

The stress response is an integral part of human physiology and one that has a long history. It not only occurs in all mammals, but also in our more primitive ancestors, such as fish. 1 It presumably evolved because it conveys a survival advantage and helps animals to avoid or vanquish an immediate threat to their well‐being by moving to a “red alert” mode that heightens vigilance and sets the stage for increased physical and mental activity. One of the first signs is an increase in heart rate and blood pressure mediated via the sympathetic nervous system (ie, the fight or flight response first described by Cannon), which increases oxygenation to the exercising muscles. There needs to be metabolic changes as well, to ensure the increased availability of glucose and other nutrients, and this is mediated by a somewhat slower activation of the hypothalamic‐pituitary‐adrenal (HPA) axis. A central tenet of stress research is that this acute response has a largely beneficial effect, but that when chronically activated it may be maladaptive or harmful. If the red alert signal fails to shut off, it is not surprising that the wear and tear on our bodies is going to increase. This chronic activation, which may be thought of as an “orange alert” situation, has been termed allostatic load by McEwen, 1 one of the leading stress researchers in this area.
Not surprisingly, most of the research on the effects of stress on blood pressure and cardiovascular disease has focused on the sympathetic nervous system, but there is another component of the stress response that may be relevant. This is the inflammatory or acute‐phase response, which is switched on more gradually than the sympathetic nervous system and the HPA axis, but which is also partly under the control of the central nervous system. Teleologically, the function of this response may be to aid the healing of wounds inflicted during the fight and flight response by activating both the blood clotting cascade and the inflammatory response. It also serves as a defense against what the body regards as intruders, such as bacteria. The cardiovascular literature has seen a sudden increase in the number of articles published on the acute‐phase response, largely because several markers of the inflammatory response have been shown to be independent predictors of cardiovascular events.
MECHANISMS OF THE INFLAMMATORY RESPONSE
The acute‐phase response is mediated by cytokines, which are proteins and peptides produced in cells throughout the body, including in the liver, endothelium, fat cells, and brain, among others. 2 Cytokines are chemical messengers that may have autocrine, paracrine, or endocrine effects. Many cytokines have been labeled as interleukins on the grounds that their principal target is the leukocyte, but this title bears little relationship to their presumed functions. The cytokine that has attracted the most interest in the study of cardiovascular disease is C‐reactive protein (CRP), which was first discovered in the serum of patients with acute inflammation and so named because it reacted with the C polysaccharide of pneumococcus. 2 Produced in the liver, CRP is a major player in the acute‐phase response. While CRP levels increase during acute inflammatory responses such as with infections, the finding of slightly high levels in patients with cardiovascular disease, and its predictive value for events, 3 was the trigger for the surge of interest in this field. The main inducer of the response, however, is another important pro‐inflammatory cytokine, interleukin 6 (IL‐6). 2 When subcutaneously administered to humans, IL‐6 provokes an acute‐phase response, which is associated with an increase in CRP level. Thus, the levels of CRP and IL‐6 are closely correlated, whereas other cytokines are not. IL‐6 largely comes from macrophages, which, when activated, not only release IL‐6, but also other cytokines such as IL‐1 and tumor necrosis factor α (TNF‐α), both of which also stimulate CRP release. Evidence for a causal role of IL‐6 comes from the observation that IL‐6 knockout mice are unable to generate a normal acute‐phase response. 4
