

Abstract
Secondary hypertension is common in clinical practice if a broad definition is applied. Various patterns of hypertension exist in the patient with an endocrine source of their disease, including new‐onset hypertension in a previously normotensive individual, a loss of blood pressure control in a patient with previously well‐controlled blood pressure, and/or labile blood pressure in the setting of either of these 2 patterns. A thorough history and physical exam, which can rule out concomitant medications, alcohol intake, and over‐the‐counter medication use, is an important prerequisite to the workup for endocrine causes of hypertension. Endocrine forms of secondary hypertension, such as pheochromocytoma and Cushing's disease, are extremely uncommon. Conversely, primary aldosteronism now occurs with sufficient frequency so as to be considered “top of the list” for secondary endocrine causes in otherwise difficult‐to‐treat or resistant hypertension. Primary aldosteronism can be insidious in its presentation since a supposed hallmark finding, hypokalemia, may be variable in its presentation. It is important to identify secondary causes of hypertension that are endocrine in nature because surgical intervention may result in correction or substantial improvement of the hypertension.
Secondary forms of hypertension must always be considered, since surgical treatment of such cases can be corrective. Secondary forms of hypertension that are endocrine in origin differ most notably from essential hypertension in that an etiology can be identified, onset is often abrupt, severity may be greater, a positive family history of hypertension is frequently missing, and there is no specific age criterion for occurrence. Specific symptoms may help guide the diagnostic evaluation. Secondary forms of endocrine hypertension can emerge in connection with such diverse endocrine disorders as primary hyperaldosteronism, Cushing's disease, or pheochromocytoma, among others. 1
DIAGNOSIS
Initial Approach
Secondary forms of hypertension are often found with preexisting hypertension. In reality, secondary forms of hypertension are culled from a lengthy list of possibilities with such a list being unique to the mindset and experience of an individual physician. For example, an endocrinologist might not think twice about diagnosing Cushing's syndrome, whereas an internist might find this a sufficient outlier diagnosis to position it much lower on his own personal list of secondary hypertension causes. Likewise, pheochromocytoma is often considered by the internist but is rarely found. In reality, many such patients with symptomatic fluctuating hypertension simply have panic‐related hypertension, which would be a diagnosis quickly made by a psychiatrist. 1
Pseudoresistance
Various endocrine forms of hypertension are marked by tachycardia (relative or absolute) and volume overload; thus, before embarking on a workup for endocrine forms of hypertension, it should be considered whether the medication regimen being used has brought about tachycardia, volume expansion, and/or renin‐angiotensin‐aldosterone system/sympathetic nervous system activation—so‐called pseudoresistance. If pseudoresistance surfaces, it often relates to plasma volume expansion, which can occur with several antihypertensive classes including peripheral α‐blockers, β‐blockers, and less commonly used direct vasodilators, such as hydralazine. This form of pseudoresistance can be present even in the absence of peripheral edema and/or concurrent diuretic therapy, particularly if dietary sodium (Na+) intake and the natriuretic response obtained from a diuretic are not matched. 2
ENDOCRINE DISORDERS
Pheochromocytoma
A pheochromocytoma is a tumor of neuroectodermal origin that produces excess quantities of catecholamines as well as numerous other physiologically active peptides. This overabundance of catecholamines causes blood pressure (BP) to increase, accompanied by a constellation of signs and symptoms that can imitate those seen with a diverse grouping of medical and surgical disorders. Early recognition, precise localization and attentive management of a benign pheochromocytoma in most instances leads to a complete cure. In up to 40% of patients with a pheochromocytoma, it is discovered during surgery, found in the course of abdominal imaging, or uncovered at the time of autopsy. These tumors can prove life‐threatening, particularly during surgical and obstetric procedures. 3
