

Abstract
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is common, with a prevalence of 1/1000 and predominantly caused by disease-causing variants in PKD1 or PKD2. Clinical diagnosis is usually by age-dependent imaging criteria, which is challenging in patients with atypical clinical features, without family history, or younger age. However, there is increasing need for definitive diagnosis of ADPKD with new treatments available. Sequencing is complicated by six pseudogenes that share 97% homology to PKD1 and by recently identified phenocopy genes. Whole-genome sequencing can definitively diagnose ADPKD, but requires validation for clinical use. We initially performed a validation study, in which 42 ADPKD patients underwent sequencing of PKD1 and PKD2 by both whole-genome and Sanger sequencing, using a blinded, cross-over method. Whole-genome sequencing identified all PKD1 and PKD2 germline pathogenic variants in the validation study (sensitivity and specificity 100%). Two mosaic variants outside pipeline thresholds were not detected. We then examined the first 144 samples referred to a clinically-accredited diagnostic laboratory for clinical whole-genome sequencing, with targeted-analysis to a polycystic kidney disease gene-panel. In this unselected, diagnostic cohort (71 males :73 females), the diagnostic rate was 70%, including a diagnostic rate of 81% in patients with typical ADPKD (98% with PKD1/PKD2 variants) and 60% in those with atypical features (56% PKD1/PKD2; 44% PKHD1/HNF1B/GANAB/ DNAJB11/PRKCSH/TSC2). Most patients with atypical disease did not have clinical features that predicted likelihood of a genetic diagnosis. These results suggest clinicians should consider diagnostic genomics as part of their assessment in polycystic kidney disease, particularly in atypical disease.
Subject terms: Genetics research, Polycystic kidney disease, Genetic testing
Introduction
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is a common monogenic condition, with a prevalence of ~1 in 1000, and carries a high disease burden [1]. Approximately 50% of all ADPKD patients develop end stage renal failure (ESRF) by 60 years [2]. ADPKD is predominantly caused by disease-causing variants in PKD1 (OMIM#601313) or PKD2 (OMIM#173910).
Traditional clinical diagnosis of ADPKD is based on patient age and kidney cyst number on ultrasound examination [3]. Although inexpensive and low-risk, ultrasonography has limitations, with ultrasound-criteria only described for those with a family history of ADPKD, thus excluding the 10–15% with de novo disease and those unaware of their family history [4]. In addition, at-risk patients cannot be excluded from having inherited the condition, using ultrasonography alone, until after the age of 40 [3]. Abdominal-MRI can be used, but is less readily available and more expensive [5]. An additional diagnostic challenge is the atypical patient with visible kidney cysts, who does not meet clinical criteria for ADPKD diagnosis. Literature on a diagnostic approach in these patients is limited [2].
Polycystic Kidney Disease (PKD) can be used to describe a broad range of conditions that cause macroscopic cystic changes in the kidney, with ADPKD being the most common cause of PKD. The landscape in PKD is rapidly changing. Increasing genetic complexity has been highlighted by recent identification of genes associated with atypical ADPKD phenotypes, such as GANAB and DNAJB11, and the increasingly recognized overlap with genes associated with an ADPKD-like phenotype, such as HNF1B [6–8]. Analyses of population genomic datasets suggest that the prevalence and expressivity of ADPKD may be broader than initially understood, further increasing the complexity of clinical diagnosis [1, 9]. Clarifying diagnosis is increasingly important as newly available therapy for ADPKD requires definitive diagnosis and prognostic information in order to select appropriate patients for treatment [10]. Genetic diagnostics, in combination with clinical features, can help predict prognosis [11].
Given the increasing complexity of atypical disease and the value of definitive diagnosis to guide therapeutics, genetic testing offers the opportunity to improve diagnostic rates and clinical care for PKD patients [12]. Genetic testing in ADPKD is complicated by six pseudogenes that share 97% sequence-similarity with PKD1, which challenge standard sequencing techniques [13, 14]. This complexity may contribute to the infrequent use of genetic diagnosis, as compared with imaging diagnostics. Yet, genetic diagnostics has added benefits—it offers diagnostic clarity in patients with atypical phenotype, provides prognostic information, informs family planning, and allows cascade testing [15].
We have previously shown that whole-genome sequencing can overcome pseudogene sequence similarity, but this technique has not previously been validated for use in a diagnostic setting [16]. As genetic results have enormous impact on clinical decision-making it is essential that any sequencing methodology be adequately validated to ensure the test is both specific and sensitive. This is particularly important in ADPKD given the challenges of pseudogene homology. An established technique for genetic diagnosis of ADPKD is by long-range PCR-amplification (LR-PCR) of PKD1 followed by Sanger or massively-parallel sequencing (MPS) of this LR-PCR-product [13, 17]. The amplification is performed to avoid inadvertently sequencing the PKD1 pseudogenes.
