

Abstract
Mucosal immunity develops in the human fetal intestine by 11–14 weeks gestation, yet whether viable microbes exist in utero and interact with the intestinal immune system is unknown. Bacterial-like morphology was identified in pockets of human fetal meconium at mid-gestation by scanning electron microscopy (n=4) and a sparse bacterial signal was detected by 16S rRNA sequencing (n=40 of 50) compared to environmental controls (n=87). Eighteen taxa were enriched in fetal meconium with Micrococcaceae (n=9) and Lactobacillus (n=6) the most abundant. Fetal intestines dominated by Micrococcaceae exhibited distinct patterns of T cell composition and epithelial transcription. Fetal Micrococcus luteus, isolated only in the presence of monocytes, grew on placental hormones, remained viable within antigen presenting cells, limited inflammation ex vivo, and possessed genomic features linked with survival in the fetus. Thus, viable bacteria are highly limited in the fetal intestine at mid-gestation, though strains with immunomodulatory capacity are detected in subsets of specimens.
Mucosal immunity is evident in the human fetal intestine by the end of the first trimester1,2. The developing intestine is populated by migrating dendritic cells capable of responding to microbial stimuli and initiating robust T cell responses3. By week 13 of gestation, memory T cells are abundant in the human fetal intestine2,4–8, possess pro-inflammatory potential6, and influence epithelial maturation7. These cells also clonally expand in response to foreign antigens8, suggesting that the presence of select microbes may influence prenatal T cells.
Recent evidence for bacterial presence in utero comes from DNA-based, culture-independent studies of the placenta9–11 and amniotic fluid10, though other studies have refuted the presence of bacteria at these sites and attributed signal to extraction kit contaminants12–14. However, whether microbes exist within the human fetal intestine and influence the earliest stages of mucosal immune development has not been examined. Neonatal meconium, the first stool of a newborn, is comprised of amniotic fluid swallowed during gestation and contains a very simple microbiota15,16. Heightened risk of chronic inflammatory disease in childhood, such as asthma, is associated with maternal lifestyle factors (e.g. farming)17 and with a distinct and perturbed neonatal meconium16, the metabolic products of which induce inflammation ex vivo18. Thus, early-life gut microbiomes have the potential to influence immunity in later life, prompting our hypothesis that specific and highly limited immunomodulatory microbes might be present in the fetal intestine and contribute to pre-natal immune priming.
RESULTS
To determine whether bacteria are present in the intestine in utero, we first performed direct visualization of human fetal intestines obtained from terminated pregnancies (Online Methods; 18–23 weeks of gestation). Terminal ileum intestinal segments were fixed and thin sectioned for light microscopy. Fluorescent in situ hybridization for eubacteria of 5 μm sections of fetal ileum suggested an extremely sparse bacterial signal (Extended Data 1a–c). Because rare signal is further diluted by thin-sectioning required for light microscopy, scanning electron microscopy (SEM) was performed on four independent fetal terminal ileum specimens; environmental exposure was minimized by ligation of the intestinal segments prior to processing (Figure 1a, Online Methods). In three of four independent fetal specimens (Figure 1b–c; Specimens 1–3), clusters of tightly packed cellular structures morphologically and proportionally consistent with bacterial cocci were observed in discrete, isolated pockets of meconium, deeply embedded within existing mucin structures in situ (Figure 1b). Specimen 4 had limited meconium in the lumen as evidenced by exposed epithelial cell structures; clusters of cocci were not observed in this specimen (Figure 1c). Confirming bacterial localization to meconium, these coccoid structures were not observed in sub-epithelial regions, such as the lamina propria or muscularis (Figure 1c-iv). Thus, discrete clusters of cellular structures consistent with coccoid bacterial morphology, embedded within isolated pockets of fetal intestinal meconium are evident during the second trimester of human gestation.
Figure 1. Rare bacterial structures in fetal meconium.

a. Schematic of sample preparation method of fetal intestines for scanning electron microscopy. b. Representative scanning electron micrographs of fetal intestinal lumen, arrowheads indicate pockets of bacterial-like morphology in meconium at 3 000 (left) and mucin-embedded structures at 50 000 (right) times magnification. c. Scanning electron micrographs of four biologically independent fetal intestinal specimens (i.) at low magnification, (ii. -iii.) two independent regions within intestinal lumen, and (iv.) sub-epithelial region outside of the lumen. Scale bars indicate size. Experiments in b-c were repeated 4 times. d. Significantly enriched taxa (Log2-fold change> 2, false discovery rate <0.05) in meconium (n=40) as compared to both kidney (n=7) and procedural swab (n=14) controls after removal of technical negative OTUs where n indicates biologically independent specimens. Dots represent differential taxa and are scaled by percent relative abundance in meconium; top abundant taxa are labeled. DESEQ2 of unnormalized reads was used to determine Log2-fold change and a two-sided false discovery rate.
We established a bank of human fetal small intestine meconium samples (n=50 subjects; n=149 samples, n=87 technical and procedural controls; Supplementary Figure 1; Supplementary Table 1; Online Methods) to quantify and identify these bacteria using molecular techniques. Irrespective of the small intestine segment sampled, and consistent with our SEM observations, total bacterial burden by 16S rRNA copy number was low and variable in fetal meconium, but significantly greater than that of extraction buffer, procedural swab, hospital room air swab, blank cotton swab, or fetal kidney controls (9 out of 13 meconium specimens were greater that the 75th percentile 16S rRNA copy number in controls; Extended Data 1d–e). These data suggested that bacterial presence, if any, was extremely low and nearing the limit of molecular detection. To enhance bacterial signal prior to V4 16S rRNA gene amplification, human mitochondrial 16S DNA (mtDNA) was depleted using Cas9 targeting (Depletion of Abundant Sequences by Hybridization, DASH; Online Methods)19; this did not alter the profile of detected bacteria as compared to band selection and gel extraction (Extended Data 2 a–c). In 16S rRNA datasets controlled for environmental and procedural contamination (Supplementary Table 1–2, Online Methods), a simple bacterial profile was identified in 40 of 50 subjects comprising a median of 23.5 operational taxonomic units (OTUs) with ≥ 5 sequence read counts per sample (Supplementary Table 1–2, Extended Data 3a). Bacterial profiles were consistent in replicate samples along the length of the intestine within subjects (n=108 samples; LME p=0.78; Extended Data 3b) and inter-sample profile distances were greater than intra-sample distances, indicating that the signal detected was unlikely due to uniform contamination (Extended Data 3c). Thus, subsequent analyses focused on the mid-segment (n=40) of the small intestine.
Eighteen taxa were significantly enriched in fetal meconium (DESEQ2; L2FC≥ 2, FDR<0.05) compared to procedural swabs and kidney controls (Figure 1d). Distinct bacterial profiles were evident and defined by the dominant organism detected (PERMANOVA R2 = 0.18, p=1e-5; Figure 2a). Lactobacillus OTU12 and Micrococcaceae OTU10 represented the two highest ranked fetal meconium taxa by relative abundance (Extended Data 3d) and the dominant taxon within distinct subsets of samples (the OTU with the greatest proportion of 16S rRNA reads in a given sample; Lactobacillus-meconium, LM; n=6, or Micrococcaceae-meconium, MM; n=9; Extended Data 3e). The remaining samples were variably dominated by other bacterial taxa (Other-meconium, OM; n=25), including distinct taxa within Lactobacillus and Micrococcaceae, as well as Bacteroides, Bifidobacteria, and Prevotella (Extended Data 3e). OM samples represented the majority of meconium studied and their 16S rRNA profiles were similar to that of biological contamination controls (Extended Data 3f–g). Only LM and MM samples (n=15) exhibited significantly distinct bacterial profiles from OM samples, procedural and kidney controls (PERMANOVA, R2=0.167, p=1e-5; Extended Data 3f–h), and from a variety of technical controls (n=48, Extended Data 3i). Lactobacillus OTU12 and Micrococcaceae OTU10 were not identified as contaminants using stringent thresholds (decontam R package; p threshold=0.6, Supplementary Table 3). Thus, the majority of human fetal intestinal samples at this stage of gestation produced a signal that could either be attributable to noise associated with molecular detection methods in low-burden samples and/or to a lack of bacteria. Nonetheless, the identification of coccoid structures in situ and the observation that approximately 30% of fetal intestinal specimens produced a bacterial profile distinct from that of biological and technical controls led us to determine whether the fetal intestinal immune context related to variance in meconium bacterial detection in paired samples.
Figure 2. Divergent immune cell phenotypes are associated with Micrococcaceae relative enrichment in fetal meconium.
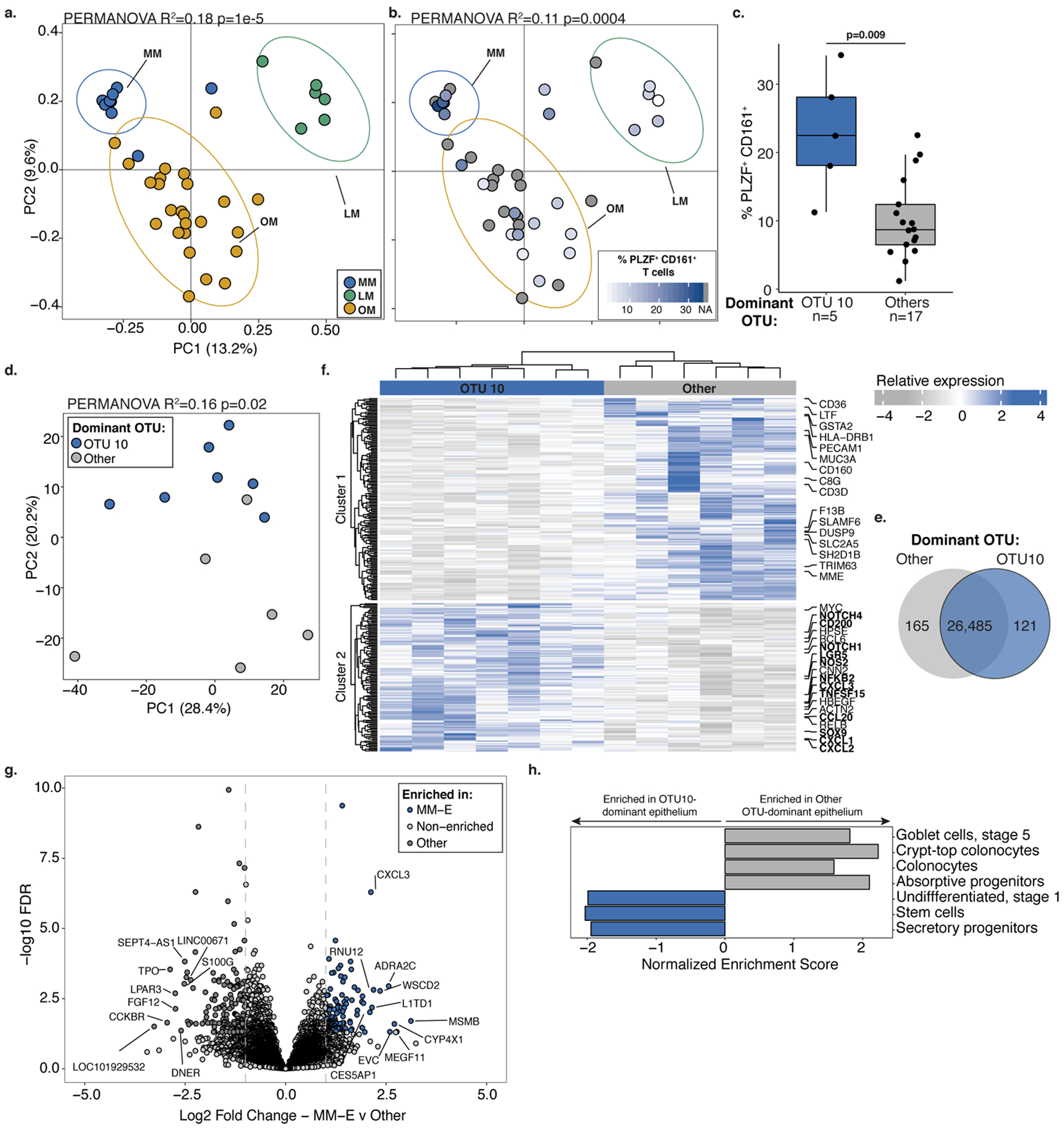
PCoA of Bray Curtis distances of 16S rRNA profiles colored by a. meconium dominated by Lactobacillus OTU12 (LM, n=6), Micrococcaceae OTU10 (MM, n=9), or other taxa (OM, n=25) or b. the proportion of PLZF+ CD161+ T cells among live, TCRβ+, Vα7.2−, CD4+ cells in intestinal lamina propria (LP) paired with LM (n=5), MM (n=5) or OM (n=12) samples indicated by ellipses at 95% confidence. c. Proportion of PLZF+ CD161+ T cells of live, CD4+ TCRβ+ Vα7.2− cells in LP among samples associated with meconium dominated by OTU10 (MM, n=5) or other OTUs (n=17). d. Principal components (PC) analysis of Euclidean distances of top 10000 variable genes (by coefficient of variation) in OTU10-dominated meconium associated epithelium (OTU10, MM-E, n=7) or other OTU-dominated meconium associated epithelium (Other, n=6) as determined by RNA sequencing. e. Venn diagram, f. heatmap with labeled immune pathway transcripts, and g. volcano plot of top differentially expressed genes between MM-E (n=7, log2 fold change >1, FDR < 0.05) and other OTU-dominated meconium associated epithelium (n=6, log2 fold change<1, FDR < 0.05). h. Normalized enrichment scores of gene set enrichment analysis of transcripts associated with epithelial cell states. For a-g, n indicates biologically independent fetal samples. PERMANOVA test for significance for a-b, d. Two-sided Wilcoxon rank sum test was used for c. DESEQ2 was used to calculate significant genes using a two-sided false discovery rate and log2 fold change. Each dot represents one independent biological replicate in a-d and one transcript in g. Boxplot indicates the median (center), the 25th and 75th percentiles, and the smallest and largest values within 1.5× the interquartile range (whiskers).
Taking advantage of intestinal immune profiling data generated at the time of specimen collection, we examined the composition of lamina propria (LP) T cells paired with meconium 16S rRNA data (n=22). Confirming recent findings6, PLZF+ CD161+ CD4+ Vα7.2− TCRαβ+ T cells were highly abundant in the fetal lamina propria in contrast to mesenteric lymph node and spleen (Supplementary Figure 2, Extended Data 4a). We noted a significant relationship between meconium bacterial profile and LP PLZF+ CD161+ T cells (PERMANOVA R2= 0.11, p=0.0004, Figure 2b). LP samples with the highest PLZF+ CD161+ T cell proportion were associated with meconium dominated by Micrococcus OTU10 (MM; Figure 2b) and MM samples exhibited significantly higher proportions of PLZF+ CD161+ T cells compared to all other samples (Figure 2c, Extended Data 4b–c).