The acute‐phase response is also an integral part of the response to external sources of stress that is mediated by the central nervous system. There is extensive evidence that during the acute stress response, cortisol, catecholamines, angiotensin, and other stress hormones, which are part of the central nervous system response, induce the liver and abdominal fat tissue to release cytokines and other inflammatory mediators, including IL‐6. 2 In general, the sympathetic nervous system increases cytokine secretion, while cortisol suppresses it. The relationship with cortisol is a 2‐way street, however, since IL‐6 acts on the hypothalamus to increase cortisol release. 5 Thus, an infusion of the β‐adrenergic agonist isoproterenol raises IL‐6 9‐fold, but has no effect on TNF‐α 6 This sympathetically mediated secretion of IL‐6 comes predominantly from adipocytes. 7 Exercise is another potent activator of the sympathetic nervous system, and it also increases IL‐6: in 1 study, a positive correlation was observed between peak plasma epinephrine or norepinephrine and IL‐6 levels after 15 minutes of aerobic exercise. 8
The acute‐phase response includes the release of other proteins besides CRP, including serum amyloid A and fibrinogen. The cytokines released as part of this response may contribute to the development of the metabolic syndrome. Thus, serum amyloid A lowers high‐density lipoprotein cholesterol levels, 9 and TNF‐α and IL‐6 both have effects that lead to insulin resistance. Furthermore, circulating IL‐6 stimulates the HPA axis, activation of which is associated with central obesity, hypertension, and insulin resistance. Adipocytes are an important source of IL‐6 and other cytokines, and the blood levels of IL‐6, CRP, TNF‐α and fibrinogen are all correlated with visceral obesity. 10 About 25% of circulating IL‐6 comes from fat cells, and visceral fat releases 2 to 3 times more IL‐6 than other fat cells. 2 The acute‐phase response also produces a prothrombotic state; in addition to increased fibrinogen levels, IL‐1 and TNF‐α increase platelet number and activity. These considerations have led Yudkin and colleagues 9 to propose that IL‐6 is a central mediator of the relationship between chronic stress, inflammation, obesity, and coronary heart disease. A schema showing some of the principal pathways by which IL‐6 might affect hypertension is shown in the Figure.
Figure.
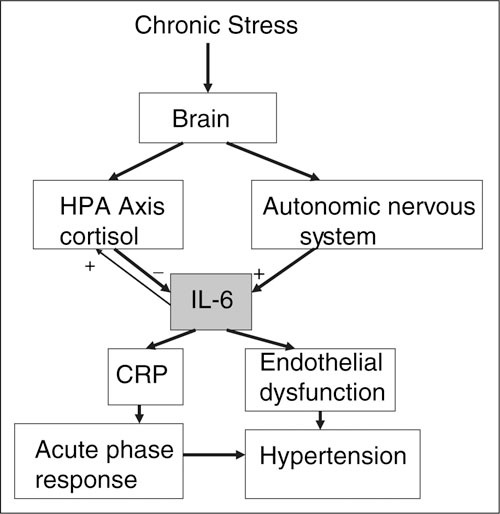
Pathways by which stress might affect hypertension that are mediated by cytokines such as C‐reactive protein (CRP) and interleukin 6 (IL‐6). HPA indicates hypothalamic‐pituitary‐adrenal.
THE ROLE OF STRESS IN ELICITING THE INFLAMMATORY RESPONSE
An emerging hypothesis proposed by neuroscientists studying experimental animal models of stress is that exposure to chronic stress can lead to a state of chronic inflammation. 11 This hypothesis is based on the finding that long‐term dysregulation of stress hormones (eg, cortisol and catecholamines) alters the patterns of cytokine release from immune cells. 12 The shift tends to favor the production of pro‐inflammatory cytokines at the expense of anti‐inflammatory cytokines, and hence induce a state of chronic low grade inflammation.
It has been shown experimentally in both animals and humans that acute mental stress can raise IL‐6 levels as well as cortisol. 5 , 13 An example is a study by Steptoe and colleagues 14 in which mental stress tasks increased IL‐6 concentration by 56% at 2 hours after the tasks (P<.05), while IL‐1Ra was increased by 12.3% (P<.01). The magnitude of blood pressure responses during tasks was correlated positively with the IL‐6 concentration after 45 minutes (r=0.70; P<.05) and with the IL‐1Ra concentration after 2 hours (r=0.63; P<.05). Increases in TNF‐α after 2 hours were correlated with heart rate responses to tasks (r=0.66; P<.05).