We validated whole-genome sequencing using a blinded cross-over method in which a cohort of ADPKD patients was sequenced by both whole-genome and Sanger sequencing. LR-PCR and Sanger sequencing was selected as the comparator as it is more sensitive and specific than MPS-based methods in ADPKD [13, 18]. Following validation, we report the results of the first 144 samples referred to an accredited national diagnostic laboratory for clinical whole-genome sequencing, with analysis targeted to a PKD-gene panel. This is the first clinical whole-genome sequencing-based diagnostic test for PKD and we report its utility in an unselected, ‘real-world’ cohort of patients with both typical and atypical PKD.
Materials and methods
Validation cohort
Study approval was obtained from the relevant Institutional Ethics Committees. ADPKD phenotype was based on standard ultrasound criteria [3]. Thirty patients initially underwent LR-PCR and Sanger sequencing of PKD1 and PKD2 in the Mayo Clinic, along with MLPA using commercial MRC-Holland Kits P351 and P352 [13]. Variant interpretation was performed using an established in-house protocol [9]. Blinded whole-genome sequencing was then performed on these 30 samples at the Garvan Institute. Concurrently, 12 patients were initially sequenced via whole-genome sequencing and then subsequent blinded LR-PCR, Sanger sequencing and MLPA of PKD1 and PKD2 (Fig. 1).
Fig. 1. Flowchart of validation cohort analysis.

Flowchart of pathway for assessment of the samples in the validation cohort. VQSR = Variant Quality Score Recalibration, MLPA = Multiplex Ligation-dependent Probe Amplification.
150-base-pair paired-end whole-genome sequencing was performed on the HiSeqX sequencing system (Illumina) after PCR-free library preparation (KAPA Hyper PCR-free kit, Roche). Raw sequencing data was aligned across the genome (hs37d5) and variant-calling performed for single nucleotide variants via a customized bioinformatics pipeline [16]. Variant-calling was optimized for detection of germline sequence variants (ref:alt limit 70:30). Seave, was used for filtering of sequence variants [19]. Variants interpretation was performed according to American College of Medical Genetics (ACMG) Guidelines and targeted to PKD1 (NM_001009944.2) and PKD2 (NM_000297.3) [20]. CNV and structural variant analysis was performed using ClinSV, an application customized to 150 bp paired-end whole-genome sequencing data (Minoche et al, in press). ClinSV combines the output of the Structural Variant caller Lumpy and CNV caller CNVator, both of which have been extensively benchmarked [21–23]. For the secondary sequencing of all samples, the reciprocal laboratory was blinded to the initial sequencing results, phenotype and family history. Reported variants have been submitted to the PKD Variant Database [24] and ClinVar (Submission ID:SUB7930405) (https://www.ncbi.nlm.nih.gov/clinvar/).
Diagnostic cohort
Referrals for diagnostic, clinically-accredited genome sequencing were accepted from Clinical Geneticists and Nephrologists across Australia by the Genome.One diagnostic laboratory (located at The Garvan Institute of Medical Research, Australia). All consecutive referrals from test implementation (May 2017) to September 2019 were included. No inclusion or exclusion criteria was applied to this cohort. The diagnostic laboratory was clinically accredited to ISO15189 by the National Association of Testing Authorities, with scope of accreditation including genomic testing for ADPKD.
Sample preparation, sequencing and bioinformatics analysis was as outlined above, with variant interpretation restricted to a virtual ‘panel’ of genes associated with a PKD phenotype. Panel curation was by a multidisciplinary-team of nephrologists, clinical geneticists, genetic counsellors, genetic pathologists, and laboratory scientists. Patient consent was obtained only for analysis of the coding regions of the genes included in the ‘panel’. The initial ‘panel’ included the genes PKD1; PKD2; GANAB; HNF1B; TSC1; TSC2; OFD1; UMOD; PKHD1. The ‘panel’ was reviewed in December 2018 and DNAJB11, DZIP1L, SEC63 and PRKCSH added. Variants were manually curated by a variant analyst and classified according to ACMG criteria. For this study (but not for clinical reporting), variants of uncertain significance were sub-classified as ‘favor pathogenic’ [25] if novel or previously reported as disease-causing with limited evidence and predicted pathogenic by in silico tools. All reportable results were confirmed by targeted LR-PCR and Sanger sequencing or MLPA. A Genetic Pathologist reviewed and authorized the clinical report. Reported variants have been submitted to Clinvar (Submission ID: SUB7645810) [https://www.ncbi.nlm.nih.gov/clinvar/].
