

Abstract
Cancer cachexia is a multifactorial syndrome that is characterised by a loss of skeletal muscle mass, is commonly associated with adipose tissue wasting and malaise, and responds poorly to therapeutic interventions. Although cachexia can affect patients who are severely ill with various malignant or non-malignant conditions, it is particularly common among patients with pancreatic cancer. Pancreatic cancer often leads to the development of cachexia through a combination of distinct factors, which, together, explain its high prevalence and clinical importance in this disease: systemic factors, including metabolic changes and pathogenic signals related to the tumour biology of pancreatic adenocarcinoma; factors resulting from the disruption of the digestive and endocrine functions of the pancreas; and factors related to the close anatomical and functional connection of the pancreas with the gut. In this review, we conceptualise the various insights into the mechanisms underlying pancreatic cancer cachexia according to these three dimensions to expose its particular complexity and the challenges that face clinicians in trying to devise therapeutic interventions.
Subject terms: Cancer metabolism, Gastrointestinal cancer
Background
Cachexia is a multifactorial syndrome defined by a loss of skeletal muscle mass, with or without a loss of fat mass, that cannot be fully reversed by conventional nutritional support and that leads to progressive functional impairment of the entire organism.1 In humans, cachexia can occur as a consequence of a number of chronic diseases, such as chronic renal failure, AIDS, advanced dementia and cancer. Cachexia associated with cancer has specific tumour-related pathomechanisms that include altered energy metabolism, the presence of pro-inflammatory signals and tumour-derived catabolic factors, sarcopenia, and adipose tissue depletion,2 and is therefore distinct from malnutrition, as well as being a prognostic indicator of poor overall survival independent of the initial body mass index (BMI) of the patient.3
Pancreatic cancer has one of the poorest prognoses of all solid tumours, with a 5-year relative survival rate below 8% across all disease stages.4 This poor prognosis is a consequence of a number of factors—the disease usually progresses rapidly, has high rates of recurrence after surgery, is commonly resistant to chemotherapy and is especially associated with the development of cachexia. In fact, as many as 63–64% of patients with pancreatic cancer develop cachexia over the course of their disease.5–7 At diagnosis, the prevalence varies more markedly, between 21.3 and 63%, which is likely to reflect heterogeneity between the patient cohorts and differences in the definition of cachexia.5,6,8,9 Pancreatic cancer patients who develop cachexia commonly suffer from poor appetite with or without concomitant fatigue, depression and cancer pain, lose muscle and fat mass despite nutritional support and frequently show signs of systemic inflammation. Additionally, different degrees of maldigestion with steatorrhoea and bloating due to poor pancreatic exocrine function, impaired glucose tolerance and other gastrointestinal complications due to the interaction of the pancreas with other organs can worsen this clinical picture. For patients with advanced pancreatic cancer, the presence of sarcopenia at diagnosis or the loss of skeletal muscle mass during chemotherapy is clinically extremely relevant and is a stronger prognostic factor of poor overall survival than an Eastern Cooperative Oncology Group (ECOG) performance status of ≥2.8 Accordingly, weight loss, digestive problems and loss of appetite significantly increase with disease progression and patients report deterioration in their overall physical quality of life (QoL) at later stages of pancreatic cancer.10 In addition, the responsibility of caregivers to assist patients with needs related to these symptoms is a major source of stress.11
The high prevalence and clinical importance of cachexia in the context of pancreatic cancer is likely to depend on distinct effects of the tumour on the organism (mediated by tumour-related systemic factors), the role of the pancreas in digestion and nutrient uptake as well as glucose homoeostasis (mediated by factors related to alterations in pancreatic function), and its close interaction with other digestive organs (mediated by digestive tract-related factors) (Fig. 1). In this review, we describe insights into the mechanisms underlying pancreatic cancer cachexia in these three dimensions and present the emerging evidence for potential clinical interventions.
Fig. 1. Conceptualisation of the three dimensions involved in the development of pancreatic cancer cachexia.
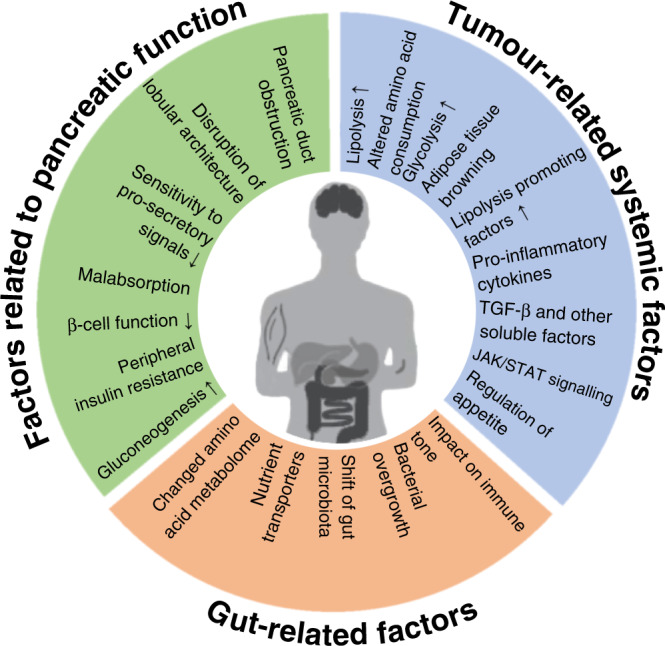
Pancreatic cancer cachexia is a complex syndrome that integrates three interdependent dimensions: tumour-related systemic factors; factors related to alterations of the pancreatic function; and factors related to the close interaction of the pancreas with other digestive organs. The systemic factors outlined are partly specific for cachexia associated with pancreatic adenocarcinoma, partly generic for that associated with solid malignancies. Factors related to the central function of the pancreas for nutritional uptake and homoeostasis, and factors related to other digestive organs are characteristic of pancreatic cancer cachexia.
Pancreatic tumour-derived factors associated with cachexia
Pancreatic cancer induces metabolic shifts, inflammatory signals and other factors which result in complex interactions with the organism. Together, they are the key mechanisms of the systemic dimension of the pathophysiology of pancreatic cancer cachexia.
KRAS-dependent metabolic changes related to pancreatic cancer
Activating mutations in the KRAS oncogene are found in up to 95% of all human pancreatic adenocarcinomas and the vast majority of these mutations cluster into only three different variants in exon 12: G12D, G12V and G12R.12 Major insights into tumour-related mechanisms of cachexia in the context of pancreatic cancer have been derived from animal models (Box 1). In mice harbouring the G12D variant, Kras-dependent signalling increases glycolysis in pancreatic cancer cells by inducing the expression of the GLUT1 glucose transporter and transcriptional upregulation of several rate-limiting glycolytic enzymes.13 Increased glycolysis is an important adaptive process in hypoxic Kras-mutant pancreatic cancer cells and promotes cancer cell survival.14 Although constituting an energetically inefficient form of glucose metabolism, a high rate of glycolysis and lactate fermentation in the cytosol, known as the Warburg effect, is likely to facilitate the proliferation of pancreatic cancer cells in the absence of oxygen,15 and heterotopic implantation of pancreatic cancer cell lines with exceptionally high levels of glycolysis has been associated with increased weight loss and the induction of skeletal-muscle proteolysis, adipose tissue lipolysis and hepatic gluconeogenesis, which are catabolic processes characteristic of cachexia (Fig. 2).16
Fig. 2. Tumour-derived factors associated with cachexia in pancreatic adenocarcinoma.

Tumours increasingly metabolise glucose through glycolysis which is increased through mutant KRAS-dependent upregulation of glycolytic enzymes (1). Other KRAS-dependent metabolic changes promote the use of other carbon sources such as glutamine and branched chain amino acids (BCAA) from the breakdown of peripheral tissue, as well as other non-essential amino acids from pancreatic stellate cell autophagy, in the TCA cycle; substrates from the TCA cycle are used for biosynthesis (2). Together, these changes increase survival in a hypoxic and nutrient-deficient environment. Tumour-derived cytokines prompt several catabolic effects in peripheral tissues (3). Interleukin (IL)-6, TNF or interferon γ (IFNg) induce the breakdown of muscle fibres through the Janus kinase (JAK)/signal transducer and activator of transcription (STAT3) pathway and nuclear factor (NF)-κB-dependent induction of nitric oxide (4) which leads to decreased myogenesis through downregulation of the myogenic regulatory factor MyoD and increased proteolysis through E3-ligase-dependent ubiquitination (5). Similarly, members of the transforming growth factor (TGF)-β superfamily activate the canonical Smad2/3 pathway which promotes ubiquitin ligase-mediated proteolysis and scleraxis-mediated muscle fibrosis and reduces Akt/mTOR-mediated myogenesis (6). Additionally, microRNAs (miRNAs) in tumour-derived extracellular vesicles induce myoblast apoptosis by Toll-like receptor 7-dependent c-Jun N-terminal kinase (JNK) signalling (7). In white adipose tissue, pro-inflammatory signals, specifically IL-6, promote lipolysis through activation of the JAK/STAT3 pathway and NF-kB in adipose tissue and induces adipocyte browning through upregulation of uncoupling protein 1 (UCP-1) (8). Lipolysis through hormone-sensitive lipase (HSL) are also mediated by adrenomedullin from tumour-derived exosomes which activates the p38 mitogen-activated protein kinase (MAPK) pathway in adipocytes. Exosomal cargoes (other than adrenomedullin) can also induce adipocyte browning through UCP-1 expression (9). Lipolysis is maintained through paracrine signals in fat tissue liked zinc-α2-glycoprotein (ZAG/LMF) and miRNAs which induce adipose triglyceride lipase (ATGL) and HSL catalyse the hydrolysis of stored triglycerides (10). In the central nervous system, pro-inflammatory cytokines, particularly IL-1β, and macrophage inhibitory cytokine (MIC)-1 (a member of the TGF-β superfamily), induce anorexia by reducing anabolic neuropeptide Y (NPY)/agouti-related protein (AgRP) and increasing catabolic proopiomelanocortin (POMC) and cocaine-and-amphetamine regulated transcript (CART) signalling (11).
KrasG12D/+ also promotes the preferential use of glycolytic metabolites in biosynthesis pathways and promotes glutamine as the main carbon source for the tricarboxylic acid (TCA) cycle instead of glucose13,17 In fact, increased glycolysis and glutamine metabolism in Kras-mutant pancreatic cancer cells are closely interlinked by some of the downstream anabolic pathways, and depend on each other to promote tumour aggressiveness and survival in the tumour’s hypoxic desmoplastic environment.14 These metabolic changes result in the increased uptake and consumption of glutamine, consistent with the observation that malignant transformation of precursor lesions in KrasG12D/+ mice (but not in non-malignant pancreatic pathologies) correlates with a reduction of free circulating glutamine levels.18 Skeletal muscle is the main reservoir of glutamine in the body, and glutamine is the primary amino acid released by catabolic breakdown of muscle mass.19 Additionally, pancreatic stellate cells of the tumour microenvironment provide other non-essential amino acids—specifically alanine—through autophagy as an alternative mitochondrial carbon source for tumour cells independent of circulating nutrients.20 This appears to be an important factor for shifting carbon from glucose towards biosynthesis and, more broadly, for supporting the altered energy metabolism related to pancreatic cancer cachexia.
Mouse models with early Kras-driven pancreatic cancers show, in addition to decreased glutamine levels, elevated levels of branched-chain amino acids (BCAAs; Fig. 2).18,21 BCAAs supply 20% of the TCA cycle carbon input in the healthy pancreas,22 but Kras-driven pancreatic cancer cell TCA cycle metabolism becomes independent of BCAA in early carcinogenesis; this contrasts with Kras-driven tumours originating in other tissues, which are dependent on the use of BCAAs.23,24 Although not crucial for pancreatic cancer cells’ energetic requirements, studies in KPC mice found generation of BCAAs through the breakdown of muscle and other body protein and in vitro studies identified them as an important source of carbon for the fatty acid biosynthesis.21,25 Detection of BCAAs in clinical samples obtained before a diagnosis of pancreatic cancer in human patients suggest that similar KRAS-dependent processes drive early proteolysis in humans.21
Box 1 Lessons from mouse models.
Most widely used in the study of pancreatic cancer are the KrasLSL-G12D/+ Trp53R172H/+ Pdx1Cre/+ (KPC) and the KrasLSL-G12D/+ Ink4a/Arffl/fl Pdx1Cre/+ mouse models, in which tumour development is driven by genetic alterations that reflect frequent equivalent somatic mutations in human pancreatic cancers.205,206 These genetically engineered mouse models develop pancreatic carcinomas with a rich desmoplastic stroma and metastatic pattern characteristic of the human disease. Unfortunately, however, the KPC mouse model does not recapitulate the clinical patterns of tissue wasting observed in human patients, and the differential gene expression in skeletal muscle samples from KPC mice was not consistent with that observed in muscle biopsy samples from cachectic pancreatic cancer patients.207 Accordingly, to more fully recapitulate the pattern and pathophysiology of the loss of muscle mass and adipose tissue observed in humans, a new mouse model incorporating tamoxifen-inducible mutant Kras and two floxed Pten-alleles, the KrasLSL-G12D/+; Ptf1aER-Cre/+; Ptenf/f (KPP) mouse, has been engineered.207 Additionally, various orthotopic and heterotopic implantation models of different murine or human pancreatic cancer cell lines and human tumour xenografts in susceptible mice have been used to study cachexia,208 although how well they reflect pancreatic cancer cachexia in humans has not been systematically assessed. Comparison of xenografts of human cancer cell lines within the flank and within the pancreas appears to suggest that certain aspects of cachexia, including the upregulation of genes associated with atrophy in the skeletal muscle, an increase of cachexia-associated cytokines and the development of significant muscle wasting, depend on the presence of the tumour in the pancreas.54 Generally, caution should be given to the potential discrepancies between cachexia in mouse models and the human disease.
