

Abstract
In contrast with the short research history of the enzymatic synthesis of nitric oxide (NO), the introduction of nitrate‐containing compounds for medicinal purposes marked its 150th anniversary in 1997. Glyceryl trinitrate (nitroglycerin) is the first compound of this category. On October 12, 1998, the Nobel Assembly awarded the Nobel Prize in Medicine or Physiology to scientists Robert Furchgott, Louis Ignarro, and Ferid Murad for their discoveries concerning NO as a signaling molecule in the cardiovascular system. NO‐mediated signaling is a recognized component in various physiologic processes (eg, smooth muscle relaxation, inhibition of platelet and leukocyte aggregation, attenuation of vascular smooth muscle cell proliferation, neurotransmission, and immune defense), to name only a few. NO has also been implicated in the pathology of many inflammatory diseases, including arthritis, myocarditis, colitis, and nephritis and a large number of pathologic conditions such as amyotrophic lateral sclerosis, cancer, diabetes, and neurodegenerative diseases. Some of these processes (eg, smooth muscle relaxation, platelet aggregation, and neurotransmission) require only a brief production of NO at low nanomolar concentrations and are dependent on the recruitment of cyclic guanosine monophosphate (cGMP)‐dependent signaling. Other processes are associated with direct interaction of NO or reactive nitrogen species derived from it with target proteins and requires a more sustained production of NO at higher concentrations but do not involve the cGMP pathway.
The vasculature undergoes considerable stress and demands such as blood stream shear force effects and the constant generation of reactive oxygen and nitrogen species. A healthy vasculature depends on the integrity of the endothelium with a dynamic autocrine and paracrine organ with vasodilator, anti‐inflammatory, and antithrombotic effects. 1 Injuries or dysfunctional endothelial cells with a loss of endothelium‐derived nitric oxide (NO) are major critical factors in the pathogenesis of vascular diseases, including hypertension, atherosclerosis, and vasospasm with compromised blood flow.
Substantial knowledge of the NO signaling pathway has been gained during the past 3 decades. In 1977, Murad's laboratory reported that “nitrovasodilators” such as nitroglycerin and nitroprusside caused smooth muscle relaxation via generation of NO that activated soluble guanylyl cyclase and increased the concentration of cyclic guanosine monophosphate (cGMP) in tissues 2 (Figure 1). NO's function as a possible messenger was first proposed in 1978. Furchgott and Zawadzki 3 further described an endothelium‐derived relaxing factor identified as NO. In addition to its discovery as a vasodilator, NO mediates several protective functions of the endothelium by inhibiting (1) neutrophil activation and adhesion, (2) platelet adhesion and aggregation, (3) vascular smooth muscle proliferation, and (4) expression of proinflammatory cytokines. Thus, a healthy endothelium with normal generation of NO acts to prevent atherosclerosis development and its complications. In contrast to the beneficial effects of NO at physiologic concentrations (nanomolar), large amounts (micromolar) of NO can be toxic and proinflammatory. For example, NO reacts with O2 •− that can form peroxynitrous acid, a very unstable and reactive oxidizing species. Involvement of peroxynitrite in inflammatory diseases has been determined by detection of nitrotyrosine formation in various inflamed tissues 4 , 5 and is the most widely studied mechanism of protein nitration. 6 Nitrite (NO2 −) is another major oxidation product derived from NO and can be oxidized by peroxidase/myeloperodixase to form reactive nitrogen intermediate(s), such as nitrogen dioxide that is capable of nitrating tyrosine. A distinct and important signaling role in mammalian biology of the anion, however, has recently been suggested. Physiologic NO2 − concentrations not only partially account for the benefit effect of NO, but also readily affect cGMP production, cytochrome P450 activities, heat shock protein 70, and heme oxygenase‐1 expression in a variety of tissues, 7 thus NO2 − homeostasis may be of great importance to NO biology.
Figure 1.

Effects of nitrovasodilators and nitric oxide (NO) on soluble guanylyl cyclase.
NO‐mediated signaling is a recognized component in various physiologic processes. Nevertheless, NO has also been implicated in the pathology of many inflammatory diseases, including arthritis, myocarditis, colitis, and nephritis and a large number of pathologic conditions such as amyotrophic lateral sclerosis, cancer, diabetes, and neurodegenerative diseases. Some of these processes (eg, smooth muscle relaxation, platelet aggregation, and neurotransmission) require only a brief production of NO at low nanomolar concentrations and are dependent on the recruitment of cGMP‐dependent signaling. Other processes are associated with direct interaction of NO or reactive nitrogen species derived from it with target proteins and requires a more sustained production of NO at higher concentrations but do not involve the cGMP pathway. Through this article, a great deal of information will be provided in a way of relevance for medical practitioners.
