

Abstract
H1 linker histones are the most abundant chromatin-binding proteins1. In vitro studies indicate that their association with chromatin determines nucleosome spacing and enables arrays of nucleosomes to fold into more compact chromatin structures. However, the in vivo roles of H1 are poorly understood2. Here we show that the local density of H1 controls the balance of repressive and active chromatin domains by promoting genomic compaction. We generated a conditional triple-H1-knockout mouse strain and depleted H1 in haematopoietic cells. H1 depletion in T cells leads to de-repression of T cell activation genes, a process that mimics normal T cell activation. Comparison of chromatin structure in normal and H1-depleted CD8+ T cells reveals that H1-mediated chromatin compaction occurs primarily in regions of the genome containing higher than average levels of H1: the chromosome conformation capture (Hi-C) B compartment and regions of the Hi-C A compartment marked by PRC2. Reduction of H1 stoichiometry leads to decreased H3K27 methylation, increased H3K36 methylation, B-to-A-compartment shifting and an increase in interaction frequency between compartments. In vitro, H1 promotes PRC2-mediated H3K27 methylation and inhibits NSD2-mediated H3K36 methylation. Mechanistically, H1 mediates these opposite effects by promoting physical compaction of the chromatin substrate. Our results establish H1 as a critical regulator of gene silencing through localized control of chromatin compaction, 3D genome organization and the epigenetic landscape.
Mammals express 11 different linker histone proteins, including 5 widely expressed subtypes, H1A–H1E (also known as H1–1 to H1–5). We previously reported that whereas deletion of one or even two H1 genes does not cause overt phenotypes3, simultaneous inactivation of H1C (H1–2), H1D (H1–3) and H1E (H1–4) leads to embryonic lethality, demonstrating that H1 is essential for mammalian development4,5, but also hindering further studies of linker histone functions in vivo. To overcome this limitation, we developed a conditional triple-H1-knockout mouse strain (H1c−/−H1dfl/flH1e−/− (H1c, H1d and H1e are also known as H1f2, H1f3 and H1f4, respectively); hereafter, H1cTKO), which enables deletion of H1C, H1D and H1E in specific lineages.
Integration of gene-expression data6 and reverse-phase high-performance liquid chromatography (RP-HPLC) of histones from 23 mouse tissues and cell types showed that the H1C, H1D and H1E subtypes constitute more than 90% of the H1 complement in B and T cells (Fig. 1a, Extended Data Fig. 1a). We therefore bred H1cTKO mice with mice containing the Vav-iCre transgene, which is expressed in haematopoietic cells (Extended Data Fig. 1b). Peripheral blood mononuclear cells from H1cTKO;Vav-iCre mice had excised the H1d gene (Extended Data Fig. 1c) and did not express H1C, H1D or H1E mRNAs (Extended Data Fig. 1d). RP-HPLC analysis of histones from thymus, bone marrow and spleen from these mice showed a complete absence of H1C, H1D and H1E proteins and a 50% reduction of total H1 levels compared with wild-type mice (Extended Data Fig. 1e–g), similar to the reduction in H1C-, H1D- and H1E-null embryos that leads to failure of normal development4,5.
Fig. 1 |. H1 depletion in the haematopoietic compartment results in reduced numbers of terminally differentiated lymphocytes in immune tissues.
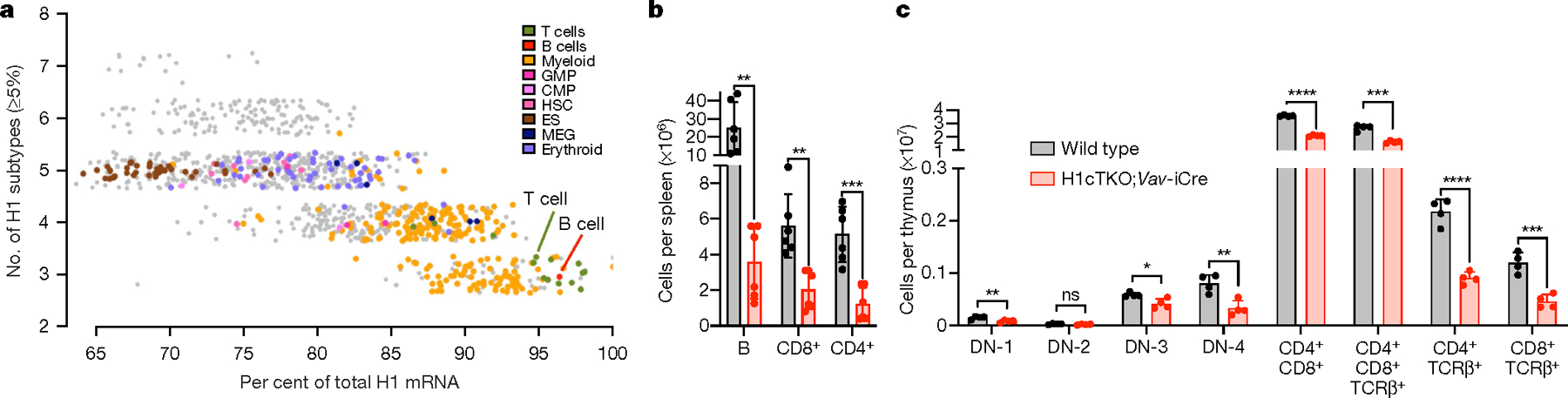
a, In silico analysis of H1 expression using mouse FANTOM56 cap analysis gene expression–sequencing (CAGE-seq) data. Total H1 was calculated as the sum of the relative log expression of normalized CAGE-seq reads mapping to the unique 5′ RNA sequences of the 11 H1 subtype transcripts. Shown is the percentage of total H1 corresponding to subtypes H1C, H1D and H1E as a function of the number of H1 subtypes expressed in each tissue. CMP, common myeloid progenitors; ES, embryonic stem cell; GMP, granulocyte–macrophage progenitors; HSC, haematopoietic stem cells; MEG, megakaryocytes. b, Number of B cells, CD4+ and CD8+ T cells in spleens (n = 6) of wild-type and H1cTKO;Vav-iCre mice, as measured by flow cytometry. c, The frequency of the indicated immature and mature T cell populations in the thymus of wild-type (n = 4) and H1cTKO;Vav-iCre (n = 4) mice was measured by flow cytometry. Absolute cell number was calculated by multiplying the relative frequency of each population by the mean thymus cellularity for wild-type and H1cTKO;Vav-iCre mice. Data are mean ± s.d.; unpaired t-test; *P ≤ 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
H1 is required for lymphocyte proliferation
Haematopoietic tissues from H1cTKO;Vav-iCre mice showed a marked reduction of white blood cells (Extended data Fig. 1h) and flow cytometry of mature haematopoietic cells revealed very large reductions in B cells and both CD4+ and CD8+ T cells (Fig. 1b, Extended Data Fig. 2e–g; flow cytometry schema are shown in Extended Data Figs. 1i, j, 2a–d), the blood lineages with the highest levels of H1C, H1D and H1E. Such reductions are not observed in mice deficient in H1C and H1D7. By contrast, cells of the erythroid and myeloid lineages, which have a more diverse H1 complement, remain mostly unaffected in the H1cTKO;Vav-iCre mice (Extended Data Fig. 2g, h). We observed reductions in the numbers of haematopoietic stem cells and some multipotent progenitor populations in the bone marrow (Extended Data Fig. 2i), but the number of common lymphoid progenitors, common myeloid progenitors and erythroid-megakaryocyte progenitors were similar to those in the wild-type mice (Extended Data Fig. 2j). Examination of the thymus revealed reductions in nearly all maturing T cell populations, with the most marked reductions in single-positive CD4 and CD8 cells (Fig. 1c). Both single-positive and double-positive CD4 and CD8 thymocytes exhibited decreased staining for the proliferation marker Ki67 (Extended Data Fig. 2k, l) but no difference in cell death (Extended Data Fig. 2m), suggesting that the decrease in thymus cellularity is owing to decreased proliferation of the highly abundant double-positive thymocytes. Measurements of B cells in bone marrow also showed the largest reductions in the most mature cell populations (Extended Data Fig. 2n, o).
We next sought to determine whether H1 depletion in mature lymphocytes causes cell-intrinsic defects in proliferation. Indeed, splenic CD4+ and CD8+ T cells and B lymphocytes from H1cTKO;Vav-iCre mice showed reduced capacities to proliferate ex vivo (Extended Data Fig. 3a–d). The reduced proliferation was accompanied by increased cell death in B cells and CD8+ T cells (Extended Data Fig. 3e). CD8+ T cells, which were most affected by H1 depletion, showed accumulation of cells in the G0–G1 phase (Extended Data Fig. 3f, g). These results are further supported by adoptive transfer experiments with splenic T cells transplanted into Rag1-knockout mice that lack mature T cells and B cells, and into congenic CD45.1 mice (Extended Data Fig. 4a). T cells from H1cTKO;Vav-iCre mice proliferated poorly in Rag1-knockout mice (Extended Data Fig. 4b–d) and did not persist in congenic CD45.1 mice (Extended Data Fig. 4e, f). We also observed upregulation of Fas expression in CD4+ and CD8+ T cells from spleens and lymph nodes of H1cTKO;Vav-iCre mice (Extended Data Fig. 4g, h). These results indicate that mature, H1-depleted B and T cells exhibit cell-intrinsic defects characterized by decreased proliferation and survival, and increased apoptosis.
H1 density regulates chromatin compaction
We examined chromatin architecture of H1-depleted lymphocytes to identify molecular changes underlying the observed defects. Transmission electron microscopy revealed grossly altered nuclear morphology in H1cTKO;Vav-iCre splenocytes, with increased nuclear size and decreased signal density within heterochromatin (Fig. 2a, Extended Data Fig. 5a, b), consistent with extensive chromatin decondensation. Hi-C analysis in CD8+ T cells showed that most A and B genomic compartments are unchanged upon H1 depletion, but affected regions primarily undergo decompaction and B-to-A compartment shifting rather than A-to-B shifting (1,021 (B to A) versus 283 (A to B)) (Fig. 2b). Similarly, H1 depletion does not affect most topologically associated domains (TADs), but TADs that do change primarily undergo decompaction and B-to-A compartment shifting (Extended Data Fig. 5c). Such unstable TADs had a smaller average size than stable TADs (Extended Data Fig. 5d). Analysis of A–B TAD borders of altered TADs showed spreading of A TADs into B TADs (Extended Data Fig. 5e). Such alterations are apparent in megabase-scale views of chromosomes (Fig. 2c, d). Aggregate analysis of regions with altered compartment scores (C-scores) in H1-depleted cells shows a global increase in local contact frequency, indicating decompaction (Extended Data Fig. 5f). H1 depletion also leads to strengthening of contacts between compartments: of the cross-compartment contacts that changed, 5,264 increased, whereas 378 decreased in intensity (Fig. 2e), such that the mean inter-compartment contact frequency of these regions increased nearly twofold (Fig. 2f). Together, these results indicate that H1 is required for maintaining condensation within both the A and B compartments and for preserving a spatially partitioned B compartment.
Fig. 2 |. H1 stoichiometry regulates chromatin compaction in spatially defined nuclear compartments.

a, Representative transmission electron microscopy of wild-type and H1cTKO;Vav-iCre spleens. b, Hi-C was performed in CD8+ T cells isolated from wild-type and H1cTKO;Vav-iCre mice and a C-score was calculated for each 100-kb genomic segment to define A and B compartments and identify compartment switching, decompaction and compaction upon H1 depletion. c, d, Representative regions illustrating local chromatin decompaction and increased chromatin contacts in H1cTKO;Vav-iCre compared with wild-type CD8+ cells. Linear genome browser views of the C-score and chromatin type (see g) at the indicated genomic loci on chromosome 18 (c) and chromosome 6 (d) are shown. e, f, Interacting chromatin regions at 100-kb resolution were identified. The number of regions exhibiting a change (adjusted P < 0.01) in interaction frequency upon H1 depletion (e) and the normalized contact frequency between compartments (f; P < 0.0001, two-tailed t-test) in wild-type and H1cTKO;Vav-iCre CD8+ T cells are shown. In box plots, the centre line shows the median, box edges represent quartiles and whiskers extend to 10th and 90th percentiles. g, ATAC-seq fragment lengths and density in wild-type and H1cTKO;Vav-iCre CD8+ T cells by chromatin type. Change in nucleosome repeat length and chromatin accessibility are shown for each chromatin state. h, CUT&Tag was performed in wild-type and H1cTKO;Vav-iCre CD8+ T cells against the linker histone H1 and core histone H3. The normalized H1 to H3 CUT&Tag read ratios and density are shown for chromatin types 1, 2 and 3.
To further understand how H1 depletion causes these changes, we performed assay for transposase-accessible chromatin using sequencing (ATAC-seq). H1 depletion resulted in alteration of 4,975 accessible sites, almost all of which had increased ATAC-seq reads (4,406 increased vs 569 decreased; adjusted P < 0.05). Integrating a chromHMM8 chromatin-state map and publicly available chromatin immunoprecipitation with sequencing (ChIP-seq) datasets (Extended Data Fig. 5g) with ATAC-seq read-length distribution revealed three distinct patterns of chromatin architectural changes, which we defined as types 1, 2 and 3 chromatin according to their response to H1 depletion (Fig. 2g, Extended Data Fig. 5h). Type 1 chromatin, encompassing 3% of the genome consisting mostly of ‘open’, nucleosome-free DNA in promoters and enhancers, is largely unaffected by H1 depletion. Type 2 chromatin, encompassing 23% of the genome and consisting of gene bodies with various levels of transcription, exhibits strong nucleosome-repeat patterns and reductions in nucleosome-repeat length (NRL) in H1-depleted CD8+ T cells. Type 3 chromatin encompasses most (74%) of the genome and includes heterochromatic chromatin states enriched for H3K27me3, H3K9me3 or ‘null’ states lacking enrichment of any specific feature. Similar to type 2 chromatin, type 3 chromatin exhibits strong H1-dependant NRLs but, uniquely, shows increased frequencies of ATAC-seq reads with loss of H1, suggesting decompaction. To our knowledge, this is the first time that H1-mediated compaction has been found to be restricted to a specific subset of chromatin. Another important distinction between type 2 and type 3 chromatin is their distributions within the Hi-C compartments: the B compartment is composed almost exclu sively (98%) of H1-compacted, type 3 chromatin, whereas the Hi-C A compartment contains nearly equal amounts of type 2 and type 3 chromatin (Extended Data Fig. 5i). Moreover, type 3 chromatin within the B compartment is marked by H3K9me3 (state E5) characteristic of constitutive heterochromatin or unmarked (null states E3 and E6), whereas the A compartment contains high levels of the facultative heterochromatin mark H3K27me3 (states E1 and E2) (Extended Data Fig. 5j). Finally, we found that decompaction and compartment shifts associated with H1 depletion occurred almost exclusively within Type 3 chromatin (Extended Data Fig. 5k, l) indicating that the spatial compartmentalization of the genome is closely linked with H1-mediated chromatin compaction.
