

Abstract
Aging is a complex physiological process associated with degenerative disorder of metabolism and immune function, which contributes to the occurrence of senile diseases. The gut microbiota affects systemic inflammation in aging processes probably through metabolism, but their relationship is still unclear. In this study, 16S-rRNA-sequencing technology, gas chromatography-time-of-flight mass spectrometry (GC-TOFMS)–based metabolic profiling, and immune factor analysis combined with advanced differential and association analysis were employed to investigate the correlation between the microbiome, metabolome, and immune factors in male Wistar rats across lifespan. Our findings showed significant changes in the ileum microbiome and serum metabolome compositions across aging process. A two-level strategy was applied to demonstrate that key metabolites associated with age such as 4-hydroxyproline, proline, and lysine were clustered together and positively correlated with beneficial microbes including Bifidobacterium, Lactobacillus, and Akkermansia. Function analysis explored association between serum metabolite class and specific gut bacteria’s metabolism pathways. Further correlation analysis on all the alteration patterns provided an interaction network of main immune factors such as IL-10, IgA, IgM, and IgG with key gut bacteria and serum metabolites. This study offers new insights into the relationship between immune factors, serum metabolome, and the gut microbiome.
Electronic supplementary material
The online version of this article (10.1007/s11357-020-00188-y) contains supplementary material, which is available to authorized users.
Keywords: Aging, Serum metabolites, Gut microbiota, Immune factor, Cytokines
Introduction
Aging is a physiological process involving complex interdependent network systems and contributes to the onset and progression of senile diseases such as cardiovascular disease, Alzheimer’s disease (AD), arthritis, and diabetes (Butler et al. 2008; Wijsman et al. 2011). Age-related decrease in gut microbiota diversity has been reported among the elderly, and this has been linked to an increase in the risk of different diseases. Bifidobacterium spp. and Lactobacillus spp. are more abundant during the early stages of life and decrease in old age (Hor et al. 2019; Singh et al. 2019). Gut flora plays a critical role of imbalance in senescence and premature death of progeria animals, and the dysbacteriosis could be significantly improved by transplantation of normal fecal bacteria or supplementation with Akk bacteria. Although the mechanisms for these changes have not been clarified, metabolic regulation may act a pivotal part in senescence regulation by intestinal flora (Bárcena et al. 2019).
From the first day of life, the gut microbiota functions in metabolism and supports growth, development, maturity, and aging (Chen et al. 2019; Korpela et al. 2019; Ticinesi et al. 2019). Gut microbial metabolites are important signaling molecules associated with the occurrence of a number of diseases such as cancer and cardiovascular diseases (Kurilshikov et al. 2019). In 2016, we explored brain metabolome, revealing gut flora–related metabolites which were detected dramatically changing in rat brain with age (Zheng et al. 2016). Subsequently, we proposed a strategy association study of brain metabolome and gut microbiome across the lifespan of rats (Chen et al. 2018). These two studies provide appropriate approaches to data analysis and association of metabolome and microbiome, expanding our understanding of the microbial-metabolite relationship.
Aging is characterized by chronic, low-grade systemic inflammation referred to as inflammaging which contributes to the pathogenesis of age-related diseases (Ferrucci and Fabbri 2018; Sansoni et al. 2008). Changes in immune cytokines have been closely related to the occurrence and development of senile diseases (Castaneda-Delgado et al. 2017), whereas the causes of this low-grade inflammation and its specific regulatory mechanism are still unclear. Recent studies suggest that the gut bacteria may play a crucial role in these age-related inflammations (Buford 2017). However, the linkage between the immune system, gut microbiota, and metabolites throughout the aging process has not been established. Identifying the relationship between the immune system and the gut microbiota can help to promote healthy aging and also provide new approaches to anti-aging.
The current study explored the relationship between immune factors, serum metabolome, and gut microbiota in male Wistar rats at different time points (weeks 1, 3, 7, 9, 12, 56, 111). The study utilized a three-pronged association study coupled with immunologic factor analysis, serum metabolic profiling, and microbiome analysis. The dual-omics data were extracted and clustered into groups with different changing characteristics (Pedersen et al. 2018). Spearman’s correlation analysis and network were used to determine the immune factor-metabolite-bacterium correlation pairs. This provides an understanding of the relationship between immune factors, serum metabolome, and gut microbiome at different aging processes.
Materials and methods
Animal handling and sample collection
All experiments were carried out strictly in accordance with recommendations on the National Institutes of Health’s Guide for Care and Use of Laboratory Animals. The experimental program was approved by the Center for Laboratory Animals of Shanghai Jiao Tong University.
The whole experimental workflow is summarized in Fig. 1. Briefly, male Wistar rats were all born by the seven mother rats purchased from Shanghai Laboratory Animal Co, Ltd. (SLAC, Shanghai, China). Six newborn rats were randomly selected from each litter and randomly assigned to each group. All laboratory rats were fed in a clean environment under the condition of 12-h light/12-h dark cycle at 20–22 °C and 45 ± 5% humidity. Also, all rats were given free sterile chow and water. The diet composition of standard chow is provided in Table S1. A total of 42 male Wistar rats were allotted to 7 groups (weeks 1, 3, 7, 9, 12, 56, and 111 after birth, represented by W1, W3, W7, W9, W12, W56, and W111, n = 6 per group). Blood, whole spleen, and intestinal content samples were collected from each rat at the end of the corresponding time point. Blood sample was extracted from the tail vein and then centrifuged at 4 °C, 3000 rpm for 20 min; the supernatant was transferred to a fresh tube. All samples were immediately stored at − 80 °C pending analysis.
Fig. 1.

Overview of the workflow integrating serum metabolome, gut microbiome, and spleen cytokines in rats among different age groups. After preprocessing raw metabolome and microbiome data, metabolites and microbiota are separately collapsed into co-abundance clusters. After which the clusters are filtered for statistically significant associations with age. Finally, the resulting clusters are taken for cross-domain association study
GC/TOF-MS sample preparation and assessment
Serum metabolomics pretreatment and measurement were performed according to the procedures established in our laboratory (Hou et al. 2015; Zhang et al. 2016). Briefly, 300 μL of a precooled mixed solvent (Vmethanol:Vchloroform = 3:1) was added to 50 μL serum in a 1.5-mL centrifuge tube, followed by vortex oscillation for 30 s. Then, the mixture was arranged in − 20 °C for 10 min and centrifuged at 1000 rpm for 10 min. A total of 300 μL supernatant was transferred into a new vial with L-2-chlorophenylalanine (0.1 mg/mL, 10 μL) as the internal standard. Afterward, the supernatant was dried under vacuum at room temperature and dissolved by 80 μL methoxamine (15 mg/mL in pyridine). After vortex oscillation for 30 s, the sample vial was stored at 30 °C for 90 min. Finally, the sample vial was added with 50 μL of bis(trimethylsilyl)trifluoroacetamide (containing 1% trimethylchlorosilane) and kept at 70 °C for 60 min. The sample vials were kept at room temperature for 1 h before mass spectrometry analysis.
