

Abstract
Glioblastoma is a universally lethal cancer driven by glioblastoma stem cells (GSCs). Here, we interrogated N6-methyladenosine (m6A) mRNA modifications in GSCs by methyl RNA-immunoprecipitation followed by sequencing (meRIP-seq) and transcriptome analysis, finding transcripts marked by m6A often upregulated compared to normal neural stem cells (NSCs). Interrogating m6A regulators, GSCs displayed preferential expression, as well as in vitro and in vivo dependency, of the m6A reader, YTHDF2, in contrast to NSCs. While YTHDF2 has been reported to destabilize mRNAs, YTHDF2 stabilized MYC and VEGFA transcripts in GSCs in an m6A-dependent manner. We identified IGFBP3 as a downstream effector of the YTHDF2-MYC axis in GSCs. The IGF1/IGF1R inhibitor, linsitinib, preferentially targeted YTHDF2-expressing cells, inhibiting GSC viability without affecting NSCs and impairing in vivo glioblastoma growth. Thus, YTHDF2 links RNA epitranscriptomic modifications and GSC growth, laying the foundation for the YTHDF2-MYC-IGFBP3 axis as a specific and novel therapeutic target in glioblastoma.
Keywords: Glioblastoma, glioblastoma stem cell, glioma stem cell, cancer stem cell, m6A, YTHDF2, MYC, IGFBP3, linsitinib, epitranscriptomics
INTRODUCTION
Glioblastoma (World Health Organization grade IV glioma) represents the most common primary, intrinsic brain tumor with inevitable recurrence, limiting the median survival of patients to little more than a year (1,2). Glioblastomas display cellular hierarchies with self-renewing glioblastoma stem cells (GSCs) at the apex, with contributions of GSCs to therapeutic resistance and tumor recurrence (3-5). Standard-of-care therapy includes surgical resection followed by combined radiation and chemotherapy, and then adjuvant chemotherapy, but treatment remains palliative (6). Given the roles of GSCs in therapeutic resistance, angiogenesis, immune escape, and invasion, clinical and preclinical observations suggest that targeting GSCs may improve tumor outcome (7).
Precision medicine efforts in neuro-oncology have demonstrated limited benefit in most patients, prompting interrogation of epigenetic cell states to inform therapeutic targeting (8). Post-transcriptional regulation of RNA, epitranscriptomics, adds to the layers of regulation in normal and neoplastic cell biology. N6-methyladenosine (m6A) is the most prevalent eukaryotic RNA modification (9) and regulates the stability and translation of modified mRNAs (10-13). Like other methylation events, m6A is dynamically regulated by three classes of proteins: a ‘writer complex’ that installs m6A marks (methyltransferase components: METTL3, METTL14, and WTAP); ‘erasers’ that remove m6A marks (demethylases: FTO and ALKBH5); and ‘readers’ and other RNA-binding proteins that recognize and bind to m6A modified transcripts and mediate the downstream signaling events (m6A binding proteins: YTHDF1-3 and YTHDC1-2) (14,15). m6A writers (16-18) and m6A erasers (19-21) are differentially regulated in cancers, including glioblastoma, but the downstream connection between RNA m6A methylation and tumor biology is poorly understood. Recently, m6A readers were implicated in tumor biology by contributing to tumor growth, proliferation and survival (22-24). m6A reader proteins contain a YTH domain (YT521-B homology), which mediates specific RNA binding (25). There are five YTH domain-containing proteins, including YTHDC1, which is predominantly nuclear and regulates post-transcriptional splicing events (26), and YTHDC2 and YTHDF1-3, which are predominantly cytosolic. Some studies have suggested that recognition of m6A by YTHDF1 and YTHDF3 facilitates protein translation (27,28), whereas YTHDF2 and YTHDF3 transport mRNAs from actively translating pool of mRNAs to the decaying mRNA pool, leading to mRNA degradation (29). In contrast, more recent reports suggest that all three YTHDF proteins function to mediate degradation of m6A-tagged mRNAs (13,30). Here, we compared the transcriptomic m6A landscapes of normal and neoplastic stem cells, investigated the roles of m6A readers in GSCs and glioblastoma, and interrogated the mechanism through which these epitranscriptomic modifications support tumor biology. We report that YTHDF2 stabilizes important oncogenic drivers in an m6A-dependent manner specific to GSCs.
RESULTS
Oncogenic transcripts upregulated in GSCs are marked by RNA m6A modification
RNA methylation is one of the most prevalent RNA modifications in human cells (31). m6A decorates messenger RNAs, long non-coding RNAs, ribosomal, and spliceosome RNAs (32). To understand the global distribution of m6A marks, we performed transcriptome-wide m6A methylation analyses in two patient-derived GSCs (4121 and 387) and two NSCs (NSC11 and HNP1) (Supplementary Fig. S1A). m6A signals were normalized to respective inputs to adjust for baseline transcript expression levels. m6A peaks discriminated GSCs from NSCs in unsupervised clustering analyses (Supplementary Fig. S1B), suggesting that GSCs demonstrate altered m6A distribution compared to their non-neoplastic counterparts, confirmed by the identification of specific m6A peaks gained in GSCs relative to NSCs (Fig. 1A; Supplementary Fig. S1C; Supplementary Table 1). Motif analysis using the top 1000 enriched peaks from GSCs and NSCs identified highly enriched, conserved consensus motifs in both cell types (Fig. 1B). m6A peak density was not globally altered between NSCs and GSCs, with approximately equal numbers of increased and decreased m6A peaks in each cell type (Supplementary Fig. S1D). Total m6A levels were quantified by ELISA after extracting total RNA (which reflects m6A from rRNA, small nuclear RNA, and mRNA) from six functionally validated human patient-derived GSCs (387, 4121, GSC23, 3565, 738, and 1919) and four human NSCs (NSC11, HNP1, ENSA, and NSC194). Total m6A RNA levels did not significantly differ between GSCs and NSCs (Supplementary Fig. S1E). m6A peaks were distributed throughout transcripts, with the majority found on the gene body (Supplementary Fig. S1F). Analysis of m6A signal distribution along mRNA transcripts confirmed an enrichment of m6A signals around stop codons, as previously reported (33,34), along with the strongest enrichment near start codons in all cell types (Supplementary Fig. S1G).
Figure 1: Cancer-specific genes, upregulated in glioblastoma stem cells are marked by RNA m6A modification. See also Figure S1.
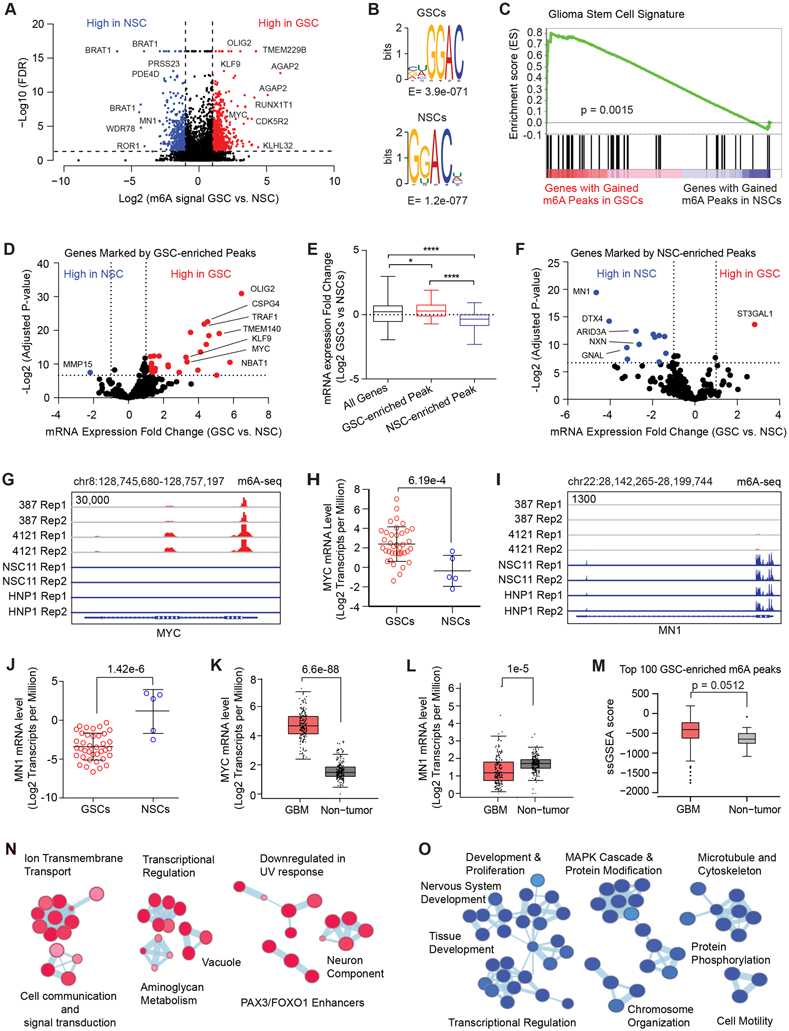
A, Volcano plot of m6A peaks detected by meRIP-seq in two GSCs and two NSCs. Red dots indicate m6A peaks gained in GSCs while blue dots indicate m6A peaks gained in NSCs. Note that multiple peaks may map to the same gene. B, (Top) Top RNA motif enriched in m6A peaks in GSCs. (Bottom) Top RNA motif enriched in m6A peaks in NSCs. C, Gene set enrichment analysis of genes with GSC-enriched m6A peaks for a “glioma stem cell” signature. D, Volcano plot showing differential mRNA expression fold change between a panel of 38 GSCs and 5 NSCs for genes marked by GSC-enriched m6A peaks. Data was derived from Mack et al. (8). E, Differential mRNA expression fold change between a panel of 38 GSCs and 5 NSCs for all genes, genes marked by GSC-enriched m6A peaks, and genes marked by NSC-enriched m6A peaks. F, Volcano plot showing differential mRNA expression fold change between a panel of 38 GSCs and 5 NSCs for genes marked by NSC-enriched m6A peaks. Data was derived from Mack et al (8). G, m6A signal in two GSCs and two NSCs at the MYC locus. H, MYC mRNA expression (Log2 transcripts per million) in a panel of 38 GSCs and 5 NSCs. The DESeq2 package was used to calculate statistical significance with multiple test correction, p = 6.19e-4. I, m6A signal in two GSCs and two NSCs at the MN1 locus. J, MN1 mRNA expression (Log2 transcripts per million) in a panel of 38 GSCs and 5 NSCs. The DESeq2 package was used to calculate statistical significance with multiple test correction, p = 1.42e-6. K, MYC mRNA expression (Log2 transcripts per million) in a panel of glioblastoma tissue specimens (n = 163) and normal brain samples (n = 207). Four-way analysis of variance (ANOVA), using sex, age, ethnicity and disease state was used to calculate statistical significance, p = 6.6e-88. Data were derived from GEPIA (80). L, MN1 mRNA expression (Log2 transcripts per million) in a panel of glioblastoma tissue specimens (n = 163) and normal brain samples (n = 207). Four-way analysis of variance (ANOVA), using sex, age, ethnicity and disease state was used to calculate statistical significance, p = 1.0e-5. Data were derived from GEPIA (80). M, A single sample gene set enrichment analysis (ssGSEA) score was calculated based on expression of the genes marked with the top 100 m6A peaks in GSCs. The signature score was compared between glioblastoma and non-tumor specimens derived from TCGA datasets. N, Pathway enrichment bubble plot of gene sets enriched among genes marked by GSC-enriched m6A peaks. O, Pathway enrichment bubble plot of gene sets enriched among genes marked by NSC-enriched m6A peaks.
We next investigated whether differential localization of m6A peaks informed GSC biology. We generated a GSC-specific gene expression signature by comparing RNA sequencing data from our cohort of 38 GSCs and 5 NSCs (8). Gene set enrichment analysis (GSEA) showed that genes with gained m6A peaks were highly enriched with the GSC signature (Fig. 1C). To assess the effect of m6A marks on gene expression, we leveraged the consensus m6A peak set to identify genes with or without m6A marks. Transcripts with m6A peaks displayed higher expression than transcripts lacking m6A marks (Supplementary Fig. S1H and S1I). This may in part reflect improved detection of m6A on more highly expressed mRNAs (35). We found genes with m6A peaks gained in GSCs were globally upregulated (Fig. 1D and E). Conversely, genes with m6A peaks lost in GSCs relative to NSCs were generally downregulated in GSCs (Fig. 1F). As detection of m6A peaks on genes expressed at low levels is technically challenging, we filtered out genes expressed at low levels across the RNA-seq cohort to minimize bias. In GSCs, m6A peaks were gained on important genes associated with cancer stem cells, including OLIG2 and MYC, concurrent with increased expression (Fig. 1G and H). MN1 (meningioma (disrupted in balanced translocation) 1) showed the highest gain in m6A signal in NSCs and was expressed at much higher levels in NSCs compared to GSCs (Fig. 1I and J). Although these changes in m6A may be due to changes in the underlying mRNA expression, these changes may also reflect a stabilizing role for m6A on mRNA in these cells.
