability and hypertension. The etiology of the association between impaired NO bioactivity and hypertension is complex, however, and has not been fully elucidated. Important mechanisms of hypertension and CV disease, in which impaired NO bioactivity plays a major role, include arterial stiffness and increased PWV, and possibly chronic SNS activation. Further clarification of the role of impaired NO bioactivity in these pathogenic mechanisms could have important implications for the management of hypertension.
Rererences
- 1. Kearney PM, Whelton M, Reynolds K, et al. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–223. [DOI] [PubMed] [Google Scholar]
- 2. Hajjar I, Kotchen TA. Trends in prevalence, awareness, treatment, and control of hypertension in the United States, 1988–2000. JAMA. 2003;290:199–206. [DOI] [PubMed] [Google Scholar]
- 3. Kearney PM, Whelton M, Reynolds K, et al. Worldwide prevalence of hypertension: a systematic review. J Hypertens. 2004;22:11–19. [DOI] [PubMed] [Google Scholar]
- 4. Wolf‐Maier K, Cooper RS, Kramer H, et al. Hypertension treatment and control in five European countries, Canada, and the United States. Hypertension. 2004;43:10–17. [DOI] [PubMed] [Google Scholar]
- 5. Lewington S, Clarke R, Qizilbash N, Et Al, and the Prospective Studies Collaboration. Age‐specific relevance of usual blood pressure to vascular mortality: a meta‐analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–1913. [DOI] [PubMed] [Google Scholar]
- 6. European Society of Hypertension‐European Society of Cardiology Guidelines Committee. 2003. European Society of Hypertension‐European Society of Cardiology guide lines for the management of arterial hypertension. J Hypertens. 2003;21:1011–1053. [DOI] [PubMed] [Google Scholar]
- 7. Chobanian AV, Bakris GL, Black HR, et al., and the National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206–1252. [DOI] [PubMed] [Google Scholar]
- 8. Turnbull F, for the Blood Pressure Lowering Treatment Trialists' Collaboration. Effects of different blood‐pressure‐lowering regimens on major cardiovascular events: results of prospectively‐designed overviews of randomised trials. Lancet. 2003;362:1527–1535. [DOI] [PubMed] [Google Scholar]
- 9. Lindholm LH, Carlberg B, Samuelsson O. Should β blockers remain first choice in the treatment of primary hyper tension? A meta‐analysis. Lancet. 2005;366:1545–1553. [DOI] [PubMed] [Google Scholar]
- 10. Brunner H, Cockcroft JR, Deanfield J, et al., on behalf of the Working Group on Endothelins and the Endothelial Factors of the European Society of Hypertension. Endothelial function and dysfunction. Part II: association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension. J Hypertens. 2005;23:233–246. [DOI] [PubMed] [Google Scholar]
- 11. Perticone F, Ceravolo R, Pujia A, et al. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation. 2001;104:191–196. [DOI] [PubMed] [Google Scholar]
- 12. Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. 2004;15:1983–1992. [DOI] [PubMed] [Google Scholar]
- 13. Lüscher TF, Vanhoutte PM. Endothelium‐dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension. 1986;8:344–348. [DOI] [PubMed] [Google Scholar]
- 14. Panza JA, Quyyumi AA, Brush JE Jr, et al. Abnormal endothelium‐dependent vascular relaxation in patients with essential hypertension. N Engl J Med. 1990;323:22–27. [DOI] [PubMed] [Google Scholar]
- 15. Huang PL, Huang Z, Mashimo H, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. [DOI] [PubMed] [Google Scholar]
- 16. Arnal JF, El Amrani AI, Chatellier G, et al. Cardiac weight in hypertension induced by nitric oxide synthase blockade. Hypertension. 1993;22:380–387. [DOI] [PubMed] [Google Scholar]
- 17. Kiowski W, Linder L, Stoschitzky K, et al. Diminished vascular response to inhibition of endothelium‐derived nitric oxide and enhanced vasoconstriction to exogenously administered endothelin‐1 in clinically healthy smokers. Circulation. 1994;90:27–34. [DOI] [PubMed] [Google Scholar]
