

Abstract
目的
探究肝细胞线粒体NDUFA13蛋白表达缺失在诱导小鼠肝脏纤维化发生中的作用及可能机制。
方法
以肝脏特异性NDUFA13杂合敲除小鼠品系(NDUFA13fl/-;Alb-Cre)作为研究对象,以同窝NDUFA13fl/fl小鼠作为对照,8只/组,进行长达两年的实验观察,分别在实验早期和晚期处死小鼠获取肝组织样本。采用HE染色、Masson染色观察不同时期小鼠的肝组织病理变化及纤维化表型。Western blot检测肝组织中NDUFA13蛋白表达水平。免疫荧光分析巨噬细胞标志物F4/80与转化生长因子TGF-β1、肿瘤坏死因子TNF-α与白介素IL-1β的表达。免疫组化分析肝星型细胞活化标志物α-SMA、基质金属蛋白酶MMP-9与基质金属蛋白酶组织抑制剂TIMP-1、胶原纤维Collagen-Ⅰ与Collagen-Ⅲ的表达情况。
结果
HE染色、Masson染色发现4周龄NDUFA13fl/-小鼠肝组织炎症浸润明显但未出现纤维化表型;2年龄NDUFA13fl/-小鼠肝组织严重损伤且伴大量胶原纤维产生。Western Blot结果显示2年龄NDUFA13fl/-小鼠肝组织中NDUFA13蛋白表达水平较对照小鼠明显降低(P < 0.05)。免疫荧光与免疫组化结果显示,NDUFA13fl/-小鼠肝组织中巨噬细胞F4/80聚集增多并伴随大量TGF-β1产生(P < 0.05),以及TNF-α、IL- 1β等炎症因子大量分泌(P < 0.05);同时,α-SMA、Collagen-Ⅰ及Collagen-Ⅲ表达明显增多(P < 0.05),基质金属蛋白酶MMP-9表达降低(P < 0.05),而其抑制剂TIMP-1表达明显增加(P < 0.05)。
结论
肝细胞线粒体NDUFA13蛋白缺陷能够诱导小鼠自发的慢性肝纤维化病理表型,可能与巨噬细胞/炎症因子信号诱导的肝星状细胞异常活化有关。
Keywords: NDUFA13缺陷, 巨噬细胞, 炎症因子, 肝星状细胞, 肝纤维化
Abstract
Objective
To investigate the role of hepatocyte mitochondrial NDUFA13 loss in the liver fibrogenesis in mice and explore the possible mechanisms.
Methods
We used liver-specific NDUFA13 heterozygous knockout mouse models (NDUFA13fl/-; Alb-Cre) established previously by intercrossing NDUFA13fl/fl and Alb-Cre mice, with their littermate control NDUFA13fl/fl mice as the control (n=8). The mice were euthanized at the age of 4 weeks and 2 years, and the liver tissues were collected for HE and Masson staining to observe the pathological changes and fibrosis phenotypes. Western blotting was performed to detect the expression of NDUFA13 protein in the liver tissues, and the infiltration of F4/80+ macrophages and the expressions of TGF-β1, TNF-α and IL-1β were analyzed by immunofluorescence assay. The expression levels of α-SMA, matrix metalloproteinase-9 (MMP-9) and tissue inhibitor of matrix metalloproteases 1 (TIMP-1), collagen-Ⅰ and collagen-Ⅲ were assayed by immunohistochemistry.
Results
HE and Masson staining showed obvious inflammatory infiltration but no significant fibrosis in the liver tissues of 4-week-old NDUFA13fl/- mice, but severe liver damage with massive fibrosis was observed in 2-year-old NDUFA13fl/- mice. NDUFA13 expression in 2-year-old NDUFA13fl/- mice markedly decreased compared with that in the control NDUFA13fl/fl mice as shown by Western blotting (P < 0.05). Immunohistochemistry showed obvious infiltration of F4/80+ macrophages in the liver tissue with a large amount of TGF-β1 production (P < 0.05) and TNF-α and IL-1β secretions in NDUFA13fl/- mice (P < 0.05). NDUFA13 knockout obviously promoted α-SMA expression (P < 0.05) and collagen-Ⅰ and collagen-Ⅲ deposition (P < 0.05) while significantly decreased MMP-9 and increased TIMP-1 expression in the liver (P < 0.05).
Conclusion
Hepatocytes-specific NDUFA13 deficiency can trigger spontaneous and chronic liver fibrosis phenotypes in mice probably in association with abnormal activation of hepatic stellate cells induced by macrophages and inflammatory factors.
