

Multilocus sequence typing (MLST) is a low-resolution but rapid genotyping method for Clostridioides difficile. Whole-genome sequencing (WGS) has emerged as the new gold standard for C. difficile typing, but cost and lack of standardization still limit broad utilization. In this study, we evaluated the potential to combine the portability of MLST with the increased resolution of WGS for a cost-saving approach to routine C. difficile typing.
KEYWORDS: Clostridium difficile, MLST, WGS, molecular subtyping
ABSTRACT
Multilocus sequence typing (MLST) is a low-resolution but rapid genotyping method for Clostridioides difficile. Whole-genome sequencing (WGS) has emerged as the new gold standard for C. difficile typing, but cost and lack of standardization still limit broad utilization. In this study, we evaluated the potential to combine the portability of MLST with the increased resolution of WGS for a cost-saving approach to routine C. difficile typing. C. difficile strains from two New York City hospitals (hospital A and hospital B) were selected. WGS single-nucleotide polymorphism (wgSNP) was performed using established methods. Sequence types (ST) were determined using PubMLST, while wgSNP analysis was performed using the Bionumerics software. An additional analysis of a subset of data (hospital A) was made comparing the Bionumerics software to the CosmosID pipeline. Cost and turnaround time to results were compared for the algorithmic approach of MLST followed by wgSNP versus direct wgSNP. Among the 202 C. difficile isolates typed, 91% (n = 185/203) clustered within the representative ST, showing a high agreement between MLST and wgSNP. While clustering was similar between the Bionumerics and CosmosID pipelines, large differences in the overall number of SNPs were noted. A two-step algorithm for routine typing results in significantly lower cost than routine use of WGS. Our results suggest that using MLST as a first step in routine typing of C. difficile followed by WGS for MLST concordant strains is a less technically demanding, cost-saving approach for performing C. difficile typing than WGS alone without loss of discriminatory power.
INTRODUCTION
Clostridioides difficile is the most common bacterial cause of hospital-associated diarrhea, with an estimated 500,000 cases in 2011 (1). In the last 10 years, several outbreaks of C. difficile infection (CDI) associated with increased disease severity and increased mortality have been reported worldwide (2, 3). Timely and precise identification of transmission events is an important step in implementing effective infection control measures. Towards that goal, many hospitals routinely employ genotyping of nosocomial isolates to track transmission within their facility.
Routine typing of C. difficile has traditionally been accomplished using a variety of methods, including PCR ribotyping, pulsed-field gel electrophoresis (PFGE), multilocus variable-number tandem repeat analysis (MLVA), and multilocus sequence typing (MLST) (4). As a sequence-based method, MLST provides discrete, easy-to-obtain, and highly reproducible data. Furthermore, MLST offers the advantage of data portability (i.e., comparison of data across laboratories) and simplicity of data interpretation compared to other typing methods (e.g., PFGE) through the use of a central database, PubMLST (http://www.pubmlst.org/cdifficile), that is expert curated, open access over the Internet, and frequently updated with new strain type schemes (4–6). However, MLST may not always provide sufficient discriminatory power to track the transmission of C. difficile over the short term. This limitation may be overcome by correlation with spatial data (patient movement), which can be labor-intensive and inadequate due to the latency between infection and symptom manifestation (7, 8). For these reasons, MLST of housekeeping genes serves the purpose of debunking transmission but cannot conclusively establish C. difficile spread in outbreak situations.
Whole-genome sequencing (WGS) has emerged as the ideal genotypic method to investigate clonal relationships among isolates (9). WGS allows for higher resolution of isolates, as data on the complete genome of an isolate can be generated in a single run. In recent years, WGS platforms have become accessible. Several studies have shown the higher discriminatory power of WGS for C. difficile compared to other typing methods (10, 11). In several studies, evolutionary relatedness of isolates was determined by either constructing a phylogenetic tree based on single-nucleotide polymorphisms (SNPs) identified by comparing raw reads to a reference genome or through de novo assembly of raw reads and comparison of genes to a reference genome (9, 11). Several barriers have to be overcome before WGS can be applied for routine bacterial typing, including cost, long turnaround time (TAT) to results, complex bioinformatic analysis, lack of established interpretive standards, and lack of portability of data generated by individual laboratories.