Fetal intestinal memory T cells, the majority of which express PLZF and CD1616, have recently been reported to support epithelial stem cell function7. Therefore, paired epithelial cell layer transcriptomes associated with MM versus other specimens were analyzed (n=7, n=6, respectively). MM-associated epithelium (MM-E) samples exhibited distinct transcriptional programs from meconium samples dominated by all other taxa (PERMANOVA p=0.02 R2=0.16, Figure 2d–g) and from LM-associated epithelium (LM-E, n=3) and OM-associated epithelium (OM-E, n=3) groups (Extended Data 4d). Gene set enrichment analysis (GSEA) identified genes associated with intestinal epithelial stem cells, transit amplifying cells, and secretory progenitors as enriched in MM-E (e.g. LGR5, SOX9, NOTCH1, NOTCH4; Figure 2g–h, Supplementary Table 4), consistent with the ability of fetal memory T cells to promote epithelial stem cell function. MUC3A was downregulated in MM-E (Figure 2f), yet transcripts associated with TLR-signaling (NFKB2, TNFSF15), phagolysosome function (NOS2), immune cell chemoattraction (CXCL1–3 and CCL20), and macrophage inhibition (CD200; Figure 2f–h) were enriched (Figure 2f–h, Supplementary Table 4). These transcripts indicated distinct programs of immune cell recruitment and regulation in the context of a nutritionally limited intestinal niche in samples with Micrococcaceae OTU10 present.
To determine whether intestinal Micrococaceae was viable, we attempted isolation from cryopreserved MM fetal meconium samples with the highest read counts for this taxon. Micrococaceae isolates could not be recovered using traditional selective media for this genus and were only obtained under culture conditions that mimicked the fetal intestinal environment (Supplementary Table 5), including addition of placental steroid hormones or THP1 human monocyte cells, suggesting they may represent microbial selective pressures in utero. Lactobacillus could not be cultured from MM samples using these culture methods or traditional selective media for the genus. Fetal bacteria isolated in monocyte co-cultures were classified as Micrococcus using full-length 16S rRNA sequence to interrogate the SILVA database (Micro36; Supplementary Table 5). Additional strains were also isolated in the presence of placental hormones (Supplementary Table 5). The V4 region of the Micrococcus isolate exhibited high homology with OTU10 (97%; Figure 3a, Extended Data 5, Supplementary Table 5).
Figure 3. Micrococcus isolate from fetal meconium exhibits adaptation to the fetal environment.
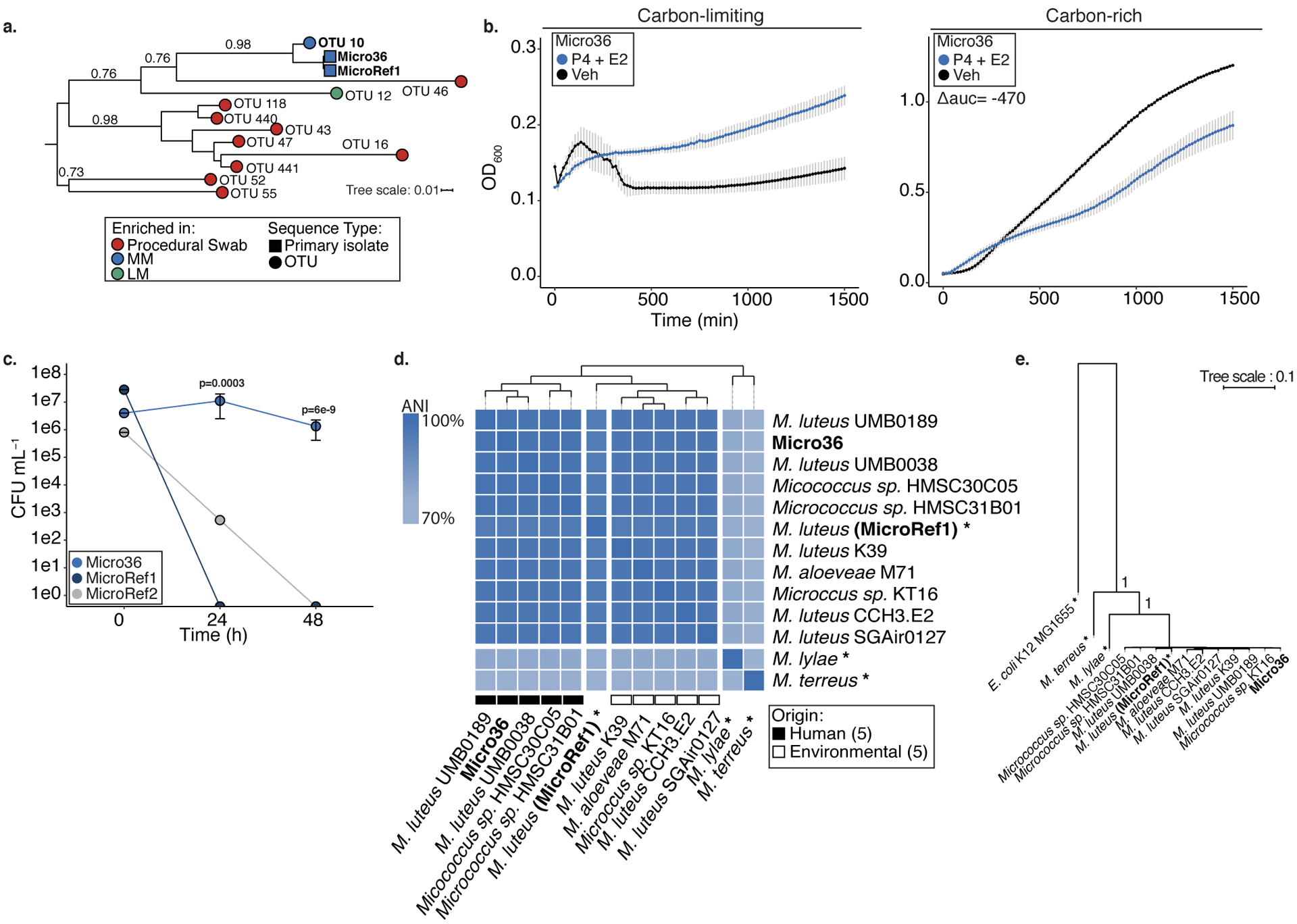
a. Phylogenetic tree of 16S V4 rRNA gene sequences from Lactobacillus-enriched meconium (LM; green), Micrococcaceae-enriched meconium (MM; blue), or procedural swab (red) enriched OTUs (circles) and primary isolate (square) from fetal meconium (Micro36 and reference strain for Micrococcus luteus (MicroRef1). b. Effects of 10−5 M progesterone (P4) and 10−6 M β-Estradiol (E2) on the growth of Micro36 compared to ethanol vehicle control in carbon-limiting media (mineral salt media, left) or carbon-rich media (brain heart infusion; right) at 37 °C. Representative growth curves of three independent experiments measured by optical density at 600nm (OD600), error bars denote standard error of the mean (SEM) from center mean between three technical experiments. For carbon-rich media conditions, integral of logistic regression model fitting was used to calculate area under the curve (auc) and difference relative to vehicle control is reported as Δauc. Intracellular survival of c. Micro36, MicroRef1, MicroRef2 in primary human antigen presenting cells isolated from the fetal intestine. Representative data of three biologically independent fetal specimens, error bars indicate SEM from center mean of three cell culture replicates. ANOVA of a generalized linear model of log(CFU+1) against MicroRef1 for each timepoint was used to calculate significance. d. Whole genome average nucleotide identity (ANI) of all available genomes in Micrococcus and Micro36 isolate. When available strain origin is represented, hierarchical clustering was performed on average nucleotide identity, asterisk (*) indicates a reference or a representative genome for the taxon. e. Phylogenetic tree of conserved single-copy genes across all publicly available genomes within Micrococcus and fetal meconium isolate Micro36 with E. coli K12 MG1655 outgroup. For a and e, branch lengths are scaled to the mean number of nucleotide substitutions per site and bootstrap values are represented for relevant nodes.
We hypothesized that fetal Micrococcus luteus exhibits a fitness advantage over phylogenetic relatives under culture conditions that mimic the intestinal environment in utero. Thus, we examined its growth and that of two reference strains MicroRef1 (ATCC 4698) and MicroRef2 (ATCC 12698) in the presence of peak third trimester cord blood concentrations20 of progesterone and/or β-estradiol. Micro36 exhibited the unique ability to grow on progesterone and β-estradiol in carbon limiting media, albeit to low cell densities (Figure 3b; Extended Data 6a–b). In carbon-rich conditions, consistent with previously reported bacteriostatic effects of steroid hormones21, progesterone and β-estradiol (but not β-estradiol alone) universally inhibited growth of all three M. luteus strains (Figure 3b; Extended Data 6c–f). These data suggest that conditions of low substrate availability coupled with pregnancy hormones permit limited growth of specific fetal bacterial isolates, offering an explanation for the control of bacterial burden in the intestine during human gestation.
The necessity of monocytes for initial Micrococcus isolation (Supplementary Table 5) suggested the capacity for survival within phagocytic cells. Isolated primary human fetal intestinal HLA-DR+ antigen presenting cells (APCs) were cleared of intracellular bacteria (Online Methods), and incubated with fetal Micrococcus isolates to permit phagocytosis followed by gentamycin protection assays. At 24h, 1×107 CFU mL−1 of Micro 36 was recovered and the fetal isolate remained viable in APCs at 48h at 1×106 CFU mL−1 (Figure 3c), indicating a capacity for prolonged intracellular survival. Control reference strains MicroRef1 and to a lesser extent MicroRef2 were non-viable under comparable conditions (Figure 3c). Similar results were obtained using a RAW264.7 macrophage cell line with an additional E. coli control (Extended Data 6g) and gentamycin resistance did not develop in the time course of either of these experiments (Extended Data 6h–i). The ability of this fetal Micrococcus strain to persist inside phagocytes offers a potential mechanism of protected entry into the fetal intestine.
Whole genome sequencing of Micro36 (Supplementary Table 6) permitted high resolution taxonomy of the isolate and identified shared and unique genomic features when compared to phylogenetic relatives. Micro36 exhibited 96.9% whole genome average nucleotide identity (ANI) to a reference genome of M. luteus and clustered by whole genome ANI with other human, but not environmental M. luteus isolates (Figure 3d, Supplementary Table 7). Pan-genomic analysis of our fetal Micrococcus and all available Micrococcus genomes identified shared single-copy genes (Extended Data 7) used to build highly resolved phylogeny (bootstrap value = 1 for relevant clade, Figure 3e). Using a 96.5% ANI speciation cut-off22, Micro36 was classified as a strain of M. luteus.
Compared to M. luteus (MicroRef1), Micro36 exhibited 425 unique genes, 256 of which were annotated (Supplementary Table 8). Genomic features of Micro36 included two sterol carrier proteins and a putative steroid ketoisomerase, which typically facilitates degradation of steroid hormones. The genome also encoded reactive oxygen and nitrogen radical reducing enzymes, and genes in the catechol pathway. While the broader prevalence of these genes is yet to be determined, these data offer plausible mechanisms by which Micro36 may grow on placental hormones23 (Figure 3b), remain viable in phagocytes24 (Figure 3c), and under conditions of hypoxia associated with elevated NOS225 in MM-associated epithelia (Figure 2f).
To determine whether fetal Micrococcus luteus isolates are found in post-natal infant samples, we utilized publicly available 16S rRNA data from three independent early-life cohorts16,18,26. Sequences exhibiting ≥97% homology to our fetal isolates were detected throughout early life (up to 12 months; Supplementary Table 9); however, sequences with the highest homology (≥99%, Supplementary Table 9) were primarily found in infant meconium (first stool) samples (Extended Data 8a). M. luteus was found in low in abundance in infant samples but highest in post-natal meconium in two independent metagenomic cohorts27,28 (Extended Data 8b–c). These species were detected on maternal chest and in vaginal introitus at delivery and were not highly abundant in maternal stool (Extended Data 8b–c). Among our fetal meconium specimens, neither the number of detected OTUs per sample nor the relative abundance of Micrococcaceae OTU10 were significantly correlated with gestational age when all samples were considered (Extended Data 8d–e). However, a positive correlation between OTU10 relative abundance and gestational age was evident within the MM group (Pearson’s r=0.5, p=0.1; Extended Data 4f). This suggests that intestinal Micro36 or highly related strains may increase during gestation, persist at least until birth and may be succeeded in the post-natal period by phylogenetically related species.
MM was associated with a distinct program of fetal epithelial gene expression (Figure 2). We thus examined the capacity of fetal M. luteus to induce characteristic features by profiling the transcriptome of primary human fetal intestinal epithelial cells (n=2) exposed to Micro36 for four hours in vitro. Transcriptional differences were observed when Micro36 exposed epithelia were compared to media controls (Figure 4a). As expected, short-term exposure to planktonic bacterial cultures in vitro did not fully recapitulate the global fetal intestinal transcriptome patterns observed in MM-E (Figure 4a). Nonetheless, Micro36 exposure induced the expression of TLR6 and its downstream regulator NKFB was enriched in MM-E (Figure 4a–b). These data suggest that even following short-term fetal bacterial exposure, fetal intestinal epithelial cells exhibit transcriptional responses to Micrococcus that partially recapitulate features observed in MM-E.
Figure 4. Fetal Micrococcus isolate promotes immunotolerance phenotypes in vitro.