To validate these observations in an independent dataset, we mined the pan-glioma TCGA dataset (36) to explore mRNA expression levels of m6A-marked genes in patient tumors. MYC, which gained multiple m6A peaks in GSCs, was overexpressed in gliomas (Fig. 1K), whereas MN1, which lost m6A peaks in GSCs, was downregulated in gliomas (Fig. 1L). The top 100 genes marked with the highest m6A signals in GSCs were overexpressed in TCGA glioblastoma tumors versus normal brain (Fig. 1M), suggesting that in GSCs m6A modification marks important genes overexpressed in glioblastomas. Genes marked by GSC-enriched m6A peaks were ranked based on both statistical significance predicting survival of patients afflicted with glioblastoma and differential duration in survival when overexpressed, relative to median mRNA levels. Several genes, including NOL3, EMILIN1, and TRAF1, portend poor prognosis when overexpressed in glioblastoma patient tumors (Supplementary Fig. S1J), suggesting that m6A peaks mark potential glioblastoma oncogenes. Furthermore, GSC-enriched m6A marks were present on transcripts essential for GSC viability by whole-genome CRISPR screening (37), including MYC, ATP2A2, and IMP4 (Supplementary Fig. S1K). Subsetting transcripts by the localization of m6A peaks revealed that the location of m6A marks was associated with differential transcript expression. Transcripts with m6A peaks on the 5’ UTR or promoter region (which may also reflect m6Am rather than m6A) were expressed at higher levels than those harboring peaks on the 3’UTR, gene body, or stop codon (Supplementary Fig. S1L and S1M). Collectively, these results indicate that both the presence and localization of m6A may impact gene expression. We performed pathway enrichment analysis on gene sets marked with cell type-specific m6A peaks. Genes with GSC-enriched m6A peaks were enriched for programs of cell communication and signal transduction, transcriptional regulation, and downregulation of UV response, among others (Fig. 1N), whereas genes with NSC-enriched m6A peaks were selectively enriched for developmental pathways, including tissue and nervous system development (Fig. 1O). Taken together, these results implicate m6A peaks in marking, and potentially regulating, critical cancer-specific genes in glioblastoma in a location and cell type-specific manner.
YTHDF2 is upregulated in GSCs and essential for GSC maintenance
As m6A deposition and localization may regulate cell-type specific processes in GSCs, we investigated the relative contributions of m6A writers, erasers, and readers in glioblastoma. In TCGA glioma patient tumor data, expression of m6A writers (METTL3 and METTL14) or m6A erasers (ALKBH5 and FTO) did not increase with tumor grade (Fig. 2A; Supplementary Fig. S2A and S2B). Of the m6A readers, expression of YTHDF2 and YTHDF3 was upregulated in high-grade gliomas (Fig. 2A). Further, YTHDF2 expression was enriched in mesenchymal glioblastomas (Supplementary Fig. S2B), which demonstrate greater therapeutic resistance and worse prognosis (38,39). We compared expression of m6A regulators in GSCs versus NSCs; GSCs expressed higher levels of the m6A writer, METTL14, and the m6A reader, YTHDF2, than NSCs, while other m6A writers and erasers were not differentially expressed between these cell types (Fig. 2B and C). To investigate the expression of METTL14 and YTHDF family members in the absence of cell culture conditions, we mined the pan-glioma TCGA dataset. Expression of YTHDF family members (YTHDF1-3) (Supplementary Fig. S2C) and METTL14 (Supplementary Fig. S2D) were elevated in glioblastoma tissues compared to the normal brain tissue.
Figure 2. The m6A reader YTHFD2 is required for maintenance of glioblastoma stem cells. See also Figure S2.
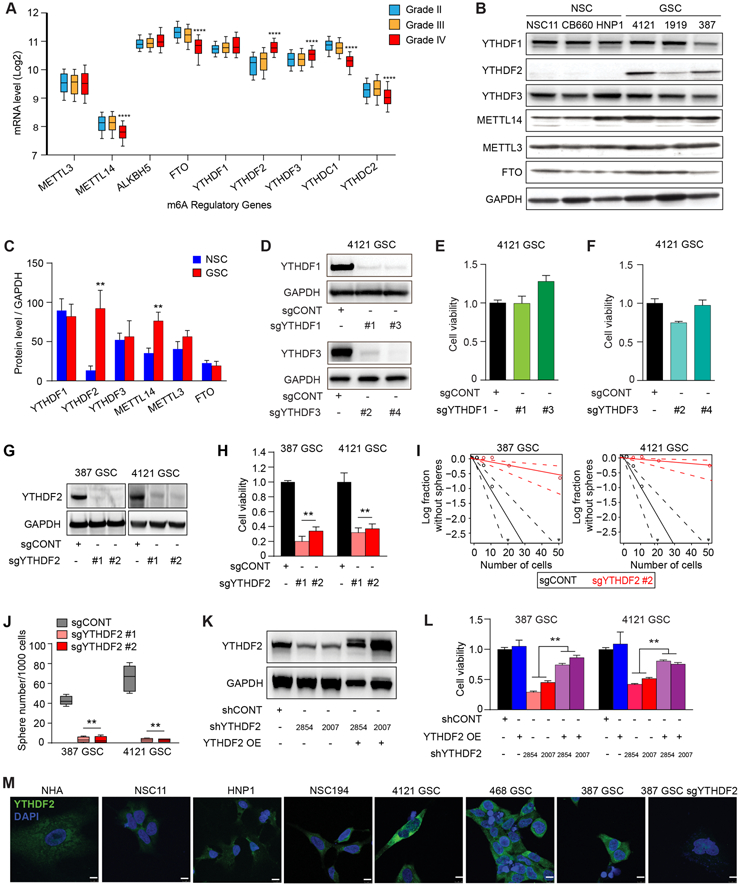
A, mRNA expression of selected m6A regulatory genes in the TCGA GBM-LGG dataset based on tumor grade. ****, p < 0.0001 for Grade IV vs either Grade II or III. B, Immunoblot assessment of YTHDF1, YTHDF2, YTHDF3, METTL14, METTL3, and FTO protein levels in NSCs and GSCs. GAPDH was used as a loading control. C, Densitometry graphs showing the protein levels of m6A readers, writers and erasers normalized to GAPDH in GSCs compared to NSCs. **, p < 0.01. Error bars show standard deviation. D, Immunoblot showing YTHDF1 and YTHDF3 levels after transduction of GSCs with a control non-targeting sgRNA sequence (sgCONT) or four independent sgRNAs targeting YTHDF1 (sgYTHDF1 #1, and sgYTHDF1 #3) and YTHDF3 (sgYTHDF3 #2 and sgYTHDF3 #4). E and F, Relative cell viability of 4121 GSCs expressing a control non-targeting sgRNA sequence (sgCONT) or two independent sgRNAs targeting YTHDF1 (sgYTHDF1 #1 and sgYTHDF1 #3) and YTHDF3 (sgYTHDF3 #2 and sgYTHDF3 #4). Error bars show standard deviation. G, Immunoblot showing YTHDF2 protein levels after transduction of two GSCs (387 and 4121), with a control non-targeting sgRNA sequence (sgCONT) or two independent sgRNAs targeting YTHDF2 (sgYTHDF2 #1 and sgYTHDF2 #2). H, Cell viability of two patient-derived GSCs (387 and 4121) following transduction with either a non-targeting control sgRNA (sgCONT) or one of two independent, non-overlapping sgRNAs targeting YTHDF2. **, p < 0.01. Error bars show standard deviation. I, Sphere formation using an extreme limiting dilution assay (ELDA) was performed with 387 and 4121 GSCs expressing sgCONT or sgYTHDF2 #2. J, Box plot shoeing the quantification of the number of spheres (per 1,000 cells) formed by GSCs. Data are presented as mean (+/−SEM) from three independent experiments. **, p < 0.01. K, Immunoblots demonstrating the knockdown and overexpression efficiency of YTHDF2 in 4121 GSCs. GAPDH is used as a loading control. L, Cell viability of GSCs transduced with one of two independent, non-overlapping shRNAs against YTHDF2 (shYTHDF2) or control shRNA (shCONT), with or without exogenous YTHDF2 overexpression. **, p < 0.01. Error bars show standard deviation. M, Cellular localization and levels of YTHDF2 in GSCs, NSCs and normal astrocytes as demonstrated by immunofluorescence staining using YTHDF2 antibody. Scale bars: 10 μm.
As YTHDF2 and METTL14 expression distinguish GSCs from NSCs and glioblastoma from normal brain, we investigated the functional roles of these molecules both in vitro and in vivo. METTL14 promotes leukemogenesis through the maintenance of hematopoietic stem cells (18). Targeting METTL14 with CRISPR-Cas9 impaired the proliferation capacity of GSCs in vitro (Supplementary Fig. S2E and S2F), but did not affect the growth of intracranial glioblastoma tumors derived from human patient-derived GSCs (Supplementary Fig. S2G), suggesting a limited role in cancer stemness.
We then investigated the functional role of the m6A YTHDF readers in glioblastoma. We first examined YTHDF1 and YTHDF3, which are typically expressed at much lower levels that YTHDF2 in most cells (40). Targeting either YTHDF1 or YTHDF3 expression with two independent sgRNAs per target gene had minimal effect on the proliferation of patient-derived GSCs relative to a control non-targeting sgRNA (Fig. 2D-F). In contrast, knockout of YTHDF2 with either of two independent sgRNAs decreased cell viability relative to a control sgRNA (Fig. 2G and H). GSC frequency and self-renewal were diminished, as assayed by extreme limiting dilution assays (ELDA), following depletion of YTHDF2 in two different GSC models (Fig. 2I). Sphere formation under serum-free conditions has served as a surrogate for self-renewal, albeit with caveats (41). YTHDF2 knockout reduced sphere formation in two GSCs relative to the control sgRNA (Fig. 2J). We next investigated whether YTHDF2 depletion induces GSC differentiation. Upon YTHDF2 knockout in GSCs, expression of stemness markers, SOX2 and OLIG2, decreased without change in differentiation marker, GFAP (Supplementary Fig. S2H and S2I). YTHDF2 functions were not selective to GSCs alone, as YTHDF2 depletion had similar effects on cell viability in matched GSCs and differentiated glioma cells (DGCs) (Supplementary Fig. S2J-S2K). To verify these phenotypes, we performed orthogonal studies using two independent shRNAs targeting YTHDF2 with rescue overexpression of an shRNA-resistant YTHDF2 (Fig. 2K). Concordant with the CRISPR studies, shRNA-mediated knockdown of YTHDF2 reduced GSC viability, which was rescued upon overexpression of shRNA-resistant YTHDF2 (Fig. 2L). YTHDF2 overexpression in GSCs was confirmed by using immunofluorescence analysis (Fig. 2M), which revealed predominantly cytosolic localization of YTHDF2. Collectively, these results establish YTHDF2 as a specific and potent regulator of glioblastoma maintenance.
YTHDF2 supports gene expression in GSCs through m6A RNA modification
To determine the mechanism by which YTHDF2 functions in GSCs, we interrogated the downstream targets of YTHDF2 using RNA sequencing (RNA-seq). Knockout of YTHDF2 induced widespread gene expression changes in GSCs (Fig. 3A; Supplementary Table 2). Programs enriched upon YTHDF2 knockdown included translational control, ribosomes and RNA processing, hypoxia response, and MYC targets (Fig. 3B and C). Transcriptional changes following YTHDF2 knockdown aligned with differential m6A peaks between GSCs and NSCs. Genes with gained m6A peaks in GSCs were more frequently downregulated upon YTHDF2 knockout, supporting YTHDF2 regulation of GSC-specific transcriptional programs in a m6A depemdent manner (Fig. 3D). Targeting YTHDF2 reduced expression of MYC targets and genes involved in hypoxia, ribosomes, glycolysis, RNA processing, and translation (Supplementary Fig. S3A). The effects of YTHDF2 knockout on decreasing mRNA levels of MYC, VEGFA, and other targets (including IGFBP3, IGFBP5, DDIT, and ribosomal genes, RPS2 and RPL13a) were validated by qPCR (Fig. 3E and 3F). Targeting YTHDF2 reduced protein levels of the GSC stemness markers, SOX2 and OLIG2 (Supplementary Fig. S3B), connecting YTHDF2 depletion and loss of GSC maintenance (Fig. 2I). MYC (42) and IGFBP3 (43) have been implicated in maintenance of tissue and cancer stem cell function. The lower transcript levels of MYC and IGFBP3 upon YTHDF2 knockout translated into decreased protein levels in GSCs, measured by immunoblot (Supplementary Fig. S3B). To predict the function of YTHDF2 in glioblastoma, we interrogated TCGA glioblastoma gene expression data and identified programs correlated with YTHDF2 expression. YTHDF2-correlated genes were positively enriched for processes of RNA metabolism and translation, cell cycle, glucose metabolism, and DNA repair signatures, whereas YTHDF2 expression negatively correlated with interferon and GPCR signaling (Fig. 3G). YTHDF2-correlated genes were highly enriched for MYC and E2F targets and for G2M checkpoint regulators and mediators of oxidative phosphorylation (Fig. 3H). These data implicate YTHDF2 as a regulator of global transcriptional programs associated with differential m6A modification.
