

Abstract
Rapamycin (RAPA) is found to have neuro-protective properties in various neuroinflammatory pathologies, including brain aging. With magnetic resonance imaging (MRI) techniques, we investigated the effect of RAPA in a lipopolysaccharide (LPS)-induced inflammaging model in rat brains. Rats were exposed to saline (control), or LPS alone or LPS combined with RAPA treatment (via food over 6 weeks). Arterial spin labeling (ASL) perfusion imaging was used to measure relative cerebral blood flow (rCBF). MR spectroscopy (MRS) was used to measure brain metabolite levels. Contrast-enhanced MRI (CE-MRI) was used to assess blood-brain barrier (BBB) permeability. Immunohistochemistry (IHC) was used to confirm neuroinflammation. RAPA restored NF-κB and HIF-1α to normal levels. RAPA was able to significantly restore rCBF in the cerebral cortex post-LPS exposure (p < 0.05), but not in the hippocampus. In the hippocampus, RAPA was able to restore total creatine (Cr) acutely, and N-acetyl aspartate (NAA) at 6 weeks, post-LPS. Myo-inositol (Myo-Ins) levels were found to decrease with RAPA treatment acutely post-LPS. RAPA was also able to significantly restore the BBB acutely post-LPS in both the cortex and hippocampus (p < 0.05 for both). RAPA was found to increase the percent change in BOLD signal in the cortex at 3 weeks, and in the hippocampus at 6 weeks post-LPS, compared to LPS alone. RAPA treatment also restored the neuronal and macro-vascular marker, EphB2, back to normal levels. These results indicate that RAPA may play an important therapeutic role in inhibiting neuroinflammation by normalizing brain vascularity, BBB, and some brain metabolites, and has a high translational capability.
Keywords: Rapamycin, Neuroinflammation, Magnetic resonance imaging (MRI), Inflammaging, Relative cerebral blood flow (rCBF), Blood-brain barrier (BBB), MR spectroscopy (MRS)
Introduction
Mammalian target of rapamycin (mTOR) is a protein kinase that is involved in controlling growth and metabolism, and is commonly dysregulated in cancer and metabolic disorders [1]. Rapamycin (RAPA) is an inhibitor of mTOR, and has been used as an anti-cancer agent [1] for over a decade. It has also been found that RAPA treatment can increase life span and health span in mice [2, 3]. Regarding neuroinflammatory-related pathologies, RAPA has been shown to have therapeutic effects in Alzheimer’s disease (AD) [4–6], cerebral ischemia (stroke) [7–9], traumatic brain injury (TBI) [10–13], intracerebral hemorrhage [14], epilepsy [15–19], Parkinson’s disease (PD) [20], multiple sclerosis (MS) [21], brain aging [22, 23], and in lipopolysaccharide (LPS)-induced encephalopathy [24–27].
For AD, RAPA was found to rescue vascular, metabolic, and learning deficits in apolipoprotein E4 transgenic mice that have pre-symptomatic-like characteristics of AD [4]. Chronic RAPA was also able to restore brain vascular integrity and function, and improve memory, in transgenic human (h)APP mice modeling AD, thought to occur via NO synthase activation [5]. In the APP/PS1 mouse model for AD, RAPA was found to regulate cholesterol biosynthesis and cytoplastic ribosomal proteins in the temporal lobe and hippocampus [6]. There is a cautionary note regarding assessing RAPA in AD mouse models, where most of these studies involved RAPA treatment before or early in the appearance of AD pathological hallmarks, and it is thought that later in the disease process when dementia occurs, that the brain’s lysosomal system is compromised, and that RAPA treatment may actually exacerbate further damage [28]. Regardless, RAPA may still provide a potential therapeutic strategy for AD if detected early, which is currently a major challenge.
Regarding stroke models, RAPA was recently found to increase collateral circulation in rodent brains following focal ischemia elicited in a middle cerebral artery occlusion (MCAo) model [8]. RAPA also was found to reduce brain metabolite alterations following transient focal ischemia (MCAo) in rats [7]. In a related intracerebral hemorrhage model involving injection of type IV collagenase into the striatum in rats, RAPA treatment improved sensorimotor deficits, and decreased brain edema and lesion volumes, which was thought to be due to suppression of microglial activation [14]. Conversely, RAPA did not have an effect on BBB permeability in early stages of cerebral ischemia in diabetic rats (streptozotocin-induced) following MCAo, although it did decrease BBB disruption in control rats subjected to MCAo [29].
In a rodent model for traumatic brain injury (TBI), RAPA was found to protect against apoptotic neuronal death and also improve neurologic function via inactivating the mTOR-p53-Bax axis [10]. In a cryogenic model for TBI, long-term RAPA treatment resulted in reducing glial scar formation [11]. In a weight-drop model of TBI in mice, RAPA was found to protect neurons from brain contusion-induced inflammatory responses via inhibiting microglial activation [12]. In a weight-drop model for TBI in rats, it was found that RAPA treatment resulted in the upregulation of autophagy and mitophagy, and decreased apoptosis, as well as improvement of cognitive and motor coordination functions [13].
RAPA has also been studied as a potential anti-eileptogenic therapy. In a kainic-acid-induced status epilepticus (SE) model in rats, RAPA was found to reduce BBB disruption, as well as seizure frequency, during the chronic epileptic phase, but at the acute phase, RAPA resulted in increased BBB permeability [15, 16]. RAPA treatment, although found to reduce seizures in this model of SE, did not affect microglial activation [17]. In a post-electrical SE rat model for temporal lobe epilepsy, RAPA did not suppress the expression of inflammatory and oxidative stress markers [18]. In a WAG/Rij rat model for epilepsy induced by intracerebral injection of LPS, RAPA was found to decrease seizures and inhibit neuroinflammatory cytokines via modulation of AMPK, AKT/mTOR pathways [19].
RAPA was found to alleviate the progression of experimental autoimmune encephalomyelitis (EAE), a model for MS, by inducing autophagy and inhibiting the immune response [21]. A combination of RAPA and trehalose treatment resulted in autophagy activation that reversed both neuronal dopaminergic and behavioral deficits in a mouse model of PD induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxicity [20].
RAPA treatment has also been studied in LPS-induced neuroinflammation. Rats intracranially injected with LPS and pre-treated with RAPA were found to have decreased expression of HIF1α (hypoxia inducible factor) and phosphorylated (p-)S6 in the cerebral cortex following immunohistochemistry analysis [24]. RAPA was also found in another LPS-induced neuroinflammation study to downregulate mTOR, p-mTOR, NF-κB, and TNF-α expression [25]. When comparing RAPA to everolimus, another mTOR inhibitor, everolimus was found to be more effective than RAPA in inhibiting iNOS and mTOR signaling pathways [26].
Regarding brain aging, RAPA-induced activation of autophagy was found to provide protection by reducing aging-induced oxidative stress, apoptotic cell death, and the expression of markers for neurodegeneration [22]. In OXYS rats, RAPA treatment was found to decrease anxiety and improve locomotor and exploratory behavior, as well as decrease demyelination and both Tau and p-Tau levels in the hippocampus regions [23].
There is no question that RAPA could be beneficial for various neuroinflammatory pathologies.
In this study, we used advanced MRI techniques, including arterial spin labeling (ASL) perfusion imaging to measure the relative cerebral blood flow (rCBF), contrast-enhanced MRI (CE-MRI) to assess BBB permeability, and MR spectroscopy (MRS) to measure brain metabolite levels, following LPS-induced encephalopathy or inflammaging over an extended period of time (up to 6 weeks), either untreated or treated with RAPA. We have previously reported on the use of these MRI techniques in LPS vs. saline-exposed rat brains [30], and in LPS-exposed rats treated with the anti-inflammatory and free radical scavenging agent, OKN-007 [31]. Immunohistochemistry (IHC) was also used to evaluate inflammatory markers, NF-κB and HIF-1α, and the neuronal and vascular marker, EphB2.
Materials and methods
Ethics statement
Animal experiments were performed with the approval and strict adherence to the policies of the Oklahoma Medical Research Foundation Institutional Animal Care and Use Committee, which specifically approved this study, with adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize suffering.