Frequency
A pheochromocytoma is a rare cause of secondary hypertension, responsible for fewer than 1% of all cases of hypertension; however, this number may be somewhat higher since pheochromocytomas are often only discovered at the time of autopsy. Although a pheochromocytoma can appear at any age, it presents most commonly during the fourth and fifth decades of life. Pheochromocytomas occur in no more than 1% or 2% of all patients with neurofibromatosis, but 5% to 25% of patients with pheochromocytoma will have neurofibromatosis; thus, patients with neurofibromatosis, symptomatic or not, should be regularly screened for pheochromocytoma. 3 , 4 , 5
Clinical Features
The predominant secretory pattern for catecholamines (norepinephrine or epinephrine and/or other neurohormones) shapes the symptom profile of an active pheochromocytoma. Symptoms, including the pattern of BP elevation, do not correlate with tumor volume per se. The triad of symptoms that typifies pheochromocytoma is headache, excessive/generalized sweating, and palpitations; however, the symptom pattern of a pheochromocytoma can be quite varied, with other common symptoms including pallor, weight loss, and feelings of panic and anxiety. There is not a specific BP pattern that could be held as the sine qua non for a pheochromocytoma. The BP patterns that can develop with a pheochromocytoma include 1) a sustained hypertensive state without BP spikes; 2) a persistent hypertensive state with intermittent hypertensive spikes potentially reaching crisis levels; and 3) a normotensive state with brief, sudden, and striking BP elevations. 1 , 3 Orthostatic hypotension is seen in more than 50% of patients with pheochromocytoma; thus, when orthostatic hypotension presents on a background of hypertension, pheochromocytoma should be a strong diagnostic consideration. 6
The symptoms that accompany a pheochromocytoma‐related hypertensive crisis include dizziness, flushing, visual disturbances, panic/anxiety, nausea, vomiting, or an epileptic aura. Although such symptoms are not pathognomonic of pheochromocytoma, they differ sufficiently from nonendocrine hypertensive crises to warrant a workup for pheochromocytoma. Hypertensive crises are truly random with this disease. They can be set off by inadvertent manipulation of the tumor as with a physical exam, exercise, postural change, or micturition and/or in the course of food intake. 3
Differential Diagnosis
A wide spectrum of diseases can masquerade as a pheochromocytoma and therein obscure its diagnosis. Some of the more important disease imitators in this regard include paroxysmal vasodilating headaches, autonomic dysfunction, anxiety and/or panic disorders, acute hypoglycemia, occult or obvious coronary artery disease, the use of sympathomimetic agents such as cocaine, and an entity termed pseudopheochromocytoma. In the case of pseudopheochromocytoma, pheochromocytoma has been excluded and there exists an augmented BP response to changes in the sympathoneural release of norepinephrine. 7
Diagnosis
Patients with signs and symptoms consistent with a pheochromocytoma should be evaluated on a priority basis. Biochemical evidence of excessive catecholamine production is a necessary step for the diagnosis of pheochromocytoma (Figure). Traditional biochemical tests include measurements of urinary and plasma catecholamines, urinary metanephrines (normetanephrine and metanephrine), and urinary vanillylmandelic acid; each of these tests has its advantages and disadvantages. 8 , 9 , 10 , 11 The assay of catecholamines and their metabolites in timed urinary samples has heretofore been considered the best methodologic approach for pheochromocytoma screening despite the difficulties inherent to accurately timed urine collections.
Figure.