To investigate the basis for the different responses of type 2 and type 3 chromatin to H1 depletion, we determined genome-wide occupancy of H1 in splenic CD8+ T cells by performing cleavage under targets and tagmentation (CUT&Tag)9. We observed increased H1 occupancy in type 3 chromatin (Fig. 2h). Similar results were obtained by analysing available H1 ChIP-seq data from mouse embryonic stem cells (Extended Data Fig. 5m). Moreover, mass spectrometry analysis of chromatin that is relatively resistant to micrococcal nuclease digestion showed higher relative levels of H1 (Extended Data Fig. 5n). These results indicate that type 3 chromatin is distinguished by having a higher H1-to-nucleosome ratio.
Collectively, these results show that H1, through increased stoichiometry in vivo, selectively confers a higher level of compaction on Type 3 chromatin. These regions correspond to physically separated regions of constitutive (null and H3K9me3; B compartment) and facultative (H3K27me3; A compartment) heterochromatin.
H1 promotes H3K27me3 and inhibits H3K36me2
We next used quantitative mass spectrometry and immunoblotting in several haematopoietic tissues to determine whether H1 depletion also led to genome-wide changes in histone post-translational modifications. We observed reduced H3K27me3 and increased H3K36me2 with H1 depletion in multiple tissues (Fig. 3a, Extended Data Fig. 6a, b). We also found that genes within altered chromatin contacts in H1-depleted cells were enriched in PRC2-related and H3K27me3 datasets, supporting a direct effect of H1-mediated chromatin structure on H3K27me3 deposition (Extended Data Fig. 6c, d). ChIP-seq for H3K27me3 and H3K36me2 in wild-type CD8+ T cells showed higher levels of H3K27me3 in type 3, H1-compacted chromatin than in type 2 chromatin. By contrast, H3K36me2 is broadly distributed in type 2 chromatin but is largely depleted from type 3 chromatin (Extended Data Fig. 6e, f). Thus, H3K27me3 and H3K36me2 exhibit an inverse spatial distribution (Fig. 3b) and marked reciprocal changes at type 2 and type 3 chromatin boundaries genome wide (Fig. 3c). Depletion of H1 led to widespread loss of H3K27me3, particularly within type 3 chromatin, and an increase in H3K36me2 almost exclusively within H1-compacted, type 3 chromatin (Fig. 3b, c). Furthermore, the observed changes of H3K27me3 and H3K36me2 at H1-compaction boundaries between type 2 and type 3 chromatin in wild-type cells is substantially diminished with loss of H1.
Fig. 3 |. H1 promotes H3K27 methylation and inhibits H3K36 methylation in vivo.
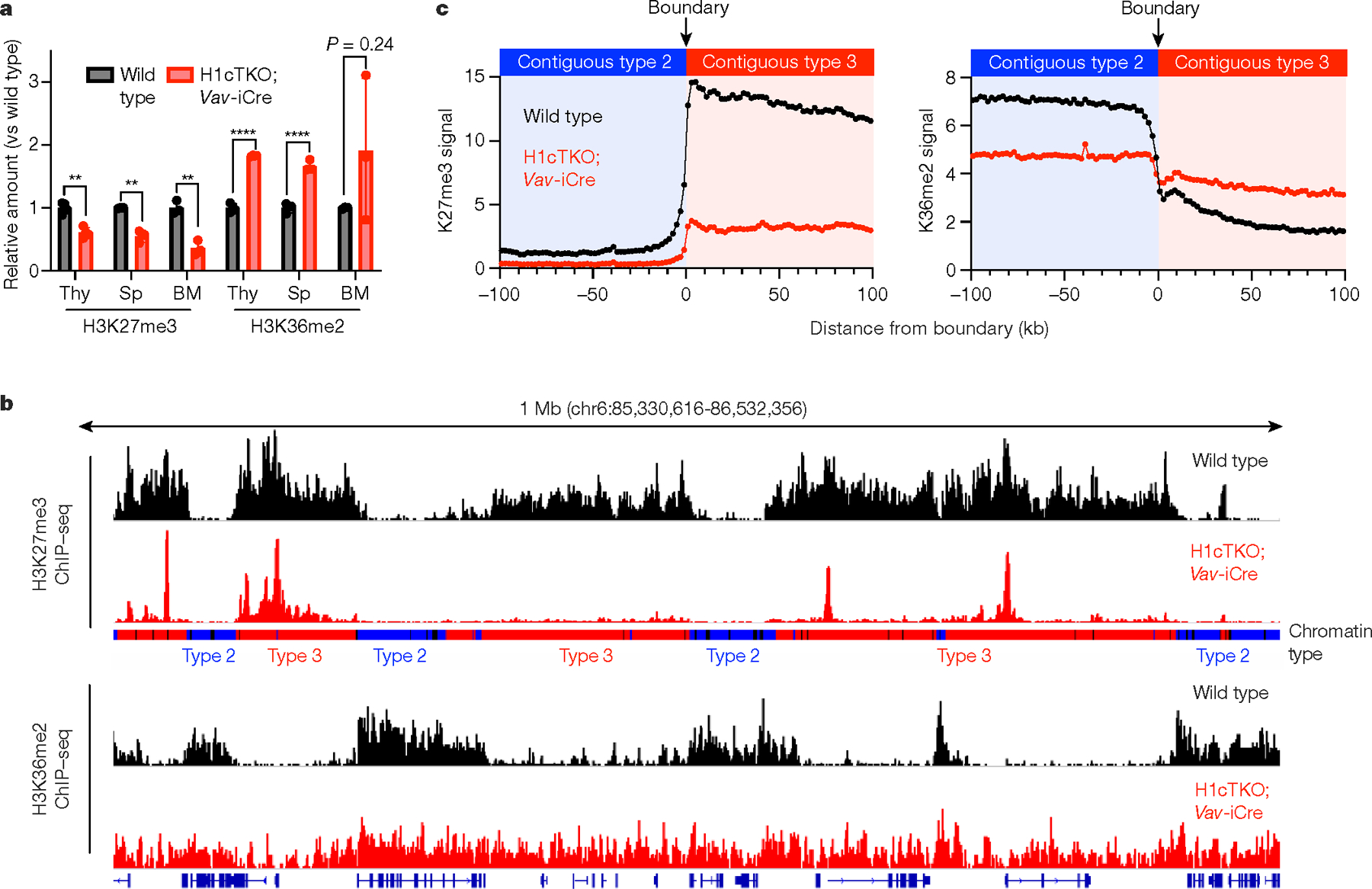
a, Acid-extracted chromatin from total mouse spleen, thymus and bone marrow was analysed by mass spectrometry. Relative abundance of peptides containing H3K36me2 and H3K27me3 from wild-type (black) and H1cTKO;Vav-iCre (red) tissues is shown. Thy, thymus; Sp, spleen; BM, bone marrow (n = 4 (spleen), 3 (thymus) and 3 (bone marrow) mice per genotype; data are mean ± s.d., unpaired t-test)), **P < 0.01, ****P < 0.0001. b, Linear genome browser views of H3K27me3 (top two tracks) and H3K36me2 (bottom two tracks) ChIP-seq signals in wild-type (black) and H1cTKO;Vav-iCre (red) CD8+ splenic T cells. Chromatin types are annotated as type 1 (black), type 2 (blue) or type 3 (red). c, Genome-wide meta-boundary analysis showing the average levels of H3K27me3 (left) and H3K36me2 (right) within a 200-kb window around the chromatin-type boundaries for all contiguous 100-kb regions of type 2 and type 3 chromatin in wild-type (black) and H1cTKO; Vav-iCre (red) CD8+ splenic T cells.
Comparing the changes in these two marks within type 2 and type 3 chromatin, we found no correlation between the magnitude of H3K27me3 and H3K36me2 changes upon H1 depletion in either chromatin type (Extended Data Fig. 6g). Indeed, whereas H3K27me3 decreases with H1 depletion in all types of chromatin, H3K36me2 increases in type 3 chromatin and decreases in type 2 chromatin. This supports the view that H3K27me3 and H3K36me2 changes are independent of each other.
To determine whether these changes observed upon H1 depletion in vivo are direct effects of H1, we assayed the activities of PRC2–AEBP2, a complex that catalyses H3K27 trimethylation, and NSD2, a H3K36 dimethyltransferase, towards in vitro-reconstituted di-nucleosomes with and without H1 (Extended Data Fig. 7a, b). We found that H1 strongly stimulates the activity of PRC2–AEBP2 (Extended Data Fig. 7c) and inhibits the activity of NSD2 (Extended Data Fig. 7d). Inhibition of NSD2 activity by H1 is observed on dinucleosomes and dodecameric nucleosome arrays, but not on mononucleosomes, suggesting that it is caused by H1-mediated chromatin compaction (Extended Data Fig. 7d–f). Since mononucleosomes are poor substrates for PRC210, we assayed the effect of a compaction-deficient, C-terminal truncated H1 on PRC2–AEBP2 activity towards dinucleosomes and observed markedly reduced stimulation (Extended Data Fig. 7g); stimulation by H1 is unlikely to be caused by allosteric effects, as a nucleosome-binding-deficient H1 (H1.2S102F) does not stimulate PRC2 activity as strongly (Extended Data Fig. 7h), and H1 has no effect on PRC2 activity towards the core histone H3 itself (Extended Data Fig. 7i).
H1 depletion phenocopies loss of EZH2
To understand how H1 depletion and the associated chromatin changes relate to defects in CD8+ T cell proliferation and survival, we performed RNA-sequencing (RNA-seq) analysis on CD8+ T cells from H1cTKO;Vav-iCre and wild-type mice. H1 depletion leads mostly to increases in expression, including in many genes that control apoptosis and negatively regulate cell proliferation (Extended Data Fig. 8a–c). About 60% of the promoters of differentially expressed genes lie in type 2 and type 3 chromatin in wild-type CD8+ T cells (Extended Data Fig. 8d), and their gene bodies exhibit decreases in NRL and increases in accessibility in H1-depleted cells (Extended Data Fig. 8e). These regions are enriched in H3K27me3 in wild-type CD8+ T cells, and this enrichment is reduced upon H1 depletion (Extended Data Fig. 8f). The most highly upregulated genes in H1-depleted cells exhibited a strong positive correlation with alterations in Hi-C chromatin contacts (normalized enrichment score = 4.604, false discovery rate < 0.001), whereas downregulated genes were negatively correlated with chromatin contact changes (normalized enrichment score = −4.725, false discovery rate < 0.001). This suggests that the chromatin architectural and epigenetic changes that accompany H1 depletion underlie the observed transcriptional alterations.
Given the specific effects of H1 depletion within H3K27me3 marked chromatin and the role of PRC2 in the control of effector functions in CD8+ T cells11, we compared the changes in gene expression that occur with H1 depletion in CD8+ T cells with the changes that occur with loss of EZH2 and T cell activation, using gene set enrichment analysis (GSEA). The results showed a substantial overlap between these gene sets (Extended Data Fig. 8g–i) suggesting that H1 depletion may cause a bias towards activated effector T cells in H1cTKO;Vav-iCre mice. This conclusion is supported by flow cytometry analysis of cells from H1cTKO;Vav-iCre mice (Extended Data Fig. 9a–c). Moreover, H1-depleted T cells exhibited markedly increased secretion of the cytokines interferon-γ (IFNγ) and interleukin-2 (IL-2) during ex vivo culture (Extended Data Fig. 9d, e). Genome browser views around the Ifng gene, as well as critical genes involved in apoptosis (Fas and Pawr) and cell cycle regulation (Cdkn2c) illustrate how chromatin changes caused by H1 depletion lead to loss of the repressive H3K27me3 mark and, in some cases, incursion of H3K36me2 into type 3 chromatin (Extended Data Fig. 9f–h).
These observations suggest that H1-mediated changes in chromatin architecture and epigenetics may have a key role during normal T cell activation. Therefore, we analysed RNA-seq and ATAC-seq data from naive and effector CD8+ T cells12. Most genes exhibiting stable expression during T cell activation reside in type 2 chromatin. By contrast, many genes that are induced or repressed during T cell activation reside in type 3 chromatin. (Extended Data Fig. 10a). Analysis of differences in NRL and ATAC-seq read frequency of genes in type 3 chromatin that are induced with T cell activation showed an average 8.5-base pair (bp) decrease in NRL and 31% increase in read frequency (Fig. 4a), changes consistent with loss of H1 and chromatin decompaction. By contrast, genes in type 2 chromatin that are repressed upon T cell activation undergo more minor, opposite changes in NRL and ATAC-seq read frequency (Extended Data Fig. 10b). Analysis of H3K27me3 enrichment within de-repressed genes residing in type 3 chromatin in naive and effector T cells13 showed that these genes lose H3K27me3 (Extended Data Fig. 10c). Both types of changes at genes within type 3 chromatin mirror changes in H1-depleted cells, suggesting that changes in H1 occupancy may be a key feature of gene regulation during T cell activation. We directly measured H1 occupancy on nucleosomes during T cell activation by performing H1 CUT&Tag9 as well as RNA-seq during ex vivo stimulation of CD8+ T cells. H1 occupancy of nucleosomes decreases on genes that are upregulated during stimulation and increases on genes that are downregulated (Fig. 4b). Moreover, there is a direct relationship (r2 = 0.89, P = 3.93 × 10−9, unpaired, two-tailed Student’s t-test) between the observed change in H1 nucleosome occupancy and the magnitude of change in gene expression.
Fig. 4 |. H1 occupancy regulates gene expression in activated and resting T cells.
