

Abstract
Introduction:
Precision drug therapy requires accounting for pertinent factors in pharmacokinetic (PK) inter-individual variability (i.e., pharmacogenetics, diseases, polypharmacy, and natural product use) that can cause sub-therapeutic or adverse effects. Although each of these individual factors can alter victim drug PK, multi-factorial interactions can cause additive, synergistic, or opposing effects. Determining the magnitude and direction of these complex multi-factorial effects requires understanding the rate-limiting redundant and/or sequential PK processes for each drug.
Areas covered:
Perturbations in drug metabolizing enzymes and/or transporters are integral to single- and multi-factorial PK interactions. Examples of single factor PK interactions presented include gene-drug (pharmacogenetic), disease-drug, drug-drug, and natural product-drug interactions. Examples of multi-factorial PK interactions presented include drug-gene-drug, natural product-gene-drug, gene-gene-drug, disease-natural product-drug, and disease-gene-drug interactions. Clear interpretation of multi-factorial interactions can be complicated by study design, complexity in victim drug PK, and incomplete mechanistic understanding of victim drug PK.
Expert opinion:
Incorporation of complex multi-factorial PK interactions into precision drug therapy requires advances in clinical decision tools, intentional PK study designs, drug metabolizing enzyme and transporter fractional contribution determinations, systems and computational approaches (e.g., physiologically-based pharmacokinetic modeling), and PK phenotyping of progressive diseases.
Keywords: complex interactions, disease-drug interaction, drug-drug interaction, multi-factorial, natural product-drug interaction, pharmacogenetics, pharmacokinetics, precision medicine, transporter-mediated drug interaction
1.1. Introduction
Precision medicine is a rapidly expanding approach to healthcare that seeks to account for all pertinent factors affecting health, including inter-individual variability (i.e., genes, environment, and lifestyle), to maximize patient benefit and minimize patient harm. Recent advances in -omics technologies and mechanistic understanding of pharmacokinetic (PK) and pharmacodynamic (PD) mediators have broadened the application of precision medicine. In 2015, the National Institutes of Health began the most expansive precision medicine effort, the Precision Medicine Initiative, with a goal of characterizing factors that affect health, including those that contribute to altered PK and PD, by recruiting at least one million people living in the United States into the All of Us Research Program [1].
The major inter-individual variability factors that can affect PK and PD include genetics (i.e., pharmacogenetics), environment (i.e., diseases and polypharmacy), and lifestyle (i.e., diet and natural product use). Additionally, other factors such as age, sex, and other physiological differences are known to affect PK and PD, however, these factors are not the focus of this article and have been thoroughly reviewed elsewhere [2–5]. Extensive evidence has demonstrated that each factor alone can alter PK and PD (Figure 1A and sections 2.1–2.4), but individual factors have not been able to explain the entirety of inter-individual variability. As our understanding of individual factors grows, evidence is emerging of multi-factorial interactions involving the combination of two or more factors with, additive, synergistic, or opposing PK effects (Figure 1B) [6,7]. The magnitude and direction of the multi-factorial effects are context dependent based on the rate-limiting PK processes for each drug. These processes typically involve functional redundant and/or sequential pathways wherein more than one metabolic enzyme or transporter is capable of metabolizing or transporting the same drug (Figure 2). This review will focus on single- and multi-factorial PK interactions involving drug metabolizing enzymes and transporters integral to PK [8–10].
Figure 1:
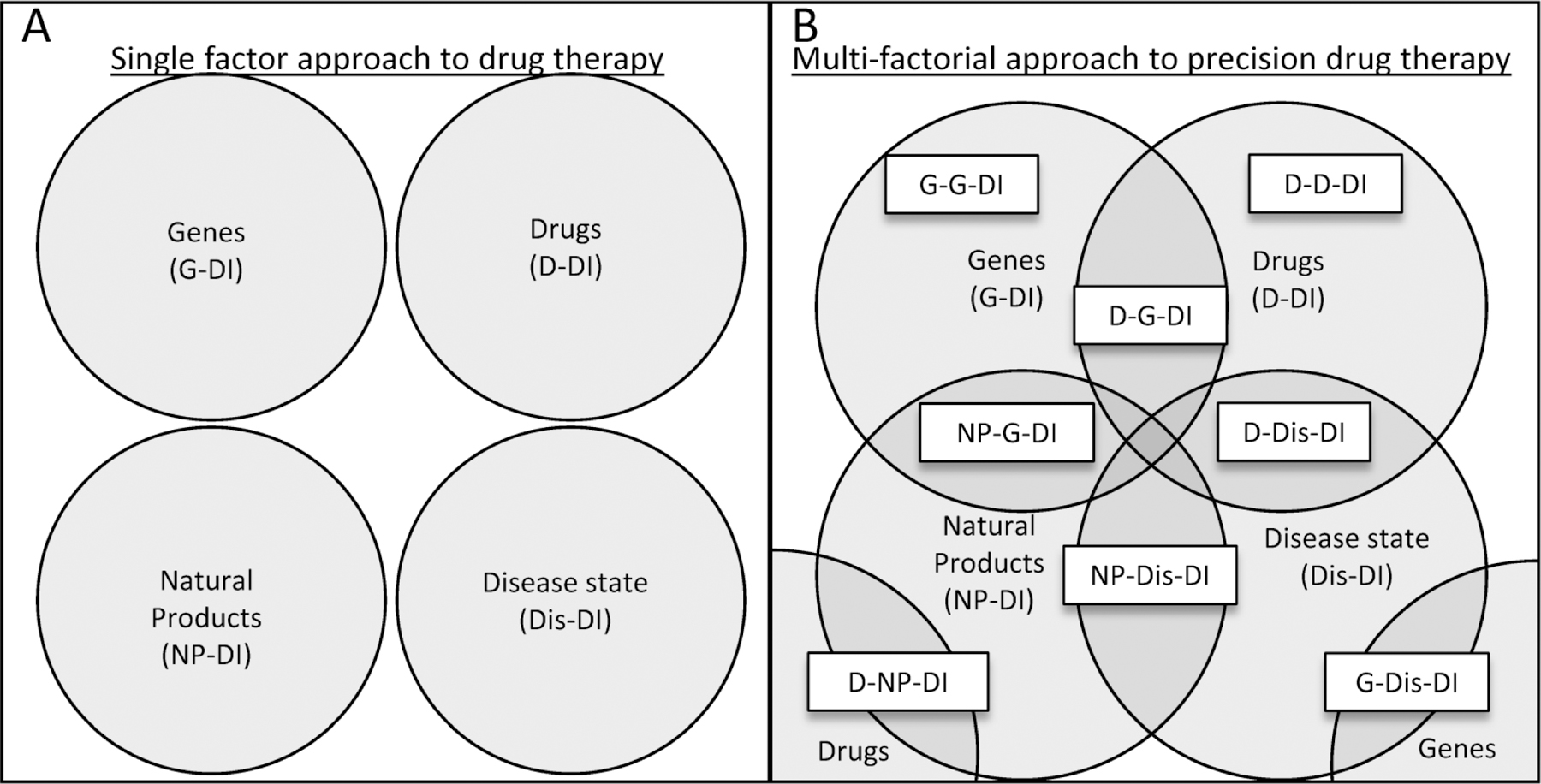
(A) Identification and testing of factors in inter-individual PK variability often take a single factor approach (i.e., genes, drugs, natural productNPs, and disease states). (B) A multi-factorial approach to precision drug therapy attempts to account for the sub-populations of patients who experience more complex PK interactions. The overlapping portions of the circles indicate all possible 2-, 3-, and 4-way interactions between factors. The 2-way interactions are shown in white textboxes at the intersection of two circles (except for G-G-DI and D-D-DI, which represent interactions between two polymorphisms in different genes and complex drug interactions, respectively). The relevance of each interaction will depend on the specific metabolizing enzymes or transporters involved and will be determined on a case-by-case basis.
Figure 2.
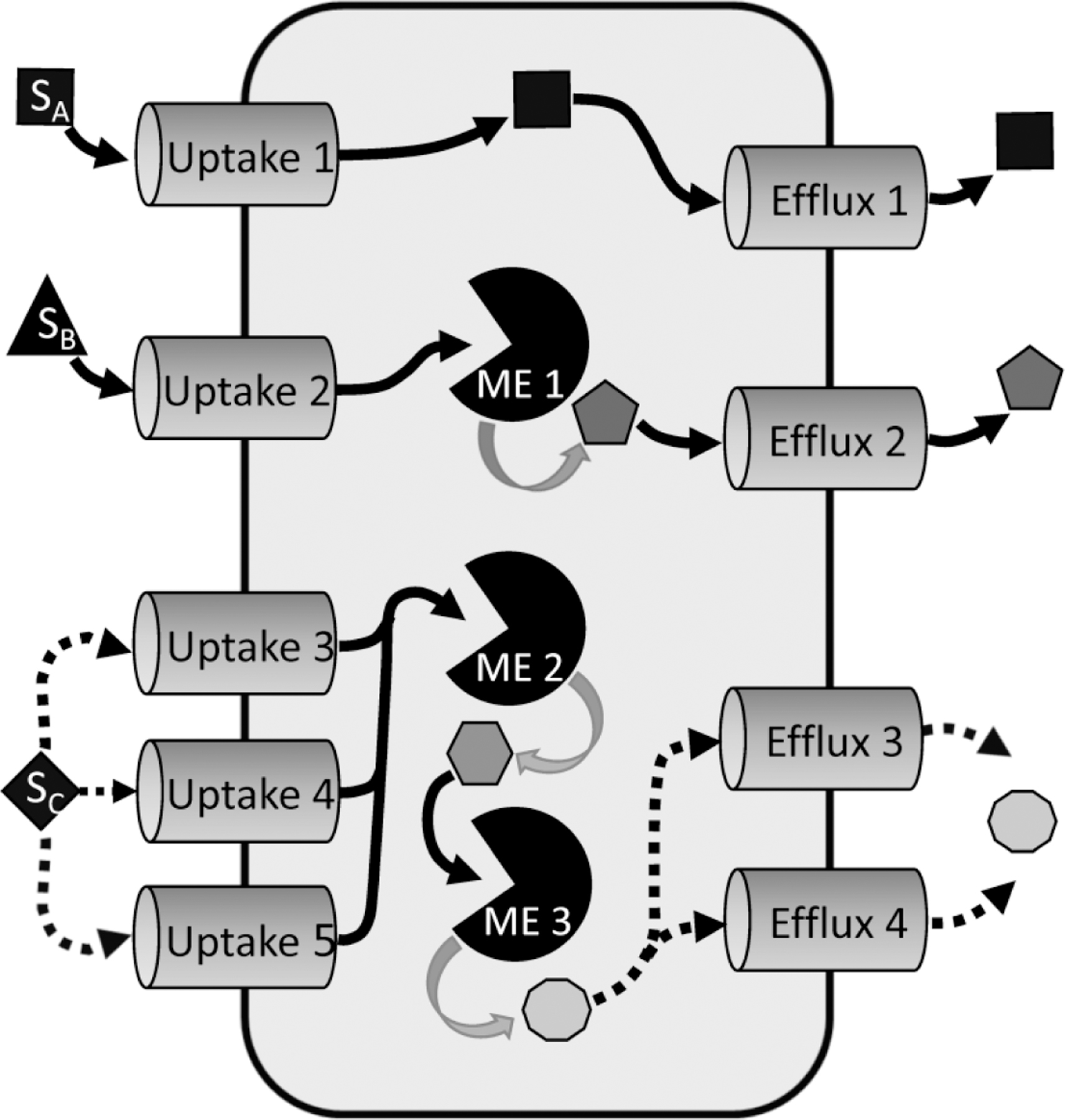
Functionally redundant and sequential PK processes are characterized by more than one PK mediator determining drug absorption, distribution, metabolism, and excretion. A drug substrate (SA) may enter a cell via a single uptake transporter (Uptake 1), then leave via a single efflux transporter (Efflux 1) without undergoing any metabolism. Another drug substrate (SB) may enter the cell via a single uptake transporter (Uptake 2), then undergo metabolism by a single metabolizing enzyme (ME 1) before export via a single efflux transporter (Efflux 2). A third drug substrate (SC) may enter the cell via functionally redundant uptake transporters (Uptake 3, Uptake 4, or Uptake 5), then undergo sequential metabolism (ME2 then ME3) before being exported from the cell via functionally redundant efflux transporters (Efflux 3 or Efflux 4). Only three scenarios are depicted, but any combination of redundant and/or sequential transport and/or metabolism is possible, and may include passive diffusion across the membrane.