All samples were randomly injected to minimize systematic deviations. Quality control (QC) samples were prepared by pooling all plasma samples and used for all analytical methods. A mixed extraction of QC sample was subjected to assessment every ten sample injections.
Metabolite profiling was acquired using gas chromatography-time-of-flight mass spectrometry (GC/TOF-MS, LECO, USA) platform and preprocessed by Chroma TOF (LECO, USA). Compounds were identified using in-house and online libraries, such as the Human Metabolome Database (HMDB) and the National Institute of Standards and Technology (NIST). The final dataset included compound name, peak area, and retention time. Annotated metabolites with zero values in more than 60% samples were excluded. The mean value of corresponding variables was used to complete the missing value. A total of 82 metabolites were obtained, which were normalized into the total peak abundance of all metabolites in each sample before statistical analysis. They were then divided into 5 metabolite types according to their chemical structure.
Ileum content microbiota assessment
The ileum contents were removed from storage. Microbial DNA was extracted using the QIAamp Stool Mini kit (Qiagen, cat. no. 51504). The V4 hypervariable region of 16SrRNA was chosen as the PCR amplified region. The bacterial forward primer was 5′-AYTGGGYDTAAAGNG-3′ and the reverse primer was 5′-TACNVGGGTATCTAATCC-3′. The PCR conditions were set as follows: one predenaturation cycle at 94 °C for 4 min, 25 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C for 45 s, elongation at 72 °C for 30 s, and one post-elongation cycle at 72 °C for 5 min. The amplified PCR products were separated on 0.8% agarose gels and extracted. The PCR products without primer dimers and contaminant bands were included for sequencing by Illumina Miseq platform.
The original data obtained by sequencing was saved in a paired-end FASTQ format and performed with QIIME 2 (version 2019.4) (Bolyen et al. 2018) with minor modification according to the official tutorials (https://docs.qiime2.org/2019.4/tutorials/). Raw reads were demultiplexed with no error in the index sequence. The DATA2 (Callahan et al. 2016) was used to obtain quality filtered, denoised, chimera-free, and merged amplicon sequence variants (ASVs). Sequences with any ambiguous based were excluded. Taxonomic annotation of ASVs was obtained in the Greengenes database (DeSantis et al. 2006). Alpha diversity metrics and beta diversity metrics were estimated using the diversity plugin. ASVs with richness in less than 0.001% total reads or with zero values more than 80% of samples were removed. In this study, 427 ASVs under 16 phyla were reserved for subsequent analysis. PICRUst2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) together with KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway library were applied to obtain predicted function matrix (Douglas et al. 2019). A total of 127 functions that related to metabolism were kept for analysis.
Measurement of immune factors and cytokines in the spleen
The levels of immune factors (IL-2, IL-4, IL-10), immunoglobulins (IgA, IgG, IgM), and NK cell in spleen samples were measured by the Rat interleukin-2 (IL-2) ELISA Kit, Rat interleukin-4 (IL-4) ELISA Kit, and Rat interleukin-10 (IL-10) ELISA Kit; Rat immunoglobulin A (IgA) ELISA Kit, immunoglobulin G (IgG) ELISA Kit, and immunoglobulin M (IgM) ELISA Kit; and NK cell ELISA Kit (Shanghai Jianglai Biotech, Shanghai, China). All steps followed the manufacturer’s instructions.
Statistical analysis
The data matrix was imported into SIMCA-P 13.0 (Umetrics, Sweden) for principal component analysis (PCA) and partial least squares discriminant analysis (PLS-DA) analyses, which were constructed using the GC-MS data as the X variable and the age as the Y variable. Also, principal coordinate analysis (PCoA) based on unweighted and weighted UniFrac OTU matrix was performed. MetaboAnalyst 3.0, GraphPad Prism 5.0 (6.0, GraphPad, USA), and R program (version 3.5, https://www.r-project.org/) were used for data analysis. Significant differences in variances among all seven groups were identified by the Kruskal-Wallis test as over 85% of the variables did not conform to normal distribution, followed by Dunn’s post hoc test using a Benjamini-Hochberg false discovery rate (FDR) for multiple testing. Data in figures were presented as mean ± SEM. All tests were two-sided, the p values were corrected using FDR, and p < 0.05 was considered statistically significant.
Association analysis
We applied a two-level strategy to determine the associations between the serum metabolome and gut microbiome. For the high-level association study, two dimensionality reduction methods were used: (1) for knowledge-driven, correlations of metabolite types and predicted metabolic function–derived bacterial data were tested; (2) for data-driven, the integration and association study of intestinal microbiome data and serum metabolome data was performed according to the computational protocol with modifications (Pedersen et al. 2018). Firstly, we collapsed co-abundant serum metabolites into metabolite clusters (labeled M01-M12) using the R package WGCNA (weighted gene co-expression network analysis) (Langfelder and Horvath 2008) (Fig. 1). Signed, weighted co-abundance correlation (biweight midcorrelations after log2 transformation) networks were calculated across all metabolites. The resulting cluster eigenvector was presented to the profile of each metabolite cluster. The parameters were set in line with the details of the polar metabolites clustering in the published paper (Pedersen et al. 2018). Secondly, the microbiota data was summarized to microbiota clusters (labeled B01-B10) using the same method of metabolites. Thirdly, in the age-filtering step, age-related clusters were filtered by Spearman’s correlation analysis with p < 0.05 and FDR < 0.1 and the resulting clusters were taken for cross-domain association analyses between metabolomics and microbiome. For the low-level association study, the associations among specified metabolite, bacterium, and cytokines were listed. Spearman’s rank correlation coefficients were calculated using the cor.test function (method = “spearman”) and were plotted with the pheatmap function in the package of R program. The significant correlation pairs were depicted in Cytoscape (3.4.0).
Results
Profiling of serum metabolites in rats by GC/TOF-MS
A total of 82 metabolites were identified by GC/TOF-MS, including 24 amino acids, 15 carbohydrates, 11 fatty acids, 19 organic acids, and 13 other small molecules. Amino acids and carbohydrates were the predominant types of metabolites, which account 62% of all metabolites (Fig. 2a). PCA was performed to characterize the global metabolomics differences among groups. The PCA score plot showed that the profile at 1 week was significantly different from other age groups (Fig. 2b). The profiles of all age groups can be clearly separated by the first and second principal components. Unsupervised analysis of serum groups showed that age was the predominant factor to the metabolic profile of healthy rats. This observation was also confirmed by the PLS-DA model at all age groups (Fig. S1a).


In order to find out the variables that contribute to the separation of serum metabolome, we further evaluated the alterations of aging on specific metabolites. Metabolites that showed significantly differences among 7 time points by the Kruskal-Wallis test were included. Several amino acids including leucine, phenylalanine, valine, and isoleucine increased while 4-hydroxyproline, lysine, and proline decreased with age (Fig. 2c). Interestingly, the levels of several carbohydrates showed a separation at the age of week 9. For example, glycerol, erythritol, and galactose decreased from W1 to W9 and increased from W9 to W111. Gluconate and glucuronate increased from W1 to W9 and decreased from W9 to W111 (Fig. 2d). The levels of tetradecanoate, dodecanoate, decanoate, and docosahexaenoate were extremely high at W1 compared with other age groups. Docosahexaenoate, elaidiate, and linoleate decreased from W1 to W9 and increased from W9 to W111 (Fig. 2e).