Figure 3. YTHDF2 supports gene expression of important cancer-specific pathways in GSCs. See also Figure S3.
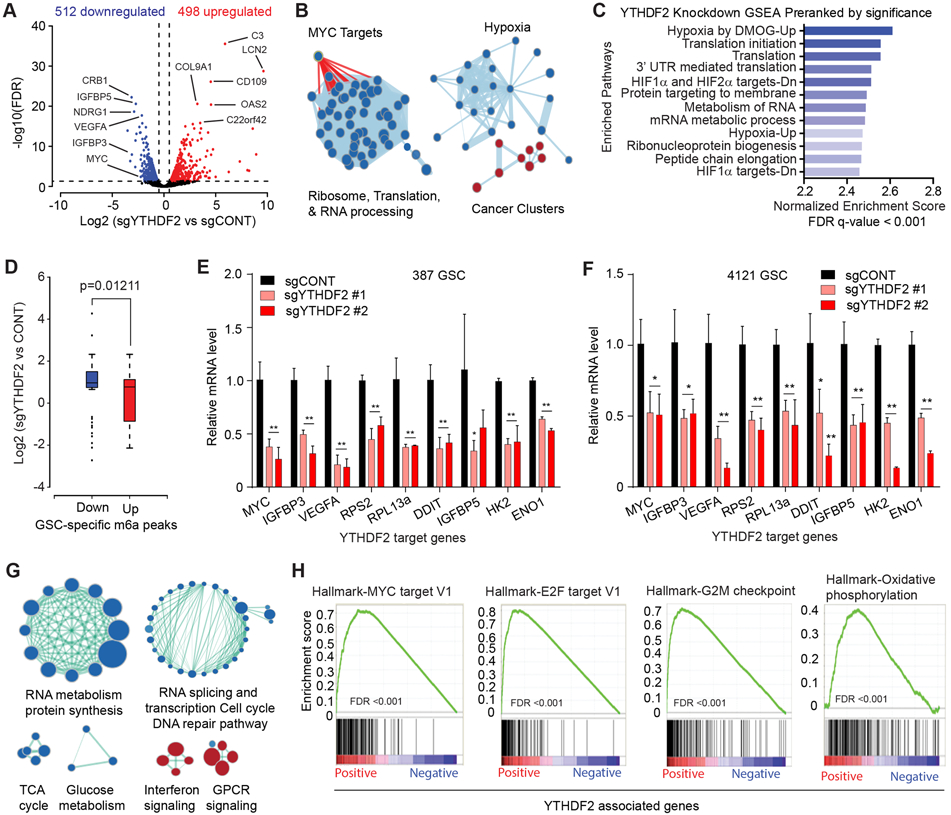
A, Volcano plot of gene expression changes in YTHDF2 knockout vs. control, obtained from 387 and 4121 GSCs. Blue indicates genes downregulated in YTHDF2 knockout at an FDR < 0.05 and log2 fold change < −1. Red indicates genes upregulated following YTHDF2 knockout at an FDR < 0.05 and log2 fold change >1. B, Gene set enrichment analysis of GO pathways enriched or depleted following YTHDF2 knockdown in 387 and 4121 GSCs. Blue indicates enrichment in genes downregulated following YTHDF2 knockdown. Red indicates programs enriched in genes upregulated after YTHDF2 knockdown. C, Gene set enrichment analysis of genes downregulated following YTHDF2 knockdown in 387 and 4121 GSCs. Enriched gene signatures are plotted with normalized enrichment score. D, Boxplot demonstrating changes in gene expression with YTHDF2 knockdown in 387 and 4121 GSCs, compared with m6A differential peaks in GSCs vs. NSCs. The x-axis indicates m6A peaks downregulated (blue) or upregulated (red) in GSCs. The y-axis represents gene expression in YTHDF2 knockdown vs. control. E, Relative mRNA expression of YTHDF2 target genes normalized to 18S mRNA level in 387 GSCs. *, p < 0.05, **, p < 0.01. Error bars show standard deviation. F, Relative mRNA expression of YTHDF2 target genes normalized to 18S mRNA level in 4121 GSCs. *, p < 0.05, **, p < 0.01. Error bars show standard deviation. G, Gene set enrichment analysis of GO pathways positively or negatively correlated with YTHDF2 expression in TCGA dataset. Blue indicates enrichment in programs positively correlated and red indicates programs enriched in negatively correlated genes with YTHDF2 expression. H, Gene set enrichment analysis of Hallmark gene sets using a pre-ranked gene list weighted by correlation of gene expression with YTHDF2 in TCGA.
To consider YTHDF2 and MYC in glioblastoma subtypes, we explored the correlation between YTHDF2 and MYC mRNA expression in all glioblastoma specimens in an independent dataset, the Chinese Glioma Genome Atlas. YTHDF2 mRNA expression correlated with MYC across glioblastoma in general, as well as proneural, mesenchymal, and classical subtypes individually (Supplementary Fig. S3C-S3F), suggesting that the correlation between MYC and YTHDF2 in our patient-derived GSCs is consistent across glioblastoma transcriptional subtypes.
YTHDF2 stabilizes MYC transcripts
Following YTHDF2 knockout, mRNA transcripts were upregulated (n = 498) and downregulated (n = 512) in almost equal proportion (Fig. 3A). As YTHDF2 is believed to bind and promote degradation of m6A-tagged transcripts (29), we asked whether genes perturbed upon YTHDF2 knockout included direct targets of YTHDF2. To identify the direct targets of YTHDF2, we mapped the binding sites of YTHDF2 using individual-nucleotide resolution crosslinking and immunoprecipitation (iCLIP) in two patient-derived GSCs (4121 and 387) and two NSCs (NSC11 and HNP1). YTHDF2 binding to RNAs in NSC11 was very low, precluding library preparation and sequencing in this NSC line (Supplementary Fig. S4A), likely as a result of low YTHDF2 levels in these cells. Motif analysis using the top enriched peaks from the two GSCs and HNP1 NSCs identified highly enriched, conserved consensus motifs in both cell types (Supplementary Fig. S4B). Motifs identified were similar to m6A consensus DRACH motifs (Supplementary Fig. S4B), indicating that YTHDF2 bound to m6A peak regions in these cells. YTHDF2 binding sites were distributed throughout transcripts with an enrichment around the stop codon and 3’ UTR region in each cell type (Supplementary Fig. S4C).
We hypothesized that critical targets of YTHDF2 would both bind to YTHDF2 (as measured by YTHDF2 iCLIP-seq) and be reduced in expression upon YTHDF2 targeting. Upon overlap of YTHDF2 iCLIP-seq data and YTHDF2-knockout RNA-seq data from GSCs, we identified 63 genes directly bound by YTHDF2 among 512 genes downregulated upon YTHDF2 knockout (Fig. 4A; Supplementary Table 3). This restricted gene set enriched for pathways associated with translation initiation, ribosome complexes, nonsense-mediated decay (NMD), and carbon metabolism in cancer (Fig. 4B). To further refine the downstream targets, we focused on the overlap between transcripts that bound YTHDF2 and had detectable m6A marks among genes downregulated upon YTHDF2 targeting, identifying 30 genes, including important oncogenes, like MYC (42,44) and VEGF (45) (Fig. 4C). In contrast, YTHDF2 bound to 33 transcripts that were upregulated upon YTHDF2 knockout (Supplementary Fig. S4D; Supplementary Table 3), of which 23 were m6A-marked (Supplementary Fig. S4E).
Figure 4. YTHDF2 stabilizes the expression of oncogenes required for survival of GSCs in a m6A dependent manner. See also Figure S4 and S5.
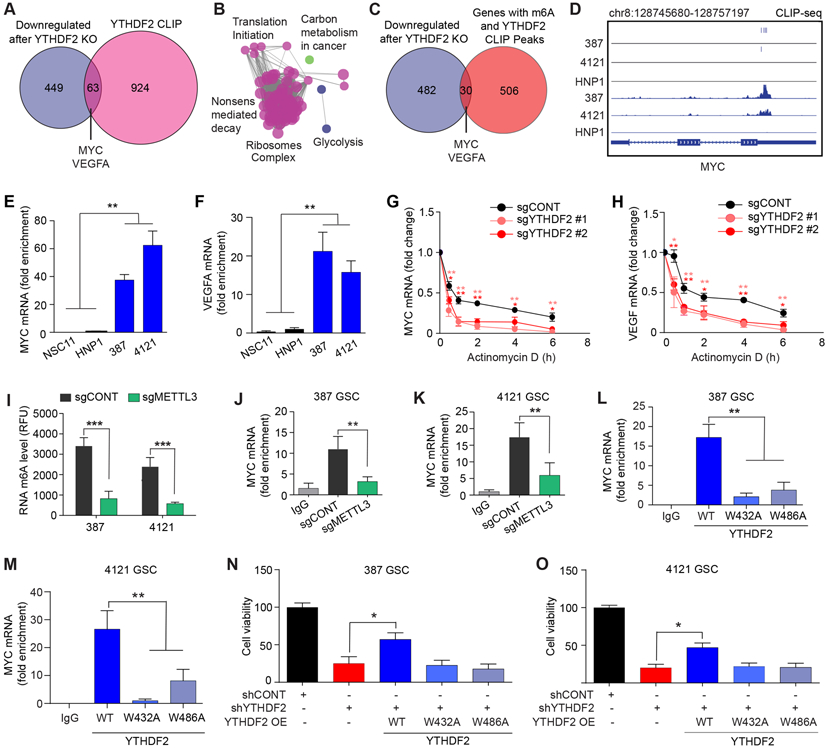
A, Overlap between mRNAs downregulated upon YTHDF2 knockout with mRNAs obtained from cross-linking immunoprecipitation (CLIP) using an anti-YTHDF2 antibody in GSCs. B, ClueGO analysis using Cytoscape to identify enriched pathways using 63 genes from Venn diagram intersection. C, Overlap between mRNAs downregulated in GSCs upon YTHDF2 knockout with mRNAs that both contain m6A modifications (m6A IP) and bind to YTHDF2 (CLIP). D, YTHDF2 binding sites identified and iCLIP signal in 387, 4121 and HNP1 at the MYC locus. E, F, Graphs showing enrichment of (E) MYC, and (F) VEGFA mRNAs in the YTHDF2 immunoprecipitated RNA fraction. **, p < 0.01. Error bars show standard deviation. G, H, Graphs showing changes in the mRNA levels of (G) MYC and (H) VEGFA, at different time points following actinomycin-D treatment in GSCs transduced with sgYTHDF2 #1, sgYTHDF2 #2 or sgCONT. *, p < 0.05 and **, p < 0.01. Error bars show standard deviation. I, Quantification of RNA m6A levels in two patient derived GSCs following knockout of METTL3. **, p < 0.01 and ***, p < 0.001. Error bars show standard deviation. J, K, Graphs showing enrichment of MYC mRNA in the YTHDF2 immunoprecipitated RNA fraction of 387 (J) and 4121 (K) GSCs following knockout of METTL3 or treatment with a non-targeting sgRNA. **, p < 0.01. Error bars show standard deviation. L, M, Graphs showing enrichment of MYC mRNA in the immunoprecipitated RNA fraction of GSCs following overexpression of either wild type (WT) YTHDF2 or m6A binding mutant YTHDF2 (W432A and W486A) in 387 (L) and 4121 (M) GSCs. **, p < 0.01. Error bars show standard deviation. N, O, Cell viability of GSCs transduced with YTHDF2 shRNAs (shYTHDF2) or control, non-targeting shRNA (shCONT), with or without exogenous overexpression of wild type (WT) or m6A binding mutant (W432A and W486A) YTHDF2 in 387 (N) and 4121 (O) GSCs. *, p < 0.05. Error bars show standard deviation.
YTHDF2 displayed selective binding to MYC mRNA in patient-derived GSCs, but not in HNP1 NSCs (Fig. 4D). To validate our findings in GSCs, we analyzed available YTHDF2 PAR-CLIP (photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation) data and m6A profiling data (29) obtained from HeLa cells and overlapped these results with our YTHDF2-knockout RNA-seq data obtained from GSCs. In agreement with our results, this study reported the presence of m6A and direct binding of YTHDF2 to MYC and VEGFA in HeLa cells (Supplementary Fig. S5A-5C). YTHDF2-bound genes in HeLa cells were enriched for pathways associated with protein translation initiation and glycolysis, providing an independent validation to our findings (Supplementary Fig. S5A and S5C).