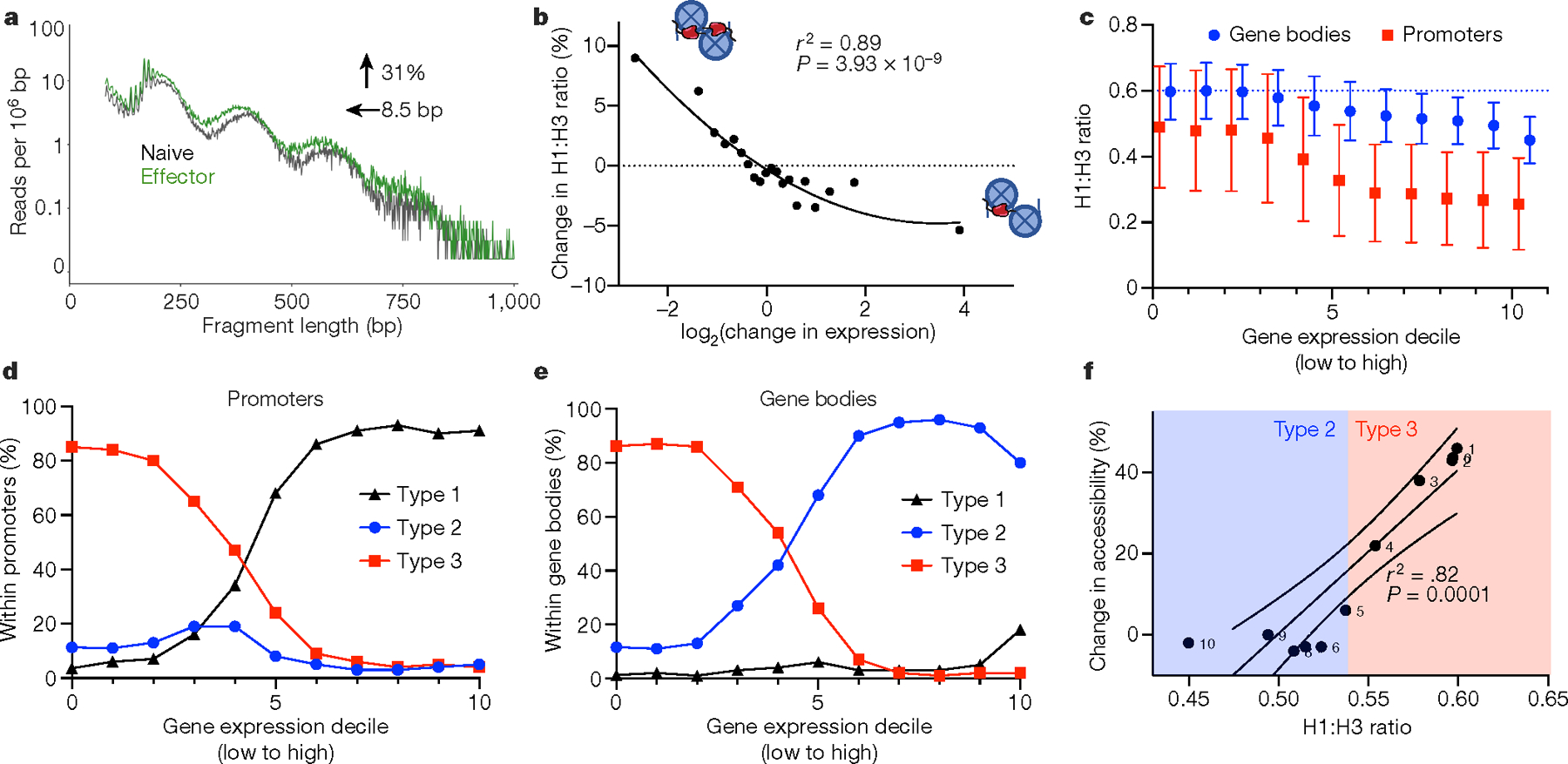
a, ATAC-seq read lengths were calculated from publicly available ATAC-seq data from naive (black) and effector (green) T cells of wild-type mice for all regions of upregulated gene bodies that fall completely within type 3 chromatin states. The data are plotted as the number of a given read length normalized to region size. The change in accessibility and NRL are shown as determined by NRLfinder. b, Change in H1 occupancy, as determined by the per cent change in the normalized H1 to H3 ratio between unstimulated and stimulated CD8+ T cells, as a function of the log2 change in gene expression between the two conditions. c, Normalized H1 occupancy in gene bodies (blue) and promoters (red) divided into deciles of gene expression in wild-type CD8+ T cells. H1 and H3 occupancy were determined using CUT&Tag and normalized as in Fig. 2h. Data are mean ± s.d. d, e, Fraction of type 1, type 2 or type 3 chromatin within promoters (d) or gene bodies (e) for each gene set, ordered by gene expression as in c. f, Alterations in accessibility as a function of the H1 to H3 ratio. Change in chromatin accessibility within each gene expression decile upon H1 depletion was calculated using NRLfinder and plotted as a function of the H1 to H3 ratio in wild-type CD8+ T cells. Type 2 and 3 chromatin designations of each gene expression decile are shown based on c. The solid line is the linear regression, and the dashed lines show the 95% confidence intervals.
We also found a strong correlation between gene expression levels and H1 occupancy in resting T cells. Combining our H1 CUT&Tag data with ATAC-seq and RNA-seq in wild-type CD8+ T cells, we observed a decrease in H1 density in both gene bodies and promoters with increasing deciles of gene expression (Fig. 4c). Promoters and gene bodies of inactive or poorly expressed genes localized almost exclusively to H1-compacted, type 3 chromatin, whereas promoters and gene bodies of highly expressed genes were primarily in type 1 and type 2 chromatin, respectively (Fig. 4d, e). We also observed an inverse relationship between H1 stoichiometry and the change in chromatin accessibility that occurs with H1 depletion (r2 = 0.82, P = 0.0001, unpaired, two-tailed Student’s t-test), suggesting that small changes in the H1-to-nucleosome ratio may lead to large changes in chromatin accessibility (Fig. 4f); a similar relationship was observed between H1 stoichiometry and nucleosome repeat length (Extended Data Fig. 10d).
Discussion
Our studies revealed that: (1) the local density of H1 regulates the degree of chromatin compaction in spatially defined genomic domains, thereby maintaining a condensed and spatially distinct chromatin B compartment and compacting regions in the A compartment with high levels of H3K27me3; (2) H1-mediated chromatin compaction promotes repressive H3K27 methylation and inhibits permissive H3K36 methylation, revealing a central role for H1 in the inverse genomic distribution of these histone marks; and (3) H1 depletion leads to de-repression of T cell activation genes, and a shift in T cells towards effector functions, a process that mimics normal T cell activation (Extended Data Fig. 10e). These findings identify H1 as a critical regulator of several aspects of the epigenetic landscape through its ability to direct localized control over the physical state of chromatin and suggest that a general feature of dynamic changes in gene expression may be chromatin decompaction and epigenetic remodelling by modulation of H1 binding. Regional differences of H1 stoichiometry are also likely to be important in disease. For example, linker histone genes are frequently mutated in cancer14,15 and the accompanying article7 reports that H1 deficiency promotes lymphomagenesis in mice. A major challenge for future work will be to decipher how regional differences in H1 stoichiometry in chromatin are established.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Mice
Ethics statement.
Animal experiments using mice were conducted under a protocol approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine (IACUC no. 20180304) in compliance with the Animal Welfare Act and other federal statutes and regulations relation to experimentation involving animals. The Albert Einstein College of Medicine animal housing facility is fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International and adheres to the principles stated in the Guide for the Care and Use of Laboratory Animals. All experimenters working with animals received training on proper technique, including euthanasia practices.
Generation of the H1cTKO mouse model and genotyping.
H1c−/−H1d+/fl H1e−/− ES cells were generated by homologous recombination of the H1d locus in WEC9.6 ES cells that contain disrupted H1c and H1e genes as previously described3,5. In brief, a H1d targeting vector similar to that described previously3,5 was modified by inserting loxP sites upstream and downstream of the H1d gene. The vector was further modified by inserting a puromycin-resistance gene (PGK-puro) flanked by Frt sites. The procedures used for detecting homologous recombination at the H1d locus, generation of chimeric mice from correctly targeted ES cells and identification of mice in which the H1dfl allele is linked to the H1c and H1e null alleles were as described previously1,3,5. The puromycin-resistance gene was removed in mice generated from correctly targeted ES cells by breeding to mice expressing Flp recombinase16. H1c−/−H1dfl/flH1e−/− mice were viable, fertile and did not exhibit any obvious abnormalities, similar to our findings with H1c−/−H1d+/+H1e−/− mice, as described previously3, 5. These mice were backcrossed for six generations to a C57BL/6 background.
Animal models, husbandry, selection and euthanasia.
All mouse models used in this study were on the C57/BL6 background (RRID: IMSR_JAX:000664). The models used in this study were: wild-type littermate controls, the H1c−/−H1dfl/flH1e−/− (hereafter referred to as H1cTKO), the H1c−/−H1dfl/flH1e−/−;Vav-iCre mice (hereafter referred to as H1cTKO;Vav-iCre mice), and the Rag1KO mice4 (RRID: IMSR_APB:4878). These mice were generated by crossing either H1c+/−H1d+/flH1e+/− females with H1c+/−H1d+/flH1e+/−;Vav1-creTg/+ males, or H1c−/−H1dfl/flH1e−/− females with H1c+/−H1d+/flH1e+/−;Vav1-creTg/+ males. All mice were housed in the Albert Einstein College of Medicine animal facility. Mice were kept in a climate and humidity-controlled facility on a 12 h diurnal light:dark cycle. Mice were fed a regular chow diet. Ear punches were taken for genotyping and numbering, and mice were gender segregated at the time of weaning, approximately 21 days of age. Adult mice were kept at 1–5 mice per cage and were provided with bedding and nesting materials.
Genotyping by PCR for colony maintenance and mouse selection for experimentation was performed as follows: genomic DNA was analysed by PCR using primers and cycling parameters according to Supplementary Table 1. Samples were analysed by 1.5–2% agarose gel electrophoresis.
For experiments, littermates were preferred, and age and gender matched mice, aged 3–6 months, were euthanized by low-flow CO2 asphyxiation, followed by cervical dislocation and immediate isolation of tissues, which could include bone marrow, peripheral blood, spleens, thymuses, lymph nodes and tail cuts for genotyping. Sample sizes were chosen in accordance with the standard number of mice in comparable studies in the field, striving for n ≥ 3. No mouse experiments were blinded or randomized.
Characterization of the H1cTKO and H1cTKO;Vav-iCre mice.
Mice were euthanized as described above and various tissues were isolated. For bone marrow, bilateral femurs and tibias were isolated by dissection, trimmed to access the marrow cavity and flushed with PBS 2% FBS 6 times using a 21G needle to an awaiting 3-mm dish on ice. Samples were passed several times through an 18G needle to disrupt any clumps and were applied to a test tube with a cell strainer cap (Corning). Bone marrow was pelleted by centrifugation at 300g for 3–5 min and was subjected to red blood cell disruption with ammonium–chloride–potassium (ACK) lysis for 3 min on ice. Cells were washed, counted by haemocytometer (0.100 mm depth; Hausser Scientific) and trypan blue exclusion (ThermoFisher), and used for downstream analysis by flow cytometry, as described in greater detail below. Whole bone marrow was also subjected to histone isolation and analysis by acrylamide gel electrophoresis, HPLC for quantitation of H1 subtypes, and post-translational modification (PTM) analysis by liquid chromatography and mass spectrometry (LC–MS), as described in greater detail below.
Peripheral blood was collected from the tail vein of live mice or by cardiocentesis from euthanized mice into heparinized capillary tubes and EDTA-coated microtainers (BD Microtainer). Complete blood counts performed on the ForCyte (Oxford Science). Otherwise, red blood cell lysis was carried out with ACK, washed with PBS, 2% FBS, and analysed by flow cytometry or fluorescence-activated cell sorting (FACS) for the isolation of primary peripheral B, and CD4+ and CD8+ T cells for cell culture. Peripheral blood was also subjected to quantitative PCR (qPCR) to assess for H1d allele excision and qPCR with reverse transcription (RT–qPCR) for gene expression of the 11 H1 subtypes, as described below.
Spleens, thymuses, and lymph nodes were isolated by dissection and were gently smashed using disposable plastic pestles in microcentrifuge tubes. Samples were passed through a syringe to disrupt clumps, filtered, subjected to ACK lysis, and cells were counted by haemocytometer. Whole spleens and thymuses were subjected to histone isolation and analysis by acrylamide gel electrophoresis, HPLC for quantitation of H1 subtypes, and PTM analysis by LC–MS. Splenic B, CD4+ and CD8+ T cells were isolated by FACS for ex vivo proliferation and functional analysis, cell cycle analysis, ATAC-seq, RNA-seq, HiC and ChIP-seq against H3K27me3 and H3K36me2. Splenic T cells were also isolated using the Dynabeads Untouched Mouse T Cell isolation kit (Life Technologies) for adoptive T cell transfer to Rag1KO and CD45.1 mice. Extended protocols for HPLC, LC–MS, ChIP-seq, RNA-seq, ATAC-seq, HiC and adoptive T cell transfer are included below.
Primary cell culture.
For the culture of primary B and T cells from wild-type, H1cTKO and H1cTKO;Vav-iCre mice, FACS sorted cells were cultured in RPMI 1640 supplemented with 15% FBS, 100 IU ml−1 penicillin and 100 μg ml−1 streptomycin, 1% glutamine, 1% sodium pyruvate, 50 μM β-mercaptoethanol, and were incubated at 37 °C with 5% CO2, with or without stimulation. For stimulation of B cells, cells were treated with mouse IL-4 (25 ng ml−1, Peprotech) and lipopolysaccharide (LPS) (50 μg ml−1) for 3 days. For stimulation of CD4+ and CD8+ T cells, cells were treated with mIL-2 (0.2 ng ml−1) and either CD3–CD28 co-stimulatory beads (20 μl per ml; ThermoFisher 11456D) or plate-bound anti-CD3ε (Clone 145–2C11) and soluble anti-CD28 antibodies (Clone 37.51).
To assay cell division, FACS-sorted B and T cells were stained with CellTrace Violet (ThermoFisher) or carboxyfluorescein succinimidyl ester (CFSE) in PBS for 20 min at 37 °C protected from light, washed, and stimulated for 3 days, as above. CellTrace dilution was analysed by flow cytometry. To assay cell number, cells were counted by flow cytometry using AccuCheck beads (Life Technologies). To assay cell cycle status, cells were stimulated and cultured for 3 days, as above. Cells were permeabilized with 20 mM HEPES-KOH pH 7.9, 10 mM KCl, 1 mM MgCl2, 0.1% Triton X-100, 20% glycerol, stained with DAPI (0.01 μg ml−1), and analysed by flow cytometry.
To assay cytokine production, CD4+ and CD8+ T cells were isolated by affinity column purification, and were co-stimulated with CD3 and CD28, as above. After 22 h, medium was collected for the assay of secreted IFNγ and IL-2 by ELISA using the ELISA MAX Deluxe Kit (Biolegend).
T cell adoptive transfer.
For adoptive transfer of wild-type and H1cTKO;Vav-iCre T cells, T cells were isolated from spleens and lymph nodes using the Dynabeads Untouched Mouse T Cell isolation kit (Life Technologies), and counted by haemocytometer. For transfer to Rag1KO or congenic C57BL/6 CD45.1 recipients, cells were stained with CellTrace CFSE (ThermoFisher), as above. One-million T cells were injected into the retro-orbital sinuses of recipient Rag1KO 34 or congenic C57BL/6 CD45.1 mice in 200 μl PBS. Recipient mice were euthanized after 7 days, spleens were isolated, and cells were counted by haemocytometer. Contribution of donor CD4+ and CD8+ T cells, and CFSE dilution was analysed by flow cytometry.
Sequencing and in silico analyses
FANTOM CAGE analysis.
For the analysis of the publicly available FANTOM5 CAGE-seq dataset6, expression data were downloaded from http://fantom.gsc.riken.jp/5/. Data analysis and visualization was carried out in R. Expression of H1 subtypes was normalized to H2B expression. For the assessment of the portion of total H1 that is comprised of subtypes H1C/D/E in various tissues represented in the dataset, we calculated the total H1 as the sum of the relative log expression-normalized reads for all expressed subtypes of H1. The percentage of total H1 of H1C, H1D and H1E is calculated as the sum of relative log expression-normalized reads of H1C, H1D and H1E divided by the total H1, and expressed as a percentage. Jitter (in R) was applied to y-values for ease of visualization.
Hi-C sample preparation and analysis.