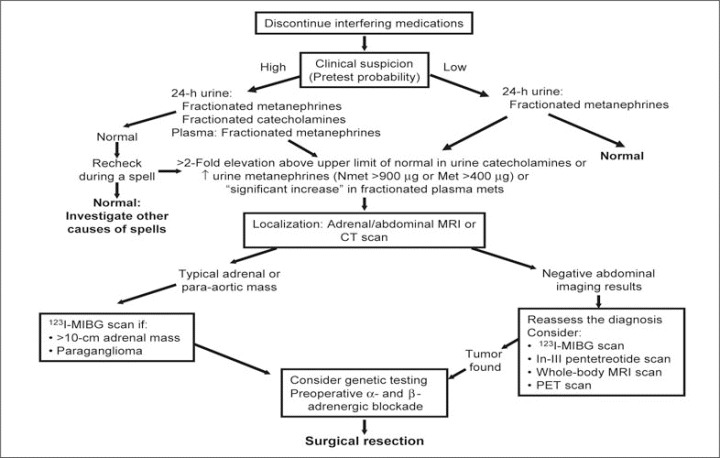
Evaluation and treatment of catecholamine‐producing tumors. Clinical suspicion is triggered by paroxysmal hypertension, family history of pheochromocytoma or associated conditions, or incidentally discovered adrenal mass. High suspicion with a 2‐fold or greater elevation in urinary metanephrines should prompt a localizing imaging study and based on findings a 123I‐MIBG scan. High suspicion with normal urinary catecholamines warrants rechecking in conjunction with a spell. Low suspicion with elevated urinary catecholamines dictates an imaging study and, if urinary catecholamines are normal, investigation for other causes of spells. MRI indicates magnetic resonance imaging; CT, computed tomography; 123I‐MIBG, 123I‐metaiodobenzylguanidine; Nmet, normetanephrine; Met, metanephrine; PET, positron emission tomography. Adapted with permission from Young. 36
Accumulating evidence, however, suggests that measurements of plasma free metanephrines or urinary fractionated metanephrines (normetanephrine and metanephrine separately) are the most sensitive tests for diagnosis and are the most suitable for reliable exclusion of pheochromocytoma. 11 In particular, plasma and urinary catecholamines can be normal when testing is carried out between spells when normotensive and asymptomatic patients with pheochromocytoma are screened for the tumor because of a hereditary predisposition or the incidental finding of an adrenal mass in the course of imaging studies. Increased sensitivity of metanephrines compared with plasma or urinary catecholamines is due to the continuous production of O‐methylated metabolites from catecholamines seeping from chromaffin stores in tumors. The generation of O‐methylated metabolites is self‐regulating and therefore independent of the highly variable release of catecholamines in tumor tissue. 12 Measurements of fractionated (and not total) metanephrines are superior to measurements of total metanephrines in that they allow tumors that produce predominantly or only one of the 3 O‐methylated metabolites to be better detected.
The majority of true‐positive biochemical results can be separated from false‐positive test results by the extent to which test results exceed reference intervals. In that regard, nearly all patients with pheochromocytoma have increases well above even the highest false‐positive results. Major diagnostic difficulties exist for patients with a clinical picture compatible with pheochromocytoma but in whom catecholamine levels fall in a range less than 2 to 3 times the upper reference limits. The significance of elevated yet indeterminate plasma catecholamine values between 1000 and 2000 pg/mL can be determined with a clonidine suppression test. This test is based on the principle that increases in catecholamines are customarily mediated through sympathetic nervous system activation. In the case of pheochromocytoma, the excess in catecholamines sidesteps the normal storage and release mechanisms; thus, clonidine would not be expected to suppress catecholamine release. 13 Failure to suppress plasma norepinephrine (ie, a reduction of <50% from basal or consistently raised basal plasma concentrations of >3 nmol/L) after clonidine therapy is highly predictive for pheochromocytoma (97%).
Abdominal computed tomography (CT) or magnetic resonance imaging (MRI) (preferred imaging mode) can be performed as a means to tumor localization in patients with clinical and biochemical features compatible with a pheochromocytoma. CT scans of the entire abdomen (including pelvis), with and without contrast, are most often used for initial localization of adrenal or possible extra‐adrenal abdominal pheochromocytomas. Because of the limited specificities of CT and MRI, a mass detected by these techniques can be further identified with of 123I‐metaiodobenzylguanidine scanning (95%–100% specificity).
Treatment
Surgical treatment is the only effective therapeutic approach for pheochromocytoma, be it benign or malignant. Adequate preoperative management includes careful attention to volume replacement (many of these patients are subclinically volume‐contracted). Drug treatment that offers combined α‐ and β‐blockade is essential for successful surgery, which is performed in most cases by laparoscopy. 14 β‐Blockade alone can result in paradoxical pressor responses. 15 If the tumor is removed, biochemical evidence of the disease disappears within 1 week; however, although prognosis after tumor resection is excellent, a significant proportion of pheochromocytomas recur, some as metastases; thus, routine follow‐up is mandatory.
