

Abstract
The diagnosis of rare diseases with multisystem manifestations can constitute a difficult process that delays the determination of the underlying cause. Whole exome sequencing (WES) provides a suitable option to examine multiple target genes associated with several disorders that display common features. In this study, we report the case of a female patient suspected of having Sotos syndrome. Screening for the initially selected genes, considering Sotos syndrome and Sotos-like disorders, did not identify any pathogenic variants that could explain the phenotype. The extended analysis, which considered all genes in the exome associated with features consistent with those shown by the studied patient, revealed a novel frameshift variant in the AMER1 gene, responsible for osteopathia striata with cranial sclerosis. WES analysis and an updated revision of previously reported disease-causing mutations, proved useful to reach an accurate diagnosis and guide further examination to identify critical abnormalities.
Keywords: AMER1, osteopathia striata with cranial sclerosis, macrocephaly, whole exome sequencing
Introduction
Whole exome sequencing (WES) constitutes a suitable option to diagnose genetic disorders. It offers an advantageous solution to evaluate genetic conditions whose clinical features overlap with numerous diseases, complicating their early and accurate diagnosis. Rare diseases with multisystem manifestations can show heterogeneous characteristics and a highly variable onset, which can pose a challenge for determining the cause of the disease. The implementation of high-throughput sequencing technologies in the diagnostic routine of genetic disorders has allowed selecting the correct management for the patient, which can otherwise be a tedious process both for patients and for health care professionals. It can also reduce the time and associated costs of unnecessary tests, therefore improving patient management. For instance, some skeletal dysplasias that manifest macrocephaly may have a similar clinical presentation to genetic diseases such as Sotos, Weaver, and Luscan–Lumish syndromes, among others.
In this case report, we described a pediatric case with clinical features of macrocephaly, dysmorphism, mild developmental delay, hypotonia, and respiratory abnormalities who was suspected of having Sotos syndrome. The analysis did not detect any pathogenic mutation in the genes initially considered for the suspected phenotype. We expanded the analysis to include other previously nonselected genes associated with overlapping conditions. Moreover, we reviewed the literature to assess the clinical features of patients with similar mutation types to our case in the AMER1 gene. This report led to a thorough study of the phenotype–genotype variation in osteopathia striata with cranial sclerosis (OSCS) cases, resulting in a precise management of this patient, thanks to being able to forecast future complications.
Material and Methods
Patient Description
The reported patient was the daughter of healthy nonconsanguineous parents with a negative family history for the studied phenotype. She was referred for neuropediatric consultation at the age of 2 years and 6 months due to motor delay in basic motor skills such as running or jumping, and mild speech delay (use of 10 words). She had undergone neonatal hospitalization for 7 days due to neonatal transient tachypnea and bilateral pneumothorax, which required orotracheal intubation. Weight and length at birth were normal. However, head circumference was above the 90th percentile, which suggested macrocephaly. She later manifested axial hypotonia, poor head control at around 4 months old, congenital muscular torticollis on the left side, and delayed sitting and social milestones. At the age of 16 months, physical examination showed clear macrocephaly (> 97 p), prominent forehead, dolichocephaly, persistent open anterior fontanelle, retromicrognathia, narrow palate, bifid uvula, hypertelorism, epicanthus, slanting palpebral fissures, low-set ears, broad nasal bridge, increased size of nasopharyngeal adenoids with open mouth appearance that led to obstructive sleep apnea syndrome (intervened adenoidectomy), and long philtrum. She presented talipes valgus and mild right hemihypertrophy without radiological findings at this stage. Biochemical test results were normal for blood levels of vitamins D, A, and B12, calcium, phosphate, folic acid, immunoglobulins, thyroid hormones, and glycosaminoglycan. Test results for amino acids in urine and organic acids in blood were also normal. Karyotype analysis, genetic testing for FMR1 , and screening for subtelomeric deletions were performed before the WES analysis, with negative results.