We validated YTHDF2 binding to its targets in GSCs by YTHDF2 RNA immunoprecipitation (RIP) coupled with qPCR. YTHDF2 binding was selectively enriched on MYC and VEGF transcripts in GSCs compared to NSCs (Fig. 4E and F). YTHDF2 binding to CBP mRNA was used as a positive control (Supplementary Fig. S5D). To determine whether YTHDF2 regulates the stability of these transcripts, we treated GSCs transduced with either control sgRNA or YTHDF2 sgRNA with actinomycin D to arrest transcription. The decay rates of MYC and VEGFA mRNAs were higher upon YTHDF2 depletion, suggesting that YTHDF2 is critical for the stabilization of these transcripts (Fig. 4G and H). Taken together, our data raise the possibility that YTHDF2 binding may not only destabilize certain transcripts (24,29), as shown by mRNAs that were m6A-tagged and upregulated upon YTHDF2 knockdown (Supplementary Fig. S4D and S4E), but also stabilize important oncogenes, such as MYC and VEGF, in an m6A-dependent manner.
To further establish the role of m6A in YTHDF2 functions, we knocked out the m6A writer, METTL3, using CRISPR-Cas9 (Supplementary Fig. S5E) and performed RIP-qPCR to measure YTHDF2 binding to MYC mRNA. As expected, METTL3 knockout decreased total m6A levels in GSCs (Fig. 4I). METTL3 knockout reduced YTHDF2 binding to MYC mRNA (Fig. 4J and K) and decreased MYC mRNA levels in GSCs (Supplementary Fig. S5F). To address the importance of m6A for YTHDF2-mediated transcript stabilization, we generated FLAG-tagged m6A binding mutant YTHDF2 constructs (W432A and W486A) and overexpressed wild-type and m6A-binding mutant YTHDF2 in GSCs. Using RIP-qPCR, we found reduced binding of mutant YTHDF2 to MYC mRNA in comparison to wild type YTHDF2 (Fig. 4L and M). These results confirmed that m6A was necessary for the effects of YTHDF2 on MYC expression. To confirm that the m6A binding function of YTHDF2 was critical to its role as a GSC dependency, we rescued YTHDF2 knockdown cells with either wildtype or m6A binding mutant YTHDF2 constructs. Only wild-type YTHDF2 rescued the viability of YTHDF2 depleted cells (Fig. 4N and O). Similarly, only wild-type YTHDF2 rescued the decreased MYC levels upon YTHDF2 depletion (Supplementary Fig. S5G and S5H). Exogenous expression of both wild type and mutant YTHDF2 clones had minimal effect on endogenous MYC mRNA levels (Supplementary Fig. S5I). Collectively, these results establish that the effects of YTHDF2 on MYC and VEGFA transcript stability and GSC viability depend on m6A.
YTHDF2 acts as a GSC-specific dependency by preserving MYC transcript stability
MYC serves as a master regulator of cancer stem cell biology, including in glioblastoma (42,44), suggesting that YTHDF2 regulates the viability and stemness of GSCs, at least in part, through MYC. Knockdown of MYC with two independent, non-overlapping shRNAs reduced mRNA levels of VEGFA, and other genes downregulated upon YTHDF2 depletion (Fig. 5A and 5B). MYC overexpression rescued GSCs from YTHDF2 knockout-induced cell death (Fig. 5C and D) and partially restored mRNA levels of YTHDF2 target genes in GSCs (Fig. 5E and F). These results establish MYC as a key downstream effector of YTHDF2 in GSCs.
Figure 5. YTHDF2 stabilizes MYC mRNA in GSCs but not in NSCs. See also Figure S6.
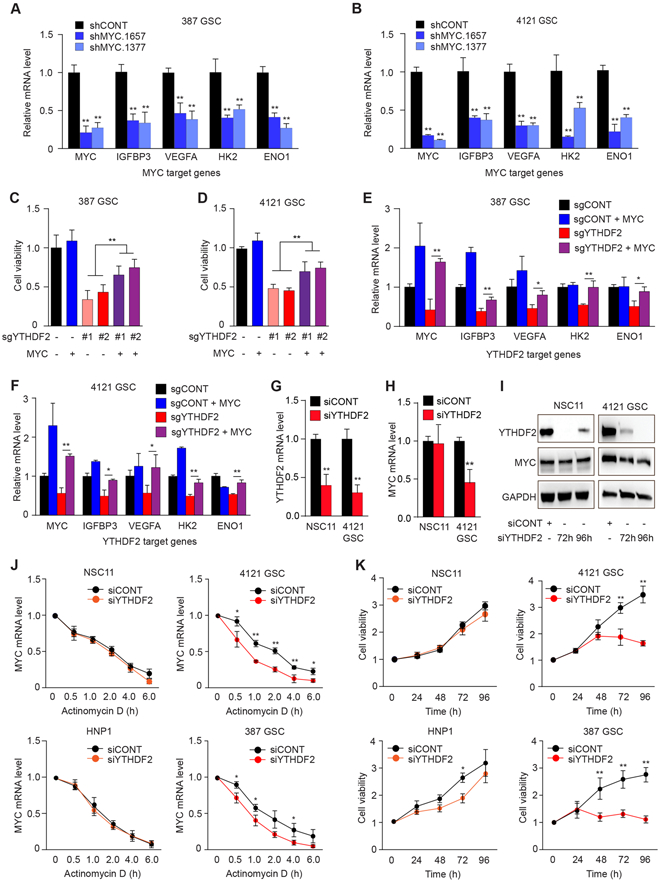
A, B, mRNA levels of MYC and MYC target genes (IGFBP3, VEGFA, HK2, and ENO1) assessed by quantitative RT-PCR following MYC knockdown with two, non-overlapping shRNAs or a non-targeting control sequence in (A) 387 and (B) 4121 GSCs. *, p<0.05, **, p < 0.01. Error bars show standard deviation. C and D, Cell viability measured in sgYTHDF2 #1-, sgYTHDF2 #2- or sgCONT-transduced GSCs in the presence or absence of MYC overexpression. **, p < 0.01. Error bars show standard deviation. E and F, Relative mRNA levels of YTHDF2 target genes normalized to 18S mRNA levels in GSCs transduced with sgYTHDF2 #2 or sgCONT, in the presence or absence of exogenous MYC overexpression. *, p < 0.05, **, p < 0.01. Error bars show standard deviation. G and H, Relative YTHDF2 and MYC mRNA level normalized to 18S mRNA levels in NSC11 and 4121 GSCs transfected with siRNA against YTHDF2 (siYTHDF2) or scrambled control siRNA (siCONT).**, p < 0.01. Error bars show standard deviation. I, Representative western blots showing the protein levels of YTHDF2 and MYC normalized to GAPDH level in NSC11 and 4121 GSCs transfected with siYTHDF2 or siCONT at 72 hours and 96 hours post transfection. J, MYC mRNA assessed over a time course following actinomycin-D treatment in siYTHDF2- or siCONT-transduced NSCs and GSCs. K, Cell viability of NSCs and GSCs transfected with siYTHDF2 or siCONT over a time course up to 96 hours post-transfection. *, p < 0.05, **, p < 0.01. Error bars show standard deviation.
To determine the specificity of YTHDF2-mediated effects on MYC in GSCs, we compared the effects of YTHDF2 depletion between NSCs and GSCs using an siRNA against YTHDF2 to avoid Cas9- and viral-mediated toxicity to NSCs. YTHDF2 knockdown in NSCs did not affect MYC mRNA levels, but reduced MYC mRNA levels in GSCs (Fig. 5G-I). A time course after actinomycin D revealed that siRNA-mediated knockdown of YTHDF2 specifically decreased MYC mRNA stability in GSCs, with little impact in NSCs (Fig. 5J). In parallel, YTHDF2 depletion reduced GSC viability without affecting NSCs (Fig. 5K). Stem cell frequency and self-renewal capacity, as assayed by extreme limiting dilution assays (ELDA), following depletion of YTHDF2 in two NSCs were not affected by YTHDF2 knockdown (Supplementary Fig. S6A and S6B). Thus, YTHDF2 represents a GSC-specific dependency that supports glioblastoma viability through GSC-specific stabilization of MYC transcripts.
IGFBP3 is a downstream target of the YTHDF2-MYC axis in GSCs
Based on the diversity of target regulation by YTHDF2, we hypothesized that molecular targets that we identified could act within a network in GSCs. IGFBP3 was one of the top downregulated genes upon YTHDF2 depletion (Fig. 3A). Pro-survival functions of IGFBP3 have been demonstrated in various cancers (43,46), but its role in glioblastoma is not well studied. Although, we detected YTHDF2 binding on IGFBP3 mRNA, YTHDF2 did not affect IGFBP3 mRNA stability (Supplementary Fig. S7A and S7B). IGFBP3 expression was downregulated following either YTHDF2 (Supplementary Fig. S3C) or MYC (Supplementary Fig. S7C) knockdown. Thus, we investigated whether IGFBP3 regulated cell viability downstream of YTHDF2-MYC axis. Supporting the functional relevance of IGFBP3 in GSCs, IGFBP3 depletion reduced GSC viability (Fig. 6A and B) and sphere formation (Fig. 6C and D). IGFBP3 knockdown has relatively milder effect on the viability of NSCs with reduction in cell viability observed only on day 5 after knockdown (Supplementary Fig. S7D and S7E). IGFBP3 overexpression rescued GSCs from YTHDF2 downregulation-mediated cell death (Fig. 6E and F). Next, we investigated the contribution of IGFBP3 to in vivo glioma growth. Analysis of the TCGA glioblastoma dataset revealed that IGFBP3 mRNA expression was upregulated in patient tumors compared to normal brain (Fig. 6G). In validation studies, we measured the expression of IGFBP3 in a cohort of 20 glioblastomas and 20 non-tumor brain tissues, observing elevated IGFBP3 mRNA expression in glioblastoma tissue (Fig. 6H). IGFBP3 expression portended poor prognosis in high grade IDH wild-type tumors in TCGA glioblastoma dataset (Fig. 6I). In the TCGA glioma patient data, IGFBP3 expression was associated with tumor grade in IDH1 wild-type tumors and was highly correlated with glucose metabolism-associated genes (Supplementary Fig. S7F). Consistent with these in silico results, IGFBP3 knockdown with either of two non-overlapping shRNAs decreased expression of the glucose metabolism genes HK2, LDHA, ENO1, and MDH2 relative to a non-targeting control shRNA (Supplementary Fig. S7G). Rescue by expression of exogenous IGFBP3 compensated for YTHDF2 depletion on metabolism-associated gene expression (except MDH2), (Supplementary Fig. S7H), indicating that IGFBP3 is a downstream effector of metabolic programs regulated by YTHDF2. The knockout and overexpression efficiency of YTHDF2 and Flag-tagged IGFBP3 was validated by immunoblotting (Supplementary Fig. S7I).
Figure 6. The YTHDF2-MYC axis regulates viability of GSCs through IGFBP3. See also Figures S7.