The test-order process requested clinicians report reason for referral, Chronic Kidney Disease (CKD) stage [26], longitudinal kidney length, presence of renal, liver and pancreatic cysts and number, diabetes, cerebral aneurysm, ethnicity and family history. Aside from age, sex and identifiers, there were no mandatory reporting requirements. Enlarged kidney length was defined as bilateral length ≥14.5 cm, >90th percentile for children or, if length not reported, where the clinician described the kidneys as enlarged [27]. Positive family history was defined as a first-degree relative with PKD.
This study was approved as a laboratory audit. As such, clinical data was restricted to that available in the test-order form. Approval was not granted to retrospectively contact referrers for additional phenotype information purely for audit purposes. For details of Statistical Analysis, see Supplementary Materials.
Results
Validation cohort
Blinded validation was performed on 42 unrelated ADPKD patients (patient characteristics—Supplementary Table S1). On initial analysis by genome sequencing of 30 samples from the Mayo Clinic, the disease-causing variant was identified in 24/30 (Fig. 1 and Supplementary Table S1). After adjusting the variant filtering stringency (while blinded), this improved to 28/30. Alteration was made to the interpretation of data from the Variant Quality Score Recalibration (VQSR) tool. VQSR annotates each variant with a likelihood of being a true variant vs being false (sequencing error). The data for this annotation is derived from a trained Gaussian mixture model in which the tool ‘learns’ the characteristic annotations of a true variant using a specified callset of high-quality variants (training set) and compares these calls to those in the callset of interest (patient data). Multiple parameters are considered, including read mapping quality (MQRankSum). MQRankSum is used to highlight variants that have markedly different mapping quality between reads that carry the reference or alternate alleles. This discrepancy is generally an indicator of sequencing error, though can also happen in regions of the genome with high sequence homology. On initial analysis, 4/30 samples had variants in PKD1 annotated by VQSR as failing due to their MQRankSum scores. As PKD1 analysis is affected by pseudogene homology, we reduced the stringency of this filter, resulting in the four variants passing the filter. Importantly, on altering this parameter across the cohort, no false-positive variants were identified and is our new default-setting for classifying PKD1 variants.
Overall, on blinded validation, the two sequencing techniques identified the same result in 40/42 patients, with a range of mutation types identified, including defining the specific boundaries of four multi-exon deletions, which had not been possible with previous methods (Figs. 1 and 2). Of the 40 concordant samples, three remained without genetic diagnosis after both methods.
Fig. 2. Variants identified in PKD1 and PKD2 across the validation and diagnostic cohorts.

A Variants identified across PKD1 in the Validation and Diagnostic cohorts, including three large deletions depicted in green arrows. B PKD2 variants identified in the Validation and Diagnostic cohorts, including a whole gene deletion. Figure developed using ProteinPaint [37].
In 2/42 patients, whole-genome sequencing did not detect mosaic variants using standard filters. In one patient (H12), visualization of the sequencing reads from the whole-genome sequencing data, after un-blinding, demonstrated the variant in 7.5% (5/67) of reads (Supplementary Fig. S1). This alternate variant allele frequency was below the threshold of detection of the whole-genome sequencing bioinformatics pathway, which was optimized for detection of germline sequence variants. In the other patient (H22), a mosaic multi-exon deletion had been detected by MLPA. There was no evidence of this deletion in genome sequencing CNV data. After exclusion of these two mosaic cases, which the genome sequencing pipeline at current depth is not designed to identify, the sensitivity and specificity of clinical whole-genome sequencing for detection of germline disease-causing variants in PKD1 and PKD2 was 100%, with a positive predictive value of 100%. For additional validation and coverage data, see Supplementary Materials, Supplementary Table S2 and Supplementary Fig. S3.
Diagnostic cohort
Following validation, which satisfied CLIA-equivalent National pathology regulatory requirements, the first 144 patients referred for diagnostic clinical whole-genome sequencing were analyzed. The cohort consisted of 71 males and 73 females. Median age at referral was 39 years (0–79 years) (Table 1). There were 12 pediatric patients (<18years old). Fifty-two percent (75/144) of patients were referred for genomic testing to clarify diagnostic uncertainty given an atypical PKD phenotype. The remaining 69 patients (48%) were referred to confirm a typical clinical diagnosis of ADPKD, including for reproductive planning or to select living-related kidney donors.
Table 1.