Information Consent
On behalf of the patient, the parents were advised upon written informed consent for genetic testing. This study was authorized by the pediatrics division of the Virgen de Altagracia Hospital (Manzanares, Spain).
High-Throughput Sequencing and Sanger
Deoxyribonucleic acid was extracted and purified from blood samples of the three family members using QIAsymphony SP (Qiagen). The proband sample was prepared for the WES protocol. The library was created using the SureSelect XT Reagent library preparation kit (Agilent) for the Illumina multiplexed sequencing paired-end. Target regions were enriched with the Agilent Clinical Research Exome V2 probe kit. Cluster preparation was performed using the cBot (Illumina; San Diego, California, United States) device, and library sequencing was performed using the Illumina HiSeq1500 platform. Bioinformatics analysis was applied through an end-to-end in-house pipeline developed by Health in Code (A Coruña, Spain), in accordance with the best WES analysis practices. At the interpretation level, we performed the analysis of variants for a panel of Sotos and Sotos-like syndrome ( APC2 , CDKN1C , EZH2 , GPC3 , NFIX , NSD1 , PTEN , SETD2 ). This panel came out negative, so we expanded the analysis of variants to the genes of the whole library, which may be associated with diseases that display overlapping phenotypes to the one observed in the patient. The coverage of the target regions for the genes of Sotos and Sotos-like syndrome, and the average coverage of the coding regions for the other genes in the exome, reached the quality parameters for the analysis. The candidate variant, detected in the WES analysis, was confirmed by bidirectional Sanger sequencing in the proband and it was absent in both parents, using a specific primer designed for the region of interest.
Results
We studied an undiagnosed female patient suspected of having Sotos syndrome with other multisystem abnormalities by WES. At the age of 2 years and 6 months, the patient was referred for genetic analysis targeting Sotos syndrome, according to the described phenotype. The main genes associated with this syndrome were screened, but no pathogenic variants that could explain the phenotype of the patient were found. In view of these results, we extended the analysis to other relevant genes that could be associated with overlapping features in our patient.
WES analysis revealed a previously undescribed heterozygous frameshift variant in AMER1 (c.956_957insT; p.Lys320Glufs*4), leading to a premature stop codon within the first APC binding domain.
Sanger sequencing confirmed the presence of the heterozygous variant in the patient and its absence on both parents, proving that it was de novo mutation ( Fig. 1 ).
Fig. 1.
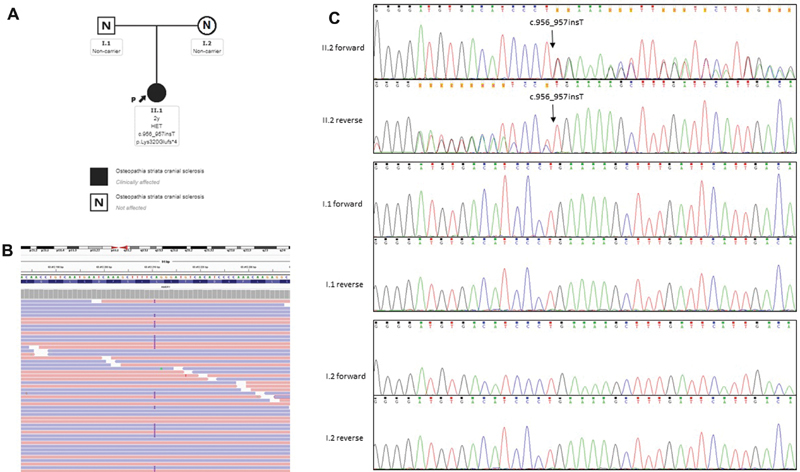
( A ) Pedigree of the studied family with the detected mutation in AMER1 . ( B ) High-throughput sequencing alignment of the sequence containing the insertion. ( C ) Sanger sequencing for the AMER1 mutation in proband and parents. II.2, index patient; I.1, father; I.2, mother.