ENDOTHELIUM‐CENTERED HEALTHY ENVIRONMENT FOR VASCULATURE
Since the study of Furchgott and Zawadzki, 3 which underlined the obligatory role of the endothelium in mediating acetylcholine‐induced vasodilatation, NO has been recognized as an endogenous nitro‐vasodilator that mediates the local regulation of basal arterial tone and is anticipated to vary in certain pathologic states. 8 , 9 For instance, Panza and colleagues 10 have indicated that patients with essential hypertension have a defect in the endothelium‐derived NO system that may at least partly account for both the increased vascular resistance under basal conditions and the impaired response to endothelium‐dependent vasodilators.
Presently, our understanding of the process that leads to hypertension is allowing us to adopt principles of therapy that may be more beneficial for patients. Hypertension, particularly in high‐risk patients, is a result of loss of balance and the absence of the ability to vasodilate normally. The interaction between the endothelial cell and the smooth muscle cell is very important in this process. The endothelium is a group of cells that produce compounds that are important in regulating vascular homeostasis by elaborating factors such as angiotensin II, NO, endothelin, and prostaglandins. Specifically, NO is found in endothelial cells responsible for smooth muscle relaxation. Gaseous NO diffuses across the endothelial cell and into the underlying smooth muscle cell, where it stimulates the pathway of guanylate cyclase to produce vasodilatation. 11 Normal endothelium maintains vascular tone and blood viscosity, prevents abnormal blood clotting and bleeding, limits inflammation of the vasculature, and suppresses smooth muscle cell proliferation. Abnormal endothelium causes increased inflammation and hypertrophy of the smooth muscle cells and promotes thrombosis and vasoconstriction, leading to the rapid growth of atherosclerotic plaques. Therefore, understanding endothelial vasculature will be imperative as researchers develop newer compounds that may enhance NO formation within the vasculature.
NO SIGNALING AND THE VASCULAR SYSTEM
The introduction of nitrate‐containing compounds for medicinal purposes marked its 150th anniversary in 1997. Glyceryl trinitrate (nitroglycerin [GTN]) is the first compound of this category and was synthesized by Sombrero in 1847. 12 Another nitrate‐containing compound, amyl nitrite, was discovered a few years later and was used by Guthrie and associates in 1859. 12 During his discovery of dynamite, Alfred Nobel experienced angina pectoris and was prescribed nitroglycerin for chest pain in 1895. Almost a century later, organic nitrates and their gaseous metabolic end product, NO, were implicated in a vast array of biologically diverse activities (Figure 2).
Figure 2.
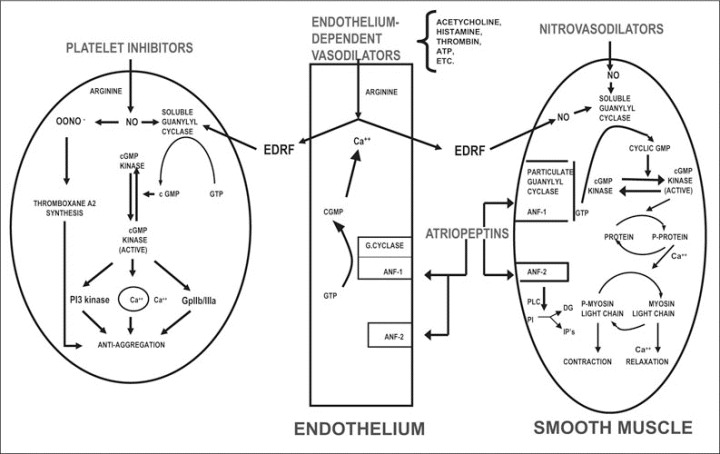
Effects of nitrovasodilators, endothelial‐dependent vasodilatators, and atriopeptins on cyclic guanosine monophosphate (cGMP) and vascular relaxation. GTP indicates guanosine‐5‐triphosphate; ANF, atrial natriuretic factor; EDRF, endothelium‐derived relaxing factor; ONOO−, peroxynitrite; NO, nitric oxide; PI3, phosphoinositide 3; Gp, glycoprotein; PLC, phospholipase; PI, phosphatidylinositol; DG, diacylglycerol; IP, inositol 3 or 4 phosphate.