LPS exposure and rapamycin treatment
Rats (Sprague-Dawley; 8–10 weeks old; male; n = 30) were exposed to a single injection of LPS (10 mg/kg in 100 μL saline; i.p.). Controls were administered saline (same volume and route of administration as for LPS). Control data was obtained from multiple studies conducted in our laboratory over several years, whereas LPS-exposed and rapamycin (RAPA)-treated rats were directly compared for the purposes of this study. RAPA was administered via the food pellets (42 ppm) (Rapamycin Holdings, San Antonio, TX). A control diet containing no RAPA was used for rats treated with saline or LPS alone.
MRI methods—contrast-enhanced MRI, perfusion imaging, and MR spectroscopy
MRI experiments were done on a Bruker Biospec 7.0 Tesla/30-cm horizontal-bore imaging system, and similar to those previously published in Towner et al. [30, 31]. Briefly, multiple brain 1H–MR image slices were taken using a RARE multi-slice (repetition time (TR) 1.3 s, echo time (TE) 9 ms, 256 × 256 matrix, 4 steps per acquisition, 4 × 4 cm2 field-of-view, 1.0-mm slice thickness) imaging sequence.
For contrast-enhanced MRI (CE-MRI), multi-slice spin echo T1-weighted images (TR = 1000.0 ms, TE = 14 ms, FOV = 4 × 4 cm2, averages = 2, slices = 16, matrix size = 256 × 256) were also performed and acquired before and 10, 20, and 30 min after intravenous contrast agent injection (Gd-DTPA, Magnevist, Bayer Inc., Wayne, NY, USA; 0.4 mmol/kg). Regional assessments were made in the following regions: cortex and hippocampus, regarding MRI signal intensity measurements.
1H–MRS was obtained using a PRESS (Point REsolved SpectroScopy) sequence with a TE of 24.0 ms, a TR of 2500.0 ms, 512 averages, and a spectral width of 4006 Hz. A non-suppressed MR spectrum was acquired beforehand by applying eddy-current correction to maximize signal intensity and decrease the peak linewidths. Water was suppressed with a VAPOR (variable power radio frequency pulses and optimized relaxation delays) suppression scheme. In all cases, the peak width (full width at half maximum) of the water peak was less than 30 Hz following localized shimming, which was conducted by using first- and second-order adjustments with Fastmap. A cubic voxel of 3.0 × 2.0 × 3.0 mm3 was positioned in the rat brain predominantly in the hippocampus region. To analyze the MRS data, an in-house Mathematica program was used (version 6.0, Wolfram Research, Champaign, IL, USA). The spectra were scaled in parts per million by calibrating against the water peak (4.78 ppm). The major brain metabolic peaks were identified as N-acetylaspartate (NAA) at 2.02 ppm, choline (Cho) at 3.22 ppm, creatine (Cr) at 3.02 ppm, and myo-inositol (Myo-Ins) at 3.53 ppm. The peak area measurements of the metabolites were used to calculate the following ratios: NAA to Cho (NAA/Cho), Cr to Cho (Cr/Cho), and Myo-Ins to Cho (Myo-Ins/Cho).
Arterial spin-labeling (ASL) perfusion maps were obtained on a single axial slice of the brain located on the point of the rostro–caudal axis where the hippocampus had the largest cross section. The imaging geometry was a 4 × 4 cm2 field-of-view (FOV) of 2 mm in thickness, with a single-shot echo-planar encoding over a 64 × 64 matrix. An TE of 13.5 ms, a TR of 18 s, and an TIR of 26.0 ms were used, and images were not submitted to time averaging. To obtain perfusion contrast, the flow alternating inversion recovery scheme was used. Briefly, inversion recovery images were acquired using a slice-selective inversion of the same geometry as the imaging slice or a nonselective inversion slice concentric with the imaging slice with a slice package margin of 5.0 mm. For each type of inversion, 22 images were acquired with inversion times evenly spaced from 26.0 to 8426.0 ms (with an increment of 400 ms between each TIR). Relative cerebral blood flow (rCBF) values were obtained by drawing circular ROIs (left and right regions) in the cortex and hippocampus regions of the brain. Negative ASL rCBF values were assumed to be zero.
Blood oxygen level-dependent (BOLD)
During the initial period of anesthesia, reference images were taken, and localized field shimming was performed. An imaging scan was taken while the animals were in this basal state to provide a baseline value for perfusion rates. At the end of the baseline perfusion imaging BOLD (T2*-weighted dynamic), acquisition was initiated, after which the carrier gas was switched to 10% CO2 in 90% carrier gas on a volume basis. The animals were allowed to acclimate to the CO2-enriched gas for an additional 12 min during which the T2*-weighted dynamic acquisition was continued. After the acclimation period, a second perfusion imaging dataset was acquired to provide data in the hypercapnia state. BOLD imaging was implemented by acquiring echo-planar imaging (EPI) with T2* contrast. An echo-planar imaging sequence was used with a repetition time of 1500 ms, an echo time of 20 ms, a flip angle of 30°, 4 excitations per slice, and 4 segments. The geometry consisted of 12 contiguous horizontal slices (as defined according to the stereotaxic reference frame of Paxinos and Watson [32]) 1.0-mm thick and 35 × 35 mm2 square. Each slice was encoded over a 128 × 96 matrix. Twenty repetitions were acquired for the initial period on medical O2, and the reaction of the animal to the increase in CO2 content was captured from another subsequent 20 repetitions to the end of the acquisition.
Immunohistochemistry (IHC)
Embedded in paraffin and mounted on HistoBond®Plus slides (Statlab Medical Products, Lewisville, TX), 5-μm-thick histological sections were rehydrated and washed in Tris buffered saline (TBS). Rabbit antibodies were used for HIF-1α (cat# ab2185, 1:1000, 1 μg/ml, Abcam, Cambridge, MA), NF-kB p65 (cat# ab16502, 1:1000, 1 μg/ml, Abcam, Cambridge, MA), and EphB2 (cat#83029, 1:200, Cell Signaling, Danvers, MA). Slides were processed for IHC using Anti-Rabbit IgG ImmPRESS® Excel Amplified Polymer kit Peroxidase (cat# MP7601, Vector Labs, Burlingame, CA). Antigen retrieval (pH 6 Citrate Antigen Unmasking Solution, cat# H-3300, Vector Labs Inc., Burlingame CA) was accomplished via 20 min in a steamer followed by 30 min of cooling at room temperature for all antibodies. Sections were treated with a peroxidase blocking reagent (Bloxall, cat# SP-6000, Vector Laboratories, Inc, Burlingame, CA) to inhibit endogenous peroxidase activity, followed by 2.5% normal horse serum blocking reagent to inhibit nonspecific binding. Appropriate washes were in TBS. Antibodies were applied to each section and following 1-h incubation at room temp in a humidified chamber, sections were washed in TBS and reagents were applied according to the manufacturer’s directions. Slides were incubated with NovaRed® (Vector Laboratories, Inc., Burlingame, CA) chromogen for visualization. Counterstaining was carried out with Hematoxylin QS Nuclear Counterstain (Vector Laboratories, Burlingame, CA). Appropriate positive and negative tissue controls were used.
Statistical analyses
Statistical analyses were performed using ANOVA with multiple comparisons, with GraphPad Prism 6 (GraphPad Prism 6 Software, San Diego, CA, USA). All p values < 0.05 were considered statistically significant. MRI signal intensities, rCBF values, metabolite peak ratios ((NAA/Cho), (Cr/Cho), and (Myo-Ins/Cho)), and IHC positivity values for NF-κB, HIF-1α, and EphB2 were reported as means ± standard deviations. For statistical analysis, one-way ANOVA tests with multiple comparisons were used to assess the differences between means of the LPS-exposed and saline-control rat brains, and those that were treated with RAPA.
Results
Rapamycin restores LPS-induced blood-brain barrier permeability to normal
CE-MRI, which detects BBB permeability alterations following administration of the contrast agent, Gd-DTPA, which does not cross the BBB in normal brain, indicated a significant increase in MRI signal intensity (SI) due to the presence of the contrast agent in LPS-exposed rat brains at 24 h post-injection in the cerebral cortex (p < 0.05) and hippocampus (p < 0.05), compared with saline-treated controls (Fig. 1D,E, respectively). At 1 week post-LPS, there was also a significant increase in BBB permeability in both the cerebral cortex and hippocampus (p < 0.01 for both regions), when compared with saline controls (Fig. 1D, E, respectively). A significantly decreased MRI SI was also observed in the LPS rat brains treated with RAPA in the cerebral cortex 1 week post-LPS (p < 0.05) (Fig. 1D), and hippocampus 1 week post-LPS (p < 0.01) (Fig. 1E). Representative T1-weighted MR images for saline-, LPS-, or LPS + RAPA-treated rat brains are shown in Fig. 1A–C, respectively.