Splenocytes from wild-type (n = 2) and H1cTKO;Vav-iCre (n = 2) mice were pooled and T cells were isolated by affinity column depletion. 1.7 × 106 −2.0 × 106 CD8+ T cells were further purified by flow cytometry. Cells were subjected to crosslinking at a concentration of 1 × 106 cells per ml in PBS, 10% FBS, 2% formaldehyde (final concentration) at room temperature for 10 min, before the reaction was quenched by adding 2.5 M glycine to a final concentration of 0.2 M and the cells were pelleted and flash frozen. Libraries were prepared using the Arima-HiC+ kit (Arima Genomics) and analysed by sequencing. Hi-C reads from separate lines were concatenated, aligned to the mouse genome (mm10) and processed with HiC-Pro (v.2.11.3-beta) using default parameters and genomic bin sizes of 25 and 100 kb. We used HiCRep36 to assess the reproducibility of our Hi-C replicates. In brief, intrachromosomal contacts were extracted for chromosome 2 using binned reads at 100-kb resolution. We then used these contacts to calculate the stratum-adjusted correlation coefficient (SCC) between wild-type and H1cTKO;Vav-iCre replicates. The SCCs for wild-type and H1cTKO;Vav-iCre samples were 0.987 and 0.986, respectively, while the inter-sample SCC comparing the first and second wild-type and H1cTKO;Vav-iCre samples were 0.909 and 0.899, respectively.
Arima restriction fragments were generated using the included digest_genome.py script. Replicates of each genotype were used as input for multiHiCcompare (https://doi.org/10.1093/bioinformatics/btz048) to determine statistically significant differences in contact frequencies between genotypes. Replicates were pooled to for A/B compartment analysis with CScoreTool (v.1.1; https://doi.org/10.1093/bioinformatics/btx802). This program was compiled from https://github.com/scoutzxb/CScoreTool, and C-scores of 100-kb bins were calculated from the intrachromosomal contact matrices and combined with the 100-kb resolution multiHiCcompare results using custom scripts. GSEA was performed as follows: C-score differences between wild-type and H1cTKO;Vav-iCre CD8+ T cells were calculated for 400-bp regions surrounding the TSS of each gene and used to generate a ranked list which was used as an input for GSEA Preranked (v.4.0.3) using the recommended settings. For overrepresentation analysis, genes within regions of B-to-A compartment shifts in H1cTKO;Vav-iCre CD8+ T cells were used as input for Enrichr17.
Analyses of topologically associated domains (TADs) in wild-type and H1cTKO;Vav-iCre CD8+ splenic T cells were performed using OnTAD18, as follows. Hi-C contact matrices were calculated for all autosomes with 50,000-bp resolution matrices (-penalty 0.1 -minsz 1 -maxsz 200 -lsize 4). Average TAD C-scores were calculated for each condition by taking the average C-score of the biological replicates (n = 2). C-scores for individual replicates were calculated with a resolution of 25,000 bp. TADs were determined to have switched compartments with loss of H1 when (1) the C-score sign changed between conditions and (2) the difference in the average C-score between conditions exceeded an absolute value of 0.25. TADs were considered to have undergone compaction or decompaction when the difference in the absolute value of the C-score exceeded 0.25 but the compartment sign was unchanged.
Analysis of TAD boundaries was performed in R using custom scripts which we have made available (Supplementary File 1, ‘Computational methods and functions’). Astacked meta-boundary plot was created showing the average C-score 200 kb upstream and 200 kb downstream of each A–B boundary for all A–B TADs where at least one of the TADs in the A–B pair was altered with loss of H1. Each A–B TAD boundary was oriented such that the TAD in the Acompartment was positioned upstream of the boundary and the TAD in the Bcompartment was downstream. The average C-scores were calculated for each position using tracks with 25 kb resolution.
ATAC-seq sample preparation and analysis.
B cells, CD4+ and CD8+ T cells were isolated from wild-type and H1cTKO;Vav-iCre mice by FACS. ATAC-seq was performed as originally described19, with digitonin for cell membrane permeabilization as per the optimized Fast-ATAC protocol20. Reads were aligned to the mouse genome (mm10) with Bowtie221. Duplicates were identified and removed with Samblaster35. Sambamba22 was used to remove mitochondrial reads and SAMtools was used to filter reads with low quality alignments23. Genrich (https://github.com/jsh58/Genrich) was used to calculate differential peak accessibility and find peaks. Coverage tracks (bigWig format) were generated with bamCoverage from deepTools24. For the calculation of nuclear repeat length, we used custom Python scripts which we have made available as an as-is public package, NRLfinder, which can be easily executed from the command-line. This package computes nuclear repeat lengths by extracting read lengths from bed files and converting read lengths into a histogram. A digital, 6th-order, Butterworth filter with zero phase shift and cut-off frequency of 0.04 cycles per read length was applied to the histogram data. This cut-off frequency was chosen heuristically to reduce histogram oscillations due to DNA winding around single histones. Minima and maxima are then extracted from the first derivative of this filtered histogram. For additional information, please see the full package, available at https://github.com/tommyjohn21/nrl_finder. For the calculation of NRL shift between wild-type and H1cTKO;Vav-iCre samples, we determine the difference of the second NRL maximum of each sample (representing two nucleosomes) and divide by two. Custom Python scripts, also available in NRLfinder, were used to calculate nucleosomal accessibility, which was calculated as the area-under-the-curve for all regions of the NRL greater than the first local minimum (for example, excluding sub-nucleosomal fragments). ATAC-seq datasets were deposited in the Sequence Read Archive.
ChIP-seq sample preparation and analysis.
CD8+ splenic T cells from wild-type and cTKO mice were isolated by flow cytometry. ChIP against H3K27me3 (1 μg; Millipore, 07–449) and H3K36me2 (1 μg; Millipore, 07–274) and sequencing library preparation was performed exactly as previously described26. Adapters were trimmed using BBTools28. Alignment to mm10 was performed using Bowtie221 and duplicate reads were marked using Picard and removed using SAMtools. Signal normalization between samples was performed using THOR30. ChIP-seq signal intensity was calculated for chromatin states using GRanges and custom R scripts (Supplementary File 1).
RNA-seq sample preparation and analysis.
A total of 5 × 105 B cells, CD4+ and CD8+ T cells were isolated from the spleens of wild-type (n = 2) and H1cTKO;Vav-iCre (n = 2) mice. RNA isolation, library preparation and sequencing were performed by Genewiz (South Plainfield, NJ). Reads were aligned to the mouse genome (mm10) using STAR31. Duplicate reads were marked using Picard and removed using Samtools. Differentially expressed genes between genotypes were determined using the R package DESeq232 (adjusted P < 0.05). All tools were run using default parameters. For GSEA, ranked gene lists were generated from DESeq2. These were used as inputs for GSEA using default parameters. Custom gene groups were used to generate enrichment plots in Fig. 4c–e. See associated text for details.
For gene expression analysis of wild-type CD8+ T cells during ex vivo stimulation, 1 × 106 cells were cultured as described above. RNA was isolated from two independent biological replicates of cells before or after stimulation. RNA isolation, library preparation and sequencing were performed by Genewiz. Read alignment and processing was carried out using the Nextflow nf-core/rnaseq (https://github.com/nf-core/rnaseq) and differential gene expression determined as above.
CUT&Tag sample preparation and analysis.
For CUT&Tag9 100,000 CD8+ T cells were isolated from the spleens of wild-type age- and sex-matched mice by flow cytometry. Additionally, cells were cultured under stimulating conditions for 3 days, as described above, and 100,000 stimulated CD8+ T cells cells were isolated by flow cytometry. The 3×Flag-pATn5 was prepared as described at https://www.protocols.io/view/3xflag-patn5-protein-purification-and-meds-loading-8yrhxv6. CUT&Tag library preparation was carried out as described at https://www.protocols.io/view/bench-top-cut-amp-tag-z6hf9b6. The following primary antibodies were used: IgG (1:50; Cell Signaling 4E-BP2), rabbit mAb anti-H3K27me3 (1:50; Cell Signaling C36B11), mouse mAb anti-H1 (1:50; Santa Cruz sc-8030 AE-4) and mouse mAb anti-H3 (1:50; Santa Cruz sc-517576). The following secondary antibodies were used: guinea pig anti-rabbit (1:100; Antibodies Online ABIN101961) and rabbit anti-mouse (1:100; Antibodies Online ABIN101785). Paired-end 150-bp sequencing was performed on an Illumina HiSeq 4000 (Genewiz). Raw fastq files were trimmed using Trim Galore (length 20, e = .1) and aligned to the mouse genome (mm10) using Bowtie2 (v.2.4.1; local, very-sensitive-local, no-unal, no-mixed, no-discordant, phred33, I = 1-, X = 700). Reads were sorted and converted to BAM format and duplicates were marked using Sambamba (v.0.6.6). Filtering, duplication removal and read down sampling to equalize reads across samples was performed using Samtools (v.1.1; F = 3340, f = 0x0002). HOMER33 was used to quantify the H1 and H3 CUT&Tag signal throughout the genome and within gene bodies. Final data analysis and visualization was performed using custom R scripts. Scripts for data processing and analysis are available in Supplementary File 1. CUT&Tag sequencing datasets were deposited in the Sequence Read Archive.
Chromatin-state modelling.
For chromatin-state modelling we used the ChromHMM software (v.1.19), which has been previously described8. The input data included ChIP-seq for H3K27me3 and K3K36me2, ATAC-seq, and RNA-seq reported in this manuscript, as described above. Additional input data including ChIP-seq for H3K4me1, H3K4me3, H3K36me3, PolII_Ser2P, CTCF, and H3K9me3, can be found in Supplementary Table 2. In brief, raw fastq files were download, replicates were combined when available, and adapters were trimmed using BBTools28. Alignment to mm10 was performed using Bowtie221 and duplicates were removed using samblaster35. The resulting signals were binarized and a new ChromHMM model was generated using the ChromHMM software default settings. Emissions parameters were visualized in R.
qPCR for H1d excision and H1 subtype expression in peripheral blood
For the measurement of H1 subtype expression in peripheral blood of H1cTKO;Vav-iCre mice, Peripheral blood mononuclear cells were isolated as described in detail above. Total RNA was isolated from peripheral blood mononuclear cells using Direct-zol isolation (Zymo). cDNA was synthesized using oligo-d(T) primers (Promega). qPCR amplification was carried out using Power Sybr (Fisher) on the Applied Biosystems Viia 7 Real Time PCR System in 384-well format. Relative quantitation calculations were performed in the ThermoFisher Cloud suite, with values adjusted to Gapdh. qPCR primers used can be found in Supplementary Table 3.
Preparation and analysis of histones by high-performance liquid chromatography
Various wild-type mouse tissues and embryos were generously provided by members of the Albert Einstein College of Medicine. Other tissues were recovered from wild-type, H1cTKO, and H1cTKO;Vav-iCre mice. Histone proteins were isolated by extraction with 0.2 N sulfuric acid, as previously described37,38. Samples were analysed by electrophoresis through a 15% SDS-polyacrylamide gel and protein was visualized by Coomassie brilliant blue staining. Otherwise, samples were applied to a Vydac 218TP C18 HPLC column using the Waters 2695 Separations Module. The effluent was monitored and peaks recorded using the Waters 996 Photodiode Array Detector at 214 nm. H1 peak areas were determined using the Waters Empower Pro software (v.2) and normalized to H2B peaks. For the wild-type tissue, data were analysed in the R statistical environment by unsupervised clustering using the pheatmap package.
Micrococcal nuclease digestion and fractionation of chromatin
Single-cell suspensions of mouse splenocytes from nine wild-type mice were isolated as described above. Nuclei were isolated using Nuclei EZ Lysis Buffer (Sigma). Chromatin from these nuclei was then digested with 0.24 units of micrococcal nuclease (New England Biolabs) for 11 min at 37 °C. The reaction was terminated by the addition of EDTA and digested chromatin was carefully layered onto a 5–40% linear sucrose gradient (10 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 2.5 mM NaCl) followed by ultracentrifugation (36,000 rpm, SW-41 rotor, 16 h at 4 °C). Fractions were collected by hand and adequate separation was confirmed by agarose gel electrophoresis of purified DNA from each fraction. Samples were analysed by mass spectrometry as described below.
Mass spectrometry
For analysis of histone H1-per-nucleosome and histone PTMs, sucrose-gradient separated polynucleosomes from wild-type spleens, or acid-extracted chromatin from wild-type and H1cTKO;Vav-iCre thymus, spleen, and bone marrow were propionylated and subjected to nano-LC-MS for comprehensive quantitation of PTMs, as previously described39. In brief, nano-LC was configured with a 75 μm internal diameter × 25 cm Reprosil-Pur C18-AQ (3 μm; Dr. Maisch) nano-column packed in-house using a Dionex RSLC Ultimate 3000 (Thermo Scientific). The HPLC gradient was as follows: 2% to 28% solvent B (A, 0.1% formic acid; B, 80% acetonitrile, 0.1% formic acid) over 45 min, from 28% to 80% solvent B in 5 min, 80% B for 10 min at a flow-rate of 300 nl min−1. nano-LC was coupled to a Q-Exactive HF mass spectrometer (Thermo Scientific). Data were acquired using a data-independent acquisition (DIA) method as described40, consisting on a full scan mass spectrum (m/z 300–1100) performed at 120,000 resolution (m/Δm for the ion at 200 m/z), followed by 16 MS/MS with windows of 50 m/z using HCD fragmentation and 15,000 resolution. For the analysis of PTMs, peptide abundance was normalized to total peptides. In brief, DIA data obtained from the nanoLC–MS/MS runs were searched using EpiProfile 2.041. The peptide relative ratio was calculated using the total area under the extracted ion chromatograms of all peptides with the same amino acid sequence (including all of its modified forms) as 100%. For isobaric peptides, the relative ratio of two isobaric forms was estimated by averaging the ratio for each fragment ion with different mass between the two species. Results confirmed by immunoblot against H3K27me3 (Active Motif, 39156) and H3K36me2 (Active Motif, 39256) carried out on acid-extracted chromatin from wild-type and H1cTKO;Vav-iCre spleens. Imaging was carried out on the LiCor Odyssey and quantitation was performed using Image Studio Lit (5.2.5).
Transmission electron microscopy
Freshly isolated spleens from wild-type and H1cTKO;Vav-iCre mice were immediately fixed in 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 M sodium cacodylate buffer. Specimens were postfixed with 1% osmium tetroxide followed by 2% uranyl acetate, dehydrated through a graded series of ethanol, and were embedded in LX112 resin (LADD Research Industries). Ultrathin sections were cut on a Leica Ultracut UCT, stained with uranyl acetate, followed by lead citrated, and viewed on a JEOL 1200EX transmission electron microscope at 80kv. ImageJ (v.1.52p) was used to measure nuclear area and density42.
Preparation of chromatin substrates and assessment of histone methyltransferase activity
Mouse recombinant histone octamers were either refolded as previously described43 or purchased (Histone Source, Colorado State University). Template DNA bearing one, two or 12 repeats of the ‘601’ strong nucleosome positioning sequence (a gift from J. Hansen (Colorado State University) and T. Richmond (ETH Zurich)) were excised from the parent plasmid by restriction digest and purified by FPLC using Source 15Q Sepharose (GE Healthcare) using an AKTA chromatography system (GE Healthcare) or gel extraction. Peak fractions were pooled, ethanol precipitated, resuspended in water and stored at −20 °C.