Organic Nitrates
Organic nitrates (RONO2) are simple nitric and nitrous acid esters of alcohols. Clinically used, RONO2 compounds include GTN, pentaerythrityltetranitrate, isosorbide dinitrate, and triethanolamine trinitrate biphosphate. Organic nitrates are susceptible to alkaline hydrolysis but are generally stable in neutral or weakly acidic solution, one of the major differences with NONOate compounds. Medical usage as well as the biological effects of organic nitrates can be attributed to their intracellular conversion to NO2 − ions and then to NO, 13 which, in turn, activates guanylate cyclase and increases the cGMP content in cells. 14 , 15 Elevated cGMP through activation of cGMP‐dependent protein kinase (protein kinase G), protein phosphorylation, and regulation of cation channels, ultimately leads to dephosphorylation of the myosin light chain, resulting in smooth muscle relaxation. It has been suggested that multiple pathways contribute to NO formation from organic nitrates in vivo; however, the metabolic mechanism is poorly understood. Chen and colleagues 16 identified a nitrate reductase known as mitochondrial aldehyde dehydrogenase (mtALDH) that specifically converts GTN to 1,2‐glyceryl dinitrate and NO2 −. The NO2 − generated within the mitochondria is metabolized further to generate NO‐based bioactivity by reduction to NO and/or by conversion to S‐nitrosothiol (SNO). These studies suggested that mechanism‐based inactivation of mtALDH is involved in the development of GTN tolerance. Several medications such as chlorpropamide analogs, chloral hydrate, and acetaminophen can pharmacologically inhibit the activity of mtALDH. Thus, patients taking organic nitrates esters for the treatment of acute ischemic syndromes and congestive heart failure will benefit from awareness of certain drugs that inhibit mtALDH activity.
S‐nitrosothiols
As early as 1981, Murad's and Ignarro's groups demonstrated that the bioactivities of certain pharmacologic nitrogen oxide donors were attributed to reactions with cellular thiols. 17 There is now a large body of literature that implicates SNO as an intermediate in NO‐dependent and guanylyl cyclase‐independent signaling processes. SNO can be synthesized from the reaction between thiols and nitrous acid in extremely acidic conditions (pH <3). Reactive protein thiols are becoming regarded as major intracellular targets of NO. 15 , 16 , 17 , 18 Although RSNOs such as S‐nitroso‐N‐acetylpenicillamine, N‐acetyl‐S‐nitrosopenicillaminyl‐S‐nitrosopenicillamine, and S‐nitrosoglutathione are commercially available, none have been used therapeutically due to the unpredictable rate of decomposition in the body. In contrast, an increasing number of proteins have been found to undergo S‐nitrosylation in vivo. The study of S‐nitrosohemoglobin is an example through which the significance of the process may be understood. Hemoglobin (Hb) is a tetrameric red blood cell protein composed of two α‐ and two β‐chains, each containing a heme prosthetic group. The ability of Hb to reversibly bind O2 is governed by a cycle of allosteric transitions between the R (relaxed, high O2 affinity) and the T (tense, low O2 affinity) conformations. In addition to its well‐recognized function of O2/CO2 transport, the allosteric transition of Hb has been proposed to capture and deliver a third gas, NO. 18 Hb contains a highly conserved cysteine residue at position β93, which is reactive toward NO in the R state and relatively unreactive in the deoxygenated T state. The allosteric transition in Hb from T to R promotes the transfer of an NO group from Hb's hemes to its thiols, forming HbCysβ93NO (SNO‐Hb). SNO‐Hb is capable of transferring the NO group to cysteins of other peptides and proteins. For example, albumin is an abundant circulating protein that may serve as an effective reservoir of NO in its nitrosated form. 19 The angiotensin‐converting enzyme (ACE) inhibitor captopril is a reduced thiol that has been reported to act as an NO donor after being nitrosated. 20 Plasminogen activator is another popular medication with the ability to undergo nitrosation. 21
REACTIVE OXYGEN/NITROGEN SPECIES AND HYPERTENSION
Arterial hypertension represents one of the most common conditions associated with endothelial dysfunction. For instance, a chronic inflammatory process associated with NO production may lead to oxidative damage of the vascular endothelium, decreasing its function. The reaction of superoxide (produced by all aerobic cells) with NO generates peroxynitrite, a potent oxidizing agent, that can also cause biological oxidative stress. Increases in reactive oxygen and/or nitrogen species following inflammatory challenge have been demonstrated in different models of organ failure 22 and hypertension. 23 , 24
PROTEIN NITRATION AND ATHEROSCLEROSIS
Protein tyrosine nitration is a post‐translational modification of proteins. Nitrated proteins are continuously produced in cells by reactive oxygen and nitrogen species even under normal physiologic conditions. 5 During periods of inflammation, protein nitration is significantly increased and may contribute to the inflammatory process or toxicity. It is well accepted that protein tyrosine nitration could be a biomarker to evaluate the extent and perhaps the cause of tissue damage in cardiovascular diseases 25 because nitrated protein tyrosines have been detected in atherosclerosis by immunohistochemistry. 26 Bian and Murad 27 have shown that tyrosine nitration can also diminish a protein's effectiveness as a substrate for tyrosine kinases. It is now well known that oxidized low‐density lipoprotein (LDL) is of central importance in triggering atherosclerosis, as Leeuwenburgh and colleagues 28 proposed that reactive nitrogen intermediates promote LDL oxidation in atherosclerosis.