Fig. 1.
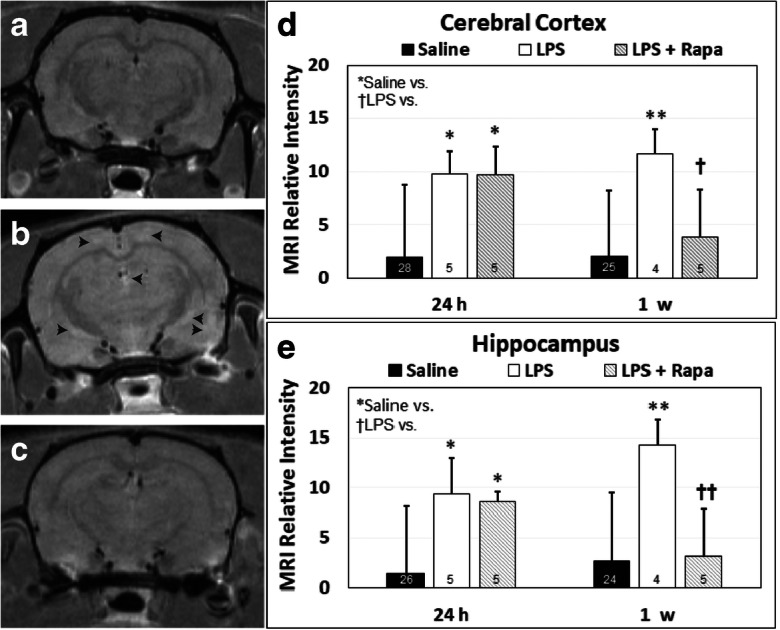
Rapamycin decreases LPS-induced BBB disruption in the cerebral cortex and hippocampus. Representative post-contrast (Gd-DTPA) MR images from brains of (A) saline, (B) LPS, or (C) LPS + rapamycin (LPS + RAPA)-treated rats at 24 h and 1 week post-LPS. In panel B, black arrowheads depict hyperintense or “bright whitened” regions of contrast enhancement, due to uptake of the Gd-DTPA through the BBB. In a T1-weighted MR image, contrast-enhanced regions depict the uptake of the MRI contrast agent, Gd-DTPA, which normally cannot pass through the BBB. Quantitative MRI relative signal intensities in the (D) cerebral cortex, and (E) hippocampus regions. CE-MRI was used to assess MRI relative intensities at 24 h and 1 week post-LPS (or saline). Saline group (n = 24–28). LPS group (n = 4–5). LPS + RAPA (n = 5). *Significant differences between saline controls and other treatment groups. †Significant differences between the LPS alone group and other treatment groups. * or † p < 0.05, ** or †† p < 0.01. There was a significant decrease in MRI relative intensities between the LPS alone group vs. the LPS + RAPA group at 1 week post-LPS in both the cerebral cortex and the hippocampus regions. Statistical analyses were performed using ANOVA with multiple comparisons
Rapamycin restores LPS-induced reduced brain tissue perfusion rates to normal
Perfusion imaging (pMRI) indicated that for the cortex and hippocampus regions, LPS-exposed rat brains had significantly decreased rCBF at all time points (1, 3, and 6 weeks post-LPS), compared with saline controls. Regarding a treatment effect from RAPA, there was a restoration of rCBF in only the cerebral cortex at 1 and 3 weeks post-LPS (Fig. 2D) for the LPS-exposed rat brains treated with RAPA, compared with LPS-exposed rat brains alone. These results strongly suggest that RAPA restores vascularity, altered by LPS exposure, back to normal in the cerebral cortex, but not the hippocampus (Fig. 2E).
Fig. 2.
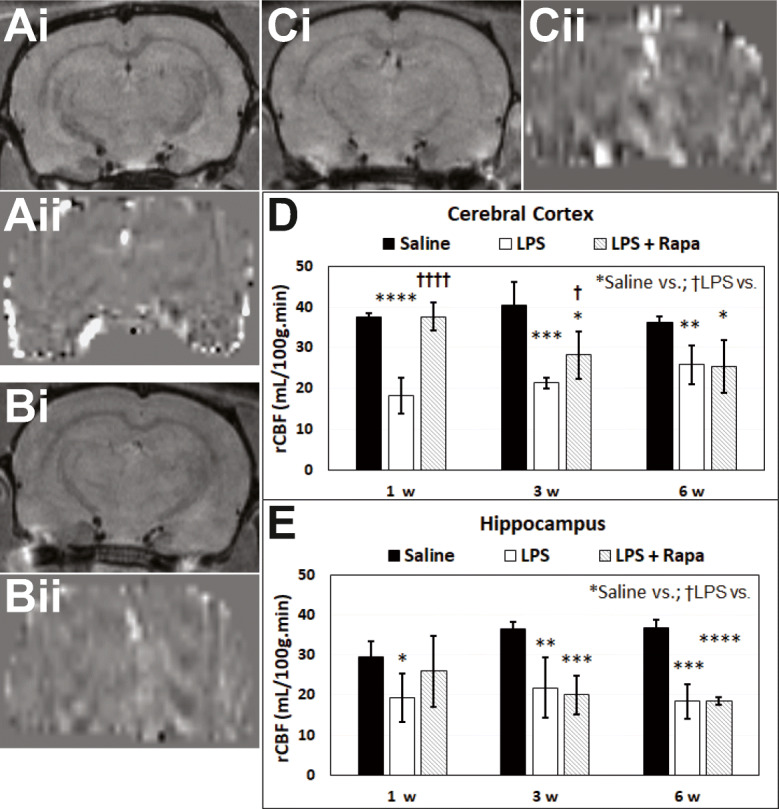
Rapamycin normalizes LPS-induced decreased rCBF in the cerebral cortex. Representative post-contrast (Gd-DTPA) MR images (i) and perfusion maps (ii) from brains of (A) saline, (B) LPS, or (C) LPS + rapamycin (LPS + RAPA)-treated rats. Note in panel Bii that the perfusion image appears darker than panel Aii or panel Cii, due to decreased rCBF. Quantitative relative cerebral blood flow (rCBF) in the (D) cerebral cortex and (E) hippocampus regions. Arterial spin labeling (ASL) perfusion imaging was used to calculate rCBF at 1, 3, and 6 weeks post-LPS (or saline). Saline group (n = 24–28). LPS group (n = 4–5). LPS + RAPA (n = 5). *Significant differences between saline controls and other treatment groups. †Significant differences between the LPS alone group and other treatment groups. * or † p < 0.05, ** or †† p < 0.01, *** or ††† p < 0.001, **** or †††† p < 0.0001. There was a significant increase in rCBF between the LPS-alone group vs. the LPS + RAPA group at 1, 3, and 6 weeks post-LPS in both the cerebral cortex, but not the hippocampus regions. Statistical analyses were performed using ANOVA with multiple comparisons
Rapamycin restores some LPS-induced reduced brain metabolite levels to normal
The MRS study showed that LPS-exposed rat brains had significantly decreased NAA/Cho metabolite ratios at 24 h (p < 0.05), 3 weeks (p < 0.05), and 6 weeks (p < 0.05) post-LPS injection; Cr/Cho ratios at 3 weeks (p < 0.01) post-LPS; and Myo-Ins/Cho ratios at 1 (p < 0.05), 3 (p < 0.0001), and 6 weeks (p < 0.05), compared with controls (Fig. 3C). RAPA treatment of LPS-exposed rat brains significantly restored some metabolites assessed, NAA at 1–6 weeks post-LPS (p < 0.05 at 1 week; p < 0.01 at 3 weeks; and p < 0.05 at 6 weeks), and Cr at 24 h post-LPS, compared with LPS-exposed animals alone. Myo-inositol (Myo-Ins) levels were found to decrease with RAPA treatment at 1 week post-LPS. These results indicate that RAPA can restore some LPS-induced depleted brain metabolites to normal levels.