Primary Hyperaldosteronism
Primary aldosteronism can occur at all ages, although in most reported series, patients fall in the 30‐to‐50–year age range. Primary aldosteronism is a common cause of resistant hypertension in black and white patients. 16 The symptoms associated with primary aldosteronism are not directly linked to excess aldosterone per se, rather they relate to the severity of the accompanying hypokalemia and/or the complications of hypertension. Primary aldosteronism should be considered as a diagnostic possibility in any patient with spontaneous hypokalemia, moderately severe hypokalemia induced by usual doses of diuretics, or refractory hypertension. 17
Frequency
Considerable dispute exists as to the true prevalence of primary aldosteronism. In contrast to previous studies, recent data suggest that primary aldosteronism is present in approximately 10% of hypertensive patients and therefore represents the most common form of secondary hypertension. 18 , 19 Much of the difference of opinion on the prevalence rate of this disorder focuses on the dependability of the screening methods (aldosterone‐renin ratios) used for diagnosis. Of note, aldosterone excess is present in only 1% or 2% of patients with incidentally discovered adrenal masses. 20
Clinical Features
Patients with primary aldosteronism can present with little more than a mild hypokalemic alkalosis and hypertension; however, if hypokalemia is severe enough, weakness, polyuria/polydipsia, and nocturia may be present. 21 , 22 Primary aldosteronism, however, does not cause edema, because of aldosterone escape in which an increase in Na+‐wasting forces offsets the Na+‐retaining effect of excess aldosterone. Although early thinking held that aldosterone‐producing adenomas caused token endorgan damage, recent evidence has proven this to be an incorrect assumption. Patients with primary aldosteronism frequently develop left ventricular hypertrophy as well as heart failure with and without preserved systolic function. 23 The myocardial susceptibility to damage in primary hyperaldosteronism may relate to aldosterone being both profibrotic and a neurohumoral substance capable of increasing oxidative stress. Treatment of primary aldosteronism is also characterized by partial reversibility of renal dysfunction and frequent return to normoalbuminuria from microalbuminuria. 24
Differential Diagnosis
Several rare syndromes produce an endocrine form of hypertension that can simulate various aspects of primary hyperaldosteronism. These hypertensive syndromes are typified by hypokalemia, metabolic alkalosis, and suppression of both plasma renin and aldosterone levels. These entities include the syndrome of apparent mineralocorticoid excess, Liddle's syndrome, and licorice ingestion. 21 Regarding the latter, the active component of licorice, glycyrrhizic acid, is hydrolyzed to glycyrrhetinic acid, which inhibits renal 11β‐hydroxysteroid dehydrogenase type 2 (a steroid metabolizing enzyme) and by that mechanism increases access of cortisol to its receptors with resultant renal Na+ retention and potassium loss. 25 Cushing's syndrome may also present with hypokalemia and hypertension, which occurs most frequently in cases of ectopic adrenocorticotropic hormone (ACTH) or adrenal carcinoma.
Diagnosis
Until relatively recently, the diagnosis of primary aldosteronism was mainly considered when hypertension and hypokalemia coexisted. It is now obvious that this approach missed many surgically correctable cases of primary aldosteronism in that hypokalemia may only occur in as few as 30% of all cases. 17 , 22 The diagnosis of primary aldosteronism can be established in the patient with a serum potassium value <3.0 mEq/L, inappropriate kaliuresis (>30 mmol of potassium excretion) in the low potassium state, a reduced plasma renin activity (PRA) value (<1.0 ng/mL/h, but typically much lower), and elevated plasma or urinary aldosterone values. Unfortunately, many cases of primary aldosteronism do not meet all such criteria. In these equivocal cases, measurement of urinary aldosterone excretion during salt loading (>250 mmol excretion of Na+) can help strengthen the diagnosis. An aldosterone excretion rate >14 mg/d following 3 days of salt loading sets apart 95% of patients with primary aldosteronism from those with essential hypertension.