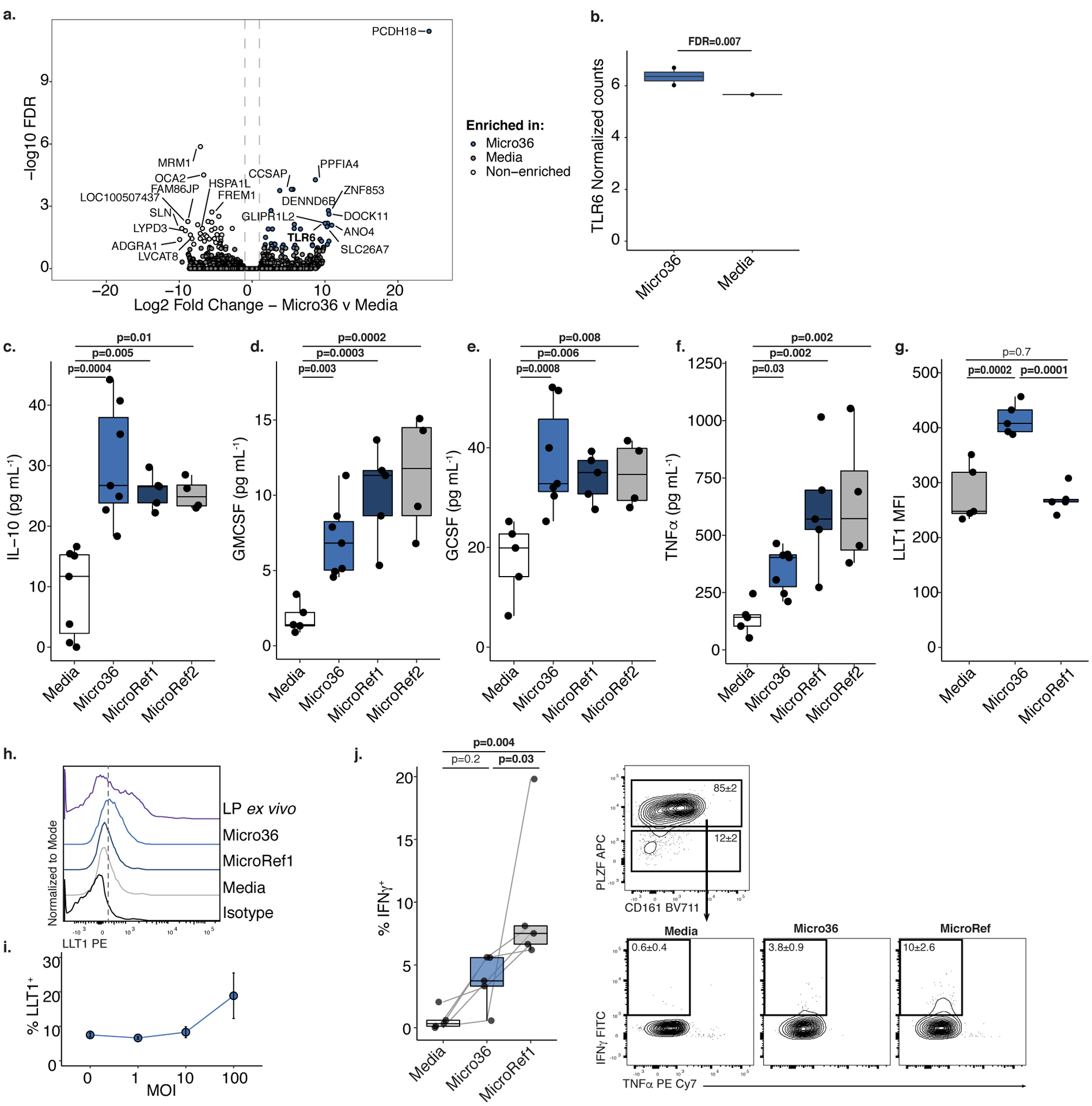
a. Volcano plot of significantly (false discovery rate, FDR, <0.05) and differentially (Log2FoldChange |1|) expressed genes and b. normalized read counts of TLR6 in primary human fetal intestinal epithelial cells after Micro36 treatment versus media control by RNAseq. For a-b, FDR-adjusted p-values were calculated using two-sided DESEQ2 algorithm, n=2 biologically independent fetal samples. Concentrations of c. IL-10, d. GM-CSF, e. G-CSF, or f. TNFα in supernatants of fetal splenic antigen presenting cells following four hours of exposure to media (n=7) or Micrococcus (Micro36 n=7, MicroRef1 n=5, MicroRef2 n=4) strains, where n represents biologically independent fetal specimens for the indicated treatment. g. Mean fluorescence intensity (MFI) and h. representative histograms of five experiments of LLT1 expression of live, lin−, CD45+, HLA-DR+ splenocytes of n=5 biologically independent fetal specimens exposed to media, Micro36, MicroRef1 or unstimulated lamina propria (LP) antigen presenting cells. i. Multiplicity of infection (MOI) of Micro36 relative to proportion of LLT1+ live, lin−, CD45+, HLA-DR+ splenocytes, each dot (center) represents mean of n=3 biologically independent fetal specimens and error bars indicate standard error of the mean. j. Intracellular INFγ production among pure intestinal effector memory T cells in mixed lymphocyte reactions with sorted, autologous lin−, CD45+, HLA-DR+ antigen presenting cells that were pre-exposed to media or Micrococcus (Micro36, MicroRef1) strains. Left, Percent IFNγ+ T cells among live, TCRβ+, CD4+, Vα7.2−, PLZF+, n=5 biologically independent fetal specimens. Right, Representative flow plots of five experiments of sorted effector memory T cells T cells (top) and intracellular cytokine, IFNγ and TNFα, expression (bottom); numbers indicate mean proportion and standard error of the mean (SEM). Lines connecting dots indicate same fetal specimen across treatments. Two-sided Satterthwaite’s method on linear mixed effects model was used to test for significance, controlling for repeated measures of cell donor, for c-g, j; Positive LME residuals are plotted for c-f. Each dot represents an independent fetal sample. Boxplots indicate the median (center), the 25th and 75th percentiles, and the smallest and largest values within 1.5× the interquartile range (whiskers).
The heightened expression of immune cell recruitment and regulatory mediators in MM-E (Figure 2f), led us to assess the capacity of Micro36 to influence primary fetal HLA-DR+ antigen presenting cell (APC) function obtained from spleen (Supplementary Figure 3). Without decreasing cell viability (Extended Data 9a), Micro36 and two reference strains induced fetal APC production of cytokines associated with maturation of intestinal macrophages (GM-CSF and G-CSF) as well as IL-10 (Figure 4c–e), which promote a tolerogenic environment29–31. Micro36 induced lower levels of TNFα compared with reference strains, indicating its ability to limit APC inflammation (Figure 4f). LLT1, the natural ligand for the fetal-specific inhibitory C-type lectin CD161, is expressed on fetal intestinal macrophages6 and can be induced upon TLR activation of APCs32. Given the capacity of fetal M. luteus to induce fetal epithelial TLR6 in vitro, we examined whether it could elicit LLT1 expression on primary human fetal splenic APCs. Compared with a phylogenetically related strain, only fetal Micro36 induced LLT1 expression and in proportion with multiplicity of infection, albeit to lower levels than observed in lamina propria APCs ex vivo (Figure 4g–i).
As ligation of CD161 inhibits IFNγ production by fetal intestinal PLZF+ CD161+ T cells6, we hypothesized that Micro36 may specifically regulate the inflammatory potential of these T cells. Sorted splenic APCs (Extended Data 9b) pre-conditioned with Micro36 or MicroRef1 were co-incubated with autologous, fetal intestinal effector memory T cells (>99% purity), the majority of which expressed PLZF and CD1616 (Extended Data 9c–d). Micro36 exposure resulted in a significant reduction of IFNγ production by these T cells as compared to MicroRef1 (Figure 4j), indicating induction of immunotolerance by fetal M. luteus. While exerting an effect on PLZF+ T cell function, Micro36 did not impact the proportional accumulation of these cells or regulatory T (Treg) cells (Extended Data 9e–f), and did not influence the production of IL17A, IL17F, GMCSF, IL-4, IL-10, IL-13, or TNFα after five days of APC-T cell co-cultures (Extended Data 9g–m). Together data suggest that fetal intestinal immune cells are capable of mounting an inflammatory response to bacteria and that fetal M. luteus may circumvent this by inducing tolerogenic APCs and inhibiting IFNγ production by fetal memory T cells.
DISCUSSION
Whether bacteria are present in utero is contentious because of the inherent limitations of molecular methods that are commonly used to identify bacteria in low-burden environments. Background noise and false positives in technical negative controls are common when bacterial burden is extremely low, therefore simple removal of all taxa detected in these controls is not deemed appropriate33. In our study, despite improving the current molecular methods to boost bacterial signal (Online Methods), 16S rRNA sequencing data remained noisy and, for the majority (70%) of samples, we identified a sparse bacterial signal that was indistinguishable from procedural or fetal kidney controls. Using a taxon filtering approach that focused on the signal detected in the majority of negative controls led to the identification of a Micrococcus taxon as one of a small number of discriminant taxa enriched in meconium samples. With this molecular data as a guide for fetal bacterial species isolation, we subsequently cultured M. luteus from parallel preserved samples that never encountered extraction buffers during the course of sample processing. The molecular bacterial signal was proportionately more sparse than the signal identified by microscopy, which indicates that our removal of contaminant taxa may have suppressed a broader bacterial signal and supports previous reports of false-positive classification of contaminants in buffer controls33. Our molecular data identified only 17 additional taxa not attributable to contamination in our cohort of 50 fetal intestinal specimens, suggesting that conditions in the fetal gut highly limit bacteria, the mechanisms of which warrant further investigation.
While there is debate regarding the best methods to address contamination in low-burden bacterial environments, our data suggest that current molecular methods alone are insufficient to support or reject the in utero sterility hypothesis. By combining molecular bacterial detection, immune correlates, microscopy, strain isolation, and ex vivo experiments, our study provides direct and indirect evidence for the presence of sparse but viable bacteria in the human fetal intestine at mid-gestation with the capacity to limit inflammatory potential by fetal immune T cell populations. While it is possible that the bacterial signal identified may arise from a contamination from a source not investigated in this study, in our judgement, the corroborating evidence suggests that restriction of bacterial entry into the human fetal intestine is not absolute. We note that the lack of clinical data associated with specimens in our cohort (as mandated by our institutional protocols) limits our ability to examine pregnancy features associated with identification of Micrococcus, an important consideration for subsequent studies.
Fetal Micrococcus most likely arises from maternal cervico-vaginal microbiomes, which commonly house this genus34,35. While our fetal Micrococcus isolate exhibited genome similarity to vaginal Micrococcus isolates, it also encoded strain-specific genes not found in these strains, which may provide survival advantages under the strong selective conditions of the fetal intestine. Indeed, fetal M. luteus, which encoded a ketoisomerase putatively involved in steroid metabolism, exhibited the unique capacity for limited growth in the presence of progesterone and β-estradiol in low-nutrient conditions. This this observation coupled with the fact that M. luteus can exist in a dormant, viable but non-culturable state under starvation conditions36, may allow it to persist under conditions of limited nutrition and pregnancy hormone exposure in the fetal intestine. Consistent with this hypothesis, intestinal epithelial transcriptome analysis suggested lower expression of microbial nutritional substrates including the mucin glycoprotein MUC3A in fetal samples in which M. luteus was found. Collectively, these observations offer a potential explanation for detection of M. luteus in specific subsets of fetal intestinal samples in which prevailing conditions of nutrient limitation and pregnancy hormones may contribute to its limited presence.
Bacterial presence may not be pervasive in the second trimester, yet in our study Micrococcus was associated with the immunological status of the intestinal epithelium and the lamina propria memory T cell compartment. Micrococcus in the fetal intestine modulates mucosal immunity and reciprocally, the immune system influences which microbes are tolerated by the host37. Thus, it is plausible that epithelial and lamina propria immunity additionally select for specific M. luteus strains. In turn Micrococcus limits the inflammatory ability of these cells, which may foster a tolerant environment that permits its survival in utero. However, we recognize that other developmental factors such as stem cell niche38, the predisposition for fetal T cells to develop into regulatory T cells39, and antigens from swallowed amniotic fluid40 also shape prenatal immunity.
Recent studies of fetal immunity have led to the hypothesis that bacterial signals in utero initiate an adaptive immune response3, including T cell activation6–8. Fetal T cells respond to non-inherited maternal- and self- antigens39 and are capable of memory formation in the intestine6–8. The presence of bacteria in the fetal intestine suggests that bacterial antigens may also contribute to T cell activation, as fetal intestinal T cells do not exclusively exhibit a tolerogenic phenotype6–8. Their ability to produce inflammatory cytokines in the absence of systemic inflammation indicates intestinal compartmentalization of immune response in utero6, which may be essential for tolerance or clearance of fetal intestinal bacteria. Micrococcus enrichment in the fetal gut associated with increased proportions of IFNγ-producing mucosal memory PLZF+ CD161+ T cells6 and only the fetal Micrococcus isolate reduced IFNγ production by these T cells. While fetal Micrococcus likely elicits a number of responses, the specific induction of LLT1 on antigen presenting cells identifies a potential bacterial mechanism of immune regulation that is unique to fetal adaptive immunity6. Thus, immunological memory to fetal Micrococcus may begin in utero.
How the fetal intestine limits bacterial presence remains underexplored, though the ability of specific bacteria to persist in nutrient limiting conditions, grow on pregnancy hormones and survive within phagocytes offer plausible mechanisms for survival in utero. The implications of in utero bacterial interactions or lack thereof on long-term health remain to be determined.
ONLINE METHODS
Human Samples and Consent
Donated human fetal tissue (small intestine, mesenteric lymph node, spleen) was obtained under the auspices of UCSF Committee on Human Research (CHR) approved protocols after written informed consent at 20 ± 2.2 gestational weeks from the Department of Obstetrics, Gynecology and Reproductive Science at San Francisco General Hospital from terminated pregnancies. Exclusion criteria were: (1) known maternal or intrauterine infection, (2) intrauterine fetal demise, and/or (3) known or suspected chromosomal abnormality6. No Human Patient Information (HPI) is associated with the data presented. All sample collection methods comply with the Helsinki Declaration principles. Samples were transported in media on ice and processed within 2 hours after collection.
Sample Collection for Fetal Meconium Cohort
Uninterrupted stomach to caecum sections (fetal intestine), kidneys, spleens, and mesenteric lymph nodes were collected by a single operator using sterile tools within 10 minutes of termination procedure and placed into sterile containers with pre-aliquoted complete RPMI (cRPMI) media composed of: RPMI media (GIBCO) without antibiotics, 10% fetal bovine serum (GIBCO), 1 mM sodium pyruvate (Life Technologies), 2 mM L-glutamine (Life Technologies), 1 × non-essential amino acids (Life Technologies), and 10 mM HEPES (Life Technologies). Sterile cotton swabs were pre-moistened with sterile 1 × phosphate-buffered saline (PBS) and stored in containers until used to vigorously sample the surgical tray for 30 seconds, thus sampling both the hospital environment and any contaminants arising from the procedure; swabs were immediately snapped off into sterile tubes containing 500 μL of pre-aliquoted, sterile RNAlater. Blank swabs were prepared as described above, but immediately snapped off into RNAlater, without sampling the surgical tray. Air swabs were prepared as described above, but held in surgical room air for 30 seconds, before immediately being snapped off into RNAlater. All specimens were immediately placed on ice and transported to the laboratory. Intestinal sections were dissected to remove the mesentery and the muscularis in a sterile petri dish in a biosafety laminar flow cabinet. Separate sterile tools were used to divide the small intestine into three equal sections and new sterile tools were used to scrape internal contents, termed fetal meconium, of each section into sterile 1 × PBS (Supplementary Figure 1). Fetal meconium was homogenized by vigorous pipetting in sterile 1 × PBS, pelleted by centrifugation at 3000 × g for 10 minutes, and re-suspended in 1 mL of sterile 1 × PBS. Half of fetal meconium suspension (by volume) was added to RNAlater (Ambion), while the remainder was re-suspended in sterile 50% (v/v) glycerol. Sterile tools were used to remove kidney capsule of the fetal kidney in a sterile petri and separate sterile tools were used to biopsy the internal kidney tissue, which was immediately placed in RNAlater. Fetal meconium samples, kidney specimens, procedural swabs, and blank swabs were cryopreserved at −80 °C, within 2 hours of the termination procedure. Additional splenic and intestinal samples were collected in the manner described above for ex vitro APC and T cell experiments. In total 77 fetal specimens were used in this study.
16S rRNA Gene Burden and Sequencing
DNA extraction.
Genomic DNA (gDNA) from fetal meconium samples, kidney specimens, procedural swabs, and blank swabs was extracted using a modified cetyltrimethylammonium bromide (CTAB)-buffer-based protocol exactly as previously described18 along with buffer controls. Buffers were prepared using HPLC-grade chemicals in a BSL2 biosafety cabinet and autoclaved before use.
16S rRNA gene burden qPCR analysis.
16S rRNA gene copy number was assessed by quantitative PCR (Q-PCR) using the 16S rRNA universal primers and TaqMan probes, as previously described41. Briefly, total 16S rRNA gene copy number was calculated against a standard curve of known 16S rRNA copy numbers (1×102−1×109). Q-PCR was performed in triplicate 20 μl reactions containing final concentrations of 1 ×TaqMan Universal Master Mix (Life Technologies), 100 ng of extracted genomic DNA, 900 nM of each primer, P891F (5’-seq-3’F) and P1033R (5’-seq-3’R) and 125 nM of UniProbe under the following conditions: 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s, and annealing and extension at 60 °C for 1 min, along with no-template control and 8 standards. Copy number was normalized either by 100ng of input DNA, when possible. When too little DNA was obtained, such as in the case of the buffers, 10μL of DNA extract was added to the PCR reaction and copy number was normalized by weight of frozen sample.