Fig. 3.
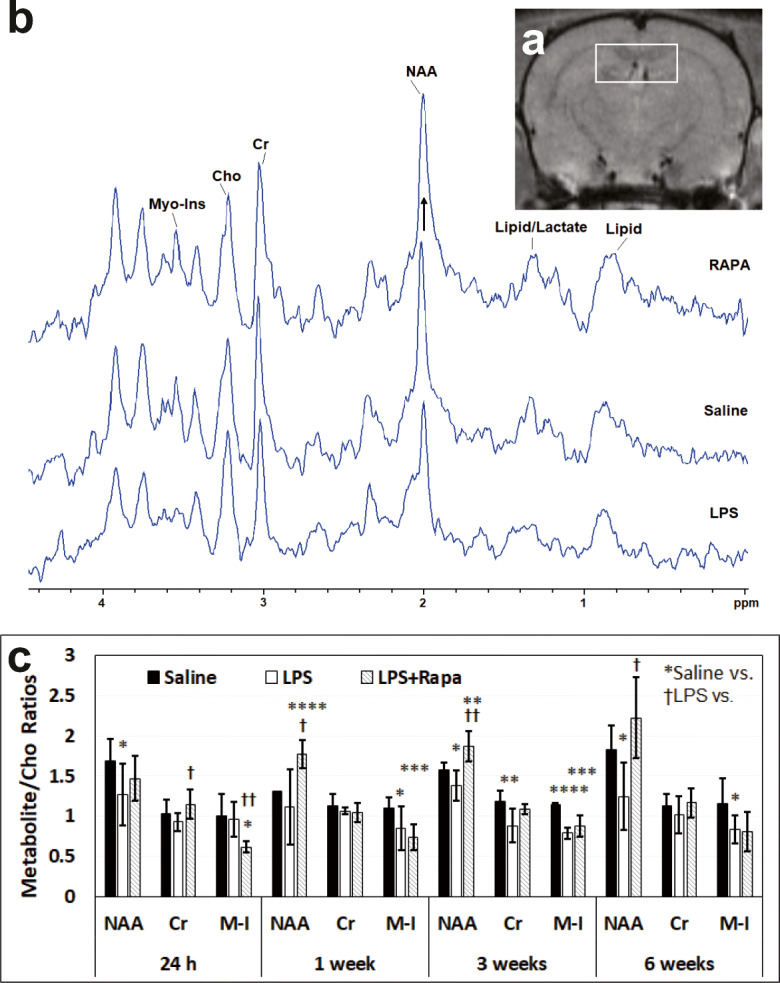
Rapamycin restores neuronal integrity from LPS-induced encephalopathy in the hippocampus. (A) Representative MRI scan with the MR spectral voxel (3 × 2 × 3 mm3) placed predominantly in the hippocampus region. (B) Representative MR spectra (0– ~4.5 ppm) from LPS, saline, and LPS + RAPA-treated rat brains in the hippocampus regions. Peak assignments: N-acetyl aspartate (NAA) at 2.02 ppm, choline (Cho) at 3.22 ppm, creatine (Cr) at 3.02 ppm, and myo-inositol (Myo-Ins) at 3.53 ppm. (C) Spectral peak area measurements of the metabolites were used to calculate the following ratios: NAA to Cho (NAA/Cho), Cr to Cho (Cr/Cho), and Myo-Ins to Cho (Myo-Ins/Cho). Note the increase in NAA in the RAPA-treated LPS-exposed rat brains (illustrated with an arrow), compared to the LPS-alone spectrum. Saline group (n = 8). LPS group (n = 5-8). LPS + RAPA (n = 5). *Significant differences between saline controls and other treatment groups. †Significant differences between the LPS-alone group and other treatment groups. * or † p < 0.05, ** or †† p < 0.01, *** or ††† p < 0.001, **** or †††† p < 0.0001. There was a significant increase in NAA between the LPS-alone group vs. the LPS + RAPA group at 1, 3, and 6 weeks post-LPS. There was a significant increase in Cr between the LPS-alone group vs. the LPS + RAPA group at 24 h. There was a significant decrease in Myo-Ins (M-I) between the LPS-alone group vs. the LPS + RAPA group at 24 h. Statistical analyses were performed using ANOVA with multiple comparisons
Rapamycin increases the percent change in BOLD signal resulting from LPS-induced encephalopathy
RAPA was found to increase the % change in BOLD signal in the LPS + RAPA treatment group in the cerebral cortex regions at 3 weeks post-LPS, and in the hippocampus at 6 weeks post-LPS, comparted to LPS alone at those time points (Fig. 4). The % change in BOLD signal in the cerebral cortex regions at 3 weeks post-LPS (Fig. 4B), which is predominantly due to perfusion of oxygenated arterial blood (see “Discussion” below), supports the rCBF data at 3 weeks post-LPS in Fig. 2D.
Fig. 4.
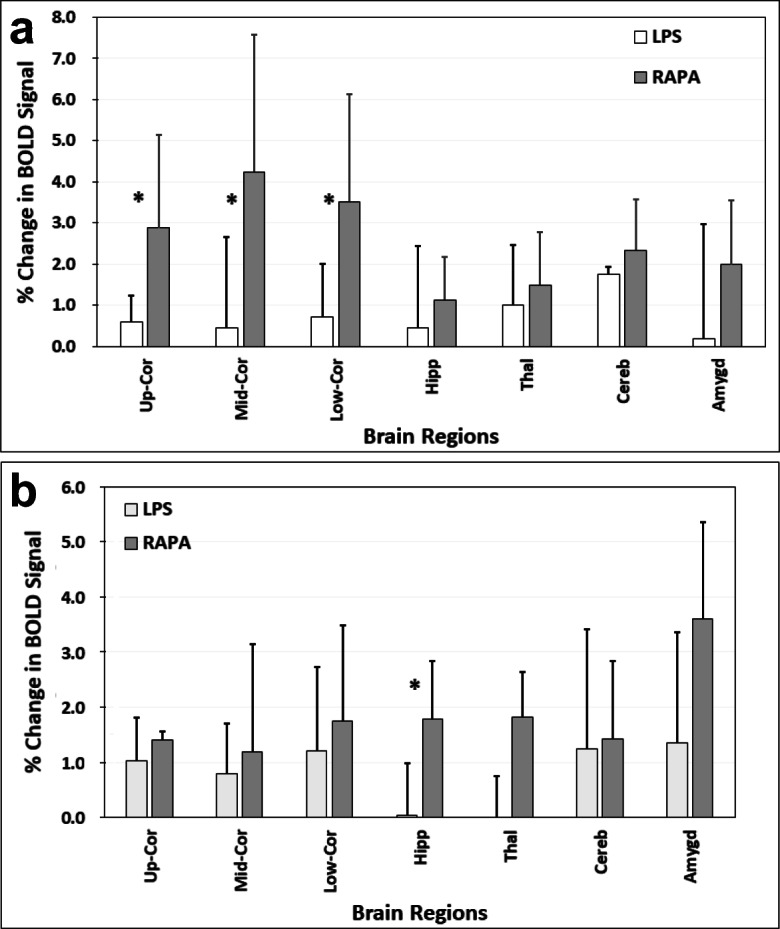
Rapamycin restores brain vascular integrity in the cerebral cortex and hippocampus regions. Percent (%) change in blood oxygen level-dependent (BOLD) signals at (A) 3 weeks, or (B) 6 weeks post-LPS in LPS alone compared to LPS + RAPA treatment groups. BOLD signals were measured in the upper, mid, and lower cerebral cortex (Up-Cor, Mid-Cor, and Low-Cor, respectively), hippocampus (Hipp), thalamus (Thal), cerebellum (Cereb), and amygdala (Amygd) brain regions. n = 6 for each group. *Significant differences between the LPS-alone group and LPS + RAPA treatment group. There was a significant increase in the % change in BOLD signal between the LPS alone and LPS + RAPA groups in all of the cerebral cortex regions assessed (*p < 0.05 for all) at 3 weeks post-LPS, and the hippocampus region (*p < 0.05) at 6 weeks post-LPS. Statistical analyses were performed using ANOVA with multiple comparisons.