Following the results of the WES analysis and the subsequent clinical report generated after genetic testing, the patient was further examined. Magnetic resonance imaging of the brain showed cranial asymmetry with left lateral ventriculomegaly. The skeletal radiographic series showed sclerosis of the cranial base and striations in the metaphyseal region of long bones, well visible in the humerus, femur, and tibia ( Fig. 2 ). She started to develop scoliosis. The patient was also referred to a pediatric cardiology unit because of apical left ventricular hypertrabeculation with normal ventricular function.
Fig. 2.
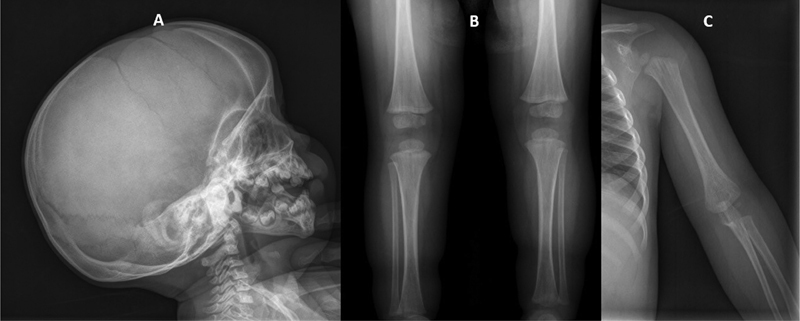
Reconstructed X-ray image of patient. ( A ) Cranial image showed sclerosis of base of skull. ( B ) Image of lower limbs showed striations in the metaphyseal region of the femur and tibia. ( C ) Image of upper limbs showed striations in the metaphyseal region of the humerus.
Review of Published Patient Series
In light of the varying severity of the established phenotypes for OSCS, we compiled information from 23 female patients, along with the female patient studied in this report, who harbored truncating mutations and large deletions in AMER1 and whose phenotype was thoroughly described in the literature ( Table 1 ). We focused this literature review on the clinical presentation of females affected by OSCS, which was characterized by the presence of major abnormalities including macrocephaly, facial dysmorphism, skull sclerosis, and longitudinal striations in the metaphyseal regions, mainly in long bones, pelvis, and scapulae, along with features that were not always expressed, such as hearing impairment, neuromuscular symptoms, or congenital heart defects. Other minor clinical manifestations affecting respiratory function and the gastrointestinal system have also been observed. We also added the clinical presentation of 12 male patients harboring truncating variants, who survived the neonatal period ( Table 2 ). These patients manifested differences in the severity of the disease.
Table 1. Clinical features of affected females truncating mutations and deletions in AMER1 .
| Patient | Variant | Zygosity | Inheritance | Dysmorphic features | Skeletal abnormalities | Neuromuscular abnormalities | Cardiovascular abnormalities | Respiratory abnormalities | Sensorineural abnormalities | Developmental delay | Other abnormalities | Source |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.194del p.Gly65Valfs*35 | Het | De novo | X | X | x | x | X | Enomoto et al 2018 17 | |||
| 2 | c.429T > A; p.Cys143* | Het | NA | X | X | Perdu et al 2011 16 | ||||||
| 3 | c.337delG; p.Gly113fs*58 | Het | NA | X | X | x | x | X | x | Perdu et al 2010 18 | ||
| 4 | c.555_556delTG; p.Ala187* |
Het | De novo | X | X | x | x | X | X | Sperotto et al 2017 10 | ||
| 5 | c.811C > T; p.Gln271* | Het | Inherited from mother | X | X | x | x | König et al 1996 19 | ||||
| 6 | c.811C > T; p.Gln271* | Het | Inherited from mother | X | X | x | x | König et al 1996 19 | ||||
| 7 | c.811C > T; p.Gln271* | Het | Inherited from mother | X | X | König et al 1996 19 | ||||||
| 8 | c.811C > T; p.Gln271* | Het | Inherited from mother | X | X | X | König et al 1996 19 | |||||
| 9 | c.654delG; p.Asp216fs*62* | Het | NA | X | X | x | Perdu et al 2011 16 | |||||
| 10 | c.827_842del16; p.(Pro276Glnfs*13) | Het | Germline mosaicism | X | X | x | x | x | x | O'Byrne et al 2016 20 | ||