Chromatin was reconstituted as follows (adapted from refs. 44,45). Optimal histone to DNA stoichiometry was determined empirically. Histone octamer and DNA template (1–5 μM each in a final volume of 50–75 μl) were combined in a high salt buffer (2 M NaCl, 10 mM Tris-HCl pH 7.5, 0.1 mM EDTA) on ice. The mixtures were transferred to 3.5-kDa MWCO dialysis cups (Pierce) and dialysed against 200 ml of NRB1 (1.4 M NaCl, 10 mM Tris-HCl pH 7.5, 0.1 mM EDTA) for 1 h at 4 °C. Low-salt buffer (350 ml) (10 mM NaCl, 10 mM Tris-HCl pH 7.5, 0.1 mM EDTA) was added using a peristaltic pump at a rate of approximately 0.5–1 ml min−1 for 6–12 h. At this step, reconstitutions were removed and heat shifted for 1 h at 37 °C. Finally, reconstitution mixtures were dialysed into low-salt buffer overnight. Mono- and di-nucleosome reconstitution mixtures were further purified by 5–20% sucrose gradient using an SW-41 rotor (Beckman Coulter) for 16 h at 36,000 rpm at 4 °C. Fractions were analysed by 0.7% agarose gel electrophoresis (0.5× TBE, 120 V for 90 min at 4 °C). Peak fractions, free of unbound template DNA or non-nucleosomal species, were pooled and concentrated. Purified nucleosomes were stored at 4 °C until use. Saturation of dodecameric nucleosome arrays was assessed using magnesium-induced self-association as described previously46.
Purified, recombinant H1s were bound to nucleosomes for 30 min on ice in binding buffer (10 mM Tris-HCl pH 8.5, 50 mM NaCl). H1 binding was assessed by non-denaturing agarose gel electrophoresis as above. Optimal H1:nucleosome ratios were determined empirically.
Methyltransferase assays were carried out as previously described47. In brief, the indicated enzymes were incubated with nucleosome substrates reconstituted with or without H1 (400 nM nucleosome) in HMT buffer (50 mM Tris-HCl pH 8.5, 10 mM NaCl, 10 mM DTT) supplemented with 100 μM S-adenosylmethionine (SAM) for 1 h at 30 °C. The reaction was terminated by the addition of 4× Laemmli buffer and incubation at 95 °C for 5 min. Reactions were separated by 15% SDS–PAGE and analysed by immunoblot for the indicated histone modification. The PRC2–AEBP2 complex48 was a gift from D. Reinberg (New York University). NSD2 was purchased from Reaction Biology (HMT-21-122).
Immunoblot
Samples were separated by SDS–PAGE and transferred (wet) to 0.2-μm nitrocellulose membrane. The membrane was blocked with Odyssey blocking buffer (Licor Biosciences) for 1 h at room temperature and incubated with primary antibody, also for 1 h at room temperature. Membranes were washed three times with TBS-T and incubated with IRDye 800 or 700CW secondary antibodies (1:10,000; LI-COR Biosciences) for 30 min at room temperature. After two additional washes with TBS-T, membranes were visualized using a LI-COR Odyssey Fc (LI-COR Biosciences) and quantitation was performed using Image Studio Lite (5.2.5). Immunoblotting was performed against H3K27me3 (1:1,000), H3K36me2 (1:1,000), H4 (1:5,000), using antibodies listed in Supplementary Table 7.
Flow cytometry
Sample preparation, staining and analysis.
Samples were stained in PBS 2% FBS. Surface markers and antibody clones used to label various cell types can be found in Supplementary Tables 4, 5, respectively. Either DAPI or 7-AAD was used for live/dead staining by dye exclusion. For measurement of intranuclear Ki67 proliferation antigen to assess cellular proliferation, cells were fixed in PBS 2% paraformaldehyde, permeabilized in PBS 0.3% Tween-20, and stained with fluorophore-conjugated anti-Ki67 antibody. For apoptosis, cells were stained with Annexin V-APC (eBioscience 88–8007) and propidium iodide (PI). To assay cell cycle status, cells were permeabilized with 20 mM HEPES-KOH pH 7.9, 10 mM KCl, 1 mM MgCl2, 0.1% Triton X-100, 20% glycerol, stained with DAPI (0.01 μg ml−1). Analysis was conducted on the LSR II (BD Biosciences). Cell sorting was conducted using the FACS Aria IIu (BD Biosciences) or the MoFlo Astrios EQ (Beckman Coulter). All flow cytometric analysis was conducted in FlowJo v.10 (BD Biosciences).
Quantification and statistical analysis
For experimental results, statistics were calculated in GraphPad Prism 8, and the unpaired, two-tailed Student’s t-test was applied. Results equal to or above a 95% confidence interval (P ≤ 0.05) were considered statistically significant. Unless P values are specifically identified, they are notated as follows: NS, P ≥ 0.05; *P = 0.01 to ≤ 0.05, **P = 0.001 to < 0.01, ***P = 0.0001 to < 0.001, ****P ≤ 0.0001. Unless otherwise specified, bar plots or the centre lines in box plots represent mean and error bars represent s.d. For all experimental results, the n is reported in the accompanying figure legend. For in silico analyses, statistics were computed in the R software environment.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Extended Data
Extended Data Fig. 1 |. Generation and characterization of H1cTKO;Vav-iCre mice.

a, Unsupervised clustering of 23 primary mouse tissues by H1 subtype protein content and percent H1c/H1d/H1e as determined by quantitative RP-HPLC of histone proteins isolated from each tissue. b, Schematic of the conditional Hist1h1d knockout in an Hist1h1c/Hist1h1e double knockout background (left). Genotyping primers are indicated. P1: 5′AAGTAGAGTGGTGCGTCCTGCTTTG P2: 5′ GCCAGAAGTCCAGCTAGAGCTGAAA. Representative genotyping results using primers which expand give a 239-bp amplicon in WT mice and a 340-bp amplicon when the Hist1h1d locus is flanked by loxP sites (right). All mice used in this study were genotyped in this manner. c, Hist1h1d conditional deletion efficiency was measured in genomic DNA isolated from peripheral blood of WT, H1cTKO and H1cTKO;Vav-iCre mice (n = 2) using primers targeting the Hist1h1d locus. d, H1 subtype mRNA transcripts in WT, H1cTKO and H1cTKO;Vav-iCre mice (n = 2) were measured by RT-qPCR and normalized to Gapdh mRNA using the ΔΔCt method. e–g, Acid extracted histones from (e) thymus (n = 2), (f) spleen (n = 4) and (g) bone marrow (n = 4) of WT and H1cTKO;Vav-iCre mice were analysed by quantitative HPLC and the relative abundance of each H1 subtype to H2B determined by dividing the area under the H1 A214 peak by one-half of the area under the H2B A214 peak. Total H1:nucleosome ratio was determined by summation of the areas under each H1 subtype peak relative to one-half the area under the H2B peak. Absorbance values of the H1 and H2B peaks were normalized to peptide bond number. h, The total number of WBCs in peripheral blood obtained from the facial vein of WT (n = 4), H1cTKO (n = 4) and H1cTKO;Vav-iCre (n = 4) was measured by automated CBC (Oxford Scientific). i, Flow cytometry strategy for separating B cells, and CD4+ and CD8+ T cells from spleens (shown) and other tissues. Corresponds to Fig. 1b, Extended Data Fig. 2e. j, Flow cytometry strategy for identifying immature and mature T cell populations in thymus. Corresponds to Fig. 1c. Data are mean ± s.d. nd, not detected; ns, not statistically significant; *P ≤ 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, unpaired t-test.
Extended Data Fig. 2 |. Analysis of haematopoietic cells in H1-depleted H1cTKO;Vav-iCre mice.
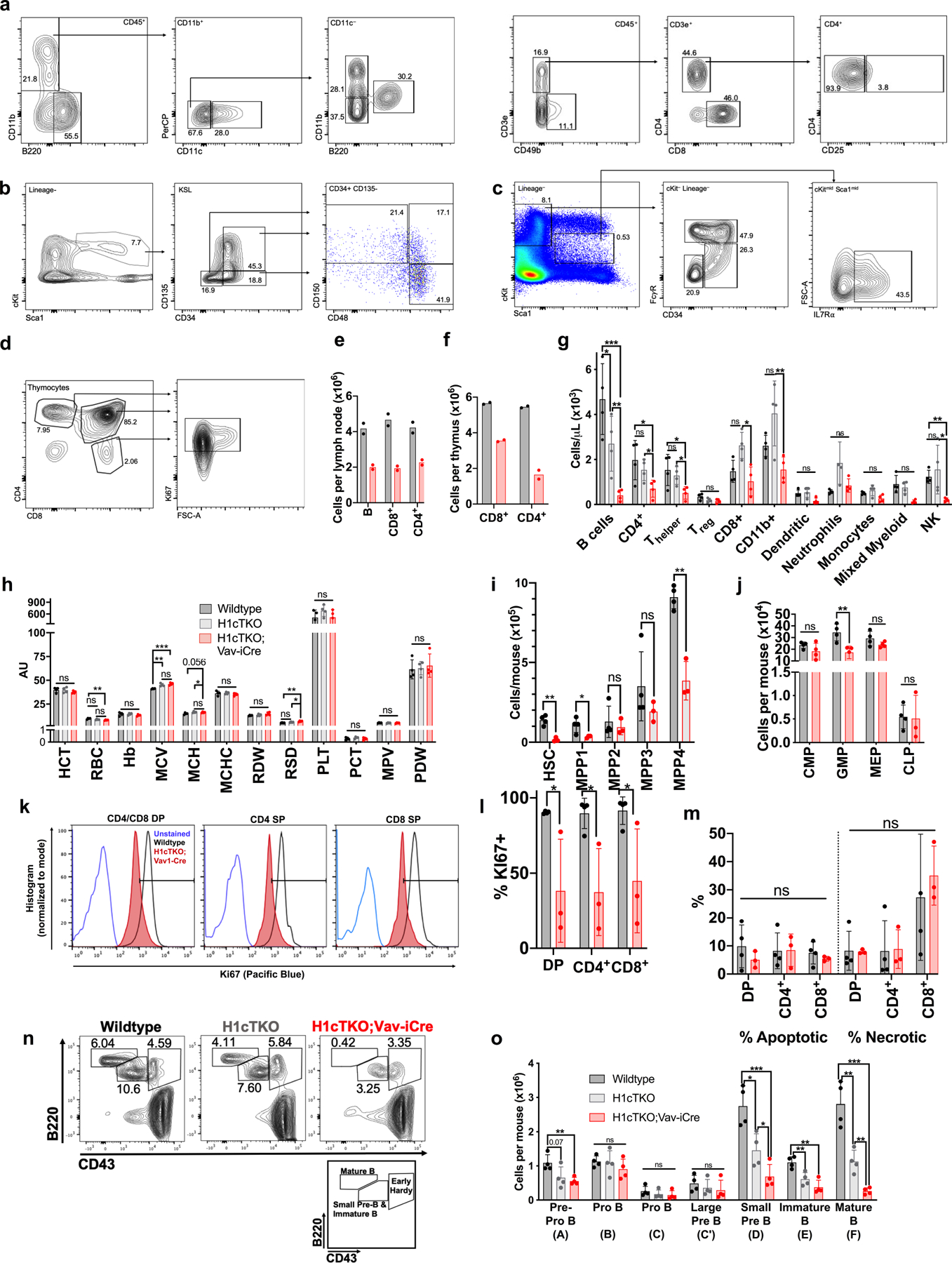
a, Flow cytometry strategy for identifying mature myeloid and lymphoid cell types in peripheral blood. Corresponds to g. Strategy was separated in two panels: B cells, dendritic cells, neutrophils, monocytes, and mixed myeloid (left), and NK and T cell populations (right). Corresponds to g. b, Flow cytometry gating strategy for the identification of HSCs and multipotent progenitors (MPPs). Corresponds to i. c, Flow cytometry gating strategy for the identification of the following lineage-specific progenitors; CMP, GMP, MEP and CLP. Corresponds to j. d, Flow cytometry gating strategy for measuring Ki67 positivity of CD4+ and CD8+ T cells. Corresponds to l. e, The total number of B cells, and CD4+ and CD8+ T cells in lymph nodes (LN) of WT and H1cTKO;Vav-iCre mice (n = 2) was measured by flow cytometry. f, The total number of CD4+ and CD8+ T cells in thymus of WT and H1cTKO;Vav-iCre mice (n = 2) was measured by flow cytometry. g, h, Peripheral blood obtained from the facial vein of WT, H1cTKO, and H1cTKO;Vav-iCre mice (n = 4) for analysis by flow cytometry and automated CBC (Oxford Scientific) showing (g) the total number of the indicated populations of mature lymphoid and myeloid cell types, and (h) normalized parameters of red blood cell and platelet count and morphology, as determined by CBC. T helper cells were defined as CD45+CD3e+CD49b−CD4+CD25−. Regulatory T (Treg) cells were defined as CD45+CD3e+CD49b−CD4+CD25+. i, Number of HSCs and various MPP populations in bone marrow of WT (n = 4) and H1cTKO;Vav-iCre (n = 3) mice. Surface markers used to define the various populations can be found in Supplementary Table 4. j, Number of myeloid and lymphoid progenitors in bone marrow of WT (n = 4) and H1cTKO;Vav-iCre (n = 4) mice. Surface markers for the identification of progenitors were as follows: CMP, CD34+ FcgR III/II− c-Kit+ Sca1− Lineage−; GMP, CD34+ FcgR III/II+ c-Kit+ Sca1− Lineage−; MEP, CD34− FcgR III/II− c-Kit+ Sca1− Lineage−; CLP, IL7R-α− (CD127)+ c-Kitmid Sca1mid Lineage−. k, l, Thymocytes were isolated from WT (n = 4) and H1cTKO;Vav-iCre (n = 3) mice and stained for the intranuclear antigen Ki67 for the examination of cellular proliferation. Representative Ki67 histogram is shown in k, quantification is shown in l. m, Thymocytes were isolated from WT (n = 4) and H1cTKO;Vav-iCre (n = 3) and stained for the surface marker Annexin V and propidium iodide for the assessment of apoptosis and necrosis. Staining was analysed by flow cytometry. n, Representative flow cytometry plots showing the number of small pre-B plus immature B cells, mature B cells, and early Hardy fraction cells as a percent of live cells in bone marrow from WT, H1cTKO, and H1cTKO;Vav-iCre mice. o, Hardy Fraction analysis of B cell maturation in bone marrow from WT (n = 4), H1cTKO (n = 4) and H1cTKO;Vav-iCre (n = 4) mice. The abundance of each population was calculated by multiplying the relative frequency of each population by bone marrow cellularity. Surface markers used to define the various populations can be found in Supplementary Table 4. Data are mean ± s.d., unpaired t-test, ns, not statistically significant; *P ≤ 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Extended Data Fig. 3 |. Ex vivo proliferation and cell cycle analysis of WT, H1cTKO and H1cTKO;Vav-iCre B and T cells.