Keywords: NDUFA13 deficiency, macrophages, inflammatory cytokines, hepatic stellate cells, hepatic fibrosis
肝纤维化是肝脏应对损伤进行自我修复的病理生理过程。各种慢性肝脏疾病过程中,肝细胞损伤与持续炎症刺激可导致肝星状细胞(HSCs)激活和细胞外基质(ECM)过度沉积加速肝纤维化病理进程[1],从而促肝硬化甚至肝癌发生。近年来,肝纤维化发病率逐年上升,但仍然缺乏有效的防治手段[2]。因此,深入探讨肝纤维化的发病机制,寻找阻滞或逆转肝纤维化的有效靶点,对肝纤维化的早期防治具有重要意义。
NDUFA13是一种核基因编码的线粒体蛋白,主要定位在线粒体内膜,是组成NADH脱氢酶Ⅰ复合体的基本亚单位,对于线粒体呼吸链膜电位及氧化磷酸化功能维持至关重要[3]。大量研究发现NDUFA13作为一种抑癌基因,在多种恶性肿瘤组织中表达紊乱,参与多种信号通路而调控肿瘤细胞增殖、凋亡及细胞侵袭与转移等过程,且与疾病的预后密切相关[3-7]。课题组前期研究揭示了NDUFA13在胃癌癌前炎症恶性进展中渐进性下调的特点[6],我们进一步证实NDUFA13蛋白失活可诱导小鼠自发性肝炎病理表型[8]。然而,NDUFA13蛋白缺失是否能够诱导小鼠肝脏纤维化的发生,仍需进一步证实。
目前,有关肝纤维化的药物或机制研究大多基于胆汁淤积、毒物诱导等干预的小鼠肝纤维化模型[9],并不能很好地模拟人类肝脏慢性纤维化的疾病状态。因此,制备能模拟人类肝炎-肝纤维化病理进程的动物模型,是探索肝纤维化疾病发生机制与治疗手段的重要基础。本研究重点关注NDUFA13蛋白失活对肝纤维化形成的影响,以期为肝纤维化治疗提供新的靶点,同时提供一种自发性、慢性肝纤维化动物模型,为肝纤维化的发病机制研究及治疗药物筛选提供新思路。
1. 材料和方法
1.1. 材料与试剂
1.1.1. 实验动物
NDUFA13fl/fl基因编辑小鼠、Alb-Cre转基因小鼠及肝脏特异性NDUFA13杂合敲除小鼠(NDUFA13fl/-;Alb-Cre)参照前期实验方法获得[8]。C57BL/6小鼠购买于重庆医科大学实验动物中心。所有小鼠均饲养于重庆医科大学附属儿童医院实验动物中心,SPF环境下(12 h/12 h光照/黑暗),自由采食与饮水。分别在4周龄、2年龄时处死小鼠,获取肝组织样本用于检测。所有操作均遵照实验动物伦理学要求(GDY1801016)进行。
1.1.2. 主要试剂
蛋白裂解液(碧云天),BCA蛋白浓度测定试剂盒(碧云天),NDUFA13小鼠抗人单克隆抗体(Santa cruz),小鼠抗β-actin抗体(sigma),Western Bright TM ECL显色试剂盒(Advansta),α-SMA小鼠抗人单克隆抗体(Bioss),Collagen-Ⅰ兔抗人多克隆抗体(Bioss),Collagen-Ⅲ兔抗人多克隆抗体(Bioss),MMP-9兔抗人多克隆抗体(Bioss),TIMP-1兔抗人多克隆抗体(Bioss),F4/80大鼠抗小鼠单克隆抗体(Biolegend),TGF-β1兔抗小鼠多克隆抗体(Bioss),TNF-α小鼠抗人单克隆抗体(Bioss),IL-1β兔抗人多克隆抗体(Bioss),即用型免疫组化(兔)试剂盒(福建迈新),即用型免疫组化(小鼠)试剂盒(福建迈新),大鼠二步法试剂盒(中杉金桥),山羊抗兔Alexa-Fluor 555荧光二抗(Bioss),山羊抗小鼠Alexa-Fluor 647荧光二抗(Bioss)。
1.1.3. 主要仪器
台式高速离心机(Thermo Fisher Scientific),凝胶电泳仪(Bio-Rad),ChemiDocTM Touch成像系统(Bio-Rad),病理切片仪(Leica),A1R激光共聚焦显微镜(Nikon)。
1.2. 实验方法
1.2.1. HE与Masson染色
经4% PFA溶液固定的小鼠肝组织,进行脱水及石蜡包埋,制成4 μm石蜡切片,于60 ℃烤箱烘烤后行二甲苯、梯度酒精脱蜡水化程序。用于HE染色的切片,分别进行苏木素染细胞核、伊红染细胞浆,再经梯度乙醇及二甲苯脱水、透明,中性树胶封片后用于镜检。用于Masson染色的切片,根据Masson染色试剂盒操作步骤依次进行Weigert铁苏木素溶液、丽春红品红溶液以及苯胺蓝溶液染色,经过相应分化与洗涤,最后脱水、透明并封固后用于镜下观察,每组小鼠取10个高倍视野(×400),进行Ishak纤维化评分分析[10]。
1.2.2. Western blot检测
取2年龄小鼠肝组织,采用蛋白提取试剂盒提取各组小鼠肝组织全蛋白,通过BCA蛋白定量试剂盒测量蛋白浓度,然后加入5×SDS PAGE上样缓冲液混合,于100 ℃加热5 min。取30 μg总蛋白上样,经10%SDS PAGE电泳后,电转至PVDF膜,利用含5%脱脂奶粉的TBST溶液封闭1 h,加入相应的一抗(NDUFA13 1∶300,内参为小鼠抗β-actin单克隆抗体1∶7000),4 ℃孵育过夜。TBST漂洗3次,加入相应二抗(辣根过氧化物酶标记的山羊抗小鼠IgG 1∶5000稀释),室温孵育1 h,TBST再次清洗3次后,滴加ECL显色剂,采用全自动化学发光成像分析系统进行检测、成像,并利用Image J软件对条带进行灰度值分析。