| 11 | c.956_957insT; p.Lys320Glufs*4 | Het | De novo | X | X | x | x | x | X | This study | ||
| 12 | c.1045C > T; p.Gln349* | Het | De novo | X | x | x | x | X | x | Fujita et al 2014 3 | ||
| 13 | c.1057C > T; p.Arg353* | Het | Inherited from mother | X | x | x | X | Zicari et al 2012 21 | ||||
| 14 | c.1057C > T; p.(Arg353*) | Het | NA | X | x | Zicari et al 2012 21 | ||||||
| 15 | c.1072C > T; p.Arg358* | Het | NA | X | x | x | x | Jenkins et al 2009 1 | ||||
| 16 | c.1072C > T; p.Arg358* | Het | NA | X | x | x | Pellegrino et al 1997 22 | |||||
| 17 | c.1072C > T; p.Arg358* | Het | NA | X | x | x | x | x | X | Bueno et al 1998 23 | ||
| 18 | c.1072C > T; p.Arg358* | Het | Inherited from father? | X | x | x | x | x | X | Ciceri et al 2013 24 | ||
| 19 | c.1267delC; p.Leu423fs*25 | Het | NA | X | x | x | x | X | Keymolen et al 1997 25 | |||
| 20 | c.1267delC; p.Leu423fs*25* | Het | Inherited from mother | X | x | x | X | Keymolen et al 1997 25 | ||||
| 21 | Whole gene deletion | Het | De novo | X | x | x | x | x | x | X | X | Herman et al 2013 2 |
| 22 | Whole gene deletion | Het | Inherited from mother | X | x | x | x | X | Savarirayan et al 1997 26 | |||
| 23 | Whole gene deletion | Het | Inherited from mother | x | Savarirayan et al 1997 26 | |||||||
| 24 | Whole gene deletion | Het | NA | x | x | x | Savarirayan et al 1997 26 |
Note: List of female patients affected by osteopathia striata with cranial sclerosis (OSCS) carrying disease-causing truncating mutations in AMER1. Variants are ordered by distance to the start codon, from the closest to the furthest position. Details of clinical presentation of some probands might not have been fully described. ?, the variant of patient 18 was suspected to be inherited from father.
Table 2. Clinical features of surviving affected males with truncating mutations in AMER1 .
| Patient | Variant | Zygosity | Inheritance | Dysmorphic features | Skeletal abnormalities | Neuromuscular abnormalities | Cardiovascular abnormalities | Respiratory abnormalities | Sensorineural abnormalities | Developmental delay | Other abnormalities | Source |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 25 | c.429T > A: p.Cys143* | Hem | Inherited from mother | x | x | x | x | x | x | x | x | Perdu et al 2011 16 |
| 26 | c.607_611delAGGCC; p.Arg203fs*2 | Mos | Somatic mosaicism | x | x | x | x | x | Hague et al 2017 7 | |||
| 27 | c.811C > T; p.Gln271* | Hem | Inherited from mother | x | x | x | x | x | König et al 1996 19 | |||
| 28 | c.811C > T; p.Gln271* | Hem | Inherited from mother | x | x | x | König et al 1996 19 | |||||
| 29 | c.654delG; p.Asp216fs*62 | Hem | Inherited from mother | x | x | x | x | Perdu et al 2011 16 | ||||
| 30 | c.1072C> T; p.Arg358* | Mos | Germline mosaicism | x | x | x | x | x | x | x | Holman et al 2011 6 | |
| 31 | c.1072C> T; p.Arg358* | Mos? | Germline mosaicism? | x | x | x | Ciceri et al 2013 24 | |||||
| 32 | c.1108G > T; p.Glu370* | Mos | Mosaicism | x | x | x | x | Joseph et al 2010 5 | ||||
| 33 | c.1275C > G; p.Tyr425* | Hem | Inherited from mother | x | x | x | x | x | Jenkins et al 2009 1 | |||
| 34 | c.1267delC; p.Leu423fs*25 | NA | NA | x | x | x | x | x | Keymolen et al 1996 25 | |||
| 35 | c.1591C> T; p.Arg531* | Hem | Inherited from mother | x | x | x | x | x | x | Holman et al 2011 6 | ||
| 36 | c.1506delA; p.Gly502fs*38 | Hem | Inherited from mother | x | x | x | Jenkins et al 2009 1 |
Abbreviations: Mos, mosaicism; NA, zygosity not available.