Tumour-related changes in adipose tissue physiology
Lipolysis is the main mechanism of adipose tissue wasting in patients with pancreatic cancer. White adipose tissue stores large amounts of energy that can be made available to the organism. In response to fasting or increased physical activity, lipolysis hydrolyses triglycerides into three fatty acids and glycerol, which are released into the blood circulation for β-oxidation into acetyl CoA by other organs and the liver (fatty acids) or for triglyceride synthesis or gluconeogenesis in the liver (glycerol). In a cohort of non-cancer patients and patients with gastrointestinal cancers, the majority of whom had pancreatic cancer, the loss of body fat significantly correlated with decreased adipocyte size and increased plasma levels of fatty acids and glycerol in the cancer patients. By contrast, other potential causes of fat loss—a decrease in the number of adipocytes, reduced lipogenesis, and inflammation in the adipose tissue—did not differ between cancer patients with or without adipose tissue loss and non-cancer patients.26 Interestingly, the increased level of lipolysis in the fat of patients with gastrointestinal cancer with weight loss does not decrease with glucose infusion, and these patients have a reduced ability to oxidise circulating fatty acids,27 indicating that the primary reason for the upregulation of lipolysis in cachectic pancreatic cancer patients might not be to fulfil the energetic requirements of the tumour. However, the clinical studies that underpin this conclusion are relatively small in size. These results are also difficult to reconcile with the observations that an increased supply of fatty acids triggers fatty acid oxidation, the formation of intracellular lipid droplets, proliferation of MiaPaCa2 human pancreatic cancer cells and the growth of larger tumours in an orthotopic implantation model.28 In the absence of a supply of additional exogenous fatty acids, mice with MiaPaCa2 implants showed a decrease in the size of inguinal and epididymal fat pads along with reduced plasma levels of fatty acids, consistent with increased fatty acid consumption.16
The results from studies in mouse models of pancreatic and other cancers suggest that upregulation of adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) in white adipose tissue is the major mechanism by which pancreatic cancer increases lipolysis (Fig. 2).16,25,29 One well-characterised mediator of increase pancreatic cancer-related lipolysis and weight loss in mice is the zinc-α2-glycoprotein (ZAG), also termed lipid-mobilising factor (LMF).30,31 It is, however, mainly expressed by adipocytes as opposed to originating from pancreatic cancer cells and does not regulate the interaction between the tumour and the adipose tissue.32,33 Similarly, overexpression of the micro-RNA 378 in human adipose tissue promotes lipolysis by regulating key lipolytic regulators, including HSL and ATGL, in cachectic patients with pancreatic cancer and other gastrointestinal tumours.34 The direct crosstalk between pancreatic tumour cells and adipocytes might depend on adrenomedullin contained in extracellular vesicles. Results from in vitro studies of human samples and studies in mice suggest that cancer-cell-derived exosomes are internalised by subcutaneous adipocytes into which they release their cargo—a specific ligand that activates the adrenomedullin receptor—leading to activation of the intracellular p38 mitogen-activated protein kinase (MAPK) signalling pathway and ultimately resulting in activation of HSL.35
In addition to increased rates of lipolysis, pancreatic cancer prompts cachexia through increased energy expenditure caused by a phenotypic shift from white adipose tissue to brown adipose tissue, termed adipose tissue browning. Adipose tissue browning occurs as a result of an increased number of mitochondria, the iron content of which confers the brown colour. The metabolic hallmark of adipose tissue browning is the uncoupling of the electron transport chain across the inner mitochondrial membrane of adipocytes mediated by the expression of uncoupling protein 1 (UCP-1; thermogenin), which generates heat by uncoupling oxidative phosphorylation from ATP production.36 In KPC and KrasLSL-G12D/+ Trp53f/f mice, adipose tissue browning linked to increased UCP-1 expression occurs even before the onset of fat wasting.37 Similarly, increased UCP-1 expression and a rise in body temperature occurs in patients with pancreatic cancer before the clinical manifestation of the disease.38,39 Interestingly, the mechanisms involved in pancreatic-cancer-induced lipolysis have also been implicated in the induction of adipose tissue browning. ZAG isolated from the urine of pancreatic cancer patients increased the expression of UCP-1 in wild-type mice, and ZAG exposure correlated with increased energy expenditure through increased UCP-1 expression in a dose-dependent manner.31,40 Extracellular vesicles released by the cancer cells have again been identified as intermediaries between the tumour and the adipose tissue; mRNA sequencing of adipocytes exposed to pancreatic cancer-derived exosomes in vitro revealed activation of pathways associated with browning in addition to promotion of lipolysis, fibrosis and acute inflammation.39
Pancreatic-cancer-related systemic inflammation
Systemic inflammation, characterised by increased levels of serum cytokines and acute-phase proteins, is exceedingly common among pancreatic cancer patients and is a well-established independent predictor of poor survival.37,41 Pro-inflammatory signals that arise from the interaction between a tumour and the immune system have been implicated in various pathomechanisms of cancer cachexia.2 In the context of pancreatic cancer, several of these signals, especially interleukin (IL)-6 and tumour necrosis factor (TNF), have been linked to increased resting energy expenditure and cachexia.41 The correlation of various serum cytokine and acute-phase protein levels with the occurrence of cachexia is, however, inconsistent across several studies,42–48 suggesting that systemic levels of inflammatory markers ineffectively capture the degree to which inflammation contributes to the development of pancreatic cancer cachexia.
A mechanistic explanation for the discrepancy between different clinical observations might be the in vitro finding that some, but not all, pancreatic cancer cells directly express IL-6, which, in turn, triggers additional, local IL-6 expression by tumour-sensitised peripheral blood mononuclear cells (PBMC) in the liver.49 Higher serum levels of IL-6 and a correlation with weight loss have also been linked to genetic polymorphisms in the IL6 gene, which might explain differences between pancreatic cancer patients in terms of cachexia susceptibility.45,50
Pancreatic cancer cells have also been identified as the source of other pro-inflammatory cytokines, including TNF, IL-1β, IL-8 and the monocyte chemoattractant protein-1 (MCP-1/CCL2).51–53 Orthotopic pancreatic cancer xenograft models that lack an adaptive immune response have been used to investigate the association between the tumour secretome and systemic cytokine production. Tumour grafts were seen to alter cytokine production in splenic tissue, and these altered cytokine levels could be linked to the expression of atrophy-associated genes and muscle wasting, but patterns were not consistent between two different studies.54,55 Additionally, the expression of TNF and MCP-1 was locally increased in adipose tissue of patients with pancreatic cancer cachexia, highlighting the importance of localised immunopathology in affected peripheral tissues.56 In summary, the immune-related mechanisms of pancreatic cachexia are complex and are likely to be heterogeneous across different patients.
Despite this complexity, dysregulation of some canonical intracellular pathways linked to cachexia underlies some of the effects that are mediated by cytokines associated with pancreatic cancer (Fig. 2). In skeletal muscle, pancreatic-cancer-derived IL-6 induces muscle fibre atrophy and exacerbates wasting through activation of the Janus kinase (JAK)/signal transducer and activator of transcription (STAT3) pathway.57 When complexed with endogenous soluble IL-6 receptor, IL-6 secreted by tumour cells induces autophagy of differentiated myocytes and, consequently, sarcopenia.58 Independent of IL-6, TNF or interferon γ (IFNγ) can activate JAK/STAT3 signalling in myocytes in vitro and in experimental mouse models, which promotes activation of the inducible nitric oxide synthase (iNOS)/nitric oxide (NO) pathway through nuclear factor (NF)-κB,59 leading to sarcopenia. Activation of iNOS reduces myogenesis by general downregulation of protein synthesis and specifically downregulates the transcription of MyoD, a regulator of muscle renewal and maintenance.60 Additionally, TNF and IFNγ promote muscle wasting by upregulating the expression of ubiquitin and muscle-specific E3 ligases, which catalyse ubiquitination of the heavy chain of the muscle motor protein myosin and its breakdown in the proteasome.61
In white adipose tissue, IL-6 secreted by infiltrating macrophages can trigger browning and moderately increased energy expenditure by inducing UCP-1. This effect is enhanced when IL-6 synergises with β-adrenergic activation, a hallmark of acute or chronic stress, linking inflammation with central stimuli.38 Some of the catabolic effects of IL-6 on white adipose tissue are also mediated through the JAK/STAT3 pathway and NF-κB, as specifically demonstrated in the KrasLSL-G12D/+ Ink4a/Arffl/fl Pdx1Cre/+ mouse model.62 Although similar to the signalling pathways involved in muscle wasting and a probable integrator of several inflammatory signals, exactly how STAT3 induces lipolysis is unknown.63
Systemic inflammation is also linked to cancer cachexia through a loss of appetite and reduced caloric intake mediated by downregulation of neuropeptide Y (NPY) and the agouti-related protein (AgRP), important anabolic signals in the central nervous system. Pro-inflammatory cytokines including TNF and IL-1β are overexpressed in the hypothalamus of cachexia models;64,65 in particular, IL-1β has been implicated in the direct inhibition of NPY expression.66 Cytokines also induce proopiomelanocortin (POMC) and cocaine-and-amphetamine regulated transcript (CART), two anorexigenic signals in hypothalamic neurons.67 The indirect pathophysiological signals that link inflammation to anorexia are complex and include the effect of cytokines on feedback loops that involve the hormones leptin and ghrelin, which regulate appetite through NPY/AgRP and POMC/CART signalling in the hypothalamus.68
Observational studies also suggest that elevated levels of pro-inflammatory cytokines, particularly IL-6, correlate with depression.69,70 Speculatively, increased levels of inflammatory signals could therefore provide a mechanism for the occurrence of depression prior to a pancreatic cancer diagnosis.71 Depression, in turn, is an important risk factor for insufficient nutritional intake and weight loss.72
TGF-β and other tumour-related soluble factors
Transforming growth factor β 1 (TGF-β1) has a key regulatory function in the tumour microenvironment of pancreatic cancer and regulates some of the hallmarks of this cellular milieu, including the induction of desmoplasia and immunosuppression through T regulatory cells.73,74 High doses of TGF-β1 can trigger experimental cachexia and systemic fibrosis in mice.75 In skeletal muscle, these effects are mediated by the induction of proteolysis through the E3 ligase atrogin-1 and induction of the transcription factor scleraxis, which stimulates fibroblast proliferation and collagen synthesis (Fig. 2).76 Blockade of TGF-β1-3 ameliorates muscle wasting in the KPC pancreatic cancer model and, of note, also improves the preservation of adipose tissue and bone density, indicating additional relevant effects of TGF-β signalling in the development of cachexia.77
Other members of the TGF-β superfamily, including activins and myostatin, have been implicated in muscle wasting through activation of the canonical Smad2/3 pathway, which decreases Akt/mTOR-mediated protein synthesis in addition to increasing ubiquitin ligase-mediated proteolysis and promoting a fibrotic response.78,79 Specific isoforms of activin are overexpressed and secreted by human pancreatic cancer cells and in the KPC pancreatic cancer model, and elevated serum levels and increased expression of activins in peripheral tissues have been found in cachectic KPC mice. Interestingly, the Smad2/3 pathway was dispensable for pancreatic-cancer-related activin-induced muscle wasting, which depended on an alternative p38 MAPK catabolic pathway instead.80
Serum levels of MIC-1 (macrophage inhibitory cytokine-1), another member of the TGF-β superfamily, are significantly elevated among patients with pancreatic cancer.81 MIC-1 triggers anorexia through the TGF-β receptor II expressed on hypothalamic neurons, as well as dysregulation of NPY/AgRP and POMC/CART using a similar effector mechanism as pro-inflammatory cytokines.82
Additional pancreatic-cancer-derived factors
In addition to pro-inflammatory cytokines and members the TGF-β superfamily, several other pancreatic-cancer-derived factors stimulate protein catabolism. Proteolysis-inducing factor (PIF) is a glycoprotein specifically associated with cancer cachexia.83 PIF activates NF-κB to upregulate the ubiquitination and proteolysis of skeletal muscle and decreases protein synthesis by phosphorylation of the transcription factor eIF2α.84–86 Serum levels of PIF are significantly increased among pancreatic cancer patients with weight loss independent of the level of inflammation markers.87 However, the secretion of PIF by pancreatic cancer cells promotes the production of IL-6, IL-8 and acute phase proteins in the liver in a STAT3- and NF-κB-dependent fashion, potentially co-operating with the detrimental effects of pancreatic-cancer-related inflammation.88
Another tumour-derived factor that contributes to pancreatic cancer cachexia is the insulin-like growth factor (IGF)-binding protein-3 (IGFBP-3). IGFBP-3 targets myoblast renewal by inhibiting the expression of MyoD and another promyogenic transcription factor, myogenin, and enhances muscle proteolysis by suppressing the PI3K/AKT pathway.89
Tumour-derived extracellular vesicles are increasingly recognised as vehicles of molecular cargo that induce skeletal muscle wasting, similar to their role in the upregulation of lipolysis. Extracellular vesicles from pancreatic cancer cells promote apoptosis in myoblasts by the intracellular delivery of miRNAs that stimulate Toll-like receptor (TLR) 7 to activate c-Jun N-terminal kinase activity90. Tumour-derived extracellular vesicles also promote muscle catabolism by releasing Hsp70 and Hsp90 extracellularly, which activate TLR4 and promote muscle wasting in a mouse model, although the relevance of this mechanism in pancreatic cancer cachexia is unknown.91
Alterations of pancreatic function
The pancreas has a dual exocrine and endocrine function. It secretes lipases, proteases and amylase, which are crucial for the breakdown of macromolecules and nutrient uptake in the gut, and controls glucose homoeostasis through the hormone’s insulin and glucagon. Pancreatic exocrine insufficiency (PEI), in which insufficient levels of pancreatic enzymes are produced, affects two-thirds of patients with pancreatic head tumours at diagnosis, and over the course of the disease this number increases to more than nine out of ten patients.92 Patients with PEI can develop malnutrition owing to insufficient caloric uptake and different deficiencies of essential amino acids and various micronutrients including lipophilic vitamins, iron and folic acid. Similarly, three-quarters of patients with pancreatic cancer have compromised endocrine function at diagnosis, characterised by impaired glucose tolerance or overt diabetes.93 Tumour surgery, especially pancreatoduodenectomy, is also an important cause of iatrogenic PEI among pancreatic cancer patients.94–96 However, in contrast to exocrine function, endocrine function frequently improves after tumour resection if the patient had poor glucose control prior to surgery.97–99
Pancreatic cancer, PEI and cachexia
Pancreatic cancer can induce PEI directly by affecting the pancreatic tissue as well as indirectly by undermining the physiological regulation of pancreatic secretion.100 Pancreatic head tumours commonly obstruct the pancreatic duct, thereby blocking the secretion of pancreatic enzymes necessary to break down nutrients in the intestine. Pancreatic cancer also induces a characteristic intra- and peri-tumoural stroma that is enriched in extracellular matrix and deficient in cells and blood vessels that are able to replace functional pancreatic tissue.101 Moreover, the microscopic architecture of the pancreatic lobules is often disrupted, especially in pancreases with tumours that arise from precursor lesions (Fig. 3).102
Fig. 3. Impaired pancreatic exocrine and endocrine function interact with alterations in the digestive tract to promote pancreatic cancer cachexia.