To clarify what is meant by “interactions” and how they are written in this review, the perpetrating factors are always listed at the beginning of the interaction and are separated by hyphens. The “drug” listed last in the interactions always represents the victim drug. Rather than suggesting that the perpetrating factors influence each other, this review is focused on how single or multiple perpetrators affect a victim drug.
1.2. Intricacies of multi-factorial interactions
To understand the intricacies of multi-factorial PK interactions, two areas of complexity require further discussion: redundant/sequential PK processes and complex inter-individual variability factors.
Functionally redundant and/or sequential PK processes are characterized by more than one enzyme or transporter (PK mediators) that determine drug absorption, distribution, metabolism, and excretion (ADME) (Figure 2). For these PK processes, a factor changing only a single PK mediator will be less likely to alter the victim drug PK. In contrast, more complex effects that alter more than one PK mediator in a redundant or sequential process may have a greater impact on PK.
Complex inter-individual variability factors are characterized by a single factor that affects multiple PK mediators. For example, a genetic polymorphism typically only affects a single PK mediator, whereas disease states may decrease expression or function of multiple PK mediators. In addition, a drug-drug interaction (D-DI) precipitated by a single drug may inhibit a few PK mediators, whereas a natural product (NP) that is a complex mixture of compounds may inhibit the function of many PK mediators, although some single drugs can elicit complex interactions [11,12]. Therefore, diseases and natural products may have a greater impact on drug disposition due to their ability to affect multiple sites of drug ADME.
Understanding these complexities and appropriately accounting for each factor will maximize drug efficacy and minimize toxicity leading to improved overall patient outcomes. This article will review single- and multi-factorial PK interactions and provide an expert opinion on the future of precision drug therapy.
2. Single factor PK interactions
Table 1 summarizes examples of single factor PK interactions.
Table 1:
Single Factor PK Interactions
| Victim Drug | Drug Class | PK-mediators | Perpetrator | Mechanism | PK effect | Ref. |
|---|---|---|---|---|---|---|
| Midazolam | Benzodiazepine | CYP3A | Fluconazole | CYP3A inhibitor | ↑ AUC 200–300% | [20] |
| Atorvastatin | Statin | CYP3A4 OATP1B | Rifampin | OATP1B inhibitor | ↑ AUC 682% | [24] |
| GFJ | CYP3A inhibitor | ↑ AUC 250% (acid) | [143] | |||
| GFJ | CYP3A inhibitor | ↑ AUC 330% (lactone) | [143] | |||
| SLCO1B1 poly. | ↓ OATP1B1 function |
↑ AUC 52% ↑ AUC 144% (acid) |
[144] | |||
| Paritaprevir | Anti-viral | CYP3A OATP1B BCRP P-gp |
Ritonavir | CYP3A4 and P-gp inhibitor | ↑ AUC 4700% | [14] |
| Cyclosporine | Potential CYP3A4 and OAT1B inhibitor | ↑ AUC 72% | [145] | |||
| Tacrolimus | NA | ↓ AUC 43% | [145] | |||
| Carbamazepine | CYP3A4 inducer | ↓ AUC 70% | [146] | |||
| Simvastatin | Statin | CYP3A4 OATP1B1 | GFJ | CYP3A4 inhibitor | ↑ AUC 1600% | [36] |
| Aripiprazole | Antipsychotic | CYP2D6 CYP3A4 | CYP2D6 poly. | ↓ CYP2D6 activity | ↑ AUC 50% | [44] |
| Paroxetine | CYP2D6 inhibitor | ↑ AUC 140% | [147] | |||
| Fluvoxamine | CYP3A4 inhibitor | ↑ AUC 60% | [147] | |||
| Itraconazole | CYP3A4 inhibitor | ↑ AUC 48% | [148] | |||
| Voriconazole | Anti-fungal | CYP2C19 CYP2C9 CYP3A4 | CYP2C19 poly. | ↑ CYP2C19 activity | ↓ AUC 48% | [45] |
| Warfarin | Anticoagulant | CYP2C9 | CYP2C9 poly. | ↓ CYP2C9 activity | ↓ clearance 90% | [47] |
| 5-fluorouracil | Anti-cancer | DPD OAT2 |
DPYD poly. | ↓ DPD activity | Accumulation | [55] |
| Cimetidine | Unknown | ↑ AUC 72% | [149] | |||
| Diflomotecan | Anti-cancer | CYP3A BCRP |
ABCG2 poly. | ↓ BCRP expression | ↑ AUC 299% | [56] |
| Repaglinide | Anti-diabetic | OATP1B1 CYP2C8 | Rifampin | CYP3A4 inducer | ↑ AUC 57% | [80] |
Abbreviations and symbols: AUC- area under the plasma concentration-time curve; PK- pharmacokinetics; Poly.- polymorphism; Ref.- reference(s);
- increased;
- decreased.
2.1. Drug-drug interactions
D-DIs are a major concern in patients experiencing polypharmacy, the concurrent use of multiple drugs, accounting for nearly 30% of all reported adverse drug reactions [13]. Between 2013 and 2016, over 51% of the 103 drugs approved by the U.S. Food and Drug Administration (FDA) were victims of at least one D-DI, 14 of which produced a ≥ 5-fold increase in area under the plasma concentration time curve (AUC) [14]. Anti-viral and chemotherapeutic drugs are the most common groups of drugs to fall victim to D-DIs, and these patient populations are particularly prone to polypharmacy [14]. As the number of approved and repurposed drugs continues to grow, the number of potential interactions involving drug metabolizing enzymes and transporters will also continue to grow.
Metabolizing enzyme-mediated D-DIs are extensively documented [15–17]. The cytochrome P450 (CYP) superfamily of metabolizing enzymes have broad substrate specificity and are majors targets for D-DIs [18]. For example, the CYP3A family, which is expressed in the liver and intestine, is involved in an approximately two-thirds of D-DIs among recently approved drugs [14]. Several anti-fungal azole drugs are metabolized by and inhibit CYP3A enzymes at plasma concentrations below 1 μM [19], and co-administration of the benzodiazepine midazolam with fluconazole increased midazolam plasma AUC 2- to 3-fold, significantly increasing PD effects as observed by the digit symbol substitution test, critical flicker fusion test, and subjective drowsiness [20]. UDP-glucuronosyltransferases (UGTs) are also involved in D-DIs, however, these interactions are less common due to the smaller number of selective inhibitors and inducers, lower inhibitor affinity, and isoform functional redundancy [10,21]. Thus, UGT-mediated D-DIs rarely produce an AUCi/AUC ratio greater than 2-fold [10].
Transporter-mediated D-DIs can affect drug distribution and excretion [21]. The FDA and the International Transporter Consortium (ITC) have highlighted multiple important transporters including organic anion transporting polypeptides (OATPs), organic anion transporters (OATs), organic cation transporters (OCTs), breast cancer resistance protein (BCRP), p-glycoprotein (P-gp), and multidrug and toxin extrusion proteins (MATEs) [21–23]. Rifampin is a canonical inhibitor of hepatic OATP1B1 and OATP1B3 uptake transporters. Intravenous infusion of 600 mg rifampin along with oral administration of 40 mg atorvastatin to healthy volunteers increased atorvastatin parent and metabolite AUC0−∞ 682 ± 241% and 167 ± 73%, respectively [24]. While transporters have traditionally garnered less attention than metabolizing enzymes in the area of D-DIs, the efforts of the FDA and ITC as well as the increasing number of review articles focusing on transporter-mediated D-DIs illustrate an increased interest and relevance in the field [23,25–27].
The above D-DI examples describe interactions between a single perpetrating drug, a single victim drug, and specific enzymes or transporters, but the pathway through the body for many drugs is often more complex and requires careful consideration to understand the D-DI mechanism. As the drug enters the body and is distributed, it can be a substrate for multiple transporters before reaching its pharmacological site of action and/or site of metabolism. Likewise, the parent drug and its metabolites can be metabolized by multiple enzymes before undergoing efflux and excretion. In addition, D-DIs can be exploited to benefit the patient. An example of this complexity is illustrated by the D-DI between the anti-viral drugs paritaprevir and ritonavir. Ritonavir is an inhibitor of, while paritaprevir is a substrate for, multiple enzymes and transporters (e.g., CYP3A, OATP1B1, OATP1B3, BCRP and P-gp). Co-administration of these two drugs increased paritaprevir AUC (47-fold), Cmax, and trough concentrations, thereby improving treatment for chronic hepatitis C infection [14,28]. Another important consideration for D-DIs is the specific molecular mechanism, which can involve different types of inhibition (e.g., competitive, uncompetitive, mechanism-based) and/or gene induction. Assiduous attention to detail is important in patients experiencing polypharmacy because the complexity of some D-DIs can create risks for adverse drug interactions and/or provide opportunities for improved pharmacotherapy.
2.2. Natural product-drug interactions
Natural product-drug interactions (NP-DIs) pose a complex challenge to the advancement of precision medicine. Natural product use continues to increase in the United States, as evident by natural product herbal supplement sales increasing 8.6% to an estimated $9.6 billion in 2019 [29]. NP use also contributes to adverse drug events with over 23,000 estimated annual emergency department visits between 2004 and 2013 reported to be associated with NP use [30]. Robust NP-DI research is complicated by inconsistent product compositions between different brands of the same NP and between different lots of the same brand because of variability in how products are sourced, processed, and formulated [31]. In contrast to D-DIs, which usually involve one specific perpetrating compound, NPs are often a complex mixture of compounds potentially capable of inhibiting multiple PK mediators. The complex nature of NP-DIs precludes a comprehensive review of this area of precision medicine, but a brief example to illustrate the challenges in this area is outlined here.
Grapefruit juice (GFJ) perpetrates a set of well-characterized NP-DIs that involve multiple perpetrating compounds, victim drugs, and PK mediators. The main perpetrating constituents in GFJ are flavonoids and furanocoumarins (FCs), which inhibit intestinal CYPs [32–34]. Thus, GFJ increased AUC of multiple statins (e.g., simvastatin by ~1,610%, atorvastatin acid by ~250%, and atorvastatin lactone by ~330%) via CYP3A4 inhibition, and irreversibly inhibited intestinal CYP2B6 and CYP3A5 [6,34–37]. GFJ flavonoids and FCs also inhibit multiple transporters such as OATPs and P-gp [38]. However, unlike the irreversible inhibition seen with the aforementioned intestinal CYPs, transporter inhibition is reversible [38]. GFJ highlights the complexity of NP-DIs because each of the multiple constituents can affect drug disposition in different ways (e.g., FCs inhibit CYP3A4 while the flavonoids inhibit intestinal OATP-mediated uptake). Several review articles provide in-depth analysis of the complex nature of GFJ-mediated NP-DIs [33,39,40].
The complex nature of NP-DIs has created often perplexing and contradictory results in the scientific literature. The Center of Excellence of Natural Product Drug Interaction Research (NaPDI Center) was formed to address these complexities and devise a series of recommended approaches for NP-DI research [41–43]. Adherence to these, or similar, recommended approaches will improve the quality of NP-DI research and facilitate incorporation of NP-DIs into multi-factorial interactions.
2.3. Pharmacogenetic-drug interactions
Pharmacogenetics, or gene-drug interactions (G-DIs), are well-documented to influence efficacious and safe pharmacotherapy, and genetic testing is a cornerstone to precision medicine. Genetic polymorphisms that alter the expression and/or function of PK mediators can involve a single nucleotide polymorphism (SNP) or multiple nucleotide polymorphisms that form a polymorphic allele. These polymorphisms can be classified into different functional phenotypes for metabolizing enzymes (i.e., poor, intermediate, extensive, and ultrarapid metabolizers) and transporters (i.e., low, intermediate, and normal function). Implementation of pharmacogenetics often involves dose adjustments or medication changes, and has been observed to reduce the number of re-hospitalizations by 53% and emergency room visits by 42%[44]. Open-source databases cataloging the characteristics of several of these pharmacogenomic interactions are established, such as Pharmacogenomic Knowledge Base (PharmGKB) and the Pharmacogene Variation (PharmVar) Consortium for example. Additionally, the Clinical Pharmacogenetics Implementation Consortium (CPIC) compiles information and data to support relevant and updated clinical decision-making guidelines to aid clinicians on known G-DIs.