Note: List of male patients affected by osteopathia striata with cranial sclerosis (OSCS) carrying disease-causing truncating mutations in AMER1 . Variants are ordered by distance to the start codon, from the closest to the furthest position. Details of clinical presentation of some probands might not have been fully described. ?, suspicion of germline mosaicism.
This procedure's purpose is to update the information on phenotype variability in females and to highlight the potential factors that could contribute to the observed differences in the clinical presentation of the disease. Among those factors, the position of the premature stop codon from truncating mutations and skewed X-inactivation have been described. 1 2 3 4 All female patients, including the one reported in this publication, showed dysmorphic features and skeletal abnormalities. Among these abnormalities, macrocephaly (or abnormal cephalic shape), skull sclerosis, and metaphyseal striations of long bones were present in more than 90% of the cohort, respectively ( some patients might not have been fully phenotyped in the original publications ). The second most frequent abnormality among affected females was hearing impairment (67%), followed by neuromuscular abnormalities (42%), developmental delay (42%), and heart defects (37%), consistent with the clinical features observed in affected surviving males. Other abnormalities such as respiratory abnormalities (17%) and other manifestations ( Supplementary Tables S1 and S2 ) were also observed. Even though we focused on the clinical presentation of affected females, there were some exceptions to the lethal phenotype in affected males who exhibited mosaic forms or somatic mutations. 5 6 7 The examination of the observed clinical data on these surviving males suggested the influence of factors other than allele dosage, which would affect the severity of the manifestations ( Table 2 ). The list factors that could affect the severity of the OSCS disease were written down in Table 3 .
Table 3. Influencing factors that were suggested to contribute in the severity of OCSC.
| Hemizygosity | Premature stop codon position | X-inactivation | Mosaicism | Large deletions, involving AMER1 | |
|---|---|---|---|---|---|
| Females | ✓ | ✓ | ✓ | ✓ | |
| Males | ✓ | ✓ | ✓ | ✓ |
Abbreviation: OSCS, osteopathia striata with cranial sclerosis.
Discussion
In this study, we described the usefulness of genetic analysis by WES and the benefits deriving from the interpretation of the results in a case of skeletal dysplasia showing overlapping features with several genetic conditions that include macrocephaly and other multisystem disorders. At the time of evaluation, the patient presented macrocephaly, dysmorphic features, skull sclerosis, and developmental delay, sharing features with several conditions such as Sotos, Weaver, and Luscan–Lumish syndromes, among other disorders. At this level of examination, reaching an accurate diagnosis was not possible in the absence of genetic testing. WES analysis ruled out the initially suspected Sotos syndrome, which further complicated the diagnosis. The flexibility of the high-throughput sequencing technology allowed expanding the analysis to include other genes associated with the patient phenotype. In addition to this advantage, it is relevant to remark the importance of reaching the quality parameters of average coverage of the exome and to identify the poor covered regions in target genes. The analysis resulted in the identification of a novel frameshift variant in AMER1 . A defective AMER1 ( WTX ) gene has been identified as the cause of X-dominant congenital OSCS, a skeletal dysplasia that is mainly produced by germline loss-of-function mutations. 1 Somatic mutations in AMER1 have also been found in Wilms tumor. 8 9 10 AMER1 has been demonstrated to function both as an inhibitor and activator of the WNT/β catenin pathway. 11 12 The modulation of this important pathway controls several cellular processes that are necessary for correct bone synthesis and bone homeostasis. AMER1 is expressed in early stages of embryogenesis in skeletal structures, neurological system, muscle, kidneys, male gonads, and lungs, inter alia. 8 13 Functional experiments using mice models have demonstrated that some abnormalities caused by defects in AMER1 can be extrapolated to the ones observed in OSCS human pathology. 14
After identifying this novel variant in AMER1 , this finding was key to guide further examinations of the patient. A detailed retrospective evaluation confirmed the presence of clinical features found in other described females affected by OSCS disease, such as striations in the metaphyseal region of the long bones and cardiovascular findings. Therefore, an accurate diagnosis allowed for screening the symptoms associated with OSCS and guided the clinical management of the patient. Genetic testing of the parents' carrier status allowed for genetic counseling to be offered to the family.