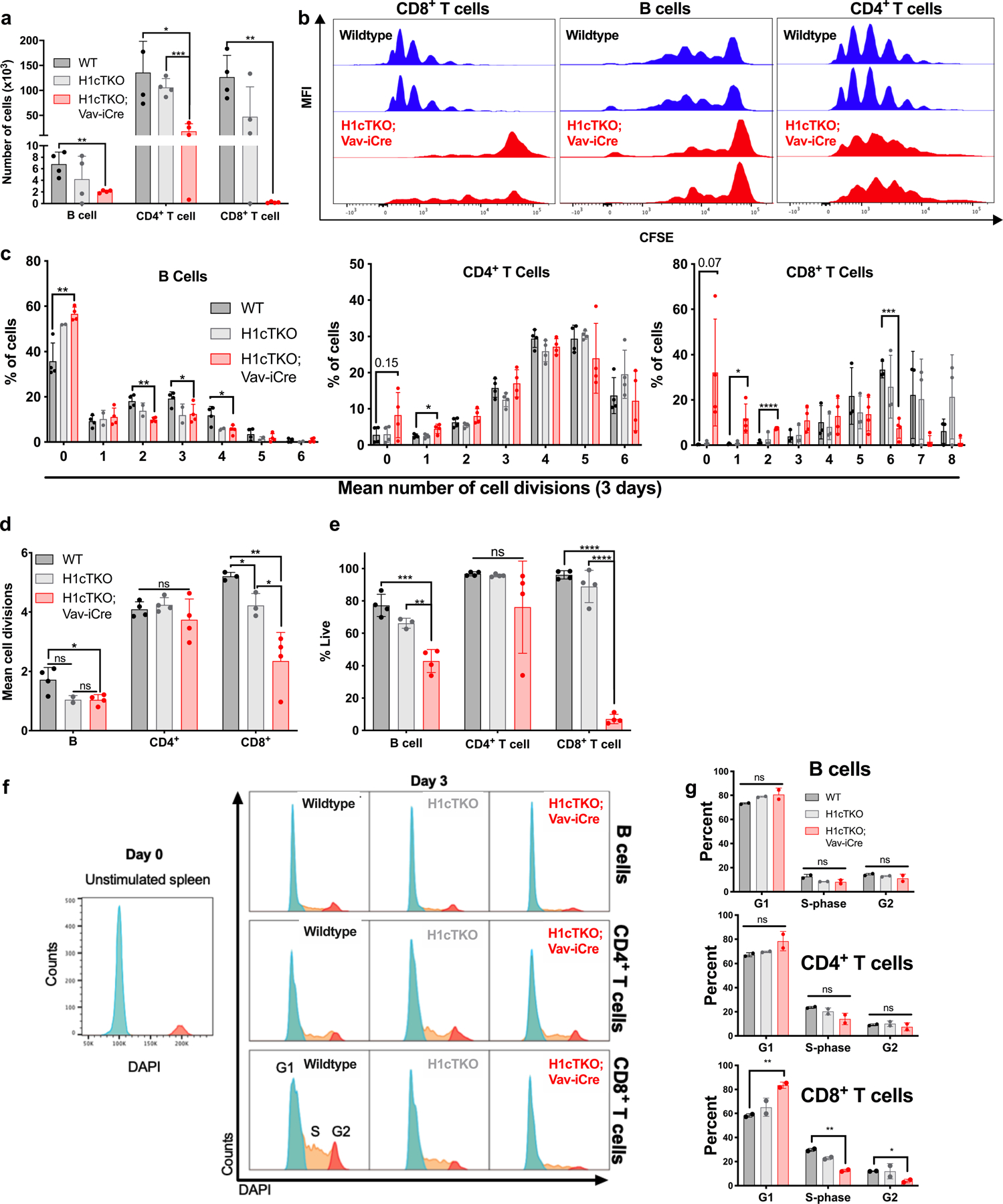
a–e, 50,000 splenic B and 25,000 splenic T cells were isolated by FACS and stained with CellTrace Violet for 30 min at 37 °C in PBS and then washed and plated in stimulation media. B cells were stimulated with IL-4 and LPS. T cells were stimulated with CD3/CD28 beads and IL-2. All cells were cultured for 3 days. Cell division was measured by flow cytometry analysis. Cells derived from n = 4 mice per genotype. a, Cell output of B cells and CD4+ and CD8+ T cells isolated from WT, H1cTKO and H1cTKO;Vav-iCre mice was determined by flow cytometry using AccuCheck beads. b, Representative plots for CD8+ T cells, B cells and CD4+ T cells. c–e, Quantitation of cell divisions (c), mean cell divisions (d) and viability (e) in B cells, and CD4+ and CD8+ T cells are shown. f, g, 2 × 106 splenic B and 1 × 106 splenic T cells were isolated by FACS and stimulated for two days before cell cycle analysis. B cells were stimulated with IL-4 and LPS. T cells were stimulated with CD3/CD28 beads and IL-2. For cell cycle analysis, cells were permeabilized, stained with DAPI and analysed by flow cytometry. f, Representative histograms of DAPI staining in WT, H1cTKO and H1cTKO;Vav-iCre B, CD4+ and CD8+ T cells. Phases of the cell cycle are indicated. g, Quantification of cell cycle phases in c in WT, H1cTKO and H1cTKO;Vav-iCre B, CD4+ and CD8+ T cells (n = 4). Data are mean ± s.d., n = 4 mice per genotype; unpaired t-test; ns, not significant, P ≤ 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Extended Data Fig. 4 |. Adoptive transfer experiments with H1-depleted splenic T cells from H1cTKO;Vav-iCre mice.

a, Schematic diagram depicting adoptive T cell transfer to Rag1KO mice, which are deficient in mature T and B cells, and to congenic CD45.1 mice. In brief, T cells were isolated from WT and H1cTKO;Vav-iCre mice by affinity Dynabead selection. In the case of transfer to Rag1KO mice the cells were stained with CellTrace CFSE. 1 × 106 cells were transplanted into recipient Rag1KO or CD45.1 mice via injection in the retro-orbital sinus. After 1 week, mice were euthanized, and adoptive T cell transfer was assessed by flow cytometry of harvested spleens. T cell, spleen and syringe images are from Servier Medical Art. b, c, Cell division of CD8+ T cells transplanted in Rag1KO mice was measured by flow cytometry (two representative traces of each genotype are displayed) after staining with CellTrace CFSE (g), and the percentages of CD4+ and CD8+ T cells that underwent <7 cell divisions were quantified (h, n = 6 mice per genotype). d, Number of CD4+ and CD8+ T cells in spleens of Rag1KO recipient mice 7 days after transfer of donor WT or H1cTKO;Vav-iCre T cells. N = 6 mice per genotype. e, f, T cells were isolated from WT and cTKO spleens and lymph nodes by column separation. 1 × 106 were injected into congenic C57BL/6 CD45.1 recipient mice. After 1 week, mice were sacrificed and adoptive T cell transfer was assessed by flow cytometry of donor CD45.2+ CD4+ and CD8+ T cells in harvested spleens. e, Representative flow cytometry plots and (f) quantification of donor-derived CD45.2+ CD4+ and CD8+ T cells in recipients of T cells from WT (n = 5) and H1cTKO;Vav-iCre (n = 5) mice is shown. Each data point represents the average of two recipients per donor. g, h, Cells were isolated from wild-type and H1cTKO;Vav-iCre spleens and LNs, and were stained for CD4, CD8, and with a fluorophore-conjugated anti-Fas antibody. Fas expression was assessed by flow cytometry. g, Representative histograms are shown alongside (h) quantification of Fas expression in CD4+ and CD8+ cells derived from WT (n = 4) and H1cTKO;Vav-iCre (n = 4) mice. Data are mean ± s.d.; unpaired t-test. ns, not statistically significant; * P ≤ 0.05; ** P < 0.01; *** P < 0.001.
Extended Data Fig. 5 |. H1 density regulates higher order chromatin structure in vivo.

a, b, Freshly isolated spleens from WT and H1cTKO;Vav-iCre mice were prepared for transmission electron microscopy (TEM) to visualize (a) nuclear size and (b) chromatin density, as described. Nuclear area and nuclear density were quantified using ImageJ. WT nuclei (n = 22) and H1cTKO;Vav-iCre nuclei (n = 17). Images were selected at random using several sections. The unpaired t-test was used. Whiskers represent minima and maxima, boxes represent the 25%-to-75%, with the mean shown. c, The average C-scores of topologically associated domains (TADs) in each condition were calculated. The Hi-C A and B compartments are characterized by a positive and negative C-score, respectively. TADs with differences in C-score between H1cTKO;Vav-iCre and WT (dCS) >0.25 were considered decompacted, and those with dCS <-0.25 compacted. TADs which changed from chromatin compartments A-to-B or B-to-A were defined as 100 kb regions with a dCS >0.25 or dCS <-0.25 and a change in the C-score sign. The rolling average change was locally estimated with a scatterplot smoothing (loess) curve. d, Change in TAD compartment score as a function of TAD size. e, Meta-analysis of A-B compartment TAD borders which were altered upon H1 depletion demonstrating A compartment spreading into B TADs. f, A genome-wide meta-region visualization of the Hi-C contact frequencies for all regions which differed between wild-type and H1cTKO;Vav-iCre CD8+ T cells. Heatmaps of the Hi-C contact frequency for wild-type (top) and H1cTKO;Vav-iCre (bottom) meta-regions were prepared using coolpup.py. g, A 15-state chromatin-state model was established in CD8+ T cells using ChromHMM. Segmentation was performed with binarized bed files for H3K36me2, RNA-seq gene expression (GE), Pol II Ser2P, H3K4me3, ATAC-seq, H3K9me3, H3K27me3, CTCF, H3K4me1, H3K36me2 (accession numbers are available in Supplementary Table 2; also see Methods). Heat maps for model parameters are shown. h, ATAC-seq was performed on WT and H1cTKO;Vav-iCre splenic CD8+ T cells and random sampling of reads was performed as needed to ensure the same number of reads between samples. Read lengths were calculated for ATAC-seq sequencing reads that fell completely within each chromatin state and are plotted as the number of reads for a given read length divided by the size of each chromatin state in base pairs. The change in nucleosome repeat length (NRL; “left shift”) and the change in chromatin accessibility (“up shift”) within nucleosomal DNA were calculated using custom Python scripts, entitled “NRLfinder,” which we have made available as an executable file (see Methods). Both the left shift, in base-pairs, and the up shift, as a percent of wildtype, are indicated on each plot along with an arrow indicating the direction of change. Plots are grouped by “Chromatin Type”, and types are determined based on their behaviour upon H1 depletion: unaffected (Type 1), left shift (Type 2), left shift and up shift (Type 3). i, Contribution of Types 1, 2, and 3 chromatin in CD8+ T cells to the whole genome, and to the A and B Hi-C compartments. j, The Hi-C C-score for each genomic region is shown, grouped by chromatin state. Chromatin states are grouped in Type 1, 2, and 3 chromatin, shown below. k, l, 100 kb genomic segments composed purely (>95%) of (k) Type 3 and (l) Type 2 were analysed. In brief, the C-score was calculated for each 100 kb genomic segment in WT and H1cTKO;Vav-iCre CD8+ T cells using the CscoreTool and analysed as in c. m, Types 1, 2, and 3 chromatin were identified in mESCs using a previously reported 15-state ChromHMM model25. The density distribution of normalized H1 ChIP signals from publicly available H1 ChIP-seq in mESCs27 in Types 1, 2, and 3 chromatin are shown. Density was calculated using geom_density (ggplot2). n, Pooled MNase-digested chromatin isolated from wild-type mouse splenocytes (n = 9 mice) was subjected to sucrose gradient ultracentrifugation to isolate mono- and polynucleosomes. Left, agarose gel demonstrating DNA size distributions within each gradient fraction. Right, samples were subjected to mass spectrometry and H1 peptides corresponding to H1c, H1d and H1e were normalized to total peptide abundance. Shown are the Z-scores for each peptide (diamond, H1c; square, H1d; triangle, H1e) and average Z-score (blue/red bar) within each fraction. Note: ns, not statistically significant; * P ≤ 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
Extended Data Fig. 6 |. H1 promotes H3K27 methylation and inhibits H3K36 methylation in vivo.
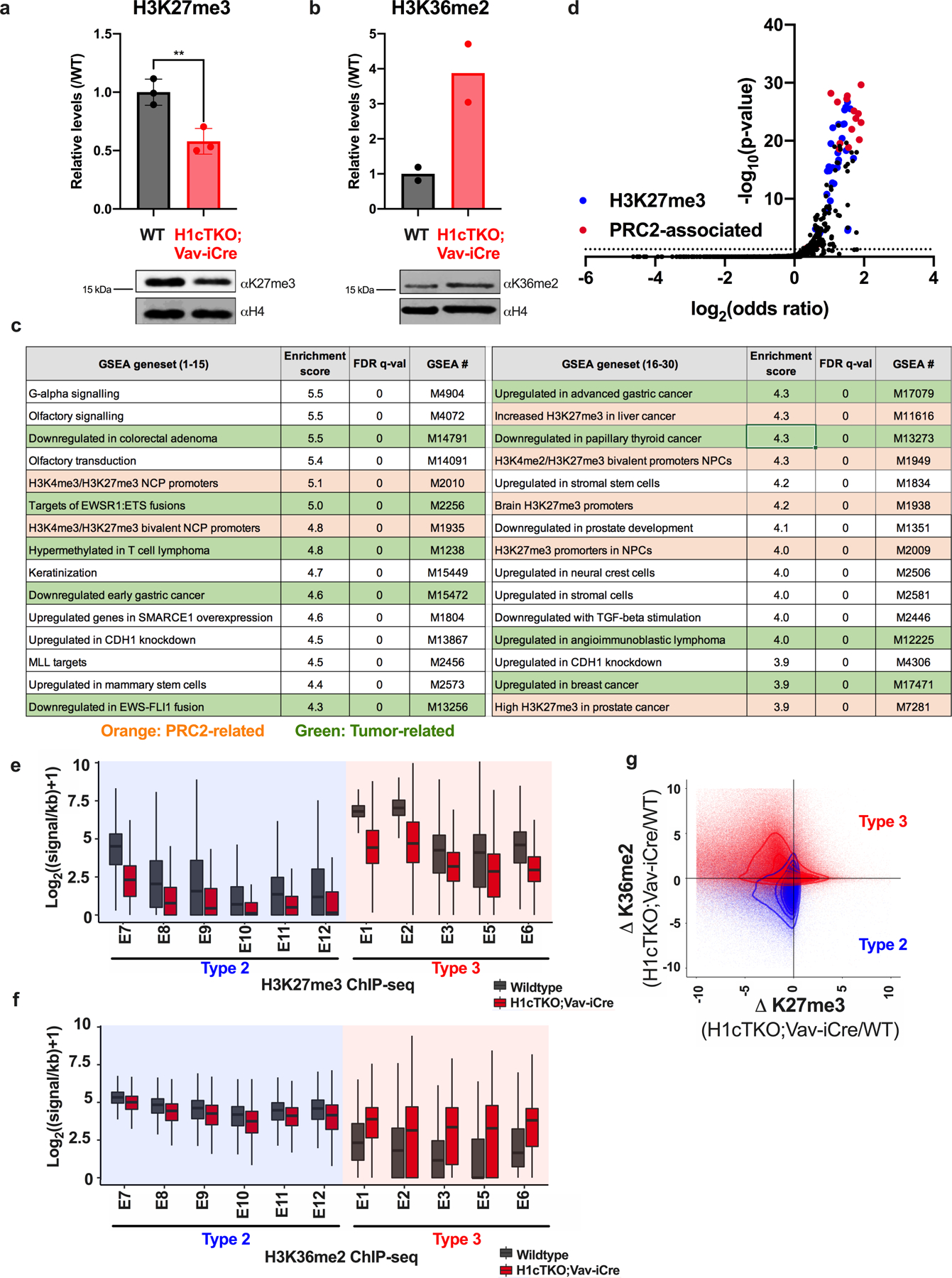
a, b, H3K27me3 and H3K36me2 levels in WT (black) and H1cTKO;Vav-iCre (red) spleens. Histone acid extracts were separated by SDS–PAGE and analysed by immunoblotting with the indicated antibodies. The intensity of H3K27me3 (a, n = 3) and H3K36me2 (b, n = 2) bands were quantified and normalized to the intensity of the corresponding H4 band Data are shown as mean ± s.d., unpaired t-test; ** P < 0.01. c, GSEA of genes with significant changes in Hi-C C-score upon H1 depletion. Gene sets associated with PRC2 are coloured in orange, and tumour-associated gene sets are in green. d, PRC2-associated and H3K27me3-associated genes were overlapped with genes contained in the chromatin compartments that decondense (B-to-A compartment change) with loss of H1 using Enrichr. The odds ratio (log2) is plotted as a function of adjusted P value (-log10). H3K27me3 and PRC2-related enrichments are colored in blue and red, respectively. e, f, ChIP-seq was performed for H3K27me3 (e) and H3K36me2 (f) in WT (black) and H1cTKO;Vav-iCre (red) CD8+ splenic T cells. Read counts were normalized between samples using THOR. The signal for each set of regions, defined by chromatin state, was calculated and the distribution of H3K27me3 and H3K36me2 signals for all regions of a given chromatin state are shown. Chromatin states are grouped into Type 2 and Type 3 chromatin, indicated below. Whiskers represent minima and maxima, boxes represent the 25%-to-75%, with the mean shown. g, Contour plot of the fold-change of H3K27me3 and H3K36me2 levels between H1cTKO;Vav-iCre and WT CD8+ splenic T cells for all regions of Type 2 (blue) and Type 3 (red) chromatin. *For gel source data, refer to Supplementary Fig. 1.