Composition of intestinal microbiota in rats by 16S rRNA
Using the 16S rRNA sequencing, a total of 73 genera and 26 species were annotated among the 427 ASVs, which were present in at least 60% of the samples. Alpha diversity metrics and beta diversity metrics were estimated in Fig. S1c. In the phylum level, Firmicutes and Proteobacteria were the most predominant bacterial phyla found in ileum, followed by Bacteroidetes, Actinobacteria, Acidobacteria, and Verrucomicrobia (Fig. 3a). The relative abundance of Firmicutes was almost equal to Proteobacteria at the infancy, and then, Firmicutes increased and Proteobacteria decreased with age. The relative abundance of Firmicutes was positively correlated with age, while other phyla negatively correlated with age, such as Bacteroidetes, Proteobacteria, and Actinobacteria (Fig. 3a).
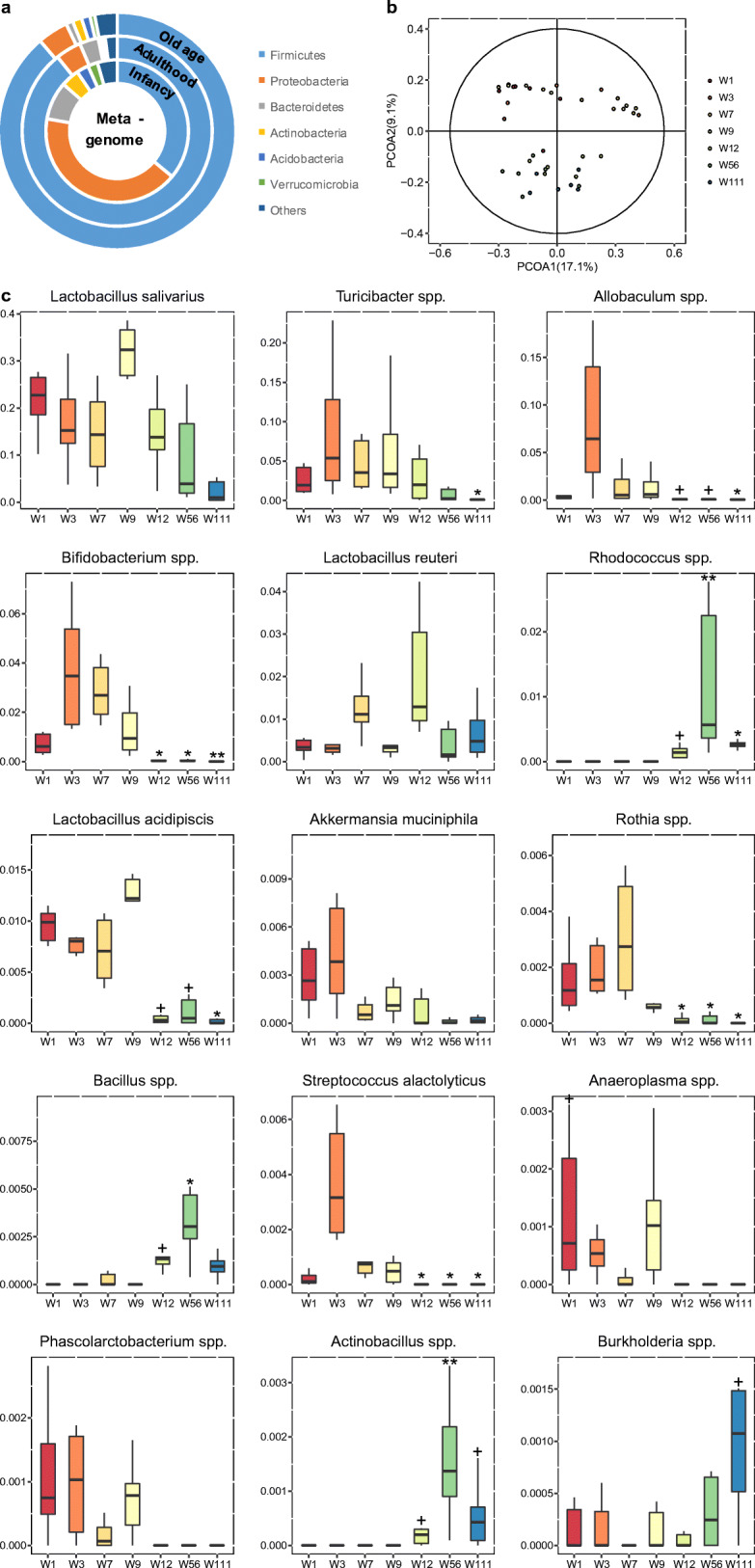
To characterize the global differences in gut microbiota composition among all seven groups, PCoA analysis was performed based on the unweighted and weighted UniFrac distances of the microbiota to visualize the clustering of all rats. From the PCoA plots shown in Fig. 3b, the composition of the ileum microbiota was dramatically changed from W1 to W111. The PCoA plot based on weighted UniFrac was depicted in Fig. S1b.
To explore the detailed gut microbiota composition, we analyzed the relative abundance of the dominant genera and species which were different in all groups (tested by the Kruskal-Wallis test with p value < 0.05). The levels of Rhodococcus spp., Actinobacillus spp., and Bacillus spp. were enriched in the aged rats. The relative abundance of Bifidobacterium spp., Lactobacillus acidipiscis, Akkermansia muciniphila, and Rothia spp. were significantly higher at the development stage (W1 to W7), while decreased from W9 and reached an extremely low level at W56 and W111 (Fig. 3c). Some genus was only higher in adulthood (W7–W12) including Turicibacter spp. and Lactobacillus reuteri.
Correlation analysis of metabolome and microbiome
In the high-level association study, we performed two different strategies for dimensionality reduction. For the knowledge-driven dimensionality reduction, the heatmap in Fig. 4a shows the correlation between metabolite types and gut microbiome functional pathways (details of each category in Table S2). The black-bordered square marks indicate the parts that are significantly related to age, and their change trend with age is shown in Fig. 4b.
Fig. 4.

Correlation analysis results of the knowledge-driven metabolite type-bacterial function across the lifespan. a The Spearman correlation coefficient for metabolite types and metabolic functions of microbiome (+p < 0.05, *p < 0.01). b The scatter plots with correlation coefficients of selected metabolite types and metabolic functions (n = 6)
For the data-driven dimensionality reduction, using the newly reported computational platform (Pedersen et al. 2018), we collapsed metabolites and gut microbiota data into 12 and 10 clusters, respectively, and the every individual metabolite and bacterium that within each cluster was summarized in Table S3 and Table S4. The metabolic and bacterial clusters that most related to age were M01 (r = − 0.870, p = 9.98 × 10−13) and B05 (r = − 0.774, p = 1.84 × 10−08), respectively. The M01 includes three amino acids proline, 4-hydroxyproline, and lysine, which were all decreased with age (Table S3). The B05 includes several probiotics, such as Rothia spp., Bifidobacterium spp., Lactobacillus acidipiscis, and Enterococcus spp., which were significantly negatively correlated with age (Table S4). Then the clusters were taken forward for cross-domain association analyses (Fig. 5). Also, the M01 was positively correlated with B05 (r = 0.651, p = 4.28 × 10−05).