It can be considered that most causative mutations for OSCS have truncating effects on the AMER1 protein. Even though these mutations belong to the same mutation type, there are differences in phenotype development within this disorder. A functional study by Jenkins et al on WTX ( AMER1 ) expression in carriers of mutant proteins 671delC (p.Pro224fs*57) and 1506delA (p.Pro260fs*16), determined that aberrant transcripts were translated, suggesting that the truncated proteins may retain some regions that conserved certain functions of WTX ( Fig. 3 ). This fact is essential to understand the influence of the position of predicted premature stop codons derived from truncating variants on the phenotypic variability of OSCS.
Fig. 3.
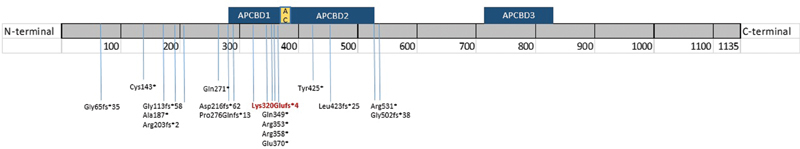
AMER1 protein sequence showing the locations of previously described truncating variants in the literature. The reported variant in this study is highlighted in red. The locations of the following domains on the protein sequence have been represented following Jenkins et al., 2008: APCBD1 (aa 280–368), APCBD2 (aa 380–531), APCBD3 (aa 717–834), and AC-acidic domain (aa 370–411).
The observed differences in the severity of the OCSC phenotype suggest the existence of influencing factors: Lethal or very severe forms have been described in affected male individuals, mainly caused by a hemizygosity condition; and milder forms have been observed in affected females and in surviving males. 2 6 7 14 The observed phenotype in affected females from published data was compared with that of the studied patient, helping to anticipate potential complications of the disease ( Table 1 ).
According to this review, phenotype–genotype correlation in females affected by OSCS is difficult to establish and it could be influenced by different factors. The position of the premature stop codon generated by truncating mutations within the protein sequence could produce aberrant translated messenger ribonucleic acids that retained certain properties of the protein; 1 Skewed X-inactivation event has been previously noted and assessed by other groups. 1 2 3 4 It should be taken into account that the pleiotropic effect of dysregulation of the Wnt pathway is due to the fact that this protein is widely expressed in developing organs. 15 Therefore, a thorough multisystem evaluation would be recommended to evaluate potential complications of the disease.
Additionally, the phenotype of surviving males showed a similar heterogeneity than the one observed in females. The factors that have been proposed to contribute in the spectrum of the disease include the position of the premature stop codon as well, and the presence of mosaicism forms that could attenuate the severity of OCSC disease in males and females. 6 7 16
In conclusion, the reported case revealed a novel frameshift mutation in AMER1 that we associated with the cause of OSCS in a female patient who was undiagnosed before genetic testing by WES.
Acknowledgments
We would like to express our gratitude to the patient's family for allowing the use of their clinical data in this study. This work and research was performed by Health in Code (A Coruña, Spain) and Virgen de Altagracia Hospital (Manzanares, Spain).
Conflict of Interest None declared.