Rapamycin reduces LPS-induced neuroinflammation (NF-κB, HIF-1α)
RAPA was found to significantly decrease LPS-induced neuroinflammatory markers at 6 weeks post-LPS in both the cerebral cortex (i) and hippocampus (ii), as determined from IHC staining for NF-κB (Fig. 5) and HIF-1α (Fig. 6). Quantitative assessment of NF-κB positivity staining in the cortex and hippocampus is shown in Fig. 5A and B, respectively. There was a significant increase in NF-κB in both the cortex (p < 0.01) and hippocampus (p < 0.0001), compared to saline controls. RAPA was able to significantly decrease NF-κB in both the cortex (p < 0.001) and hippocampus (p < 0.0001), when compared to LPS-alone groups. Representative IHC tissue sections for NF-κB staining for saline-, LPS alone-, or LPS + RAPA treatments are shown in Fig. 5C–H, in the cortex and hippocampus, respectively. Quantitative assessment of HIF-1α positivity staining in the cortex and hippocampus is shown in Fig. 6A and B, respectively. There was a significant increase in HIF-1α in the cortex of LPS-alone treatment, compared to saline controls (p < 0.001); however, regarding HIF-1α in the hippocampus, the level in the LPS-alone group was not quite significantly increased (increasing trend; p = 0.0501) compared to saline controls. RAPA was able to significantly decrease HIF-1α in both the cortex (p < 0.0001) and hippocampus (p < 0.05), when compared to LPS-alone groups. Representative IHC tissue sections for HIF-1α staining for saline-, LPS alone-, or LPS + RAPA treatments in the cortex (i) and hippocampus (ii) are shown in Fig. 6C–H, in the cortex and hippocampus, respectively.
Fig. 5.

Rapamycin restores inflammatory signaling molecule, NF-κB, levels to normal in the cerebral cortex and hippocampus regions. Quantitative assessment of positivity staining for NF-κB in the cortex (A) or hippocampus (B) regions of rat brains treated with either saline (n = 4), LPS (n = 3), or LPS + RAPA (n = 5). There was a significant increase in NF-κB levels in LPS-treated rat brains, compared to saline controls, and a significant decrease in NF-κB levels in the LPS + RAPA-treated rat brains, compared to LPS-alone. Statistical analyses were performed using ANOVA with multiple comparisons. IHC staining for NF-κB is shown in representative tissue slices in either the cortex (middle panel) or hippocampus (lower panel) regions for saline- (C or F, respectively), LPS- (D or G, respectively), or LPS + RAPA- (E or H, respectively) treated rat brains at 6 weeks post-LPS
Fig. 6.

Rapamycin restores inflammatory signaling molecule, HIF-1α, levels to normal in the cerebral cortex and hippocampus regions. Quantitative assessment of positivity staining for HIF-1α in the cortex (A) or hippocampus (B) regions of rat brains treated with either saline (n = 4), LPS (n = 3), or LPS + RAPA (n = 5). There was a significant increase in HIF-1α levels in LPS-treated rat brains compared to saline controls, and a significant decrease in HIF-1α levels in the LPS + RAPA-treated rat brains, compared to LPS-alone. Statistical analyses were performed using ANOVA with multiple comparisons. IHC staining for HIF-1α is shown in representative tissue slices in either the cortex (middle panel) or hippocampus (lower panel) regions for saline- (C or F, respectively), LPS- (D or G, respectively), or LPS + RAPA- (E or H, respectively) treated rat brains at 6 weeks post-LPS
Rapamycin decreases an LPS-induced increase in EphB2
RAPA was found to significantly decrease an LPS-induced increase in EphB2, Ephrin receptor 2, a transmembrane receptor protein involved in Ephrin receptor signaling (Fig. 7). Quantitative assessment of EphB2 positivity staining in the cortex and hippocampus is shown in Fig. 7A and B, respectively. There was a significant increase in EphB2 in both the cortex (p < 0.01) and hippocampus (p < 0.05), compared to saline controls. RAPA was able to significantly decrease EphB2 in both the cortex (p < 0.001) and hippocampus (p < 0.01), when compared to LPS-alone groups. Representative IHC tissue sections for EphB2 staining for saline-, LPS alone-, or LPS + RAPA treatments are shown in Fig. 7C–H, in the cortex and hippocampus, respectively.
Fig. 7.

Rapamycin restores neuronal Ephrin receptor, EphB2, levels to normal in the cerebral cortex and hippocampus regions. Quantitative assessment of positivity staining for EphB2 in the cortex (A) or hippocampus (B) regions of rat brains treated with either saline (n = 4), LPS (n = 3), or LPS + RAPA (n = 5). There was a significant increase in EphB2 levels in LPS-treated rat brains, compared to saline controls, and a significant decrease in EphB2 levels in the LPS + RAPA-treated rat brains compared to LPS-alone. Statistical analyses were performed using ANOVA with multiple comparisons. IHC staining for EphB2 is shown in representative tissue slices in either the cortex (middle panel) or hippocampus (lower panel) regions for saline- (C or F, respectively), LPS- (D or G, respectively), or LPS+RAPA- (E or H, respectively) treated rat brains at 6 weeks post-LPS
Discussion
Advanced MRI techniques, including CE-MRI, ASL pMRI, and MRS, were able to demonstrate that RAPA has the capacity to restore BBB integrity in both the cerebral cortex and hippocampus, particularly at 1 week post-LPS exposure; restore vascularity, as measured from rCBF, to normal in the cerebral cortex at 1 and 3 weeks post-LPS; restore vascularity integrity, as measured from BOLD imaging, in the cerebral cortex 3 weeks post-LPS and in the hippocampus 6 weeks post-LPS; and restore NAA at longer time points (1–6 weeks post-LPS), as well as Cr at an early time point (24 h post-LPS), respectively. Further studies are being planned in the future to assess longer time points with RAPA treatment regarding BBB effect.
RAPA has been previously shown to reduce BBB leakage after status epilepticus, particularly in the chronic epileptic phase [15, 16]. CE-MRI was used to assess MRI signal intensities from gadobutrol (another type of MRI contrast agent that also does not cross the BBB in normal brain) leakage found to be widespread, particularly in the piriform cortex and amygdala after status epilepticus [15, 16]. Conversely, RAPA treatment was found to increase BBB leakage during the acute epileptic phase [15, 16]. In a pre-clinical study for an apolipoprotein E4 (apo-E4) transgenic mouse model for AD, RAPA treatment was found to restore BBB integrity, as measured by manganese (Mn)-enhanced MRI (MEMRI), where Mn also does not penetrate BBB under normal conditions [4]. In our study, Gd-DTPA, a MRI contrast agent, which normally does not pass through the BBB, was used. The LPS-exposed rat brains demonstrated an increase in BBB permeability due to an enhanced contrast signal intensity depicted by hyperintensity or “bright” regions, whereas RAPA was able to restore BBB integrity. This finding has also been confirmed by others in some models of neuroinflammation (as discussed above).
Age-associated inflammation, also referred to as inflammaging, has been studied as a major player in shortened life span and chronic disease [33]. LPS is known to accelerate the inflammaging process, and could be used as an inflammaging model [34]. In a normal aging wild-type (WT) mouse model, BBB permeability or impaired BBB function has also been found to occur in aging mice, compared to young mice [35, 36]. Likewise, in rats, BBB permeability is also increased in aged animals, compared to a younger cohort [37]. Regarding BBB, the LPS-induced inflammaging model seems to emulate normal aging.
In the apo-E4 model for AD, RAPA was able to restore rCBF, as measured by ASL pMRI [4] in a similar approach to what we used in our study. In another model for AD, transgenic human (h)APP mice, rCBF was also restored following RAPA treatment [5]. In our study, we found that rCBF was reduced by ~ 50% in the cerebral cortex and by ~ 30% in the hippocampus. In another normal aging mouse study (C57BL/6N WT mice), there was a ~ 50% reduction in CBF (as measured by MRI) in old mice compared to young mice, both in whole brain and the hippocampus [36]. In another more recent study by Liu et al., they found a significant decrease (p < 0.05; ~ 60%) in the percent difference in rCBF (using laser Doppler flowmetry) between old and young WT (C57BL/6) mouse brains [38]. In another study using a rat aging model (Fisher-344), there was an average 17% reduction in rCBF for 24-month-old rats compared to 12-month-old rats [39]. Regarding rCBF, the LPS-induced inflammaging model also seems to resemble normal aging, to some extent more so in the aging mouse study, than the aging rat study. In our rat study, RAPA was found to restore rCBF levels back to those seen in the saline controls. It seems that RAPA can restore brain vascular integrity in both the LPS-induced neuroinflammatory model, as well as preclinical models for AD.