Pancreatic cancer induces pancreatic exocrine insufficiency (PEI) (1) through obstruction of the pancreatic duct (2) or disruption of the organ architecture (3) as well as an altered physiological function, i.e. a reduced sensitivity to the pro-secretory signals secretin and cholecystokinin (CCK) resulting in an impaired priming of the pancreatic (4). PEI results in malabsorption of nutrients including lipophilic vitamins and n-3 fatty acids which results in insufficient caloric uptake and a lack of anabolic or anti-catabolic signals (5). PEI, in combination with changes of the histological architecture of the pancreas, also leads to excess bacterial growth in the duodenum and the translocation of bacteria and bacterial components into the tumour with implications for the immune tone (6). Pancreatic cancer is associated with specific changes of the gut microbiota and outgrowth of Enterobacteriaceae (7), particularly Klebsiella, is associated with increased intestinal permeability and systemic inflammation linked to the development of cachexia (8). Pancreatic enzyme replacement therapy (PERT) counteracts cancer-related dysbiosis and promotes the abundance of Lactobacillus reuteri and Akkermansia muciniphila which potentially reduce muscle wasting and increase the anti-tumoural immune response, respectively (9). Changes of the endocrine function of the pancreas comprise tumour-derived local adrenomedullin that reduces the Insulin secretion from islet cells in the pancreas (10) and an increase of peripheral insulin resistance through pancreatic-cancer-induced secretion of the islet amyloid polypeptide (IAPP) from β-cells and likely an increase of the S-100A8 N-terminal peptide (11). Endocrine dysregulation is also associated with increased gluconeogenesis (12), which depletes peripheral tissues to maintain the altered glucose utilisation.
Pancreatic secretion is initiated through the parasympathetic nervous system during food intake.103 Following food intake, distention of the duodenal wall and the passage of nutrients into the duodenal lumen trigger the release of secretin and cholecystokinin (CCK), which stimulate the secretion of alkali from pancreatic ducts and enzymes from pancreatic acinar cells.104,105 Perineural invasion is a histopathological hallmark of pancreatic cancer, and invasion of sympathetic nervous fibres by tumour tissue is central to cancer pain.106 Analogously, parasympathetic denervation due to tumour infiltration could contribute to PEI through disruption of the physiological priming of the pancreas. Histological studies of healthy pancreatic tissue and specimens from patients with pancreatic cancer, however, showed that parasympathetic innervation was largely unchanged despite severe a reduction in the number of sympathetic fibres.107 This suggests a minor role for neural remodelling as a result of tumour infiltration in tumour-associated PEI.
In contrast to the relatively minor impact of pancreatic tumours on parasympathetic innervation and, consequently, pancreatic secretion, disruption of hormonal regulation appears to affect pancreatic secretion, and patients with pancreatic ductal adenocarcinoma express alternatively spliced secretin receptor isoforms with reduced secretin-binding capacity.108 Similarly, plasma levels of CCK are comparable between patients with pancreatic cancer and non-cancer patients but CCK receptors are differentially expressed in the healthy pancreas and pancreatic cancer.109,110 Although the functional activity of CCK signalling in healthy humans is controversial,111 these differences suggest that reduced tissue sensitivity to pro-secretory signals rather than altered hormone levels due to anatomical alterations of the GI-tract contribute to cancer-associated PEI (Fig. 3).
Regardless of the underlying mechanisms of PEI, compromised pancreatic exocrine function in the context of pancreatic cancer is associated with a significantly increased loss of muscle mass. In a prospective radiological cohort study of 132 patients with pancreatic disease, of whom 59 had pancreatic cancer, sarcopenia was strongly associated with PEI.112 A similar effect exists in mice, which is dependent on the presence of the tumour in the pancreas, and heterotopic tumour transplants failed to initiate cachexia. In this model, tissue wasting could be attenuated by pancreatic enzyme replacement, which indicates a direct link between the pancreatic tumour, decreased exocrine function and cachexia.113
A lack of digestive enzymes causes malabsorption, thereby reducing the uptake of macronutrients to provide energy, and is characterised by bloating and diarrhoea, which further reduce appetite and oral intake.114 Moreover, among the nutrients that are taken up—the extent of the uptake of which is determined by the exocrine pancreatic function—essential fatty acids and lipophilic vitamins have additional significance for pancreatic cancer cachexia.100 In a small cross-sectional study, the levels of several essential n-3 polyunsaturated fatty acids in the plasma of patients with newly diagnosed pancreatic cancer were reduced compared with those in patients with gastro-oesophageal or non-small cell lung cancer, unless these patients had weight loss.115 In accordance with this observation and the early occurrence of other metabolic changes, altered plasma levels of n-3 polyunsaturated fatty acids were identified as potential predictors of the risk of developing pancreatic cancer.116 In addition, n-3 polyunsaturated fatty acids also appear to have direct anabolic and anti-catabolic effects on peripheral tissues, including improved insulin sensitivity as well as a reduced acute-phase response and inhibition of the ubiquitin-proteasome proteolytic pathway, which counteracts skeletal muscle wasting.117 Thus, the reduced levels of n-3 polyunsaturated fatty acids in the plasma of patients with newly diagnosed pancreatic cancer might be expected to bring about catabolic and anti-anabolic effects. These effects have, however, not been studied in detail in the context of pancreatic cancer and results from Phase 3 clinical trials call into question whether or not n-3 polyunsaturated fatty acids are relevant for the survival of cancer patients.118
Observational studies of patients with chronic pancreatitis have shown associations between PEI and decreased serum levels of α-tocopherol, the main component of vitamin E, and of vitamin D metabolites.119,120 However, the underlying cause of PEI might be relevant here because patients with pancreatic cancer showed altered serum levels of only vitamin D, not of vitamin E.121 Unfortunately, the 103 patients in this cohort study were heterogeneous across various disease stages, which might have confounded the results. Moreover, vitamin D deficiency is highly prevalent among cachectic patients with other primary tumours.122 Therefore, it is unclear whether or not some of the mechanisms that contribute to dysregulation of lipophilic vitamin homoeostasis are specific for pancreatic cancer. Reduced levels of vitamin D have been implicated in reduced muscle strength with underlying mechanisms that might promote cachexia in rodents.123 Briefly, reduced vitamin D levels are likely to trigger the overexpression of the vitamin D receptor (VDR) on muscle cells, which is associated with compromised muscle regeneration.124 However, relatively little is known about the regulation of this process and it has not been studied specifically for pancreatic cancer. Vitamin E has been implicated in the pathogenesis of cachexia because of its antioxidative properties, which limit inflammation, but the evidence is generally weak.125
Alterations of pancreatic endocrine function
The development of diabetes mellitus, accompanied by weight loss, often precedes pancreatic cancer by months to years.126 Analogous to Type 2 diabetes, pancreatic-cancer-induced diabetes develops as a consequence of impaired β-cell function and peripheral insulin resistance.127 These effects are considered to be mediated by tumour-cell-secreted paraneoplastic factors. The ensuing metabolic changes associated with pancreatic cancer promote weight loss, but this weight loss is not associated with the improvement in glucose control that usually occurs in Type 2 diabetes patients who lose weight.127 The underlying mechanisms of this observation are likely to be complex, but paradoxical weight loss might at least be partially mediated by tumour-secreted adrenomedullin, which both stimulates lipolysis and induces β-cell dysfunction (Fig. 3).128,129
Peripheral insulin resistance has been suggested to be mediated by the S-100A8 N-terminal peptide, which was identified in the plasma proteome of pancreatic cancer patients with diabetes.130,131 In vitro, this peptide reduced glucose catabolism in myoblasts but its exact connection to peripheral insulin resistance is still unclear.132 Pancreatic-cancer-induced secretion of the islet amyloid polypeptide (IAPP) from β-cells is another potential mediator of peripheral insulin resistance,133,134 reducing insulin sensitivity in vivo and glycogen synthesis in vitro. Irrespective of the particular factor that mediates pancreatic-cancer-induced peripheral insulin resistance, the mechanisms associated with it involve altered gene expression downstream of the insulin receptor, which decreases glycogen synthesis and storage despite a normal expression of glucose transporters.135 In the liver, lack of insulin from β-cells and insulin resistance, in addition to depletion of glycogen storage, also lead to a reduced inhibition of gluconeogenesis which, through the Cori cycle, consumes pyruvate from increased fat oxidation and depletion of adipose tissue.136
Digestive-tract-related factors
The pancreas is located anatomically close to the gastrointestinal tract and linked to it through an essential role for digestion. Pancreatic cancer can affect the structure of other digestive organs or indirectly alter the physiology of the gut—factors that are increasingly recognised to contribute to pancreatic cancer cachexia.
Gastric outlet obstruction
The development of a pancreatic tumour can affect adjacent digestive organs through infiltrative growth or external compression. Some studies investigating surgical and interventional strategies to manage gastric outlet obstruction, usually of the duodenum, have estimated a prevalence of 5–10% in patients with newly diagnosed pancreatic cancer. Gastric outlet obstruction can cause nausea and vomiting and other consequences of impaired oral intake including dehydration, malnutrition and weight loss.137,138 In a proportion of patients, these effects might contribute to the cachexia phenotype through poor nutrition.
Microbiota and pancreatic cancer cachexia
The pancreas and gastrointestinal tract reciprocally influence each other even without direct tumour involvement, and major changes in the systemic metabolism result from this cross-talk. The microbiota is a central mediator of this interaction and its role in cancer and has been studied extensively in various tumour types.139 The pancreas is located at the point at which the oral microbiota and the gut microbiota meet, and dysbiosis in either microbial population has been implicated in the development of pancreatic cancer.140–142 Significantly increased differential abundance of the genus Klebsiella and other Enterobacteriaceae characterises certain patterns of the human gut microbiome that are associated with pancreatic cancer and favour its progression.142,143 Enteric bacteria can also translocate into the pancreas and are present in healthy pancreatic tissue, pancreatic cysts and tumours,144,145 albeit as different populations of different species—accordingly, characterisation of bacterial 16S ribosomal DNA identified distinct microbial patterns that distinguished cancer from healthy tissue.144 Enterobacteriaceae and other bacteria of the phylum Proteobacteria are also major constituents of the intratumoural microbiome in human pancreatic tumours, although substantial variation does exist between individuals.142,144 Inflammation that results from the outgrowth of the Gram-negative Enterobacteriaceae family in the gut (particularly Klebsiella oxytoca), has been associated with increased intestinal permeability, bacterial translocation to mesenteric lymph nodes, increased systemic inflammation markers and the development of cachexia in mice.146,147 However, in KPC mice with metastatic tumours, the increased abundance of likely resistant Enterobacteriaceae occurring after treatment with broad-spectrum antibiotics was associated with a polarisation of the tumour microenvironment towards helper T (TH1) cells/cytotoxic (Tc1) cells and attenuation of tumour growth.143 Moreover, Enterobacteriaceae also appear to be characteristic of the gut microbiome of naturally slow-progressing cases of pancreatic cancer in KPC mice.142 Therefore, the exact significance of the microbiome changes for pancreatic cancer cachexia in the context of overall disease progression as well as related immune mechanisms are still unclear.