1.2.3. 免疫荧光染色
取2年龄小鼠新鲜肝脏组织,经OCT快速冷冻包埋,在冰冻切片机中切成8 μm冰冻切片,切片迅速置于4%PFA溶液固定0.5 h。PBS洗涤3次,加山羊血清室温封闭1 h,甩干后滴加稀释好的一抗溶液(F4/80 1∶300,TGF-β1 1∶200,TNF-α 1∶300,IL-1β 1∶300),置于4 ℃冰箱孵育过夜。PBS洗涤后,滴加相应二抗溶液,室温孵育1 h,再次PBS洗涤,滴加DAPI溶液染细胞核,最后洗涤并采用抗荧光淬灭封片剂封片,用于激光共聚焦检测成像。每组小鼠选取10个高倍视野(×400),进行平均荧光强度分析[11-13]。
1.2.4. 免疫组化染色
2年龄小鼠肝组织石蜡切片经脱蜡水化后,采用0.01 mol/L pH6.0枸橼酸钠缓冲液微波加热修复15 min,室温冷却后经PBS洗涤,滴加3%H2O2溶液避光孵育20 min,再次洗涤后,加5%BSA溶液室温封闭30 min。轻轻甩干,然后滴加稀释好的一抗溶液(α-SMA1∶800,MMP-91∶400,TIMP-11∶400,Collagen-Ⅰ 1∶400,Collagen-Ⅲ1∶600)于4 ℃孵育过夜。PBS洗涤3次,滴加相应二抗溶液覆盖组织,室温孵育1 h,再次洗涤后,加新鲜配制的DAB显色液,镜下控制显色时间。最后脱水透明并封片,用于显微镜镜检。每组小鼠选取10个高倍视野(×400),结合阳性细胞百分比与着色强度进行免疫组化评分分析[11-13]。
1.2.5. 统计学分析
采用GraphPad Prism 7.0和SPSS 18.0软件进行统计分析,组间差异行配对t检验或单因素方差分析,P < 0.05为差异具有统计学意义。
2. 结果
2.1. NDUFA13fl/-小鼠肝组织损伤及肝纤维化表型
小鼠肝组织石蜡切片用于HE染色、Masson染色分析。图 1结果显示,相比同年龄对照小鼠,4周龄NDUFA13fl/-小鼠肝组织小叶结构不清,肝细胞胞核皱缩坏死,Masson染色未见明显的胶原纤维生成(图 1A、C);2年龄对照小鼠肝组织结构基本清晰,大部分肝细胞形态正常,少许细胞胞浆出现衰老性水样变性,而同年龄NDUFA13fl/-小鼠肝组织肝小叶排列紊乱,中央静脉及汇管区域出现大量浅蓝色胶原沉积(P < 0.001,图 1B、C),大部分肝细胞发生肿胀坏死,且可见炎症细胞浸润。
1.
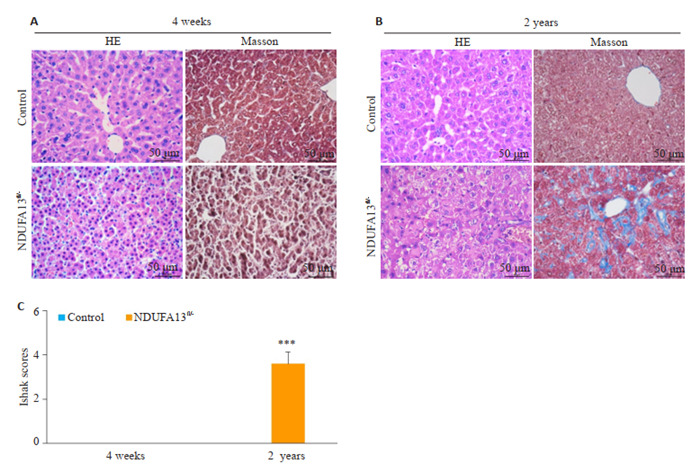
HE染色、Masson染色检测小鼠肝组织损伤及纤维化情况
Pathological and fibrotic changes in liver tissues of the mice revealed by HE staining and Masson staining (Original magnification: ×400). A: HE staining and Masson staining of 4-week-old mice liver tissues. B: HE staining and Masson staining of 2-year-old mice liver tissues. C: Ishak scores of Masson staining. ***P < 0.001 vs control.
2.2. NDUFA13fl/-小鼠肝组织中NDUFA13蛋白的表达情况
2年龄小鼠肝组织提取全蛋白,用于Western blot检测NDUFA13蛋白表达水平。图 2结果显示,相比对照小鼠,NDUFA13fl/-小鼠肝组织中的NDUFA13蛋白表达明显减少(P < 0.001,图 2A、B)。
2.
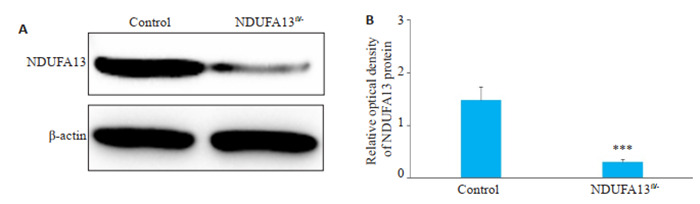
NDUFA13fl/-小鼠肝组织NDUFA13蛋白表达检测
NDUFA13 protein expression in the liver tissues detected by Western blotting. A: Western blots. B: Relative density of NDUFA13 expression. ***P < 0.001 vs control.
2.3. NDUFA13缺陷诱导巨噬细胞及炎症因子活化
2年龄小鼠肝组织用于免疫荧光检测,图 3结果显示,与对照小鼠相比,NDUFA13fl/-小鼠肝组织中F4/80巨噬细胞浸润明显增多,伴随TGF-β1分泌显著增加(P < 0.001,图 3A);且TNF-α与IL-1β炎症因子的表达也明显增强(P < 0.001,图 3B)。
3.