In this study, we evaluated a two-tiered approach that combines the simplicity and portability of MLST with the high resolution of WGS as a more cost-effective and practical approach for routine typing of C. difficile compared to WGS alone.
(This study was presented in part at the 28th European Congress for Clinical Microbiology and Infectious Diseases [ECCMID], Madrid, Spain, April 2018.)
MATERIALS AND METHODS
Study settings.
This study was conducted at two tertiary care medical centers in New York City, hospital A, a 478-bed tertiary care cancer center, and hospital B, an 862-bed academic medical center. The study was approved by the Institutional Review Boards at both institutions.
C. difficile strains.
C. difficile strains included in this study were recovered between 2014 and 2017. Testing for CDI was accomplished using a commercially available nucleic acid amplification test (Cepheid Xpert C. difficile; Sunnyvale, CA) at both institutions. At hospital A, bacterial culture for the recovery and typing of C. difficile isolates was attempted on all stool samples positive for C. difficile in real time. As MLST is routinely performed at hospital A, C. difficile isolates for this study were selected to represent the most common STs (endemic strains) and a few sporadic strains (<3% of all STs) (12). Stool specimens from hospital B were collected in 2017, stored at −80°C, and sent to hospital A for bacterial culture and typing. All recovered isolates were included in the study.
C. difficile toxigenic culture.
Toxigenic culture was performed as previously described, with some modifications (13). Brain heart infusion agar supplemented (BHIS) with 5 g yeast extract per liter, 0.1% l-cysteine, 0.1% taurocholate (TA) (BHIS-TA), 16 g/liter cefoxitin sodium, and 62.5 g/liter d-cycloserine were prepared in-house and reduced in an anaerobic chamber for 24 h prior to use. Stool samples were added to 4 parts 100% ethanol, vortexed, and incubated at room temperature for 1 to 2 h. The solution was centrifuged at 1,200 × g for 10 min, ethanol was removed, and the stool pellet was inoculated onto reduced BHIS-TA agar plates. The plates were incubated for 48 h under anaerobic conditions. Colonies resembling C. difficile (pale yellow to yellow) were subcultured on sheep blood agar (SBA) plates (Remel, Lenexa, KS), and their identity was further confirmed by matrix-assisted laser desorption-time of flight mass spectrometry (bioMérieux Vitek MS; Durham, NC).
Xpert C. difficile Epi PCR.
The Xpert C. difficile Epi PCR (Xpert PCR) assay is a multiplex real-time PCR that detects the toxin B gene (tcdB), the binary toxin gene (cdt), and the tcdC gene deletion at nucleotide 117. Xpert PCR was performed according to the manufacturer’s instructions.
C. difficile MLST.
MLST was performed on culture isolates as previously described (5). The allelic profile and strain type identification for each sample was completed using the PubMLST database (http://www.pubmlst.org/cdifficile).
C. difficile DNA isolation.
For MLST, C. difficile isolates recovered from BHIS-TA agar were added to 200 μl of Instagene matrix (Bio-Rad, Hercules, CA) and processed per the manufacturer’s instructions, with the resulting supernatant used for PCRs. For WGS, C. difficile isolates were cultured on SBA and incubated anaerobically for 48 h. Genomic DNA (gDNA) was extracted using the QIAamp bacterial suspension protocol (Qiagen, Germantown, MD), with modifications described by Sim and colleagues (14). DNA concentration was measured using the double-stranded DNA high-sensitivity kit protocol on a Qubit 2.0 fluorometer (ThermoFisher Scientific, Waltham, MA).
Library construction and sequencing.