Clinical features and overall results of diagnostic cohort.
| Clinical feature | Diagnostic cohort n = 144 |
Typical n = 69 |
Atypical n = 75 |
P value |
|---|---|---|---|---|
| Median age at referral (yr) | 39 (0–79) | 37 (15–75) | 43 (0–79) | 0.63 |
| M:F | 71:73 | 32:37 | 39:36 | 0.50 |
| Family history of PKDc | 64d (45%) | 45 (67%) | 19 (26%) | <0.0001 |
| Median CKD stage | 1b | 1 | 2 | 0.35 |
| Enlarged kidney length (≥14.5 cm bilaterally) | 34a | 23 | 11 | 0.003 |
| Results: | ||||
| Overall reportable results | 101 (70%) | 56 (81%) | 45 (60%) | |
| Pathogenic | 40 (28%) | 23 (33%) | 17 (23%) | |
| Likely pathogenic | 29 (20%) | 22 (32%) | 7 (9%) | |
| Uncertain significance | 32 (22%) | 11 (16%) | 21 (28%) | |
PKD polycystic kidney disease.
aNot reported in 36 (25%) patients.
bNot reported in 32 (22%) patients.
cFirst-degree relative with PKD.
dBiological family history unknown to three patients.
Typical and atypical cases were distinguished based on the clinician’s reason for referral, with patients categorized with typical disease if referred to clarify a typical clinical diagnosis of ADPKD [3]. Conversely, patients categorized with atypical disease were referred because the clinician was uncertain of the diagnosis based on imaging and clinical features alone. This information was requested from the clinician as the ‘reason for referral’.
As patients were referred for diagnostic testing, the laboratory did not enforce mandatory submission of phenotypic features. ‘Reason for referral’ and family history were universally reported. All patients had multiple kidney cysts. CKD stage was reported in 77% and kidney length in 75% (Table 1). Additional clinical features are detailed in Supplementary Table S4.
Seventy percent (101/144) of patients had a clinically reportable result (Fig. 3A; Supplementary Table S3). Only variants classified as ‘Pathogenic’, ‘Likely Pathogenic’ or ‘Variant of Uncertain Significance’ (VUS) were reported to clinicians.
Fig. 3. Overall Results in the Diagnostic Cohort.

A Overall results across all patients in the diagnostic cohort; B Overall results stratified by the clinical features of the diagnostic cohort (typical vs atypical disease); C Number of reportable variants identified per gene across the PKD-gene panel in the diagnostic cohort; D Number of reportable variants identified per gene across the PKD-gene panel in the diagnostic cohort, stratified by clinical features (typical vs atypical disease).
When data were stratified by reason for referral, 81% (56/69) of patients referred with typical ADPKD and 60% (45/75) referred with atypical disease had a reportable result (Fig. 3C). Of the patients with typical ADPKD and a reportable result, all but one had PKD1 or PKD2 variants, with the other having a PKHD1 variant (Fig. 3D, Fig. 4). The patients with atypical disease had variants reported in eight different genes, though the majority (55%) had PKD1 or PKD2 variants (Fig. 3D, Fig. 4). Five patients had variants in newly described PKD-genes, GANAB and DNAJB11. All three DNAJB11-patients were referred with atypical PKD and a parent with ESRF. There were also five patients with atypical disease and pathogenic HNF1B-variants, with mild-to-moderate renal impairment (CKD stages 1–3)(Supplementary Tables S3 and S4).
Fig. 4. Diagnostic Cohort Results by Clinical Features and Gene.
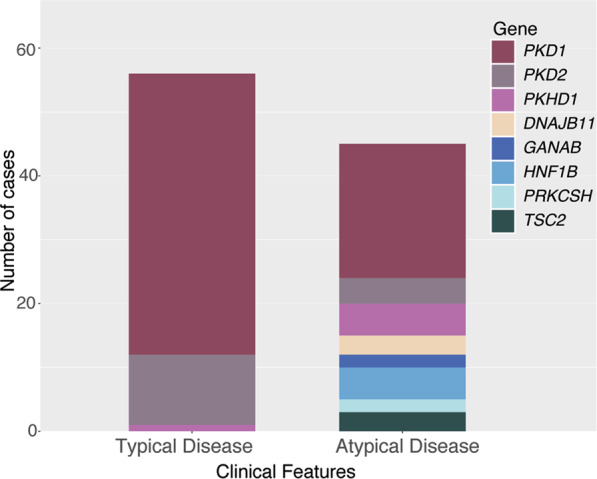
Overall results from diagnostic cohort stratified by clinical features (typical vs atypical) and the gene in which the reportable variant was identified.
Of the 13 patients in the typical group with negative results, only one had a family history of PKD. These apparent de novo patients suggest the possibility of germline mosaicism. Patients with positive family history (p = < 0.01) or enlarged kidneys (p = 0.03) were more likely to have a reportable result. In the atypical disease sub-group, neither family history (p = 0.43) or enlarged kidney size (p = 0.11) increased the likelihood of a reportable result (Table 1).