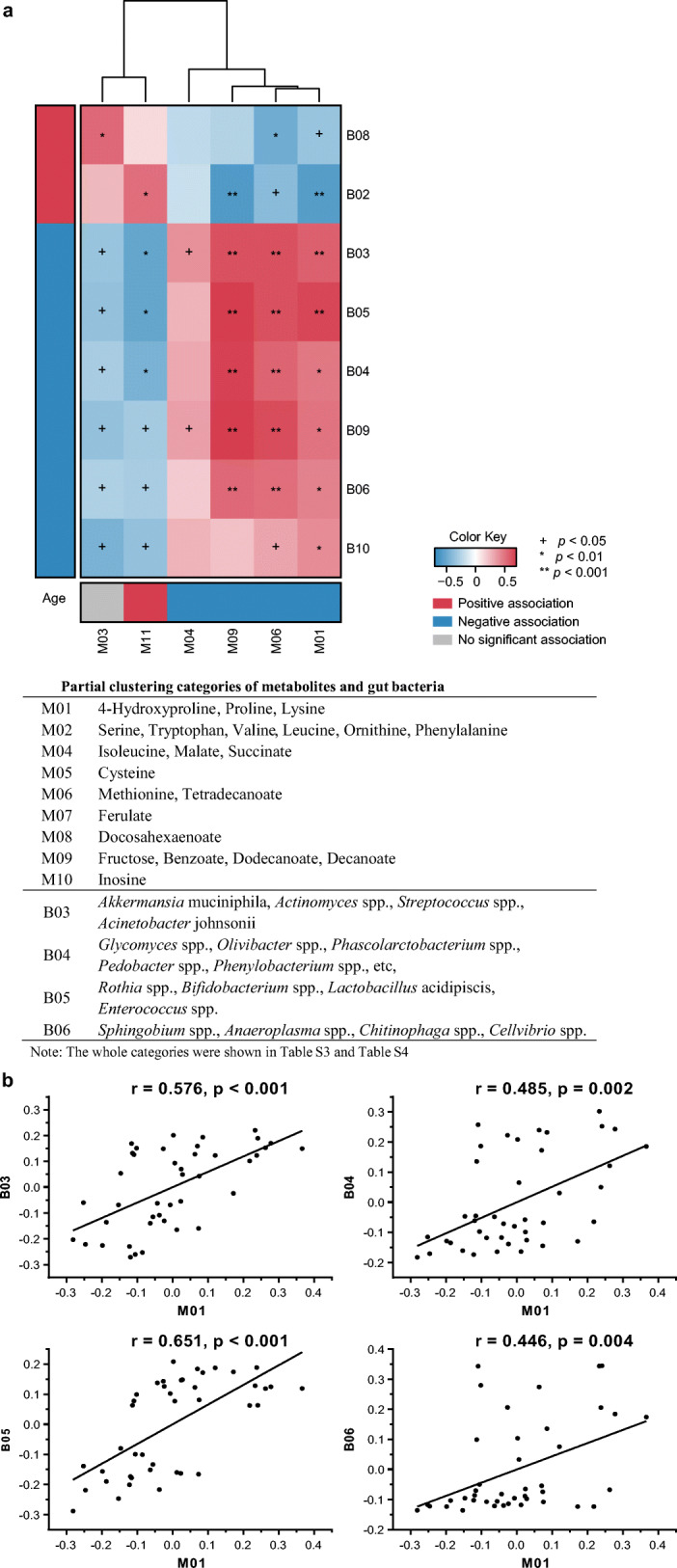
In the low-level association study, the association between individual metabolite and gut microbiota was performed and is shown in Fig. S2. Several associations between metabolites and microbiota were identified at an FDR of 0.05. In this heatmap, a variety of age-related metabolites, including 4-hydroxyproline, tetradecanoate, dodecanoate, decanoate, and benzoate, were significantly correlated with gut genus. Tetradecanoate, dodecanoate, and decanoate were positively correlated with Glycomyces spp., Olivibacter spp., Phascolarctobacterium spp., Pedobacter spp., Phenylobacterium spp., etc. in B04; and Sphingobium spp., Anaeroplasma spp., Chitinophaga spp., and Cellvibrio spp. in B06.
Correlation analysis on metabolites, bacterium, and immune factors
The alterations of immune factors and cytokines among the lifespan are illustrated in Fig. S3; they are represented by “factors” hereby. In addition, cytokines and immune indicator levels did not change a lot with age, except for IL-2 which showed a decreasing trend with age (Fig. S3). At the last time point, a few factors showed significant differences within group, indicating differences in the immune status of the elderly (Fig. S3).
The percentage of two related variable pairs (bacteria-metabolites, immune factor-bacteria, and immune factor-metabolites) is shown in Fig. 6a, showing that the correlations between immune factors and metabolome declined with age. Details of age-specific correlations are listed in Fig. S4, indicating amino acids, fatty acids, and TCA participants showed high correlations with immune factors in childhood period.
Fig. 6.

Correlation analysis results of the low-level dual-omics data. a The percentage line chart of significantly correlated pairs with age. Infancy: W1 + W3; adulthood: W7 + W9 + W12; old age: W56 + W111. b The interactions between meta significant correlated pairs of metabolite-microbiota-factors. Lines in red and blue represent positive and negative correlations, respectively; upwards arrow: positively related to age; downwards arrow: negatively related to age
The associated relationship between immune factors and metabolites, as well as gut microbiota in whole samples (whole ages), was conducted and is displayed in Fig. S2. The complex network in Fig. 6b summarized complicated relationships among the changes of key variables, showing that IL-10, IgM, IgA, 4-hydroxyproline, and tetradecanoate were crucial nodes, including local correlation groups such as tetradecanoate-decanoate-dodecanoate, IgG-cysteine, and 4-hydroxyproline-IgA. In general, amino acids and long-chain fatty acids showed a stronger association with immune factors and cytokines. Succinate, malate, fructose, benzoate, and long-chain fatty acids including tetradecanoate, dodecanoate, and decanoate were positively correlated with IgM, but most of them were negatively correlated with IL-10 (Fig. S2). IgA is proportional to inosine and inversely proportional to 4-hydroxyproline and proline (Figs. 6b and S2).