- 18. Joannides R, Richard V, Haefeli WE, et al. Role of basal and stimulated release of nitric oxide in the regulation of radial artery caliber in humans. Hypertension. 1995;26:327–331. [DOI] [PubMed] [Google Scholar]
- 19. Higashi Y, Sasaki S, Kurisu S, et al. Regular aerobic exercise augments endothelium‐dependent vascular relaxation in normotensive as well as hypertensive subjects: role of endothelium‐derived nitric oxide. Circulation. 1999;100:1194–1202. [DOI] [PubMed] [Google Scholar]
- 20. Podjarny E, Hasdan G, Bernheim J, et al. Effect of chronic tetrahydrobiopterin supplementation on blood pressure and proteinuria in 5/6 nephrectomized rats. Nephrol Dial Transplant. 2004;19:2223–2227. [DOI] [PubMed] [Google Scholar]
- 21. Fortepiani LA, Reckelhoff JF. Treatment with tetrahydrobiopterin reduces blood pressure in male SHR by reducing testosterone synthesis. Am J Physiol Regul Integr Comp Physiol. 2005;288:R733–R736. [DOI] [PubMed] [Google Scholar]
- 22. Shinozaki K, Nishio Y, Okamura T, et al. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aortas of insulin‐resistant rats. Circ Res. 2000;87:566–573. [DOI] [PubMed] [Google Scholar]
- 23. Ihlemann N, Rask‐Madsen C, Perner A, et al. Tetrahydrobiopterin restores endothelial dysfunction induced by an oral glucose challenge in healthy subjects. Am J Physiol Heart Circ Physiol. 2003;285:H875–H882. [DOI] [PubMed] [Google Scholar]
- 24. Maier W, Cosentino F, Lutolf RB, et al. Tetrahydrobiopterin improves endothelial function in patients with coronary artery disease. J Cardiovasc Pharmacol. 2000;35:173–178. [DOI] [PubMed] [Google Scholar]
- 25. Hambrecht R, Hilbrich L, Erbs S, et al. Correction of endothelial dysfunction in chronic heart failure: additional effects of exercise training and oral L‐arginine supplementation. J Am Coll Cardiol. 2000;35:706–713. [DOI] [PubMed] [Google Scholar]
- 26. McEniery CM, Schmitt M, Qasem A, et al. Nebivolol increases arterial distensibility in vivo. Hypertension. 2004;44:305–310. [DOI] [PubMed] [Google Scholar]
- 27. Hornig B, Landmesser U, Kohler C, et al. Comparative effect of ace inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease: role of superoxide dismutase. Circulation. 2001;103:799–805. [DOI] [PubMed] [Google Scholar]
- 28. Haefeli WE, Linder L, Lüscher TF. Quinaprilat induces arterial vasodilation mediated by nitric oxide in humans. Hypertension. 1997;30:912–917. [DOI] [PubMed] [Google Scholar]
- 29. Hishikawa K, Lüscher TF. Felodipine inhibits free‐radical production by cytokines and glucose in human smooth muscle cells. Hypertension. 1998;32:1011–1015. [DOI] [PubMed] [Google Scholar]
- 30. Mason RP. Mechanisms of plaque stabilization for the dihydropyridine calcium channel blocker amlodipine: review of the evidence. Atherosclerosis. 2002;165:191–199. [DOI] [PubMed] [Google Scholar]
- 31. Zhou M‐S, Jaimes EA, Raij L. Inhibition of oxidative stress and improvement of endothelial function by amlodipine in angiotensin II‐infused rats. Am J Hypertens. 2004;17:167–171. [DOI] [PubMed] [Google Scholar]
- 32. Cai H, Harrison DG. Endothelial dysfunction in cardio vascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. [DOI] [PubMed] [Google Scholar]
- 33. Dominiczak AF, Bohr DF. Nitric oxide and its putative role in hypertension. Hypertension. 1995;25:1202–1211. [DOI] [PubMed] [Google Scholar]
- 34. Taddei S, Virdis A, Mattei P, et al. Defective L‐arginine‐nitric oxide pathway in offspring of essential hypertensive patients. Circulation. 1996;94:1298–1303. [DOI] [PubMed] [Google Scholar]
- 35. Benjamin EJ, Larson MG, Keyes MJ, et al. Clinical correlates and heritability of flow‐mediated dilation in the community: the Framingham Heart Study. Circulation. 2004;109:613–619. [DOI] [PubMed] [Google Scholar]