Depletion of Abundant Sequences by Hybridization (DASH).
Depletion of human 16S mitochondrial DNA (mtDNA) using single guide RNA (sgRNA) targeting of Cas9 was performed as previously described19. Briefly, 54 sgRNAs targeting the human mtDNA were transcribed from pooled sgRNA templates using custom T7 RNA polymerase generously provided by the DeRisi laboratory at UCSF. sgRNAs were purified and concentrated using a column-based RNA purification kit with DNAse treatment (Zymo) and incubated with purified Cas9 (Berkeley Macrolab) for 10 minutes at 37°C. sgRNA-loaded Cas9 was incubated with either meconium genomic DNA (gDNA) or pooled library of 16S rRNA V4 amplicon (see below) for 2 hours at 37°C. Cas9 was deactivated by boiling the in vitro reaction at 98°C for 10 minutes and Ampure XP beads (Agencourt) were used to purify the amplicon DNA. To test the effects of DASH on bacterial community composition, a subset of meconium samples from our bank (n=10) was depleted of mtDNA either from individual meconium gDNA (individual DASH) prior to 30-cycle amplification or from the pooled library of 30-cycle amplicons (pooled DASH). DASH bacterial profiles were compared to 30-cycle or 35-cycle amplicons that were depleted of mtDNA by gel extraction, using a gel extraction kit (Quiagen). For sequencing of the entire bank of fetal meconium gDNA, individual DASH was implemented on all samples including buffer blanks and contamination swabs.
Sequencing preparation.
The V4 region of the depleted genomic DNA was amplified using primers designed by Caporaso et al42 using PCR conditions and protocol as described in Fujimura et al18. Briefly, samples were amplified in heptuplicate from a single mastermix per template, aliquoted into 384-well plates, and included a negative control reaction for each template mastermix and each reverse barcoded primer. PCR reactions were performed in 25μL volumes using 0.025 U Takara Hot Start ExTaq (Takara Mirus Bio Inc.), 1X Takara buffer with MgCl2, 0.4 pmol μl-1 of F515 and barcoded R806 primers, 0.56 mg/ml of bovine serum albumin (BSA; Roche Applied Science), 200 μM of dNTPs and 10 ng of DASH gDNA. PCR conditions were: initial denaturation (98 °C, 2 min), 30 cycles of 98 °C (20 s), annealing at 50 °C (30 s), extension at 72 °C (45 s) and final extension at 72 °C (10 min), except in validation of DASH protocol (see above), where 35 cycles of amplification were also used. Amplicons were pooled and verified using a 2% TBE agarose e-gel (Life Technologies), purified using AMPure SPRI beads (Beckman Coulter), quality checked using Bioanalyzer DNA 1000 Kit (Agilent) and quantified using the Qubit 2.0 Fluorometer and the dsDNA HS Assay Kit (Life Technologies). Amplicons were pooled at equimolar amounts to create the sequencing library, with the exception of buffer controls, which did not yield enough amplicon and were pooled at the average volume. A mock community (BEI Resources HM-277D) composed of equal genomic concentration of bacterial genomic DNA was sequenced for each amplification plate to monitor and standardize data between amplification plates. Denatured libraries were diluted to 2 nM and were loaded onto the Illumina MiSeq cartridge at 5 pM with 15% (v/v) denatured 12.5 pM PhiX spike-in for sequencing. Complete fetal meconium bank of samples was sequenced on one 250 × 250 base pair Illumina MiSeq run.
Sequence data processing and quality control.
Paired-end reads were assembled using FLASH v1.2.1143 requiring a minimum base pair overlap of 200 and de-multiplexed by barcode using QIIME (Quantitative Insights Into Microbial Ecology, v1.9.1)44. Quality filtering was accomplished using USEARCH v8.0.1623 to remove reads with >2 expected errors45. Quality reads were de-replicated at 100% sequence identity, clustered at 97% sequence identity into operational taxonomic units (OTUs), filtered of chimeric sequences, and mapped back to resulting OTUs using USEARCH. Taxonomy was assigned to the OTUs using SILVA database.
Fetal meconium data analysis.
OTUs detected in greater than 50% of extraction buffer, blank swab, and air swab controls were removed from all samples prior to further filtering. OTUs comprising fewer than 5 reads and fewer than 0.0001% of the total read counts across all samples were removed. Additional buffer contaminants were identified using decontam package46 in R. Resulting sequence reads were normalized by multiply rarefying to 1,000 reads per sample as previously described, to assure reduced data were representative of the fuller data for each sample18. Dominant taxa were identified for each rarefied sample by determining the OTU with the greatest number of reads per sample.
Post-natal meconium data analysis.
16S rRNA gene V4 amplicon sequencing profiles of meconium collected at birth was obtained from the European Nucleotide Archive (ENA) under accession number PRJEB20766 and post-processed as described above for fetal meconium. OTUs were re-picked with combined fetal and post-natal meconium datasets combined. Infant stool samples with high identify to fetal isolates were identified by first trimming the appropriate variable region (depending on study) from full-length 16S rRNA gene Micro36 sequences. These sequences were then aligned using BLASTn to publicly available infant stool cohorts16,18,26 with accession numbers PRJEB13896, PRJEB20766, PRJEB8463; sequences with >97% identity and >99% coverage were identified.
Immune Cell Isolation
Uninterrupted stomach to caecum sections of the fetal small intestine were dissected in cold 1x PBS (see above). The intestine was cut into 1cm sections and washed three times with 1mM DTT in 1x PBS for 10 minutes at 37°C to remove mucus. The epithelial layer was dissociated with three washes of 1mM EDTA in 1x PBS for 20 minutes at 37 °C and the latter wash was preserved in RNAlater (Ambion) at −80°C for RNAseq. The remaining lamina propria cells were dissociated with freshly prepared 1mg/mL Collagenase IV (Gibco) and 10mg mL−1 DNAse (Roche) in cRPMI for 30 minutes at 37°C, in a shaking water bath at 200 rpm. Mesenteric lymph node and spleen cells were isolated by a 30-minute digestion in Collagenase IV media as described above and then gently pressed through a 70μm strainer. Cells were separated in a 20%-40%-80% Percoll density gradient at 400 × g for 40 minutes: T cells were recovered at the 40–80% interface, while antigen presenting cells were recovered at the 20–40% interface. All cells were washed twice with cRPMI media. Viability was measured with propidium iodide (Sigma Aldrich) and AQUA dye (Invitrogen) using flow cytometry.
Epithelial Cell RNA Sequencing
Cryopreserved epithelial cell layers (in RNAlater, Ambion) were lysed using QIAshredder (QIAGEN) columns and RNA was extracted using RNAqueous kit (ThermoFisher). RNA was quantified using Qubit RNA HS Assay (ThermoFisher), normalized, and converted to cDNA using SMARTer cDNA Synthesis Kit (Takara Bio) using 7 cycles of amplification. RNA and cDNA quality was determined by Bioanalyzer (Agilent). cDNA was fragmented, ligated with Illumina adapters using Nextera XT kit (Illumina), following manufacturer’s instructions, and sequenced on NovaSeq6000 sequencer using two lanes. Paired-end 100 by 100 bp reads were obtained, demultiplexed, quality filtered, removed of Illumina adapters using TrimGalore (github.com/FelixKrueger/TrimGalore), and aligned to the human genome (Hg38 release) using STAR47 with ENCODE recommended parameters. Features were assigned to transcripts using featureCounts48, normalized using DESEQ249. Differential expression was evaluated using DESEQ2 genes with at least 20 reads per gene in respective sample grouping. Log-normalized read counts were obtained from DESEQ2 package, genes were filtered for presence in 75% of samples per comparison group, top variable genes were identified by the coefficient of variance and used to calculate principal components of Euclidean distances.
Fluorescence In Situ Hybridization
Murine and human fetal terminal ileum was fixed in Carnoy fixative to preserve the mucous layer50, embedded in Tissue-Tek OCT (VWR) medium, and cryosectioned to 5 μm sections using a cryostat. Sections were thawed, were post-fixed with acetone for 15 minutes, and rinsed with 1x PBS. Slides were incubated with sterile-filtered 100μL of probe solution containing 35% formamide, as previously described50. Hybridizations were performed for 10 hours at 48°C, followed by a washing step for one hour at the same temperature, as previously described50. Hybridization probes were utilized at 0.5 μM final concentration and included fluorescently-labeled oligos eubacterial (EUB) /5Cy3/GC TGC CTC CCG TAG GAG T/3Cy3Sp/51 or non-targeting (NEUB) /5Cy3/AC TCC TAC GGG AGG CAG C/3Cy3Sp/51. Slides were mounted in Vectashield with DAPI (Vector Laboratories) and imaged at 400x and 1000x magnification using epifluorescence Keyence Microscope BZ-X700. Quantification of images was performed in ImageJ software using the set scale function to calibrate pixels to μm units, freehand selection tool was used to trace the perimeter of each villi, and tracing lengths were measured and summed for each section. The point tool was used to manually count EUB or NEUB signal.
Electron Microscopy
Terminal ileum of fetal intestines was dissected and ligated with sterile suture to prevent contamination of the internal lumen. Ligated samples were immediately immersed in 2.5% (v/v) electron microscopy (EM) grade glutaraldehyde fixative (Sigma Aldrich) in 1x PBS solution and incubated overnight at room temperature with agitation. Samples were washed twice with 1x PBS for 15 minutes and dehydrated with a series of ethanol baths. Samples were then critical point dried (Tousimisautosamdri-815), sliced open with a clean razorblade, mounted in conductive silver epoxy (Ted Pella, Inc.), and coated with 15–30 nm of iridium (Cressington 208-HR sputter coater). Electron micrographs were recorded using a Carl Zeiss ULTRA55 FE-SEM at accelerating voltages in the range 1.24–3.9 keV, working distances of 4.8–9.2 mm, and 20–60 μm diameter apertures with high-current mode. Post-processing of images was not performed. Specimens were stored in a vacuum chamber to avoid contamination between imaging sessions.
Bacterial Isolation
Punch biopsies were taken from three samples of cryopreserved meconium with highest read counts for Micrococcus using a sterile surgical punch biopsy tool (Integra Miltex, Plainsboro, NJ) in clean biosafety cabinet. Three independent fetal meconium samples were used for isolation. Punch biopsies of Micrococcus enriched meconium were incubated in antibiotic-free cRPMI with or without 2 ×106 THP1 human monocyte feeder cells for 48 hours at 37 °C in ambient atmospheric stationary conditions. Single colonies were isolated after transfer to brain heart infusion (BHI; TekNova) agar plates and single colonies were picked. Colony sequencing (Quintara Biosciences) was performed using the full length 16S rRNA gene using primer pairs 27F (5’-seq-3’) and 1492R (5’-seq-3’)52. Full-length gene was assembled using Clustal Omega and taxonomy was determined by SINA53 against the curated SILVA database. Reference strains were obtained from American Type Culture Collection for Micrococcus luteus (MicroRef1, ATCC 4698; MicroRef2 ATCC 12698) and grown by ATCC’s protocol.
Bacterial Whole Genome Sequencing and Comparative Genomics
Whole genome sequencing and assembly.
Twenty-four-hour cultures of Micro36 were obtained in media and culture conditions as described above, and DNA was extracted using CTAB-based protocol as described above. Genomic DNA (gDNA) was fragmented and Illumina adapters were ligated using Nextera XT (Illumina) kit following manufacturer’s instructions. gDNA library quality was verified by gel-electrophoresis Bioanalyzer (Agilent) and was sequenced on Illumina MiSeq using a MiSeq Reagent Kit v3 (Illumina) with 300 × 300bp paired-end reads. Reads were removed of adapters and quality filtered using TrimGalore. When possible, paired-end reads were assembled using FLASh43 for use as a single-ended library for assembly using SPAdes54 genome assembler. Genome assembly quality was determined by QUAST55 and genomes were submitted NCBI Prokaryotic Genome Annotation Pipeline (PGAP). Annotation was performed locally using NCBI COG database in anvi’o package56.
Comparative genomics.
Micrococcus genomes were downloaded from NCBI using NCBI genome download tool (github.com/kblin/ncbi-genome-download) and imported into anvi’o pangenome analysis environment56. Average nucleotide identity and coverage was calculated using ANIb within pyani package (widdowquinn.github.io/pyani/)57. Single copy genes58 were identified for all relevant genomes within anvi’o environment, aligned using MUSCLE59, phylogenetic trees were constructed using FastTree260, and visualized in iTOL61.
Post-natal data analysis.
A custom kraken262 database was created by adding Micro36 genome contigs to the standard database. Maternal and infant stool and various body site bacterial metagenomic reads27,28 and public metadata were obtained from NCBI SRA in FASTQ format using accession numbers PRJNA475246 and PRJNA352475. Percent relative abundance of M. luteus per sample was obtained using kraken2 software was used to classify metagenomic reads against the custom database using a minimum base quality threshold of 20 and a confidence threshold of 95%.
Bacterial Growth Curves
Liquid cultures of Micrococcus strains were grown for 24–48 hours at 37°C in BHI. Cultures were normalized to 0.05 optical density at A600nm (OD600) and incubated with indicated molar concentrations of progesterone (Tocris Bioscience) and 17β-estradiol (Tocris Bioscience) or equal volume of absolute ethanol vehicle (Sigma Aldrich), in respective culture media (see above). To test whether bacterial isolates were capable of growth with progesterone and 17β-estradiol as the sole carbon source, bacterial growth curves were performed in freshly prepared mineral salt media63 supplemented with 1×10−5M progesterone and 1×10−6M 17β-estradiol or equal volume of absolute ethanol vehicle at a normalized starting OD600 of 0.1. Bacterial cultures were then incubated in a Cytation3 spectrophometer (BioTek) at 37°C for 35 hours, and OD600 was recorded every 15 minutes.
Gentamycin Protection Assay
Intracellular lifestyle of bacterial isolates was determined by gentamycin protection assays as described previously64. Primary human antigen presenting cells from fetal spleen were enriched by negative selection using Easy Step Human Biotin Isolation kit (STEMCELL Technologies) and biotin-conjugated mouse anti-human mAbs for CD3, CD56, CD19, and CD20. Isolated cells were incubated for 24h in cRPMI with penicillin and streptomycin at 4°C. Fetal antigen presenting cells or RAW 264.7 macrophage cells (ATCC) were seeded in each well of a 96-well plate and incubated for two hours at 37°C 5% CO2 with bacterial isolate overnight cultures at a multiplicity of infection (MOI) of 10. Non-adherent bacteria were removed by washing three times with 1x PBS and incubating for 30 minutes with cRPMI supplemented with 50μg mL−1 gentamycin. Cells were then incubated with 10μg mL−1 gentamycin supplemented cRPMI for 3, 24, 40, 48 or 50 hours at 37°C 5% CO2. Intracellular bacteria were recovered by lysing eukaryotic cells with sterile 1% (v/v) Triton X (Sigma Aldrich) solution for 10 minutes, with lysis was visually confirmed by light microscope. CFUs were counted from serial dilutions of lysate, grown on either BHI (see above) agar plates for Micrococcus exposed cells. Escherichia coli strain DH10B was used as a negative control. Lysate was plated on respective media agar plates with 10μg mL−1 gentamycin to determine acquisition of antibiotic resistance.