Of the reported results, 40% (40/101) were pathogenic variants, 29% (29/101) likely pathogenic and 32% (32/101) VUS (Fig. 3A). 59% (19/32) of the uncertain variants favored pathogenicity (Supplementary Table S4). Patients with atypical phenotype were more likely to have a VUS (p = 0.004). All different mutation types were identified, including four large deletions (three whole-gene HNF1B deletions and a multi-exon PKD2 deletion). (Fig. 2; Supplementary Fig. S2). Reportable variants were identified in eight of the 13 genes on the panel (Fig. 3B). All reportable variants were confirmed by Sanger sequencing or MLPA, without false-positive results.
Discussion
We report the results of whole-genome sequencing in 186 patients with PKD and show that clinical whole-genome sequencing provides the basis for a new diagnostic test for PKD. Whole-genome sequencing is able to overcome pseudogene homology and identify all types of variants in the PKD1 gene. The technique also has the sensitivity and specificity to meet specifications for a diagnostic test. Importantly, it enables the analysis of multiple genes associated with PKD, along with CNV detection.
Clinical whole-genome sequencing identified a clinically reportable result in 70% of an unselected diagnostic PKD cohort, with variants identified in eight different PKD-related genes. When results are stratified by reason for referral, 60% of patients with atypical clinical features (i.e., patients in which the referring clinician was uncertain as to the diagnosis) had a clinically reportable result, demonstrating that this is an important group of patients in whom genomic testing has utility. This atypical group reflects patients that are challenging to diagnose in clinical practice and are a group in whom there is limited current literature.
PKD1 was the most common cause of disease in both the typical and atypical groups. In patients with atypical clinical features, making a diagnosis of PKD1-related ADPKD has important prognostic and management implications, given PKD1 is associated with increased likelihood of ESRF [28]. Alternatively, GANAB-mediated disease has not been reported causing ESRF, therefore confirming this diagnosis could provide reassuring information [6, 28]. HNF1B-mediated disease, which was identified in five atypical patients, has associated clinical features (including diabetes and reproductive tract malformations) that are important to screen for [8]. Importantly the atypical patients did not have a clinical feature (such as family history or enlarged kidney size) that predicted the likelihood of identifying a genetic result, highlighting that these diagnoses could not have been distinguished using clinical and imaging features alone. This demonstrates that genetic investigation is a valuable tool in patients with atypical PKD, to clarify their diagnosis.
Eighty-one percent of patients referred with typical ADPKD features had a clinically reportable result and all but one of these patients had PKD1 or PKD2 variants. This is similar to the 85–90% diagnostic rate published in carefully pre-selected research ADPKD cohorts [4, 16, 17]. Although these patients had a clinical diagnosis, genetic testing was sought to clarify living-related kidney donor selection, screen young-adult family members and for reproductive planning. As guidelines suggest, though genetic counselling should be offered to all families with ADPKD, genetic testing is not indicated in all patients with typical ADPKD [29]. However, for families who would utilize genetic information for reproductive planning or family screening, validated, diagnostic-testing methods are required [29]. Genetic diagnosis can also assist in predicting prognosis and in selecting patients most likely to benefit from therapies, which is increasingly relevant with the availability of tolvaptan treatment [10, 11]. Future treatments may be most valuable in patients yet to manifest clinical disease, thus genetic diagnostics may be required to identify at-risk patients prior to them developing macroscopic cysts [15, 30, 31].
This is the first report using whole-genome sequencing with a virtual panel-based approach to diagnose ADPKD in a clinical setting. Whole-genome sequencing is being increasingly utilized in clinical care across the disease spectrum—the importance of validating whole-genome sequencing in PKD is highlighted by the National Health Service England planning to phasein the technique for PKD1 diagnostic sequencing [32]. Other commonly used techniques, such as whole exome-sequencing, cannot be used for ADPKD diagnosis because of pseudogene homology to PKD1 [33]. In our study, PKD1 was the most common gene implicated, reiterating the importance of PKD1 being accurately sequenced in any PKD-focused diagnostic test. There are other sequencing approaches tested for PKD sequencing. Bullich et al. utilized custom-designed capture probes targeted to a cystic disease panel with a diagnostic rate of 88% in clinically-suspected ADPKD [34]. A significant limitation of this technique is that adding new genes to the panel requires redesigning and revalidating the entire custom library-preparation process. In contrast, whole-genome sequencing allows analysis of genes with phenotypic overlap with ADPKD and addition of new genes as understanding evolves [6–8] (Table 2). The value of rapid panel re-curation is highlighted by adding DNAJB11 to our diagnostic panel and the patients subsequently diagnosed.
Table 2.