- 36. Schlaich MP, Parnell MM, Ahlers BA, et al. Impaired L‐arginine transport and endothelial function in hypertensive and genetically predisposed normotensive subjects. Circulation. 2004;110:3680–3686. [DOI] [PubMed] [Google Scholar]
- 37. Cardillo C, Kilcoyne CM, Cannon RO III, et al. Impairment of the nitric oxide‐mediated vasodilator response to mental stress in hypertensive but not in hypercholesterolemic patients. J Am Coll Cardiol. 1998;32:1207–1213. [DOI] [PubMed] [Google Scholar]
- 38. Panza JA, García CE, Kilcoyne CM, et al. Impaired endothelium‐dependent vasodilation in patients with essential hypertension: evidence that nitric oxide abnormality is not localized to a single signal transduction pathway. Circulation. 1995;91:1732–1738. [DOI] [PubMed] [Google Scholar]
- 39. Higashi Y, Sasaki S, Nakagawa K, et al. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am J Hypertens. 2002;15:326–332. [DOI] [PubMed] [Google Scholar]
- 40. Achan V, Broadhead M, Malaki M, et al. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler Thromb Vasc Biol. 2003;23:1455–1459. [DOI] [PubMed] [Google Scholar]
- 41. Hyndman ME, Parsons HG, Verma S, et al. The T‐786→C mutation in endothelial nitric oxide synthase is associated with hypertension. Hypertension. 2002;39:919–922. [DOI] [PubMed] [Google Scholar]
- 42. Kato N, Sugiyama T, Morita H, et al. Lack of evidence for association between the endothelial nitric oxide synthase gene and hypertension. Hypertension. 1999;33:933–936. [DOI] [PubMed] [Google Scholar]
- 43. Lacolley P, Gautier S, Poirier O, et al. Nitric oxide synthase gene polymorphisms, blood pressure and aortic stiffness in normotensive and hypertensive subjects. J Hypertens. 1998;16:31–35. [DOI] [PubMed] [Google Scholar]
- 44. Dzau VJ. Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37:1047–1052. [DOI] [PubMed] [Google Scholar]
- 45. Blake GJ, Rifai N, Buring JE, et al. Blood pressure, C‐reactive protein, and risk of future cardiovascular events. Circulation. 2003;108:2993–2999. [DOI] [PubMed] [Google Scholar]
- 46. Sesso HD, Buring JE, Rifai N, et al. C‐reactive protein and the risk of developing hypertension. JAMA. 2003;290:2945–2951. [DOI] [PubMed] [Google Scholar]
- 47. Schillaci G, Pirro M, Gemelli F, et al. Increased C‐reactive protein concentrations in never‐treated hypertension: the role of systolic and pulse pressures. J Hypertens. 2003;21:1841–1846. [DOI] [PubMed] [Google Scholar]
- 48. Bautista LE, Lopez‐Jaramillo P, Vera LM, et al. Is C‐reactive protein an independent risk factor for essential hyper tension? J Hypertens. 2001;19:857–861. [DOI] [PubMed] [Google Scholar]
- 49. Cao JJ, Thach C, Manolio TA, et al. C‐reactive protein, carotid intima‐media thickness, and incidence of ischemic stroke in the elderly: the Cardiovascular Health Study. Circulation. 2003;108:166–170. [DOI] [PubMed] [Google Scholar]
- 50. Danesh J, Wheeler JG, Hirschfield GM, et al. C‐reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med. 2004;350:1387–1397. [DOI] [PubMed] [Google Scholar]
- 51. Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 52. Engström G, Janzon L, Berglund G, et al. Blood pressure increase and incidence of hypertension in relation to inflammation‐sensitive plasma proteins. Arterioscler Thromb Vasc Biol. 2002;22:2054–2058. [DOI] [PubMed] [Google Scholar]
- 53. Mahmud A, Feely J. Arterial stiffness is related to systemic inflammation in essential hypertension. Hypertension. 2005;46:1118–1122. [DOI] [PubMed] [Google Scholar]
- 54. Davey Smith G, Lawlor DA, Harbord R, et al. Association of C‐reactive protein with blood pressure and hypertension: life course confounding and mendelian randomization tests of causality. Arterioscler Thromb Vasc Biol. 2005;25:1051–1056. [DOI] [PubMed] [Google Scholar]
- 55. Alexander RW. Hypertension and the pathogenesis of atherosclerosis: oxidative stress and the mediation of arterial inflammatory response: a new perspective. Hypertension. 1995;25:155–161. [DOI] [PubMed] [Google Scholar]