CYPs are highly polymorphic (e.g., CYP2D6 has over 100 variant alleles), and the number of clinically actionable CYP polymorphisms continues to grow [45]. CYP2D6, CYP2C9, and CYP2C19 represent the most common metabolizing enzymes with respect to polymorphic expression, thus are of greatest regulatory interest when assessing potential G-DIs [46]. For example, CYP2D6 polymorphisms are responsible for significant variations in exposure to the antipsychotic aripiprazole, where poor metabolizers saw a 50% increase in AUC compared to extensive metabolizers [47]. Such increases suggest a dose adjustment may be necessary for some populations, however the FDA does not currently mandate CYPD2D6 genotyping [47]. CYP2C19 polymorphisms are also known to affect exposure and efficacy of the anti-fungal voriconazole. Voriconazole AUC was 48% and 85% lower in healthy male volunteers expressing an ultra-rapid metabolizer phenotype compared to the extensive metabolizer and poor metabolizers, respectively [48]. This is particularly notable because trough concentrations of voriconazole < 1 mg/L are associated with sub-therapeutic effects, and may warrant dose adjustments or medication changes [48,49]. Another established G-DI involves CYP2C9 and the anticoagulant warfarin. Warfarin is administered in a racemic mixture comprising both the S- and R-enantiomers, with the S-enantiomer producing a 3- to 5-fold higher anticoagulant effect [50]. Elimination of the S-enantiomer is mediated primarily by CYP2C9. Therefore, CYP2C9 poor metabolizers are prone to increased systemic exposure, and can experience uncontrolled internal bleeding [50,51]. Comprehensive CYP-based G-DI reviews are available in the literature [52–55].
Some metabolizing enzyme polymorphisms affect specific classes of drugs. For example, the dihydropyrimidine dehydrogenase (DPYD) gene, which encodes for dihydropyrimidine dehydrogenase (DPD) and is responsible for the catabolism of the anti-cancer agent 5-fluorouracil (5-FU), has multiple variant alleles that decrease DPD activity [56]. 5-FU has serious dose-dependent toxicities that occur due to decreased DPD activity and increased 5-FU plasma accumulation [57]. These variants have three times greater prevalence in African populations compared to Caucasian populations [56,58]. The CPIC has established guidelines for 5-FU dose adjustments in patients expressing DPYD variant alleles [57].
Polymorphisms in uptake and efflux transporters can directly affect the absorption, distribution, and excretion of a victim drug, and may indirectly affect the metabolism of that drug when transport is the rate-limiting step of its elimination [8]. For example, the c.421C>A nonsynonymous SNP in ATP-binding cassette G2 (ABCG2), the gene encoding BCRP, significantly increased the exposure of the investigational anti-cancer agent diflomotecan by 299% in patients heterozygous for the SNP [59]. The underlying mechanism for the c.421C>A SNP dysfunction is believed to be a result of decreased protein expression [60,61]. The clinical importance of transporter polymorphisms on PK is expected to grow as functional characterization of polymorphic transporters expands [62–65].
G-DIs are distinct from D-DIs and NP-DIs because polymorphisms generally affect a single metabolizing enzyme or transporter, whereas D-DIs and NP-DIs have a greater potential to affect multiple ADME processes. Thus, drug interactions are most impactful for drugs that are dependent on a single drug metabolizing enzyme or transporter in the ADME of the drug. In contrast, drug substrates with broader selectivity may be impacted in multi-factorial PK interactions when the redundant mechanisms that compensate for the dysfunctional polymorphic protein are also perturbed. The importance of complex inter-individual variability factors is discussed further in the multi-factorial sections (see Sections 3.1–3.4).
2.4. Disease-drug interactions
Diseases of the major ADME organs (i.e., intestine, liver, and kidney) can alter PK through mechanisms involving organ structure and/or function. The liver will be the focus of this section to illustrate examples of disease-drug interactions (Dis-DIs). The FDA has issued a guidance document for assessing PK in patients with hepatic impairment and held a workshop in October to discuss ways to improve these assessments [66]. Conditions that elicit an immune response can alter CYP expression and activity. For example, interferons decreased CYP expression [67], and interleukin-6 production in the days following surgery decreased CYP3A4 activity 20–60% [68]. Other biological insults that elicit similar immune responses, such as autoimmune diseases, acute infections, and severe trauma, also decreased hepatic CYP3A4 activity [69]. Likewise, hepatic CYP3A activity decreased in patients with advanced malignancies, which is noteworthy considering the number of chemotherapeutic agents metabolized by the CYP3A family and the often narrow therapeutic window for these drugs [70]. In fact, altered CYP3A activity is believed to account for some of the interindividual variability in chemotherapy efficacy [70]. Dysfunction of other CYP enzymes have also been reported in chronic liver diseases [71,72].
Transporters are also subject to liver disease-mediated changes in function. For example, NTCP and OATP1B3 protein expression decreased in livers from nonalcoholic steatohepatitis (NASH) patients. In contrast, MRP2 protein expression increased in NASH [11], although MRP2 was shown to undergo a mislocalization event in NASH, resulting in reduced MRP2 function [73]. The changes in OATP1B3 and MRP2 function decreased 99mTc-mebrofenin uptake clearance from the blood to liver and clearance from liver to bile [74]. Protein expression of other efflux transporters such as MRP1, MRP3, P-gp, and BCRP also increased in NASH [73]. Hepatitis C (HCV) is also reported to affect hepatic transporter expression. Uptake transporters OCT1, OATP1B1, and OATP1B3, and efflux transporters MATE1, MDR1, MRP1, MRP2, MRP4, and BCRP all had elevated mRNA levels in cirrhotic HCV livers compared to healthy livers [75]. The increase in transporter transcription could be associated with the increased transcription of tumor necrosis factor-alpha (TNF-α) or possibly nuclear factor erythroid 2-related factor (Nrf2), both of which have been shown to affect transcription of transporters [76,77]. Interestingly, NASH and HCV are also reported to alter NP PK. For example, silymarin flavonolignan AUC and Cmax were increased in NASH and HCV patients [78,79]. A review of liver disease effects on hepatic transporter mRNA and protein expression can be found elsewhere [80].
Diseases of ADME organs frequently occur as co-morbidities and affect multiple steps in sequential and/or redundant PK processes. Although hepatic impairment was the focus of this section, impaired renal function can have similar effects on drug metabolism and transport and is the subject of a recent FDA guidance document [66]. As stated above, diseases often affect more than a single gene or protein. However, as noted by the ITC, broad specificity of potential victim drugs may mitigate the PK-mediated effects associated with diseases through redundant transport processes [81]. Thus, Dis-DIs may be most impactful in a multi-factorial PK interaction.
3. Multi-factorial PK interactions
Table 2 summarizes examples of multi-factorial PK interactions.
Table 2:
Multi-factorial PK Interactions
| Victim Drug | Drug Class | PK-mediators | Perpetrator | Mechanism | Single PK effect | Combined PK effect | Ref. |
|---|---|---|---|---|---|---|---|
| Repaglinide | Anti-diabetic | OATP1B1 CYP2C8 | SLCO1B1 poly. | ↓ OATP1B1 function | ↑ AUC 180% | ↑ AUC 1110% | 80 |
| Gemfibrozil | CYP2C8 inhibitor | ↑ AUC 820% | |||||
| Simvastatin | Statin | CYP3A4 OATP1B1 | Amlodipine | CYP3A4 inhibitor |
↑ AUC 80% ↑ AUC 40% (acid) |
↑ AUC 90% | 83 |
| SLCO1B1 poly. (hetero.) | ↓ OATP1B1 function | ↑ AUC 40% (acid) | |||||
| Pravastatin | Statin | OATP1B1 | SLCO1B1 poly. | ↓ OATP1B1 function | ↑ AUC 88%1 | ↑ AUC 531%1 | 91 |
| Cyclosporine | OATP1B1 inhibitor | ↑ AUC 500% | |||||
| CP-I | Endogenous biomarker | OATP1B1 | Cyclosporine2 | OATP1B1 inhibitor | ↑ AUC 171% | ↑ AUC 400%1 | 91 |
| SLCO1B1 poly.2 | ↓ OATP1B1 function | ↑ AUC 158%1 | |||||
| Rosuvastatin | Statin | OATP1B1 CYP2C9 | Baicalin | ↑ OATP1B1 function | ↓ AUC 42% | NS | 98 |
| SLCO1B1 poly.3 | ↓OATP1B1 function | ↑ AUC 45%1 | |||||
| Fexofendaine | Antihistamine | OATP2B1 | Apple juice | OATP2B1 inhibitor | ↓ AUC 20% | ↓ AUC 83% | 102 |
| SLCO2B1 poly. | ↓ OATP2B1 function | ↓ AUC 37% |
Abbreviations and symbols: AUC- area under the plasma concentration-time curve; CP-I- coproporphyrin-I; Hetero.- heterozygous; NS- not significant; PK- pharmacokinetics; Poly.- polymorphism; Ref.- reference(s);
- increased;
- decreased.
Calculation based on published data without determination of statistical significance.
Pravastatin was also administered to the subjects in this study.
Comparison of homozygous SLCO1B1*1b and homozygous SLCO1B1*15.
3.1. Drug-gene-drug interaction and natural product-gene-drug interaction
Multi-factorial interactions between exogenous compounds (e.g., drugs or natural products) and genetic polymorphisms have several layers of complexity that determine clinical effects. Central to this complexity is the sequential and/or redundant nature of the specific PK processes for the victim drug. Several examples of multi-factorial effects on sequential or redundant PK processes are reviewed in this section. In addition, the polymorphism effect on intrinsic clearance (i.e., Vmax/Km) and/or inhibition kinetics (i.e., IC50 or Ki) can influence the clinical multi-factorial effect. The details for each victim drug, polymorphism, and perpetrating compound(s) should be considered to determine the risk of these multi-factorial interactions.
Sequential PK processes involving the combination of transporter polymorphisms and D-DIs can alter victim drug plasma concentrations greater than each factor alone through a drug-gene-drug interaction (D-G-DI). For example, repaglinide is a substrate for OATP1B1 and CYP2C8. The SLCO1B1 c.521T>C SNP encoding the SLCO1B1*5 polymorphism (homozygous) increased repaglinide plasma AUC 1.8-fold, while inhibition of CYP2C8 via gemfibrozil increased repaglinide plasma AUC 8.2-fold [82]. The combination of SLCO1B1*5 (homozygous) and gemfibrozil-mediated CYP2C8 inhibition increased repaglinide plasma AUC 11.1-fold [82]. These data suggest that patients with the SLCO1B1*5 allele who experience CYP2C8 inhibition will have an elevated risk of increased exposure to repaglinide, potentially compromising glycemic control [82].
Another example of a sequential D-G-DI involves CYP3A4 inhibition and OATP1B1 and P-gp polymorphisms on the disposition of simvastatin. Simvastatin is metabolized into its active metabolite, simvastatin acid, by CYP3A4, which undergoes hepatic uptake via OATP1B1 [83,84]. Pre-treatment with amlodipine alone, a weak CYP3A4 inhibitor, increased simvastatin and simvastatin acid AUC 80% and 40%, respectively [85]. Subjects heterozygous for the SLCO1B1*5 allele had a 40% increase in simvastatin acid AUC [85]. The combination of amlodipine and heterozygous SLCO1B1*5 caused a 90% increase in simvastatin acid AUC relative to control groups homozygous for the reference SLCO1B1 allele [85]. No combined effect was observed in the presence of the MDR1 c.1236T>C, 2677G>T(A), or 3435C>T polymorphisms, suggesting P-gp is not as integral to simvastatin acid disposition, however conflicting data exist [85,86]. Perturbations in simvastatin PK may cause elevated levels of simvastatin acid leading to myopathy and potential termination of statin treatment, increasing the risk of cardiovascular disease [86–90].
Redundant PK processes involving the combination of transporter polymorphisms and D-DIs can have divergent effects based on substrate specificity. For example, hexadecanedioate (HDA), tetradecanedioate (TDA), coproporphyrin I (CP-I), and coproporphyrin III (CP-III) are considered endogenous biomarkers of hepatic OATP1B1/OATP1B3 function [91,92]. Polymorphic SLCO1B1 increased plasma AUC of all four substrates, but compounded PK effects were only observed for CP-I and CP-III in homozygous SLCO1B1*5 subjects [93]. The lack of combined effects of the polymorphism and the D-DI may be due to the fact that HDA and TDA are also substrates of OAT1 and OAT3, whereas CP-I and CP-III are primarily substrates of hepatic OATPs [94]. Another study co-administrated fevipiprant with either rosuvastatin or simvastatin in patients expressing the SLCO1B1*5 allele, and reported an additive effect only for simvastatin acid Cmax [95]. This may be explained by the fact that rosuvastatin is a substrate for multiple transporters, including NTCP, whereas simvastatin acid is more dependent on OATP1B1 for hepatic uptake [88]. Importantly, both studies described here suffered from small sample sizes for the individuals carrying the polymorphism and require further investigation to confirm the results.