Overview of different sequencing techniques for polycystic kidney disease diagnosis.
| Long-range PCR, Sanger sequencing and MLPA of PKD1 and PKD2 | Custom capture next-generation sequencing of PKD gene-panel | Whole genome sequencing with analysis targeted to PKD gene-panel |
|---|---|---|
| Requires extensive laboratory preparation specific to PKD testing | Requires laboratory preparation specific to PKD testing | Able to ‘batch’ specimens with other disease-groups undergoing the same sequencing pathway |
| Specific to only targeted genes | Specific to only targeted genes | Analysis can be expanded to entire genome, with consent |
| Technical expertize required for long-range PCR and potential for amplification bias | Custom capture probes require redesigning when gene-panel requires updating | Flexibility to vary gene panel based on patient phenotype |
| Requires MLPA for detecting copy number variants and challenging to detect structural variants | Less robust for detecting copy number variants and challenging to detect structural variants | Able to detect both copy number and structural variants within same diagnostic test |
| Limited data storage challenges | Reduced data quantity reduces analysis and storage costs | Large data quantity increases analysis and storage costs |
| Able to detect some mosaicism of single nucleotide variants | Improved detection of low-frequency mosaic single nucleotide variants | Reduced coverage makes detection of mosaicism less robust |
MLPA Multiplex Ligation-dependent Probe Amplification.
The use of a targeted virtual panel (rather than analysis of the entire genome) reduces variant analysis burden and cost, and dramatically reduces possibility of incidental findings. The panel was specifically targeted to ADPKD-like genes because, for patients with a macrocystic kidney disease phenotype (as opposed to microcysts in nephronophthisis), the yield in other ciliopathy genes is low [34]. For a diagnostic laboratory in the Australian-population context, this whole-genome sequencing methodology is cost-effective, compared with other sequencing techniques for ADPKD. This is based on being able to ‘batch’ PKD samples with other disease groups also undergoing the same whole-genome sequencing pathway. A known challenge of whole-genome sequencing is the increased analysis and storage costs. For our laboratory, this is offset by avoiding a separate laboratory technique purely for sequencing PKD1. For a larger population-base, other techniques may also be cost-effective. However, whole-genome sequencing offers the most seamless method to add new genes and, if patients remain undiagnosed after diagnostic analysis, allows transition to broader research analysis, after appropriate research consent. The whole-genome sequencing PKD diagnostic panel was offered for ~$1500USD, through the diagnostic laboratory. Given the limited literature on clinically-accredited (CLIA-equivalent), validated diagnostic methods for PKD and the international differences between health systems, it is difficult to compare this cost. Detailed health-economic analysis is required for true cost-analysis, which is outside the scope of this diagnostic laboratory dataset.
In this study, clinically reportable results included variants reported as pathogenic, likely pathogenic or uncertain by ACMG criteria [20]. Uncertain variants were included as they have potential clinical relevance and require consideration for family studies, future re-analysis or functional analysis [25]. 32% of the reportable results were classified of uncertain significance, with 59% favoring pathogenicity. These uncertain variants highlight the challenges of variant-classification in ADPKD. Most ADPKD families have novel variants unique to their family, which precludes classifying variants using previous reports [17, 20]. In addition, in adult-onset disease, segregation analysis is difficult as older family members are often deceased or uncontactable [35]. Interestingly, VUSs were more likely in patients with atypical clinical features. This highlights the variant-interpretation challenges in these patients, but also suggests patients with atypical disease may have ‘atypical’ or previously unreported variants contributing to disease. Study of large population datasets suggests ADPKD may be more prevalent, with more variable penetrance—patients with atypical clinical features may be part of this wider disease-spectrum [1, 9]. These uncertain variants require further study to clarify pathogenicity.
A limitation of this whole-genome sequencing technique is in detecting mosaic variants. While altering the bioinformatics pipeline could enable detection of low allele-frequency variants, further validation is required before diagnostic application. An important distinction of this study, as compared with most ADPKD literature, is the use of an unselected diagnostic cohort, rather than a pre-selected research cohort. The diagnostic cohort allows analysis of patients that are challenging in clinical practice, rather than only those that meet pre-defined study criteria. However, the diagnostic consent did not allow retrospective collection of additional phenotype data or further analysis of genomic data. An important finding of this study was the need to adjust the genome sequencing variant-filtering stringency to avoid false-negative results. This highlights the importance of validating any new sequencing methodology against a reference cohort prior to diagnostic application. Complex analytics are applied in interpreting raw MPS data with a multi-faceted filtering process. This filtration process has the potential to discard true disease-causing variants. Previous groups have shown that if different variant filtration pathways are applied to the same MPS dataset, different true variants are identified by the two approaches, demonstrating the need for caution when assessing the true sensitivity of any MPS-based diagnostic test [36].
ADPKD is a common monogenic disease that has challenged genetic diagnostics due to pseudogene homology and recently identified phenocopy genes [6, 7, 16]. Whole-genome sequencing is rapidly being integrated into clinical care and we demonstrate its validity as a diagnostic test for PKD, including over homologous regions. We also demonstrate the utility of genomic testing in making a diagnosis in patients with atypical cystic kidney disease phenotypes, in which clinical and imaging features alone were not sufficient to clarify diagnosis. Clinicians should consider diagnostic genomics in the assessment of patients with PKD, particularly in atypical disease.