Antibodies and Flow Cytometry
Extracellular staining of isolated cells was performed in 2% FBS in PBS with 1mM EDTA (staining buffer) with human Fc blocking antibody (STEMCELL Technologies) and with fluorochrome-conjugated antibodies against surface markers, as previously described6. Intracellular protein were detected in fixed, permeabilized cells using the Foxp3/Transcription Factor Staining Buffer set (Tonbo Biosciences), as previously described6. Mouse anti-human monoclonal antibodies used in this study: TCRβ PerCP Cy5.5 (Clone IP26, eBioscience Cat. No. 46-9986-42, dilution 1:100), Vα7.2 BV605 (Clone 3C10, BioLegend Cat. No. 351720, dilution 1:100), CD4 APC H7 (Clone L200, BD Pharmingen Cat. No. 560837, dilution 1:100), CD8α FITC and PE Cy7(Clone B7–1, BD Pharmingen Cat. No. 347313, dilution 1:100), CD45RA PE Cy7 (Clone HI100, BD Pharmingen Cat. No. 555489, dilution 1:100), CCR7 PE (Clone G043H7, BioLegend Cat. No. 353208, dilution 1:100), PLZF-APC (Clone 6318100, R&D Cat. No. IC2944A, dilution 1:50), CD161-BV711 (Clone DX12, BD Biosciences Cat. No. 563865, dilution 1:50), CD25 FITC (Clone 2A3, BD Biosciences Cat. No. 347643, 1:100), FoxP3 PE (Clone PCH101, eBioscience Cat. No. 12-4776-42, 1:50), IFNγ-FITC (Clone 25723.11, BD Biosciences Cat. No. 340449, 1:50), TNFα-PE Cy7 (Clone MAB11, BD Pharmingen Cat. No. 557647, 1:50), CD45 APC (Clone HI30, Tonbo Cat. No. 20–0459, 1:100), HLA-DR APC-R700 (Clone G46–6, BD Cat. No. 565127, dilution 1:100), CD3 biotin (Clone OKT3, eBioscience Cat. No. 13-0037-82, dilution 1:100), CD19 biotin (Clone HIB19, BioLegend Cat. No. 203304, dilution 1:100), CD20 biotin (Clone 2H7, eBioscience Cat. No. 13-0209-82, dilution 1:100), CD56 biotin (Clone NCAM16.2, BD Cat. No. 555515, dilution 1:100), LLT1 PE (Clone 402659 R&D Cat. No. FAB3480P, dilution 1:50)6. Biotin antibodies were detected with streptavidin conjugated to BV711 (BD Biosciences Cat. No. 563262, 1:200), as previously described6. Dead cells were excluded from analysis using Aqua LIVE/DEAD Fixable Dead Cell Stain Kit (Invitrogen) stain. All data were acquired with BD LSR/Fortessa Dual SORP using FACS Diva software (BD Biosciences) and analyzed with FlowJo (TreeStar) software.
Ex vivo Intestinal Epithelial Cell Transcriptomics after Bacterial Isolates Exposure
EDTA washes containing fetal intestinal epithelial cells (see above) were washed with 1x PBS, passed through 40 μm strainer, and plated on Collagen I coated 96-well plates (Corning) in cRPMI containing 5 ng/mL epidermal growth factor (Gibco). Cells were incubated overnight at 37°C 5%CO2 4% O2, to mimic hypoxic conditions in the fetal intestine65 and non-adherent cells were removed. Cells were allowed to differentiate for five days or until 80% confluence, with media replacement every two days. Cells were incubated with a multiplicity of infection of 10 of bacterial isolates in cRPMI for 4 at 37°C 5%CO2 4% O2. After 4h, cells were preserved in RNAlater and RNA was prepared for sequencing as described above.
Ex vivo Antigen Presenting Cell Activation with Bacterial Isolates
Antigen presenting cells from fetal spleen were enriched by negative selection using Easy Step Human Biotin Isolation kit (STEMCELL Technologies) as described above. Cells were seeded into 96-well plates and incubated with multiplicity of infection of 10 of bacterial isolates in cRPMI for 4 hours at 37°C 5%CO2 4% O2, to mimic hypoxic conditions in the fetal intestine65 and normalize for bacterial growth.
Ex vivo Autologous Mixed Lymphocyte Reactions
Lamina propria T cells were enriched using Easy Sep Human T cell isolation kit (STEMCELL Technologies), effector memory cells were sorted to >99% purity (Extended Data 8i) using BD Aria Fusion SORP, and cells were labeled with cell trace violet (Invitrogen). Splenic antigen presenting cells autologous to isolated T cells were enriched as described above, sorted to >96% purity (Extended Data 8j), and exposed to bacterial isolates as described above. Bacteria were removed with three washes of cRPMI supplemented with penicillin and streptomycin. Sorted, labeled effector memory T cells were incubated with pre-exposed antigen presenting cells in a 2:1 ratio in cRPMI with supplemented with 10ng/mL IL-2 (PeproTech), 10ng/mL IL-7 (PeproTech), 2 μg/mL purified anti-CD28 (Clone CD28.2, BD Pharmigen Cat. No. 555725), 2 μg/mL purified anti-CD49d (Clone, BD Pharmingen Cat No. 555501), and 10 μg/mL gentamycin for three days at 37°C 5% CO2 4% O2. Cells were incubated with 10μg/mL Brefeldin A (Sigma Aldrich) in the same media for 4 hours at 37°C 5% CO2 4% O2 and were subsequently stained for intracellular cytokine production as described above. Mixed lymphocyte reactions as described above were extended to 5 days with enriched T cells and antigen presenting cells, and T cell proportions were measured using flow cytometry as described above.
Statistical Analysis
Shannon’s diversity index was calculated in Qiime and student’s, Welch’s, or Wicoxon t-tests were calculated in R, depending on the distribution. Bray Curtis distance matrices were calculated in QIIME to assess compositional dissimilarity between samples and visualized using principal coordinates analysis (PCoA) plots in R. Permutational multivariate analysis of variance (PERMANOVA) was performed using Adonis function of vegan package66 in R to determine factors that significantly (p<0.05) explained variation in microbiota β-diversity. In cases where replicates were included, linear mixed effects modeling was used to determine significance using the R package lmerTest67. Ranked abundance curve fit to geometric or log-series functions was determined by Bayesian Information Criterion (BIC) to evaluate models generated from fitsad function in vegan R package. To determine which OTUs differed in relative abundance between contamination swab and meconium, unnormalized read counts were transformed using DESEQ2 in QIIME to identify log-fold change enrichment and corrected for multiple hypothesis testing using the false-discovery rate (q<0.05). Growth curves were modeled using a logistic regression in R package growthcurver68, integral of the best fit regression was used to calculate the area under the curve (auc), and auc of vehicle was subtracted from hormone treatment controls according to the following formula:
Significance in gentamycin protection assays was evaluated by transforming colony forming unit (CFU) counts using log10(CFU + 1) and applying a generalized linear model to transformed data. Significance in ex vivo immune cell assays was evaluated using linear mixed effect modeling to account for cell donor correlations and where indicated, residuals are plotted. Except where indicated, all analyses were performed using R statistical programming language in the Jupyter Notebook environment.
DATA AVAILABILITY STATEMENT
All sequencing data associated with this study has been made publicly available. 16S rRNA bacterial profiling data generated in this study is available in the EMBLI-EBI ENA repository accession #PRJEB25779 (https://www.ebi.ac.uk/ena). De novo assembled genomes were deposited at DDBJ/ENA/GenBank under the accession number VFQL00000000 for Micro36. The genome version described in this paper is version VFQL01000000 for Micro36. Raw sequence reads used for genome assembly were deposited in NCBI SRA under BioProject accession # PRJNA498337 for Micro36, respectively. RNA sequencing dataset is available in NCBI under PRJNA506292 accession. All additional datasets and materials are available from the corresponding author upon request; requests are promptly reviewed by the University of California San Francisco Innovation Office to verify if the request is subject to any intellectual property or confidentiality obligations. Any data and materials that can be shared will be released via a Material Transfer Agreement. Filtered and unfiltered OTU tables as well as metadata are provided as Supplemental Table 2 to this manuscript. Source data is provided for all figures and extended data.
Extended Data
Extended Data 1. Low-burden bacterial signal in fetal meconium.
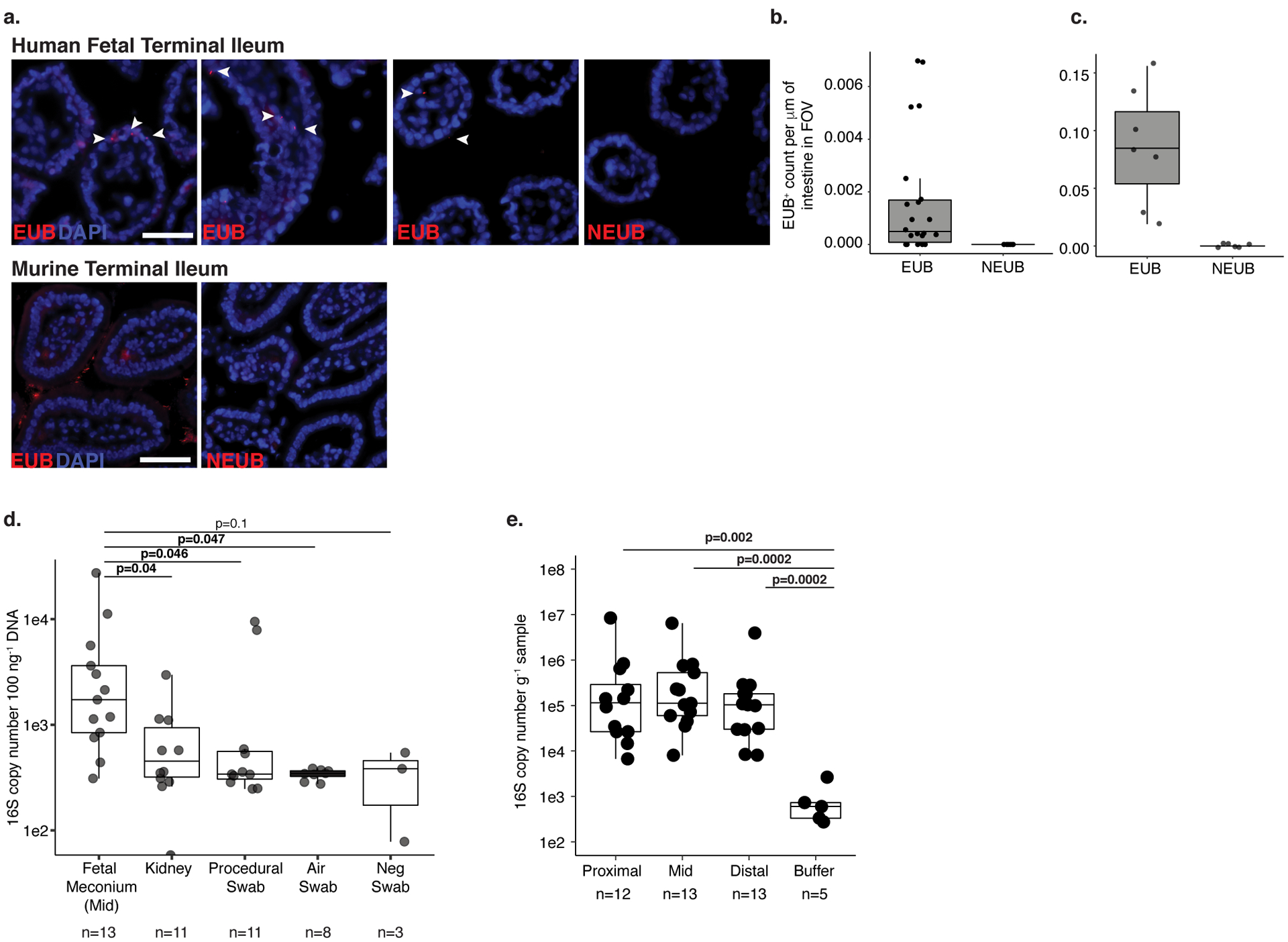
a. Fluorescent in situ hybridization probes targeting eubacteria (EUB) or non-targeting probe (NEUB) in 5 μm cryosections of human fetal (top panel) or murine (bottom panel) terminal ileum at 400x magnification. Arrowheads indicate EUB-positive findings in fetal sections. Scale bar corresponds to 50 mm. Quantification of independent fields of view (FOV) per mm within a representative b. human fetal (n=28) or c. murine (n=13) intestinal segments, where n is an independent segment of the intestine. For a-b, representative images of three fetal specimens and three mice. d. Total 16S copy number per 100 ng gDNA in meconium from mid-section of the fetal small intestine (n=13), fetal kidney (n=11), and procedural (n=11), air (n=8), or blank (n=3) swab was quantified by qPCR of DNA extracts using a standard curve where n represents biologically independent samples; two-sided Satterthwaite’s method on linear mixed effects model to test for significance. e. Total 16S copy number per gram frozen sample in meconium from proximal (n=12), mid (n=13), and distal (n=13) sections of the fetal small intestine where n represents independent biological samples across the length of the intestine or extraction buffer (n=5, n represents biologically independent samples) was quantified by qPCR of DNA extracts using a standard curve; two-sided Wilcoxon rank sum test for significance compared to buffer control. Boxplots indicate the median (center), the 25th and 75th percentiles, and the smallest and largest values within 1.5× the interquartile range (whiskers).
Extended Data 2. Depletion of mtDNA by Cas9 does not alter bacterial composition after 30 cycles of amplification.
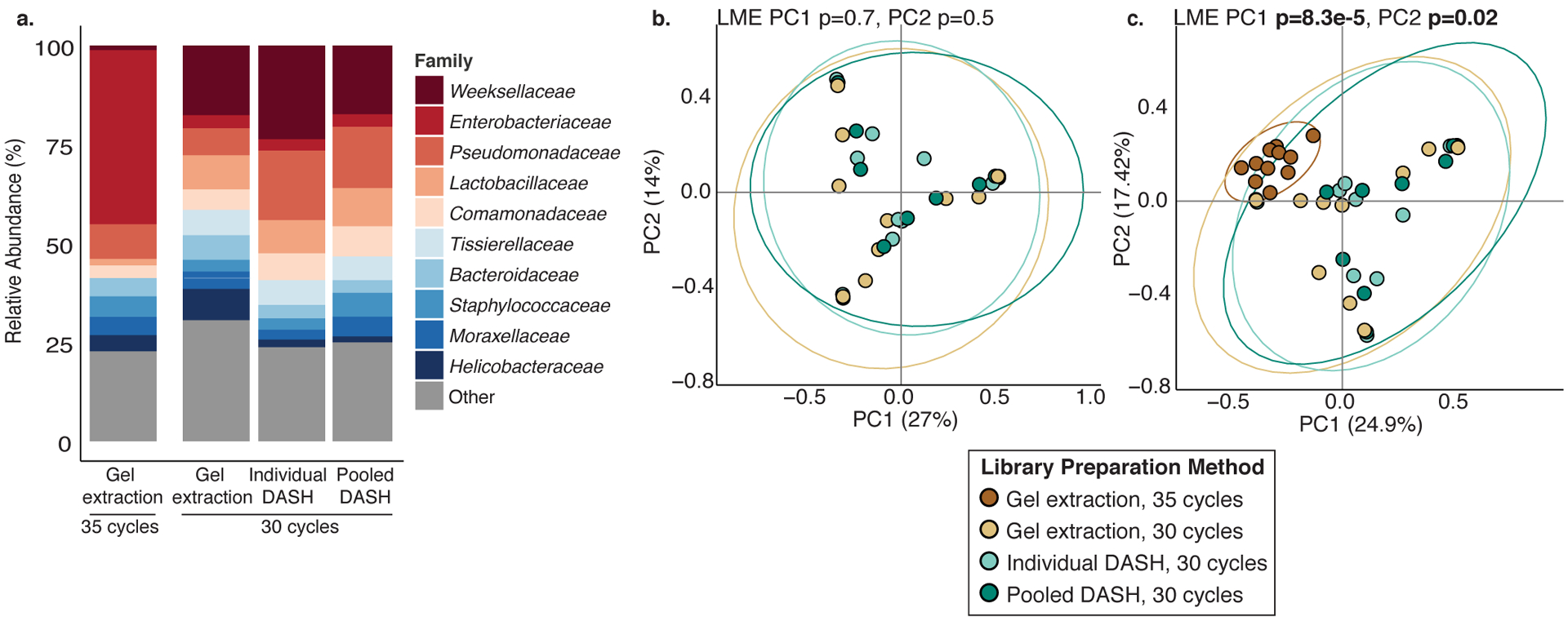
16S rRNA V4 profiling of a subset (n=10) of banked fetal meconium samples using different library preparation methods: gel extraction and 30 or 35 cycles of amplification, or 30 cycles combined with DASH performed on individual samples (Individual DASH) or on the library pool (Pooled DASH). a. Expansion in Enterobacteriaceae family is detected in 35-cycle amplification method, while small expansion of Pseudomonadaceae is detected post-DASH. Principal coordinates analysis of Bray Curtis distances of libraries using b. 30 cycles of amplification or c. 30 and 35 cycles of amplification, latter to provide an outgroup known to skew bacterial composition. Ellipses indicate 95% confidence intervals. All p-values were calculated using two-sided Satterthwaite’s method on Linear Mixed Effects (LME) modeling to correct for n=10 paired samples that underwent multiple library preparation methods.