Physiologically based pharmacokinetic (PBPK) modeling has been used to evaluate multi-factorial PK interactions. For example, the modeling of pitavastatin and atorvastatin exposure in the context of SCLO1B1*5 polymorphisms in combination with multiple known OATP inhibitors (itraconazole, erythromycin, and gemfibrozil) provided encouraging results [96]. However, the model continued to under-predict the statin exposure even after adjustments to scaling factors were applied [96]. It is believed that the under-predictions were due, in part, to inconsistencies in the inhibition kinetic data between in vitro and in vivo systems [96]. The authors noted similar under-predictions reported by other groups modeling similar systems [96–98]. The reduction in Ki values of the inhibitors corrected some of the prediction, but highlights the need for quality in vivo data to validate PBPK models [96].
Some polymorphisms reduce the effect of the perpetrating drug or NP on victim drug PK. An example of this is the interaction between baicalin and rosuvastatin. Hyperbilirubinemia occurs in individuals suffering from total or substantial loss of OATP1B1 and OATP1B3 function, a rare disease known as Rotor’s Syndrome [99]. The NP baicalin can partially rescue SLCO1B1 polymorphic transporter dysfunction by decreasing bilirubin levels, suggesting potential induction of OATPs by baicalin [100]. A PK NP-gene-drug interaction (NP-G-DI) study investigated the effect of baicalin on rosuvastatin disposition in subjects with SLCO1B1 polymorphisms. SLCO1B1*15/*15 subjects had a modest increase in rosuvastatin AUC0−∞ compared to SLCO1B1*1b/*1b subjects. Administration of 50 mg baicalin three times daily for 14 days reduced rosuvastatin AUC0−∞ in SLCO1B1*1b/*1b subjects by 41.9% and SLCO1B1*1b/*15 subjects by 23.9%, whereas baicalin did not affect rosuvastatin AUC0−∞ in SLCO1B1*15/*15 subjects [100]. The authors suggest hepatic OATP induction may explain reduced rosuvastatin AUC, although no data to support this claim were presented. Alternatively, baicalin inhibits OATP2B1, which could reduce rosuvastatin AUC after baicalin administration [101]. The mechanism for reduced baicalin effect in subjects carrying one or two copies of SLCO1B1*15 is unclear and requires further investigation. A limitation to this study is the absence of reference allele participants (SLCO1B1*1a/*1a). This study demonstrates the importance of identifying the perpetrator target transporter(s), the major PK processes for the victim drug, and where the two intersect in order to clearly characterize the multi-factorial interaction.
As with hepatic PK mediators, intestinal metabolizing enzymes and transporters are also susceptible to multi-factorial NP-G-DIs involving fruit juices, such as grapefruit, orange, and apple [102]. For example, co-administration of apple juice with fexofenadine reduced fexofenadine AUC and increased t1/2 in all SLCO2B1 genotypes, although to a slightly lesser extent in individuals with the c.1457C>T allele, suggesting reduced inhibitory effect in the presence of the polymorphism and greater dependence on OATP2B1 for fexofenadine uptake [103]. In contrast, apple juice decreased atenolol AUC in a dose-dependent manner, but the SLCO2B1 c.1457C>T polymorphism had no effect, suggesting that an OATP2B1 redundant transporter may be involved in atenolol uptake in the intestine [104]. Inconsistent genotype-dependent effects emphasize the importance of substrate specificity and redundant processes with respect to these intestinal transporters.
An additional layer of complexity to potential multi-factorial interactions between exogenous compounds and polymorphisms is altered perpetrator inhibition kinetics for polymorphic alleles. Polymorphisms in CYP2D6 and CYP3A4 increased or decreased IC50 values for several inhibitors in a polymorphism specific manner. For example, CYP2D6*2 decreased terbinafine IC50 for the substrate venlafaxine (607 nM to 93 nM) [105]. Decreased IC50 values will increase the risk of an NP-DI or D-DI, while increased IC50 values will decrease the risk of an NP-DI or D-DI. These changes in CYP IC50 values are relevant because a retrospective analysis of 1,143 individuals reported 217 individuals (19%) may experience a multi-factorial D-G-DI involving CYP2C9, CYP2C19, or CYP2D6 [106]. More research is needed to determine how these changes in inhibition kinetics effect multi-factorial PK interactions.
These data highlight the complexity of metabolizing enzyme and transporter specificities, and the relevance of redundant and/or sequential uptake, metabolism, and efflux processes. To account for NP use in precision drug therapy, clinicians first need to accurately capture the quantity and content of NP consumption, which requires full patient disclosure and accurate product characterization. Additionally, this requires an understanding of which NPs are potentially clinically relevant for the multitude of ADME processes of the drugs taken by the patient. Application of pharmacogenomic testing, especially in polypharmacy populations, as part of the clinical decision making process has been shown to decrease rehospitalization [107]. Therefore, by expanding pharmacogenetic testing and increasing our understanding of these discrete PK processes, better clinical decisions can be made to maximize precision drug therapy.
3.2. Gene-gene-drug interactions
Genetic polymorphisms in two or more drug metabolizing enzymes or transporters occur in a predictable manner based on allele copy number frequencies. These complex genetic interactions differ from other multi-factorial interactions because these will never involve a ‘double hit’ on a single rate-limiting PK mediator, whereas the combination of a natural product inhibitor and a polymorphism could impact the same metabolizing enzyme or transporter. Thus, gene-gene-drug interactions (G-G-DIs) will always involve sequential and/or redundant PK processes.
An example of such an interaction is SN-38, the active anti-cancer metabolite of the prodrug irinotecan, which is a sequential substrate of OATP1B1 and UGT1A1 (metabolism is an inactivation step) [108]. SLCO1B1 and UGT1A1 polymorphisms create an additive PK effect, increasing plasma SN-38 and causing severe toxicity [109–112]. Notably, individuals heterozygous for SLCO1B1*5 and UGT1A1*28 had toxicity risks similar to patients that are homozygous for either polymorphism, and patients with two or more SLCO1B1*5 and UGT1A1*28 polymorphisms had an odds ratio of 4.15 for grade 3/4 neutropenia compared to reference patients [109]. Another study found that these polymorphisms were associated with severe irinotecan toxicity, which necessitated termination of cancer treatment. These data demonstrate that additive PK effects can have a deleterious effect on patient outcomes [110]. The parent drug, irinotecan, is hydrolyzed by carboxylesterases (primarily CES2) into its metabolite SN-38, which is 100–1000-fold more toxic than the parent drug [108]. While CES2 polymorphisms have been investigated, data suggests the polymorphic effect on irinotecan metabolism is not significant [113–115].
The anticonvulsant phenytoin exhibits a small therapeutic index with substantial inter-individual variability requiring dose monitoring to avoid serious toxicities. Phenytoin is primarily metabolized by CYP2C9 and CYP2C19 and is a substrate for P-gp, therefore involving both redundant and sequential PK processes. In one study CYP2C9*3 and MDR1 c.3435C>T polymorphisms were associated with altered phenytoin plasma concentrations, but CYP2C19*2 had no effect [116]. CYP2C19*3, which increased phenytoin exposure in another study, was not tested because it was not present in the study participants [116]. Phenytoin plasma concentrations were associated with the number of polymorphic alleles (i.e., homozygous reference < heterozygous < homozygous polymorphic). Likewise, phenytoin metabolite to parent ratio was associated with the number of polymorphic alleles (i.e., homozygous reference: 1.83 ± 0.67, heterozygous: 1.34 ± 0.38, and homozygous: 0.76 ± 0.48 polymorphic) [116]. Inclusion of both CYP2C9*3 and MDR1 c.3435C>T alleles improved prediction of phenytoin plasma concentration variability from 9.2% to 15.4% [116].
One limitation of these studies is the inability to genotype and account for every metabolizing enzyme and transporter involved in the victim drug’s disposition, thereby disregarding how the genotypes of redundant processes contributed to the observed changes in drug disposition. Genetic testing is already a mainstay in precision drug therapy, and will continue to play a major role as our knowledge of important polymorphisms grows and patient genotyping becomes more common. In addition, as genomic data continue to become more affordable and accessible, retrospective investigation of potential G-G-DIs will become more practical. Access to massive datasets, such as the All of Us research program, will facilitate this process and open research opportunities. In addition, continued refinement of mechanistic PK processes will allow for hypothesis testing for specific sequential and/or redundant PK processes.
3.3. Disease-natural product-drug interactions
NPs are marketed with structure-function claims that target specific disease populations, suggesting that patients with diseases may experience disease-NP-drug interactions (Dis-NP-DIs). Unfortunately, most NP-DI studies are completed in healthy volunteers because designing and executing NP-DI studies involving patients with a specific disease can be logistically and ethically challenging [117]. Thus, there are limited clinical data for these multi-factorial PK interactions. As an alternative to clinical studies, animal models can recapitulate certain diseases, however, not every disease condition is able to be replicated in animal models, and interspecies variability in transporters and metabolizing enzymes is a significant limitation when translating findings to clinical populations. Nonetheless, animal models can provide valuable PK interaction data if the preclinical models are selected judiciously and the data are interpreted and translated with caution. As covered in Sections 2.2 and 2.4, disease states can alter the structure and/or function of ADME organs, and NPs are complex mixtures of potential NP-DI perpetrators. Thus, the combination of a disease with a NP can potentially have a multi-factorial PK effect.
Multi-factorial PK interactions can also have opposing effects, wherein the effect of each factor is negated when the factors are combined. An example of this is illustrated by the individual and combined effects of Nisha Amalaki (NA), which is a formulation of Curcuma longa and Phyllanthus emblica, and streptozotocin-induced diabetes on metformin PK in Wistar rats. The NP-DI between NA and metformin caused a significant increase in metformin Cmax (70.0%), tmax (43.8%), and AUC (53.0%) in healthy animals. The authors attributed the increased metformin systemic exposure to inhibition of renal Oct2 [117]. Diabetes alone increased metformin Cmax (164%), tmax (67.8%), and AUC (60.3%) compared to healthy rats [117]. The authors speculated that the diabetes-associated increase in metformin systemic exposure was due to altered hepatic CYP2C11 metabolism, however, metformin is minimally metabolized and this is unlikely to explain the increase in exposure. Rather, various streptozotocin doses and treatment times are reported to alter intestinal, hepatic, and renal transporter expression [118–120]. There are two probable transporter changes that could explain increased metformin exposure in the diabetic group; either decreased renal and/or hepatic uptake transporters, or increased intestinal uptake transporters. Interestingly, the combination of NA and diabetes decreased metformin Cmax (62.1%) and AUC (42.7%) when compared to diabetic rats receiving only metformin, and were comparable to metformin Cmax and AUC in healthy rats [117]. The first probable scenario for the effect of diabetes described above is not supported by the effect of NA on metformin in diabetes; rather, induction of an intestinal transporter that is susceptible to NA inhibition could explain the combined effect of NA and diabetes on metformin exposure. Unfortunately, these mechanisms were not explored, and more research is needed for this multi-factorial interaction in patient populations. This example illustrates the complexity and sometimes counteractive nature of multi-factorial PK interactions involving NPs.
The multi-factorial effect of silymarin and NASH on pitavastatin disposition illustrates a potential additive Dis-NP-DI. Pitavastatin plasma concentrations are heavily dependent on hepatic OATP1B1/OATP1B3 transporters. NASH is known to affect the expression and/or glycosylation of OATP1B1 and OATP1B3 in patients and Oatp1b2 in rats [11,12], and clinically relevant silymarin flavonolignan concentrations are known to inhibit OATP activity [121,122]. In this study, the combination of silymarin and methionine and choline deficient diet-induced NASH altered intravenous pitavastatin PK in rats [12]. According to a two-way ANOVA test, NASH and silymarin had significant effects on pitavastatin AUC0–120min without having a significant interaction effect. The latter effect suggests an additive interaction between NASH and silymarin [12]. Pitavastatin is a widely prescribed drug to reduce the risk of cardiovascular disease and it is used as a probe drug to represent a larger class of drugs dependent on hepatic OATP1B1/OATP1B3 uptake. Thus, these data demonstrate a potential risk of a hepatic OATP-mediated Dis-NP-DI, although more research is needed to demonstrate this interaction in patient populations.