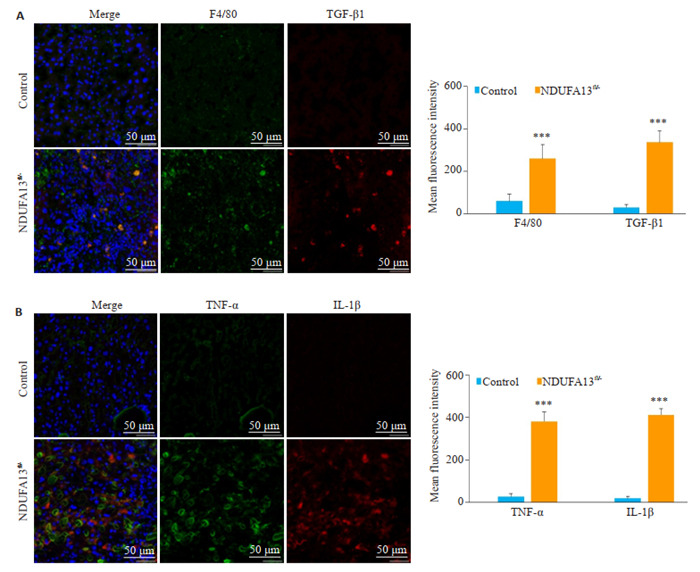
NDUFA13缺陷诱导巨噬细胞及炎症因子活化
NDUFA13 loss promotes activation of macrophages and inflammatory cytokines (Original magnification: ×400). A: F4/80 and TGF-β1 expression in control and NDUFA13fl/- mice detected by immunofluorescence assay. B: TNF-α and IL-1β expressions in control and NDUFA13fl/- mice detected by immunofluorescence assay. ***P < 0.001 vs control.
2.4. NDUFA13缺陷诱导纤维化相关因子激活
2.4.1. 肝星状细胞活化标志物α-SMA检测
2年龄小鼠肝组织用于免疫组化检测,结果显示对照小鼠肝脏组织未见明显的α-SMA阳性表达,而NDUFA13fl/-小鼠肝脏肝小叶内可见明显的胞浆棕褐色颗粒沉积,α-SMA阳性细胞显著增加(P < 0.001,图 4A、B)。
4.
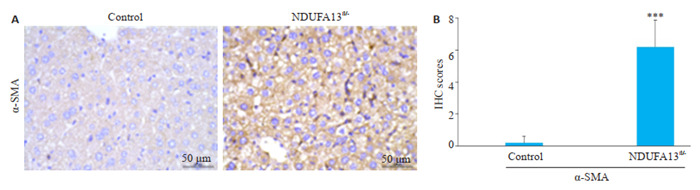
NDUFA13缺陷诱导肝星状细胞α-SMA激活
NDUFA13 deficiency induces α-SMA activation in hepatic stellate cells (× 400). A: α-SMA expression in control and NDUFA13fl/- mice revealed by IHC staining. B: Statistical analysis of α-SMA in control and NDUFA13fl/- mice. ***P < 0.001 vs control.
2.4.2. 肝脏组织MMP-9、TIMP-1检测
2年龄小鼠肝组织用于免疫组化检测(图 5),对照组小鼠肝组织可见散在的棕褐色颗粒MMP-9表达,而NDUFA13fl/-小鼠肝组织MMP-9的表达明显减少(P < 0.001,图 5A、B);对照组小鼠肝组织中仅见少许TIMP-1阳性分布,而NDUFA13fl/-小鼠肝组织TIMP-1表达显著增强(P < 0.001,图 5A、C)。
5.
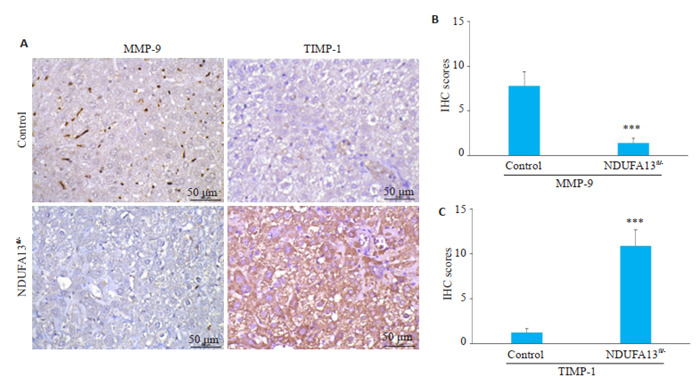
NDUFA13缺陷诱导MMP-9/TIMP-1表达失衡
NDUFA13 inactivation results in abnormal MMP-9 and TIMP-1 expression (×400). A: MMP-9 and TIMP-1 expression in Control and NDUFA13fl/- mice analyzed by IHC staining. B, C: Statistical analysis of MMP-9 and TIMP-1 in control and NDUFA13fl/- mice. ***P < 0.001 vs control.
2.4.3. 肝脏组织胶原纤维Collagen-Ⅰ、Collagen-Ⅲ检测
2年龄小鼠肝组织用于免疫组化检测,对照组小鼠肝组织未见明显的胶原沉积,而NDUFA13fl/-小鼠肝脏汇管区、中央静脉区及肝小叶内均有大量棕褐色聚集,Collagen-Ⅰ、Collagen-Ⅲ表达显著增加(P < 0.001,图 6A~C)。
6.