- 56. The Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin‐converting‐enzyme inhibitor, ramipril, on cardiovascular events in high‐risk patients. N Engl J Med. 2000;342:145–153. [DOI] [PubMed] [Google Scholar]
- 57. European Trial on Reduction of Cardiac Events With Perindopril in Stable Coronary Artery Disease Investigators. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double‐blind, placebo‐controlled, multicentre trial (the EUROPA study). Lancet. 2003;362:782–788. [DOI] [PubMed] [Google Scholar]
- 58. Dahlöf B, Devereux RB, Kjeldsen SE, et al., for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003. [DOI] [PubMed] [Google Scholar]
- 59. Lithell H, Hansson L, Skoog I, et al., for the SCOPE Study Group. The Study on Cognition and Prognosis in the Elderly (SCOPE): principal results of a randomized double‐blind intervention trial. J Hypertens. 2003;21:875–886. [DOI] [PubMed] [Google Scholar]
- 60. Julius S, Kjeldsen SE, Weber M, et al, for the VALUE trial group. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial. Lancet. 2004;363:2022–2031. [DOI] [PubMed] [Google Scholar]
- 61. Perticone F, Ceravolo R, Candigliota M, et al. Obesity and body fat distribution induce endothelial dysfunction by oxidative stress: protective effect of vitamin C. Diabetes. 2001;50:159–165. [DOI] [PubMed] [Google Scholar]
- 62. Taddei S, Virdis A, Ghiadoni L, et al. Vitamin C improves endothelium‐dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation. 1998;97:2222–2229. [DOI] [PubMed] [Google Scholar]
- 63. Weber T, Auer J, O'Rourke MF, et al. Arterial stiffness, wave reflections, and the risk of coronary artery disease. Circulation. 2004;109:184–189. [DOI] [PubMed] [Google Scholar]
- 64. Cohn JN, Quyyumi AA, Hollenberg NK, et al. Surrogate markers for cardiovascular disease: functional markers. Circulation. 2004;109(suppl IV):IV31–IV46. [DOI] [PubMed] [Google Scholar]
- 65. Laurent S, Boutouyrie P, Asmar R, et al. Aortic stiffness is an independent predictor of all‐cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37:1236–1241. [DOI] [PubMed] [Google Scholar]
- 66. Cruickshank K, Riste L, Anderson SG, et al. Aortic pulse‐wave velocity and its relationship to mortality in diabetes and glucose intolerance: an integrated index of vascular function? Circulation. 2002;106:2085–2090. [DOI] [PubMed] [Google Scholar]
- 67. Blacher J, Guerin AP, Pannier B, et al. Impact of aortic stiffness on survival in end‐stage renal disease. Circulation. 1999;99:2434–2439. [DOI] [PubMed] [Google Scholar]
- 68. O'Rourke MF, Gallagher DE. Pulse wave analysis. J Hypertens Suppl. 1996;14:S147–S157. [PubMed] [Google Scholar]
- 69. Meaume S, Benetos A, Henry OF, et al. Aortic pulse wave velocity predicts cardiovascular mortality in subjects >70 years of age. Arterioscler Thromb Vasc Biol. 2001;21:2046–2050. [DOI] [PubMed] [Google Scholar]
- 70. Dahlöf B, Sever PS, Poulter NR, et al, for the ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo‐Scandinavian Cardiac Outcomes Trial‐Blood Pressure Lowering Arm (ASCOT‐BPLA): a multicentre randomised controlled trial. Lancet. 2005;366:895–906. [DOI] [PubMed] [Google Scholar]
- 71. The CAFE Investigators. Differential impact of blood pressure‐lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation. 2006;113:1213–1225. [DOI] [PubMed] [Google Scholar]
- 72. Van Vliet BN, Chafe LL, Montani J‐P. Characteristics of 24 h telemetered blood pressure in eNOS‐knockout and C57Bl/6J control mice. J Physiol. 2003;549:313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kinlay S, Creager MA, Fukumoto M, et al. Endothelium‐derived nitric oxide regulates arterial elasticity in human arteries in vivo. Hypertension. 2001;38:1049–1053. [DOI] [PubMed] [Google Scholar]