In contrast to the diagnostic usefulness of elevated urinary aldosterone excretion under conditions of salt loading, plasma aldosterone values are of limited usefulness in that 60% of patients with primary aldosteronism have plasma aldosterone values that fall within the range for essential hypertension. Interpretation of plasma aldosterone values can prove difficult in that they are influenced by a diurnal rhythm (highest in the morning), the presence of hypokalemia (low potassium suppresses production), and concurrent medications (angiotensin‐converting enzyme inhibitors and β‐blockers tend to reduce values). 1
Finally, PRA values are increasingly used as a way to index the appropriateness of a plasma aldosterone value (plasma aldosterone‐PRA ratio). This ratio assumes some diagnostic significance when it is >30 and the plasma aldosterone value is elevated. Although this computation is viewed as the screening test of choice for primary aldosteronism, there are drawbacks with its use. First, there is inherent variability in PRA and plasma aldosterone values, even in the presence of a tumor; second, PRA values remain suppressed or stimulated for some time after medication discontinuation, which can complicate interpretation of this calculation; and finally, extremely low PRA values can drive this ratio to >30 with a small change in the absolute PRA value (eg, with a plasma aldosterone of 15 ng/dL and a PRA value of 0.5 ng/mL/h, the ratio is 30; however, a plasma aldosterone value of 15 ng/dL and a PRA value of 0.3 ng/mL/h computes to a ratio of 45) (Table). 1
Table.
Diagnostic Considerations for Primary Aldosteronism
| Test | Comment |
|---|---|
| Plasma K+ levels | In the hypertensive patient, 2 to 3 plasma K+ levels should be checked taking care to avoid artifactual effects from poor sampling and/or harvesting technique. Primary aldosteronism is more likely if hypokalemia exists, but it only occurs in one‐third of cases. |
| 24‐Hour K+ excretion | >30 mmol/d in the presence of hypokalemia indicates K+ wasting. |
| Plasma aldosterone‐renin ratio | Can be measured randomly but is best assessed in the morning (when aldosterone is naturally highest) and in the absence of antihypertensive agents (particularly spironolactone). A ratio >30 is suggestive of primary aldosteronism. |
| Fludrocortisone test | Fludrocortisone increases extracellular fluid volume; plasma aldosterone levels remain elevated in primary aldosteronism. |
| Computed tomography | Adrenal scanning to determine the presence of unilateral or bilateral disease. |
| Adrenal vein sampling | Useful if imaging is negative/equivocal and surgery is being considered. |
Treatment
The various types of primary aldosteronism need to be distinguished because the best management practices can differ. Unilateral adrenal adenomas are best treated surgically, either by an open or preferably a laparoscopic procedure. In most but not all instances, adenoma removal alleviates or substantially improves the hypertension. Older age, longer duration of hypertension, and the presence of other conditions associated with hypertension (obesity, sleep abnormalities, etc) predict a less favorable response to surgical intervention. Medical therapy is indicated in patients with bilateral adrenal hyperplasia or adenomas and in those patients with adenomas in whom there exists a high operative risk. Medical therapy with the aldosterone receptor antagonist spironolactone is generally effective in reversing the biochemical abnormalities of primary aldosteronism, but additional antihypertensive medication may be required for full BP control. In particular, sustained salt and water depletion with aggressive diuretic therapy noticeably improves the BP‐reducing effect of spironolactone. 26 Calcium channel blockers do not suppress aldosterone production, as is commonly believed; therefore, they fall short in correcting the primary metabolic abnormalities of this disease. The dosage of spironolactone may be limited by symptoms of gynecomastia and impotence. 27 Eplerenone is a newer aldosterone receptor antagonist with fewer endocrine side effects than spironolactone and can be used in its place; however, it is less potent than spironolactone on a milligram‐for‐milligram basis. 27
Cushing's Syndrome
Cushing's syndrome of a noniatrogenic nature can be separated into ACTH‐dependent and ACTH‐independent causes. In ACTH‐dependent states, most typically an ACTH‐secreting adenoma (also known as Cushing's disease), the excess of cortisol originates from direct adrenal stimulation. This represents about 80% of the ACTH‐dependent causes of Cushing's disease. Nonpituitary ectopic sources of ACTH, such as an oat cell, small‐cell lung carcinoma, or carcinoid tumor of the bronchus or thymus, cause the balance of ACTH‐dependent disease. Most ACTH‐independent cases of Cushing's disease result from an adrenal adenoma, carcinoma, or macronodular or micronodular hyperplasia.