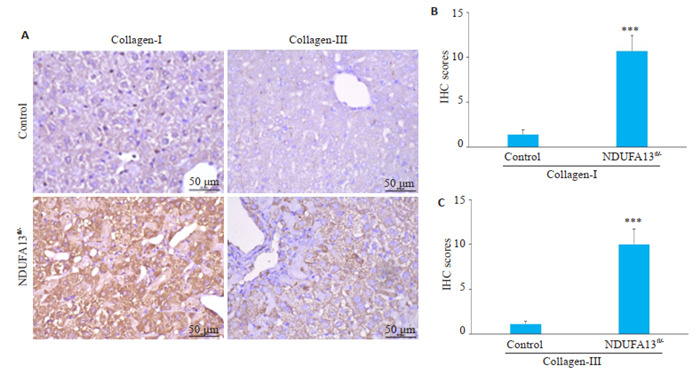
NDUFA13缺陷诱导胶原纤维沉积
NDUFA13 inactivation causes collagen deposition in the liver (×400). A: Collagen-Ⅰ and collagen-Ⅲ expression in control and NDUFA13fl/- mice analyzed by IHC staining. B, C: Statistical analysis of collagen-Ⅰ and collagen-Ⅲ in control and NDUFA13fl/- mice. ***P < 0.001 vs control.
3. 讨论
肝纤维化是慢性肝损伤向肝硬化甚至肝癌转变的必经病理路径,能否在此阶段阻遏或逆转纤维化进程,是慢性肝疾病防控的难点与重点[14-15]。目前,有关肝纤维化的细胞与分子机制成为研究的热点,包括肌成纤维细胞激活、炎症信号转导、细胞自噬与衰老及细胞器功能失调等[16-17]。其中,线粒体作为细胞能量代谢和氧化应激调节的关键场所,其结构或功能的紊乱是多种急慢性肝病的重要病理特征,如线粒体肿胀、氧化磷酸化功能减弱及活性氧异常释放等[17],引起肝细胞代谢障碍和炎症激活[15]。此外,线粒体应激和损伤还可通过钙离子释放、融合与分裂动力学改变以及介导胞内信号转导等方式,调节肝脏细胞的自我更新、增殖与凋亡等过程[15]。探寻改善线粒体结构与功能稳态的治疗药物或靶点,可能是缓解肝炎与肝纤维化疾病的有效途径[14, 18]。
NDUFA13作为一种线粒体呼吸链组分蛋白,对小鼠器官发育极其重要。有文献报道NDUFA13基因整体敲除可导致小鼠胚胎死亡[19],而我们实验发现NDUFA13纯合敲除的小鼠几乎无法存活,这提示NDUFA13蛋白在肝脏发育中的重要作用。近期NDUFA13作为一种新型的抑癌基因而备受关注,在多种恶性肿瘤中表达紊乱[7]。最新研究发现,NDUFA13蛋白可抑制STAT3信号介导的炎症反应而减轻DSS诱导的结肠炎[20],并可抑制类风湿关节炎成纤维细胞样滑膜细胞的增殖和侵袭[21],在炎症调控中发挥重要作用。课题组前期揭示了NDUFA13在慢性萎缩性胃炎病理进程中表达渐进性下调的特点[6],后续我们证实了NDUFA13失活可通过激活ROS/NF-κB/NLRP3炎症通路,募集炎症细胞并分泌炎症因子,从而诱导小鼠自发性肝炎[7]。本研究进一步探讨了肝细胞NDUFA13缺陷在诱导小鼠自发性肝纤维化中的作用及初步机制,发现NDUFA13缺失可诱导巨噬细胞活化并分泌TGF-β1等促纤维化炎性因子,进而激活HSCs,导致胶原沉积而促成肝纤维化。
细胞外基质(ECM)的过度沉积是慢性肝损伤发生纤维化转变的直接原因,而HSCs作为胶原纤维的主要生产者,其增殖与活化是肝纤维化病理的核心环节[16]。多种炎症和纤维化相关的信号通路均可激活HSCs[16]。其中,转化生长因子TGF-β家族涉及各种纤维化疾病的发生发展过程[22],能与多种信号分子如活性氧(ROS)、血小板源生长因子(PDGF)、结缔组织生长因子(cTGF)等相互作用,被认为是驱动HSCs活化与ECM沉积的关键因子[23]。作为研究最广泛的一类转化生长因子,TGF-β1主要由肝组织内Kupffer巨噬细胞表达和释放,通过激活Smad2/3信号通路诱导HSCs内Collagen-Ⅰ、Collagen-Ⅲ胶原转录[16],还可介导脂质富集的肝细胞死亡[24],并诱导肝细胞结缔组织生长因子产生[25],促进肝纤维化发生。但鉴于TGF-β1广泛的生物学作用,其对肝纤维化及慢性肝病也存在其他多种调控机制与途径[23]。除了分泌TGF-β1等促纤维化因子,肝脏巨噬细胞也释放促肝细胞凋亡介质,募集炎症细胞并激活肌成纤维细胞[26]。此外,巨噬细胞还能促进HSCs的增殖与迁移[27]。我们的研究结果发现,NDUFA13表达缺失的小鼠肝组织中,巨噬细胞趋化聚集并分泌大量TGF-β1,刺激并激活静息HSCs转化为可分泌α-SMA的肌成纤维细胞。这提示NDUFA13缺陷可诱导肝脏巨噬细胞活化与TGF-β1释放,为HSCs的活化提供诱因。
炎症因子的分泌伴随慢性肝炎-肝纤维化病理的全过程,在纤维化的起始和延续中发挥重要作用。IL-1β与TNF-α作为强效炎症细胞因子,可由多种细胞分泌产生如巨噬细胞或活化HSCs等。IL-1β在炎症小体NLRP3的作用下剪切成熟与释放,触发炎症反应,参与多种因素诱导的肝纤维化病理过程[27]。TNF-α可促进肝细胞凋亡、免疫细胞活化和抑制星状细胞凋亡。研究报道了NLRP3通过IL-1β、TNF-α等促炎因子的活化而诱导肝细胞损伤与肝纤维化[28-29]。此外,IL-1β、TNF-α均可上调基质金属蛋白酶组织抑制剂TIMP-1的表达,并介导巨噬细胞对星状细胞NF-κB信号的活化,但它们并不直接活化HSCs,而是通过促进活化星状细胞在体内外的存活而发挥作用[30]。结合我们前期的研究结果,NDUFA13失活诱导活性氧ROS异常释放而活化NF-κB/NLRP3炎症通路,引起IL-1β、TNF-α等炎症因子的分泌与释放,进一步促进HSCs增殖与活力,推动纤维化进程。
肝脏ECM的动态平衡主要由基质金属蛋白酶/基质金属蛋白酶组织抑制剂(MMPs/TIMPs)调节和维持。MMPs可降解胶原蛋白,其活性受TIMPs的抑制。在活化的HSCs中,MMPs/TIMPs合成与分泌失衡,导致ECM降解减少而异常沉积,促进纤维化形成[28]。文献报道了纤维细胞因子TGF-β1和炎性细胞因子IL-1β、TNF-α等均可调控MMPs/TIMPs蛋白的表达与活性[27, 31]。这与我们的实验结果相一致,NDUFA13缺陷的小鼠肝脏中,活化的巨噬细胞分泌产生大量TGF-β1及IL-1β、TNF-α,通过影响MMP-9表达下调、TIMP-1表达上调,促进组织内Collagen-Ⅰ、Collagen-Ⅲ等胶原纤维的沉积,最终形成肝纤维化。
综上所述,小鼠肝脏NDUFA13缺陷可以诱导自发性的慢性肝纤维化病理表型,其可能机制为NDUFA13缺失诱导巨噬细胞活化并分泌TGF-β1及IL-1β、TNF-α等炎症相关因子,进一步激活HSCs并扰乱MMPs/ TIMPs平衡,促进Collagen-Ⅰ、Collagen-Ⅲ等胶原纤维的分泌与沉积。本研究立足于前期有关“小鼠肝脏NDUFA13失活通过调控ROS/NF-κB/NLRP3炎症通路诱导自发性肝炎病理进程”的研究[8],进一步明确了NDUFA13在后续肝纤维化中的调控作用与初步机制,对慢性肝病及肝纤维化恶性进展的早期防治具有重要意义,并为慢性肝炎-肝纤维化相关药物或机制研究提供一种有利的动物模型。后续我们将采用诱导型敲除小鼠模型,实现在小鼠出生后的肝脏特异性敲除,获得NDUFA13杂合和纯合敲除小鼠,继而深入探究NDUFA13缺陷活化肝星状细胞的具体分子机制,揭示其相关区域的免疫学特征。
Biography
徐小惠,硕士研究生,助理实验师,E-mail: 1049168422@qq.com
Funding Statement
国家自然科学基金(30701004);重庆市渝中区基础研究与前沿探索项目(20190106)
Supported by National Natural Science Foundation of China (30701004)
Contributor Information
徐 小惠 (Xiaohui XU), Email: 1049168422@qq.com.
黄 轶 (Yi HUANG), Email: yihuang828@foxmail.com.
References
- 1.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411. doi: 10.1038/nrgastro.2017.38. [Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation[J]. Nat Rev Gastroenterol Hepatol, 2017, 14(7): 397-411.] [DOI] [PubMed] [Google Scholar]