Disease populations experience polypharmacy and are more likely to take one or more dietary supplement [123,124]. A proactive approach needs to be taken to identify potential at-risk populations to ensure proper monitoring of medications and NP use and avoid Dis-NP-DIs. More research is needed to define how the multi-factorial mechanisms combine to elicit additive, synergistic, or opposing PK effects.
3.4. Disease-gene-drug interactions
Multi-factorial disease-gene-drug interactions (Dis-G-DIs) can complicate precision drug therapy. As previously discussed, a polymorphism in a single PK mediator can alter victim drug disposition, whereas a disease can impact multiple proteins involved in PK. In addition, diseases are typically progressive, and often have heterogeneity in PK mediator function across and within disease stages [125]. Thus, the combination of these two factors can have divergent effects depending on the different PK mediators involved in each step of victim drug PK.
Multiple polymorphic CYPs exhibited a genotype-driven modulation in in vitro substrate kinetics. A distinct genotype effect was observed to alter many, but not all, substrate kinetic parameters (Km, Vmax, and CLint) in both healthy and hepatocellular carcinoma (HCC) human liver microsomes [126]. Although no statistical comparison was attempted between the healthy and HCC groups, examination of the data suggests increased CYP2D6 reference allele Vmax in the HCC samples compared to healthy samples of the same genotype. CYP2D6*10 appeared to counteract the HCC-associated increase in Vmax because the polymorphism decreased Vmax only in the HCC samples. Further statistical tests are required to test this hypothesis. In addition, this study did not provide clinical data and noted inconsistencies with previously published data, suggesting a lack of uniformity in the selection criteria for the HCC patients [126].
Dis-D-DIs can involve transporters in redundant and/or sequential processes. For example, the combination of methionine and choline deficient diet-induced NASH and Slco1b2 knockout in mice produced a synergistic increase in pravastatin plasma AUC, potentially due to changes in redundant transporters [127]. In this study, NASH alone did not alter pravastatin PK, while Slco1b2 knockout had a modest effect. A potential mechanism for the synergistic interaction is the combined effects of genetic loss of a primary transporter along with downregulation of multiple redundant hepatic Oatp transporters. A similar study was performed in healthy versus NASH patients with or without the SLCO1B1*15 polymorphism. NASH increased 99mTc mebrofenin AUC in the blood and the liver potentially due to decreased expression or function of the sequential hepatic transporters OATP1B3 and MRP2 [74]. Hepatic uptake clearance was progressively lower from healthy subjects with normal OATP1B1 function to healthy subjects with intermediate/low OATP1B1 function to NASH subjects with normal OATP1B1 function. Unfortunately, this study was not able to confirm the synergistic effect observed in rodents due to insufficient NASH subjects with intermediate/low OATP1B1 function. Another example of redundant transporter processes in Dis-G-DIs involves the combination of methionine and choline deficient diet-induced NASH and Bcrp knockout on SN-38 PK. SN-38 is the active metabolite of the anti-cancer agent irinotecan, and is an established substrate of the hepatic efflux transporter BCRP [128]. However, SN-38 biliary AUC did not change in Bcrp knockout animals, suggesting a redundant canalicular efflux transporter contributes to SN-38 biliary elimination [129]. Although Bcrp expression increased in NASH, Bcrp wild-type NASH rodents had no change in SN-38 biliary disposition [129]. Interestingly, although each factor in isolation did not change SN-38 disposition, the combination of the Bcrp knockout and NASH significantly decreased SN-38 biliary efflux by 68.1% [129]. These data illustrate the complex nature of Dis-G-DIs, and emphasize the importance of understanding all transporters involved in each step of a specific drug’s disposition.
The complexity of Dis-G-DIs is highlighted by the multiple examples provided. As illustrated, the combination of two factors, that may or may not have clinical impact in isolation, can cause significant changes in victim drug PK. Another challenge for these complex interactions is accurately categorizing these multi-factorial patient populations. Identifying genetic polymorphisms is becoming more affordable and common, but disease diagnosis and staging can be imprecise, although diagnostic methods continue to improve. Consideration of liver or renal impairment has been included in precision drug therapy for many years and will continue to be integral when accounting for inter-individual variability in PK and improving patient outcomes. Finally, it is important to note that polymorphisms in metabolizing enzymes and transporters increase susceptibility or severity of liver disease, thus potentially placing these specific disease populations at a unique risk for exacerbated or idiosyncratic liver toxicity [130–132].
4. Expert opinion
Incorporation of multi-factorial PK interactions into precision drug therapy requires better mechanistic understanding of redundant and sequential PK processes, deconvolution of complex factors, characterization of combined effects, and advancing methodological and clinical approaches (Figure 3).
Figure 3.
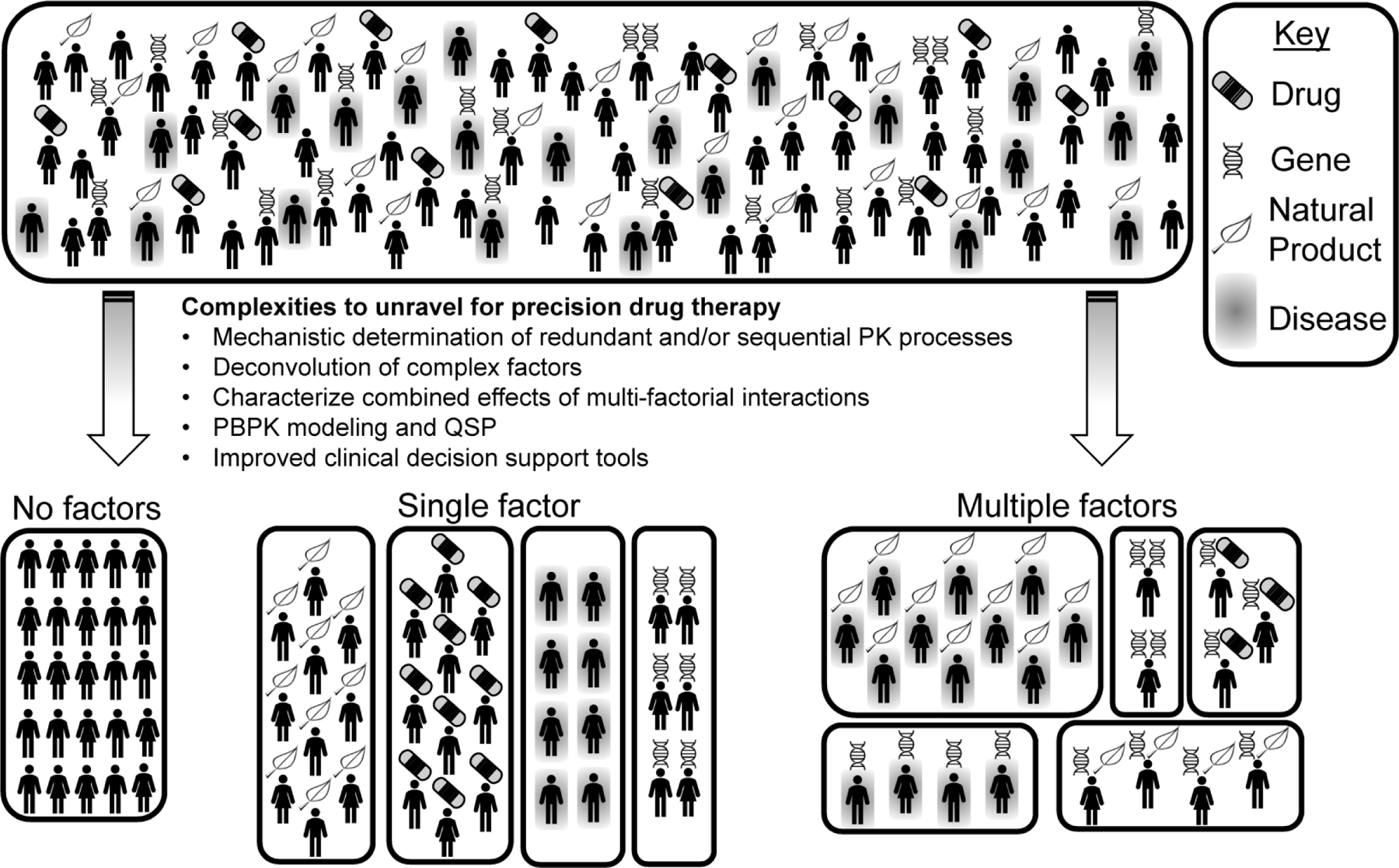
A hypothetical population of patients is depicted (top). Unraveling the complexities involved in pharmacotherapy such as accounting for redundant and sequential PK processes, deconvoluting complex factors, characterizing combined effects, improving physiologically-based pharmacokinetic (PBPK) modeling and quantitative systems pharmacology (QSP), determining the combined effects of multiple factor, and utilizing advanced clinical decision support tools will facilitate division of patients into unique sub-populations (bottom) and administration of precision drug therapy.
4.1. Mechanistic determination of redundant and/or sequential PK processes
Knowing the substrate specificity and fractional contribution of each metabolizing enzyme or transporter to rate-limiting PK processes is central to predicting complex PK interactions. Fraction metabolized (fm) for drug metabolizing enzymes and fraction transported (ft), and relative activity factors (RAF) or relative expression factors (REF) for transporters are important fractional contribution metrics. Critical areas for continued improvement to these metrics include expansion of specific inhibitors and substrates, advances in in vitro models (e.g., sandwich cultured hepatocytes and microfluidics cultures), and advances in protein quantification methods [133–135]. A limitation to RAF calculations include not accounting for changes in protein expression, which can be influenced by polymorphisms and disease. In contrast, REF calculations do account for relative protein expression, but have a limitation of not distinguishing between functioning and non-functioning transporters due to technical limitations of protein quantification. Both approaches provide useful data in select contexts, but neither alone are universally accepted for determining relative contribution [135]. Therefore, prudence should be exercised when selecting the appropriate method.
Protein quantification for transporters is further complicated by functional regulation through post-translational modifications, plasma membrane localization, and oligomerization [136]. Transporter phosphorylation has also been suggested to alter localization and function of ABC transporters, however the direction and magnitude of the effect is inconsistent making PK interaction predictions challenging [137]. Thus, further research is needed to determine the mechanism of phosphorylation-mediated transporter regulation, and whether these changes will impact clinical PK (e.g., pharmacological phosphorylation modulators). Likewise, transporter glycosylation affects transporter localization and function, but unlike phosphorylation, impaired glycosylation consistently decreases transporter plasma membrane localization and function [138]. For example, transporter glycosylation decreased in liver tissue of NASH patients, potentially causing decreased uptake transporter function and contributing to decreased hepatic uptake clearance of 99mTc-mebrofenin as described in Section 2.4. Accounting for post-translational regulation of transporter function is challenging because current LC-MS/MS methods do not capture transporter phosphorylation or glycosylation without performing specific assays [138]. As research tools and our knowledge of these molecular processes continue to expand, better predictions can be made for single- and multi-factorial PK interactions to improve patient outcomes.
4.2. Deconvolution of complex factors
The major complex factors discussed in this review are NPs and diseases in ADME organs. These complex factors make identification of the perpetrating compound or perturbed physiological process more difficult because it may not be readily apparent which factor is primarily mediating the interaction. Each factor carries a unique set of challenges and opportunities that need to be addressed in unique and deliberate ways to appropriately incorporate them into precision drug therapy. For NPs, the important information includes a complete knowledge of all NP consumption for each patient, knowing the NP composition and dose, and following robust and reproducible experimental approaches. This is complicated by under-reporting of NP use and by variability in NP composition, formulations, and dosing. For diseases in ADME organs, complexities include mechanistic understanding of underlying disease states and accurately and consistently diagnosing the disease stage. As a disease progresses, changes in PK mediators may be heterogeneous within and across diagnostic categories. Additionally, subjectivity in diagnostic procedures further complicates the ability to accurately and consistently stage disease progression. For example, the gold standard for NAFLD diagnosis and staging utilizes pathologist scoring of liver biopsy for histological features such as steatosis, lobular inflammation, and ballooning [139]. Liver biopsy is limited by a small sample volume collected via an invasive procedure, and histological scoring carries an inherent level of variability [140]. Improved diagnosis and PK phenotyping of progressive diseases will enable clearer predictions and prescribing practices in these patients.