Extended Data 3. Sparse bacterial signal distinct from background detected in fetal meconium.
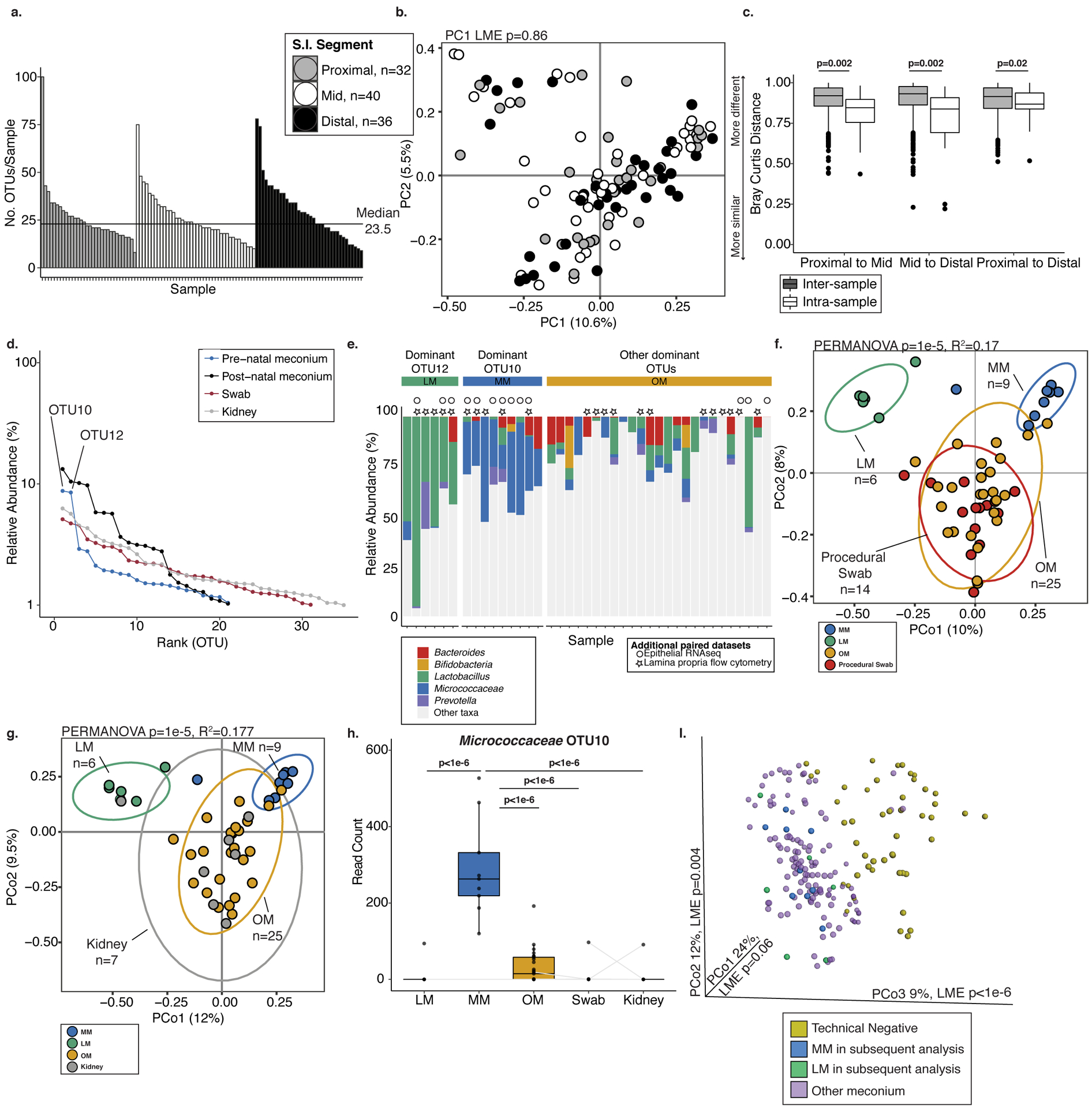
a. Histogram of number of operational taxonomic units (OTUs) per sample detected in fetal meconium from proximal-, mid-, or distal-segments of the small intestine after technical control filtering. b. Principal coordinates analysis (PCoA) of Bray Curtis distances of rareified bacterial profiles of proximal- (n=32), mid- (n=40), and distal- (n=36) sections of the intestine. c. Inter- and intra-sample Bray Curtis distances between indicated comparisons of proximal- (n=32), mid- (n=40), and distal- (n=36) intestinal sections. For a-b, n represents biologically independent fetal samples across the length of the intestine. d. Bacterial abundance ranks in fetal meconium, post-natal meconium, procedural swab, and kidney control. e. Relative abundance of select genera among samples dominated by OTU12, OTU10, or other OTUs. Symbols indicate samples with paired immunological datasets. PCoA of Bray Curtis distances of Lactobacillus-meconium (LM, n=6), Micrococcaceae-meconium (MM, n=9), or Other-meconium (OM, n=25) compared to f. procedural swab (n=14) or g. fetal kidney (n=7) control, where n represents biologically independent fetal samples. h. Normalized read counts for Micrococcocaceae OTU10 in LM (n=6), MM (n=9), OM (n=25), swab (n=14), and fetal kidney (n=7) control samples, where n represents biologically independent fetal samples; samples collected from the same fetus indicated by grey line, when possible. i. PCoA of Bray Curtis distances of unrareified and unfiltered bacterial profiles of meconium (n=138 biologically independent fetal samples across three segments of the intestine) with technical negative controls (n=48 biologically independent samples including extraction buffer, room air swab, pre-moistened swabs). LM (n=6) and MM (n=9) samples identified in later analyses are highlighted; significance was measured using two-sided Satterthwaite’s method on linear mixed effects model to test for significance and correct for repeated measures in b,h; ANOVA of linear mixed effects model was used to test for significance and correct for repeated measures in i, two-sided t-test was used for c, PERMANOVA was used in f-g. Boxplots indicate the median (center), the 25th and 75th percentiles, and the smallest and largest values within 1.5× the interquartile range (whiskers).
Extended Data 4. Divergent epithelial transcriptome and lamina propria T cells in samples associated with LM, MM, or OM.
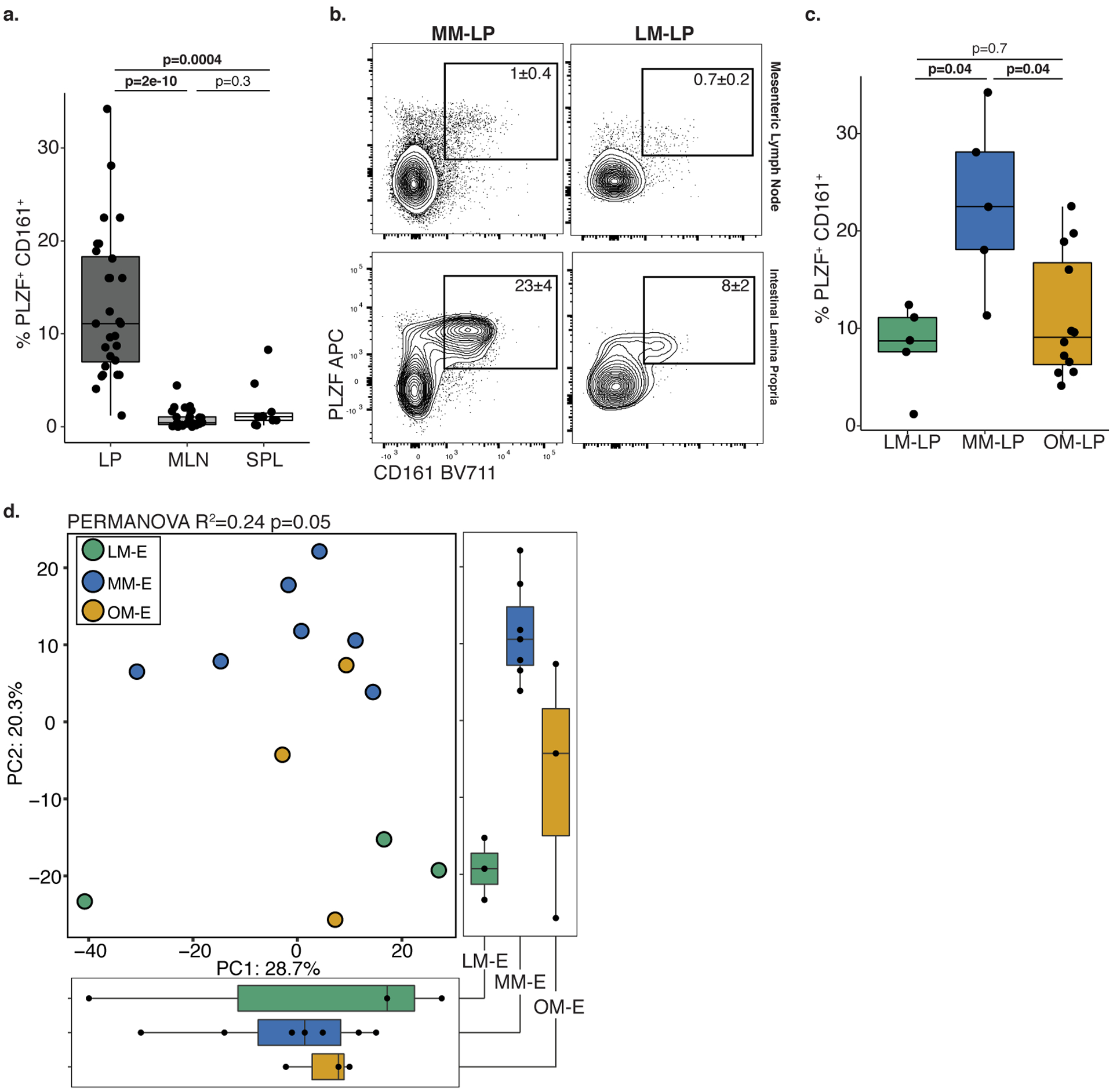
a. Proportion of PLZF+ CD161+ T cells among live, TCRβ+, Vα7.2−, CD4+ cells in intestinal lamina propria (LP, n=28), mesenteric lymph node (MLN, n=27), and spleen (SPL, n=10) where n is a biologically independent fetal sample. b. Representative flow plots of mesenteric lymph node (top panel, gating control) or intestinal lamina propria (bottom panel) associated with MM and LM. Experiments were repeated 5 independent times for each LM and MM associated samples with similar results. c. Proportion of PLZF+ CD161+ T cells in intestinal lamina propria paired with LM, MM, or OM (LM-LP, n=5; MM-LP, n=5; OM-LP, n=12) among live, TCRβ+, Vα7.2−, CD4+ cells. d. Principal components (PC) analysis of Euclidean distances of top 10000 variable genes (by coefficient of variation) in LM associated epithelium (LM-E, n=3), MM associated epithelium (MM-E, n=7), or OM associated epithelium (OM-E, n=3) as determined by RNA sequencing where n represents a biologically independent fetal sample. Kruskal-Wallis ANOVA, with Dunnet’s correction for multiple comparisons was used for a, c. PERMANOVA test for significance in d. Each dot represents a biological replicate. Boxplots indicate the median (center), the 25th and 75th percentiles, and the smallest and largest values within 1.5× the interquartile range (whiskers).
Extended Data 5. Micrococcus fetal isolate exhibits high 16S rRNA V4 sequence identity to fetal meconium OTUs.
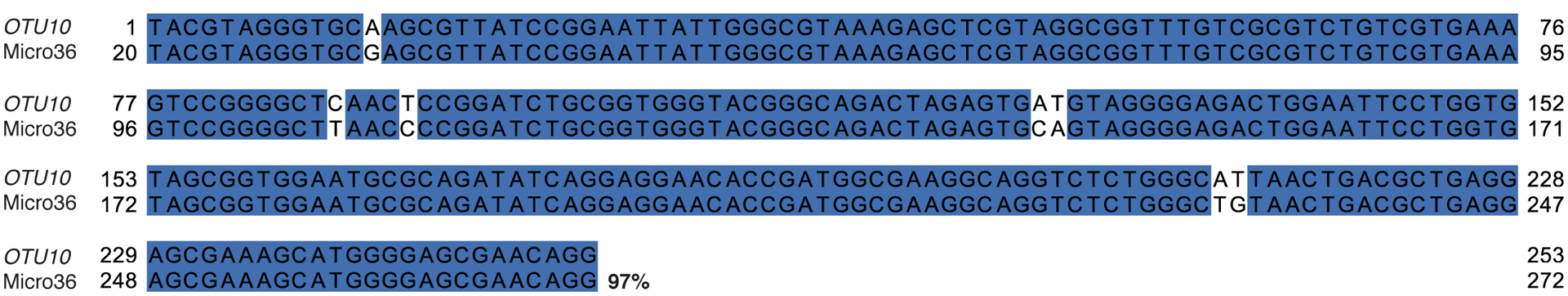
Alignment of 16S V4 rRNA gene sequences of Micro36 to OTU10. Percentage indicates identity to representative OTU sequence.
Extended Data 6. Fetal meconium Micrococcus isolate exhibits adaptation to the fetal environment.