Ethical Approval
This study was approved by the Ethics Committee of Research and Drugs from the hospital institution (Virgen de Altagracia Hospital, Manzanares, Spain).
Supplementary Material
References
- 1.Jenkins Z A, van Kogelenberg M, Morgan T. Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat Genet. 2009;41(01):95–100. doi: 10.1038/ng.270. [DOI] [PubMed] [Google Scholar]
- 2.Herman S B, Holman S K, Robertson S P, Davidson L, Taragin B, Samanich J. Severe osteopathia striata with cranial sclerosis in a female case with whole WTX gene deletion. Am J Med Genet A. 2013;161A(03):594–599. doi: 10.1002/ajmg.a.35716. [DOI] [PubMed] [Google Scholar]
- 3.Fujita A, Ochi N, Fujimaki H. A novel WTX mutation in a female patient with osteopathia striata with cranial sclerosis and hepatoblastoma. Am J Med Genet A. 2014;164A(04):998–1002. doi: 10.1002/ajmg.a.36369. [DOI] [PubMed] [Google Scholar]
- 4.Holman S K, Morgan T, Baujat G. Osteopathia striata congenita with cranial sclerosis and intellectual disability due to contiguous gene deletions involving the WTX locus. Clin Genet. 2013;83(03):251–256. doi: 10.1111/j.1399-0004.2012.01905.x. [DOI] [PubMed] [Google Scholar]
- 5.Joseph D J, Ichikawa S, Econs M J. Mosaicism in osteopathia striata with cranial sclerosis. J Clin Endocrinol Metab. 2010;95(04):1506–1507. doi: 10.1210/jc.2009-2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holman S K, Daniel P, Jenkins Z A. The male phenotype in osteopathia striata congenita with cranial sclerosis. Am J Med Genet A. 2011;155A(10):2397–2408. doi: 10.1002/ajmg.a.34178. [DOI] [PubMed] [Google Scholar]
- 7.Hague J, Delon I, Brugger K. Male child with somatic mosaic Osteopathia striata with cranial sclerosis caused by a novel pathogenic AMER1 frameshift mutation. Am J Med Genet A. 2017;173(07):1931–1935. doi: 10.1002/ajmg.a.38261. [DOI] [PubMed] [Google Scholar]
- 8.Rivera M N, Kim W J, Wells J. The tumor suppressor WTX shuttles to the nucleus and modulates WT1 activity. Proc Natl Acad Sci U S A. 2009;106(20):8338–8343. doi: 10.1073/pnas.0811349106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camp N D, James R G, Dawson D W. Wilms tumor gene on X chromosome (WTX) inhibits degradation of NRF2 protein through competitive binding to KEAP1 protein. J Biol Chem. 2012;287(09):6539–6550. doi: 10.1074/jbc.M111.316471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sperotto F, Bisogno G, Opocher E. Osteopathia striata with cranial sclerosis and Wilms tumor: coincidence or consequence? Clin Genet. 2017;92(06):674–675. doi: 10.1111/cge.13082. [DOI] [PubMed] [Google Scholar]
- 11.Tanneberger K, Pfister A S, Brauburger K. Amer1/WTX couples Wnt-induced formation of PtdIns(4,5)P2 to LRP6 phosphorylation. EMBO J. 2011;30(08):1433–1443. doi: 10.1038/emboj.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanneberger K, Pfister A S, Kriz V, Bryja V, Schambony A, Behrens J. Structural and functional characterization of the Wnt inhibitor APC membrane recruitment 1 (Amer1) J Biol Chem. 2011;286(22):19204–19214. doi: 10.1074/jbc.M111.224881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grohmann A, Tanneberger K, Alzner A, Schneikert J, Behrens J.AMER1 regulates the distribution of the tumor suppressor APC between microtubules and the plasma membrane J Cell Sci 2007120(Pt 21):3738–3747. [DOI] [PubMed] [Google Scholar]