Supplementary information
Acknowledgements
This work was supported by the PKD Foundation of Australia, PKD Foundation USA and The Lewis Foundation. ACM was supported by the RACP Jacquot Foundation. The Mayo Translational PKD Center (DK090728) is thanked for Sanger sequencing and exchange of samples and Sravanthi Lavu is thanked for providing data on the validation cohort.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41431-020-00796-4) contains supplementary material, which is available to authorized users.
References
- 1.Lanktree M, Haghighi A, Guiard E, Harris PC, Paterson AD, Pei Y. Prevalence Estimates of Polycystic Kidney and Liver Disease by Population Sequencing. J Am Soc Nephrol. 2018;29:2593–600. doi: 10.1681/ASN.2018050493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cornec-Le Gall E, Torres VE, Harris PC. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J Am Soc Nephrol. 2018;29:13–23. doi: 10.1681/ASN.2017050483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, et al. Unified Criteria for Ultrasonographic Diagnosis of ADPKD. J Am Soc Nephrol. 2009;20:205–12. doi: 10.1681/ASN.2008050507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iliuta I-A, Kalatharan V, Wang K, Cornec-Le Gall E, Conklin J, Pourafkari M, et al. Polycystic Kidney Disease without an Apparent Family History. J Am Soc Nephrol. 2017;28:2768–76. doi: 10.1681/ASN.2016090938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pei Y, Hwang YH, Conklin J, Sundsbak JL, Heyer CM, Chan W, et al. Imaging-Based Diagnosis of Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol. 2015;26:746–53. doi: 10.1681/ASN.2014030297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porath B, Gainullin VG, Gall EC-L, Dillinger EK, Heyer CM, Hopp K, et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am J Hum Genet. 2016;98:1193–207. doi: 10.1016/j.ajhg.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, Smith JM, et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am J Hum Genet. 2018;102:832–44. doi: 10.1016/j.ajhg.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B-associated renal and extra-renal disease—an expanding clinical spectrum. Nat Rev Nephrol. 2014;11:102–12. doi: 10.1038/nrneph.2014.232. [DOI] [PubMed] [Google Scholar]
- 9.Mallawaarachchi AC, Furlong TJ, Shine J, Harris PC, Cowley MJ. Population data improves variant interpretation in autosomal dominant polycystic kidney disease. Genet Med. 2019;21:1425–34. [DOI] [PMC free article] [PubMed]
- 10.Gansevoort RT, Arici M, Benzing T, Birn H, Capasso G, Covic A, et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA Working Groups on Inherited Kidney Disorders and European Renal Best Practice. Nephrol Dial Transpl. 2016;31:337–48. doi: 10.1093/ndt/gfv456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cornec-Le Gall E, Audrezet MP, Rousseau A, Hourmant M, Renaudineau E, Charasse C, et al. The PROPKD Score: a New Algorithm to Predict Renal Survival in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol. 2016;27:942–51. doi: 10.1681/ASN.2015010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanktree MB, Iliuta IA, Haghighi A, Song X, Pei Y. Evolving role of genetic testing for the clinical management of autosomal dominant polycystic kidney disease. Nephrol Dial Transpl. 2019;34:1453–60. doi: 10.1093/ndt/gfy261. [DOI] [PubMed] [Google Scholar]
- 13.Rossetti S, Hopp K, Sikkink RA, Sundsbak JL, Lee YK, Kubly V, et al. Identification of Gene Mutations in Autosomal Dominant Polycystic Kidney Disease through Targeted Resequencing. J Am Soc Nephrol. 2012;23:915–33. doi: 10.1681/ASN.2011101032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bogdanova N, Markoff A, Gerke V, McCluskey M, Horst J, Dworniczak B. Homologues to the First Gene for Autosomal Dominant Polycystic Kidney Disease Are Pseudogenes. Genomics. 2001;74:333–41. doi: 10.1006/geno.2001.6568. [DOI] [PubMed] [Google Scholar]
- 15.Ong ACM, Devuyst O, Knebelmann B, Walz G. Autosomal dominant polycystic kidney disease:the changing face of clinical management. Lancet. 2015;385:1993–2002. doi: 10.1016/S0140-6736(15)60907-2. [DOI] [PubMed] [Google Scholar]
- 16.Mallawaarachchi AC, Hort Y, Cowley MJ, McCabe MJ, Minoche A, Dinger ME, et al. Whole-genome sequencing overcomes pseudogene homology to diagnose autosomal dominant polycystic kidney disease. Eur J Hum Genet. 2016;24:1584–90. doi: 10.1038/ejhg.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Audrézet M-P, Cornec-Le Gall E, Chen J-M, Redon S, Quere I, Creff J, et al. Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat. 2012;33:1239–50. doi: 10.1002/humu.22103. [DOI] [PubMed] [Google Scholar]