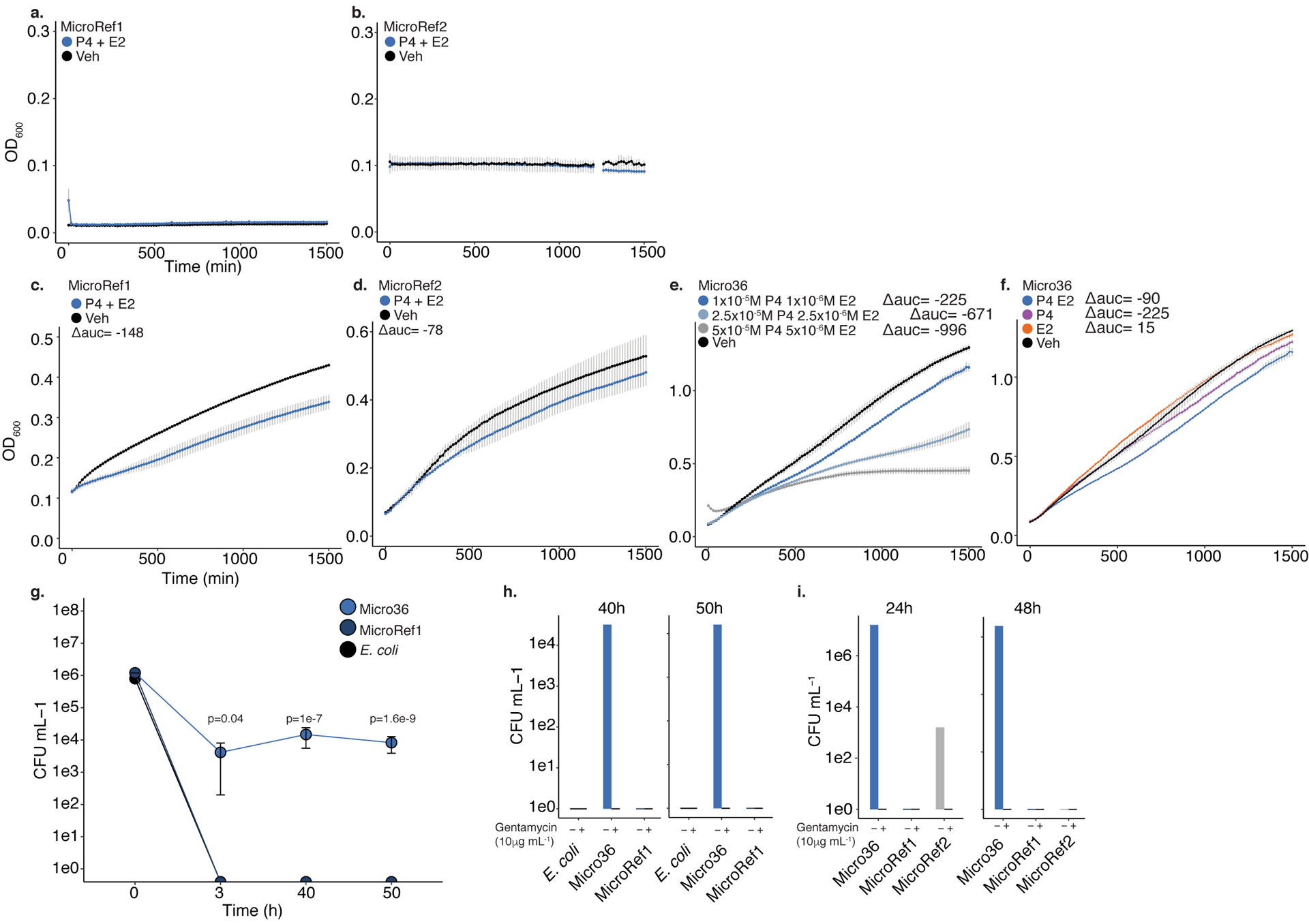
Effects of 10−5 M progesterone (P4) and 10−6 M β-Estradiol (E2) on the growth a. MicroRef1 or b. MicroRef2 in carbon limiting media or of c. MicroRef1, d. MicroRef2, e. Micro36 with indicated concentrations of P4 and E2 or f. combinations of hormones compared to ethanol vehicle control, in carbon-rich media at 37 °C. Representative growth curves of three independent experiments measured by optical density at 600nm (OD600), error bars denote standard error of the mean (SEM) between three technical experiments. For carbon-rich media conditions, integral of logistic regression model fitting was used to calculate area under the curve (auc) and change with respect to vehicle control is reported as Δauc. g. Intracellular survival of Micro36 or MicroRef1 or E. coli in RAW264.3 cells. ANOVA of generalized linear model of log(CFU+1) against E. coli for each timepoint was used to calculate significance. Error bars indicate SEM around center mean of n=3 independent cell culture experiments. Growth of indicated strains on media with (+) or without (−) gentamycin (10μg mL−1) following 24–50 hours of intracellular growth in h. RAW264.7 cells or i. primary human fetal intestinal antigen presenting cells.
Extended Data 7. Genomic features of fetal Micrococcus isolate.
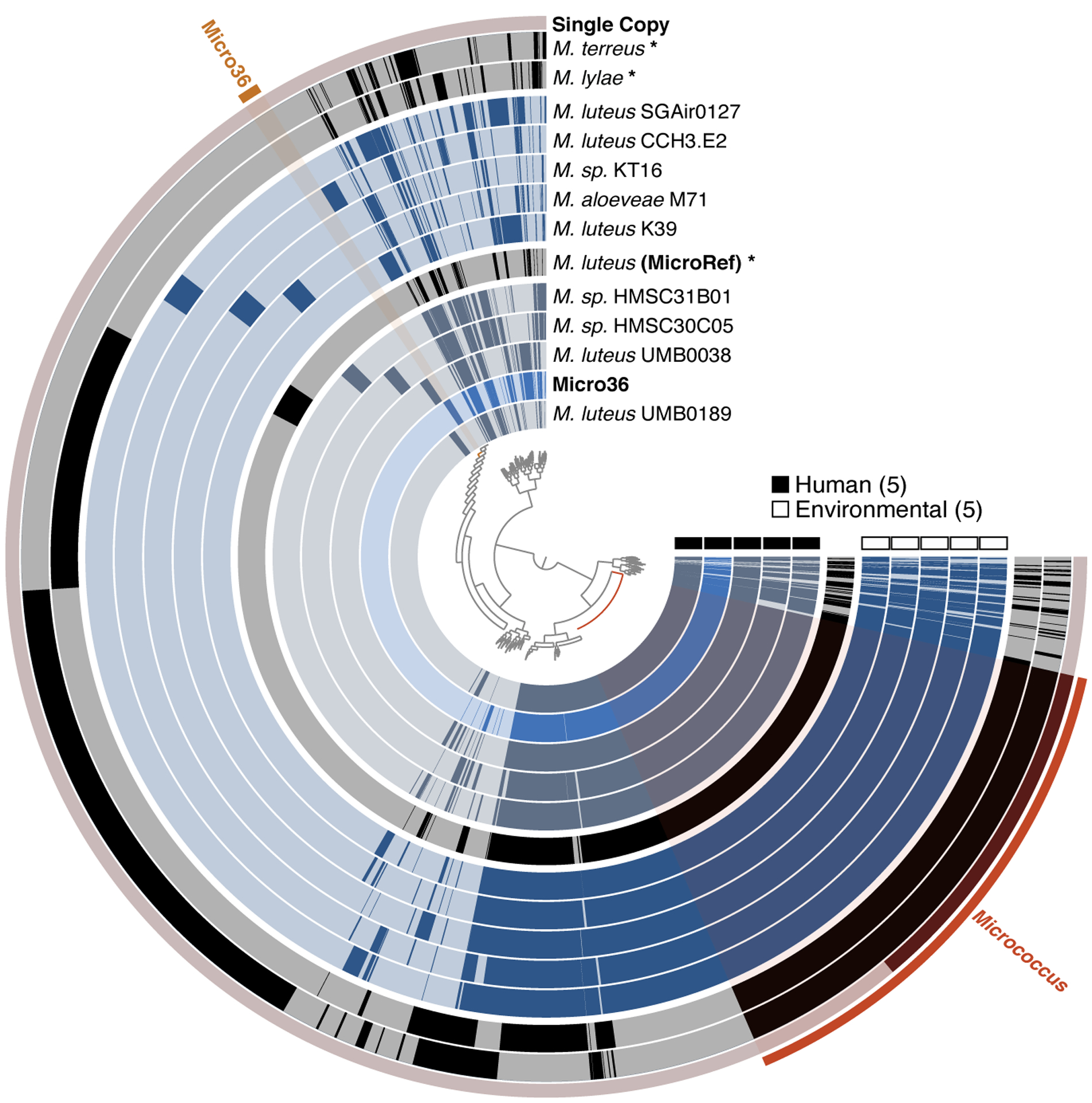
Alignment of all publicly available Micrococus genomes; single copy Micrococcus genes used for phylogeny (inset) and genes unique to Micro36 isolate are highlighted. Figure was generated using the Anvi’o package; each radial layer represents a genome; representative or reference genomes are colored in black indicated with asterisk; inner dendrogram represents hierarchical clustering of amino acid sequences based on their sequence composition and distribution across genomes; genomes are organized based on gene clusters they share using Euclidian distance and Ward ordination; outer ring represents single copy genes predicted using hidden markov model in Anvi’o package.
Extended Data 8. Prevalence of M. luteus in infants and mothers.
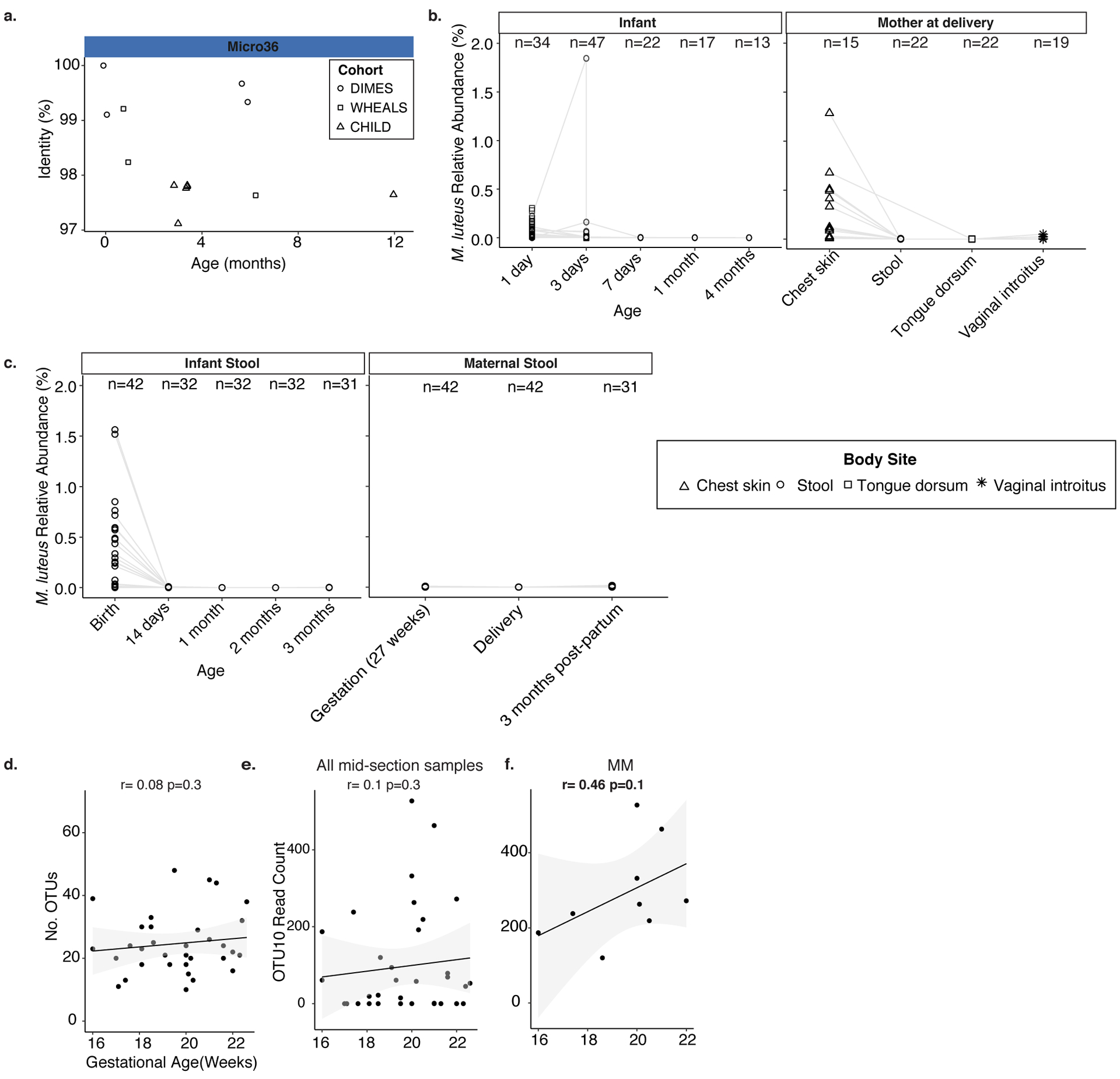
a. Percent identity of samples to 16S rRNA gene of Micro36 in three independent infant stool cohorts. Each symbol represents a sample with a positive hit (>97% sequence identity); symbol shape indicates cohort. Relative abundance of Micrococcus luteus in metagenomic sequencing cohorts across b. body sites at delivery in mother and infant within four months after birth, or c. in maternal stool around delivery and infant stool within the first three months of life. Metagenomic sequences obtained from two independent studies were classified using a custom kraken2 database including the fetal M. luteus Micro 36 genome. Correlation of gestational age with d. total number of OTUs or e. Micrococcaceae OTU10 count in mid-section meconium samples (n= 35 biologically independent fetal specimens) or f. among Micrococaceae meconium (MM, n=9 biologically independent fetal specimens). Pearson’s product-moment correlation coefficient and a one-sided t-distribution p-value is reported for d-f.
Extended Data 9. Fetal Micrococcus isolate promotes distinct APC and T cell phenotypes.