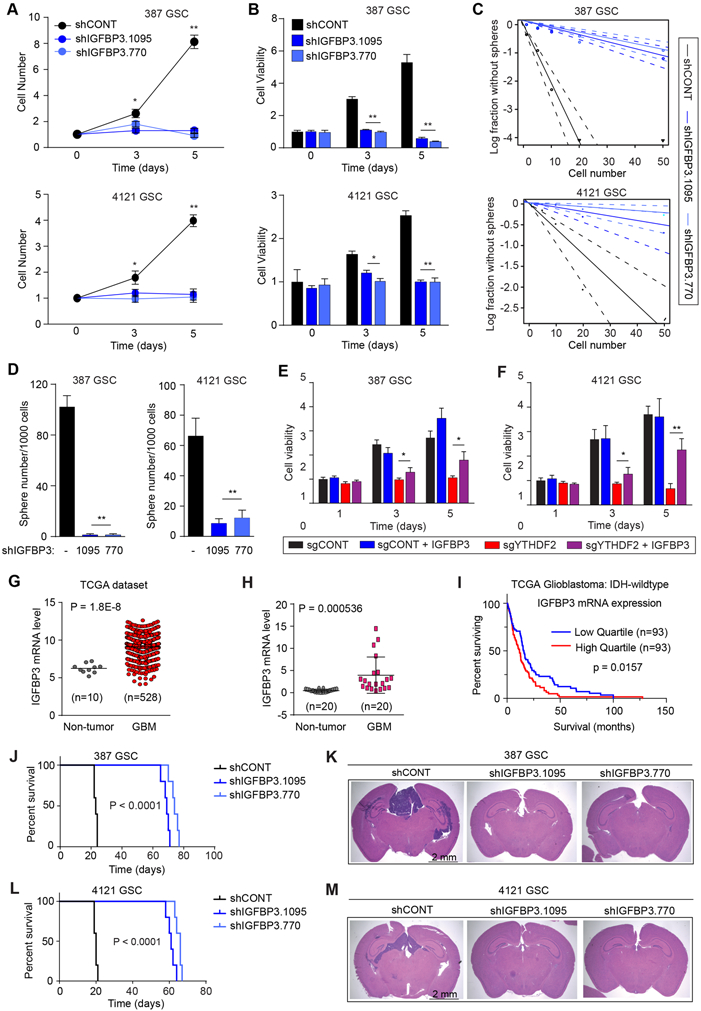
A, Growth of 387 and 4121 GSCs expressing shCONT, shIGFBP3.1095 or shIGFBP3.770 as measured by direct cell count (fold change). **, p < 0.01. Error bars show standard deviation. B, Cell viability of 387 and 4121 GSCs over 5 days post-transduction with shCONT, shIGFBP3.1095 or shIGFBP3.770, as measured by CellTiter-Glo reagent. **, p < 0.01. Error bars show standard deviation. C, Sphere formation using an extreme limiting dilution assay (ELDA) was performed with 387 and 4121 GSCs expressing shCONT, shIGFBP3.1095 or shIGFBP3.770. D, Quantification of the number of spheres (per 1,000 cells) formed by 387 and 4121 GSCs following transduction with shCONT, shIGFBP3.1095 or shIGFBP3.770. **, p < 0.01. Error bars show standard deviation. E and F, Cell viability of sgYTHDF2- or sgCONT-transduced GSCs, in the presence or absence of IGFBP3 overexpression, up to 5 days post-transduction. **, p < 0.01. Error bars show standard deviation. G, mRNA expression of IGFBP3 in normal brain (n=10) and glioblastoma tissues (n=528) derived from the TCGA glioblastoma dataset. H, qRT-PCR quantification of IGFBP3 mRNA levels in glioblastoma and normal brain tissue. I, Kaplan-Meier curve showing patient survival based on IGFBP3 mRNA expression in IDH-wildtype glioblastoma patients from the TCGA dataset, p = 0.0157. J, Kaplan-Meier survival curves of immunocompromised mice bearing intracranial 387 GSC transduced with shCONT, shIGFBP3.1095 or shIGFBP3.770. K, Representative images of hematoxylin and eosin-stained sections of tumor-bearing brains. Tumors were derived from 387 GSC transduced with shIGFBP3.1095, shIGFBP3.770 or shCONT. Brains were harvested after the presentation of first neurological sign in any cohort. Scale bar: 2 mm. L, Kaplan-Meier survival curves of immunocompromised mice bearing intracranial 4121 GSC transduced with shCONT, shIGFBP3.1095 or shIGFBP3.770. M, Representative images of hematoxylin and eosin-stained sections of tumor-bearing brains. Tumors were derived from 4121 GSC transduced with shIGFBP3.1095, shIGFBP3.770 or shCONT. Brains were harvested after the presentation of first neurological sign in any cohort. Scale bar: 2 mm.
To examine the functional contributions of IGFBP3 to glioma growth, we knocked down IGFBP3 expression in GSCs, and implanted these cells in the brains of immunocompromised mice. IGFBP3 knockdown in vivo led to increased survival of tumor-bearing mice and reduced tumor size (Fig. 6J-M).
To assess the contribution of IGFBP3 in YTHDF2 mediated regulation of MYC mRNA levels and stability and to rule out a potential indirect feedback effect, we measured the MYC mRNA levels upon IGFBP3 perturbations. IGFBP3 knockdown showed no effect on MYC levels in GSCs (Supplementary Fig. S7J). Further, rescue by expression of exogenous IGFBP3 failed to compensate for YTHDF2 depletion mediated MYC expression (Supplementary Fig. S7K). Collectively, these results establish IGFBP3 as a key downstream effector of the YTHDF2-MYC signaling axis in GSCs.
The YTHDF2-MYC-IGFBP3 axis promotes in vivo tumor growth
To address the potential benefit of therapeutic targeting of YTHDF2 in vivo, mice bearing orthotopic xenografts from patient-derived GSCs were transduced with a non-targeting, control sgRNA (sgCONT) or one of two sgRNAs targeting YTHDF2. Knockout of YTHDF2 prolonged tumor latency, as measured by time to onset of neurological signs, and reduced tumor volumes compared to mice bearing GSCs transduced with a control sgRNA (Fig. 7A-D). YTHDF2 knockout thus impaired the in vivo tumor formation capacity of GSCs. To further understand the clinical significance and pan-cancer dependency on YTHDF2, we interrogated the Cancer Dependency Map (www.depmap.org). Across a panel of 558 cell lines profiled using whole genome CRISPR-Cas9 screening, several cancer types showed a dependency on YTHDF2 (Supplementary Fig. S8A). YTHDF2 expression correlated with poor prognosis in the TCGA glioblastoma and CGGA datasets (Supplementary Fig. S8B and S8C). Given the ability of IGFBP3 to rescue the proliferative effects of YTHDF2 depletion in vitro, we investigated whether IGFBP3 could reverse the effects of YTHDF2 knockdown in vivo. IGFBP3 overexpression restored in vivo tumor formation capacity of YTHDF2-depleted GSCs (Fig. 7E-H).
Figure 7. YTHDF2-MYC-IGFBP3 axis promotes in vivo tumor growth and has therapeutic potential in GSCs. See also Figure S8 and S9.
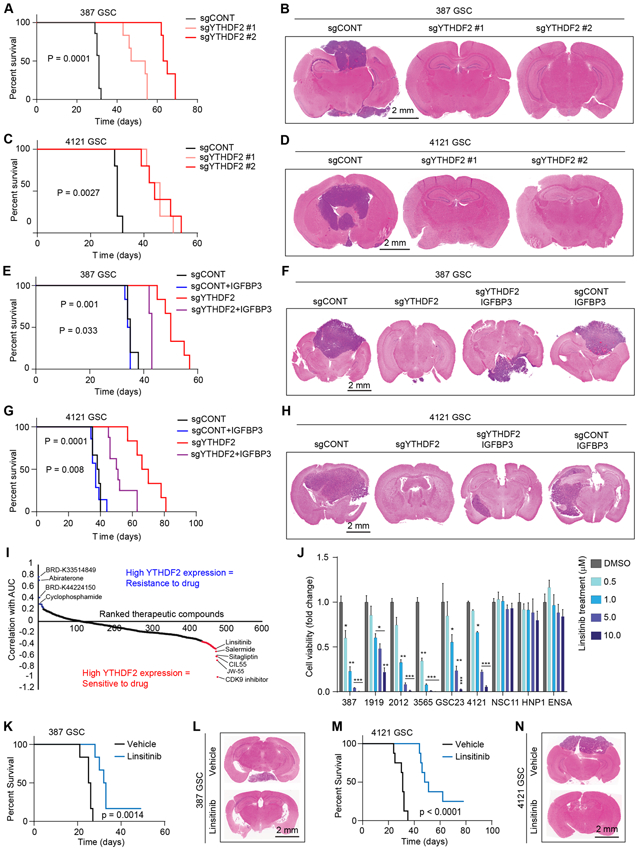
A, Kaplan-Meier survival curves of immunocompromised mice bearing intracranial 387 GSC transduced with sgCONT, sgYTHDF2 #1 or sgYTHDF2 #2. B, Representative images of hematoxylin and eosin-stained sections of tumor-bearing brains. Tumors were derived from 387 GSC transduced with sgYTHDF2#1, sgYTHDF2#2 or sgCONT. Brains were harvested after the appearance of first neurological sign in any cohort. Scale bar: 2 mm. C, Kaplan-Meier survival curves of immunocompromised mice bearing intracranial 4121 GSC transduced with sgCONT, sgYTHDF2 #1 or sgYTHDF2 #2. D, Representative images of hematoxylin and eosin-stained sections of tumor-bearing brains. Tumors were derived from 4121 GSC transduced with sgYTHDF2#1, sgYTHDF2#2 or sgCONT. Brains were harvested after the appearance of first neurological sign in any cohort. Scale bar: 2 mm. E, Kaplan-Meier survival curves of immunocompromised mice bearing intracranial 387 GSC transduced with sgYTHDF2 or sgCONT, in the presence or absence of IGFBP3 overexpression. F, Representative images of hematoxylin and eosin-stained sections of tumor-bearing brains. Tumors were derived from 387 GSC transduced with sgYTHDF2 or sgCONT, in the presence or absence of IGFBP3 overexpression. Brains were harvested after the presentation of first neurological sign in any cohort. Scale bar: 2 mm. G, Kaplan-Meier survival curves of immunocompromised mice bearing intracranial 4121 GSC transduced with sgYTHDF2 or sgCONT, in the presence or absence of IGFBP3 overexpression. H, Representative images of hematoxylin and eosin-stained sections of tumor-bearing brains. Tumors were derived from 4121 GSC transduced with sgYTHDF2 or sgCONT, in the presence or absence of IGFBP3 overexpression. Brains were harvested after the presentation of first neurological sign in any cohort. Scale bar: 2 mm. I, Therapeutic efficacy prediction of all drugs in brain cancer cells in the CTRP dataset based on mRNA expression of YTHDF2. Plot shows ranked therapeutic compounds based on correlation between YTHDF2 expression and area under the curve (AUC) of each drug. J, Cell viability of five patient-derived GSCs (387, 1919, 2012, 3565, GSC23 and 4121) and three NSCs (NSC11, HNP1 and ENSA) following a five day treatment of vehicle control (DMSO) and various concentrations of linsitinib. *, p < 0.05, **, p < 0.01, ***, p < 0.001, Error bars show standard deviation. K, Kaplan-Meier survival curves of immunocompromised mice bearing intracranial 387 GSC, which received orally, vehicle or linsitinib (50 mg/kg body weight). L, Representative images of hematoxylin and eosin-stained sections of tumor-bearing brains. Brains were harvested after the presentation of first neurological sign in any cohort. Tumors were derived from 387 GSC and mice were treated orally with either vehicle or linsitinib (50 mg/kg body weight). Scale bar: 2 mm. M, Kaplan-Meier survival curves of immunocompromised mice bearing intracranial 4121 GSC, which received orally, vehicle or linsitinib (50 mg/kg body weight). N, Representative images of hematoxylin and eosin-stained sections of tumor-bearing brains. Brains were harvested after the presentation of first neurological sign in any cohort. Tumors were derived from 4121 GSC and mice were treated orally with either vehicle or linsitinib (50 mg/kg body weight). Scale bar: 2 mm.
We then sought to identify YTHDF2-based druggable therapeutic dependencies. We leveraged the Cancer Response Therapeutics Portal (CTRP) and Cancer Cell Line Encyclopedia (CCLE) datasets, which contain mRNA expression and drug screening data from 481 compounds in 860 cancer cell lines. Correlation of drug sensitivity (area under the curve, AUC) with YTHDF2 mRNA expression in brain cancer cell lines revealed that high YTHDF2 expression correlated with sensitivity to a CDK9 inhibitor, a SIRT1 inhibitor (salermide), and an IGF1/IGF1R inhibitor (linsitinib), among others (Fig. 7I). We prioritized linsitinib for further investigation based upon the role of the IGF pathway member, IGFBP3, as a downstream effector of YTHDF2 in GSCs. Linsitinib treatment reduced the cell viability of a panel of GSCs in a concentration-dependent manner without affecting the viability of a panel of NSCs (Fig. 7J). Linsitinib treatment blocked the phosphorylation of IGF receptor (IGF1R) and downstream signaling molecules, AKT, S6K, and S6RP in 387 and 4121 GSCs in a concentration-dependent manner (Supplementary Fig. S9). In in vivo studies, oral administration of linsitinib (50 mg/kg body weight) prolonged tumor latency as measured by time to onset of neurological signs, and reduced tumor volumes compared to mice bearing intracranial xenografts of two patient-derived GSC models (Fig. 7K-N).
Collectively, these studies demonstrate that the m6A reader, YTHDF2, and its downstream effector, IGFBP3, are promising therapeutic targets in glioblastoma and that linsitinib serves as an effective agent with a broad therapeutic window.
DISCUSSION
Although discovered in early 1970s, the biological significance of mRNA modifications has only recently been appreciated, owing to the discoveries of important regulator proteins and advances in transcriptome-wide m6A mapping techniques. Although many RNA-binding proteins have been implicated in cancer development, the functional importance of m6A modifiers in the glioblastoma hierarchy remains an open area of investigation. Here, we interrogated m6A localization and its regulators by mapping m6A methylated RNAs in GSCs and their normal counterpart, NSCs. Identification of YTHDF2 as a GSC-specific m6A effector implicates the m6A pathway as a critical dependency in glioblastoma.