4.3. Characterizing the combined effects of multi-factorial interactions
Multi-factorial PK interactions cannot necessarily be predicted based on the effect of each factor alone. Examples presented in this review demonstrate that multi-factorial interactions can have combined (i.e., additive or synergistic) or opposing effects. Additive and opposing effects are the most intuitive, but synergistic effects, where one or more factor individually has no significant effect but there is a greater combined effect, are more difficult to explain. Thus, it is critical that the potential combined effects are accounted for and utilized in prediction models and clinical trials.
4.4. Future directions
The future of precision drug therapy must go beyond single factor interaction studies, and take deliberate steps to incorporate pertinent multi-factorial interactions. This can be accomplished by accounting for the complexities discussed above, advancing quantitative systems pharmacology (QSP), and developing clinical decision support tools. QSP is an emerging field of systems biology incorporating exposure level parameters with PD target biology. PBPK modeling is an established and integral tool in drug development and can be used to predict drug interactions. Similar to preclinical and clinical studies, these models have traditionally focused on single factor interactions, but as the granularity and accuracy of PBPK input parameters improve, multi-factorial interaction models are becoming more common [141,142]. Therefore, continued advancements in understanding how each inter-individual variability factor, discrete pathway, and redundant/sequential process integrate to affect PK for individual patients are integral to the incorporation of PBPK and QSP and into precision drug therapy. Clinical decision support tools are designed to incorporate pertinent inter-individual variability factors discussed throughout this review into patient care. Important improvements to clinical decision support tools include expanded pharmacogenomic testing, improved disease staging, and accurate reporting of NP content and use. The immediate future of multi-factorial PK interaction research and its clinical implementation will hinge tightly on employment of the concepts discussed. Unraveling the complexities described here will inevitably lead to more questions but will also advance precision drug therapy.
Article highlights.
Genetics (i.e., pharmacogenetics), environment (i.e., diseases and polypharmacy), and lifestyle (i.e., diet and natural product use) are factors that contribute to inter-individual variability in pharmacotherapy.
Combinations of these factors cause additive, synergistic, or opposing effects on drug PK.
Functionally redundant and/or sequential PK processes dictate the direction and magnitude of these effects on drug disposition.
Advances in clinical decision tools, drug metabolizing enzyme and transporter fractional contribution determinations, PBPK modeling, and PK phenotyping of progressive diseases will facilitate precision drug therapy.
Funding
JD Clarke was supported in part by National Institutes of Health National Center for Complementary and Integrative Health (NCCIH) grants U54 AT008909, R21 AT011101, R56 AT010650 and National Institute of Environmental Health Sciences grant R00 ES024455. BJ Bechtold was supported in part by NCCIH grant R21 AT011101.
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Articles of special interest have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- [1].Precision Medicine Initiative (PMI) Working Group. The precision medicine initiative cohort program – building a research foundation for 21st century medicine. Precis. Med. Initiat. Work. Gr. Rep. to Advis. Comm. to Dir. NIH 2015.
- [2].Mangoni AA, Jackson SHD. Age-related changes in pharmacokinetics and pharmacodynamics: basic principles and practical applications. Br J Clin Pharmacol 2004;57:6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Waxman DJ, Holloway MG. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol 2009;76:215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Farkouh A, Riedl T, Gottardi R, et al. Sex-Related Differences in Pharmacokinetics and Pharmacodynamics of Frequently Prescribed Drugs: A Review of the Literature. Adv Ther 2020;37:644–655. [DOI] [PubMed] [Google Scholar]
- [5].Bartelink IH, Rademaker CMA, Schobben AFAM, et al. Guidelines on paediatric dosing on the basis of developmental physiology and pharmacokinetic considerations. Clin Pharmacokinet 2006;45:1077–1097. [DOI] [PubMed] [Google Scholar]
- [6].Hu M, Mak VWL, Yin OQP, et al. Effects of grapefruit juice and SLCO1B1 388A>G polymorphism on the pharmacokinetics of Pitavastatin. Drug Metab Pharmacokinet 2013;28:104–108. [DOI] [PubMed] [Google Scholar]
- [7].Caesar LK, Cech NB. Synergy and antagonism in natural product extracts: when 1 + 1 does not equal 2. Nat Prod Rep 2019;36:869–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shitara Y, Horie T, Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. Eur J Pharm Sci 2006;27:425–446. [DOI] [PubMed] [Google Scholar]
- [9].Guengerich FP. Cytochrome P450 and chemical toxicology. Chem Res Toxicol 2008;21:70–83. [DOI] [PubMed] [Google Scholar]
- [10].Williams JA, Hyland R, Jones BC, et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUC 1/AUC) ratios. Drug Metab Dispos 2004;32:1201–1208. [DOI] [PubMed] [Google Scholar]
- [11].Clarke JD, Novak P, Lake AD, et al. Impaired N-linked glycosylation of uptake and efflux transporters in human non-alcoholic fatty liver disease. Liver Int 2017;37:1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Montonye ML, Tian DD, Arman T, et al. A pharmacokinetic natural product-disease-drug interaction: A double hit of silymarin and nonalcoholic steatohepatitis on hepatic transporters in a rat model. J Pharmacol Exp Ther 2019;371:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Iyer SV, Harpaz R, LePendu P, et al. Mining clinical text for signals of adverse drug-drug interactions. J Am Med Informatics Assoc 2014;21:353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yu J, Zhou Z, Tay-Sontheimer J, et al. Risk of Clinically Relevant Pharmacokinetic-Based Drug-Drug Interactions with Drugs Approved by the U.S. Food and Drug Administration Between 2013 and 2016. Drug Metab Dispos 2018;46:835–845. [DOI] [PubMed] [Google Scholar]
- [15].Guengerich FP. Cytochrome P-450 3A4: Regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol 1999;39:1–17. [DOI] [PubMed] [Google Scholar]
- [16].Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet 2000;38:41–57. [DOI] [PubMed] [Google Scholar]
- [17].Cicali EJ, Smith DM, Duong BQ, et al. A Scoping Review of the Evidence Behind Cytochrome P450 2D6 Isoenzyme Inhibitor Classifications. Clin Pharmacol Ther 2020;108:116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev 2002;34:17–35. [DOI] [PubMed] [Google Scholar]
- [19].Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol 1998;38:389–430. [DOI] [PubMed] [Google Scholar]
- [20].Ahonen J, Olkkola KT, Neuvonen PJ. Effect of route of administration of fluconazole on the interaction between fluconazole and midazolam. Eur J Clin Pharmacol 1997;51:415–419. [DOI] [PubMed] [Google Scholar]
- [21].Yoshida K, Zhao P, Zhang L, et al. In Vitro–In Vivo Extrapolation of Metabolism- and Transporter-Mediated Drug–Drug Interactions—Overview of Basic Prediction Methods. J Pharm Sci 2017;106:2209–2213. [DOI] [PubMed] [Google Scholar]
- [22].Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers | FDA [Internet] 2020. [cited 2020 Aug 20]. Available from: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers.
- [23].Hillgren KM, Keppler D, Zur AA, et al. Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther 2013;94:52–63. [DOI] [PubMed] [Google Scholar]
- [24].Lau YY, Huang Y, Frassetto L, et al. Effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther 2007;81:194–204. [DOI] [PubMed] [Google Scholar]
- [25].Shitara Y, Sugiyama Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: Drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther 2006;112:71–105. [DOI] [PubMed] [Google Scholar]
- [26].Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol 2009;158:693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Keogh JP. Membrane Transporters in Drug Development [Internet]. Adv. Pharmacol 2012. [cited 2020 Sep 7]. p. 1–42. Available from: http://web.a.ebscohost.com/ehost/pdfviewer/pdfviewer?sid=7fe8b1bb-56a6-4c4f-b87d-02118619a084@sessionmgr4001&vid=2&hid=4114. [DOI] [PubMed]
- [28].Hézode C, Asselah T, Reddy KR, et al. Ombitasvir plus paritaprevir plus ritonavir with or without ribavirin in treatment-naive and treatment-experienced patients with genotype 4 chronic hepatitis C virus infection (PEARL-I): A randomised, open-label trial. Lancet 2015;385:2502–2509. [DOI] [PubMed] [Google Scholar]
- [29].Smith T, May G, Eckl V, et al. Market report. Off. Bot. Mark. Austin (TX); 2020.
- [30].Geller AI, Shehab N, Weidle NJ, et al. Emergency department visits for adverse events related to dietary supplements. N Engl J Med 2015;373:1531–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fenclova M, Novakova A, Viktorova J, et al. Poor chemical and microbiological quality of the commercial milk thistle-based dietary supplements may account for their reported unsatisfactory and non-reproducible clinical outcomes. Sci Rep 2019;9:11118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Girennavar B, Poulose SM, Jayaprakasha GK, et al. Furocoumarins from grapefruit juice and their effect on human CYP 3A4 and CYP 1B1 isoenzymes. Bioorganic Med Chem 2006;14:2606–2612. [DOI] [PubMed] [Google Scholar]
- [33].Hanley MJ, Cancalon P, Widmer WW, et al. The effect of grapefruit juice on drug disposition. Expert Opin Drug Metab Toxicol 2011;7:267–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lin HL, Kent UM, Hollenberg PF. The grapefruit juice effect is not limited to cytochrome P450 (P450) 3A4: Evidence for bergamottin-dependent inactivation, heme destruction, and covalent binding to protein in P450s 2B6 and 3A5. J Pharmacol Exp Ther 2005;313:154–164. [DOI] [PubMed] [Google Scholar]
- [35].Lilja JJ, Kivistö K, Neuvonen P. Grapefruit juice increases serum concentrations of atorvastatin and has no effect on pravastatin. Clin Pharmacol Ther 1999;66:118–127. [DOI] [PubMed] [Google Scholar]
- [36].Lilja JJ, Kivistö KT, Neuvonen PJ. Grapefruit juice-simvastatin interaction: effect on serum concentrations of simvastatin, simvastatin acid, and HMG-CoA reductase inhibitors. Clin Pharmacol Ther 1998;64:477–483. [DOI] [PubMed] [Google Scholar]
- [37].Kantola T, Kivistö KT, Neuvonen PJ. Grapefruit juice greatly increases serum concentrations of lovastatin and lovastatin acid. Clin Pharmacol Ther 1998;63:397–402. [DOI] [PubMed] [Google Scholar]
- [38].Glaeser H, Bailey DG, Dresser GK, et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin Pharmacol Ther 2007;81:362–370. [DOI] [PubMed] [Google Scholar]
- [39].Bailey DG, Dresser GK. Interactions Between Grapefruit Juice and Cardiovascular Drugs. Am J Cardiovasc Drugs 2004;4:281–297. [DOI] [PubMed] [Google Scholar]
- [40].Kirby BJ, Unadkat JD. Grapefruit juice, a glass full of drug interactions? Clin Pharmacol Ther 2007;81:631–633. [DOI] [PubMed] [Google Scholar]
- [41].Johnson EJ, González-Peréz V, Tian DD, et al. Selection of priority natural products for evaluation as potential precipitants of natural product-drug interactions: A NaPDI center recommended approach. Drug Metab Dispos 2018;46:1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kellogg JJ, Paine MF, McCune JS, et al. Selection and characterization of botanical natural products for research studies: A NaPDI center recommended approach. Nat Prod Rep 2019;36:1196–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Paine MF, Shen DD, McCune JS. Recommended approaches for pharmacokinetic natural product-drug interaction research: A NaPDI center commentary. Drug Metab Dispos 2018;46:1041–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Elliott LS, Henderson JC, Neradilek MB, et al. Clinical impact of pharmacogenetic profiling with a clinical decision support tool in polypharmacy home health patients: A prospective pilot randomized controlled trial. PLoS One 2017;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].PharmVar [Internet] [cited 2020 Aug 24]. Available from: https://www.pharmvar.org/gene/CYP2D6.