Neuro-metabolites assessed with MRS include the neuronal marker N-acetylasparate (NAA), as well as the glial marker myo-inositol (Myo-Ins) [40]. The concentrations of these metabolites typically correspond to disease severity and often correlate well with clinical variables in various brain disorders [40]. Neuroinflammation with activated astrocytes and microglia in brain disorders is often associated with elevated Myo-Ins, and to a lesser extent elevated total creatine (Cr) and choline-containing compounds (Cho), which are found in higher concentrations in glia than neurons, while neuronal injury is indicated by lower than normal levels of NAA [40]. The spectral peak at 2.02 ppm associated with NAA and NAA-glutamyl (NAAG) is widely used as a marker of neuronal density since NAA is found within mature neurons, while the resonance peak for myo-inositol (Myo-Ins at 3.56 ppm) is a putative glial marker since it is primarily present in glial cells [40]. The peak for choline compounds (Cho at 3.2 ppm) includes soluble choline-containing compounds and is a marker for cell membrane metabolism and cellular turnover [40].
In the LPS-induced rat model of neuroinflammation, we compared the metabolites, NAA, Cr, and Myo-Ins, to a Cho reference, as we found that Cho levels did not significantly change between saline and LPS-exposure groups [30]. In that study, we previously found that although NAA/Cho was decreased, Myo-Ins and Cr to Cho ratios were also decreased. It was interesting that the Myo-Ins/Cho ratio was found to decrease with RAPA treatment at 1 week post-LPS, as in previous studies we found that the Myo-Ins/Cho ratio in saline controls was increased compared to rat brains following LPS exposure [30, 31]. We are unsure at this time what this result signifies regarding RAPA treatment. In previous studies assessing intracerebral hemorrhage in rats [14] or TBI in mice [12], RAPA treatment was found to suppress microglial activation, which may contribute to increased Cho, and therefore may reduce the Myo-Ins/Cho ratio that we observed. In an aging mouse study, we found that levels of Myo-Ins are elevated ~ 37% in spinal cord from old mice, which we speculated could influence the levels of secondary messengers, as Myo-Ins is a precursor for inositol triphosphate [41]. Myo-Ins is an osmotically active molecule, and changes in its concentration could potentially disrupt ionic balance in the cell and favor cell death [41].
In a normal aging mouse model, NAA has also been found to be significantly decreased in old mice compared to young mice [42]. In a rat aging model, there was a significant decrease in hippocampal concentrations of NAA in old rats compared to young rats [43]. With respect to NAA levels, the LPS-induced inflammaging model seems to be similar to a normal aging model.
It is important to note that NAA, a neuronal-specific marker, was restored with RAPA treatment in this study for 1–6 weeks post-LPS (Fig. 3). In a TBI pre-clinical study in mice, RAPA treatment was able to protect against apoptotic neuronal death [10]. In a focal ischemia model in rats, RAPA was found to also restore NAA levels, as well as Myo-Ins and Cr levels [7]. Our results, along with that of the TBI study mentioned above, indicate that RAPA may be neuronal-protective.
RAPA seems to increase the % change in BOLD signal in the cerebral cortex regions at 3 weeks post-LPS, and in the hippocampus at 6 weeks post-LPS. BOLD MRI images reflect local changes in oxyhemoglobin levels by virtue of the increased spin relaxation rate of the deoxyhemoglobin environment compared to the oxyhemoglobin environment. As a result, the signal decreases with higher tissue metabolism of oxygen, but increases with the perfusion of oxygenated arterial blood [44, 45]. As we used hypercapnia for the stimulus, the primary contributor to the BOLD signal would be changes in arterial blood perfusion, since the signal under the hypercapnia conditions is considered isometabolic [46]. The 3-week post-LPS data of increase % change in BOLD signal in the cerebral cortical regions (Fig. 4A) seems to support the rCBF data at 3 weeks post-LPS in the cerebral cortex region (Fig. 2D). RAPA has also been found to regulate cholesterol biosynthesis and cytoplasmic ribosomal proteins in the hippocampus of APP/PS1 mice [6]. Possibly, by week 6 post-LPS (Fig. 4B), RAPA is affecting similar pathways in the hippocampus, as detected in the AD mouse model. Further studies would need to be conducted to verify this finding. Regardless, the arterial blood perfusion, as measured by BOLD imaging, indicates that RAPA seems to restore vascular integrity in the hippocampus at 6 weeks post-LPS. The amygdala seems also to have a trending increase in % change in BOLD signal; however, this was not found significant.
LPS is known to induce neuroinflammation via microglial activation by activating the NF-κB signaling pathway [47]. In our study, we were able to demonstrate NF-κB protein expression at 6 weeks post-LPS in both the cerebral cortex and hippocampus brain regions (Fig. 5), which was significantly reduced by RAPA treatment. It is also known that LPS induces the expression of HIF-1α at early stages of neuroinflammation [24]. In that same study, RAPA treatment was found to decrease the LPS-induced levels of HIF-1α in the cerebral cortex, as determined from IHC [24]. In our study, we established that HIF-1α was elevated also at later stages of neuroinflammation in both the cerebral cortex and hippocampus regions from a single LPS exposure, and that RAPA was able to reduce the LPS-induced levels of HIF-1α at 6 weeks post-LPS (Fig. 6).
LPS was recently found to increase both mRNA expression and protein expression of EphB2 in the hypothalamus region of mice [48]. Eph receptors in the nervous system are thought to regulate aspects of axon growth [49] and axon guidance [50], responsible for dendritic spine formation and synaptic maturation in hippocampal neurons [51], and facilitate neuronal activation and structural plasticity in learning-associated neurons involved in memory [52]. The signaling pathways of the Eph receptors include molecules known to regulate neurite outgrowth (e.g., neural cell adhesion molecules of the L1 family) [49]. In a DBA/2J glaucoma mouse model, EphB2 was found to be upregulated and coincided with axon loss [53]. It has also been found that there is a connection between TNF-α, NF-κB, and EphB2, where when tissue injury occurs then the activation of EphB2 may be involved in promoting cellular repair in damaged regions, but if the surrounding environment is unfavorable for regrowth, the apoptosis occurs [54]. Although heavily associated with neurons, EphB2 is also expressed in microvascular endothelial cells [55]. In our study, we observed that EphB2 is elevated following LPS exposure (6 weeks post-LPS) in both the cortex and hippocampus regions (Fig. 7Bi and ii). We found that RAPA was found to return EphB2 protein levels to normal (same as saline controls; Fig. 7D). Perhaps the effect of RAPA on neuronal EphB2 supports the MR spectroscopy data where NAA levels were also restored with RAPA treatment (Fig. 3), or additionally support the rCBF vascular data (Fig. 2). To understand the mechanism of action more fully, i.e., whether this is related to neurons or cerebral microvasculature, this will need to be further investigated in future studies.
With respect to BBB, rCBF, and NAA levels, we found that our LPS-induced inflammaging model had similar effects in the brains of normal aging models. Future studies however should directly compare the LPS-induced inflammaging model with normal aging.
RAPA was found to restore BBB, rCBF, and NAA to those similar to the saline-treated controls, in the LPS-induced inflammaging model.
Conclusions
RAPA has the capability to inhibit neuroinflammation in a LPS-induced inflammaging model, via decreasing BBB permeability, normalizing brain vasculature, normalizing inflammatory markers (NF-κB and HIF-1α), and restoring neuronal integrity. RAPA should be considered as a possible therapeutic strategy for various neuroinflammatory diseases. The translatable capability for RAPA into humans is very high.