Libraries for each isolate were prepared using the Illumina Nextera XT DNA library preparation kit (Illumina, San Diego, CA). An input of 0.2 ng of total gDNA was used to construct libraries. The quality of each library was assessed prior to sequencing using a high-sensitivity DNA kit on a Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA). Libraries were indexed and sequencing performed on a MiSeq instrument using the MiSeq reagent kit v3 (150-cycle) (Illumina, San Diego, CA).
wgSNP analysis.
Whole-genome single-nucleotide polymorphism (wgSNP) analysis was performed using Bionumerics version 7.6 software (bioMérieux Inc., Austin, TX). Within the Bionumerics software, imported FASTQ files were mapped to a C. difficile reference genome, strain 630 (RT012), a multidrug-resistant C. difficile strain isolated from a Swiss patient with pseudomembranous colitis in 1982 and fully sequenced in 2006 (15, 16). SNP analysis was performed using the default SNP filtering function with the following adjustments to the filters: all sequence positions with more than 20% unreliable bases were removed, each retained SNP position had a minimum 5× absolute coverage and was covered at least once in both forward and reverse directions, reads with more than 20% ambiguous bases were removed, and all noninformative SNPs were removed. Cluster analysis was performed using the categorical (SNP) similarity coefficient and the complete linkage method.
CosmosID pipeline.
Raw paired reads were trimmed at a q-score of >20 and minimum length of 25 bp per read using bbduk of the BBMap suite. Trimmed reads were de novo assembled by the SPAdes assembler (17) using default parameters. Contigs of fewer than 200 nucleotides were excluded from subsequent analyses. Bioinformatic analyses of draft assemblies were performed by CosmosID in-house using the cloud bioinformatics pipeline (https://app.cosmosid.com) (18). Draft genomes of the sequenced C. difficile isolates, along with additional publicly available C. difficile reference genomes, were analyzed using the parsnp program from Harvest suite (19) to identify core genomes across these isolates and build a maximum likelihood tree using core genome SNPs (cgSNPs). The generated Newick-format tree was then visualized using MEGA7 (20). Sequence types (ST) of these genomes were also determined using MLST with the MLSTcheck and pubMLST databases (http://pubmlst.org/cdifficile/) as described elsewhere (5).
Discriminatory power.
The discriminatory power was calculated for wgSNP and MLST for both cohorts of patients using the discriminatory power calculator based on the Simpson’s index of diversity (21, 22).
Data availability.
All C. difficile raw sequence reads generated in this study have been deposited in the Sequence Read Archive under BioProject no. PRJNA672122.
RESULTS
C. difficile isolates.
A total of 245 isolates (108 from hospital A and 137 from hospital B) were recovered in culture for typing. These isolates included both epidemiologically related and sporadic isolates. Based on historical data, the most common STs at hospital A were ST-2, ST-42, ST-1, ST-8, and ST-11. A total of 108 clinical isolates with known ST types were selected (Table 1). For hospital B, MLST was performed de novo on 137 isolates recovered from 167 PCR-positive stool samples (80.8% recovery) collected over a period of 12 months. Overall, 36 STs were identified from hospital B samples. The most frequent STs identified were ST-8 (10%), ST-42 (10%), ST-1 (10%), ST-2 (5%), and ST-11 (5%). The distribution of STs identified at hospital B during the study is shown in Fig. S1 in the supplemental material. The top 10 most common STs recovered at both institutions and some infrequently recovered ST types were selected for further WGS (Table 1).
TABLE 1.
MLST C. difficile strains included in the study
| No. of strains from: |
||
|---|---|---|
| MLST strain | Hospital A | Hospital B |
| 1 | 15 | 15 |
| 2 | 20 | 8 |
| 3 | 0 | 5 |
| 6 | 0 | 4 |
| 8 | 10 | 17 |
| 11 | 9 | 9 |
| 14 | 5 | 0 |
| 28 | 0 | 3 |
| 35 | 0 | 3 |
| 42 | 20 | 13 |
| 44 | 1 | 6 |
| 53 | 0 | 8 |
| 54 | 5 | 0 |
| 55 | 5 | 0 |
| 58 | 5 | 0 |
| 110 | 6 | 4 |
| Infrequent ST | 5a | 1b |
| Total no. of strains analyzed | 106 | 96 |
ST-26, ST-37, ST-49, ST-98, and ST-190, one of each.