- 2.Nordström A, Bergman J, Björk S, et al. A multiple risk factor program is associated with decreased risk of cardiovascular disease in 70-year-olds: a cohort study from Sweden. PLoS Med. 2020;17(6):e1003135. doi: 10.1371/journal.pmed.1003135. [Nordström A, Bergman J, Björk S, et al. A multiple risk factor program is associated with decreased risk of cardiovascular disease in 70-year-olds: a cohort study from Sweden[J]. PLoS Med, 2020, 17 (6): e1003135.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nallar SC, Kalvakolanu DV. GRIM-19: a master regulator of cytokine induced tumor suppression, metastasis and energy metabolism. Cytokine Growth Factor Rev. 2017;33:1–18. doi: 10.1016/j.cytogfr.2016.09.001. [Nallar SC, Kalvakolanu DV. GRIM-19: a master regulator of cytokine induced tumor suppression, metastasis and energy metabolism[J]. Cytokine Growth Factor Rev, 2017, 33: 1-18.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hao H, Liu J, Liu G, et al. Depletion of GRIM-19 accelerates hepatocellular carcinoma invasion via inducing EMT and loss of contact inhibition. J Cell Physiol. 2012;227(3):1212–9. doi: 10.1002/jcp.24025. [Hao H, Liu J, Liu G, et al. Depletion of GRIM-19 accelerates hepatocellular carcinoma invasion via inducing EMT and loss of contact inhibition[J]. J Cell Physiol, 2012, 227(3): 1212-9.] [DOI] [PubMed] [Google Scholar]
- 5.Huang Y, Yang M, Yang H, et al. Upregulation of the GRIM-19 gene suppresses invasion and metastasis of human gastric cancer SGC-7901 cell line. Exp Cell Res. 2010;316(13):2061–70. doi: 10.1016/j.yexcr.2010.05.010. [Huang Y, Yang M, Yang H, et al. Upregulation of the GRIM-19 gene suppresses invasion and metastasis of human gastric cancer SGC-7901 cell line[J]. Exp Cell Res, 2010, 316(13): 2061-70.] [DOI] [PubMed] [Google Scholar]
- 6.Huang Y, Yang M, Hu H, et al. Mitochondrial GRIM-19 as a potential therapeutic target for STAT3-dependent carcinogenesis of gastric cancer. http://www.ncbi.nlm.nih.gov/pubmed/27167343. Oncotarget. 2016;7(27):41404–20. doi: 10.18632/oncotarget.9167. [Huang Y, Yang M, Hu H, et al. Mitochondrial GRIM-19 as a potential therapeutic target for STAT3-dependent carcinogenesis of gastric cancer[J]. Oncotarget, 2016, 7(27): 41404-20.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Ye T, Xue B, et al. Mitochondrial GRIM-19 deficiency facilitates gastric cancer metastasis through oncogenic ROS-NRF2-HO-1 axis via a NRF2-HO-1 loop. Gastric Cancer. 2021;24(1):117–32. doi: 10.1007/s10120-020-01111-2. [Wang X, Ye T, Xue B, et al. Mitochondrial GRIM-19 deficiency facilitates gastric cancer metastasis through oncogenic ROS-NRF2-HO-1 axis via a NRF2-HO-1 loop[J]. Gastric Cancer, 2021, 24(1): 117-32.] [DOI] [PubMed] [Google Scholar]
- 8.徐 小惠, 李 锐, 曾 欣, et al. 线粒体蛋白NDUFA13失活诱导自发性肝炎病理进程的作用及机制. http://www.j-smu.com/oa/darticle.aspx?type=view&id=20210155. 南方医科大学学报. 2021;41(1):55–63. doi: 10.12122/j.issn.1673-4254.2021.01.07. [徐小惠, 李锐, 曾欣, 等. 线粒体蛋白NDUFA13失活诱导自发性肝炎病理进程的作用及机制[J]. 南方医科大学学报, 2021, 41(1): 55-63.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.王梓睿, 田飞鸿, 饶木艳, 等. 小鼠肝纤维化造模方法研究进展[J]. 中国现代医学杂志, 2021, [Epub ahead of print].
- 10.Goodman ZD. Grading and staging systems for inflammation and fibrosis in chronic liver diseases. J Hepatol. 2007;47(4):598–607. doi: 10.1016/j.jhep.2007.07.006. [Goodman ZD. Grading and staging systems for inflammation and fibrosis in chronic liver diseases[J]. J Hepatol, 2007, 47(4): 598-607.] [DOI] [PubMed] [Google Scholar]