Oncogenic functions for m6A ‘writers’ (34) and ‘erasers’ (21) indicate that target-specific m6A modifications contribute to glioblastoma tumor maintenance. The m6A writer METTL3 is reported to have both oncogenic (17) and tumor suppressive (47) functions in glioblastoma, highlighting the complexity of the functional role of m6A RNA modifications. Although RNA and DNA methylation appear distinctly regulated, lessons gained from DNA methylation studies caution against oversimplifying methylation events as solely oncogenic or tumor suppressive. Methylation can have diverse effects in DNA regulation, and our results support a similarly nuanced function of RNA methylation. By overlapping the m6A profiling data with RNA-seq in our GSC models, we observed that the steady-state expression of m6A tagged transcripts was higher than non-m6A transcripts. YTHDF2 has been reported to destabilize m6A tagged transcripts by activating mRNA degradation pathways (29,48,49). Although our findings are largely consistent with the proposed role of YTHDF2 in mRNA destabilization, we also observe that YTHDF2 stabilizes certain important transcripts in GSCs, suggesting that the precise nature of the interaction between YTHDF2 and transcripts and its outcome might be controlled by additional unknown factors which might function in a cell type specific manner. Alternatively, YTHDF2 may destabilize mRNAs that encode suppressors of other mRNAs, thus allowing YTHDF2 to stabilize certain transcripts through an indirect pathway. We also determined that YTHDF2 expression was elevated in GSCs as well as in glioblastoma tumors. Moreover, when we overlapped RNA-seq data from YTHDF2-knockout GSCs with m6A profiling data, m6A-tagged mRNAs tended to be downregulated in comparison to non-m6A tagged mRNAs. These observations emphasize the complexity of the functional outcome of m6A-YTHDF2 interaction in regulating gene expression.
MYC is overexpressed in approximately 70% of malignancies (50) and informs poor prognosis in glioblastoma patients. MYC is also a central regulator of GSCs (42). Direct targeting of MYC has been a challenge owing to its “undruggable” protein structure. Thus, several studies have aimed to identify regulators of MYC as an alternative therapeutic strategy for multiple cancer types including glioblastoma. We observed a cell-type specific effect of YTHDF2 on MYC where it selectively stabilizes MYC transcripts in GSCs, but not in NSCs, and establishes a GSC-specific dependency. These findings indicate towards the involvement of a cell type-specific factors that may convert general mRNA destabilizing function of YTHDF2 to the mRNA stabilizing function and warrants further investigation. Moreover, it will be important to interrogate whether certain element(s) in MYC and other mRNAs contribute to such functional alteration. Recent studies have found that YTHDF1 and YTHDF3, which are typically expressed at low levels, show minimal effects upon knockout due to compensation by the more abundant YTHDF2 (13). Nevertheless, YTHDF1 and YTHDF3 can partially compensate for the depletion of the more highly expressed YTHDF2 paralog (13). Depletion of all three YTHDF paralogs may produce even more robust effects on MYC expression and tumor growth suppression. Thus, YTHDF2, or the YTHDF family of proteins, may represent a target with a broad therapeutic index to perturb MYC signaling in glioblastoma.
We established IGFBP3 as an indirect target of YTHDF2, as YTHDF2 depletion downregulated IGFBP3 mRNA and protein levels, without affecting its mRNA stability. YTHDF2 regulated IGFBP3 levels via MYC in GSCs. IGFBP3 is a member of the family of insulin-like growth factor (IGF) binding proteins, which promote several malignancies (51,52). To address the translational potential of YTHDF2, we predicted available therapeutic compounds with efficacy in YTHDF2-overexpressing cells. We identified the IGF1/IGF1R inhibitor, linsitinib, as a selective inhibitor of GSC growth and tumor formation, which further validates our finding that IGFBP3 is a downstream effector of YTHDF2. Linsitinib has been used in several clinical trials and has been well tolerated (53,54), although it has not yet been tested in glioblastoma patients. Our results support further investigation into the clinical utility of linsitinib and other IGF1R inhibitors in glioblastoma.
Through a combination of in vitro and in vivo studies in GSCs, we elucidated the functions of m6A mediators in GSCs and identified YTHDF2 as a GSC-specific dependency that regulates glucose metabolism in GSCs through stabilization of MYC transcripts. These findings reveal new therapeutic opportunities for targeting previously undruggable, essential nodes in glioblastoma.
METHODS
GSC derivation
Glioblastoma tissues were obtained from excess surgical resection samples from patients at Duke University after review by neuropathology with written informed consent and in accordance with an IRB-approved protocol (090401). All patient studies were conducted in accordance with the Declaration of Helsinki. GSC387 and GSC4121 were derived by our laboratory and transferred via a material transfer agreement from Duke University. GSC23 was transferred via a material transfer agreement from MD Anderson Cancer Center. All GSCs were cultured in Neurobasal media (Invitrogen) supplemented with B27 without vitamin A (Invitrogen), EGF, and bFGF (20 ng/ml each; R&D Systems), sodium pyruvate, and glutamax. To decrease the incidence of cell culture-based artifacts, patient derived xenografts were produced and propagated as a renewable source of tumor cells for study. Short Tandem Repeat (STR) analyses were performed to authenticate the identity of each tumor model used in this article on a yearly basis. Cells were frozen and stored at −196°C (liquid nitrogen) when not being actively cultured. Mycoplasma testing was performed by qPCR cellular supernatants on a yearly basis. Cells were grown for fewer than 20 in vitro passages from xenografts.
Other cell models
The non-malignant neural stem cell models ENSA, HNP1 and NSC11, as well as normal human astrocytes (NHA) were used in this study. ENSA (ENStem-A) is a human embryonic stem derived neural progenitor cell (Millipore Sigma, Cat SCC003). NSC11 is a human induced pluripotent derived neural progenitor cell (Alstem, Cat # hNSC11). HNP1 (STEMEZ HNP1) is a human neural progenitor cell (Neuromics, Cat # HN60001).
In vivo tumorigenesis
For YTHDF2 sgRNA and IGFBP3 shRNA and IGFBP3 over-expression experiments, intracranial xenografts were generated by implanting 5000 human derived GSCs into the right cerebral cortex of NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, The Jackson Laboratory, RRID:IMSR_JAX:005557) at a depth of 3.5 mm under a University of California, San Diego Institutional Animal Care and Use Committee (IACUC) approved protocol. Brains were harvested and fixed in 4% formaldehyde, cryopreserved in 30% sucrose, and then cryosectioned. Hematoxylin and eosin (H&E) staining was performed on sections for histological analysis. In parallel survival experiments, mice were observed until the development of neurological signs. For in vivo drug treatment studies, intracranial xenografts were generated by implanting 5000 patient-derived GSCs (387 and 4121) into the right cerebral cortex of NSG mice as described above. Mice recovered for 7 days were randomly assigned into drug vs. treatment group by a blinded investigator. Mice were then treated daily with either vehicle (25 mM Tartaric acid) or 50 mg/kg linsitinib by oral gavage.
Healthy, wild-type male or female mice of NSG background, 4–6 weeks old, were randomly selected and used in this study for intracranial injection. Mice had not undergone prior treatment or procedures. Mice were maintained in 14 hours light/10 hours dark cycle by animal husbandry staff with no more than 5 mice per cage. Experimental animals were housed together. Housing conditions and animal status were supervised by a veterinarian. Animals were monitored until neurological signs were observed, at which point they were sacrificed. Neurological signs or signs of morbidity included hunched posture, gait changes, lethargy and weigh loss. Brains were harvested and fixed in 4% formaldehyde for 48 hours, stored at 4°C in 70% ethanol, and sectioned. Hematoxylin and eosin (H&E) staining was performed on sections for histological analysis.
Plasmids and cloning
Lentiviral constructs expressing non-overlapping shRNAs directed against YTHDF2 (TRCN0000254410 and TRCN0000168751), MYC (TRCN0000039640 and TRCN0000039642), IGFBP3 (TRCN0000072508 and TRCN0000072512) or a non-targeting control shRNA (TRCN0000231489) with no targets in the human genome were obtained from Sigma-Aldrich. All shRNAs were assayed for knockdown efficiency by qPCR and were then used for all following experiments. For CRISPR-Cas9 experiments, sgRNAs were cloned into the lentiCRISPR v2 plasmid (Addgene #52961). sgRNAs used in this study were sgYTHDF1 #1: 5’-AGTTTCAAAGCCGACCTCGT-3’, sgYTHDF1 #3: 5’-TTGGCACCTGGGATAACAAG-3’, sgYTHDF2 #1: 5’-TGGGTAGCACCTCGGAACCG-3’, sgYTHDF2 #2: 5’-GTCCATTACTAGTAACATCG-3’, sgYTHDF3 #2: 5’- TGGTGTATTTAGTCAACCTG-3’, sgYTHDF3 #4: CAACCGAAACTTAAACCCAA-3’, sgMETTL3: 5’- ATTCTGTGACTATGGAACCA-3’, sgMETTL14 #2: 5’- GTCCAGTGTCTACAAAATGT-3’, sgMETTL14 #3: 5’- TTGAAGAATATCCTAAACTG-3’. Human YTHDF2 cDNA and a control vector were obtained from Vectorbuilder (YTHDF2: VB170515-1074cyz, control: VB171005-1094yhn). Lentiviral overexpression plasmid for human MYC and IGFBP3 were generated by cloning the respective ORFs with a N-terminal Flag tag into the pCDH-MCS-T2A-Puro-MSCV vector (System Biosciences) using In-Fusion® HD Cloning Kit (Clontech, 638920). All plasmids were verified by Sanger sequencing. 293T cells (ATCC Cat# CRL-3216, RRID:CVCL_0063) were used to generate lentiviral particles by co-transfecting the packaging vectors psPAX2 and pMD2.G using LipoD293 transfection reagent (SignaGen, SL100668) following manufacturer’s instructions.
Proliferation and neurosphere formation assays
Cell proliferation experiments were conducted by plating cells of interest at a density of 3,000 cells per well in a 96-well plate with 8 replicates. CellTiter-glo (Thermo Fisher Scientific) was used to measure cell viability. Data is presented as mean +/− standard deviation. Neurosphere formation was measured by in vitro limiting dilution assay, as previously reported (55). Briefly, decreasing numbers of cells per well (50, 20, 10, 5 and 1) were plated into 96-well plates. The presence and number of neurospheres in each well were recorded seven days after plating. Extreme limiting dilution analysis was performed using software available at http://bioinf.wehi.edu.au/software/elda, as previously described (55,56).
Immunoblotting
Cells were collected and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.5; 150 mM NaCl; 0.5% NP-40; 50 mM NaF with protease inhibitors) and incubated on ice for 1 hour. Lysates were centrifuged at 4°C for 20 minutes at 14,000 rpm, and supernatant was collected. The BCA assay (Bio-Rad Laboratories, RRID:SCR_008426) was utilized for determination of protein concentration. Equal amounts of protein samples were mixed with SDS Laemmli loading buffer, boiled for 5 minutes, and electrophoresed using NuPAGE Bis-Tris Gels, then transferred onto Nitrocellulose membranes. TBS-T supplemented with 5% non-fat dry milk was used for blocking for a period of 1 hour followed by blotting with primary antibodies and appropriate dilution at 4°C for 16 hours. Blots were washed five times for 5 minutes each with TBS-T and then incubated with appropriate secondary antibodies in 5% non-fat milk in TBS-T for 1 hour. For all western immunoblot experiments, blots were imaged using Bio-Rad Image Lab software (RRID:SCR_008426) and subsequently processed using Adobe Illustrator (RRID:SCR_010279) to create the figures.
Immunofluorescence staining and imaging
For immunofluorescence microscopy, cells were plated on matrigel coated coverslips. After 24 hours, cells were fixed twice using 4% PFA (15 min each time) and processed as described previously (57). Briefly, fixed cells were washed in PBS, neutralized (10 min; 50 mM glycine in PBS), blocked in PBS with 1 mg/ml BSA (blocking buffer) for 10 min and permeabilized in blocking buffer containing 0.05% saponin. Cells were incubated with YTHDF2 antibody (Proteintech Cat# 24744-1-AP, RRID:AB_2687435) at a 1:200 dilution in blocking buffer at 4°C overnight. Next day, the cells were washed three times with blocking buffer and incubated with donkey anti-rabbit secondary antibody (Molecular Probes Cat# A-21206, RRID:AB_2535792). Cells were washed three times in blocking buffer and coverslips were mounted using Prolong Gold Antifade (Life Technologies). Optical sections Z-stacks were imaged using 60x Magnification on Leica Confocal SPE (Sanford Consortium, UCSD facility) and processed using ImageJ software (NIH, Bethesda, MD, RRID:SCR_003070).