In the correlation analysis between gut bacteria and factors, we only found that IL-10, IgM, IgA, IgG, and NK were significantly correlated to gut bacteria (Figs. 6 and S2). Specifically, IL-10 was positively related to a variety of bacteria, such as Lactobacillus spp., Lactobacillus reuteri, Bacillus spp., and Flavobacterium succinicans, and negatively related to most of gut genera, especially Cellvibrio spp., Clostridium spp., and Clostridium celatum. IgA in the spleen was positively correlated with Flavobacterium succinicans, Actinobacillus spp., Cupriavidus spp., Rhizobium spp., Rhodococcus spp., and Salinispora spp. and negatively correlated with Rothia spp., Bifidobacterium spp., Enterococcus spp., and Candidatus Arthromitus, indicating that immune factors are highly related to ileal flora.
Discussion
The gut microbiota is associated with the immune system factors which are important contributing factors to age-related degenerative diseases (Biragyn and Ferrucci 2018). Compared with mice, experimental rat gut metagenome showed a higher pairwise overlap with humans than that between mice and humans at the gene level (Pan et al. 2018). In our previous studies, we demonstrated metabolic deviation in different brain regions due to aging (Zheng et al. 2016) and investigated the correlation between intestinal flora and brain metabolites in rats (Chen et al. 2018). Brain metabolism showed significant shifts in different life stages of rats. Similar results were also reported in the current study. Serum metabolite levels variated differently at early ages (W1–W9) compared with those at an older age (W12, W56, and W111) (Fig. 2). These changes at 9 weeks were associated with the rat’s maturation. In infancy, minimal carbohydrate (saccharides and glycols) levels are detected, and then, they reach normal levels in adulthood and remain unchanged or slightly increased in old age (Fig. 2d). However, fatty acids were shown to be at higher levels in the early stages of life (Fig. 2e). Glycerol and fatty acids are significantly higher in the early stages of life compared with older stages, and this has been associated with the breast milk diet (Dessì et al. 2018). Among the three most abundant phyla in the ileum, Firmicutes have been shown to gradually increased with age, while the others showed different trends (Fig. 3a). It can be inferred that the difference in diet is one of the reasons for the differences in gut flora at infancy. In addition, disorders associated with flora disturbance result in the regulation of energy metabolism and the accumulation of excessive fat in old age (Maier et al. 2017).
Serum amino acid level displayed a positive correlation with nucleic acid and lipid metabolism; amino acids had a negative correlation with amino acid and vitamin metabolism (Fig. 4), indicating that gut bacteria might have complementary effect on amino acid production (Dodd et al. 2017).There is a similar relationship between ureas and multiple pathways of gut bacteria, as it was proven that the supplement with prebiotics and probiotics significantly increased uric acid and decreased urea and urea nitrogen levels in blood (Firouzi and Haghighatdoost 2018). This correlation results in Fig. 4 illustrated an overall interaction of circulating metabolites and gut flora’s metabolism function.
Our results showed 4-hydroxyproline having the most significant changes among the metabolites due to the significant decline after aging (Fig. 2c), and also an important node in the correlation network (Fig. 6b). One of the significant characteristics of old age is the loss of collagen, while 4-hydroxyproline is a decomposition product of collagen. Furthermore, the results showed that serum 4-hydroxyproline levels gradually decreased with age. Age-related probiotics in the B05 bacterial group (Fig. 5, details in Fig. S2 and Table S4) including Rothia spp., Bifidobacterium spp., Lactobacillus acidipiscis, and Enterococcus spp. were shown to be positively correlated with 4-hydroxyproline. It has been reported that 4-hydroxyproline metabolic enzymes exist widely in the gut microbiota, which participate its metabolism with the host (Huang et al. 2018). So it could be inferred that the collagen metabolism is partly impacted by aging caused by changes in the microbiota. In the cluster M01 (Fig. 5, details in Fig. S2 and Table S3), proline and lysine showed similar trends with 4-hydroxyproline (Fig. 2c). These two amino acids are also closely related to aging and inflammation (Nepal et al. 2018). While in cluster M11, gluconate and glucuronate decreased in old age. In addition, infants’ serum levels of myo-inositol and citrate were found to be significantly lower than those in adults (Fig. 2d). This indicates that energy metabolism is slowed down by aging.
Cysteine, a precursor of the anti-oxidant glutathione with multiple physiological functions, was found to be significantly declined in both infancy and old age (Fig. 2c). Oxidative stress induces abnormal cysteine oxidation in aging and neurodegenerative diseases and also affects protein function (Gu and Robinson 2016). This confirms the need for cysteine supplementation for infants, old adults, and even people with certain metabolic diseases (McCarty and DiNicolantonio 2015). In this study, cysteine was positively correlated with Bifidobacterium spp., Desulfovibrio spp., and Streptococcus alactolyticus (Fig. 6, details in Fig. S2). Studies have shown that the expressions of cysteine biosynthetic genes are very important for Bifidobacteria growth (Ferrario et al. 2015). A recent study suggested an antagonistic relationship between sulfur-reducing bacteria and Bifidobacterium spp., which compete for cysteine (Malmuthuge et al. 2019). In this study, Bifidobacterium sp. content and cysteine were low during the first week of life. With the increase in abundance of Bifidobacterium spp., cysteine was also increased, and then, they were found to both decrease in old age. Therefore, it could be inferred that supplementation of Bifidobacterium spp. or cysteine may promote each other and help improve metabolic disorders in old age.
The levels of an aromatic amino acid (including phenylalanine, tyrosine, and tryptophan) and branched-chain amino acid (BCAA, including isoleucine, leucine, and valine) were shown to be increased from W9 to W111 (Fig. 2c), whereas higher levels of circulating BCAA were associated with insulin resistance and the development of diabetes and have been reported as predictors of cardiovascular diseases (Chen et al. 2016; Magnusson et al. 2013; Wang et al. 2011). Correlation analysis results revealed that branched-chain amino acids and their metabolites, ornithine and phenylalanine, were found to be positively correlated with IL-10 (Figs. 6b and S2). It was reported that branched-chain amino acid affects immune functions and even the concentration of IL-10 (De Simone et al. 2013). IL-10 is believed to regulate aging-related inflammation by affecting insulin resistance, while maintaining high levels of IL-10 may help to enhance body vitality and resist aging (Dagdeviren et al. 2017; Jankord and JEMIOLO 2004). In this correlation network diagram (Fig. 6b), IL-10 is located at a crucial node of the network, which is associated with multiple metabolites and enteric bacteria. Recent studies have reported that inflammatory factors are regulated by a variety of metabolites, such as methionine and serine (Yu et al. 2019). Methionine was found to be positively correlated with IgG, while ferulate was positively whereas serine was negatively correlated with IgG and NK (Figs. 6 and S2).