Funding
Grant funding was provided by the National Institutes of Health (NIH) grants R01 NS092458 and S10 OD023508.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10(11):868–880. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 2.Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, Woodward MA, Miller RA. Rapamycin slows aging in mice. Aging Cell. 2012;11(4):675–682. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bitto A, Ito TK, Pineda VV, LeTexier NJ, Huang HZ, Sutlief E, Tung H, Vizzini N, Chen B, Smith K, Meza D, Yajima M, Beyer RP, Kerr KF, Davis DJ, Gillespie CH, Snyder JM, Treuting PM, Kaeberlein M. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife. 2016;5:e16351. doi: 10.7554/eLife.16351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin AL, Jahrling JB, Zhang W, DeRosa N, Bakshi V, Romero P, Galvan V, Richardson A. Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J Cereb Blood Flow Metab. 2017;37(1):217–226. doi: 10.1177/0271678X15621575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin AL, Zheng W, Halloran JJ, Burbank RR, Hussong SA, Hart MJ, Javors M, Shih YY, Muir E, Solano Fonseca R, Strong R, Richardson AG, Lechleiter JD, Fox PT, Galvan V. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J Cereb Blood Flow Metab. 2013;33(9):1412–1421. doi: 10.1038/jcbfm.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Xia W, Li K, Zhang Y, Ge W, Ma C. Rapamycin regulates cholesterol biosynthesis and cytoplasmic ribosomal proteins in hippocampus and temporal lobe of APP/PS1 mouse. J Neurol Sci. 2019;399:125–139. doi: 10.1016/j.jns.2019.02.022. [DOI] [PubMed] [Google Scholar]
- 7.Chauhan A, Sharma U, Jagannathan NR, Gupta YK. Rapamycin ameliorates brain metabolites alterations after transient focal ischemia in rats. Eur J Pharmacol. 2015;757:28–33. doi: 10.1016/j.ejphar.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Lin X, Mu Z, Shen F, Zhang L, Xie Q, Tang Y, Wang Y, Zhang Z, Yang GY. Rapamycin increases collateral circulation in rodent brain after focal ischemia as detected by multiple modality dynamic imaging. Theranostics. 2019;9(17):4923–4934. doi: 10.7150/thno.32676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chi OZ, Mellender SJ, Barsoum S, Liu X, Damito S, Weiss HR. Effects of rapamycin pretreatment on blood-brain barrier disruption in cerebral ischemia-reperfusion. Neurosci Lett. 2016;620:132–136. doi: 10.1016/j.neulet.2016.03.053. [DOI] [PubMed] [Google Scholar]
- 10.Ding K, Wang H, Wu Y, Zhang L, Xu J, Li T, Ding Y, Zhu L, He J. Rapamycin protects against apoptotic neuronal death and improves neurologic function after traumatic brain injury in mice via modulation of the mTOR-p53-Bax axis. J Surg Res. 2015;194(1):239–247. doi: 10.1016/j.jss.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 11.Fan YY, Nan F, Guo BL, Liao Y, Zhang MS, Guo J, Niu BL, Liang YQ, Yang CH, Zhang Y, Zhang XP, Pang XF. Effects of long-term rapamycin treatment on glial scar formation after cryogenic traumatic brain injury in mice. Neurosci Lett. 2018;678:68–75. doi: 10.1016/j.neulet.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Song Q, Xie D, Pan S, Xu W, et al. Mol Med Rep. 2015;12(5):7203–7210. doi: 10.3892/mmr.2015.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang C, Hu Z, Zou Y, Xiang M, Jiang Y, Botchway BOA, Huo X, Du X, Fang M. The post-therapeutic effect of rapamycin in mild traumatic brain-injured rats ensuing in the upregulation of autophagy and mitophagy. Cell Biol Int. 2017;41(9):1039–1047. doi: 10.1002/cbin.10820. [DOI] [PubMed] [Google Scholar]
- 14.Li D, Liu F, Yang T, Jin T, Zhang H, Luo X, Wang M. Rapamycin protects against neuronal death and improves neurological function with modulation of microglia after experimental intracerebral hemorrhage in rats. Cell Mol Biol (Noisy-le-grand) 2016;62(11):67–75. [PubMed] [Google Scholar]
- 15.van Vliet EA, Otte WM, Wadman WJ, Aronica E, Kooij G, de Vries HE, Dijkhuizen RM, Gorter JA. Blood-brain barrier leakage after status epilepticus in rapamycin-treated rats II: potential mechanisms. Epilepsia. 2016;57(1):70–78. doi: 10.1111/epi.13245. [DOI] [PubMed] [Google Scholar]
- 16.van Vliet EA, Otte WM, Wadman WJ, Aronica E, Kooij G, de Vries HE, Dijkhuizen RM, Gorter JA. Blood-brain barrier leakage after status epilepticus in rapamycin-treated rats I: magnetic resonance imaging. Epilepsia. 2016;57(1):59–69. doi: 10.1111/epi.13246. [DOI] [PubMed] [Google Scholar]
- 17.van Vliet EA, Forte G, Holtman L, den Burger JC, Sinjewel A, de Vries HE, Aronica E, Gorter JA. Inhibition of mammalian target of rapamycin reduces epileptogenesis and blood-brain barrier leakage but not microglia activation. Epilepsia. 2012;53(7):1254–1263. doi: 10.1111/j.1528-1167.2012.03513.x. [DOI] [PubMed] [Google Scholar]
- 18.Drion CM, van Scheppingen J, Arena A, Geijtenbeek KW, Kooijman L, van Vliet EA, Aronica E, Gorter JA. Effects of rapamycin and curcumin on inflammation and oxidative stress in vitro and in vivo - in search of potential anti-epileptogenic strategies for temporal lobe epilepsy. J Neuroinflammation. 2018;15(1):212. doi: 10.1186/s12974-018-1247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russo E, Andreozzi F, Iuliano R, Dattilo V, Procopio T, Fiume G, Mimmi S, Perrotti N, Citraro R, Sesti G, Constanti A, De Sarro G. Early molecular and behavioral response to lipopolysaccharide in the WAG/Rij rat model of absence epilepsy and depressive-like behavior, involves interplay between AMPK, AKT/mTOR pathways and neuroinflammatory cytokine release. Brain Behav Immun. 2014;42:157–168. doi: 10.1016/j.bbi.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 20.Pupyshev AB, Tikhonova MA, Akopyan AA, Tenditnik MV, Dubrovina NI, Korolenko TA. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacol Biochem Behav. 2019;177:1–11. doi: 10.1016/j.pbb.2018.12.005. [DOI] [PubMed] [Google Scholar]
- 21.Xu L, Zhang C, He D, Jiang N, Bai Y, Xin Y. Rapamycin and MCC950 modified gut microbiota in experimental autoimmune encephalomyelitis mouse by brain gut axis. Life Sci. 2020;253:117747. doi: 10.1016/j.lfs.2020.117747. [DOI] [PubMed] [Google Scholar]
- 22.Singh AK, Singh S, Tripathi VK, Bissoyi A, Garg G, Rizvi SI. Rapamycin confers neuroprotection against aging-induced oxidative stress, mitochondrial dysfunction, and neurodegeneration in old rats through activation of autophagy. Rejuvenation Res. 2019;22(1):60–70. doi: 10.1089/rej.2018.2070. [DOI] [PubMed] [Google Scholar]
- 23.Kolosova NG, Vitovtov AO, Muraleva NA, Akulov AE, Stefanova NA, Blagosklonny MV. Rapamycin suppresses brain aging in senescence-accelerated OXYS rats. Aging (Albany NY) 2013;5(6):474–484. doi: 10.18632/aging.100573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mengke NS, Hu B, Han QP, Deng YY, Fang M, Xie D, Li A, Zeng HK. Rapamycin inhibits lipopolysaccharide-induced neuroinflammation in vitro and in vivo. Mol Med Rep. 2016;14(6):4957–4966. doi: 10.3892/mmr.2016.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu YC, Gao XX, Chen L, You XQ. Rapamycin suppresses Aβ25-35- or LPS-induced neuronal inflammation via modulation of NF-κB signaling. Neuroscience. 2017;355:188–199. doi: 10.1016/j.neuroscience.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 26.Yang MT, Lin YC, Ho WH, Liu CL, Lee WT. Everolimus is better than rapamycin in attenuating neuroinflammation in kainic acid-induced seizures. J Neuroinflammation. 2017;14(1):15. doi: 10.1186/s12974-017-0797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dello Russo C, Lisi L, Tringali G, Navarra P. Involvement of mTOR kinase in cytokine-dependent microglial activation and cell proliferation. Biochem Pharmacol. 2009;78(9):1242–1251. doi: 10.1016/j.bcp.2009.06.097. [DOI] [PubMed] [Google Scholar]