Chronic stress may also activate the inflammatory response, although here the causal links are less clear. It has been proposed that stress‐related activation of the HPA axis can cause accumulation of abdominal fat, 15 which, unlike fat cells in other parts of the body, contains glucocorticoid receptors. Stimulation of these receptors enhances fatty acid uptake by adipocytes, and adrenalectomy reduces visceral fat. 2 In cynomolgus monkeys, the stress of social subordination leads to insulin resistance with hyperglycemia, central obesity, dyslipidemia, and hypertension. 16
Several studies have shown that low socioeconomic status, which is often considered to be a source of chronic stress, is associated with increased levels of cytokines. IL‐6 levels are much higher in Indian slum dwellers than people living in the rural areas. 17 A survey of women in the Dominican Republic found a significant correlation between abdominal obesity and internalized racism, 18 and the Whitehall study of British civil servants reported that men of lower socioeconomic status had enhanced responses of IL‐6 to psychological stress. 19
RELEVANCE FOR HYPERTENSION AND CARDIOVASCULAR DISEASE
As mentioned above, the reason for the interest in this field is the ever‐growing number of reports that have shown that blood levels of cytokines are independent predictors of cardiovascular events. This has been shown for CRP in several studies. 3 In a case‐control study of 202 men with a myocardial infarction and 202 matched controls, all of whom were participants in the Physicians' Health Study, IL‐6 was also found to independently predict events of other risk factors, including CRP. 20
Several studies have reported an association between CRP levels and hypertension, 10 but their interpretation is somewhat difficult for 2 reasons: (1) CRP correlates with several other related risk factors such as age, sex, body mass index, and smoking, which may confound the relationship with blood pressure; and (2) it is not clear whether the hypertension is the cause or the consequence of the CRP elevation. 21 There is a linear relationship between blood pressure and CRP, and the nondipping of diurnal blood pressure change is more common in patients with the highest CRP levels. 22 In the ATTICA study, 23 a population survey of Greeks, 3 cytokines were evaluated: CRP, TNF‐α, and serum amyloid A, all of which showed a progressive increase from normotensives, prehypertensives, and hypertensives. Another classic marker of inflammation, the white blood cell count, also showed parallel increases. IL‐6 has also been correlated with hypertension in several studies. 10
One prospective study with a follow‐up duration of 11 years found, however, that initially normotensive men with high CRP levels were nearly 3 times more likely to develop hypertension than those with low levels, even after adjusting for other known predictors. 24 Additional evidence for a causal role of IL‐6 comes from several sources. IL‐6 knockout mice show a significantly smaller blood pressure increase when exposed to acute psychological stress (moving the mice to a cage previously inhabited by another mouse), 25 without there being any differences in plasma renin or norepinephrine. There was a second but smaller peak of blood pressure that occurred about 5 hours after the acute response, which was completely absent in the knockout mice. In human studies, people exposed to chronic psychosocial stress 16 have increased IL‐6 levels. A study that measured the blood pressure and biochemical responses to an acute laboratory stressor found that the stress‐related increase of both fibrinogen and IL‐6 predicted the ambulatory blood pressure measured 3 years later. 20
TNF‐α is another cytokine that may be involved in hypertension. The literature on TNF‐α levels and blood pressure is conflicting, however. One study found that the ratio of soluble TNF‐α receptor 1 to TNF‐α receptor 2, which is thought to be a marker of the activation of the TNF‐α system, was significantly related to blood pressure and also found that lowering the blood pressure reduced the ratio. 26
Several mechanisms have been postulated to explain how cytokines could contribute to hypertension. A crucial component may be the imbalance between endothelium‐derived relaxing and contracting factors such as nitric oxide, endothelium derived hyperpolarizing factor, and prostacyclin. Both CRP and IL‐6 have been associated with endothelial dysfunction in correlational studies, and CRP and TNF‐α have both been shown experimentally to reduce nitric oxide synthesis. 10 Oxidative stress may also be involved, since reactive oxygen species can cause vasoconstriction both by decreasing nitric oxide and by increasing vasoconstrictor prostaglandins. A convenient way of studying the causal links between cytokines and cardiovascular physiology is to study the response to vaccination. In a study of the response to vaccination against typhoid, Clapp and associates 27 found an increase of 2 cytokines (IL‐6 and IL‐1Ra), which was associated with a fall in total antioxidant status, impaired endothelial function, and increased urinary albumin excretion. The authors concluded that inflammation causes widespread endothelial dysfunction, reduces vascular nitric oxide bioavailability, and increases oxidative stress.