CARDIOVASCULAR SYSTEM: A MAJOR TARGET FOR PROTEIN NITRATION
The biological role of heme has been extensively reviewed. 29 The presence of heme and peroxides modulate vascular cell survival and oxidative stress adaptation, providing a possible mechanism for the evolution of atherosclerotic lesions. 30 We have tested the hypothesis that both free heme and iron play a key role in nitrotyrosine formation and further demonstrated that isolated heme and free metals are capable of tyrosine nitration in the presence of hydrogen peroxide and NO2 −. Our data show that hemoprotein‐rich tissues such as cardiac muscle are vulnerable to protein nitration in pathologic conditions characterized by the overproduction of H2O2 and NO2 − or NO. 31 Therefore, our studies offer important mechanisms that underlie the heme/iron catalyzed nitration. Further clarification of the behavior of the heme (iron) and reaction with NO2 −/H2O2 system is essential to the development of therapeutic methods that will focus on the prevention of oxidation and nitration.
CARDIOVASCULAR THERAPEUTIC AGENTS AND NO SIGNALING
β‐Blockers
β‐Blockers have historically been considered an effective and safe option for the initial treatment of hypertension. Evidence suggests that β‐blockers work at the vascular biology level to produce NO release. β‐Blockers differ in terms of their β receptor selectivity, intrinsic sympathomimetic activity, and benefit/risk in diabetes and insulin sensitivity. Nebivolol, the newest of the β‐blockers, is long‐acting and the most cardioselective β1‐blocker currently available. 32 Nebivolol‐induced endothelium‐dependent vasodilation associated with activation of the l‐arginine/NO pathway may confer benefits to patients. The combined mechanisms of β‐adrenoceptor antagonism and NO‐mediated vasodilation may potentiate the blood pressure‐lowering effect of this agent and confer a broader favorable metabolic profile, which may be clinically relevant for hypertensive patients. The antioxidant effects of nebivolol and its neutral or even favorable effects on both carbohydrate and lipid metabolism are well documented. 33 These effects consistently differentiate nebivolol from nonvasodilating β‐blockers such as atenolol, metoprolol, and bisoprolol. The risk for diabetes is lower, the metabolic effects are lower, and persons with diabetes who have clear NO dysfunction may have particular benefits from this agent. Therefore, third‐generation β‐blockers, such as labetolol, carvedilol, bucindolol, and nebivolol, vasodilate by different mechanisms, behaving differently than traditional β‐blockers and offering different benefits. Compared with conventional β‐blockers, vasodilating β‐blockers have beneficial hemodynamic effects including decreased pressure wave reflection from the periphery, leading to decreases in central aortic blood pressure. Larger trials are needed, however, to determine whether reduced central pressure will translate into improved cardiovascular outcomes compared with nonvasodilating β‐blockers. 34
ACE Inhibitors or AT1 Receptor Blockers
Several studies have shown that hypertension is associated with enhanced LDL oxidation and oxidative stress in the bloodstream and arterial tissue. 35 The reduction of exceeding oxidative stress after intervention with antihypertensive drugs is achieved in experimental models 35 , 36 and in hypertensive patients. 37 Among antihypertensive drugs, sulfhydryl ACE inhibitors reduce LDL oxidation, formation of oxidation‐specific epitopes in the arterial wall, and atherogenic lesion formation in experimental models. The increased level of oxidant stress (ie, generation of oxygen radicals such as superoxide anion) is involved in the expression of hypertension in genetic and experimental (eg, angiotensin II‐induced) forms of hypertension. 