- 14.Comai G, Boutet A, Tanneberger K. Genetic and molecular insights into genotype-phenotype relationships in osteopathia striata with cranial sclerosis (OSCS) through the analysis of novel mouse Wtx mutant alleles. J Bone Miner Res. 2018;33(05):875–887. doi: 10.1002/jbmr.3387. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Akyildiz S, Xiao Y. Structures of the APC-ARM domain in complexes with discrete Amer1/WTX fragments reveal that it uses a consensus mode to recognize its binding partners. Cell Discov. 2015;1:15016. doi: 10.1038/celldisc.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perdu B, Lakeman P, Mortier G, Koenig R, Lachmeijer A M, Van Hul W. Two novel WTX mutations underscore the unpredictability of male survival in osteopathia striata with cranial sclerosis. Clin Genet. 2011;80(04):383–388. doi: 10.1111/j.1399-0004.2010.01553.x. [DOI] [PubMed] [Google Scholar]
- 17.Enomoto Y, Tsurusaki Y, Harada N, Aida N, Kurosawa K. Novel AMER1 frameshift mutation in a girl with osteopathia striata with cranial sclerosis. Congenit Anom (Kyoto) 2018;58(04):145–146. doi: 10.1111/cga.12258. [DOI] [PubMed] [Google Scholar]
- 18.Perdu B, de Freitas F, Frints S G. Osteopathia striata with cranial sclerosis owing to WTX gene defect. J Bone Miner Res. 2010;25(01):82–90. doi: 10.1359/jbmr.090707. [DOI] [PubMed] [Google Scholar]
- 19.König R, Dukiet C, Dörries A, Zabel B, Fuchs S. Osteopathia striata with cranial sclerosis: variable expressivity in a four generation pedigree. Am J Med Genet. 1996;63(01):68–73. doi: 10.1002/(SICI)1096-8628(19960503)63:1<68::AID-AJMG14>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 20.O'Byrne J J, Phelan E, Steenackers E, van Hul W, Reardon W. Germline mosaicism in osteopathia striata with cranial sclerosis--recurrence in siblings. Clin Dysmorphol. 2016;25(02):45–49. doi: 10.1097/MCD.0000000000000116. [DOI] [PubMed] [Google Scholar]
- 21.Zicari A M, Tarani L, Perotti D. WTX R353X mutation in a family with osteopathia striata and cranial sclerosis (OS-CS): case report and literature review of the disease clinical, genetic and radiological features. Ital J Pediatr. 2012;38:27. doi: 10.1186/1824-7288-38-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pellegrino J E, McDonald-McGinn D M, Schneider A, Markowitz R I, Zackai E H. Further clinical delineation and increased morbidity in males with osteopathia striata with cranial sclerosis: an X-linked disorder? Am J Med Genet. 1997;70(02):159–165. [PubMed] [Google Scholar]
- 23.Bueno A L, Ramos F J, Bueno O, Olivares J L, Bello M L, Bueno M. Severe malformations in males from families with osteopathia striata with cranial sclerosis. Clin Genet. 1998;54(05):400–405. doi: 10.1111/j.1399-0004.1998.tb03753.x. [DOI] [PubMed] [Google Scholar]
- 24.Ciceri S, Cattaneo E, Fossati C, Radice P, Selicorni A, Perotti D. First evidence of vertical paternal transmission of osteopatia striata with cranial sclerosis. Am J Med Genet A. 2013;161A(05):1173–1176. doi: 10.1002/ajmg.a.35813. [DOI] [PubMed] [Google Scholar]
- 25.Keymolen K, Bonduelle M, De Maeseneer M, Liebaers I. How to counsel in osteopathia striata with cranial sclerosis. Genet Couns. 1997;8(03):207–211. [PubMed] [Google Scholar]
- 26.Savarirayan R, Nance J, Morris L, Haan E, Couper R. Osteopathia striata with cranial sclerosis: highly variable phenotypic expression within a family. Clin Genet. 1997;52(04):199–205. doi: 10.1111/j.1399-0004.1997.tb02547.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.