- 11.Ye T, Yang M, Huang D, et al. MicroRNA-7 as a potential therapeutic target for aberrant NF-κB-driven distant metastasis of gastric cancer. J Exp Clin Cancer Res. 2019;38(1):55. doi: 10.1186/s13046-019-1074-6. [Ye T, Yang M, Huang D, et al. MicroRNA-7 as a potential therapeutic target for aberrant NF-κB-driven distant metastasis of gastric cancer[J]. J Exp Clin Cancer Res, 2019, 38(1): 55.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang Y, Ma C, Zhang Q, et al. CD4+ and CD8+T cells have opposing roles in breast cancer progression and outcome. Oncotarget. 2015;6(19):17462–78. doi: 10.18632/oncotarget.3958. [Huang Y, Ma C, Zhang Q, et al. CD4+ and CD8+T cells have opposing roles in breast cancer progression and outcome[J]. Oncotarget, 2015, 6(19): 17462-78.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu RH, Yang MH, Xiang H, et al. Depletion of OLFM4 gene inhibits cell growth and increases sensitization to hydrogen peroxide and tumor necrosis factor-alpha induced-apoptosis in gastric cancer cells. J Biomed Sci. 2012;19:38. doi: 10.1186/1423-0127-19-38. [Liu RH, Yang MH, Xiang H, et al. Depletion of OLFM4 gene inhibits cell growth and increases sensitization to hydrogen peroxide and tumor necrosis factor-alpha induced-apoptosis in gastric cancer cells[J]. J Biomed Sci, 2012, 19: 38.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang JW, Hong JM, Lee SM. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J Pineal Res. 2016;60(4):383–93. doi: 10.1111/jpi.12319. [Kang JW, Hong JM, Lee SM. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis[J]. J Pineal Res, 2016, 60(4): 383-93.] [DOI] [PubMed] [Google Scholar]
- 15.Abdellah Mansouri, Charles-Henry Gattolliat, Tarik Asselah. Mito-chondrial dysfunction and signaling in chronic liver diseases. Gastroenterology. 2018;155(3):629–47. doi: 10.1053/j.gastro.2018.06.083. [Abdellah Mansouri, Charles-Henry Gattolliat, Tarik Asselah. Mito-chondrial dysfunction and signaling in chronic liver diseases[J]. Gastroenterology, 2018, 155(3): 629-47.] [DOI] [PubMed] [Google Scholar]
- 16.Seki E, Brenner DA. Recent advancement of molecular mechanisms of liver fibrosis. J Hepatobiliary Pancreat Sci. 2015;22(7):512–8. doi: 10.1002/jhbp.245. [Seki E, Brenner DA. Recent advancement of molecular mechanisms of liver fibrosis[J]. J Hepatobiliary Pancreat Sci, 2015, 22(7): 512-8.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grattagliano I, Russmann S, Diogo C, et al. Mitochondria in chronic liver disease. Curr Drug Targets. 2011;12(6):879–93. doi: 10.2174/138945011795528877. [Grattagliano I, Russmann S, Diogo C, et al. Mitochondria in chronic liver disease[J]. Curr Drug Targets, 2011, 12(6): 879-93.] [DOI] [PubMed] [Google Scholar]
- 18.Turkseven S, Bolognesi M, Brocca A, et al. Mitochondria-targeted antioxidant mitoquinone attenuates liver inflammation and fibrosis in cirrhotic rats. Am J Physiol Gastrointest Liver Physiol. 2020;318(2):G298–304. doi: 10.1152/ajpgi.00135.2019. [Turkseven S, Bolognesi M, Brocca A, et al. Mitochondria-targeted antioxidant mitoquinone attenuates liver inflammation and fibrosis in cirrhotic rats[J]. Am J Physiol Gastrointest Liver Physiol, 2020, 318(2): G298-304.] [DOI] [PubMed] [Google Scholar]
- 19.Kalakonda S, Nallar SC, Jaber S, et al. Monoallelic loss of tumor suppressor GRIM-19 promotes tumorigenesis in mice. PNAS. 2013;110(45):E4213–22. doi: 10.1073/pnas.1303760110. [Kalakonda S, Nallar SC, Jaber S, et al. Monoallelic loss of tumor suppressor GRIM-19 promotes tumorigenesis in mice[J]. PNAS, 2013, 110(45): E4213-22.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim JK, Lee SH, Lee SY, et al. Grim19 attenuates DSS induced colitis in an animal model. PLoS One. 2016;11(6):e0155853. doi: 10.1371/journal.pone.0155853. [Kim JK, Lee SH, Lee SY, et al. Grim19 attenuates DSS induced colitis in an animal model[J]. PLoS One, 2016, 11(6): e0155853.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao D, Jiang Z, Wang Z, et al. Retinoid interferon-induced mortality19 (GRIM19) inhibits proliferation and invasion in rheumatoid arthritis fibroblast-like synoviocytes. Biomed Pharmacother. 2018;98:719–725. doi: 10.1016/j.biopha.2017.12.114. [Zhao D, Jiang Z, Wang Z, et al. Retinoid interferon-induced mortality19 (GRIM19) inhibits proliferation and invasion in rheumatoid arthritis fibroblast-like synoviocytes[J]. Biomed Pharmacother, 2018, 98: 719-725.] [DOI] [PubMed] [Google Scholar]