38 This increased oxidant stress promotes oxidation of cysteine residues (ie, enhanced disulfide bond formation) in functional proteins. The increased activity of the renin‐angiotensin system plays a role in the expression of hypertension. It is likely that increased activity of the renin‐angiotensin system elicits hypertension by generating oxidant stress since ACE inhibitors or angiotensin AT1 receptor blockers, normalize blood pressure and endothelium‐dependent vasodilation in hypertensive models, including spontaneously hypertensive rats 39 and reduce oxidant stress in these rats thereby improving the redox status of cysteine residues in proteins in cells and on cell membranes. The oxidative stress in patients with essential hypertension is improved by chronic administration of therapeutic doses of the sulfhydryl ACE inhibitor zofenopril but not with the nonsulfhydryl ACE inhibitor enalapril. This effect was coupled with an improvement of the NO/asymmetric dimethyl‐l‐arginine balance. Sulfhydryl ACE inhibition induces sustained reduction of systemic oxidative stress and improves the NO pathway in patients with essential hypertension. 40
In addition to exerting various beneficial actions on vascular structure and function beyond their blood pressure‐lowering effects, data from experimental studies show that ACE inhibitors can attenuate the development of atherosclerosis in a wide range of species. The postulated mechanisms of this atheroprotective effect are the antioxidant actions of ACE inhibitors and their enhancement of the endothelial elaboration of bioactive NO. Using bovine aortic endothelial cells in culture, Scribner and colleagues 40 assessed the comparative effects of 3 ACE inhibitors on endothelial NO production and action and endothelial oxidative stress. Results showed that the sulfhydryl ACE inhibitor zofenopril has unique effects compared with captopril and enalapril. This compound improves NO production and bioactivity and does so in conjunction with decreased endothelial cell oxidant stress. The biochemical basis for this protective mechanism is not entirely clear; however, these actions suggest that zofenopril may reduce endothelial effects of risk factors for atherothrombotic disease.
CONCLUSIONS
NO research has expanded rapidly in the past 20 years, and the role of NO in physiology and pathology have been extensively studied. The pathways of NO synthesis, signaling, and metabolism in vascular biological systems have been a major area of research resulting in the 1998 Nobel Prize in Medicine or Physiology. As a gas and free radical with an unshared electron, NO participates in various biological processes. The rapid growth of NO research has generated more than 80,000 publications in the field of NO signaling. The interaction between NO and proteins may be roughly divided into 2 categories. In many instances, NO mediates its biological effects by activating guanylyl cyclase and increases intracellular cGMP synthesis from guanosine‐5 ‐triphosphate. The list of cGMP‐independent effects of NO, however, is also growing at a rapid rate. In this review, a brief history of the medical usage of NO is introduced. The importance and relevance of overproduction of NO in cardiovascular pathology has been stressed. The utilization of intact cell cultures, tissues, and cell‐free preparations with the use of pharmacologic, biochemical, and molecular biological approaches to characterize, purify, and reconstitute these NO regulatory pathways should lead to the development of new therapies for various pathologic conditions characterized by an unbalanced production of NO.
Disclosures:
Financial support of our research was provided by the John S. Dunn Foundation, Harold and Leila Mathers, the Robert A. Welch Foundation (AU‐1437), the National Institutes of Health (GM061731), and the Department of Defense (T5).