ST-13.
Comparison of wgSNP analysis to conventional MLST.
Of the 245 C. difficile isolates submitted for WGS, 202 isolates (106 from hospital A and 96 from hospital B) passed all the quality control requirements (e.g., median coverage, median contig number, median N50, and number of core genome loci) for further analysis by wgSNP.
The discriminatory power of MLST was 0.89 and 0.90 for hospital A and hospital B, respectively, compared to 0.99 and 0.98 for wgSNP (P < 0.05). The 106 hospital A C. difficile isolates represented 16 distinct STs, as identified by MLST. Based on wgSNP analysis, 87 distinct clusters or genetically distinct strains were identified (≤30 SNP differences per cluster) (Fig. 1). Agreement between MLST and WGS was 91% (97/106) (Fig. 2A). By wgSNP analysis, 9 isolates clustered differently than expected based on their ST types. One ST-98 strain clustered with the ST-11 strains, and the six ST-2 strains clustered separately from the other 14 ST-2 isolates. Further subclustering could be observed within each ST. ST-2 strains demonstrated the greatest diversity (54 to 143 SNPs), and the least diversity was observed within the ST-54 strains (8 to 66 SNPs). Of note, two ST-1 strains isolated from the same patient 53 days apart clustered separately from the other ST-1 strains in this group. The number of SNPs between strains of different STs ranged from 43 to 57 (ST-98/ST-11) to >7,700 SNPs (Table S1).
FIG 1.

Phylogenetic tree generated from whole-genome sequencing of hospital A C. difficile strains previously characterized by MLST. The tree was generated using the SNP analysis the categorical SNP module with a scaling factor of 100 and complete linkage cluster method.
FIG 2.
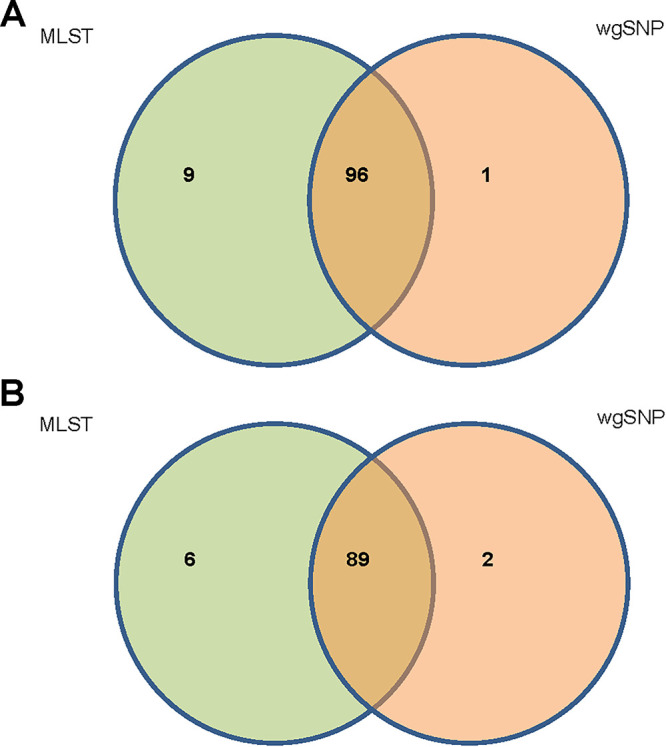
Venn diagram of isolate clustering with MLST and wgSNP analysis. (A) Hospital A isolates. wgSNP analysis confirmed clustering of 96 isolates within their correct ST types. (B) Hospital B isolates. wgSNP analysis confirmed clustering of 89 isolates within their correct ST types.