The way in which pancreatic cancer influences the gut microbiome largely involves the exocrine function of the pancreas. PEI is associated with small intestinal bacterial overgrowth, a syndrome that comprises excess bacterial growth in the duodenum, jejunum and ileum, microbial imbalance and the translocation of bacteria into the pancreas.148 In mice, supplementation of pancreatic enzymes alters the composition of the intestinal microbiome and promotes the abundance of Lactobacillus reuteri and Akkermansia muciniphila.149 L. reuteri attenuates muscle wasting when fed to ApcMin/+ mice that are prone to cancer cachexia (albeit not a pancreatic cancer model) and A. muciniphila is linked to the systemic immune tone in cancer patients by inducing, educating and tuning the immunological balance between responsiveness and sustained tolerance towards increased effectiveness of programmed cell death protein (PD)-1/programmed death-ligand (PD-L)-1 through increased recruitment of TH1 cells to the tumour.150,151 Other evidence suggests that antibacterial peptides secreted from the acini, rather than pancreatic enzymes, are involved in shaping the gut microbiome and limiting bacterial outgrowth; loss of acini function leads to gut inflammation, problems with weight gain and death (Fig. 3).152
Notably, nonspecific disruption of the gut microbiota of cancer-free mice can result in decreased physical endurance.153 However, the animals show no morphological signs of muscle loss and the phenomenon appears to involve impaired expression of intestinal nutrient transporters and reduced glycogen availability in the muscle.153 Alterations of the gut microbiota also broadly affect the blood metabolome, especially amino acid metabolites.154 Although not well-characterised in the context of weight loss, microbiomes from obese donors differed from those of lean donors specifically in the abundance of genes related to glutamine/glutamate transport and BCAA degradation, which corresponded to reduced serum glutamine levels and higher serum concentrations of BCAA. In addition, gavage of mice with Bacteroides thetaiotaomicron (which is dominant in the human gut microbiota but decreased in obese individuals) increased the expression of genes involved in lipolysis and fatty acid oxidation of white adipose tissue in the host.155 Conclusions for cancer cachexia should only be made with great care, but the knowledge that pathogenic microbiomes can directly affect fat tissue through metabolic pathways that are also crucial to the pathophysiology of the pancreatic-cancer-associated loss of body mass is exciting.
Implications for clinical interventions
A large spectrum of therapeutic approaches that target various aspects of the different dimensions of pancreatic cancer cachexia have been suggested, some of which are outlined below. Unfortunately, failure of cachexia-directed treatment is common, and amelioration or reversal of pancreatic cancer cachexia remains a major clinical challenge.156,157
Nutritional strategies to counter metabolic changes
Nutritional counselling to ensure adequate energy and protein intake and, if indicated, supplemental or complete artificial nutrition is the mainstay of cancer cachexia therapy.158 Modifications of volitional nutrition constitute the typical primary interventions against cancer cachexia and the most common class of cachexia treatment in clinical trials.2 Although provision of sufficient caloric intake is a generic strategy to address cachexia, several nutritional supplements also target the specific metabolic changes that are involved in pancreatic cancer cachexia, as outlined below.
A high-fat, low-carbohydrate diet (a ketogenic diet) that generates ketone bodies might act against cancer cachexia by inducing systemic metabolic changes, and treatment with ketone bodies (sodium hydroxybutyrate and lithium acetoacetate) was able to reduce glycolytic flux and glutamine catabolism in a pancreatic cancer model.159 Supplementation with glutamine together with arginine and β-hydroxy-β-methylbutyrate is likely to shift the metabolic equilibrium away from proteolysis as a source of glutamine and positively support protein synthesis, and this supplementation significantly increased fat-free mass in a cohort of patients with advanced cancers, among them a minority of pancreatic cancer patients.160 However, supplementation with excess quantities of glutamine is generally not recommended because of concerns regarding potential adverse effects of glutamine on other biochemical pathways such as compromised distribution of other amino acids that use the same transporter and excess glutamate and ammonia production.158,161 The ketogenic BCAA leucin and its metabolite β-hydroxy-β-methylbutyrate, which are often used in conjunction with glutamine supplementation, can stimulate muscle protein synthesis through anabolic signalling mediated by the mTOR pathway and inhibition of proteolysis by the reduced expression of proteasome components and ubiquitin ligases.162–164 Accordingly, results from several prospective trials indicated that both BCAA and β-hydroxy-β-methylbutyrate improved the protein metabolism of cachectic patients.160,165,166 Unfortunately, a Phase 3 trial of β-hydroxyl β-methyl butyrate, glutamine and arginine suffered from poor protocol adherence and failed to reach its primary endpoint of an improvement of lean body mass.167
Another nutritional intervention that has been extensively investigated is supplementation with n-3 fatty acids, which inhibit proteolytic pathways, preserve protein biosynthesis and inhibit lipolysis.168 In small non-randomised cohorts of pancreatic cancer patients, n-3 fatty acid supplementation was associated with a moderate but persistent weight gain.169,170 This observation was corroborated in a randomised, double-blind trial in which sufficient intake of n-3 fatty acid resulted in weight gain, lean body mass and improved quality of life.171
Targeting of pancreatic cancer cachexia-related inflammatory pathways
Insufficient evidence exists to support the unimodal blockade of inflammatory pathways to alleviate pancreatic cancer cachexia. Nevertheless, an improved understanding of the immunopathology of cachexia as well as studies exploring the combination of immunomodulation in a multimodal approach might warrant re-evaluation.2
Through the synthesis of prostanoids, cyclo-oxygenase-2 (COX-2) induces IL-6 and regulates the expression of TNF,172,173 and COX-2 metabolites have been directly implicated in the regulation of muscle turnover.174 In two small Phase 2 trials, cachexia patients with different malignancies, the majority of which were upper gastrointestinal cancers, were treated with the selective COX-2 inhibitor celecoxib, resulting in weight gain and improved QoL scores.175,176 Another Phase 2 trial that predominantly included patients with pancreatic cancer with weight loss (49/73) reported that the combination of megestrol acetate, a synthetic derivative of progesterone, to stimulate appetite, and the nonselective COX-inhibitor ibuprofen reduced weight loss and improved QoL.177 Megestrol acetate alone has been extensively studied as an appetite stimulant to counter anorexia in patients with hormone-insensitive tumours but has not been shown to affect body weight in patients with gastrointestinal cancer.178 Analogous to COX-2 inhibition, thalidomide, an inhibitor of TNF synthesis, has been shown to reduce weight loss and loss of lean body mass in patients with cachexia and advanced pancreatic cancer.179 By contrast, however, a placebo-controlled Phase 2 trial of infliximab, a monoclonal antibody against TNF, did not demonstrate any significant improvements in lean body mass, endurance or survival.180 Similarly, an exploratory analysis of body weight and QoL in two randomised trials of the JAK1/JAK2 inhibitor ruxolitinib were terminated after interim analyses failed to uncover any clinically relevant benefits in the inhibition of the JAK/STAT pathway.181
In a multimodal feasibility study of patients with pancreatic cancer cachexia, supplementation with n-3 polyunsaturated fatty acids plus celecoxib, together with exercise, was associated with a trend towards better preservation of body mass, and this approach is currently being tested in an adequately powered Phase 3 trial.182,183 Physical exercise alone has previously been shown to improve muscle strength in patients with pancreatic cancer but appears to have little effect on body weight without additional interventions.184
Targeting other mediators of pancreatic cancer cachexia
Anamorelin, an agonist of the ghrelin receptor, improves anorexia and addresses the hypothalamic effects of systemic inflammation, and was found to improve appetite, lean body mass and body weight in patients with gastrointestinal cancer in a small non-randomised trial.185 Similarly, cannabinoids paradoxically activate hypothalamic POMC neurons through cannabinoid receptor 1 (CB1R), which results in a selective increase in the expression and release of β-endorphin and an appetite-stimulating effect.186 The orally active cannabinoid Δ-9-tetrahydrocannabinol (dronabinol) was shown to mitigate anorexia in a randomised Phase 3 trial of a mixed population of cachectic cancer patients containing a substantial proportion of individuals with gastrointestinal cancers. However, the effects of dronabinol were inferior to those of megestrol acetate, and dronabinol did not confer an additional benefit when both drugs were combined.187
Similarly, targeting members of the TGF-β superfamily has not been effective. In a Phase 2 trial of patients with advanced pancreatic cancer, the combination of a myostatin antibody with standard-of-care chemotherapy for advanced pancreatic cancer failed to confer additional clinical benefits (the primary endpoint was overall survival).188
To address the potential significance of altered neurosignalling or depression for pancreatic cancer cachexia, different classes of antidepressants could hypothetically be considered but have, to our knowledge, not been systematically evaluated. Mirtazapine, an atypical tetracyclic antidepressant, has been evaluated exclusively because of its capability to improve appetite with some signs of efficacy to stabilise weight in a small non-randomised trial of nondepressed patients with pancreatic cancer.189
Pancreatic enzyme replacement therapy for PEI
Pancreatic enzyme replacement therapy (PERT) is the oral substitution of pancreatic enzymes to mitigate malabsorption and ensuing symptoms related to PEI. However, in a New Zealand retrospective study of 129 patients with metastatic pancreatic cancer who had symptoms that could be attributed to malabsorption, only 21% of patients were found to have received pancreatic enzyme replacement therapy.190 A lack of adequate management of PEI and resulting dietary issues adversely affected the QoL of patients with pancreatic cancer, with a lack of routine dietary consultation, a perceived unwillingness of clinicians to prescribe enzyme therapy and insufficient clarity of dosing instructions being reported.114
This lack of clinical awareness is concerning, as several placebo-controlled studies have demonstrated that PERT improves parameters such as fat absorption and the frequency of bowel movements and supports weight stabilisation of patients with advanced pancreatic cancer.191–193 Moreover, PERT has also been associated with prolonged survival.194 Accordingly, UK multi-society guidelines for the management of patients with pancreatic cancer periampullary and ampullary carcinomas,195 US NCCN guidelines on pancreatic adenocarcinoma,196 the European Society for Clinical Nutrition and Metabolism (ESPEN) guidelines on nutrition in cancer patients158 and the United European Gastroenterology (UEG) recommendations for PEI197 support the use of PERT for pancreatic cancer patients.
Management of endocrine insufficiency
Patients with pancreatic-cancer-related diabetes commonly require insulin therapy, and concomitant metformin therapy is encouraged to address peripheral insulin resistance.198 In the liver, metformin also reduces hepatic gluconeogenesis through inhibition of mitochondrial glycerophosphate dehydrogenase, which limits the conversion of lactate and glycerol into glucose.199 This effect might mitigate tumour-induced peripheral tissue wasting and metformin was able, in fact, to reverse sarcopenia in an experimental cancer cachexia model.200 It is therefore surprising that metformin treatment was associated with a trend towards a slightly more prevalent weight loss (12 versus 8%) and anorexia (37 versus 20%) and had no effect on overall survival in a double-blind placebo-controlled Phase 2 trial of 121 patients with advanced pancreatic cancer.201
Microbiome
The microbiota has not been the target of strategies in clinical trials to modulate pancreatic cancer cachexia, although experimental approaches to minimise the overall gut microbiome with antibiotics143 or to modulate its composition or diversity with probiotics202 or faecal transplants203 to target pancreatic cancer exist. In a clinical setting, a more targeted elimination of detrimental bacteria than broad-spectrum antibiotics would likely be necessary to counter harmful effects of a broad depletion of the microbiota which has frequently been associated with poorer response of various cancers to treatment and shorter patient survival. Based on the available preclinical evidence supplementation of probiotics, especially Lactobacillus, might be an interesting approach to ameliorate pancreatic cancer cachexia but well-planned prospective trials would be needed to inform clinical practice.204
Conclusion
Pancreatic cancer cachexia is an exceedingly common problem that arises independent of the stage of pancreatic cancer. Reflecting the particular systemic effects of pancreatic cancer and the central role of the pancreas in the digestive tract, pancreatic cancer cachexia has three interacting dimensions that make up a complex syndrome: systemic metabolic changes and signals, often associated with KRAS-mutations; an impaired exocrine and endocrine pancreatic function; and alterations of the intestinal tract with a particular impact on the microbiome. A number of approaches to target some aspects of the different dimensions of pancreatic cancer cachexia, such as nutritional strategies, targeting inflammatory and other mediators of pancreatic cancer cachexia, and PERT, have been or are being studied. The interdependent nature of these dimensions also needs to be considered in the clinical management of patients with pancreatic cancer cachexia. There are several points of convergence between the three different dimensions—for example, immune state and exocrine insufficiency—that are attractive targets for development of therapeutic strategies and future research. Addressing these links—as exemplified by substituting exocrine pancreatic function with PERT—has significant potential to ameliorate cachexia and consequently improve the quality of life and prognosis of patients with pancreatic cancer.
Acknowledgements
The authors would like to thank Rainer Heuchel for his critical comments and Erika Nieser for language editing.
Author contributions
Conception and design: M.K. and J.M.L. Manuscript writing: M.K. and J.M.L. Final approval of manuscript: all authors. Accountable for all aspects of the work: all authors.
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Data availability
Not applicable.
Competing interests
M.K.: Alligator Bioscience (Consulting), Roche (Consulting); L.L.: no disclosures; L.E.: no disclosures; J.M.L.: Abbot (Honoraria), Mylan (Honoraria).