- 22.Dooley S, ten Dijke P. TGF-β in progression of liver disease. Cell Tissue Res. 2012;347(1):245–56. doi: 10.1007/s00441-011-1246-y. [Dooley S, ten Dijke P. TGF-β in progression of liver disease[J]. Cell Tissue Res, 2012, 347(1): 245-56.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dewidar B, Meyer C, Dooley S, et al. TGF-β in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells. 2019;8(11):1419. doi: 10.3390/cells8111419. [Dewidar B, Meyer C, Dooley S, et al. TGF-β in hepatic stellate cell activation and liver fibrogenesis-updated 2019[J]. Cells, 2019, 8(11): 1419.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L, Kwon J, Popov Y, et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology. 2014;146(5):1339–50. doi: 10.1053/j.gastro.2014.01.061. [Yang L, Kwon J, Popov Y, et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice[J]. Gastroenterology, 2014, 146(5): 1339-50.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.郭 冶, 卢 凤美, 刘 东璞. 肝纤维化发病机制及大黄酸对肝纤维化的作用. 医学信息. 2020;33(12):27–32, 39. doi: 10.3969/j.issn.1006-1959.2020.12.010. [郭冶, 卢凤美, 刘东璞. 肝纤维化发病机制及大黄酸对肝纤维化的作用[J]. 医学信息, 2020, 33(12): 27-32, 39.] [DOI] [Google Scholar]
- 26.Lodder J, Denaës T, Chobert MN, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11(8):1280–92. doi: 10.1080/15548627.2015.1058473. [Lodder J, Denaës T, Chobert MN, et al. Macrophage autophagy protects against liver fibrosis in mice[J]. Autophagy, 2015, 11(8): 1280-92.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61(3):1066–79. doi: 10.1002/hep.27332. [Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways[J]. Hepatology, 2015, 61(3): 1066-79.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wree A, Eguchi A, McGeough MD, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59(3):898–910. doi: 10.1002/hep.26592. [Wree A, Eguchi A, McGeough MD, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice[J]. Hepatology, 2014, 59(3): 898-910.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wree A, McGeough MD, Inzaugarat ME, et al. NLRP3 inflamm-asome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology. 2018;67(2):736–49. doi: 10.1002/hep.29523. [Wree A, McGeough MD, Inzaugarat ME, et al. NLRP3 inflamm-asome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice[J]. Hepatology, 2018, 67(2): 736-49.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pradere JP, Kluwe J, De Minicis S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58(4):1461–73. doi: 10.1002/hep.26429. [Pradere JP, Kluwe J, De Minicis S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice[J]. Hepatology, 2013, 58(4): 1461-73.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robert S, Gicquel T, Bodin A, et al. Characterization of the MMP/ TIMP imbalance and collagen production induced by IL-1β or TNF-α release from human hepatic stellate cells. PLoS One. 2016;11(4):e0153118. doi: 10.1371/journal.pone.0153118. [Robert S, Gicquel T, Bodin A, et al. Characterization of the MMP/ TIMP imbalance and collagen production induced by IL-1β or TNF-α release from human hepatic stellate cells[J]. PLoS One, 2016, 11(4): e0153118.] [DOI] [PMC free article] [PubMed] [Google Scholar]