One of the stimuli to cytokine release from endothelial cells may be increased shear stress. There is thus a chicken‐and‐egg dilemma here, but the fact that lowering blood pressure does not necessarily reverse the endothelial dysfunction or the increased level of cytokines suggests that the inflammatory response may indeed be contributing to the hypertension. 10
Another mechanism by which inflammation and cytokines may affect blood pressure is through increased arterial stiffness. Several observational studies have shown a relationship between the two, 28 but the most direct evidence for a causal relationship comes from a study of healthy individuals before and after vaccination for typhoid. Vaccination produced an acute rise of IL‐6 and CRP levels and also increased pulse wave velocity. In a second group of participants, pretreatment with aspirin blocked the increases of cytokines and the increased stiffness. 28
CONCLUSIONS
The measurement of blood levels of cytokines is relatively straightforward and could be easily incorporated into clinical practice as well as research studies. The picture that is emerging is that subtle increases of several cytokines are independent predictors of clinical outcomes, that both acute and chronic stress may increase the release of cytokines, and that they may not be just markers, but also mediators of processes such as endothelial dysfunction and atherosclerosis. If these statements prove to be correct, they open up new avenues not only for research, but also for the prevention and treatment of cardiovascular disease. Anti‐inflammatory drugs might seem to be appropriate, but so far have got a bad rap in terms of their cardiovascular effects. Therefore, perhaps the answer is to continue to recommend aspirin.
References
- 1. McEwen BS, Lasley EN, Lasley E. The End of Stress As We Know It. Washington, DC: National Academies Press; 2002. [Google Scholar]
- 2. Black PH. The inflammatory response is an integral part of the stress response: implications for atherosclerosis, insulin resistance, type II diabetes and metabolic syndrome X. Brain Behav Immun. 2003;17(5):350–364. [DOI] [PubMed] [Google Scholar]
- 3. Albert MA, Ridker PM. C‐reactive protein as a risk predictor: do race/ethnicity and gender make a difference? Circulation. 2006;114(5):e67–e74. [DOI] [PubMed] [Google Scholar]
- 4. Inui A. Cytokines and sickness behavior: implications from knockout animal models. Trends Immunol. 2001;22(9):469–473. [DOI] [PubMed] [Google Scholar]
- 5. Baker DG, Ekhator NN, Kasckow JW, et al. Plasma and cerebrospinal fluid interleukin‐6 concentrations in posttraumatic stress disorder. Neuroimmunomodulation. 2001;9(4):209–217. [DOI] [PubMed] [Google Scholar]
- 6. Mohamed‐Ali V, Bulmer K, Clarke D, et al. beta‐Adrenergic regulation of proinflammatory cytokines in humans. Int J Obes Relat Metab Disord. 2000;24(suppl 2):S154–S155. [DOI] [PubMed] [Google Scholar]
- 7. Mohamed‐Ali V, Flower L, Sethi J, et al. β‐Adrenergic regulation of IL‐6 release from adipose tissue: in vivo and in vitro studies. J Clin Endocrinol Metab. 2001;86(12):5864–5869. [DOI] [PubMed] [Google Scholar]
- 8. Papanicolaou DA, Petrides JS, Tsigos C, et al. Exercise stimulates interleukin‐6 secretion: inhibition by glucocorticoids and correlation with catecholamines. Am J Physiol. 1996;271(3 pt 1):E601–E605. [DOI] [PubMed] [Google Scholar]
- 9. Yudkin JS, Kumari M, Humphries SE, et al. Inflammation, obesity, stress and coronary heart disease: is interleukin‐6 the link? Atherosclerosis. 2000;148(2):209–214. [DOI] [PubMed] [Google Scholar]
- 10. Bautista LE. Inflammation, endothelial dysfunction, and the risk of high blood pressure: epidemiologic and biological evidence. J Hum Hypertens. 2003;17(4):223–230. [DOI] [PubMed] [Google Scholar]