Funding information
This work was supported through a clinician-scientist grant from Region Stockholm to M.K. and the Lars Vesterlund minnesfond to J.M.L.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12:489–495. doi: 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- 2.Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer-associated cachexia. Nat. Rev. Dis. Primers. 2018;4:17105. doi: 10.1038/nrdp.2017.105. [DOI] [PubMed] [Google Scholar]
- 3.Martin L, Birdsell L, MacDonald N, Reiman T, Clandinin MT, McCargar LJ, et al. Cancer cachexia in the age of obesity: skeletal muscle depletion Is a powerful prognostic factor, independent of body mass index. J. Clin. Oncol. 2013;31:1539–1547. doi: 10.1200/JCO.2012.45.2722. [DOI] [PubMed] [Google Scholar]
- 4.Minicozzi P, Cassetti T, Vener C, Sant M. Analysis of incidence, mortality and survival for pancreatic and biliary tract cancers across Europe, with assessment of influence of revised European age standardisation on estimates. Cancer Epidemiol. 2018;55:52–60. doi: 10.1016/j.canep.2018.04.011. [DOI] [PubMed] [Google Scholar]
- 5.Hendifar AE, Chang JI, Huang BZ, Tuli R, Wu BU. Cachexia, and not obesity, prior to pancreatic cancer diagnosis worsens survival and is negated by chemotherapy. J. Gastrointest. Oncol. 2018;9:17–23. doi: 10.21037/jgo.2017.11.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitsunaga S., Kasamatsu E. & Machii K. Incidence and frequency of cancer cachexia during chemotherapy for advanced pancreatic ductal adenocarcinoma. Support Care Cancer28, 5271–5279 (2020) [DOI] [PMC free article] [PubMed]
- 7.Kays JK, Shahda S, Stanley M, Bell TM, O’Neill BH, Kohli MD, et al. Three cachexia phenotypes and the impact of fat-only loss on survival in FOLFIRINOX therapy for pancreatic cancer. J. Cachexia Sarcopenia Muscle. 2018;9:673–684. doi: 10.1002/jcsm.12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi Y, Oh DY, Kim TY, Lee KH, Han SW, Im SA, et al. Skeletal muscle depletion predicts the prognosis of patients with advanced pancreatic cancer undergoing palliative chemotherapy, independent of body mass index. PLoS ONE. 2015;10:e0139749. doi: 10.1371/journal.pone.0139749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachmann J, Heiligensetzer M, Krakowski-Roosen H, Buchler MW, Friess H, Martignoni ME. Cachexia worsens prognosis in patients with resectable pancreatic cancer. J. Gastrointest. Surg. 2008;12:1193–1201. doi: 10.1007/s11605-008-0505-z. [DOI] [PubMed] [Google Scholar]
- 10.Bauer MR, Bright EE, MacDonald JJ, Cleary EH, Hines OJ, Stanton AL. Quality of life in patients with pancreatic cancer and their caregivers. Pancreas. 2018;47:368–375. doi: 10.1097/MPA.0000000000001025. [DOI] [PubMed] [Google Scholar]
- 11.Hagensen A, London AE, Phillips JJ, Helton WS, Picozzi VJ, Blackmore CC. Using experience-based design to improve the care experience for patients with pancreatic cancer. J. Oncol. Pract. 2016;12:e1035–e1041. doi: 10.1200/JOP.2016.011213. [DOI] [PubMed] [Google Scholar]
- 12.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guillaumond F, Leca J, Olivares O, Lavaut M-N, Vidal N, Berthezène P, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl Acad. Sci. USA. 2013;110:3919–3924. doi: 10.1073/pnas.1219555110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Cairns R, Papandreou I, Koong A, Denko NC. Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PLoS ONE. 2009;4:e7033. doi: 10.1371/journal.pone.0007033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang F, Liu H, Hu L, Liu Y, Duan Y, Cui R, et al. The Warburg effect in human pancreatic cancer cells triggers cachexia in athymic mice carrying the cancer cells. BMC Cancer. 2018;18:360. doi: 10.1186/s12885-018-4271-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roux C, Riganti C, Borgogno SF, Curto R, Curcio C, Catanzaro V, et al. Endogenous glutamine decrease is associated with pancreatic cancer progression. Oncotarget. 2017;8:95361–95376. doi: 10.18632/oncotarget.20545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vasseur S, Tomasini R, Tournaire R, Iovanna JL. Hypoxia induced tumor metabolic switch contributes to pancreatic cancer aggressiveness. Cancers (Basel) 2010;2:2138–2152. doi: 10.3390/cancers2042138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536:479–483. doi: 10.1038/nature19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 2014;20:1193–1198. doi: 10.1038/nm.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neinast MD, Jang C, Hui S, Murashige DS, Chu Q, Morscher RJ, et al. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metabolism. 2019;29:417–429.e414. doi: 10.1016/j.cmet.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science. 2016;353:1161–1165. doi: 10.1126/science.aaf5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JH, Cho YR, Kim JH, Kim J, Nam HY, Kim SW, et al. Branched-chain amino acids sustain pancreatic cancer growth by regulating lipid metabolism. Exp. Mol. Med. 2019;51:1–11. doi: 10.1038/s12276-019-0350-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333:233–238. doi: 10.1126/science.1198973. [DOI] [PubMed] [Google Scholar]
- 26.Ryden M, Agustsson T, Laurencikiene J, Britton T, Sjolin E, Isaksson B, et al. Lipolysis–not inflammation, cell death, or lipogenesis–is involved in adipose tissue loss in cancer cachexia. Cancer. 2008;113:1695–1704. doi: 10.1002/cncr.23802. [DOI] [PubMed] [Google Scholar]
- 27.Shaw JH, Wolfe RR. Fatty acid and glycerol kinetics in septic patients and in patients with gastrointestinal cancer. The response to glucose infusion and parenteral feeding. Ann. Surg. 1987;205:368–376. doi: 10.1097/00000658-198704000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang F, Kumagai-Braesch M, Herrington MK, Larsson J, Permert J. Increased lipid metabolism and cell turnover of MiaPaCa2 cells induced by high-fat diet in an orthotopic system. Metabolism. 2009;58:1131–1136. doi: 10.1016/j.metabol.2009.03.027. [DOI] [PubMed] [Google Scholar]
- 29.Agustsson T, Ryden M, Hoffstedt J, van Harmelen V, Dicker A, Laurencikiene J, et al. Mechanism of increased lipolysis in cancer cachexia. Cancer Res. 2007;67:5531–5537. doi: 10.1158/0008-5472.CAN-06-4585. [DOI] [PubMed] [Google Scholar]
- 30.Russell ST, Tisdale MJ. Effect of a tumour-derived lipid-mobilising factor on glucose and lipid metabolism in vivo. Br. J. Cancer. 2002;87:580–584. doi: 10.1038/sj.bjc.6600493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russell ST, Zimmerman TP, Domin BA, Tisdale MJ. Induction of lipolysis in vitro and loss of body fat in vivo by zinc-alpha2-glycoprotein. Biochim. Biophys. Acta. 1636;59–68:2004. doi: 10.1016/j.bbalip.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Bao Y, Bing C, Hunter L, Jenkins JR, Wabitsch M. Trayhurn P. Zinc-α2-glycoprotein, a lipid mobilizing factor, is expressed and secreted by human (SGBS) adipocytes. FEBS Lett. 2005;579:41–47. doi: 10.1016/j.febslet.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 33.Bing C, Bao Y, Jenkins J, Sanders P, Manieri M, Cinti S, et al. Zinc-α2-glycoprotein, a lipid mobilizing factor, is expressed in adipocytes and is up-regulated in mice with cancer cachexia. Proc. Natl Acad. Sci. USA. 2004;101:2500–2505. doi: 10.1073/pnas.0308647100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulyte A, Lorente-Cebrian S, Gao H, Mejhert N, Agustsson T, Arner P, et al. MicroRNA profiling links miR-378 to enhanced adipocyte lipolysis in human cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2014;306:E267–E274. doi: 10.1152/ajpendo.00249.2013. [DOI] [PubMed] [Google Scholar]
- 35.Sagar G, Sah RP, Javeed N, Dutta SK, Smyrk TC, Lau JS, et al. Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut. 2016;65:1165–1174. doi: 10.1136/gutjnl-2014-308350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rohm M., Zeigerer A., Machado J. & Herzig S. Energy metabolism in cachexia. EMBO Rep. 20, e47258 (2019) [DOI] [PMC free article] [PubMed]
- 37.Mitsunaga S, Ikeda M, Shimizu S, Ohno I, Furuse J, Inagaki M, et al. Serum levels of IL-6 and IL-1beta can predict the efficacy of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer. 2013;108:2063–2069. doi: 10.1038/bjc.2013.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014;20:433–447. doi: 10.1016/j.cmet.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 39.Sah RP, Sharma A, Nagpal S, Patlolla SH, Sharma A, Kandlakunta H, et al. Phases of metabolic and soft tissue changes in months preceding a diagnosis of pancreatic ductal adenocarcinoma. Gastroenterology. 2019;156:1742–1752. doi: 10.1053/j.gastro.2019.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bing C, Russell ST, Beckett EE, Collins P, Taylor S, Barraclough R, et al. Expression of uncoupling proteins-1, -2 and -3 mRNA is induced by an adenocarcinoma-derived lipid-mobilizing factor. Br. J. Cancer. 2002;86:612–618. doi: 10.1038/sj.bjc.6600101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Falconer JS, Fearon KC, Plester CE, Ross JA, Carter DCCytokines. the acute-phase response, and resting energy expenditure in cachectic patients with pancreatic cancer. Ann. Surg. 1994;219:325–331. doi: 10.1097/00000658-199404000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzalez-Bulnes A, Fujiwara Y, Kobayashi T, Chayahara N, Imamura Y, Toyoda M, et al. Metabolomics evaluation of serum markers for cachexia and their intra-day variation in patients with advanced pancreatic cancer. PLoS ONE. 2014;9:e113259. doi: 10.1371/journal.pone.0113259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bye A, Wesseltoft-Rao N, Iversen PO, Skjegstad G, Holven KB, Ulven S, et al. Alterations in inflammatory biomarkers and energy intake in cancer cachexia: a prospective study in patients with inoperable pancreatic cancer. Med. Oncol. 2016;33:54. doi: 10.1007/s12032-016-0768-2. [DOI] [PubMed] [Google Scholar]
- 44.Bachmann J, Buchler MW, Friess H, Martignoni ME. Cachexia in patients with chronic pancreatitis and pancreatic cancer: impact on survival and outcome. Nutr. Cancer. 2013;65:827–833. doi: 10.1080/01635581.2013.804580. [DOI] [PubMed] [Google Scholar]
- 45.Talar-Wojnarowska R, Gasiorowska A, Smolarz B, Romanowicz-Makowska H, Kulig A, Malecka-Panas E. Clinical significance of interleukin-6 (Il-6) gene polymorphism and Il-6 serum level in pancreatic adenocarcinoma and chronic pancreatitis. Dig. Dis. Sci. 2008;54:683–689. doi: 10.1007/s10620-008-0390-z. [DOI] [PubMed] [Google Scholar]
- 46.Miura T, Mitsunaga S, Ikeda M, Shimizu S, Ohno I, Takahashi H, et al. Characterization of patients with advanced pancreatic cancer and high serum interleukin-6 levels. Pancreas. 2015;44:756–763. doi: 10.1097/MPA.0000000000000335. [DOI] [PubMed] [Google Scholar]
- 47.Talbert EE, Lewis HL, Farren MR, Ramsey ML, Chakedis JM, Rajasekera P, et al. Circulating monocyte chemoattractant protein-1 (MCP-1) is associated with cachexia in treatment-naive pancreatic cancer patients. J. Cachexia Sarcopenia Muscle. 2018;9:358–368. doi: 10.1002/jcsm.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hou Y-C, Wang C-J, Chao Y-J, Chen H-Y, Wang H-C, Tung H-L, et al. Elevated serum interleukin-8 level correlates with cancer-related cachexia and sarcopenia: An indicator for pancreatic cancer outcomes. J. Clin. Med. 2018;7:502. doi: 10.3390/jcm7120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martignoni ME. Role of mononuclear cells and inflammatory cytokines in pancreatic cancer-related cachexia. Clin. Cancer Res. 2005;11:5802–5808. doi: 10.1158/1078-0432.CCR-05-0185. [DOI] [PubMed] [Google Scholar]
- 50.Zhang D, Zhou Y, Wu L, Wang S, Zheng H, Yu B, et al. Association of IL-6 gene polymorphisms with cachexia susceptibility and survival time of patients with pancreatic cancer. Ann. Clin. Lab. Sci. 2008;38:113–119. [PubMed] [Google Scholar]
- 51.Egberts JH, Cloosters V, Noack A, Schniewind B, Thon L, Klose S, et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008;68:1443–1450. doi: 10.1158/0008-5472.CAN-07-5704. [DOI] [PubMed] [Google Scholar]
- 52.de Matos-Neto EM, Lima JDCC, de Pereira WO, Figuerêdo RG, Riccardi DMDR, Radloff K, et al. Systemic inflammation in cachexia - Is tumor cytokine expression profile the culprit? Front. Immunol. 2015;6:629–629. doi: 10.3389/fimmu.2015.00629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimada M, Andoh A, Araki Y, Fujiyama Y, Bamba T. Ligation of the Fas antigen stimulates chemokine secretion in pancreatic cancer cell line PANC-11. J. Gastroenterol. Hepatol. 2001;16:1060–1067. doi: 10.1046/j.1440-1746.2001.02583.x. [DOI] [PubMed] [Google Scholar]