Notably, the W1 group had higher abundance of all fatty acids detected in the serum than other age groups (Fig. 2e), indicating that fatty acids participate in the development of rats at infant stage. The decline in fatty acids from W1 to W7 revealed rapid consumption from infant to adulthood. In our results, the abundance of docosahexaenoate (DHA), elaidiate, and linoleate gradually increased from W9 to W111. Previous study reported that elaidiate is elevated in obese individuals and those with metabolic syndrome (Gil-Campos et al. 2008; Kim et al. 2013). Circulating PUFAs affect host inflammation state (Perreault et al. 2014; Steffen et al. 2012). It has been proven that DHA have an effect on IL-2 signaling pathways (Gorjão et al. 2013). Our results show a positive association between docosahexaenoate (DHA) and inflammatory IL-2, and both of them were isolated from the whole network (Fig. 6b), while other long-chain fatty acids such as tetradecanoate, dodecanoate, and decanoate were positively correlated with Phascolarctobacterium spp. in B04, Anaeroplasma spp. and Cellvibrio spp. in B06, and Akkermansia muciniphila in B03. Akkermansia muciniphila was reported to regulate metabolism balance and immune tolerance (Greer et al. 2016; Plovier et al. 2017). Recent studies have shown that Phascolarctobacterium spp. produce propionate and are associated with dietary intervention among vulnerable elderly people (O'Hara et al. 2018; Tran et al. 2019). Anaeroplasma was also associated with polyunsaturated fatty acid feeding and positively correlates with acetic acid level in rat brains (Nguyen et al. 2019; Robertson et al. 2017). Furthermore, tetradecanoate, dodecanoate, and decanoate were all associated with IgM and “tetradecanoate-decanoate-dodecanoate-IL-10-IgM” formed a local close connection in Fig. 6b, reflecting the possible interaction between immune factors and metabolites.
The line chart of correlated pairs in Fig. 6a revealed that the percentage of significantly correlated factor-metabolite pairs is higher in the early stages of life declined with age (infancy). In particular, amino acids, fatty acids, and TCA participants such as citrate and malate showed higher correlations (Fig. S4). This is probably due to the fact that infants require an external diet intake to support the establishment of a mature immune system (Rijkers et al. 2010). It should be noted that all the pair (factor-bacteria, factor-metabolites, bacteria-metabolites) correlations are decreased in old age (Fig. S4), which may be the result of too large variation within the group due to the inconsistent aging process of individuals.
Previous studies have shown that intestinal flora regulates some peripheral circulating cytokines, which also influence the structure of gut flora (Biancheri and Watson 2017). The result showed that spleen IL-10 was positively related to a variety of bacteria, such as Lactobacillus spp. and Lactobacillus reuteri (Figs. 6b and S2), since IL-10 has been implicated in the maintenance of gut homeostasis (Ray et al. 2015). Probably it does this by regulating probiotics including Lactobacillus. As for IgA, studies have shown that secretory IgA affects mucosal bacterial communities (Donaldson et al. 2018). Recent research also demonstrates that plasma IgA reacts brings about different gut flora structures (Grosserichter-Wagener et al. 2019). Our result showed that IgA in the spleen was positively correlated with Actinobacillus spp. and Rhodococcus spp. and negatively correlated with Rothia spp., Bifidobacterium spp., and Enterococcus spp., indicating that immune factors are highly related to ileal flora as well. Although IgM could help IgA to promote highly diverse commensal communities of gut mucosa, spleen IgM was not directly related to a certain group of bacteria in this study (Magri et al. 2017).
In summary, this comprehensive dual-omics aging study identified key metabolites, gut bacteria, immune factors, and cytokines varying across the lifespan. Our findings provide new insights into the interactions between circulating metabolites and gut bacteria functional metabolism pathways. A two-level strategy associate study establishes network links of key metabolites (as 4-hydroxyproline, cysteine); several beneficial gut bacteria including Bifidobacterium, Lactobacillus, and Akkermansia; and IgA, IgM, and IL-10 during the growth, development, and aging of rats.
Electronic supplementary material
(DOCX 2007 kb)
Funding information
This work was financially supported by Medicine and Engineering Interdisciplinary Research Fund of Shanghai Jiao TongUniversity (YG2016MS40, YG2017MS28), the National Nature Science Foundation of China (30901997 and 31972935), and the National Basic Research Program of China (2012CB910102).
Compliance with ethical standards
All experiments were carried out strictly in accordance with recommendations on the National Institutes of Health’s Guide for Care and Use of Laboratory Animals. The experimental program was approved by the Center for Laboratory Animals of Shanghai Jiao Tong University.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xia Zhang, Yuping Yang, and Juan Su equally contributed to this study as first authors.
Contributor Information
Tianlu Chen, Email: chentianlu@sjtu.edu.cn.
Wei Jia, Email: weijia@sjtu.edu.cn.
Xiaoyan Wang, Email: cathywxy@sjtu.edu.cn.
References
- Bárcena C, Valdés-Mas R, Mayoral P, Garabaya C, Durand S, Rodríguez F, Fernández-García MT, Salazar N, Nogacka AM, Garatachea N, Bossut N, Aprahamian F, Lucia A, Kroemer G, Freije JMP, Quirós PM, López-Otín C. Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat Med. 2019;25:1234–1242. doi: 10.1038/s41591-019-0504-5. [DOI] [PubMed] [Google Scholar]
- Biancheri P, Watson AJM. The relative contributions of the gut microbiome, host genetics, and environment to cytokine responses to microbial stimulation. Gastroenterology. 2017;152:2068–2070. doi: 10.1053/j.gastro.2017.04.037. [DOI] [PubMed] [Google Scholar]
- Biragyn A, Ferrucci L. Gut dysbiosis: a potential link between increased cancer risk in ageing and inflammaging. Lancet Oncol. 2018;19:e295–e304. doi: 10.1016/S1470-2045(18)30095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E et al. (2018) QIIME 2: reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Preprints, [DOI] [PMC free article] [PubMed]
- Buford TW. (Dis) Trust your gut: the gut microbiome in age-related inflammation, health, and disease. Microbiome. 2017;5:80. doi: 10.1186/s40168-017-0296-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler RN, Miller RA, Perry D, Carnes BA, Williams TF, Cassel C, Brody J, Bernard MA, Partridge L, Kirkwood T, Martin GM, Olshansky SJ. New model of health promotion and disease prevention for the 21st century. Bmj. 2008;337:a399. doi: 10.1136/bmj.a399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda-Delgado JE, et al. Differences in cytokine production during aging and its relationship with antimicrobial peptides production. Immunol Investig. 2017;46:48–58. doi: 10.1080/08820139.2016.1212873. [DOI] [PubMed] [Google Scholar]
- Chen T, Ni Y, Ma X, Bao Y, Liu J, Huang F, Hu C, Xie G, Zhao A, Jia W, Jia W. Branched-chain and aromatic amino acid profiles and diabetes risk in Chinese populations. Sci Rep. 2016;6:20594. doi: 10.1038/srep20594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, You Y, Xie G, Zheng X, Zhao A, Liu J, Zhao Q, Wang S, Huang F, Rajani C, Wang C, Chen S, Ni Y, Yu H, Deng Y, Wang X, Jia W. Strategy for an association study of the intestinal microbiome and brain metabolome across the lifespan of rats. Anal Chem. 2018;90:2475–2483. doi: 10.1021/acs.analchem.7b02859. [DOI] [PubMed] [Google Scholar]
- Chen Y, Li Z, Tye KD, Luo H, Tang X, Liao Y, Wang D, Zhou J, Yang P, Li Y, Su Y, Xiao X. Probiotics supplementation during human pregnancy affects the gut microbiome and immune status. Front Cell Infect Microbiol. 2019;9:254. doi: 10.3389/fcimb.2019.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagdeviren S, Young Jung D, Friedline RH, Noh HL, Kim JH, Patel PR, Tsitsilianos N, Inashima K, Tran DA, Hu X, Loubato MM, Craige SM, Kwon JY, Lee KW, Kim JK. IL-10 prevents aging-associated inflammation and insulin resistance in skeletal muscle. FASEB J. 2017;31:701–710. doi: 10.1096/fj.201600832R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone R, Vissicchio F, Mingarelli C, De Nuccio C, Visentin S, Ajmone-Cat MA, Minghetti L. Branched-chain amino acids influence the immune properties of microglial cells and their responsiveness to pro-inflammatory signals. Biochim Biophys Acta. 2013;1832:650–659. doi: 10.1016/j.bbadis.2013.02.001. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessì A, Briana D, Corbu S, Gavrili S, Cesare Marincola F, Georgantzi S, Pintus R, Fanos V, Malamitsi-Puchner A. Metabolomics of breast milk: the importance of phenotypes. Metabolites. 2018;8:79. doi: 10.3390/metabo8040079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd D, Spitzer MH, van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, le A, Cowan TM, Nolan GP, Fischbach MA, Sonnenburg JL. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature. 2017;551:648–652. doi: 10.1038/nature24661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson G et al. (2018) Gut microbiota utilize immunoglobulin A for mucosal colonization. Science (80- ) 360:795–800. [DOI] [PMC free article] [PubMed]
- Douglas GM et al. (2019) PICRUSt2: an improved and extensible approach for metagenome inference. BioRxiv:672295.