- 18.Trujillano D, Bullich G, Ossowski S, Ballarín J, Torra R, Estivill X, et al. Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1and PKD2 targeted next-generation sequencing. Mol Genet Genom Med. 2014;2:412–21. doi: 10.1002/mgg3.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gayevskiy V, Roscioli T, Dinger ME, Cowley MJ. Seave: a comprehensive web platform for storing and interrogating human genomic variation. Bioinformatics. 2019;35:122–5. doi: 10.1093/bioinformatics/bty540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:1–19. doi: 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–84. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trost B, Walker S, Wang Z, Thiruvahindrapuram B, MacDonald JR, Sung WWL, et al. A Comprehensive Workflow for Read Depth-Based Identification of Copy-Number Variation from Whole-Genome Sequence Data. Am J Hum Genet. 2018;102:142–55. doi: 10.1016/j.ajhg.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gout AM, Martin NC, Brown AF, Ravine D. PKDB: Polycystic Kidney Disease Mutation Database-a gene variant database for autosomal dominant polycystic kidney disease. Hum Mutat. 2007;28:654–9. doi: 10.1002/humu.20474. [DOI] [PubMed] [Google Scholar]
- 25.McLaughlin HM, Ceyhan-Birsoy O, Christensen K, Kohane I, Green RC, Rehm HL, et al. A systematic approach to the reporting of medically relevant findings from whole genome sequencing. BMC Med Genet. 2014;15:134–48. doi: 10.1186/s12881-014-0134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int Supplements. 2013: 1–163.
- 27.Irazabal MV, Rangel LJ, Bergstralh EJ, Osborn SL, Harmon AJ, Sundsbak JL, et al. Imaging Classification of Autosomal Dominant Polycystic Kidney Disease: a Simple Model for Selecting Patients for Clinical Trials. J Am Soc Nephrol. 2015;26:160–72. doi: 10.1681/ASN.2013101138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cornec-Le Gall E, Audrezet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, et al. Type of PKD1 Mutation Influences Renal Outcome in ADPKD. J Am Soc Nephrol. 2013;24:1006–13. doi: 10.1681/ASN.2012070650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rangan GK, Alexander SI, Campbell KL, Dexter MA, Lee VW, Lopez-Vargas P, et al. KHA-CARI guideline recommendations for the diagnosis and management of autosomal dominant polycystic kidney disease. Nephrology. 2016;21:705–16. doi: 10.1111/nep.12658. [DOI] [PubMed] [Google Scholar]
- 30.De Rechter S, Breysem L, Mekahli D. Is Autosomal Dominant Polycystic Kidney Disease Becoming a Pediatric Disorder? Front Pediatrics. 2017;5:332. doi: 10.3389/fped.2017.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gimpel C, Bergmann C, Bockenhauer D, Breysem L, Cadnapaphornchai MA, Cetiner M, et al. International consensus statement on the diagnosis and management of autosomal dominant polycystic kidney disease in children and young people. Nat Rev Nephrol. 2019;15:713–26. doi: 10.1038/s41581-019-0155-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.National Genomic Test Directory for rare and inherited diseasehttps://www.england.nhs.uk/publication/national-genomic-test-directories/. (Accessed: 2nd February 2020)
- 33.Ali H, Al-Mulla F, Hussain N, Naim M, Asbeutah AM, AlSahow A, et al. PKD1 Duplicated regions limit clinical Utility of Whole Exome Sequencing for Genetic Diagnosis of Autosomal Dominant Polycystic Kidney Disease. Sci Rep. 2019;9:4141. doi: 10.1038/s41598-019-40761-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bullich G, Domingo-Gallego A, Vargas I, Ruiz P, Lorente-Grandoso L, Furlano M, et al. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 2018;94:363–71. doi: 10.1016/j.kint.2018.02.027. [DOI] [PubMed] [Google Scholar]
- 35.Bayrak-Toydemir P, McDonald J, Mao R, Phansalkar A, Gedge F, Robles J, et al. Likelihood ratios to assess genetic evidence for clinical significance of uncertain variants: hereditary hemorrhagic telangiectasia as a model. Exp Mol Pathol. 2008;85:45–9. doi: 10.1016/j.yexmp.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 36.Miller NA, Farrow EG, Gibson M, Willig LK, Twist G, Yoo B, et al. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med. 2015;7:100–16. doi: 10.1186/s13073-015-0221-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou X, Edmonson MN, Wilkinson MR, Patel A, Gang W, Liu Y, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet. 2016;48:4–6. doi: 10.1038/ng.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.