- 54.Delitto D, Judge SM, Delitto AE, Nosacka RL, Rocha FG, DiVita BB, et al. Human pancreatic cancer xenografts recapitulate key aspects of cancer cachexia. Oncotarget. 2017;8:1177–1189. doi: 10.18632/oncotarget.13593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gerber MH, Underwood PW, Judge SM, Delitto D, Delitto AE, Nosacka RL, et al. Local and systemic cytokine profiling for pancreatic ductal adenocarcinoma to study cancer cachexia in an era of precision medicine. Int. J. Mol. Sci. 2018;19:3836. doi: 10.3390/ijms19123836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haugen F, Labori KJ, Noreng HJ, Buanes T, Iversen PO, Drevon CA. Altered expression of genes in adipose tissues associated with reduced fat mass in patients with pancreatic cancer. Arch. Physiol. Biochem. 2011;117:78–87. doi: 10.3109/13813455.2011.560609. [DOI] [PubMed] [Google Scholar]
- 57.Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2012;303:E410–E421. doi: 10.1152/ajpendo.00039.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pettersen K, Andersen S, Degen S, Tadini V, Grosjean J, Hatakeyama S, et al. Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci. Rep. 2017;7:2046. doi: 10.1038/s41598-017-02088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma JF, Sanchez BJ, Hall DT, Tremblay AK, Di Marco S, Gallouzi IE. STAT3 promotes IFNgamma/TNFalpha-induced muscle wasting in an NF-kappaB-dependent and IL-6-independent manner. EMBO Mol. Med. 2017;9:622–637. doi: 10.15252/emmm.201607052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hall DT, Ma JF, Di Marco S, Gallouzi I-E. Inducible nitric oxide synthase (iNOS) in muscle wasting syndrome, sarcopenia, and cachexia. Aging. 2011;3:702–715. doi: 10.18632/aging.100358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, et al. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J. Clin. Invest. 2004;114:370–378. doi: 10.1172/JCI200420174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gilabert M, Calvo E, Airoldi A, Hamidi T, Moutardier V, Turrini O, et al. Pancreatic cancer-induced cachexia Is Jak2-dependent in mice. J. Cell Physiol. 2014;229:1437–1443. doi: 10.1002/jcp.24580. [DOI] [PubMed] [Google Scholar]
- 63.Zimmers TA, Fishel ML, Bonetto A. STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell. Dev. Biol. 2016;54:28–41. doi: 10.1016/j.semcdb.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lira FS, Yamashita AS, Rosa JC, Tavares FL, Caperuto E, Carnevali LC, Jr., et al. Hypothalamic inflammation is reversed by endurance training in anorectic-cachectic rats. Nutr. Metab. 2011;8:60–60. doi: 10.1186/1743-7075-8-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Plata-Salamán CR, Ilyin SE, Gayle D. Brain cytokine mRNAs in anorectic rats bearing prostate adenocarcinoma tumor cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1998;275:R566–R573. doi: 10.1152/ajpregu.1998.275.2.R566. [DOI] [PubMed] [Google Scholar]
- 66.Inui A, Neuropeptide Y. a key molecule in anorexia and cachexia in wasting disorders? Mol. Med. Today. 1999;5:79–85. doi: 10.1016/S1357-4310(98)01395-1. [DOI] [PubMed] [Google Scholar]
- 67.Reyes TM, Sawchenko PE. Involvement of the arcuate nucleus of the hypothalamus in interleukin-1-induced anorexia. J. Neurosci. 2002;22:5091–5099. doi: 10.1523/JNEUROSCI.22-12-05091.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Amitani M, Asakawa A, Amitani H, Inui A. Control of food intake and muscle wasting in cachexia. Int. J. Biochem. Cell Biol. 2013;45:2179–2185. doi: 10.1016/j.biocel.2013.07.016. [DOI] [PubMed] [Google Scholar]
- 69.Ji YB, Bo CL, Xue XJ, Weng EM, Gao GC, Dai BB, et al. Association of inflammatory cytokines with the symptom cluster of pain, fatigue, depression, and sleep disturbance in chinese patients with cancer. J. Pain Symptom Manage. 2017;54:843–852. doi: 10.1016/j.jpainsymman.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 70.Breitbart W, Rosenfeld B, Tobias K, Pessin H, Ku GY, Yuan J, et al. Depression, cytokines, and pancreatic cancer. Psychooncology. 2014;23:339–345. doi: 10.1002/pon.3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yaskin JC. Nervous symptoms as earliest manifestations of carcinoma of the pancreas. JAMA. 1931;96:1664–1668. doi: 10.1001/jama.1931.02720460010003. [DOI] [Google Scholar]
- 72.Morley JE. Anorexia of aging: physiologic and pathologic. Am. J. Clin. Nutr. 1997;66:760–773. doi: 10.1093/ajcn/66.4.760. [DOI] [PubMed] [Google Scholar]
- 73.Moo-Young TA, Larson JW, Belt BA, Tan MC, Hawkins WG, Eberlein TJ, et al. Tumor-derived TGF-beta mediates conversion of CD4+Foxp3+ regulatory T cells in a murine model of pancreas cancer. J. Immunother. 2009;32:12–21. doi: 10.1097/CJI.0b013e318189f13c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Löhr M, Schmidt C, Ringel J, Kluth M, Müller P, Nizze H, et al. Transforming Growth Factor-β1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001;61:550–555. [PubMed] [Google Scholar]
- 75.Zugmaier G, Paik S, Wilding G, Knabbe C, Bano M, Lupu R, et al. Transforming Growth Factor β1 induces cachexia and szystemic fibrosis without an antitumor effect in nude mice. Cancer Res. 1991;51:3590–3594. [PubMed] [Google Scholar]
- 76.Mendias CL, Gumucio JP, Davis ME, Bromley CW, Davis CS, Brooks SV. Transforming growth factor-beta induces skeletal muscle atrophy and fibrosis through the induction of atrogin-1 and scleraxis. Muscle Nerve. 2012;45:55–59. doi: 10.1002/mus.22232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Greco SH, Tomkotter L, Vahle AK, Rokosh R, Avanzi A, Mahmood SK, et al. TGF-beta blockade reduces mortality and metabolic changes in a validated murine model of pancreatic cancer cachexia. PLoS ONE. 2015;10:e0132786. doi: 10.1371/journal.pone.0132786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, et al. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–1488. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]
- 79.Chen JL, Walton KL, Winbanks CE, Murphy KT, Thomson RE, Makanji Y, et al. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 2014;28:1711–1723. doi: 10.1096/fj.13-245894. [DOI] [PubMed] [Google Scholar]
- 80.Zhong X, Pons M, Poirier C, Jiang Y, Liu J, Sandusky GE, et al. The systemic activin response to pancreatic cancer: implications for effective cancer cachexia therapy. J. Cachexia Sarcopenia Muscle. 2019;10:1083–1101. doi: 10.1002/jcsm.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koopmann J, Buckhaults P, Brown DA, Zahurak ML, Sato N, Fukushima N, et al. Serum macrophage inhibitory cytokine 1 as a marker of pancreatic and other periampullary cancers. Clin. Cancer Res. 2004;10:2386–2392. doi: 10.1158/1078-0432.CCR-03-0165. [DOI] [PubMed] [Google Scholar]
- 82.Johnen H, Lin S, Kuffner T, Brown DA, Tsai VW, Bauskin AR, et al. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat. Med. 2007;13:1333–1340. doi: 10.1038/nm1677. [DOI] [PubMed] [Google Scholar]
- 83.Todorov P, Cariuk P, McDevitt T, Coles B, Fearon K, Tisdale M. Characterization of a cancer cachectic factor. Nature. 1996;379:739–742. doi: 10.1038/379739a0. [DOI] [PubMed] [Google Scholar]
- 84.Whitehouse AS, Tisdale MJ. Increased expression of the ubiquitin-proteasome pathway in murine myotubes by proteolysis-inducing factor (PIF) is associated with activation of the transcription factor NF-kappaB. Br. J. Cancer. 2003;89:1116–1122. doi: 10.1038/sj.bjc.6601132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wyke SM, Tisdale MJ. NF-κB mediates proteolysis-inducing factor induced protein degradation and expression of the ubiquitin–proteasome system in skeletal muscle. Br. J. Cancer. 2005;92:711–721. doi: 10.1038/sj.bjc.6602402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eley HL, Tisdale MJ. Skeletal muscle atrophy, a link between depression of protein synthesis and increase in degradation. J. Biol. Chem. 2007;282:7087–7097. doi: 10.1074/jbc.M610378200. [DOI] [PubMed] [Google Scholar]
- 87.Wigmore SJ, Todorov PT, Barber MD, Ross JA, Tisdale MJ, Fearon KCH. Characteristics of patients with pancreatic cancer expressing a novel cancer cachectic factor. Br. J. Surg. 2000;87:53–58. doi: 10.1046/j.1365-2168.2000.01317.x. [DOI] [PubMed] [Google Scholar]
- 88.Watchorn TM, Waddell ID, Dowidar N, Ross JA. Proteolysis-inducing factor regulates hepatic gene expression via the transcription factors NF-κΒ and STAT3. FASEB J. 2001;15:562–564. doi: 10.1096/fj.00-0534fje. [DOI] [PubMed] [Google Scholar]
- 89.Huang X-Y, Huang Z-L, Yang J-H, Xu Y-H, Sun J-S, Zheng Q, et al. Pancreatic cancer cell-derived IGFBP-3 contributes to muscle wasting. J Exp. Clin. Cancer Res. 2016;35:46. doi: 10.1186/s13046-016-0317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.He WA, Calore F, Londhe P, Canella A, Guttridge DC, Croce CM. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc. Natl Acad. Sci USA. 2014;111:4525–4529. doi: 10.1073/pnas.1402714111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang, G., Liu, Z., Ding, H., Zhou, Y., Doan, H. A., Sin, K. W. T. et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat. Commun8, 589 (2017) [DOI] [PMC free article] [PubMed]
- 92.Sikkens ECM, Cahen DL, de Wit J, Looman CWN, van Eijck C, Bruno MJ. A prospective assessment of the natural course of the exocrine pancreatic function in patients with a pancreatic head tumor. J. Clin. Gastroenterol. 2014;48:e43–e46. doi: 10.1097/MCG.0b013e31829f56e7. [DOI] [PubMed] [Google Scholar]
- 93.Permert J, Ihse I, Jorfeldt L, von Schenck H, Arnqvist HJ, Larsson J. Pancreatic cancer is associated with impaired glucose metabolism. Eur. J. Surg. 1993;159:101–107. [PubMed] [Google Scholar]
- 94.Lim P-W, Dinh KH, Sullivan M, Wassef WY, Zivny J, Whalen GF, et al. Thirty-day outcomes underestimate endocrine and exocrine insufficiency after pancreatic resection. HPB (Oxford) 2016;18:360–366. doi: 10.1016/j.hpb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maignan A, Ouaissi M, Turrini O, Regenet N, Loundou A, Louis G, et al. Risk factors of exocrine and endocrine pancreatic insufficiency after pancreatic resection: A multi-center prospective study. J. Visc. Surg. 2018;155:173–181. doi: 10.1016/j.jviscsurg.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 96.Speicher JE, Traverso LW. Pancreatic exocrine function is preserved after distal pancreatectomy. J. Gastroenterol. Surg. 2010;14:1006–1011. doi: 10.1007/s11605-010-1184-0. [DOI] [PubMed] [Google Scholar]
- 97.Beger HG, Poch B, Mayer B, Siech M. New onset of diabetes and pancreatic exocrine insufficiency after pancreaticoduodenectomy for benign and malignant tumors. Ann. Surg. 2018;267:259–270. doi: 10.1097/SLA.0000000000002422. [DOI] [PubMed] [Google Scholar]
- 98.Kang MJ, Jung HS, Jang J-Y, Jung W, Chang J, Shin YC, et al. Metabolic effect of pancreatoduodenectomy: resolution of diabetes mellitus after surgery. Pancreatology. 2016;16:272–277. doi: 10.1016/j.pan.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 99.Wu J-M, Kuo T-C, Yang C-Y, Chiang P-Y, Jeng Y-M, Huang P-H, et al. Resolution of diabetes after pancreaticoduodenectomy in patients with and without pancreatic ductal cell adenocarcinoma. Ann. Surg. Oncol. 2013;20:242–249. doi: 10.1245/s10434-012-2577-y. [DOI] [PubMed] [Google Scholar]
- 100.Vujasinovic M, Valente R, Del Chiaro M, Permert J, Löhr J-M. Pancreatic exocrine insufficiency in pancreatic cancer. Nutrients. 2017;9:183. doi: 10.3390/nu9030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schober M, Jesenofsky R, Faissner R, Weidenauer C, Hagmann W, Michl P, et al. Desmoplasia and chemoresistance in pancreatic cancer. Cancers (Basel) 2014;6:2137–2154. doi: 10.3390/cancers6042137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brune K, Abe T, Canto M, O’Malley L, Klein AP, Maitra A, et al. Multifocal neoplastic precursor lesions associated with lobular atrophy of the pancreas in patients having a strong family history of pancreatic cancer. Am. J. Surg. Pathol. 2006;30:1067–1076. [PMC free article] [PubMed] [Google Scholar]
- 103.Anagnostides A, Chadwick V, Selden A, Maton P. Sham feeding and pancreatic secretion: evidence for direct vagal stimulation of enzyme output. Gastroenterology. 1984;87:109–114. doi: 10.1016/0016-5085(84)90132-X. [DOI] [PubMed] [Google Scholar]
- 104.White T, McAlexander R, Magee D. The effect of gastric distension on duodenal aspirates in man. Gastroenterology. 1963;44:48–51. doi: 10.1016/S0016-5085(63)80117-1. [DOI] [PubMed] [Google Scholar]
- 105.Watanabe S, Shiratori K, Takeuchi T, Chey W, You C, Chang T-M. Release of cholecystokinin and exocrine pancreatic secretion in response to an elemental diet in human subjects. Dig. Dis. Sci. 1986;31:919–924. doi: 10.1007/BF01303211. [DOI] [PubMed] [Google Scholar]
- 106.Bapat AA, Hostetter G, Von Hoff DD, Han H. Perineural invasion and associated pain in pancreatic cancer. Nat. Rev. Cancer. 2011;11:695–707. doi: 10.1038/nrc3131. [DOI] [PubMed] [Google Scholar]
- 107.Ceyhan GO, Demir IE, Rauch U, Bergmann F, Muller MW, Buchler MW, et al. Pancreatic neuropathy results in “neural remodeling” and altered pancreatic innervation in chronic pancreatitis and pancreatic cancer. Am. J. Gastroenterol. 2009;104:2555–2565. doi: 10.1038/ajg.2009.380. [DOI] [PubMed] [Google Scholar]