Frequency
Endogenous Cushing's syndrome is an infrequent cause of hypertension in that it affects fewer than 0.1% of the population, or 5 to 25 cases/million/y. However, hypertension occurs with considerable regularity in Cushing's syndrome; it is seen in some 80% of affected patients. The peak incidence of Cushing's syndrome, whether due to an adrenal or pituitary adenoma, occurs from age 25 to 40. 28
Clinical Features
The clinical features in Cushing's disease relate to excessive cortisol production and, in some patients, an excess of adrenal androgens. The typical clinical presentation of Cushing's syndrome is that of truncal obesity including a buffalo hump, hypertension, plethoric moon facies, proximal muscle weakness/fatigue, hirsutism, emotional disturbances, and skin abnormalities (acne, purple skin striae, easy bruising). 29 Insulin resistance or diabetes, a prothrombotic state, amenorrhea, loss of libido, osteoporosis and/or spontaneous bone fractures may also be encountered; however, few patients have all of these features. These associated abnormalities determine an increased cardiovascular risk not only during the active phase of the disease but also for some time after the “biomedical remission.” 30 The hypertension of Cushing's syndrome can be explained by the oversupply of cortisol. 31 Patients with ectopic ACTH excess may not exhibit the classic manifestations of cortisol excess; instead they may present with skin hyperpigmentation (secondary) to overproduction of melanocyte‐stimulating hormone), severe hypertension, and obvious hypokalemic alkalosis. 32
Differential Diagnosis
The most common cause of Cushing's syndrome remains iatrogenic, occurring as a consequence of exogenous steroid administration. Often, the obese hypertensive patient with the metabolic syndrome and a Cushingoid physical appearance can also have an incorrect diagnosis of true Cushing's syndrome. Chronic alcohol excess can be accompanied by clinical or biochemical features or both of Cushing's syndrome, with disappearance of the same with discontinuation of alcohol intake. 33
Diagnosis
The diagnosis of Cushing's syndrome can prove difficult. Although it is cumbersome to perform, determination of 24‐hour urinary free cortisol is the best available test for documenting endogenous hypercortisolism. This test is highly sensitive and very specific, with a level >100 mg/d being highly discriminant for Cushing's syndrome when highly sensitive assays are used; however, because plasma free cortisol undergoes glomerular filtration and partial tubular reabsorption, the amount of free cortisol appearing in the urine is dependent on the glomerular filtration rate and is significantly reduced in states of moderate to severe renal failure. 34
False‐positive results for urinary free cortisol may, however, be obtained in non‐Cushing hypercortisolemic states such as stress, malnutrition, chronic strenuous exercise, polycystic ovary syndrome, and depression. The dexamethasone suppression test is a widely used screening test for Cushing's syndrome. In this test, a single dose of dexamethasone (1 mg) is given at midnight, and plasma cortisol is measured the next morning at 6 am. In healthy patients, cortisol values will be suppressed to <2 µg/dL. If dexamethasone suppression testing is used for screening, urinary free cortisol should be measured as a confirmatory test.
Treatment
The 5‐year survival of untreated Cushing's syndrome is only 50% and relates primarily to the effects of excess glucocorticoids. The preferred approach in Cushing's syndrome is selective excision of the pituitary adenoma by transsphenoidal surgery, with preservation of as much pituitary function as is possible.
Adrenalectomy (unilateral or bilateral) is indicated in the case of adrenal adenomas, micronodular or macronodular hyperplasia, and carcinoma.
Medical management of hypercortisolism is reserved for extensive and inoperable disease, such as in the case of ectopic ACTH or metastatic adrenal carcinoma. Inhibitors of steroidogenesis reduce cortisol production by blocking one (metyrapone, trilostane) or several (aminoglutethimide, ketoconazole, fluconazole, etomidate) enzymes involved in steroid biosynthesis. Ketoconazole probably is the most effective of these agents for long‐term use and usually is the agent of choice. Mitotane is a steroidogenesis inhibitor with adrenolytic properties. Mifepristone blocks glucocorticoid receptor activation without modifying cortisol synthesis. 35
Although the main mechanism of hypertension in Cushing's syndrome is overstimulation of the nonselective mineralocorticoid receptor by cortisol, other causes, such as insulin resistance and sleep apnea, are major contributors to hypertension in patients with Cushing's disease. 31 Hypertension generally remits with corrective surgery of Cushing's syndrome unless exposure to cortisol has been sufficiently prolonged to establish a structural basis for more permanent hypertension. Antihypertensive drug therapy in Cushingoid patients should avoid exacerbating preexisting hypokalemia, as would be the case with diuretic therapy and/or worsening depression with β‐blockers. Loop diuretics, in particular, should be avoided in patients with Cushing's syndrome since they are calciuretic and tend to further worsen the negative calcium balance state seen in this disease. Potassium‐sparing diuretics (amiloride or spironolactone) alone or in combination sometimes control BP, reduce edema, and correct hypokalemia in these patients.