Extended Data Fig. 7 |. H1 promotes PRC2-mediated H3K27 trimethylation and inhibits NSD2-mediated H3K36 dimethylation in vitro.
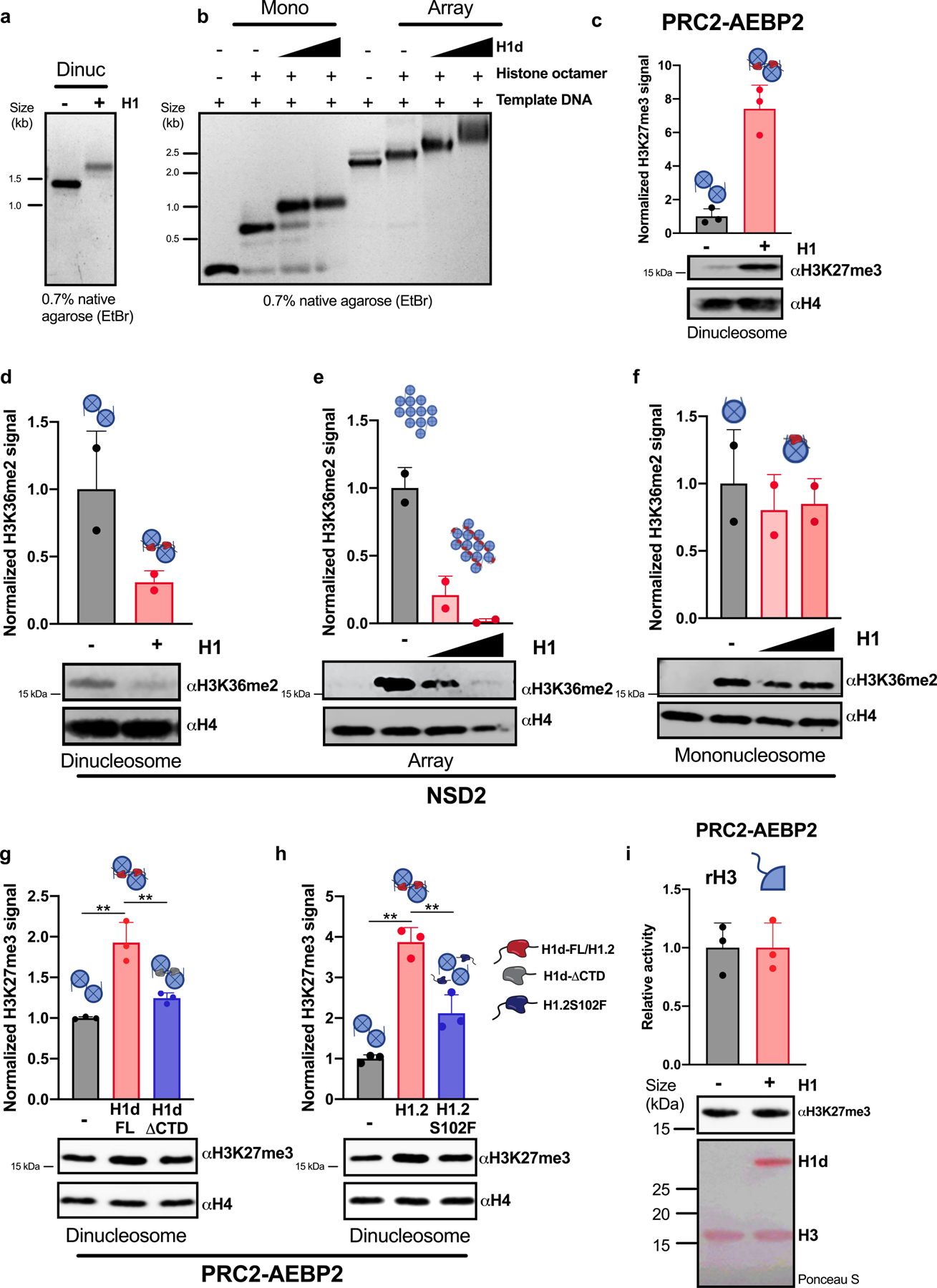
a, b, Chromatin was reconstituted in vitro using DNA templates bearing one, two or twelve repeats of the synthetic ‘601’ nucleosome positioning sequence and recombinant histone octamers either in the presence or absence of H1 proteins. Linker histone incorporation was verified by non-denaturing agarose gel electrophoresis followed by ethidium bromide staining. Representative gels are shown. Optimal H1:nucleosome ratios were determined empirically. In a, a 0.8 H1d:nucleosome ratio was used. In b, H1d:nucleosome ratios of 0.7:1 and 1.3:1 were used. c–h, In vitro histone methyltransferase (HMT) assays with reconstituted chromatin in the presence or absence of the indicated H1 proteins: (H1: M. musculus H1d; H1.2: H. sapiens H1.2; H1.2S102F; H1d-FL: H1d(1–221); H1d-ΔCTD: H1d(1–114). Chromatin was reconstituted in vitro using a DNA template bearing one (f), two (c, d, g, h) or twelve (e) repeats of the synthetic ‘601’ nucleosome positioning sequence and recombinant histone octamers either in the presence or absence of H1; optimal H1:nucleosome ratios were determined empirically. Chromatin substrates were incubated with the indicated HMT enzyme under conditions described in the Methods. HMT activity was detected by immunoblotting for the indicated histone modifications. Immunoblots of H4 are shown as a loading control. H3K27me3 and H3K36me2 signals were quantified by densitometry and normalized to the intensity of the corresponding H4 band; quantification is shown as relative activity compared to substrates in the absence of H1. i, In vitro HMT assay of histone H3 in the presence or absence of H1d. H3 was incubated with PRC2-AEBP2 complex in the presence or absence of equimolar quantities of H1d. HMT activity was detected by immunoblotting for H3K27me3. Above, quantification of activity of 3 independent HMT reactions relative to reactions lacking H1d. Middle, representative immunoblot demonstrating enzymatic activity. Below, Ponceau S stain of transferred proteins showing equal quantities of H3. Quantification of 2 (d–f) or 3 (c, g–i) independent experiments is shown above each immunoblot. Data are presented as mean values ± s.d., unpaired t-test; ns, not statistically significant; * P ≤ 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. *For gel source data, refer to Supplementary Fig. 1.
Extended Data Fig. 8 |. H1 depletion phenocopies loss of Ezh2 and H1 is required for compaction and epigenetic marking of genes controlling T cell activation.
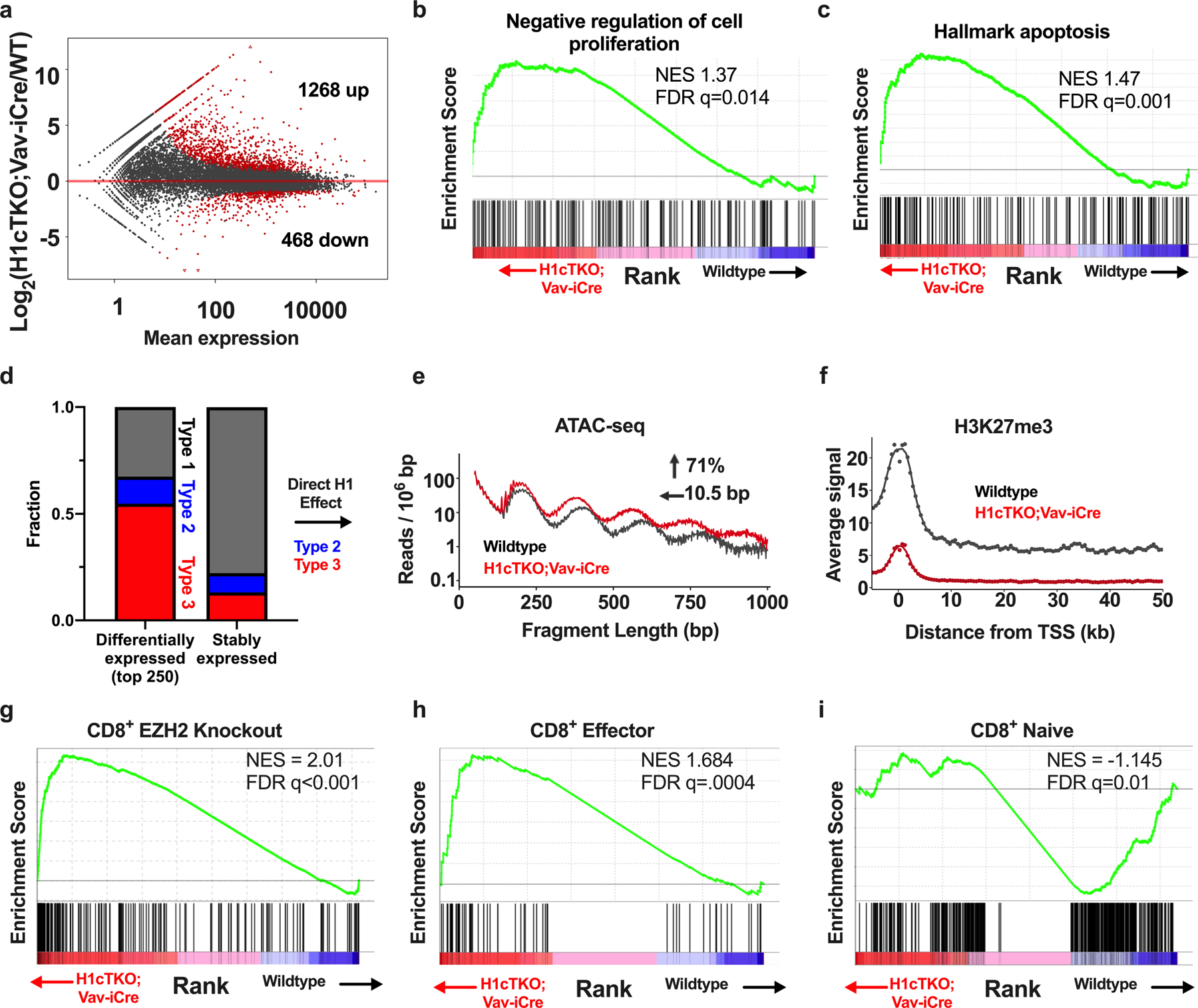
a, Poly-A+ RNA-seq was performed on splenic CD8+ T cells from WT and H1cTKO;Vav-iCre mice. Sequencing reads were processed (see Methods) and quantified using DESeq2 and custom R scripts. Mean expression as a function of the fold change is plotted for each gene. Differentially expressed transcripts, indicated in red, were defined as those with an adjusted P value of <0.05. b, c, Quantified transcripts from poly-A+ RNA-seq of wild-type and H1cTKO;Vav-iCre CD8+ T cells were used as inputs for GSEA to determine significant overlaps between genes that are differentially regulated upon H1 depletion and gene sets within the GSEA database. Shown is an enrichment plot and scores of differentially expressed genes in a set of genes associated with negative regulation of cell proliferation (b) and apoptosis (c). d, RNA-seq was performed on CD8+ T cells from wild-type and H1cTKO;Vav-iCre mice and differentially expressed genes and genes unchanged in expression were identified using DESeq2. The 250 most upregulated genes compared with genes not exhibiting altered expression were selected using the adjusted P value. The chromatin “Type” of the TSS of each gene was determined by identifying the ChromHMM regions from our 15-state map that fell within a ± 200 bp window of each TSS and annotating each of these states with the assigned chromatin type. The fractional composition of chromatin types 1 (grey), 2 (blue) and 3 (red) was compared between the 250 most upregulated genes in H1cTKO;Vav-iCre compared to wild-type cells and genes that had unchanged expression. e, ATAC-seq was performed on wild-type (black) and H1cTKO;Vav-iCre (red) CD8+ T cells. ATAC-seq reads that fell within H1-dependent chromatin (Type 2 and Type 3) of the top 250 most differentially expressed genes were identified. ATAC-seq read lengths (bp) were calculated and the data are plotted as the number of reads for a given read length normalized to region size. The change in NRL and accessibility (area-under-the-curve) were calculated using NRLfinder. f, Regions surrounding the TSS of each of the top 250 most differentially expressed genes (analysed in a) were identified and stacked to create a “metagene plot.” The average ChIP-seq signal intensity for H3K27me3 in wild-type (black) and H1cTKO;Vav-iCre (red) CD8+ cells is shown for the metagene. g–i, Quantified transcripts from poly-A+ RNA-seq of wild-type and H1cTKO;Vav-iCre CD8+ T cells were used as inputs for GSEA to determine significant overlaps between genes that are differentially regulated upon H1 depletion and gene sets within the GSEA database. Shown are enrichment plots and scores of differentially expressed genes in a set of (g) EZH2 regulated genes in CD8+ T cells, (h) CD8+ effector T cell genes, and (i) CD8+ naive T cell genes. GSEA plots are shown with normalized enrichment scores and q-values. CD8+ effector and naive T cell gene sets are from Pace et al.29.