- 108.Körner M, Hayes GM, Rehmann R, Zimmermann A, Friess H, Miller LJ, et al. Secretin receptors in normal and diseased human pancreas: marked reduction of receptor binding in ductal neoplasia. Am. J. Patho. 2005;167:959–968. doi: 10.1016/S0002-9440(10)61186-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Weinberg DS, Ruggeri B, Barber MT, Biswas S, Miknyocki S, Waldman SA, Cholecystokinin A. and B receptors are differentially expressed in normal pancreas and pancreatic adenocarcinoma. J. Clin. Invest. 1997;100:597–603. doi: 10.1172/JCI119570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weinberg DS, Heyt GJ, Cavanagh M, Pitchon D, McGlynn KA, London WT. Cholecystokinin and gastrin levels are not elevated in human pancreatic adenocarcinoma. Cancer Epidemiol. Biomarkers Prev. 2001;10:721–722. [PubMed] [Google Scholar]
- 111.Ji B, Bi Y, Simeone D, Mortensen RM, Logsdon CD. Human pancreatic acinar cells lack functional responses to cholecystokinin and gastrin. Gastroenterology. 2001;121:1380–1390. doi: 10.1053/gast.2001.29557. [DOI] [PubMed] [Google Scholar]
- 112.Shintakuya R, Uemura K, Murakami Y, Kondo N, Nakagawa N, Urabe K, et al. Sarcopenia is closely associated with pancreatic exocrine insufficiency in patients with pancreatic disease. Pancreatology. 2017;17:70–75. doi: 10.1016/j.pan.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 113.Danai LV, Babic A, Rosenthal MH, Dennstedt EA, Muir A, Lien EC, et al. Altered exocrine function can drive adipose wasting in early pancreatic cancer. Nature. 2018;558:600–604. doi: 10.1038/s41586-018-0235-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gooden HM, White KJ. Pancreatic cancer and supportive care—pancreatic exocrine insufficiency negatively impacts on quality of life. Support Care Cancer. 2013;21:1835–1841. doi: 10.1007/s00520-013-1729-3. [DOI] [PubMed] [Google Scholar]
- 115.Zuijdgeest-Van Leeuwen SD, Van Der Heijden MS, Rietveld T, Van Den Berg JWO, Tilanus HW, Burgers JA, et al. Fatty acid composition of plasma lipids in patients with pancreatic, lung and oesophageal cancer in comparison with healthy subjects. Clin. Nutr. 2002;21:225–230. doi: 10.1054/clnu.2001.0530. [DOI] [PubMed] [Google Scholar]
- 116.Matejcic M, Lesueur F, Biessy C, Renault AL, Mebirouk N, Yammine S, et al. Circulating plasma phospholipid fatty acids and risk of pancreatic cancer in a large European cohort. Int. J. Cancer. 2018;143:2437–2448. doi: 10.1002/ijc.31797. [DOI] [PubMed] [Google Scholar]
- 117.Murphy RA, Yeung E, Mazurak VC, Mourtzakis M. Influence of eicosapentaenoic acid supplementation on lean body mass in cancer cachexia. Br. J. Cancer. 2011;105:1469–1473. doi: 10.1038/bjc.2011.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dewey, A., Baughan, C., Dean, T. P., Higgins, B. & Johnson I. Eicosapentaenoic acid (EPA, an omega‐3 fatty acid from fish oils) for the treatment of cancer cachexia. Cochrane Database Syst. Rev. CD004597 (2007) [DOI] [PMC free article] [PubMed]
- 119.Haaber AB, Rosenfalck AM, Hansen B, Hilsted J, Larsen S. Bone mineral metabolism, bone mineral density, and body composition in patients with chronic pancreatitis and pancreatic exocrine insufficiency. Int. J. Pancreatol. 2000;27:21–27. doi: 10.1385/IJGC:27:1:21. [DOI] [PubMed] [Google Scholar]
- 120.Nakamura T, Takebe K, Imamura K, Tando Y, Yamada N, Arai Y, et al. Fat-soluble vitamins in patients with chronic pancreatitis (pancreatic insufficiency) Acta Gastroenterol. Belg. 1996;59:10–14. [PubMed] [Google Scholar]
- 121.Klapdor S, Richter E, Klapdor R. Vitamin D status and per-oral vitamin D supplementation in patients suffering from chronic pancreatitis and pancreatic cancer disease. Anticancer Res. 2012;32:1991–1998. [PubMed] [Google Scholar]
- 122.Dev R, Del Fabbro E, Schwartz GG, Hui D, Palla SL, Gutierrez N, et al. Preliminary report: vitamin D deficiency in advanced cancer patients with symptoms of fatigue or anorexia. Oncologist. 2011;16:1637–1641. doi: 10.1634/theoncologist.2011-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Garcia M, Seelaender M, Sotiropoulos A, Coletti D, Lancha AH. Vitamin D, muscle recovery, sarcopenia, cachexia, and muscle atrophy. Nutrition. 2019;60:66–69. doi: 10.1016/j.nut.2018.09.031. [DOI] [PubMed] [Google Scholar]
- 124.Camperi A, Pin F, Costamagna D, Penna F, Menduina ML, Aversa Z, et al. Vitamin D and VDR in cancer cachexia and muscle regeneration. Oncotarget. 2017;8:21778–21793. doi: 10.18632/oncotarget.15583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mantovani G, Madeddu C, Maccio A. Cachexia and oxidative stress in cancer: an innovative therapeutic management. Curr. Pharm. Des. 2012;18:4813–4818. doi: 10.2174/138161212803216889. [DOI] [PubMed] [Google Scholar]
- 126.Huang, B. Z., Pandol, S. J., Jeon, C. Y., Chari, S. T., Sugar, C. A., Chao, C. R. et al. New-onset diabetes, longitudinal trends in metabolic markers, and risk of pancreatic cancer in a heterogeneous population. J. Gastroenterol. Hepatol. 18, 1812–1821.e7 (2019). [DOI] [PMC free article] [PubMed]
- 127.Sah RP, Nagpal SJS, Mukhopadhyay D, Chari ST. New insights into pancreatic cancer-induced paraneoplastic diabetes. Nat. Rev. Gastroenterol. Hepatol. 2013;10:423–433. doi: 10.1038/nrgastro.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Aggarwal G, Ramachandran V, Javeed N, Arumugam T, Dutta S, Klee GG, et al. Adrenomedullin is up-regulated in patients with pancreatic cancer and causes insulin resistance in beta cells and mice. Gastroenterology. 2012;143:1510–1517.e1511. doi: 10.1053/j.gastro.2012.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Armstrong EA, Beal EW, Chakedis J, Paredes AZ, Moris D, Pawlik TM, et al. Exosomes in Pancreatic Cancer: from Early Detection to Treatment. J. Gastrointest. Surg. 2018;22:737–750. doi: 10.1007/s11605-018-3693-1. [DOI] [PubMed] [Google Scholar]
- 130.Wang WS, Liu XH, Liu LX, Jin DY, Yang PY, Wang XL. Identification of proteins implicated in the development of pancreatic cancer-associated diabetes mellitus by iTRAQ-based quantitative proteomics. J. Proteomics. 2013;84:52–60. doi: 10.1016/j.jprot.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 131.Basso D, Greco E, Fogar P, Pucci P, Flagiello A, Baldo G, et al. Pancreatic cancer-associated diabetes mellitus: an open field for proteomic applications. Clin. Chim. Acta. 2005;357:184–189. doi: 10.1016/j.cccn.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 132.Basso D, Greco E, Fogar P, Pucci P, Flagiello A, Baldo G, et al. Pancreatic cancer-derived S-100A8 N-terminal peptide: a diabetes cause? Clin. Chim. Acta. 2006;372:120–128. doi: 10.1016/j.cca.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 133.Permert J, Larsson J, Westermark GT, Herrington MK, Christmanson L, Pour PM, et al. Islet amyloid polypeptide in patients with pancreatic cancer and diabetes. N. Engl. J. Med. 1994;330:313–318. doi: 10.1056/NEJM199402033300503. [DOI] [PubMed] [Google Scholar]
- 134.Ding X, Flatt PR, Permert J, Adrian TE. Pancreatic cancer cells selectively stimulate islet β cells to secrete amylin. Gastroenterology. 1998;114:130–138. doi: 10.1016/S0016-5085(98)70641-9. [DOI] [PubMed] [Google Scholar]
- 135.Liu J, Knezetic JA, Strömmer L, Permert J, Larsson JR, Adrian TE. The intracellular mechanism of insulin resistance in pancreatic cancer patients. J. Clin. Endocrinol. Metab. 2000;85:1232–1238. doi: 10.1210/jcem.85.3.6400. [DOI] [PubMed] [Google Scholar]
- 136.Yoshikawa T, Noguchi Y, Doi C, Makino T, Okamoto T, Matsumoto A. Insulin resistance was connected with the alterations of substrate utilization in patients with cancer. Cancer Lett. 1999;141:93–98. doi: 10.1016/S0304-3835(99)00086-5. [DOI] [PubMed] [Google Scholar]
- 137.Watanapa P, Williamson RC. Surgical palliation for pancreatic cancer: developments during the past two decades. Br. J. Surg. 1992;79:8–20. doi: 10.1002/bjs.1800790105. [DOI] [PubMed] [Google Scholar]
- 138.Wong YT, Brams DM, Munson L, Sanders L, Heiss F, Chase M, et al. Gastric outlet obstruction secondary to pancreatic cancer: surgical vs endoscopic palliation. Surgical Endosc. 2002;16:310–312. doi: 10.1007/s00464-001-9061-2. [DOI] [PubMed] [Google Scholar]
- 139.Dzutsev A, Badger JH, Perez-Chanona E, Roy S, Salcedo R, Smith CK, et al. Microbes and cancer. Annu. Rev. Immunol. 2017;35:199–228. doi: 10.1146/annurev-immunol-051116-052133. [DOI] [PubMed] [Google Scholar]
- 140.Torres PJ, Fletcher EM, Gibbons SM, Bouvet M, Doran KS, Kelley ST. Characterization of the salivary microbiome in patients with pancreatic cancer. Peer J. 2015;3:e1373. doi: 10.7717/peerj.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Farrell JJ, Zhang L, Zhou H, Chia D, Elashoff D, Akin D, et al. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut. 2012;61:582–588. doi: 10.1136/gutjnl-2011-300784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 2018;8:403–416. doi: 10.1158/2159-8290.CD-17-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sethi V, Kurtom S, Tarique M, Lavania S, Malchiodi Z, Hellmund L, et al. Gut microbiota promotes tumor growth in mice by modulating immune response. Gastroenterology. 2018;155:33–37 e36. doi: 10.1053/j.gastro.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357:1156–1160. doi: 10.1126/science.aah5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li S, Fuhler GM, Bn N, Jose T, Bruno MJ, Peppelenbosch MP, et al. Pancreatic cyst fluid harbors a unique microbiome. Microbiome. 2017;5:147. doi: 10.1186/s40168-017-0363-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bindels LB, Neyrinck AM, Loumaye A, Catry E, Walgrave H, Cherbuy C, et al. Increased gut permeability in cancer cachexia: mechanisms and clinical relevance. Oncotarget. 2018;9:18224–18238. doi: 10.18632/oncotarget.24804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pötgens SA, Brossel H, Sboarina M, Catry E, Cani PD, Neyrinck AM, et al. Klebsiella oxytoca expands in cancer cachexia and acts as a gut pathobiont contributing to intestinal dysfunction. Sci. Rep. 2018;8:12321. doi: 10.1038/s41598-018-30569-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bures J, Cyrany J, Kohoutova D, Forstl M, Rejchrt S, Kvetina J, et al. Small intestinal bacterial overgrowth syndrome. World J. Gastroenterol. 2010;16:2978–2990. doi: 10.3748/wjg.v16.i24.2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nishiyama H, Nagai T, Kudo M, Okazaki Y, Azuma Y, Watanabe T, et al. Supplementation of pancreatic digestive enzymes alters the composition of intestinal microbiota in mice. Biochem. Biophys. Res. Commun. 2018;495:273–279. doi: 10.1016/j.bbrc.2017.10.130. [DOI] [PubMed] [Google Scholar]
- 150.Varian BJ, Gourishetti S, Poutahidis T, Lakritz JR, Levkovich T, Kwok C, et al. Beneficial bacteria inhibit cachexia. Oncotarget. 2016;7:11803–11816. doi: 10.18632/oncotarget.7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science. 2018;359:91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 152.Ahuja M, Schwartz DM, Tandon M, Son A, Zeng M, Swaim W, et al. Orai1-mediated antimicrobial secretion from pancreatic acini shapes the gut microbiome and regulates gut innate immunity. Cell Metab. 2017;25:635–646. doi: 10.1016/j.cmet.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nay K, Jollet M, Goustard B, Baati N, Vernus B, Pontones M, et al. Gut bacteria are critical for optimal muscle function: a potential link with glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 2019;317:E158–E171. doi: 10.1152/ajpendo.00521.2018. [DOI] [PubMed] [Google Scholar]
- 154.Oliphant K, Allen-Vercoe E. Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome. 2019;7:91. doi: 10.1186/s40168-019-0704-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu R, Hong J, Xu X, Feng Q, Zhang D, Gu Y, et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017;23:859–868. doi: 10.1038/nm.4358. [DOI] [PubMed] [Google Scholar]
- 156.Biswas AK, Acharyya S. Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer. 2020;20:274–284. doi: 10.1038/s41568-020-0251-4. [DOI] [PubMed] [Google Scholar]
- 157.Yakovenko A, Cameron M, Trevino JG. Molecular therapeutic strategies targeting pancreatic cancer induced cachexia. World J. Gastrointest. Surg. 2018;10:95–106. doi: 10.4240/wjgs.v10.i9.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Arends J, Bachmann P, Baracos V, Barthelemy N, Bertz H, Bozzetti F, et al. ESPEN guidelines on nutrition in cancer patients. Clin. Nutr. 2017;36:11–48. doi: 10.1016/j.clnu.2016.07.015. [DOI] [PubMed] [Google Scholar]