- [46].Medhasi S, Pasomsub E, Vanwong N, et al. Clinically relevant genetic variants of drug-metabolizing enzyme and transporter genes detected in thai children and adolescents with autism spectrum disorder. Neuropsychiatr Dis Treat 2016;12:843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Belmonte C, Ochoa D, Román M, et al. Influence of CYP2D6, CYP3A4, CYP3A5 and ABCB1 Polymorphisms on Pharmacokinetics and Safety of Aripiprazole in Healthy Volunteers. Basic Clin Pharmacol Toxicol 2018;122:596–605. [DOI] [PubMed] [Google Scholar]
- [48].Wang G, Lei H-P, Li Z, et al. The CYP2C19 ultra-rapid metabolizer genotype influences the pharmacokinetics of voriconazole in healthy male volunteers. Eur J Clin Pharmacol 2009;65:281–285. [DOI] [PubMed] [Google Scholar]
- [49].Pascual A, Calandra T, Bolay S, et al. Voriconazole therapeutic drug monitoring in patients with invasive mycoses improves efficacy and safety outcomes. Clin Infect Dis 2008;46:201–211. [DOI] [PubMed] [Google Scholar]
- [50].Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin. Pharmacokinet Adis International Ltd; 2001. p. 587–603. [DOI] [PubMed] [Google Scholar]
- [51].Aithal GP, Day CP, Kesteven PJL, et al. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 1999;353:717–719. [DOI] [PubMed] [Google Scholar]
- [52].Ingelman-Sundberg M Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J 2005;5:6–13. [DOI] [PubMed] [Google Scholar]
- [53].Rodriguez-Antona C, Ingelman-Sundberg M. Cytochrome P450 pharmacogenetics and cancer. Oncogene 2006;25:1679–1691. [DOI] [PubMed] [Google Scholar]
- [54].Aka I, Bernal CJ, Carroll R, et al. Clinical pharmacogenetics of cytochrome P450-associated drugs in children. J Pers Med 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Isvoran A, Louet M, Vladoiu DL, et al. Pharmacogenomics of the cytochrome P450 2C family: impacts of amino acid variations on drug metabolism. Drug Discov. Today Elsevier Ltd; 2017. p. 366–376. [DOI] [PubMed] [Google Scholar]
- [56].Saif MW, Lee AM, Offer SM, et al. A DPYD variant (Y186C) specific to individuals of African descent in a patient with life-threatening 5-FU toxic effects: potential for an individualized medicine approach. Mayo Clin Proc 2014;89:131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lee AM, Shi Q, Pavey E, et al. DPYD variants as predictors of 5-fluorouracil toxicity in adjuvant colon cancer treatment (NCCTG N0147). J Natl Cancer Inst 2014;106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mattison LK, Fourie J, Desmond RA, et al. Increased prevalence of dihydropyrimidine dehydrogenase deficiency in African-Americans compared with Caucasians. Clin Cancer Res 2006;12:5491–5495. [DOI] [PubMed] [Google Scholar]
- [59].Sparreboom A, Gelderblom H, Marsh S, et al. Diflomotecan pharmacokinetics in relation to ABCG2 421C>A genotype. Clin Pharmacol Ther 2004;76:38–44. [DOI] [PubMed] [Google Scholar]
- [60].Imai Y, Nakane M, Kage K, et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol Cancer Ther 2002;1:611–616. [PubMed] [Google Scholar]
- [61].Kobayashi D, Ieiri I, Hirota T, et al. Functional assessment of ABCG2 (BRCP) gene polymorphisms to protein expression in human placenta. Drug Metab Dispos 2005;33:94–101. [DOI] [PubMed] [Google Scholar]
- [62].Nebert DW. Pharmacogenetics and pharmacogenomics: why is this relevant to the clinical geneticist? Clin Genet 1999;56:247–258. [DOI] [PubMed] [Google Scholar]
- [63].Tamai I Oral drug delivery utilizing intestinal OATP transporters. Adv Drug Deliv Rev 2012;64:508–514. [DOI] [PubMed] [Google Scholar]
- [64].Bluth M, Li J. Pharmacogenomics of drug metabolizing enzymes and transporters: implications for cancer therapy. Pharmgenomics Pers Med 2011;4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ahmed S, Zhou Z, Zhou J, et al. Pharmacogenomics of Drug Metabolizing Enzymes and Transporters: Relevance to Precision Medicine. Genomics, Proteomics Bioinforma 2016;14:298–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].FDA. Guidance for Industry- Pharmacokinetics in Patients with Impaired Hepatic Function: Study Design, Data Analysis, and Impact on Dosing. Food Drug Adm. Publ 2020.
- [67].Renton KW, Mannering GJ. Depression of the hepatic cytochrome P 450 mono oxygenase system by administered tilorone (2,7 bis[2 (diethylamino)ethoxy]fluoren 9 one dihydrochloride). Drug Metab Dispos 1976;4:223–231. [PubMed] [Google Scholar]
- [68].Haas CE, Kaufman DC, Jones CE, et al. Cytochrome P450 3A4 activity after surgical stress. Crit Care Med 2003;31:1338–1346. [DOI] [PubMed] [Google Scholar]
- [69].Morgan ET. Regulation of cytochromes P450 during inflammation and infection. Drug Metab Rev 1997;29:1129–1188. [DOI] [PubMed] [Google Scholar]
- [70].Rivory LP, Slaviero KA, Clarke SJ. Hepatic cytochrome P450 3A drug metabolism is reduced in cancer patients who have an acute-phase response. Br J Cancer 2002;87:277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Fisher CD, Lickteig AJ, Augustine LM, et al. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos 2009;37:2087–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Villeneuve J-P, Pichette V. Cytochrome P450 and Liver Diseases. Curr Drug Metab 2005;5:273–282. [DOI] [PubMed] [Google Scholar]
- [73].Hardwick RN, Fisher CD, Canet MJ, et al. Variations in ATP-binding cassette transporter regulation during the progression of human nonalcoholic fatty liver disease. Drug Metab Dispos 2011;39:2395–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ali I, Slizgi JR, Kaullen JD, et al. Transporter-Mediated Alterations in Patients With NASH Increase Systemic and Hepatic Exposure to an OATP and MRP2 Substrate. Clin Pharmacol Ther 2018;104:749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ogasawara K, Terada T, Toshiya K, et al. Hepatitis c virus-related cirrhosis is a major determinant of the expression levels of hepatic drug transporters. Drug Metab Pharmacokinet 2010;25:190–199. [DOI] [PubMed] [Google Scholar]
- [76].Le Vee M, Lecureur V, Stieger B, et al. Regulation of drug transporter expression in human hepatocytes exposed to the proinflammatory cytokines tumor necrosis factor-alpha or interleukin-6. Drug Metab Dispos 2009;37:685–693. [DOI] [PubMed] [Google Scholar]
- [77].Maher JM, Dieter MZ, Aleksunes LM, et al. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology 2007;46:1597–1610. [DOI] [PubMed] [Google Scholar]
- [78].Schrieber SJ, Wen Z, Vourvahis M, et al. The pharmacokinetics of silymarin is altered in patients with hepatitis C virus and nonalcoholic Fatty liver disease and correlates with plasma caspase-3/7 activity. Drug Metab Dispos 2008;36:1909–1916. [DOI] [PubMed] [Google Scholar]
- [79].Schrieber SJ, Hawke RL, Wen Z, et al. Differences in the disposition of silymarin between patients with nonalcoholic fatty liver disease and chronic hepatitis C. Drug Metab Dispos 2011;39:2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Thakkar N, Slizgi JR, Brouwer KLR. Effect of Liver Disease on Hepatic Transporter Expression and Function. J Pharm Sci 2017;106:2282–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Evers R, Piquette-Miller M, Polli JW, et al. Disease-Associated Changes in Drug Transporters May Impact the Pharmacokinetics and/or Toxicity of Drugs: A White Paper From the International Transporter Consortium. Clin Pharmacol Ther 2018;104:900–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kalliokoski A, Backman JT, Kurkinen KJ, et al. Effects of gemfibrozil and atorvastatin on the pharmacokinetics of repaglinide in relation to SLCO1B1 polymorphism. Clin Pharmacol Ther 2008;84:488–496.* Study reports the individual effects of transporter polymorphism and drug interaction along with their combined effect, effectively illustrating the complexity of multi-factorial PK interactions on sequential processes.
- [83].Prueksaritanont T, Ma B, Yu N. The human hepatic metabolism of simvastatin hydroxy acid is mediated primarily by CYP3A, and not CYP2D6. Br J Clin Pharmacol 2003;56:120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Brunham LR, Lansberg PJ, Zhang L, et al. Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin. Pharmacogenomics J 2012;12:233–237. [DOI] [PubMed] [Google Scholar]
- [85].Jiang F, Choi J-Y, Lee J-H, et al. The influences of SLCO1B1 and ABCB1 genotypes on the pharmacokinetics of simvastatin, in relation to CYP3A4 inhibition. Pharmacogenomics 2017;18:459–469. [DOI] [PubMed] [Google Scholar]
- [86].World Health Organization. The top 10 causes of death [Internet] 2014. [cited 2020 Sep 2]. Available from: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
- [87].Pasanen MK, Neuvonen M, Neuvonen PJ, et al. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics 2006;16:873–879. [DOI] [PubMed] [Google Scholar]
- [88].Meade T, Sleight P, Collins R, et al. SLCO1B1 variants and statin-induced myopathy - A genomewide study. N Engl J Med 2008;359:789–799. [DOI] [PubMed] [Google Scholar]
- [89].CDC FastStats - Leading Causes of Death [Internet] [cited 2020 Sep 2]. Available from: https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm.
- [90].Xu J, Murphy SL, Kochanek KD, et al. Mortality in the United States, 2018 Key findings Data from the National Vital Statistics System Hyattsville (MD); 2020. [Google Scholar]
- [91].Yee SW, Giacomini MM, Hsueh C-H, et al. Metabolomic and Genome-wide Association Studies Reveal Potential Endogenous Biomarkers for OATP1B1. Clin Pharmacol Ther 2016;100:524–536.* Results show that endogenous biomarker plasma concentrations track closely with probe drug substrate for the OATP1B family, suggesting future PK interaction studies for OATP1B1 may be executed without the need to co-adminster exogenous probe substrates that may interfere with the investigated compound.
- [92].Lai Y, Mandlekar S, Shen H, et al. Coproporphyrins in Plasma and Urine Can Be Appropriate Clinical Biomarkers to Recapitulate Drug-Drug Interactions Mediated by Organic Anion Transporting Polypeptide Inhibition. J Pharmacol Exp Ther 2016;358:397–404. [DOI] [PubMed] [Google Scholar]
- [93].Yee SW, Giacomini MM, Shen H, et al. Organic Anion Transporter Polypeptide 1B1 Polymorphism Modulates the Extent of Drug–Drug Interaction and Associated Biomarker Levels in Healthy Volunteers. Clin Transl Sci 2019;12:388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Shen H, Chen W, Drexler DM, et al. Comparative evaluation of plasma bile acids, dehydroepiandrosterone sulfate, hexadecanedioate, and tetradecanedioate with coproporphyrins I and III as markers of OATP inhibition in healthy subjects. Drug Metab Dispos 2017;45:908–919. [DOI] [PubMed] [Google Scholar]
- [95].Poller B, Woessner R, Barve A, et al. Fevipiprant has a low risk of influencing co-medication pharmacokinetics: Impact on simvastatin and rosuvastatin in different SLCO1B1 genotypes. Pulm Pharmacol Ther 2019;57:101809. [DOI] [PubMed] [Google Scholar]
- [96].Duan P, Zhao P, Zhang L. Physiologically Based Pharmacokinetic (PBPK) Modeling of Pitavastatin and Atorvastatin to Predict Drug-Drug Interactions (DDIs). Eur J Drug Metab Pharmacokinet 2017;42:689–705. [DOI] [PubMed] [Google Scholar]
- [97].Varma MVS, Lai Y, Feng B, et al. Physiologically based modeling of pravastatin transporter- Mediated hepatobiliary disposition and Drug-Drug interactions. Pharm Res 2012;29:2860–2873.* Improvements of PBPK modeling in the field of multi-factorial PK interactions serves to better predict clincally relevant PK interactions in populations without the need to conduct laborious and expensive clinical trials.