CONCLUSIONS
Secondary forms of hypertension can be subtle in their presentation and in many cases present as refractory hypertension. Endocrine etiologies are always important considerations, particularly in patients with refractory hypertension. The workup for an endocrine etiology of hypertension centers on evaluations for pheochromocytoma, aldosterone abnormalities, and alterations in cortisol homeostasis. Simple and fairly accurate screening tests exist for each of these system abnormalities. There is a very low yield with routine screening for these abnormalities in a hypertensive patient other than for aldosterone abnormalities. Endocrine forms of hypertension are for the most part surgically correctable; however, in instances where surgery is ill advised, customized treatments exist.
References
- 1. Sica DA. Secondary forms of hypertension. In: Hypertension. Philadelphia, PA: American College of Physicians; 2005:167–188. [Google Scholar]
- 2. Izzo J, Sica DA. Antihypertensive drugs: pharmacologic principles and dosing effects. In: Hypertension Primer. 3rd ed. Izzo J, Black H, eds. Dallas, TX: American Heart Association; 2003:405–408. [Google Scholar]
- 3. Lenders JW, Eisenhofer G, Mannelli M, et al. Phaeochromocytoma. Lancet. 2005;366:665–675. [DOI] [PubMed] [Google Scholar]
- 4. Zelinka T, Eisenhofer G, Pacak K. Pheochromocytoma as a catecholamine producing tumor: implications for clinical practice. Stress. 2007;10:195–203. [DOI] [PubMed] [Google Scholar]
- 5. Pacak K, Eisenhofer G, Ahlman H, et al; International Symposium on Pheochromocytoma . Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab. 2007;3:92–102. [DOI] [PubMed] [Google Scholar]
- 6. Streeten DH, Anderson GH Jr. Mechanisms of orthostatic hypotension and tachycardia in patients with pheochromocytoma. Am J Hypertens. 1996;9:760–769. [DOI] [PubMed] [Google Scholar]
- 7. Sharabi Y, Goldstein DS, Bentho O, et al. Sympathoadrenal function in patients with paroxysmal hypertension: pseudopheochromocytoma. J Hypertens. 2007;25:2286–2295. [DOI] [PubMed] [Google Scholar]
- 8. Kudva YC, Sawka AM, Young WF Jr. Clinical review 164: The laboratory diagnosis of adrenal pheochromocytoma: the Mayo Clinic experience. J Clin Endocrinol Metab. 2003;88:4533–4539. [DOI] [PubMed] [Google Scholar]
- 9. Sawka AM, Prebtani AP, Thabane L, et al. A systematic review of the literature examining the diagnostic efficacy of measurement of fractionated plasma free metanephrines in the biochemical diagnosis of pheochromocytoma. BMC Endocr Disord. 2004;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scholz T, Schulz C, Klose S, et al. Diagnostic management of benign and malignant pheochromocytoma. Exp Clin Endocrinol Diabetes. 2007;115:155–159. [DOI] [PubMed] [Google Scholar]
- 11. Lenders JW, Pacak K, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287:1427–1434. [DOI] [PubMed] [Google Scholar]
- 12. Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004;56:331–349. [DOI] [PubMed] [Google Scholar]
- 13. Lenz T, Ross A, Schumm‐Draeger P, et al. Clonidine suppression test revisited. Blood Press. 1998;7:153–159. [DOI] [PubMed] [Google Scholar]
- 14. Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92:4069–4079. [DOI] [PubMed] [Google Scholar]
- 15. Eisenhofer G, Rivers G, Rosas AL, et al. Adverse drug reactions in patients with phaeochromocytoma: incidence, prevention and management. Drug Saf. 2007;30:1031–1062. [DOI] [PubMed] [Google Scholar]