- 74. Wilkinson IB, Qasem A, McEniery CM, et al. Nitric oxide regulates local arterial distensibility in vivo. Circulation. 2002;105:213–217. [DOI] [PubMed] [Google Scholar]
- 75. Stewart AD, Millasseau SC, Kearney MT, et al. Effects of inhibition of basal nitric oxide synthesis on carotid‐femoral pulse wave velocity and augmentation index in humans. Hypertension. 2003;42:915–918. [DOI] [PubMed] [Google Scholar]
- 76. Joannides R, Richard V, Haefeli WE, et al. Role of nitric oxide in the regulation of the mechanical properties of peripheral conduit arteries in humans. Hypertension. 1997;30:1465–1470. [DOI] [PubMed] [Google Scholar]
- 77. Girerd X, Giannattasio C, Moulin C, et al. Regression of radial artery wall hypertrophy and improvement of carotid artery compliance after long‐term antihypertensive treatment in elderly patients. J Am Coll Cardiol. 1998;31:1064–1073. [DOI] [PubMed] [Google Scholar]
- 78. Nashar K, Nguyen JP, Jesri A, et al. Angiotensin receptor blockade improves arterial distensibility and reduces exercise‐induced pressorresponses in obese hypertensive patients with the metabolic syndrome. Am J Hypertens. 2004;17:477–482. [DOI] [PubMed] [Google Scholar]
- 79. Ghiadoni L, Magagna A, Versari D, et al. Different effect of antihypertensive drugs on conduit artery endothelial function. Hypertension. 2003;41:1281–1286. [DOI] [PubMed] [Google Scholar]
- 80. Neutel JM, Smith DH, Weber MA. Effect of antihypertensive monotherapy and combination therapy on arterial distensibility and left ventricular mass. Am J Hypertens. 2004;17:37–42. [DOI] [PubMed] [Google Scholar]
- 81. Taddei S, Virdis A, Ghiadoni L, et al. Effect of calcium antagonist or beta blockade treatment on nitric oxide‐dependent vasodilation and oxidative stress in essential hypertensive patients. J Hypertens. 2001;19:1379–1386. [DOI] [PubMed] [Google Scholar]
- 82. Lewis SJ, Ohta H, Machado B, et al. Microinjection of Snitrosocysteine into the nucleus tractus solitarii decreases arterial pressure and heart rate via activation of soluble guanylate cyclase. Eur J Pharmacol. 1991;202:135–136. [DOI] [PubMed] [Google Scholar]
- 83. Lepori M, Sartori C, Trueb L, et al. Haemodynamic and sympathetic effects of inhibition of nitric oxide synthase by systemic infusion of N(G)‐monomethyl‐Larginine into humans are dose dependent. J Hypertens. 1998;16:519–523. [DOI] [PubMed] [Google Scholar]
- 84. Sartori C, Lepori M, Scherrer U. Interaction between nitric oxide and the cholinergic and sympathetic nervous system in cardiovascular control in humans. Pharmacol Ther. 2005;106:209–220. [DOI] [PubMed] [Google Scholar]
- 85. Noll G, Wenzel RR, Schneider M, et al. Increased activation of sympathetic nervous system and endothelin by mental stress in normotensive offspring of hypertensive parents. Circulation. 1996;93:866–869. [DOI] [PubMed] [Google Scholar]
- 86. Victor RG, Leimbach WN Jr, Seals DR, et al. Effects of the cold pressor test on muscle sympathetic nerve activity in humans. Hypertension. 1987;9:429–436. [DOI] [PubMed] [Google Scholar]
- 87. Anderson EA, Sinkey CA, Lawton WJ, et al. Elevated sympathetic nerve activity in borderline hypertensive humans. Evidence from direct intraneural recordings. Hypertension. 1989;14:177–183. [DOI] [PubMed] [Google Scholar]
- 88. Esler M, Kaye D. Sympathetic nervous system activation in essential hypertension, cardiac failure and psychosomatic heart disease. J Cardiovasc Pharmacol. 2000;35(suppl 4):S1–S7. [DOI] [PubMed] [Google Scholar]
- 89. Matsukawa T, Mano T, Gotoh E, et al. Elevated sympathetic nerve activity in patients with accelerated essential hypertension. J Clin Invest. 1993;92:25–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sander M, Hansen J, Victor RG. The sympathetic nervous system is involved in the maintenance but not initiation of the hypertension induced by Nω‐nitro‐L‐arginine methyl ester. Hypertension. 1997;30:64–70. [DOI] [PubMed] [Google Scholar]