- Ferrario C, et al. Exploring amino acid auxotrophy in Bifidobacterium bifidum PRL2010. Front Microbiol. 2015;6:1331. doi: 10.3389/fmicb.2015.01331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505–522. doi: 10.1038/s41569-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firouzi S, Haghighatdoost F. The effects of prebiotic, probiotic, and synbiotic supplementation on blood parameters of renal function: a systematic review and meta-analysis of clinical trials. Nutrition. 2018;51:104–113. doi: 10.1016/j.nut.2018.01.007. [DOI] [PubMed] [Google Scholar]
- Gil-Campos M, del Carmen R-TM, Larque E, Linde J, Aguilera CM, Canete R, Gil A. Metabolic syndrome affects fatty acid composition of plasma lipids in obese prepubertal children. Lipids. 2008;43:723–732. doi: 10.1007/s11745-008-3203-4. [DOI] [PubMed] [Google Scholar]
- Gorjão R, Hirabara S, Cury-Boaventura M, de Lima T, Passos M, Levada-Pires A, Curi R. Signaling pathways involved in the effects of different fatty acids on interleukin-2 induced human lymphocyte proliferation. J Clin Cell Immunol. 2013;4:171. [Google Scholar]
- Greer RL, Dong X, Moraes ACF, Zielke RA, Fernandes GR, Peremyslova E, Vasquez-Perez S, Schoenborn AA, Gomes EP, Pereira AC, Ferreira SRG, Yao M, Fuss IJ, Strober W, Sikora AE, Taylor GA, Gulati AS, Morgun A, Shulzhenko N. Akkermansia muciniphila mediates negative effects of IFNγ on glucose metabolism. Nat Commun. 2016;7:13329. doi: 10.1038/ncomms13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosserichter-Wagener C, Radjabzadeh D, van der Weide H, Smit KN, Kraaij R, Hays JP, van Zelm MC. Differences in systemic IgA reactivity and circulating Th subsets in healthy volunteers with specific microbiota enterotypes. Front Immunol. 2019;10:341. doi: 10.3389/fimmu.2019.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Robinson RA. Proteomic approaches to quantify cysteine reversible modifications in aging and neurodegenerative diseases. Proteomics Clin Appl. 2016;10:1159–1177. doi: 10.1002/prca.201600015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hor YY, et al. Lactobacillus sp. improved microbiota and metabolite profiles of aging rats. Pharmacol Res. 2019;146:104312. doi: 10.1016/j.phrs.2019.104312. [DOI] [PubMed] [Google Scholar]
- Hou Y, Wang X, Lei Z, Ping J, Liu, Ma Z, Zhang Z, Jia C, Jin M, Li X, Li X, Chen S, Lv Y, Gao Y, Jia W, Su J. Heat-stress-induced metabolic changes and altered male reproductive function. J Proteome Res. 2015;14:1495–1503. doi: 10.1021/pr501312t. [DOI] [PubMed] [Google Scholar]
- Huang YY, Martinez-Del Campo A, Balskus EP. Anaerobic 4-hydroxyproline utilization: discovery of a new glycyl radical enzyme in the human gut microbiome uncovers a widespread microbial metabolic activity. Gut Microbes. 2018;9:437–451. doi: 10.1080/19490976.2018.1435244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankord R, JEMIOLO B. Influence of physical activity on serum IL-6 and IL-10 levels in healthy older men. Med Sci Sports Exerc. 2004;36:960–964. doi: 10.1249/01.mss.0000128186.09416.18. [DOI] [PubMed] [Google Scholar]
- Kim OY, Lim HH, Lee MJ, Kim JY, Lee JH. Association of fatty acid composition in serum phospholipids with metabolic syndrome and arterial stiffness. Nutr Metab Cardiovasc Dis. 2013;23:366–374. doi: 10.1016/j.numecd.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Korpela K, Dikareva E, Hanski E, Kolho K-L, De Vos WM, Salonen A. Cohort profile: Finnish Health and Early Life Microbiota (HELMi) longitudinal birth cohort. BMJ Open. 2019;9:e028500. doi: 10.1136/bmjopen-2018-028500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurilshikov A, van den Munckhof ICL, Chen L, Bonder MJ, Schraa K, Rutten JHW, Riksen NP, de Graaf J, Oosting M, Sanna S, Joosten LAB, van der Graaf M, Brand T, Koonen DPY, van Faassen M, LifeLines DEEP Cohort Study, BBMRI Metabolomics Consortium. Slagboom PE, Xavier RJ, Kuipers F, Hofker MH, Wijmenga C, Netea MG, Zhernakova A, Fu J. Gut microbial associations to plasma metabolites linked to cardiovascular phenotypes and risk. Circ Res. 2019;124:1808–1820. doi: 10.1161/CIRCRESAHA.118.314642. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson M, Lewis GD, Ericson U, Orho-Melander M, Hedblad B, Engström G, Östling G, Clish C, Wang TJ, Gerszten RE, Melander O. A diabetes-predictive amino acid score and future cardiovascular disease. Eur Heart J. 2013;34:1982–1989. doi: 10.1093/eurheartj/ehs424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magri G, et al. Human secretory IgM emerges from plasma cells clonally related to gut memory B cells and targets highly diverse commensals. Immunity. 2017;47:118–134. e118. doi: 10.1016/j.immuni.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier TV, Lucio M, Lee LH, VerBerkmoes NC, Brislawn CJ, Bernhardt J, et al. Impact of dietary resistant starch on the human gut microbiome, metaproteome, and metabolome. MBio. 2017;8. [DOI] [PMC free article] [PubMed]
- Malmuthuge N, Liang G, Griebel PJ, Guan LL. Taxonomic and functional compositions of the small intestinal microbiome in neonatal calves provide a framework for understanding early life gut health. Appl Environ Microbiol. 2019;85. [DOI] [PMC free article] [PubMed]