Quantitative RT-PCR
Trizol reagent (Sigma Aldrich) was used to isolate total cellular RNA from cell pellets according to the manufacturer’s instructions. The high-capacity cDNA reverse transcription Kit (ThermoFisher scientific, catalog 4368814) was used for reverse transcription into cDNA. Quantitative real-time PCR was performed using Applied Biosystems 7900HT cycler using Radiant Green Hi-ROX qPCR kit (catalog number QS2050). qPCR primers used in this study were SOX2-fwd: 5’-TACAGCATGTCCTACTCGCAG-3’, SOX2-rev: 5’-GAGGAAGAGGTAACCACAGGG-3’, OLIG2-fwd: 5’-TGGCTTCAAGTCATCCTCGTC-3’, OLIG2-rev: 5’-ATGGCGATGTTGAGGTCGTG-3’, GAPDH-fwd: 5’-GGAGCGAGATCCCTCCAAAAT-3’, GAPDH-rev: 5’-ATGGCGATGTTGAGGTCGTG-3’, 18S-fwd: 5’-GGCCCTGTAATTGGAATGAGTC-3’, 18S-rev: 5’-CCAAGATCCAACTACGAGCTT-3’, MYC-fwd: 5’-GGCTCCTGGCAAAAGGTCA-3’, MYC-rev: 5’-CTGCGTAGTTGTGCTGATGT-3’, YTHDF2-fwd: 5’-TAGCCAACTGCGACACATTC-3’, YTHDF2-rev: 5’-CACGACCTTGACGTTCCTTT-3’, IGFBP3-fwd: 5’-AGAGCACAGATACCCAGAACT-3’, IGFBP3-rev: 5’-GGTGATTCAGTGTGTATTCCATT-3’, VEGFA-fwd: 5’-ATCTGCATGGTGATGTTGGA-3’, VEGFA-rev: 5’-GGGCAGAATCATCACGAAGT-3’, HK2-fwd: 5’-TGCCACCAGACTAAACTAGACG-3’, HK2-rev: 5’-CCCGTGCCCACAATGAGAC-3’, LDHA-fwd: 5’-TTGACCTACGTGGCTTGGAAG-3’, LDHA-rev: 5’-GGTAACGGAATCGGGCTGAAT-3’, MDH2-fwd: 5’-GCCATGATCTGCGTCATTGC-3’, MDH2-rev: 5’-CCGAAGATTTTGTTGGGGTTGT-3’, ENO1-fwd: 5’-TGGTGTCTATCGAAGATCCCTT-3’, ENO1-rev: 5’-CCTTGGCGATCCTCTTTGG-3’, RPS2-fwd: 5’-GGCCTCTCTCAAGGATGAGGT-3’, RPS2-rev: 5’-GTCCCCGATAGCAACAAATGC-3’, RPL13a-fwd: 5’-GCCATCGTGGCTAAACAGGTA-3’, RPL13a-rev: 5’-GTTGGTGTTCATCCGCTTGC-3’, DDIT-fwd: 5’-TGAGGATGAACACTTGTGTGC-3’, DDIT-rev: 5’-CCAACTGGCTAGGCATCAGC-3’, CBP-fwd: 5’-ACCGGTGTAAGGAAAGGCTG-3’, CBP-rev: 5’-TCAGGTGTTGGGAAGATGGC-3’.
Patient database bioinformatics
For survival analyses, TCGA data (58) was downloaded using the “TCGA2STAT” R package (59). The Kaplan-Meier survival analysis with the log-rank analysis was used to assess prognostic significance of every gene in the TCGA GBM HG-U133A microarray and GBM or GBMLGG RNA-seq datasets (1). The processed UCSC TOIL analysis of TCGA and GTEx RNA-seq data were used to determine genes that were differentially expressed between glioblastoma specimens and normal brain specimens (60). The Cox Proportional Hazards model and log-rank analysis were used to assess prognostic significance of every gene in the TCGA GBM HG-U133A microarray dataset regardless of IDH mutation status.
RNA-seq and data analysis
Trizol reagent (Sigma Aldrich) was used to isolate total cellular RNA from cell pellets according to the manufacturer’s instructions. RNA was purified and subjected to RNA-sequencing. FASTQ sequencing reads were trimmed using Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and transcript quantification was performed using Salmon (RRID:SCR_017036) in the quasi-mapping mode (61). Salmon “quant” files were converted using Tximport (RRID:SCR_016752 ) (https://bioconductor.org/packages/release/bioc/html/tximport.html) and differential expression analysis was performed using DESeq2 (DESeq, RRID:SCR_000154) (62). Gene set enrichment analysis was performed by selecting differentially expressed genes (FDR-corrected p-value < 0.05), generating a pre-ranked list, and inputting a pre-ranked list into the GSEA desktop application (http://software.broadinstitute.org/gsea/downloads.jsp) (63,64). Pathway enrichment bubble plots were generated using the Bader Lab Enrichment Map Application (65) and Cytoscape (RRID:SCR_003032) (http://www.cytoscape.org).
N6-methyladenine RNA-immunoprecipitation followed by next generation sequencing (meRIP-seq)
meRIP was performed as described previously (66,67). Briefly, total RNA was extracted using Trizol reagent (Invitrogen) and treated with RNase-free DNase I (Roche) to deplete DNA contamination. PolyA RNA was purified using a GenElute mRNA Miniprep Kit (Sigma-Aldrich) per manufacturer instructions and fragmented using an RNA fragmentation kit (Ambion). 200 μg of fragmented RNA was incubated with 3 μg anti-m6A (Synaptic Systems, Cat#202 003) in RIP buffer (150 mM NaCl, 10 mM Tris and 0.1% NP-40) for 2 hours at 4 °C, followed by addition of washed protein A/G magnetic beads (Millipore) and incubation at 4 °C for further 2 hours. Beads were washed 6 times in RIP buffer and incubated with 50 μl immunoprecipitation buffer containing 0.5 mg/ml m6AMP (Sigma-Aldrich) to elute RNA. Immunoprecipitated RNA was extracted with phenol/chloroform, and RNA samples were sent for next-generation sequencing.
meRIP-seq alignment and peak calling
FASTQ files were processed using Trim Galore v0.4.5 (RRID:SCR_011847) and cutadapt v2.1 (RRID:SCR_011841) to trim adaptors. FASTQ files were aligned using STAR v2.7 (RRID:SCR_015899) to hg19 and sorted and indexed using SAMtools v1.3 (RRID:SCR_002105). Indexed bams were processed to generate individual and differential peaksets using m6a Viewer v1.6. Peaks were detected using default settings comparing meRIP-seq to input at a minimum 2-fold enrichment and false discovery rate (FDR) ≤ 0.05 using Benjamini-Hotchberg correction. To generate consensus peaksets for each cell model, overlapping peaks were used. Differential peaks between NSCs and GSCs were called using the R package MeTDiff with an FDR cutoff of 0.05 and differential fold enrichment of 1. m6A metagene plots were generated using the “makeMetaGeneProfile.pl” function in HOMER.
RNA immunoprecipitation (RIP)-qPCR analysis
RIP for YTHDF2 was performed using 2 μg of Anti-YTHDF2 (Cat# 24744-1-AP, ProteinTech), as described earlier (68). Briefly, cell extracts from 10 × 106 cells were prepared as described and 10% volume was taken out as input at this stage. Remaining cell extracts were incubated with Anti-YTHDF2 antibody coated with magnetic Protein A/G beads for 12-16 hours for immunoprecipitation (IP). After IP, RNA was isolated from Input and IP fractions using phenol/chloroform extraction. cDNA was prepared using high-capacity cDNA reverse transcription Kit (ThermoFisher scientific, catalog 4368814). Quantitative real-time PCR was performed using Applied Biosystems 7900HT cycler using Radiant Green Hi-ROX qPCR kit (catalog number QS2050).
Construction of YTHDF2 iCLIP libraries
YTHDF2 iCLIP was performed as previously described (69,70) with the following modifications. Crosslinked cell pellets were resuspended in 100 μL of cold 1% SDS + 10mM DTT + 10X Halt protease inhibitor cocktail (Thermo Fisher) solution to dissociate YTHDF2 complexes, and sonicated using a Branson Digital Sonifier Model 450 fitted with 3.125 mm tapered micro tip probe on ice at 10% amplitude (2s ON, 10s OFF cycle, total 20s). The pellet was topped up to 1mL of 1X RIPA buffer (50 mM Tris-HCl pH 7.5, 200 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS), and sonicated again at 10% amplitude (2s ON, 10s OFF cycle, total 20s). Protein concentration was measured using the Pierce BCA Protein Assay Kit. DNase I (New Englamnd Biolabs) and RNase I (Thermo Fisher) was used to digest DNA and RNA in the cell pellet (high RNase I dilution = 1:5, low RNase I dilution = 1:50 or 1:100 depending on cell line). 4 μg of YTHDF2 antibody (Aviva Systems Biology, ARP67917) was used for immunoprecipitation of each 1mg of cell lysate. Equivalent amounts of normal rabbit IgG (Cell Signalling Technology) were used for immunoprecipitation as a negative control. YTHDF2 antibodies or Rabbit IgG were first bound to Pierce Protein A/G Magnetic Beads (Thermo Fisher) in RIPA buffer for 1 hour at 4 °C. Immunoprecipitation was performed in RIPA buffer at 4 °C overnight. RNA bound to YTHDF2 was ligated to adaptors and purified as previously described (69). SuperScript IV and primers with unique barcodes were used for reverse transcription to tag each replicate. The medium cDNA fraction (80 – 100 nucleotides) was extracted by gel purification, and sequenced on the Illumina HiSeq4000 (paired end, 150 bases). Due to low levels of YTHDF2-bound RNA in the cell line NSC11, YTHDF2 iCLIP libraries were not sequenced for the NSC11 cell line.
Analysis of YTHDF2 iCLIP libraries
Low-quality bases and adaptor sequences were trimmed using FLEXBAR (71) to remove reads with PHRED (RRID:SCR_001017) quality score < 30. Reads were demultiplexed using the pyCRAC software’s pyBarcodeFilter (72), and aligned to hg19 using the bwa alignment tool (73) (bwa aln -n 0.06). Paired-end alignments were pooled as previously described to allow downstream analysis without duplicate sequence names (74). For calling sites with truncations, only forward reads were used. Duplicates were pooled for analysis using the CTK pipeline (75) for identification of CITS sites and CIMS deletion sites (FDR < 0.05). For motif analysis, CITS and CIMS deletion sites were pooled, and sites in genic regions were extracted. Motifs were called from DNA sequences +/−10 nucleotides of these sites using HOMER (RRID:SCR_010881). To identify the positions of the YTHDF2 iCLIP sites along an mRNA, metagene analysis was performed on the pooled CITS and CIMS deletions sites using metaplotR (76).
RNA stability assay
Actinomycin D (Sigma-Aldrich) at 5 μg/ml was added to GSCs either or transfected with YTHDF2 siRNA or transduced with CRISPR/Cas9 targeting YTHDF2 using two non-overlapping sgRNAs or non-targeting control sgRNAs. After 0, 0.5, 1, 3 and 6 hours of incubation, cells were collected and RNAs were isolated for qPCR.
Drug sensitivity prediction
Therapeutic sensitivity and gene expression data were accessed through the Cancer Therapeutics Response Portal (https://portals.broadinstitute.org/ctrp/) (77-79). Gene signature scores were calculated for each cell line in the dataset using the single sample Gene Set Enrichment Analysis Projection (ssGSEAProjection) module on GenePattern (RRID:SCR_003201) (https://cloud.genepattern.org). Gene signature score was then correlated with area under the curve (AUC) values for drug sensitivity for each compound tested. Correlation r-value was plotted and statistical analyses were corrected for multiple test correction.
Quantification and statistical analysis
All statistical analyses are described in the figure legends. For TCGA glioblastoma vs normal brain RNA-seq calculations, four-way ANOVA controlling for sex, age, and ethnicity with Benjamini and Hochberg false discovery rate (FDR) method was used for statistical analysis. For survival analyses, the Cox proportional hazards and log-rank analyses were used. For qPCR analyses, Student t-test was used to assess statistical significance, when appropriate. Two-way repeated measures ANOVA was used for statistical analysis with Dunnett’s multiple hypothesis test correction. For proliferation assays, two-way repeated measures ANOVA was used for statistical analysis with Dunnett’s multiple hypothesis test correction.
Data and software availability
All newly generated raw sequencing data related to this study (related to YTHDF2 knockout RNA sequencing, RNA m6A profiling in NSCs and GSCs, and iCLIP-seq) is available on GEO using the accession number GSE158742. All data accessed from external sources and prior publications have been referenced in the text and corresponding figure legends. Additional data will be made available upon request.
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Jeremy N. Rich (drjeremyrich@gmail.com).
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Epitranscriptomics promotes cellular heterogeneity in cancer. RNA m6A landscapes of cancer and neural stem cells identified cell-type-specific dependencies and therapeutic vulnerabilities. The m6A reader, YTHDF2, stabilized MYC mRNA specifically in cancer stem cells. Given the challenge of targeting MYC, YTHDF2 presents a therapeutic target to perturb MYC signaling in glioblastoma.