Extended Data Fig. 9 |. H1 depletion leads to de-repression of genes associated with T cell activation and apoptosis.
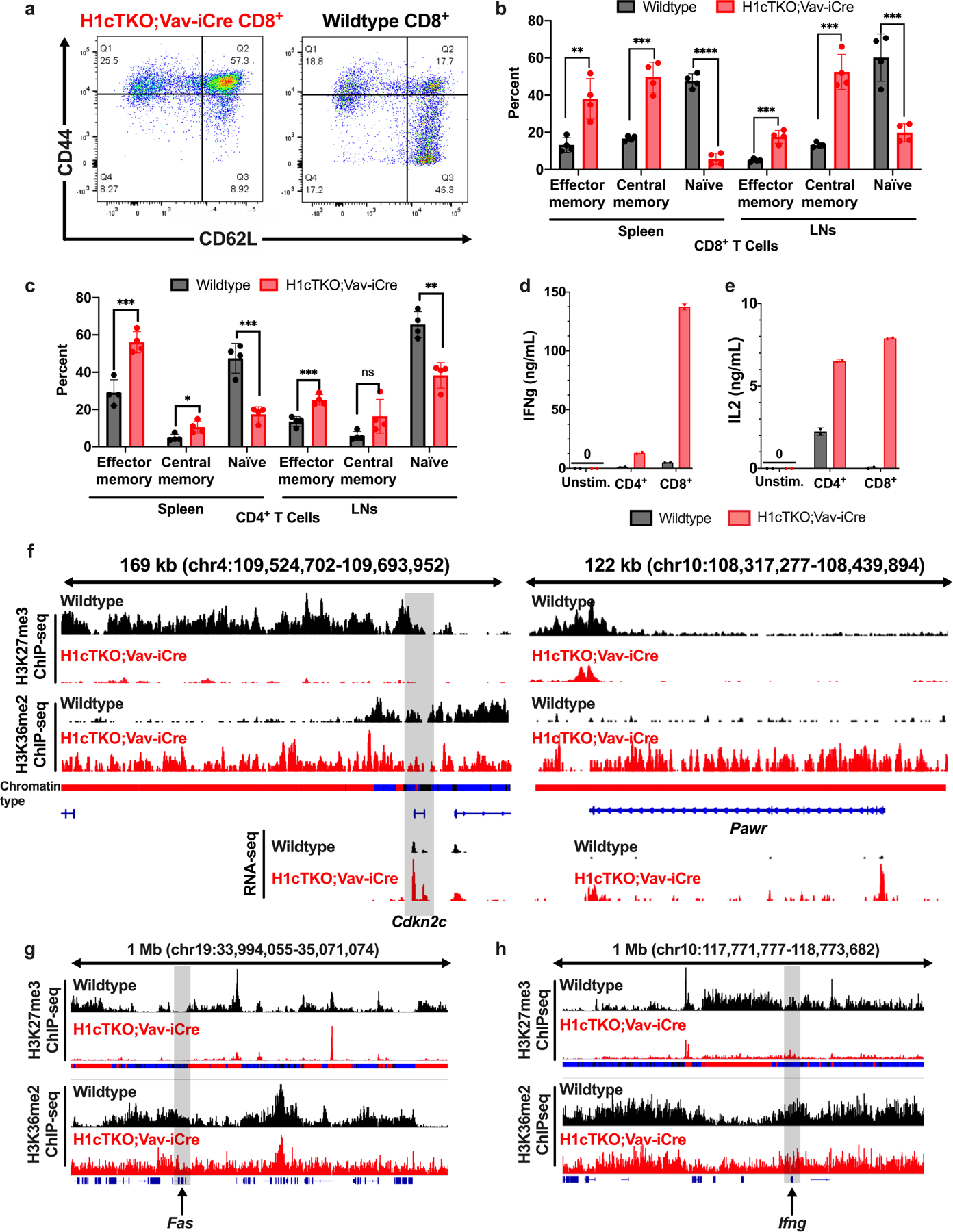
a, A single cell suspension was prepared from spleens of wild-type (right) and H1cTKO;Vav-iCre (left) mice and ACK lysis was performed to remove red blood cells. Cells were stained for CD8+ T cell markers (CD45+CD3e+CD8+) as well as markers for T cell activation, CD44 and CD62L, and analysed by flow cytometry. Representative flow plots are shown. b, c, Analysis of (b) CD8+ and (c) CD4+ lymphocyte cell types from spleen and LN of WT and H1cTKO;Vav-iCre mice. Shown is each cell type calculated as a percent of CD45+CD3e+CD8+ or CD45+CD3e+CD4+ (n = 4 for each genotype). d, e, CD8+ T cells were isolated from wild-type (n = 2 mice, black) and H1cTKO;Vav-iCre (n = 2 mice, red) mice by column purification. Cells were then stimulated in T cell medium containing anti-CD3e/28. After 22 h the medium was collected and the secretion of (d) IFNγ and (e) IL-2 was determined by ELISA. Data in b–e is mean ± s.d., unpaired t-test; ns, not statistically significant; *P ≤ 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. f–h, H3K27me3 (top two tracks) and H3K36me2 (bottom two tracks) ChIP-seq signals in WT (black) and H1cTKO;Vav-iCre (red) CD8+ splenic T cells are shown for IGV genome browser views (mm10 genome assembly) containing the (f) Cdkn2c (left) and Pawr (right), Fas (g) and Ifng (h) genes. Shown are chromatin states (bars below the top two tracks) in wild-type CD8+ splenic T cells annotated as Type 1 (black), Type 2 (blue) and Type 3 (red), and quantified transcripts from poly-A+ RNA-seq (lowest two tracks). g, h, H3K27me3 (top two tracks) and H3K36me2 (bottom two tracks) ChIP-seq signals in WT (black) and H1cTKO;Vav-iCre (red) CD8+ splenic T cells are shown for a 1 Mb IGV genome browser view (mm10 genome assembly) containing the (g) Fas and (h) Ifng genes.
Extended Data Fig. 10 |. Chromatin remodelling and epigenetic changes associated with H1 depletion also occur during normal T cell activation.

a, RNA-seq data12 from effector CD8+ T cells was compared with naive T cells and the 250 most upregulated genes, the 250 most downregulated genes and 250 genes not exhibiting altered expression were selected using the adjusted P value produced by DESeq2. The chromatin state of each gene was determined by identifying the ChromHMM regions from our 15-state map that fell within a window of -200 bp of each TSS through the gene body. The relative abundance of each chromatin type within each gene set is shown as a bar plot and the chromatin types are annotated as Type 1 (black), Type 2 (blue) and Type 3 (red). The set of all genes is shown as a reference. b, ATAC-seq read lengths (bp) were calculated from publicly available ATAC-seq data12 from naive (black) and effector (green) T cells of wild-type mice for all regions of upregulated gene bodies that fall completely within Type 3 chromatin states. The data are plotted as the number of a given read length normalized to region size in base pairs. The change in accessibility (%) and NRL (bp) are shown as determined by NRLfinder. c, Regions surrounding the TSS of genes identified as in b were identified and stacked to create a “metagene plot.” The average ChIP-seq signal intensity for H3K27me3 in naive and effector T cells is shown (reanalysed from data produced and stored in the Gene Expression Omnibus repository GSE111902). d, Transcripts from WT CD8+ T cells were divided into 10 categories based on expression (with “0” representing an additional category of genes with little or no expression). The average NRL of gene bodies within each expression category are plotted against the average CUT&Tag H1 to H3 ratio within the same regions. Type 2 (blue) and Type 3 (red) chromatin designations of each gene expression category (based on Fig. 4c) are shown. A linear regression with 95% confidence interval is shown. e, Top, a linear genome browser view of chr2:11,774,283–13,172,671 in summary of the data presented herein. Shown from top to bottom are the following from WT (black) and H1cTKO;Vav-iCre (red) CD8+ T cells: ChIP-seq for H3K27me3 (tracks 1–2), ChIP-seq for H3K36me2 (tracks 3–4), RNA-seq reads (tracks 5–6), defined Chromatin Types 1 (black), 2 (blue) and 3 (red) (track 7), and C-score-defined Hi-C chromatin compartments A and B (tracks 8–9). Bottom, a summary figure of the proposed roles for differences in H1 stoichiometry in regulating local genome architecture, the deposition of core-histone post-translational modifications by modification of the chromatin substrate, and changes in gene expression identified from observed differences in wild-type and H1cTKO;Vav-iCre CD8+ T cells.
Supplementary Material
Acknowledgements
We thank the Albert Einstein College of Medicine Analytical Imaging Facility for their assistance in preparing samples for transmission electron microscopy and acquiring images, and the Flow Cytometry Core Facility (NCI P30CA013330). We thank D. Reinberg for preparations of the PRC2–AEBP complex; M. Gamble, C. Query and all members of the Skoultchi laboratory for stimulating scientific discussions; G. Yu, B. Will, K. Gritsman, A. Emelyanov, D. Fyodorov and M. Scharff for reagents and expert advice. This work was supported in part by funds from the NIGMS (R01GM116143 to A.I.S.; GM110104 to B.A.G.), the National Cancer Institute (F30CA210539 to S.E.H.; CA196539 to B.A.G.) and the National Institute of Diabetes and Digestive and Kidney Diseases (F30DK108532 to C.N.W.; F30DK107182 to M.A.W.) and the National Institute of Allergy and Infectious Disease (AI118891 to B.A.G.). E.C. and A.M.M. are funded through NIH/NCI R01 CA234561 and STARR I9-A9-062. In addition, this work is supported by an NIH, NIGMS MSTP Training Grant T32GM007288 (S.E.H., C.N.W. and M.A.W., under M. Akabas (programme director)).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-020-3032-z.
Code availability
The code for the NRLfinder generated and used in this study is available at https://github.com/tommyjohn21/nrl_finder and in Supplementary File 3. To facilitate the reproducibility of this work, and the utilization of the data and analyses in future studies, we have made the full suite of tools and custom-code used for the processing and analysis of the CUT&Tag, RNA-seq, ATACseq, ChIPseq and Hi-C data, as well as the methods for the development of the ChromHMM model used for chromatin-state modelling in this manuscript available in Supplementary File 1.
Competing interests The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41586-020-3032-z.
Peer review information Nature thanks Iannis Aifantis, Ralph Stadhouders and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
The sequencing datasets generated in this study are available at the Gene Expression Omnibus under accession code GSE141187. The nano liquid chromatography–mass spectrometry data generated in this study are available in Supplementary Table 6 and Supplementary File 2. Publicly available data used in this study is listed in Supplementary Table 7.
References
- 1.Woodcock CL, Skoultchi AI & Fan Y Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res 14, 17–25 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Fyodorov DV, Zhou BR, Skoultchi AI & Bai Y Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol 19, 192–206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan Y, Sirotkin A, Russell RG, Ayala J & Skoultchi AI Individual somatic H1 subtypes are dispensable for mouse development even in mice lacking the H10 replacement subtype. Mol. Cell. Biol 21, 7933–7943 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan Y et al. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123, 1199–1212 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Fan Y et al. H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. Mol. Cell. Biol 23, 4559–4572 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The FANTOM Consortium and the RIKEN PMI and CLST (DGT). A promoter-level mammalian expression atlas. Nature 507, 462–470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yusufova N et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature (in the press). [DOI] [PMC free article] [PubMed]
- 8.Ernst J & Kellis M ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1930 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin C, Cao R & Zhang Y Substrate preferences of the EZH2 histone methyltransferase complex. J. Biol. Chem 281, 8365–8370 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y et al. The polycomb repressive complex 2 governs life and death of peripheral T cells. Blood 124, 737–749 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Philip M et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu B et al. Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation. Nat. Immunol 18, 573–582 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reddy A et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell 171, 481–494 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landau DA et al. Mutations driving CLL and their evolution in progression and relapse. Nature 526, 525–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raymond CS & Soriano P ROSA26Flpo deleter mice promote efficient inversion of conditional gene traps in vivo. Genesis 48, 603–606 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44 (W1), W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.An L et al. OnTAD: hierarchical domain structure reveals the divergence of activity among TADs and boundaries. Genome Biol 20, 282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 2015, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corces MR et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet 48, 1193–1203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ & Prins P Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramrez F, Dndar F, Diehl S, Grning BA & Manke T deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bogu GK et al. Chromatin and RNA maps reveal regulatory long noncoding RNAs in mouse. Mol. Cell. Biol 36, 809–819 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brind’Amour J et al. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun 6, 6033 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Cao K et al. High-resolution mapping of h1 linker histone variants in embryonic stem cells. PLoS Genet 9, e1003417 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bushnell B, Rood J & Singer E BBMerge-accurate paired shotgun read merging via overlap. PLoS ONE 12, e0185056 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pace L et al. The epigenetic control of stemness in CD8+ T cell fate commitment. Science 359, 177–186 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Allhoff M, Ser K, Pires JF, Zenke M & G Costa I Differential peak calling of ChIP-seq signals with replicates with THOR. Nucleic Acids Res 44, e153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mombaerts P et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877 (1992). [DOI] [PubMed] [Google Scholar]
- 35.Faust GG & Hall IM SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang T et al. HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res 27, 1939–1949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin Q et al. Reductions in linker histone levels are tolerated in developing spermatocytes but cause changes in specific gene expression. J. Biol. Chem 279, 23525–23535 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Sirotkin AM et al. Mice develop normally without the H1(0) linker histone. Proc. Natl Acad. Sci. USA 92, 6434–6438 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sidoli S & Garcia BA Characterization of Individual Histone Posttranslational Modifications and Their Combinatorial Patterns by Mass Spectrometry-Based Proteomics Strategies vol. 1528 (Springer, 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sidoli S, Simithy J, Karch KR, Kulej K & Garcia BA Low resolution data-independent acquisition in an LTQ-Orbitrap allows for simplified and fully untargeted analysis of histone modifications. Anal. Chem 87, 11448–11454 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan ZF et al. EpiProfile 2.0: A computational platform for processing epi-proteomics mass spectrometry data. J. Proteome Res 17, 2533–2541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dyer P-N et al. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol 375, 23–44 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Mishra LN & Hayes J A nucleosome-free region locally abrogates histone H1-dependent restriction of linker DNA accessibility in chromatin. J. Biol. Chem 293, 19191–19200 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mishra LN, Pepenella S, Rogge R, Hansen JC & Hayes JJ Acetylation mimics within a single nucleosome alter local DNA accessibility in compacted nucleosome arrays. Sci. Rep 6, 34808 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Debelouchina GT, Gerecht K & Muir TW Ubiquitin utilizes an acidic surface patch to alter chromatin structure. Nat. Chem. Biol 13, 105–110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Healton SE et al. H1 linker histones silence repetitive elements by promoting both histone H3K9 methylation and chromatin compaction. Proc. Natl Acad. Sci. USA 117, 14251–14258 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee C-H et al. Allosteric activation dictates PRC2 activity independent of its recruitment to chromatin. Mol. Cell 70, 422–434 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing datasets generated in this study are available at the Gene Expression Omnibus under accession code GSE141187. The nano liquid chromatography–mass spectrometry data generated in this study are available in Supplementary Table 6 and Supplementary File 2. Publicly available data used in this study is listed in Supplementary Table 7.