- 159.Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, Radhakrishnan P, et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014;2:18. doi: 10.1186/2049-3002-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.May PE, Barber A, D’Olimpio JT, Hourihane A, Abumrad NN. Reversal of cancer-related wasting using oral supplementation with a combination of β-hydroxy-β-methylbutyrate, arginine, and glutamine. Am. J. Surg. 2002;183:471–479. doi: 10.1016/S0002-9610(02)00823-1. [DOI] [PubMed] [Google Scholar]
- 161.Holecek M. Side effects of long-term glutamine supplementation. JPEN J. Parenter Enteral. Nutr. 2012;37:607–616. doi: 10.1177/0148607112460682. [DOI] [PubMed] [Google Scholar]
- 162.Smith HJ, Mukerji P, Tisdale MJ. Attenuation of proteasome-induced proteolysis in skeletal muscle by β-hydroxy-β-methylbutyrate in cancer-induced muscle loss. Cancer Res. 2005;65:277–283. doi: 10.1158/0008-5472.CAN-05-0169. [DOI] [PubMed] [Google Scholar]
- 163.Wilkinson DJ, Hossain T, Hill DS, Phillips BE, Crossland H, Williams J, et al. Effects of leucine and its metabolite β-hydroxy-β-methylbutyrate on human skeletal muscle protein metabolism. J. Physiol. 2013;591:2911–2923. doi: 10.1113/jphysiol.2013.253203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Eley Helen L, Russell Steven T, Tisdale, Michael J. Effect of branched-chain amino acids on muscle atrophy in cancer cachexia. Biochem. J. 2007;407:113–120. doi: 10.1042/BJ20070651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tayek JA, Bistrian BR, Hehir DJ, Martin R, Moldawer LL, Blackburn GL. Improved protein kinetics and albumin synthesis by branched chain amino acid‐enriched total parenteral nutrition in cancer cachexia: a prospective randomized crossover trial. Cancer. 1986;58:147–157. doi: 10.1002/1097-0142(19860701)58:1<147::AID-CNCR2820580126>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 166.Deutz NE, Safar A, Schutzler S, Memelink R, Ferrando A, Spencer H, et al. Muscle protein synthesis in cancer patients can be stimulated with a specially formulated medical food. Clin. Nutr. 2011;30:759–768. doi: 10.1016/j.clnu.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Berk L, James J, Schwartz A, Hug E, Mahadevan A, Samuels M, et al. A randomized, double-blind, placebo-controlled trial of a beta-hydroxyl beta-methyl butyrate, glutamine, and arginine mixture for the treatment of cancer cachexia (RTOG 0122) Support Care Cancer. 2008;16:1179–1188. doi: 10.1007/s00520-008-0403-7. [DOI] [PubMed] [Google Scholar]
- 168.Malta FAPS, Estadella D, Gonçalves DC. The role of omega 3 fatty acids in suppressing muscle protein catabolism: a possible therapeutic strategy to reverse cancer cachexia? J. Funct. Foods. 2019;54:1–12. doi: 10.1016/j.jff.2018.12.033. [DOI] [Google Scholar]
- 169.Wigmore SJ, Barber MD, Ross JA, Tisdale MJ, Fearon KCH. Effect of oral eicosapentaenoic acid on weight loss in patients with pancreatic cancer. Nutr Cancer. 2000;36:177–184. doi: 10.1207/S15327914NC3602_6. [DOI] [PubMed] [Google Scholar]
- 170.Abe K, Uwagawa T, Haruki K, Takano Y, Onda S, Sakamoto T, et al. Effects of ω-3 fatty acid supplementation in patients with bile duct or pancreatic cancer undergoing chemotherapy. Anticancer Res. 2018;38:2369–2375. doi: 10.21873/anticanres.12732. [DOI] [PubMed] [Google Scholar]
- 171.Fearon KCH, von Meyenfeldt MF, Moses AGW, van Geenen R, Roy A, Gouma DJ, et al. Effect of a protein and energy dense n-3 fatty acid enriched oral supplement on loss of weight and lean tissue in cancer cachexia: a randomised double blind trial. Gut. 2003;52:1479–1486. doi: 10.1136/gut.52.10.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hinson RM, Williams JA, Shacter E. Elevated interleukin 6 is induced by prostaglandin E2 in a murine model of inflammation: possible role of cyclooxygenase-2. Proc. Natl Acad. Sci. USA. 1996;93:4885–4890. doi: 10.1073/pnas.93.10.4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kunkel SL, Spengler M, May MA, Spengler R, Larrick J, Remick D. Prostaglandin E2 regulates macrophage-derived tumor necrosis factor gene expression. J. Biol. Chem. 1988;263:5380–5384. doi: 10.1016/S0021-9258(18)60727-6. [DOI] [PubMed] [Google Scholar]
- 174.Thompson MG, Pascal M, Mackie SC, Thom A, Morrison KS, Colette Backwell FR, et al. Evidence that protein kinase C and mitogen activated protein kinase are not involved in the mechanism by which insulin stimulates translation in L6 myoblasts. Biosci. Rep. 1995;15:37–46. doi: 10.1007/BF01200213. [DOI] [PubMed] [Google Scholar]
- 175.Mantovani G, Maccio A, Madeddu C, Serpe R, Antoni G, Massa E, et al. Phase II nonrandomized study of the efficacy and safety of COX-2 inhibitor celecoxib on patients with cancer cachexia. J. Mol. Med. (Berl) 2010;88:85–92. doi: 10.1007/s00109-009-0547-z. [DOI] [PubMed] [Google Scholar]
- 176.Lai V, George J, Richey L, Kim HJ, Cannon T, Shores C, et al. Results of a pilot study of the effects of celecoxib on cancer cachexia in patients with cancer of the head, neck, and gastrointestinal tract. Head Neck. 2008;30:67–74. doi: 10.1002/hed.20662. [DOI] [PubMed] [Google Scholar]
- 177.McMillan DC, Wigmore SJ, Wigmore KCH, O’Gorman P, Wright CE, McArdle CS. A prospective randomized study of megestrol acetate and ibuprofen in gastrointestinal cancer patients with weight loss. Br J Cancer. 1999;79:495–500. doi: 10.1038/sj.bjc.6690077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.McMillan DC, Simpson JM, Preston T, Watson WS, Fearon KCH, Shenkin A, et al. Effect of megestrol acetate on weight loss, body composition and blood screen of gastrointestinal cancer patients. Clin. Nutr. 1994;13:85–89. doi: 10.1016/0261-5614(94)90065-5. [DOI] [PubMed] [Google Scholar]
- 179.Gordon JN, Trebble TM, Ellis RD, Duncan HD, Johns T, Goggin PM. Thalidomide in the treatment of cancer cachexia: a randomised placebo controlled trial. Gut. 2005;54:540–545. doi: 10.1136/gut.2004.047563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wiedenmann B, Malfertheiner P, Friess H, Ritch P, Arseneau J, Mantovani G, et al. A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia. J. Support. Oncol. 2008;6:18–25. [PubMed] [Google Scholar]
- 181.Hurwitz H, Van Cutsem E, Bendell J, Hidalgo M, Li C-P, Salvo MG, et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Invest. New Drugs. 2018;36:683–695. doi: 10.1007/s10637-018-0580-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Solheim TS, Laird BJA, Balstad TR, Stene GB, Bye A, Johns N, et al. A randomized phase II feasibility trial of a multimodal intervention for the management of cachexia in lung and pancreatic cancer. J. Cachexia Sarcopenia Muscle. 2017;8:778–788. doi: 10.1002/jcsm.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Solheim TS, Laird BJ, Balstad TR, Bye A, Stene G, Baracos V, et al. Cancer cachexia: rationale for the MENAC (Multimodal—Exercise, Nutrition and Anti-inflammatory medication for Cachexia) trial. BMJ Support. Palliat. Care. 2018;8:258–265. doi: 10.1136/bmjspcare-2017-001440. [DOI] [PubMed] [Google Scholar]
- 184.Wiskemann J, Clauss D, Tjaden C, Hackert T, Schneider L, Ulrich CM, et al. Progressive resistance training to impact physical fitness and body weight in pancreatic cancer patients: A randomized controlled trial. Pancreas. 2019;48:257–266. doi: 10.1097/MPA.0000000000001221. [DOI] [PubMed] [Google Scholar]
- 185.Hamauchi S, Furuse J, Takano T, Munemoto Y, Furuya K, Baba H, et al. A multicenter, open-label, single-arm study of anamorelin (ONO-7643) in advanced gastrointestinal cancer patients with cancer cachexia. Cancer. 2019;125:4294–4302. doi: 10.1002/cncr.32406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Koch M, Varela L, Kim JG, Kim JD, Hernández-Nuño F, Simonds SE, et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature. 2015;519:45–50. doi: 10.1038/nature14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Jatoi A, Windschitl HE, Loprinzi CL, Sloan JA, Dakhil SR, Mailliard JA, et al. Dronabinol versus megestrol acetate versus combination therapy for cancer-associated anorexia: a North Central Cancer Treatment Group study. J. Clin. Oncol. 2002;20:567–573. doi: 10.1200/JCO.2002.20.2.567. [DOI] [PubMed] [Google Scholar]
- 188.Golan T, Geva R, Richards D, Madhusudan S, Lin BK, Wang HT, et al. LY2495655, an antimyostatin antibody, in pancreatic cancer: a randomized, phase 2 trial. J. Cachexia Sarcopenia Muscle. 2018;9:871–879. doi: 10.1002/jcsm.12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Riechelmann RP, Burman D, Tannock IF, Rodin G, Zimmermann C. Phase II trial of mirtazapine for cancer-related cachexia and anorexia. Am J Hosp. Palliat. Med. 2010;27:106–110. doi: 10.1177/1049909109345685. [DOI] [PubMed] [Google Scholar]
- 190.Landers A, Muircroft W, Brown H. Pancreatic enzyme replacement therapy (PERT) for malabsorption in patients with metastatic pancreatic cancer. BMJ Support. Palliat. Care. 2016;6:75–79. doi: 10.1136/bmjspcare-2014-000694. [DOI] [PubMed] [Google Scholar]
- 191.Bruno M, Haverkort E, Tijssen G, Tytgat G, Van, Leeuwen D. Placebo controlled trial of enteric coated pancreatin microsphere treatment in patients with unresectable cancer of the pancreatic head region. Gut. 1998;42:92–96. doi: 10.1136/gut.42.1.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Dominguez-Munoz JE, Nieto-Garcia L, Lopez-Diaz J, Larino-Noia J, Abdulkader I, Iglesias-Garcia J. Impact of the treatment of pancreatic exocrine insufficiency on survival of patients with unresectable pancreatic cancer: a retrospective analysis. BMC Cancer. 2018;18:534. doi: 10.1186/s12885-018-4439-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Woo SM, Joo J, Kim SY, Park SJ, Han SS, Kim TH, et al. Efficacy of pancreatic exocrine replacement therapy for patients with unresectable pancreatic cancer in a randomized trial. Pancreatology. 2016;16:1099–1105. doi: 10.1016/j.pan.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 194.Iglesia DDL, Avci B, Kiriukova M, Panic N, Bozhychko M, Sandru V, et al. Pancreatic exocrine insufficiency and pancreatic enzyme replacement therapy in patients with advanced pancreatic cancer: a systematic review and meta-analysis. United European Gastroenterol. J. 2020;8:1115–1125. doi: 10.1177/2050640620938987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Johnson C. Guidelines for the management of patients with pancreatic cancer periampullary and ampullary carcinomas. Gut. 2005;54:v1–v16. doi: 10.1136/gut.2004.048132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Tempero MA, Malafa MP, Al-Hawary M, Asbun H, Bain A, Behrman SW, et al. Pancreatic adenocarcinoma, version 2.2017, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Canc Netw. 2017;15:1028–1061. doi: 10.6004/jnccn.2017.0131. [DOI] [PubMed] [Google Scholar]
- 197.Löhr JM, Dominguez-Munoz E, Rosendahl J, Besselink M, Mayerle J, Lerch MM, et al. United European Gastroenterology evidence-based guidelines for the diagnosis and therapy of chronic pancreatitis (HaPanEU) United European Gastroenterol. J. 2017;5:153–199. doi: 10.1177/2050640616684695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Andersen DK, Korc M, Petersen GM, Eibl G, Li D, Rickels MR, et al. Diabetes, pancreatogenic diabetes, and pancreatic cancer. Diabetes. 2017;66:1103–1110. doi: 10.2337/db16-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–546. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Oliveira AG, Gomes-Marcondes MCC. Metformin treatment modulates the tumour-induced wasting effects in muscle protein metabolism minimising the cachexia in tumour-bearing rats. BMC Cancer. 2016;16:418. doi: 10.1186/s12885-016-2424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kordes S, Pollak MN, Zwinderman AH, Mathôt RA, Weterman MJ, Beeker A, et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16:839–847. doi: 10.1016/S1470-2045(15)00027-3. [DOI] [PubMed] [Google Scholar]
- 202.Chen S-M, Chieng W-W, Huang S-W, Hsu L-J, Jan M-S. The synergistic tumor growth-inhibitory effect of probiotic Lactobacillus on transgenic mouse model of pancreatic cancer treated with gemcitabine. Sci. Rep. 2020;10:20319. doi: 10.1038/s41598-020-77322-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell. 2019;178:795–806.e712. doi: 10.1016/j.cell.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Cheng WY, Wu C-Y, Yu J. The role of gut microbiota in cancer treatment: friend or foe? Gut. 2020;69:1867–1876. doi: 10.1136/gutjnl-2020-321153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 206.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Talbert EE, Cuitiño MC, Ladner KJ, Rajasekerea PV, Siebert M, Shakya R, et al. Modeling human cancer-induced cachexia. Cell Rep. 2019;28:1612–1622.e1614. doi: 10.1016/j.celrep.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Henderson SE, Makhijani N, Mace TA. Pancreatic cancer–induced cachexia and relevant mouse models. Pancreas. 2018;47:937–945. doi: 10.1097/MPA.0000000000001124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.