References
- 1. Willerson JT, Kereiakes DJ. Endothelial dysfunction. Circulation. 2003;108:2060–2061. [DOI] [PubMed] [Google Scholar]
- 2. Katsuki S, Arnold W, Mittal C, et al. Stimulation of guanylate cyclase by sodium nitroprusside, nitroglycerin and nitric oxide in various tissue preparations and comparison to the effects of sodium azide and hydroxylamine. J Cyclic Nucleotide Res. 1977;3:23–35. [PubMed] [Google Scholar]
- 3. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. [DOI] [PubMed] [Google Scholar]
- 4. Bian K, Davis K, Kuret J, et al. Nitrotyrosine formation with endotoxin‐induced kidney injury detected by immunohistochemistry. Am J Physiol. 1999;277:F33–F40. [DOI] [PubMed] [Google Scholar]
- 5. Ishii N, Patel KP, Lane PH, et al. Nitric oxide synthesis and oxidative stress in the renal cortex of rats with diabetes mellitus. J Am Soc Nephrol. 2001;12:1630–1639. [DOI] [PubMed] [Google Scholar]
- 6. Beckman JS. Protein tyrosine nitration and peroxynitrite. FASEB J. 2002;16:1144. [DOI] [PubMed] [Google Scholar]
- 7. Bryan NS, Fernandez BO, Bauer SM, et al. Nitrite is a signaling molecule and regulator of gene expression in mammalian tissues. Nat Chem Biol. 2005;1(5):290–297. [DOI] [PubMed] [Google Scholar]
- 8. Lockette W, Otsuka Y, Carretero O. The loss of endothelium‐dependent vascular relaxation in hypertension. Hypertension. 1986;8:II61–II66. [DOI] [PubMed] [Google Scholar]
- 9. Calver A, Collier J, Vallance P. Inhibition and stimulation of nitric oxide synthesis in the human forearm arterial bed of patients with insulin‐dependent diabetes. J Clin Invest. 1992;90:2548–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Panza JA, Casino PR, Kilcoyne CM, et al. Role of endothelium‐derived nitric oxide in the abnormal endothelium‐dependent vascular relaxation of patients with essential hypertension. Circulation. 1993;87:1468–1474. [DOI] [PubMed] [Google Scholar]
- 11. Murad F. Cyclic guanosine monophosphate as a mediator of vasodilation. J Clin Invest. 1986;78:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marsh N, Marsh A. A short history of nitroglycerine and nitric oxide in pharmacology and physiology. Clin Exp Pharmacol Physiol. 2000;27:313–319. [DOI] [PubMed] [Google Scholar]
- 13. Ignarro LJ, Lippton H, Edwards JC, et al. Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: evidence for the involvement of S‐nitrosothiols as active intermediates. J Pharmacol Exp Ther. 1981;218:739–749. [PubMed] [Google Scholar]
- 14. Bennett BM, Schroder H, Hayward LD, et al. Effect of in vitro organic nitrate tolerance on relaxation, cyclic GMP accumulation, and guanylate cyclase activation by glyceryl trinitrate and the enantiomers of isoidide dinitrate. Circ Res. 1988;63:693–701. [DOI] [PubMed] [Google Scholar]
- 15. Papapetropoulos A, Go CY, Murad F, et al. Mechanisms of tolerance to sodium nitroprusside in rat cultured aortic smooth muscle cells. Br J Pharmacol. 1996;117:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci U S A. 2002;99:8306–8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ignarro LJ, Kadowitz PJ, Baricos WH. Evidence that regulation of hepatic guanylate cyclase activity involves interactions between catalytic site ‐SH groups and both substrate and activator. Arch Biochem Biophys. 1981;208:75–86. [DOI] [PubMed] [Google Scholar]
- 18. Patel RP. Biochemical aspects of the reaction of hemoglobin and NO: implications for Hb‐based blood substitutes. Free Radic Biol Med. 2000;28:1518–1525. [DOI] [PubMed] [Google Scholar]
- 19. Jourd'heuil D, Hallen K, Feelisch M, et al. Dynamic state of S‐nitrosothiols in human plasma and whole blood. Free Radic Biol Med. 2000;28:409–417. [DOI] [PubMed] [Google Scholar]
- 20. Jia L, Blantz RC. The effects of S‐nitrosocaptopril on renal filtration and blood pressure in rats. Eur J Pharmacol. 1998;354:33–41. [DOI] [PubMed] [Google Scholar]