The 96 hospital B C. difficile isolates represented 13 distinct STs, as identified by MLST. Greater diversity was observed based on wgSNP analysis with 79 distinct clusters, and genetically distinct strains were identified (≤30 SNP differences per cluster) (Fig. 3). The agreement between MLST and WGS for establishing relatedness or lack of relatedness among the strains typed was 91.7% (Fig. 2B). By wgSNP analysis, 8 isolates clustered differently than expected based on their STs. Similar to data obtained with the hospital A strains, 1 ST-110 strain clustered with the ST-2 cluster. There were 5 ST-3 strains isolated from hospital B; 3 clustered with ST-6 strains and 2 clustered with ST-2 strains. Additional subclustering could be observed within each ST. Similar to hospital A strains, the greatest diversity was observed within ST-2 strains (54 to 1,359 SNPs), and the least diversity was observed within the ST-11 strains (66 to 145 SNPs) (Table S2).
FIG 3.

Phylogenetic tree generated from whole-genome sequencing of hospital B C. difficile strains characterized by MLST. The tree was generated using the SNP analysis the categorical SNP module with a scaling factor of 100 and complete linkage cluster method.
Comparison of different pipelines to conventional MLST.
We further determined agreement for WGS data obtained using different bioinformatics pipelines on the hospital A data set. A side-by-side comparison of phylogenetic trees, one generated in-house using the Bionumerics software (wgSNP) and the other generated by the CosmosID pipeline (cgSNP), showed that both tools resulted in similar clustering of isolates (Fig. 1 and 4 and Table S3). However, the range of SNPs identified by different methods varied significantly, with larger SNP differences observed with the Bionumerics pipeline (P < 0.0) (Table 2). The discriminatory power was 0.99 for the in-house Bionumerics analysis and 0.96 for the CosmosID pipeline (P < 0.05).
FIG 4.

Phylogenetic trees generated from whole-genome sequencing of hospital A C. difficile strains previously characterized by MLST. Maximum likelihood tree generated using core genome single-nucleotide polymorphisms (cgSNPs) analysis by CosmosID.
TABLE 2.
Comparison of SNP differences between two kinds of analysis software on hospital A strains
| ST | Bionumerics (range of wgSNPs) | CosmosID (range of cgSNPs) |
|---|---|---|
| 1 | 37–109 | 12–62 |
| 11 | 37–684 | 10–879 |
| 110 | 31–70 | 13–31 |
| 14 | 23–63 | 17–23 |
| 2 | 54–143 | 36–92 |
| 42 | 11–105 | 8–66 |
| 54 | 8–66 | 1–51 |
| 55 | 23–89 | 8–26 |
| 58 | 37–150 | 12–96 |
| 8 | 9–96 | 1–78 |
Cost analysis and turnaround time to results.
We evaluated the reagent cost savings that may be associated with a two-step algorithm compared to one-step WGS for routine surveillance for hospital A. For MLST, each isolate requires Sanger sequencing reactions for an overall cost of approximately $50 per isolate. WGS cost, including DNA extraction, library preparation, and sequencing, is approximately $165 per isolate (Fig. 5). With hospital A positivity rates, between 6 and 15 C. difficile strains are available for genotyping weekly, with a range of 2 to 4 ST concordant isolates per week. Performing MLST on all isolates comes to a maximum of $39,000/year compared to $129,000/year for WGS. Limiting WGS to ST concordant cases would cost between $74,000 and $111,000/year and result in savings ranging from approximately $18,000 to $55,000 per year compared to WGS of all isolates. Furthermore, while MLST data analysis is simple and free to perform on the PubMLST site, analysis of WGS data requires either a trained bioinformatician, investment in a tool like Bionumerics, or use of a commercial service like CosmosID, which can add between $100 and $150 per isolate to analyze.
FIG 5.

Cost and time to result analysis of a one-step versus the two-step algorithm for routine surveillance of C. difficile. (A) Proposed two-step algorithm where only isolates with identical strain types (ST) are further analyzed by whole-genome sequencing and single-nucleotide polymorphism (wgSNP). The overall time to results and cost (*) associated with the algorithm depend on the number of isolates with similar or different ST. (B) One-step testing where all isolates are analyzed by wgSNP. The time to results and overall cost associated with that approach are shown.