- 91. Liu J‐L, Murakami H, Zucker IH. Angiotensin II‐nitric oxide interaction on sympathetic outflow in conscious rabbits. Circ Res. 1998;82:496–502. [DOI] [PubMed] [Google Scholar]
- 92. Sander M, Hansen PG, Victor RG. Sympathetically mediated hypertension caused by chronic inhibition of nitric oxide. Hypertension. 1995;26:691–695. [DOI] [PubMed] [Google Scholar]
- 93. Sakuma I, Togashi H, Yoshioka M, et al. NG‐methyl‐L‐arginine, an inhibitor of L‐arginine‐derived nitric oxide synthesis, stimulates renal sympathetic nerve activity in vivo. A role for nitric oxide in the central regulation of sympathetic tone? Circ Res. 1992;70:607–611. [DOI] [PubMed] [Google Scholar]
- 94. Sakima A, Teruya H, Yamazato M, et al. Prolonged NOS inhibition in the brain elevates blood pressure in normotensive rats. Am J Physiol Regul Integr Comp Physiol. 1998;275:R410–R417. [DOI] [PubMed] [Google Scholar]
- 95. Horn T, Smith PM, McLaughlin BE, et al. Nitric oxide actions in paraventricular nucleus: cardiovascular and neurochemical implications. Am J Physiol Regul Integr Comp Physiol. 1994;266:R306–R313. [DOI] [PubMed] [Google Scholar]
- 96. Hansen J, Jacobsen TN, Victor RG. Is nitric oxide involved in the tonic inhibition of central sympathetic outflow in humans? Hypertension. 1994;24:439–444. [DOI] [PubMed] [Google Scholar]
- 97. Cui J, Zhang R, Wilson TE, et al. Nitric oxide synthase inhibition does not affect regulation of muscle sympathetic nerve activity during head‐up tilt. Am J Physiol Heart Circ Physiol. 2003;285:H2105–H2110. [DOI] [PubMed] [Google Scholar]
- 98. Owlya R, Vollenweider L, Trueb L, et al. Cardiovascular and sympathetic effects of nitric oxide inhibition at rest and during static exercise in humans. Circulation. 1997;96:3897–3903. [DOI] [PubMed] [Google Scholar]
- 99. Sander M, Chavoshan B, Victor RG. A large blood pressure‐raising effect of nitric oxide synthase inhibition in humans. Hypertension. 1999;33:937–942. [DOI] [PubMed] [Google Scholar]
- 100. Ramchandra R, Barrett CJ, Malpas SC. Nitric oxide and sympathetic nerve activity in the control of blood pressure. Clin Exp Pharmacol Physiol. 2005;32:440–446. [DOI] [PubMed] [Google Scholar]
- 101. Lepori M, Sartori C, Duplain H, et al. Sympathectomy potentiates the vasoconstrictor response to nitric oxide synthase inhibition in humans. Cardiovasc Res. 1999;43:739–743. [DOI] [PubMed] [Google Scholar]
- 102. Johansson M, Elam M, Rundqvist B, et al. Differentiated response of the sympathetic nervous system to angiotensin‐converting enzyme inhibition in hypertension. Hypertension. 2000;36:543–548. [DOI] [PubMed] [Google Scholar]
- 103. Béchir M, Enseleit F, Chenevard R, et al. Effect of losartan on muscle sympathetic activity and baroreceptor function in systemic hypertension. Am J Cardiol. 2005;95:129–131. [DOI] [PubMed] [Google Scholar]
- 104. Binggeli C, Corti R, Sudano I, et al. Effects of chronic calcium channel blockade on sympathetic nerve activity in hypertension. Hypertension. 2002;39:892–896. [DOI] [PubMed] [Google Scholar]
- 105. Wenzel RR, Bruck H, Noll G, et al. Antihypertensive drugs and the sympathetic nervous system: imadazolidine 11 receptor agonists and the sympathetic nervous system in cardiovascular and metabolic diseases. J Cardiovasc Pharmacol. 2000;35(suppl 4):S43–S52. [DOI] [PubMed] [Google Scholar]
- 106. Corti R, Binggeli C, Sudano I, et al. The beauty and the beast: aspects of the autonomic nervous system. News Physiol Sci. 2000;15:125–129. [DOI] [PubMed] [Google Scholar]
- 107. Lüscher TF, Spieker LE, Noll G, et al. Vascular effects of newer cardiovascular drugs: focus on nebivolol and ACE‐inhibitors. J Cardiovasc Pharmacol. 2001;38(suppl 3): S3–S11. [DOI] [PubMed] [Google Scholar]
- 108. Joannides R, Haefeli WE, Linder L, et al. Nitric oxide is responsible for flow‐dependent dilatation of human peripheral conduit arteries in vivo. Circulation. 1995;91:1314–1319. [DOI] [PubMed] [Google Scholar]