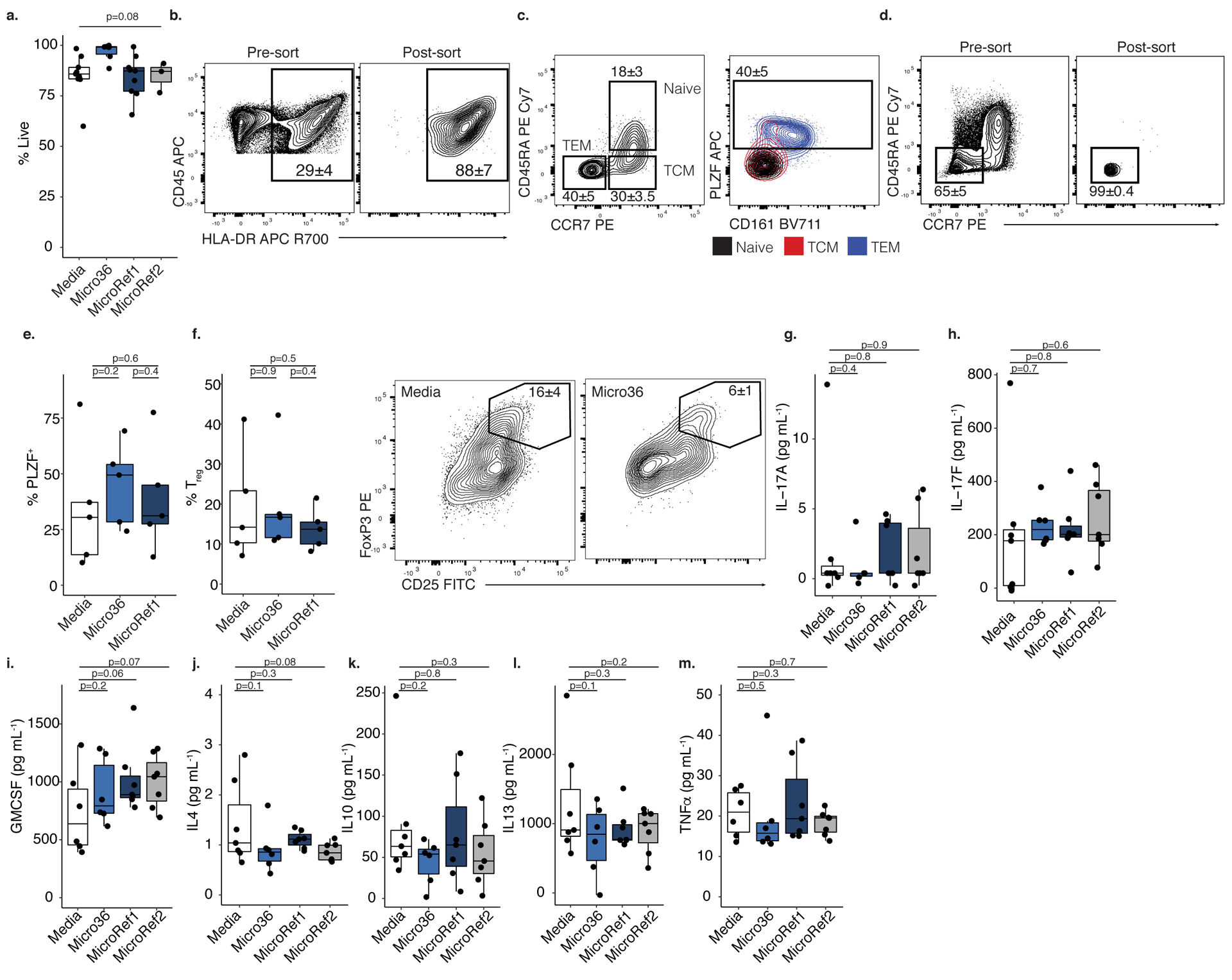
a. Proportion of live cells after treatment with media (n=9) or Micrococcus (Micro36 n=6, MicroRef1 n=9, MicroRef2 n=3) strains, where n represents biologically independent fetal specimens for the indicated treatment. ANOVA test for significance. b. HLA-DR+ CD45+ lin− cells pre- (left) and post- (right) fluorescence activated cell sorting (FACS). c. Proportion of naïve (CD45RA+ CCR7+), central memory (TCM, CD45RA− CCR7+), and effector memory T cells (TEM, CD45RA− CCR7−) among live, TCRβ+, CD4+ cells (left panel) and PLZF and CD161 expression among memory subsets, numbers indicate proportion in TEM (right panel). d. Pre- (left) and post- (right) FACS of effector memory T cells. e. Proportion of PLZF+ T cells or f. left, proportion of CD25hi FoxP3+ regulatory T cells (Tregs) and right, representative flow plots of FoxP3 and CD25 expression among intestinal live, TCRβ+, CD4+, Vα7.2−, cells after exposure to splenic APCs pretreated with media or Micrococcus (Micro36, MicroRef1) strains for n=5 biologically independent fetal specimens. Concentration of g. IL-17A, h. IL-17F, i. GM-CSF, j. IL-4, k. IL-10, l. IL-13, m. TNFα in culture supernatants of lamina propria T cell co-cultures with splenic antigen presenting cells pre-exposed to media (n=7) or Micrococcus (Micro36 n=6, MicroRef1 n=7, MicroRef2 n=7) strains, where n represents biologically independent fetal specimens for the indicated treatment. For b-d, f numbers indicate mean proportion and standard error of the mean (SEM) representative of five independent experiments. For e-f, g-m two-sided Satterthwaite’s method on linear mixed effects model was used to test for significance between strains, controlling for repeated measures of cell donor. Positive LME residuals are plotted for g-m. Each dot represents an independent fetal sample, unless otherwise indicated. Boxplots indicate the median (center), the 25th and 75th percentiles, and the smallest and largest values within 1.5× the interquartile range (whiskers).
Supplementary Material
ACKNOWLEDGEMENTS
We express gratitude to the tissue donors. Thanks to M. Ng, H. Boushey and A. Molofsky for thoughtful critique of this manuscript. UCSF flow core provided instrumentation assistance, which was supported by National Institutes of Health (NIH) grant no. P30 DK063720 and NIH Shared Instrument Grant no. 1S10OD021822-01. The FE-SEM and supporting facilities were obtained under National Science Foundation (NSF)-MRI award no. 0821619 and NSF-EAR award no. 0949176, respectively. This study was supported in part by the UCSF Clinical and Translational Science Institute Pilot Award for Basic and Translational Investigators grant no. 2014908 to JH. ER was supported by NSF Graduate Research Fellowship grant no. 1650113 and by the National Institute of Allergy and Infectious Diseases (NIAID) of the NIH F31AI136336. JH was supported by NIH NIAID grant no. K08 AI128007. SVL was supported by NIH NIAID grant no. AI114271 and UG30D023282. We thank C. Domingo for support of the electron microscopy core facility at SFSU and D. Mars for assistance with FE-SEM sample preparation. We thank J. L. DeRisi at UCSF for generously sharing custom-made T7 RNA polymerase with our group. We express gratitude to E. Stapleton for illustration assistance and to J. M. Rackaitis for support during these studies. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSF or the NIH.
Footnotes
Competing Interests
SVL is co-founder of Siolta Therapeutics Inc. and serves as both a consultant and a member of its Board of Directors. The Regents of the University of California, San Francisco have filed a patent application (PCT/US2019/045354) on behalf of S.V.L and E.R. relating to the methods and compositions of fetal bacteria.
REFERENCES
- 1.Spencer J, Dillon SB, Isaacson PG & Macdonald TT T cell subclasses in fetal ileum. Clin. Exp. Immunol 65, 553–558 (1986). [PMC free article] [PubMed] [Google Scholar]
- 2.Howie D et al. Extrathymic T Cell Differentiation in the Human Intestine Early in Life. J. Immunol 161, 5862–5872 (1998). [PubMed] [Google Scholar]
- 3.McGovern N et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature (2017). doi: 10.1038/nature22795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spencer J, MacDonald TT, Finn T & Isaacson PG The development of gut associated lymphoid tissue in the terminal ileum of fetal human intestine. Clin. Exp. Immunol 64, 536–43 (1986). [PMC free article] [PubMed] [Google Scholar]
- 5.Podd BS et al. T Cells in Cryptopatch Aggregates Share TCR Variable Region Junctional Sequences with T Cells in the Small Intestinal Epithelium of Mice. J. Immunol 176, 6532–6542 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Halkias J et al. CD161 contributes to prenatal immune suppression of IFNγ-producing PLZF+ T cells. J. Clin. Invest (2019). doi: 10.1172/JCI125957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schreurs RRCE et al. Human Fetal TNF-α-Cytokine-Producing CD4+ Effector Memory T Cells Promote Intestinal Development and Mediate Inflammation Early in Life. Immunity 0, 1–15 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Li N et al. Memory CD4+ T cells are generated in the human fetal intestine. Nat. Immunol (2019). doi: 10.1038/s41590-018-0294-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aagaard K et al. The placenta harbors a unique microbiome. Sci. Transl. Med 6, 237ra65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collado MC, Rautava S, Aakko J, Isolauri E & Salminen S Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep 6, 23129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steel JH et al. Bacteria and Inflammatory Cells in Fetal Membranes Do Not Always Cause Preterm Labor. Pediatr. Res 57, 404–411 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Lim ES, Rodriguez C & Holtz LR Amniotic fluid from healthy term pregnancies does not harbor a detectable microbial community. 4–11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lauder AP et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 1–11 (2016). doi: 10.1186/s40168-016-0172-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Goffau MC et al. Human placenta has no microbiome but can contain potential pathogens. Nature (2019). doi: 10.1038/s41586-019-1451-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu DM et al. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med 23, 314–326 13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durack J et al. Delayed gut microbiota development in high-risk for asthma infants is temporarily modifiable by Lactobacillus supplementation. Nat. Commun 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu J et al. Maternal exposure to farming environment protects offspring against allergic diseases by modulating the neonatal TLR-Tregs-Th axis. Clin. Transl. Allergy 8, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujimura KE et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat. Med 22, 1187–1191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu W, Crawford ED, O’Donovan BD, Wilson MR, Chow ED, Retallack H, D. J. Depletion of Abundant Sequences by Hybridization (DASH): Using Cas9 to remove unwanted high- abundance species in sequencing libraries and molecular counting applications. Genome Biol. 17, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trotter A, Maier L, Grill H-J, Wudy SA & Pohlandt F 17β-Estradiol and Progesterone Supplementation in Extremely Low-Birth-Weight Infants. Pediatr. Res 45, 489–493 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Yotis W & Stanke R Bacteriostatic action of progesterone on staphylococci and other microorganisms. J. Bacteriol 92, 1285–1289 (1966). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varghese NJ et al. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–6771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benach J et al. Structure of bacterial 3β/17β-hydroxysteroid dehydrogenase at 1.2 Å resolution: A model for multiple steroid recognition. Biochemistry 41, 14659–14668 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Hillas PJ, Soto Del Alba F, Oyarzabal J, Wilks A & Ortiz De Montellano PR The AhpC and AhpD antioxidant defense system of Mycobacterium tuberculosis. J. Biol. Chem 275, 18801–18809 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Dons LE et al. Role of the Listeria monocytogenes 2-Cys peroxiredoxin homologue in protection against oxidative and nitrosative stress and in virulence. Pathog. Dis 70, 70–74 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Arrieta M-C et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med 7, 307ra152–307ra152 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Ferretti P et al. Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 24, 133–145.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yassour M et al. Strain-Level Analysis of Mother-to-Child Bacterial Transmission during the First Few Months of Life. Cell Host Microbe 24, 146–154.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Egea L, Hirata Y & Kagnoff MF GM-CSF: A role in immune and inflammatory reactions in the intestine. Expert Rev. Gastroenterol. Hepatol 4, 723–731 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krause P et al. IL-10-producing intestinal macrophages prevent excessive antibacterial innate immunity by limiting IL-23 synthesis. Nat. Commun 6, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denning TL, Wang YC, Patel SR, Williams IR & Pulendran B Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol 8, 1086–1094 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Rosen DB et al. Functional Consequences of Interactions between Human NKR-P1A and Its Ligand LLT1 Expressed on Activated Dendritic Cells and B Cells. J. Immunol 180, 6508–6517 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minich JJ et al. Quantifying and Understanding Well-to-Well Contamination in Microbiome Research. mSystems 4, e00186–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martín R et al. Characterization of indigenous vaginal lactobacilli from healthy women as probiotic candidates. Int. Microbiol 11, 261–266 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Chen C et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun 8, 875 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mukamolova GV et al. The rpf gene of Micrococcus luteus encodes an essential secreted growth factor. Mol. Microbiol 46, 611–621 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Duerkop BA, Vaishnava S & Hooper LV Immune Responses to the Microbiota at the Intestinal Mucosal Surface. Immunity 31, 368–376 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Mold JE et al. Fetal and Adult Hematopoietic Stem Cells Give Rise to Distinct T Cell Lineages in Humans. Science (80-.) 330, 1695–1699 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mold JE et al. Maternal Alloantigens Promote the Development of Tolerogenic Fetal Regulatory T Cells in Utero. Science (80-.) 322, 1562–1565 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pastor-vargas C et al. Detection of major food allergens in amniotic fluid : initial allergenic encounter during pregnancy. 27, 716–720 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Iwai S et al. The lung microbiome of Ugandan HIV-infected pneumonia patients is compositionally and functionally distinct from that of San Franciscan patients. PLoS One 9, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caporaso JG et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci 108, 4516–4522 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magoč T & Salzberg SL FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caporaso JG et al. correspondence QIIME allows analysis of high- throughput community sequencing data Intensity normalization improves color calling in SOLiD sequencing. Nat. Publ. Gr 7, 335–336 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edgar RC & Flyvbjerg H Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 31, 3476–3482 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Davis NM, Proctor DM, Holmes SP, Relman DA & Callahan BJ Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobin A et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao Y, Smyth GK & Shi W FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johansson MEV & Hansson GC Preservation of Mucus in Histological Sections, Immunostaining of Mucins in Fixed Tissue, and Localization of Bacteria with FISH. in Mucins: Methods and Protocols (eds. McGuckin MA & Thornton DJ) 229–235 (Humana Press, 2012). doi: 10.1007/978-1-61779-513-8_13 [DOI] [PubMed] [Google Scholar]
- 51.Vaishnava S et al. The Antibacterial Lectin RegIII. Science (80-.) 334, 255–258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weisburg WG, Barns SM, Pelletier DA & Lane DJ 16S ribosomal DNA amplification for phylogenetic study. Weisburg, WG 173, 697–703 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pruesse E, Peplies J & Glöckner FO SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bankevich A et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gurevich A, Saveliev V, Vyahhi N & Tesler G QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eren AM et al. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pritchard L, Glover RH, Humphris S, Elphinstone JG & Toth IK Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24 (2016). [Google Scholar]
- 58.Rinke C et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Edgar RC MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Price MN, Dehal PS & Arkin AP FastTree 2 - Approximately maximum-likelihood trees for large alignments. PLoS One 5, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Letunic I & Bork P Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wood DE & Salzberg SL Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sekar S, Mahadevan S, Kumar SSD & Mandal AB Thermokinetic responses of the metabolic activity of Staphylococcus lentus cultivated in a glucose limited mineral salt medium. J. Therm. Anal. Calorim 104, 149–155 (2011). [Google Scholar]
- 64.Subashchandrabose S, Smith SN, Spurbeck RR, Kole MM & Mobley HLT Genome-Wide Detection of Fitness Genes in Uropathogenic Escherichia coli during Systemic Infection. PLoS Pathog. 9, 1–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ducsay CA et al. Gestational Hypoxia and Developmental Plasticity. Physiol. Rev 98, 1241–1334 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oksanen J et al. Vegan: Community Ecology Package. R Package Version. 2.0–10. CRAN (2013). [Google Scholar]
- 67.Kuznetsova A, Brockhoff PB & Christensen RHB lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw 82, (2017). [Google Scholar]
- 68.Sprouffske K & Wagner A Growthcurver: An R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinformatics 17, 17–20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data associated with this study has been made publicly available. 16S rRNA bacterial profiling data generated in this study is available in the EMBLI-EBI ENA repository accession #PRJEB25779 (https://www.ebi.ac.uk/ena). De novo assembled genomes were deposited at DDBJ/ENA/GenBank under the accession number VFQL00000000 for Micro36. The genome version described in this paper is version VFQL01000000 for Micro36. Raw sequence reads used for genome assembly were deposited in NCBI SRA under BioProject accession # PRJNA498337 for Micro36, respectively. RNA sequencing dataset is available in NCBI under PRJNA506292 accession. All additional datasets and materials are available from the corresponding author upon request; requests are promptly reviewed by the University of California San Francisco Innovation Office to verify if the request is subject to any intellectual property or confidentiality obligations. Any data and materials that can be shared will be released via a Material Transfer Agreement. Filtered and unfiltered OTU tables as well as metadata are provided as Supplemental Table 2 to this manuscript. Source data is provided for all figures and extended data.