ACKNOWLEDGMENTS
We appreciate Dr. Xiang-Dong Fu and members of the Rich lab laboratory for critical reading of the manuscript and helpful discussions. Control and m6A binding mutant YTHDF2 constructs were generously provided by Dr. Bin Shen from Nanjing Medical University, Nanjing, China. We appreciate the Histology Core at UCSD for their work on histologic experiments and analysis. Finally, we would like to thank our funding sources: The National Institutes of Health grants CA217066 (B.C.P.), CA217065 (R.C.G.), CA203101 (L.J.Y.K.), CA186702 (S.R.J.), GM110090, GM132292 (J.C.Z.), CA197718, CA238662, and NS103434 (J.N.R.). P.H. is supported by the Danish Cancer Society/Kraeftens Bekaempelses Videnskabelige Udvalg Foundation, Knaek Cancer, and the Novo Nordisk Foundation. H.X.P. is supported by Agency for Science, Technology and Research (A*STAR).
Footnotes
Conflict of interest statement: The authors declare no competing financial interests.
REFERENCES
- 1.Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016;164(3):550–63 doi 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell 2012;149(1):36–47 doi 10.1016/j.cell.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444(7120):756–60 doi 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 4.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer research 2003;63(18):5821–8. [PubMed] [Google Scholar]
- 5.Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nature reviews Drug discovery 2009;8(10):806–23 doi 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- 6.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology 2009;10(5):459–66 doi 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 7.Gimple RC, Bhargava S, Dixit D, Rich JN. Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes & development 2019;33(11-12):591–609 doi 10.1101/gad.324301.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mack SC, Singh I, Wang X, Hirsch R, Wu Q, Villagomez R, et al. Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. The Journal of experimental medicine 2019;216(5):1071–90 doi 10.1084/jem.20190196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012;485(7397):201–6 doi 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 10.Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science 2018;361(6409):1346–9 doi 10.1126/science.aau1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5' UTR m(6)A Promotes Cap-Independent Translation. Cell 2015;163(4):999–1010 doi 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, He C. Dynamic RNA modifications in posttranscriptional regulation. Molecular cell 2014;56(1):5–12 doi 10.1016/j.molcel.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zaccara S, Jaffrey SR. A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell 2020;181(7):1582–95 e18 doi 10.1016/j.cell.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer KD, Jaffrey SR. Rethinking m(6)A Readers, Writers, and Erasers. Annual review of cell and developmental biology 2017;33:319–42 doi 10.1146/annurev-cellbio-100616-060758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu B, Li L, Huang Y, Ma J, Min J. Readers, writers and erasers of N(6)-methylated adenosine modification. Current opinion in structural biology 2017;47:67–76 doi 10.1016/j.sbi.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 16.Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Molecular cell 2016;62(3):335–45 doi 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene 2018;37(4):522–33 doi 10.1038/onc.2017.351. [DOI] [PubMed] [Google Scholar]
- 18.Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell stem cell 2018;22(2):191–205 e9 doi 10.1016/j.stem.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 2018;172(1-2):90–105 e23 doi 10.1016/j.cell.2017.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer cell 2017;31(1):127–41 doi 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer cell 2017;31(4):591–606 e6 doi 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai Y, Yang C, Wu R, Huang L, Song S, Li W, et al. YTHDF1 Regulates Tumorigenicity and Cancer Stem Cell-Like Activity in Human Colorectal Carcinoma. Frontiers in oncology 2019;9:332 doi 10.3389/fonc.2019.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lobo J, Costa AL, Cantante M, Guimaraes R, Lopes P, Antunes L, et al. m(6)A RNA modification and its writer/reader VIRMA/YTHDF3 in testicular germ cell tumors: a role in seminoma phenotype maintenance. Journal of translational medicine 2019;17(1):79 doi 10.1186/s12967-019-1837-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, et al. Targeting the RNA m(6)A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell stem cell 2019. July 3;25(1):137–148.e6. doi 10.1016/j.stem.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z, Theler D, Kaminska KH, Hiller M, de la Grange P, Pudimat R, et al. The YTH domain is a novel RNA binding domain. The Journal of biological chemistry 2010;285(19):14701–10 doi 10.1074/jbc.M110.104711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasowitz SD, Ma J, Anderson SJ, Leu NA, Xu Y, Gregory BD, et al. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS genetics 2018;14(5):e1007412 doi 10.1371/journal.pgen.1007412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015;161(6):1388–99 doi 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell research 2017;27(3):315–28 doi 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014;505(7481):117–20 doi 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lasman L, Krupalnik V, Viukov S, Mor N, Aguilera-Castrejon A, Schneir D, et al. Context-dependent functional compensation between Ythdf m(6)A reader proteins. Genes & development 2020. September 17 doi 10.1101/gad.340695.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilbert WV, Bell TA, Schaening C. Messenger RNA modifications: Form, distribution, and function. Science 2016;352(6292):1408–12 doi 10.1126/science.aad8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nature reviews Genetics 2014;15(5):293–306 doi 10.1038/nrg3724. [DOI] [PubMed] [Google Scholar]
- 33.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 2012;149(7):1635–46 doi 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Visvanathan A, Patil V, Abdulla S, Hoheisel JD, Somasundaram K. N(6)-Methyladenosine Landscape of Glioma Stem-Like Cells: METTL3 Is Essential for the Expression of Actively Transcribed Genes and Sustenance of the Oncogenic Signaling. Genes 2019;10(2) doi 10.3390/genes10020141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McIntyre ABR, Gokhale NS, Cerchietti L, Jaffrey SR, Horner SM, Mason CE. Limits in the detection of m(6)A changes using MeRIP/m(6)A-seq. Scientific reports 2020;10(1):6590 doi 10.1038/s41598-020-63355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-oncology 2017;19(1):139–41 doi 10.1093/neuonc/now247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacLeod G, Bozek DA, Rajakulendran N, Monteiro V, Ahmadi M, Steinhart Z, et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell reports 2019;27(3):971–86 e9 doi 10.1016/j.celrep.2019.03.047. [DOI] [PubMed] [Google Scholar]
- 38.Bhat KPL, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer cell 2013;24(3):331–46 doi 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344(6190):1396–401 doi 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patil DP, Pickering BF, Jaffrey SR. Reading m(6)A in the Transcriptome: m(6)A-Binding Proteins. Trends in cell biology 2018;28(2):113–27 doi 10.1016/j.tcb.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pastrana E, Silva-Vargas V, Doetsch F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell stem cell 2011;8(5):486–98 doi 10.1016/j.stem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, et al. c-Myc is required for maintenance of glioma cancer stem cells. PloS one 2008;3(11):e3769 doi 10.1371/journal.pone.0003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D'Addio F, La Rosa S, Maestroni A, Jung P, Orsenigo E, Ben Nasr M, et al. Circulating IGF-I and IGFBP3 Levels Control Human Colonic Stem Cell Function and Are Disrupted in Diabetic Enteropathy. Cell stem cell 2015;17(4):486–98 doi 10.1016/j.stem.2015.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. Pten and p53 converge on c-Myc to control differentiation, self-renewal, and transformation of normal and neoplastic stem cells in glioblastoma. Cold Spring Harbor symposia on quantitative biology 2008;73:427–37 doi 10.1101/sqb.2008.73.047. [DOI] [PubMed] [Google Scholar]
- 45.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer research 2006;66(16):7843–8 doi 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 46.Grkovic S, O'Reilly VC, Han S, Hong M, Baxter RC, Firth SM. IGFBP-3 binds GRP78, stimulates autophagy and promotes the survival of breast cancer cells exposed to adverse microenvironments. Oncogene 2013;32(19):2412–20 doi 10.1038/onc.2012.264. [DOI] [PubMed] [Google Scholar]
- 47.Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell reports 2017;18(11):2622–34 doi 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018;67(6):2254–70 doi 10.1002/hep.29683. [DOI] [PubMed] [Google Scholar]
- 49.Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nature communications 2016;7:12626 doi 10.1038/ncomms12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012;151(1):56–67 doi 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nature reviews Cancer 2014;14(5):329–41 doi 10.1038/nrc3720. [DOI] [PubMed] [Google Scholar]
- 52.Martin JL, Lin MZ, McGowan EM, Baxter RC. Potentiation of growth factor signaling by insulin-like growth factor-binding protein-3 in breast epithelial cells requires sphingosine kinase activity. The Journal of biological chemistry 2009;284(38):25542–52 doi 10.1074/jbc.M109.007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barata P, Cooney M, Tyler A, Wright J, Dreicer R, Garcia JA. A phase 2 study of OSI-906 (linsitinib, an insulin-like growth factor receptor-1 inhibitor) in patients with asymptomatic or mildly symptomatic (non-opioid requiring) metastatic castrate resistant prostate cancer (CRPC). Investigational new drugs 2018;36(3):451–7 doi 10.1007/s10637-018-0574-0. [DOI] [PubMed] [Google Scholar]
- 54.von Mehren M, George S, Heinrich MC, Schuetze SM, Yap JT, Yu JQ, et al. Linsitinib (OSI-906) for the Treatment of Adult and Pediatric Wild-Type Gastrointestinal Stromal Tumors, a SARC Phase II Study. Clinical cancer research : an official journal of the American Association for Cancer Research 2019. April 15;26(8):1837–1845 doi 10.1158/1078-0432.CCR-19-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nature neuroscience 2013;16(10):1373–82 doi 10.1038/nn.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. Journal of immunological methods 2009;347(1-2):70–8 doi 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 57.Patwardhan A, Bardin S, Miserey-Lenkei S, Larue L, Goud B, Raposo G, et al. Routing of the RAB6 secretory pathway towards the lysosome related organelle of melanocytes. Nature communications 2017;8:15835 doi 10.1038/ncomms15835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155(2):462–77 doi 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wan YW, Allen GI, Liu Z. TCGA2STAT: simple TCGA data access for integrated statistical analysis in R. Bioinformatics 2016;32(6):952–4 doi 10.1093/bioinformatics/btv677. [DOI] [PubMed] [Google Scholar]
- 60.Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, et al. Toil enables reproducible, open source, big biomedical data analyses. Nature biotechnology 2017;35(4):314–6 doi 10.1038/nbt.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017;14(4):417–9 doi 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34(3):267–73 doi 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 65.Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 2010;5(11):e13984 doi 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nature cell biology 2014;16(2):191–8 doi 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler-Reya RJ, et al. N(6)-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nature neuroscience 2018;21(2):195–206 doi 10.1038/s41593-017-0057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keene JD, Komisarow JM, Friedersdorf MB. RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nature protocols 2006;1(1):302–7 doi 10.1038/nprot.2006.47. [DOI] [PubMed] [Google Scholar]
- 69.Huppertz I, Attig J, D'Ambrogio A, Easton LE, Sibley CR, Sugimoto Y, et al. iCLIP: protein-RNA interactions at nucleotide resolution. Methods 2014;65(3):274–87 doi 10.1016/j.ymeth.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016;537(7620):369–73 doi 10.1038/nature19342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dodt M, Roehr JT, Ahmed R, Dieterich C. FLEXBAR-Flexible Barcode and Adapter Processing for Next-Generation Sequencing Platforms. Biology 2012;1(3):895–905 doi 10.3390/biology1030895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Webb S, Hector RD, Kudla G, Granneman S. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome biology 2014;15(1):R8 doi 10.1186/gb-2014-15-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grozhik AV, Linder B, Olarerin-George AO, Jaffrey SR. Mapping m(6)A at Individual-Nucleotide Resolution Using Crosslinking and Immunoprecipitation (miCLIP). Methods in molecular biology 2017;1562:55–78 doi 10.1007/978-1-4939-6807-7_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shah A, Qian Y, Weyn-Vanhentenryck SM, Zhang C. CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 2017;33(4):566–7 doi 10.1093/bioinformatics/btw653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Olarerin-George AO, Jaffrey SR. MetaPlotR: a Perl/R pipeline for plotting metagenes of nucleotide modifications and other transcriptomic sites. Bioinformatics 2017;33(10):1563–4 doi 10.1093/bioinformatics/btx002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013;154(5):1151–61 doi 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nature chemical biology 2016;12(2):109–16 doi 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seashore-Ludlow B, Rees MG, Cheah JH, Cokol M, Price EV, Coletti ME, et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer discovery 2015;5(11):1210–23 doi 10.1158/2159-8290.CD-15-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic acids research 2017;45(W1):W98–W102 doi 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All newly generated raw sequencing data related to this study (related to YTHDF2 knockout RNA sequencing, RNA m6A profiling in NSCs and GSCs, and iCLIP-seq) is available on GEO using the accession number GSE158742. All data accessed from external sources and prior publications have been referenced in the text and corresponding figure legends. Additional data will be made available upon request.