- 28.Carosi JM, Sargeant TJ. Rapamycin and Alzheimer disease: a double-edged sword? Autophagy. 2019;15(8):1460–1462. doi: 10.1080/15548627.2019.1615823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chi OZ, Kiss GK, Mellender SJ, Liu X, Weiss HR. Rapamycin decreased blood-brain barrier permeability in control but not in diabetic rats in early cerebral ischemia. Neurosci Lett. 2017;654:17–22. doi: 10.1016/j.neulet.2017.06.021. [DOI] [PubMed] [Google Scholar]
- 30.Towner RA, Saunders D, Smith N, Towler W, Cruz M, Do S, Maher JE, Whitaker K, Lerner M, Morton KA. Assessing long-term neuroinflammatory responses to encephalopathy using MRI approaches in a rat endotoxemia model. Geroscience. 2018;40(1):49–60. doi: 10.1007/s11357-018-0009-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Towner RA, Saunders D, Smith N, Gulej R, McKenzie T, Lawrence B, Morton KA. Anti-inflammatory agent, OKN-007, reverses long-term neuroinflammatory responses in a rat encephalopathy model as assessed by multi-parametric MRI: implications for aging-associated neuroinflammation. Geroscience. 2019;41(4):483–494. doi: 10.1007/s11357-019-00094-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Burlington: Academic Press; 2007. [Google Scholar]
- 33.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 34.Kim KA, Jeong JJ, Yoo SY, Kim DH. Gut microbiota lipopolysaccharide accelerates inflamm-aging in mice. BMC Microbiol. 2016;16:9. doi: 10.1186/s12866-016-0625-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee P, Kim J, Williams R, Sandhir R, Gregory E, Brooks WM, Berman NE. Effects of aging on blood brain barrier and matrix metalloproteases following controlled cortical impact in mice. Exp Neurol. 2012;234(1):50–61. doi: 10.1016/j.expneurol.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffman JD, Parikh I, Green SJ, Chlipala G, Mohney RP, Keaton M, Bauer B, Hartz AMS, Lin AL. Age drives distortion of brain metabolic, vascular and cognitive functions, and the gut microbiome. Front Aging Neurosci. 2017;9:298. doi: 10.3389/fnagi.2017.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei BB, Liu MY, Zhong X, Yao WF, Wei MJ. Increased BBB permeability contributes to EGCG-caused cognitive function improvement in natural aging rats: pharmacokinetic and distribution analyses. Acta Pharmacol Sin. 2019;40(11):1490–1500. doi: 10.1038/s41401-019-0243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu D, Ahmet I, Griess B, Tweedie D, Greig NH, Mattson MP. Age-related impairment of cerebral blood flow response to KATP channel opener in Alzheimer’s disease mice with presenilin-1 mutation. J Cereb Blood Flow Metab. 2020:271678X20964233. 10.1177/0271678X20964233. [DOI] [PMC free article] [PubMed]
- 39.Ohata M, Sundaram U, Fredericks WR, London ED, Rapoport SI. Regional cerebral blood flow during development and ageing of the rat brain. Brain. 1981;104(2):319–332. doi: 10.1093/brain/104.2.319. [DOI] [PubMed] [Google Scholar]
- 40.Chang L, Munsaka SM, Kraft-Terry S, Ernst T. Magnetic resonance spectroscopy to assess neuroinflammation and neuropathic pain. J NeuroImmune Pharmacol. 2013;8(3):576–593. doi: 10.1007/s11481-013-9460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piekarz KM, Bhaskaran S, Sataranatarajan K, Street K, Premkumar P, Saunders D, Zalles M, Gulej R, Khademi S, Laurin J, Peelor R, Miller BF, Towner R, Van Remmen H. Molecular changes associated with spinal cord aging. Geroscience. 2020;42(2):765–784. doi: 10.1007/s11357-020-00172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu M, Akimana C, Wang E, Ng CK. 1H-MRS Quantitation of age-dependent taurine changes in mouse brain. Mol Imaging Biol. 2019;21(5):812–817. doi: 10.1007/s11307-019-01333-6. [DOI] [PubMed] [Google Scholar]
- 43.Shi L, Olson J, D'Agostino R, Jr, Linville C, Nicolle MM, Robbins ME, Wheeler KT, Brunso-Bechtold JK. Aging masks detection of radiation-induced brain injury. Brain Res. 2011;1385:307–316. doi: 10.1016/j.brainres.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoge RD, Atkinson J, Gill B, Crelier GR, Marrett S, Pike GB. Investigation of BOLD signal dependence on cerebral blood flow and oxygen consumption: the deoxyhemoglobin dilution model. Magn Reson Med. 1999;42(5):849–863. doi: 10.1002/(sici)1522-2594(199911)42:5<849::aid-mrm4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 45.Mitschelen M, Garteiser P, Carnes BA, Farley JA, Doblas S, Demoe JH, Warrington JP, Yan H, Nicolle MM, Towner R, Sonntag WE. Basal and hypercapnia-altered cerebrovascular perfusion predict mild cognitive impairment in aging rodents. Neuroscience. 2009;164(3):918–928. doi: 10.1016/j.neuroscience.2009.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bulte DP, Drescher K, Jezzard P. Comparison of hypercapnia-based calibration techniques for measurement of cerebral oxygen metabolism with MRI. Magn Reson Med. 2009;61(2):391–398. doi: 10.1002/mrm.21862. [DOI] [PubMed] [Google Scholar]
- 47.Zhao J, Bi W, Xiao S, Lan X, Cheng X, Zhang J, Lu D, Wei W, Wang Y, Li H, Fu Y, Zhu L. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci Rep. 2019;9(1):5790. doi: 10.1038/s41598-019-42286-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu Y, Wei Z, Li Y, Wei C, Li Y, Cheng P, Xu H, Li Z, Guo R, Qi X, Jia J, Jia Y, Wang W, Gao X. Perturbation of Ephrin receptor signaling and glutamatergic transmission in the hypothalamus in depression using proteomics integrated with metabolomics. Front Neurosci. 2019;13:1359. doi: 10.3389/fnins.2019.01359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zisch AH, Pasquale EB. The Eph family: a multitude of receptors that mediate cell recognition signals. Cell Tissue Res. 1997;290(2):217–226. doi: 10.1007/s004410050926. [DOI] [PubMed] [Google Scholar]
- 50.Robichaux MA, Chenaux G, Ho HY, Soskis MJ, Greenberg ME, Henkemeyer M, Cowan CW. EphB1 and EphB2 intracellular domains regulate the formation of the corpus callosum and anterior commissure. Dev Neurobiol. 2016;76(4):405–420. doi: 10.1002/dneu.22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Henkemeyer M, Itkis OS, Ngo M, Hickmott PW, Ethell IM. Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippocampus. J Cell Biol. 2003;163(6):1313–1326. doi: 10.1083/jcb.200306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Talebian A, Henkemeyer M. EphB2 receptor cell-autonomous forward signaling mediates auditory memory recall and learning-driven spinogenesis. Commun Biol. 2019;2:372. doi: 10.1038/s42003-019-0625-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Du J, Tran T, Fu C, Sretavan DW. Upregulation of EphB2 and ephrin-B2 at the optic nerve head of DBA/2J glaucomatous mice coincides with axon loss. Invest Ophthalmol Vis Sci. 2007;48(12):5567–5581. doi: 10.1167/iovs.07-0442. [DOI] [PubMed] [Google Scholar]
- 54.Pozniak PD, White MK, Khalili K. TNF-α/NF-κB signaling in the CNS: possible connection to EPHB2. J NeuroImmune Pharmacol. 2014;9(2):133–141. doi: 10.1007/s11481-013-9517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shirai T, Fujii H, Ono M, Watanabe R, Shirota Y, Saito S, Ishii T, Nose M, Harigae H. A novel autoantibody against ephrin type B receptor 2 in acute necrotizing encephalopathy. J Neuroinflammation. 2013;10:128. doi: 10.1186/1742-2094-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]