- 16. Calhoun DA, Nishizaka MK, Zaman MA, et al. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension. 2002;40:892–896. [DOI] [PubMed] [Google Scholar]
- 17. Young WF. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol (Oxf). 2007;66:607–618. [DOI] [PubMed] [Google Scholar]
- 18. Fardella C, Mosso L. Prevalence of primary aldosteronism in unselected hypertensive populations: screening and definitive diagnosis. J Clin Endocrinol Metab. 2001;86:4003–4004. [DOI] [PubMed] [Google Scholar]
- 19. Mulatero P, Stowasser M, Loh KC, et al. Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J Clin Endocrinol Metab. 2004;89:1045–1050. [DOI] [PubMed] [Google Scholar]
- 20. Young WF Jr. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007;356:601–610. [DOI] [PubMed] [Google Scholar]
- 21. Nussberger J. Investigating mineralocorticoid hypertension. J Hypertens Suppl. 2003;21:S25–S30. [DOI] [PubMed] [Google Scholar]
- 22. Young WF Jr. Mini‐review: primary aldosteronism: changing concepts in diagnosis and treatment. Endocrinology. 2003;144:2208–2213. [DOI] [PubMed] [Google Scholar]
- 23. Catena C, Colussi G, Lapenna R, et al. Long‐term cardiac effects of adrenalectomy or mineralocorticoid antagonists in patients with primary aldosteronism. Hypertension. 2007;50:911–918. [DOI] [PubMed] [Google Scholar]
- 24. Sechi LA, Novello M, Lapenna R, et al. Long‐term renal outcomes in patients with primary aldosteronism. JAMA. 2006;295:2638–2645. [DOI] [PubMed] [Google Scholar]
- 25. Olukoga A, Donaldson D. Liquorice and its health implications. J R Soc Health. 2000;120:83–88. [DOI] [PubMed] [Google Scholar]
- 26. Bravo El. Dustan HP, Tarazi RC. Spironolactone as a nonspecific treatment for primary aldosteronism. Circulation. 1973;48:491–498. [DOI] [PubMed] [Google Scholar]
- 27. Sica DA. The risks and benefits of aldosterone antagonists. Curr Heart Fail Rep. 2005;2:65–71. [DOI] [PubMed] [Google Scholar]
- 28. Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab. 2003;88:5593–5602. [DOI] [PubMed] [Google Scholar]
- 29. Shibli‐Rahhal A, Van Beek M, Schlechte JA. Cushing's syndrome. Clin Dermatol. 2006;24:260–265. [DOI] [PubMed] [Google Scholar]
- 30. Arnaldi G, Mancini T, Polenta B, et al. Cardiovascular risk in Cushing's syndrome. Pituitary. 2004;7:253–256. [DOI] [PubMed] [Google Scholar]
- 31. Magiakou MA, Smyrnaki P, Chrousos GP. Hypertension in Cushing's syndrome. Best Pract Res Clin Endocrinol Metab. 2006;20:467–482. [DOI] [PubMed] [Google Scholar]
- 32. Torpy DJ, Mullen N, Ilias I, et al. Association of hypertension and hypokalemia with Cushing's syndrome caused by ectopic ACTH secretion: a series of 58 cases. Ann N Y Acad Sci. 2002;970:134–144. [DOI] [PubMed] [Google Scholar]
- 33. Veldman RG, Meinders AE. On the mechanism of alcohol‐induced pseudo‐Cushing's syndrome. Endo Rev. 1996;17:262–268. [DOI] [PubMed] [Google Scholar]
- 34. Chan KC, Lit LC, Law EL, et al. Diminished urinary free cortisol excretion in patients with moderate and severe renal impairment. Clin Chem. 2004;50:757–759. [DOI] [PubMed] [Google Scholar]
- 35. Diez JJ, Iglesias P. Pharmacological therapy of Cushing's syndrome: drugs and indications. Mini Rev Med Chem. 2007;7:467–480. [DOI] [PubMed] [Google Scholar]
- 36. Young WF Jr. Pheochromocytoma: 1926–1993. New York, NY: Elsevier Science Inc.; 1993:122. [Google Scholar]