- [98].Jamei M, Bajot F, Neuhoff S, et al. A mechanistic framework for in vitro-in vivo extrapolation of liver membrane transporters: Prediction of drug-drug interaction between rosuvastatin and cyclosporine. Clin Pharmacokinet 2014;53:73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].van de SE, Stranecky V, Hartmannova H, et al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J ClinInvest 2012;122:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Fan L, Zhang W, Guo D, et al. The effect of herbal medicine baicalin on pharmacokinetics of rosuvastatin, substrate of organic anion-transporting polypeptide 1B1. Clin Pharmacol Ther 2008;83:471–476. [DOI] [PubMed] [Google Scholar]
- [101].Karlgren M, Vildhede A, Norinder U, et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): Influence of protein expression on drug-drug interactions. J Med Chem 2012;55:4740–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Dresser GK, Bailey DG, Leake BF, et al. Fruit juices inhibit organic anion transporting polypeptide-mediated drug uptake to decrease the oral availability of fexofenadine. Clin Pharmacol Ther 2002;71:11–20. [DOI] [PubMed] [Google Scholar]
- [103].Imanaga J, Kotegawa T, Imai H, et al. The effects of the SLCO2B1 c.1457C>T polymorphism and apple juice on the pharmacokinetics of fexofenadine and midazolam in humans. Pharmacogenet Genomics 2011;21:84–93.* Findings show a combined effect of a natural product and gene polymorphism on the absroption of compounds dependent upon active uptake of a transporter in the drug’s disposition.
- [104].Jeon H, Jang IJ, Lee S, et al. Apple juice greatly reduces systemic exposure to atenolol. Br J Clin Pharmacol 2013;75:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Dong AN, Ahemad N, Pan Y, et al. Functional and structural characterisation of common cytochrome P450 2D6 allelic variants—roles of Pro34 and Thr107 in catalysis and inhibition. Naunyn Schmiedebergs Arch Pharmacol 2019;392:1015–1029. [DOI] [PubMed] [Google Scholar]
- [106].Verbeurgt P, Mamiya T, Oesterheld J. How common are drug and gene interactions? Prevalence in a sample of 1143 patients with CYP2C9, CYP2C19 and CYP2D6 genotyping. Pharmacogenomics 2014;15:655–665. [DOI] [PubMed] [Google Scholar]
- [107].Bartsch H, Nair U, Risch A, et al. Genetic polymorphism of CYP genes, alone or in combination, as a risk modifier of tobacco-related cancers. Cancer Epidemiol Biomarkers Prev 2000;9:3–28. [PubMed] [Google Scholar]
- [108].Paulík A, Grim J, Filip S. Predictors of irinotecan toxicity and efficacy in treatment of metastatic colorectal cancer. Acta medica (Hradec Kral 2012;55:153–159. [DOI] [PubMed] [Google Scholar]
- [109].Teft WA, Welch S, Lenehan J, et al. OATP1B1 and tumour OATP1B3 modulate exposure, toxicity, and survival after irinotecan-based chemotherapy. Br J Cancer 2015;112:857–865.* Results illustrate that double heterozygote [SLCO1B1*5 and UGT1A1*28] in sequential processes have similar PK effects as the patients that are homozygous for either polymorphism further exemplifying the need to better understand sequential ADME processes.
- [110].Takane H, Kawamoto K, Sasaki T, et al. Life-threatening toxicities in a patient with UGT1A1*6/*28 and SLCO1B1*15/*15 genotypes after irinotecan-based chemotherapy. Cancer Chemother Pharmacol 2009;63:1165–1169. [DOI] [PubMed] [Google Scholar]
- [111].Sakaguchi S, Garcia-Bournissen F, Kim R, et al. Prolonged neutropenia after irinotecan-based chemotherapy in a child with polymorphisms of UGT1A1 and SLCO1B1. Arch Dis Child 2009;94:981–982. [DOI] [PubMed] [Google Scholar]
- [112].Sai K, Saito Y, Maekawa K, et al. Additive Effects of drug transporter genetic polymorphisms on irinotecan pharmacokinetics/pharmacodynamics in Japanese cancer patients. Cancer Chemother Pharmacol 2010;66:95–105. [DOI] [PubMed] [Google Scholar]
- [113].Smith NF, Figg WD, Sparreboom A. Pharmacogenetics of irinotecan metabolism and transport: An update. Toxicol Vitr 2006;20:163–175. [DOI] [PubMed] [Google Scholar]
- [114].Kim SR, Sai K, Tanaka-Kagawa T, et al. Haplotypes and a novel defective allele of CES2 found in a Japanese population. Drug Metab Dispos 2007;35:1865–1872. [DOI] [PubMed] [Google Scholar]
- [115].Rouits E, Charasson V, Pétain A, et al. Pharmacokinetic and pharmacogenetic determinants of the activity and toxicity of irinotecan in metastatic colorectal cancer patients. Br J Cancer 2008;99:1239–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kerb R, Aynacioglu AS, Brockmöller J, et al. The predictive value of MDR1, CYP2C9, and CYP2C19 polymorphisms for phenytoin plasma levels. Pharmacogenomics J 2001;1:204–210. [DOI] [PubMed] [Google Scholar]
- [117].Shengule S, Kumbhare K, Patil D, et al. Herb-drug interaction of Nisha Amalaki and Curcuminoids with metformin in normal and diabetic condition: A disease system approach. Biomed Pharmacother 2018;101:591–598. [DOI] [PubMed] [Google Scholar]
- [118].Thomas MC, Tikellis C, Kantharidis P, et al. The role of advanced glycation in reduced organic cation transport associated with experimental diabetes. J Pharmacol Exp Ther 2004;311:456–466. [DOI] [PubMed] [Google Scholar]
- [119].Zhai T, Wang J, Sun L, et al. The Effect of Streptozotocin and Alloxan on the mRNA Expression of Rat Hepatic Transporters In Vivo. AAPS PharmSciTech 2015;16:767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Wang Z, Yang H, Xu J, et al. Prediction of atorvastatin pharmacokinetics in high-fat diet and low-dose streptozotocin-induced diabetic rats usin a semiphysiologically based pharmacokinetic model involving both enzymes and transporters. Drug Metab Dispos 2019;47:1066–1079. [DOI] [PubMed] [Google Scholar]
- [121].Köck K, Xie Y, Hawke RL, et al. Interaction of silymarin flavonolignans with organic anion-transporting polypeptides. Drug Metab Dispos 2013;41:958–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hawke RL, Schrieber SJ, Soule TA, et al. Silymarin ascending multiple oral dosing phase I study in noncirrhotic patients with chronic hepatitis C. J Clin Pharmacol 2010;50:434–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Dickinson A, MacKay D. Health habits and other characteristics of dietary supplement users: a review. Nutr J 2014;13:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Roe AL, Paine MF, Gurley BJ, et al. Assessing Natural Product-Drug Interactions: An End-to-End Safety Framework. Regul Toxicol Pharmacol 2016;76:1–6. [DOI] [PubMed] [Google Scholar]
- [125].Lake AD, Novak P, Fisher CD, et al. Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos 2011;39:1954–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Zhou J, Wen Q, Li SF, et al. Significant change of cytochrome P450s activities in patients with hepatocellular carcinoma. Oncotarget 2016;7:50612–50623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Clarke JD, Hardwick RN, Lake AD, et al. Synergistic interaction between genetics and disease on pravastatin disposition. J Hepatol 2014;61:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Houghton PJ, Germain GS, Harwood FC, et al. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res 2004;64:2333–2337. [DOI] [PubMed] [Google Scholar]
- [129].Toth EL, Li H, Dzierlenga AL, et al. Gene-by-environment interaction of Bcrp−/− and methionine-and choline-deficient diet-induced nonalcoholic steatohepatitis alters SN-38 disposition. Drug Metab Dispos 2018;46:1478–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Silvestri L, Sonzogni L, De Silvestri A, et al. CYP enzyme polymorphisms and susceptibility to HCV-related chronic liver disease and liver cancer. Int J Cancer 2003;104:310–317. [DOI] [PubMed] [Google Scholar]
- [131].Zeng T, Guo F-F, Zhang C-L, et al. Roles of Cytochrome P4502E1 Gene Polymorphisms and the Risks of Alcoholic Liver Disease: A Meta-Analysis. PLoS One 2013;8:e54188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Sookoian S, Castaño G, Gianotti TF, et al. Polymorphisms of MRP2 (ABCC2) are associated with susceptibility to nonalcoholic fatty liver disease. J Nutr Biochem 2009;20:765–770. [DOI] [PubMed] [Google Scholar]
- [133].Izumi S, Nozaki Y, Kusuhara H, et al. Relative Activity Factor (RAF)-Based Scaling of Uptake Clearance Mediated by Organic Anion Transporting Polypeptide (OATP) 1B1 and OATP1B3 in Human Hepatocytes. Mol Pharm 2018;15:2277–2288. [DOI] [PubMed] [Google Scholar]
- [134].Nordell P, Winiwarter S, Hilgendorf C. Resolving the distribution-metabolism interplay of eight OATP substrates in the standard clearance assay with suspended human cryopreserved hepatocytes. Mol Pharm 2013;10:4443–4451. [DOI] [PubMed] [Google Scholar]
- [135].Li CY, Gupta A, Gáborik Z, et al. Organic anion transporting polypeptide-mediated hepatic uptake of glucuronide metabolites of androgens. Mol Pharmacol 2020;98:234–242. [DOI] [PubMed] [Google Scholar]
- [136].Zhang Y, Ruggiero M, Hagenbuch B. OATP1B3 expression and function is modulated by coexpression with OCT1, OATP1B1, and NTCP. Drug Metab Dispos 2020;48:622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Crawford RR, Potukuchi PK, Schuetz EG, et al. Beyond Competitive Inhibition: Regulation of ABC Transporters by Kinases and Protein-Protein Interactions as Potential Mechanisms of Drug-Drug Interactions. Drug Metab Dispos 2018;46:567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Clarke JD, Novak P, Lake AD, et al. Impaired N-linked glycosylation of uptake and efflux transporters in human non-alcoholic fatty liver disease. Liver Int 2017;37:1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Hashimoto E, Tokushige K, Ludwig J. Diagnosis and classification of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis: Current concepts and remaining challenges. Hepatol Res 2015;45:20–28. [DOI] [PubMed] [Google Scholar]
- [140].Jennison E, Patel J, Scorletti E, et al. Diagnosis and management of non-alcoholic fatty liver disease. Postgrad Med J 2019;95:314–322. [DOI] [PubMed] [Google Scholar]
- [141].Türk D, Hanke N, Wolf S, et al. Physiologically Based Pharmacokinetic Models for Prediction of Complex CYP2C8 and OATP1B1 (SLCO1B1) Drug–Drug–Gene Interactions: A Modeling Network of Gemfibrozil, Repaglinide, Pioglitazone, Rifampicin, Clarithromycin and Itraconazole. Clin Pharmacokinet 2019;58:1595–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Xu R, Ge W, Jiang Q. Application of physiologically based pharmacokinetic modeling to the prediction of drug-drug and drug-disease interactions for rivaroxaban. Eur J Clin Pharmacol 2018;74:755–765. [DOI] [PubMed] [Google Scholar]
- [143].Lilja JJ, Kivistö K, Neuvonen P. Grapefruit juice increases serum concentrations of atorvastatin and has no effect on pravastatin. Clin Pharmacol Ther 1999;66:118–127. [DOI] [PubMed] [Google Scholar]
- [144].Pasanen MK, Fredrikson H, Neuvonen PJ, et al. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther 2007;82:726–733. [DOI] [PubMed] [Google Scholar]
- [145].Badri P, Dutta S, Coakley E, et al. Pharmacokinetics and dose recommendations for cyclosporine and tacrolimus when coadministered with ABT-450, ombitasvir, and dasabuvir. Am J Transplant 2015;15:1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Menon RM, Badri PS, Wang T, et al. Drug-drug interaction profile of the all-oral anti-hepatitis C virus regimen of paritaprevir/ritonavir, ombitasvir, and dasabuvir. J Hepatol 2015;63:20–29. [DOI] [PubMed] [Google Scholar]
- [147].Azuma J, Hasunuma T, Kubo M, et al. The relationship between clinical pharmacokinetics of aripiprazole and CYP2D6 genetic polymorphism: Effects of CYP enzyme inhibition by coadministration of paroxetine or fluvoxamine. Eur J Clin Pharmacol 2012;68:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Kubo M, Koue T, Inaba A, et al. Influence of itraconazole co-administration and CYP2D6 genotype on the pharmacokinetics of the new antipsychotic ARIPIPRAZOLE. Drug Metab Pharmacokinet 2005;20:55–64. [DOI] [PubMed] [Google Scholar]
- [149].Harvey V, Slevin M, Dilloway M, et al. The influence of cimetidine on the pharmacokinetics of 5‐fluorouracil. Br J Clin Pharmacol 1984;18:421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]