- 21. Stamler JS, Simon DI, Jaraki O, et al. S‐nitrosylation of tissue‐type plasminogen activator confers vasodilatory and antiplatelet properties on the enzyme. Proc Natl Acad Sci U S A. 1992;89:8087–8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Motoyama T, Okamoto K, Kukita I, et al. Possible role of increased oxidant stress in multiple organ failure after systemic inflammatory response syndrome. Crit Care Med. 2003;31:1048–1052. [DOI] [PubMed] [Google Scholar]
- 23. Akiba Y, Yamaguchi N, Amano H, et al. Role of nitric oxide in the control of blood pressure in young and adult spontaneously hypertensive rats. Clin Exp Pharmacol Physiol Suppl. 1995;22:S142–S143. [DOI] [PubMed] [Google Scholar]
- 24. Gil‐Longo J, Fernandez‐Grandal D, Alvarez M, et al. Study of in vivo and in vitro resting vasodilator nitric oxide tone in normotensive and genetically hypertensive rats. Eur J Pharmacol. 1996;310:175–183. [DOI] [PubMed] [Google Scholar]
- 25. Turko IV, Murad F. Protein nitration in cardiovascular diseases. Pharmacol Rev. 2002;54:619–634. [DOI] [PubMed] [Google Scholar]
- 26. Beckmann JS, Ye YZ, Anderson PG, et al. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe Seyler. 1994;375:81–88. [DOI] [PubMed] [Google Scholar]
- 27. Bian K, Murad F. Nitric oxide (NO)‐biogeneration, regulation, and relevance to human diseases. Front Biosci. 2003;8:d264–d278. [DOI] [PubMed] [Google Scholar]
- 28. Leeuwenburgh C, Hardy MM, Hazen SL, et al. Reactive nitrogen intermediates promote low density lipoprotein oxidation in human atherosclerotic intima. J Biol Chem. 1997;272:1433–1436. [DOI] [PubMed] [Google Scholar]
- 29. Paoli M, Marles‐Wright J, Smith A. Structure‐function relationships in heme‐proteins. DNA Cell Biol. 2002;21:271–280. [DOI] [PubMed] [Google Scholar]
- 30. Asatryan L, Ziouzenkova O, Duncan R, et al. Heme and lipid peroxides in hemoglobin‐modified low‐density lipoprotein mediate cell survival and adaptation to oxidative stress. Blood. 2003;102:1732–1739. [DOI] [PubMed] [Google Scholar]
- 31. Bian K, Gao Z, Weisbrodt N, et al. The nature of heme/iron‐induced protein tyrosine nitration. Proc Natl Acad Sci U S A. 2003;100:5712–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reidenbach C, Schwinger RH, Steinritz D, et al. Nebivolol induces eNOS activation and NO‐liberation in murine corpus cavernosum. Life Sci. 2007;80:2421–2427. [DOI] [PubMed] [Google Scholar]
- 33. Pedersen ME, Cockcroft JR. The Vasodilatory Beta‐blockers. Curr Hypertens Rep. 2007;9:269–277. [DOI] [PubMed] [Google Scholar]
- 34. Agabiti Rosei E, Rizzoni D. Metabolic profile of nebivolol, a beta‐adrenoceptor antagonist with unique characteristics. Drugs. 2007;67:1097–1107. [DOI] [PubMed] [Google Scholar]
- 35. Napoli C, Sica V, De Nigris F, et al. Sulfhydryl angiotensin‐converting enzyme inhibition induces sustained reduction of systemic oxidative stress and improves the nitric oxide pathway in patients with essential hypertension. Am Heart J. 2004;148:e5. [DOI] [PubMed] [Google Scholar]
- 36. Shou I, Fukui M, Tomino Y. Efficacy of ACE inhibitor (captopril) on glomerular antioxidant enzyme activity and hypertension in diabetic hypertensive rats. Contrib Nephrol. 2001;134:74–78. [DOI] [PubMed] [Google Scholar]
- 37. Taddei S, Virdis A, Ghiadoni L, et al. Effect of calcium antagonist or beta blockade treatment on nitric oxide‐dependent vasodilation and oxidative stress in essential hypertensive patients. J Hypertens. 2001;19:1379–1386. [DOI] [PubMed] [Google Scholar]
- 38. Wilson SK. Role of oxygen‐derived free radicals in acute angiotensin II‐induced hypertensive vascular disease in the rat. Circ Res. 1990;66:722–734. [DOI] [PubMed] [Google Scholar]
- 39. Lewis SJ, Hashmi‐Hill MP, Owen JR, et al. ACE inhibition restores the vasodilator potency of the endothelium‐derived relaxing factor, L‐S‐nitrosocysteine, in conscious Spontaneously Hypertensive rats. Vascul Pharmacol. 2006;44:491–507. [DOI] [PubMed] [Google Scholar]
- 40. Scribner AW, Loscalzo J, Napoli C. The effect of angiotensin‐converting enzyme inhibition on endothelial function and oxidant stress. Eur J Pharmacol. 2003;482:95–99. [DOI] [PubMed] [Google Scholar]