- McCarty MF, DiNicolantonio JJ. An increased need for dietary cysteine in support of glutathione synthesis may underlie the increased risk for mortality associated with low protein intake in the elderly. Age (Dordr) 2015;37:96. doi: 10.1007/s11357-015-9823-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepal M, Ma C, Xie G, Jia W, Fei P. Fanconi Anemia complementation group C protein in metabolic disorders. Aging (Albany NY) 2018;10:1506–1522. doi: 10.18632/aging.101487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TD, Prykhodko O, Fak Hallenius F, Nyman M. Monovalerin and trivalerin increase brain acetic acid, decrease liver succinic acid, and alter gut microbiota in rats fed high-fat diets. Eur J Nutr. 2019;58:1545–1560. doi: 10.1007/s00394-018-1688-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara E, Kelly A, McCabe MS, Kenny DA, Guan LL, Waters SM. Effect of a butyrate-fortified milk replacer on gastrointestinal microbiota and products of fermentation in artificially reared dairy calves at weaning. Sci Rep. 2018;8:14901. doi: 10.1038/s41598-018-33122-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Guo R, Zhu J, Wang Q, Ju Y, Xie Y, et al. A gene catalogue of the Sprague-Dawley rat gut metagenome. Gigascience. 2018;7. [DOI] [PMC free article] [PubMed]
- Pedersen HK, Forslund SK, Gudmundsdottir V, Petersen AØ, Hildebrand F, Hyötyläinen T, Nielsen T, Hansen T, Bork P, Ehrlich SD, Brunak S, Oresic M, Pedersen O, Nielsen HB. A computational framework to integrate high-throughput ‘-omics’ datasets for the identification of potential mechanistic links. Nat Protoc. 2018;13:2781–2800. doi: 10.1038/s41596-018-0064-z. [DOI] [PubMed] [Google Scholar]
- Perreault M, Zulyniak MA, Badoud F, Stephenson S, Badawi A, Buchholz A, Mutch DM. A distinct fatty acid profile underlies the reduced inflammatory state of metabolically healthy obese individuals. PLoS One. 2014;9:e88539. doi: 10.1371/journal.pone.0088539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plovier H, Everard A, Druart C, Depommier C, van Hul M, Geurts L, Chilloux J, Ottman N, Duparc T, Lichtenstein L, Myridakis A, Delzenne NM, Klievink J, Bhattacharjee A, van der Ark KCH, Aalvink S, Martinez LO, Dumas ME, Maiter D, Loumaye A, Hermans MP, Thissen JP, Belzer C, de Vos WM, Cani PD. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med. 2017;23:107–113. doi: 10.1038/nm.4236. [DOI] [PubMed] [Google Scholar]
- Ray A, Basu S, Gharaibeh RZ, Cook LC, Kumar R, Lefkowitz EJ, Walker CR, Morrow CD, Franklin CL, Geiger TL, Salzman NH, Fodor A, Dittel BN. Gut microbial dysbiosis due to helicobacter drives an increase in marginal zone B cells in the absence of IL-10 signaling in macrophages. J Immunol. 2015;195:3071–3085. doi: 10.4049/jimmunol.1500153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijkers GT, Niers L, Stasse-Wolthuis M, Rombouts FM (2010) Nutrition, the infant and the immune system. In: Dietary Components and Immune Function. Springer, pp 3–23.
- Robertson RC, Seira Oriach C, Murphy K, Moloney GM, Cryan JF, Dinan TG, Ross RP, Stanton C. Deficiency of essential dietary n-3 PUFA disrupts the caecal microbiome and metabolome in mice. Br J Nutr. 2017;118:959–970. doi: 10.1017/S0007114517002999. [DOI] [PubMed] [Google Scholar]
- Sansoni P, Vescovini R, Fagnoni F, Biasini C, Zanni F, Zanlari L, Telera A, Lucchini G, Passeri G, Monti D, Franceschi C, Passeri M. The immune system in extreme longevity. Exp Gerontol. 2008;43:61–65. doi: 10.1016/j.exger.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Singh H, Torralba MG, Moncera KJ, DiLello L, Petrini J, Nelson KE, Pieper R (2019) Gastro-intestinal and oral microbiome signatures associated with healthy aging. Geroscience. [DOI] [PMC free article] [PubMed]
- Steffen BT, Steffen LM, Tracy R, Siscovick D, Hanson NQ, Nettleton J, Tsai MY. Obesity modifies the association between plasma phospholipid polyunsaturated fatty acids and markers of inflammation: the Multi-Ethnic Study of Atherosclerosis. Int J Obes. 2012;36:797–804. doi: 10.1038/ijo.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticinesi A, Nouvenne A, Tana C, Prati B, Meschi T. Gut microbiota and microbiota-related metabolites as possible biomarkers of cognitive aging. Adv Exp Med Biol. 2019;1178:129–154. doi: 10.1007/978-3-030-25650-0_8. [DOI] [PubMed] [Google Scholar]
- Tran TT, et al. Prebiotic supplementation in frail older people affects specific gut microbiota taxa but not global diversity. Microbiome. 2019;7:39. doi: 10.1186/s40168-019-0654-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, O'Donnell CJ, Carr SA, Mootha VK, Florez JC, Souza A, Melander O, Clish CB, Gerszten RE. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17:448–453. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijsman CA, Rozing MP, Streefland TCM, le Cessie S, Mooijaart SP, Slagboom PE, Westendorp RGJ, Pijl H, van Heemst D, On behalf of the Leiden Longevity Study group Familial longevity is marked by enhanced insulin sensitivity. Aging Cell. 2011;10:114–121. doi: 10.1111/j.1474-9726.2010.00650.x. [DOI] [PubMed] [Google Scholar]
- Yu W, et al. One-carbon metabolism supports S-adenosylmethionine and histone methylation to drive inflammatory macrophages. Mol Cell. 2019;75:1147–1160.e1145. doi: 10.1016/j.molcel.2019.06.039. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wang X, Wang J, Jia Z, Liu Y, Xie X, Wang C, Jia W. Metabonomics approach to assessing the metabolism variation and endoexogenous metabolic interaction of ginsenosides in cold stress rats. J Proteome Res. 2016;15:1842–1852. doi: 10.1021/acs.jproteome.6b00015. [DOI] [PubMed] [Google Scholar]
- Zheng X, Chen T, Zhao A, Wang X, Xie G, Huang F, Liu J, Zhao Q, Wang S, Wang C, Zhou M, Panee J, He Z, Jia W. The brain metabolome of male rats across the lifespan. Sci Rep. 2016;6:24125. doi: 10.1038/srep24125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 2007 kb)