- 11. Black PH, Garbutt LD. Stress, inflammation and cardiovascular disease. J Psychosom Res. 2002;52(1):1–23. [DOI] [PubMed] [Google Scholar]
- 12. Elenkov IJ, Iezzoni DG, Daly A, et al. Cytokine dysregulation, inflammation and well‐being. Neuroimmunomodulation. 2005;12(5):255–269. [DOI] [PubMed] [Google Scholar]
- 13. Kunz‐Ebrecht SR, Mohamed‐Ali V, Feldman PJ, et al. Cortisol responses to mild psychological stress are inversely associated with proinflammatory cytokines. Brain Behav Immun. 2003;17(5):373–383. [DOI] [PubMed] [Google Scholar]
- 14. Steptoe A, Willemsen G, Owen N, et al. Acute mental stress elicits delayed increases in circulating inflammatory cytokine levels. Clin Sci (Lond). 2001;101(2):185–192. [PubMed] [Google Scholar]
- 15. Rosmond R, Dallman MF, Bjorntorp P. Stress‐related cortisol secretion in men: relationships with abdominal obesity and endocrine, metabolic and hemodynamic abnormalities. J Clin Endocrinol Metab. 1998;83(6):1853–1859. [DOI] [PubMed] [Google Scholar]
- 16. Shively CA, Clarkson TB. Regional obesity and coronary artery atherosclerosis in females: a non‐human primate model. Acta Med Scand Suppl. 1988;723:71–78. [DOI] [PubMed] [Google Scholar]
- 17. Yudkin JS, Yajnik CS, Mohamed‐Ali V, et al. High levels of circulating proinflammatory cytokines and leptin in urban, but not rural, Indians. A potential explanation for increased risk of diabetes and coronary heart disease. Diabetes Care. 1999;22(2):363–364. [DOI] [PubMed] [Google Scholar]
- 18. Butler C, Tull ES, Chambers EC, et al. Internalized racism, body fat distribution, and abnormal fasting glucose among African‐Caribbean women in Dominica, West Indies. J Natl Med Assoc. 2002;94(3):143–148. [PMC free article] [PubMed] [Google Scholar]
- 19. Brydon L, Edwards S, Mohamed‐Ali V, et al. Socioeconomic status and stress‐induced increases in interleukin‐6. Brain Behav Immun. 2004;18(3):281–290. [DOI] [PubMed] [Google Scholar]
- 20. Ridker PM, Rifai N, Stampfer MJ, et al. Plasma concentration of interleukin‐6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101(15):1767–1772. [DOI] [PubMed] [Google Scholar]
- 21. Brydon L, Steptoe A. Stress‐induced increases in interleukin‐6 and fibrinogen predict ambulatory blood pressure at 3‐year follow‐up. J Hypertens. 2005;23(5):1001–1007. [DOI] [PubMed] [Google Scholar]
- 22. Bellelli G, Rozzini R, Battista Frisoni G, et al. Is C‐reactive protein an independent risk factor for essential hypertension? J Hypertens. 2001;19(11):2107. [DOI] [PubMed] [Google Scholar]
- 23. Chrysohoou C, Pitsavos C, Panagiotakos DB, et al. Association between prehypertension status and inflammatory markers related to atherosclerotic disease: The ATTICA Study. Am J Hypertens. 2004;17(7):568–573. [DOI] [PubMed] [Google Scholar]
- 24. Niskanen L, Laaksonen DE, Nyyssonen K, et al. Inflammation, abdominal obesity, and smoking as predictors of hypertension. Hypertension. 2004;44(6):859–865. [DOI] [PubMed] [Google Scholar]
- 25. Lee DL, Leite R, Fleming C, et al. Hypertensive response to acute stress is attenuated in interleukin‐6 knockout mice. Hypertension. 2004;44(3):259–263. [DOI] [PubMed] [Google Scholar]
- 26. Fernandez‐Real JM, Lainez B, Vendrell J, et al. Shedding of TNF‐alpha receptors, blood pressure, and insulin sensitivity in type 2 diabetes mellitus. Am J Physiol Endocrinol Metab. 2002;282(4):E952–E959. [DOI] [PubMed] [Google Scholar]
- 27. Clapp BR, Hingorani AD, Kharbanda RK, et al. Inflammation‐induced endothelial dysfunction involves reduced nitric oxide bioavailability and increased oxidant stress. Cardiovasc Res. 2004;64(1):172–178. [DOI] [PubMed] [Google Scholar]
- 28. Vlachopoulos C, Dima I, Aznaouridis K, et al. Acute systemic inflammation increases arterial stiffness and decreases wave reflections in healthy individuals. Circulation. 2005;112(14):2193–2200. [DOI] [PubMed] [Google Scholar]