In terms of TAT, for MLST a colony of C. difficile can be used and DNA extracted without further growth. With WGS, additional growth in broth is necessary to improve the quality of sequencing, resulting in an additional day prior to DNA extraction. The TAT for Sanger sequencing is approximately 28 h for 12 samples, and data analysis is, on average, 1 h, for an overall TAT from colony recovery to results of 1.5 days. For WGS, the TAT, including library preparation, quality control check, and sequencing, is, on average, 6 days. Data analysis using the Bionumerics software is, on average, 1 to 2 h for 12 samples. This results in an average of 6 days from colony recovery to results (Fig. 5).
DISCUSSION
In this study, we evaluated a two-tiered approach that combined the simplicity and portability of MLST with the high resolution of WGS as a cost-saving and practical approach compared to WGS of all isolates. Using a large number of C. difficile isolates from two medical centers, we showed that MLST performed well in identifying unrelated strains based on differences in STs. Clustering by MLST was greater than 90% concordant with WGS. As previously reported and confirmed in our study, greater differences within ST clusters were observed by WGS owing to its greater discriminatory power (23, 24). Therefore, if two isolates are distinguishable by MLST, it is likely that they will similarly be distinguishable by WGS. Given that WGS is still more costly and computationally more challenging, an approach to limit its application to isolates with identical STs may be a viable, cost-effective option for laboratories performing routine typing to inform timely infection control practices. We showed that a two-tiered approach could result in up to $55,000 of savings compared to WGS of all isolates, with a more rapid TAT. This approach would be most beneficial for hospital laboratories performing or considering routine surveillance where the number of isolates is not significantly high. This approach would not be ideal for large reference laboratories, public health laboratories, or during an outbreak investigation. As Sanger sequencing is still more accessible than WGS to many laboratories, taking advantage of the portability of MLST as a first step in identifying isolates with similar STs might also help facilitate multicenter studies.
We also evaluated two workflows, one utilizing an external vendor for the computational analysis and the other utilizing commercially available software for in-house data analysis to determine the impact of the pipeline used for data analysis given the lack of standardization of wgSNP analysis. While we observed differences in the overall number of SNPs detected among isolates with identical STs, the overall clustering was similar by both methods. However, given that relatedness among strains is based on the number of SNP differences, e.g., ≤2 SNPs for undistinguishable C. difficile strains (25), more attention should be paid to the fact that different algorithms and analysis filters (e.g., number of core genes used) results in different overall SNP numbers. These differences are likely attributes of two distinct SNP typing methods being used, i.e., wgSNP typing by Bionumerics and cgSNP typing by CosmosID. In cgSNPs, SNPs occurred only in the core genome, i.e., vertically inherited essential genes that are present in all strains are used in inferring the phylogeny, whereas wgSNP includes SNPs occurring both in core and accessory genome sequences. Therefore, cgSNPs are often considered more robust for phylogenetic inferences, whereas wgSNPs offer higher resolution for strain discriminations (19, 26, 27). As these methods assess genetic variation in two distinct ways, they should be viewed as complementary to each other and can be used in concert to achieve high-resolution phylogenetic inference with high confidence (26, 27).
Our results are similar to those from Zhou and colleagues, showing concordance between MLST and WGS, with isolates with similar STs clustering (23). As stated by the authors, limitations of that study were the small number of isolates (n = 46) and the single-center design. Our study overcomes these limitations with its multicenter design and a significantly larger number of isolates. As suggested in our comparison of different pipelines, the specific number of SNPs identified in each ST could vary when different pipelines and/or filters are used. For example, in Zhou et al., the number of SNP differences within ST-1 (n = 6) ranged between 99 and 656 SNPs, while our study showed that SNP differences within hospital A ST-1 (n = 15) were between 37 and 109 wgSNPs using the Bionumerics software and 12 and 62 cgSNPs using the CosmosID pipeline. Similarly, in an analysis of 486 C. difficile strains collected over a 4-year period in the United Kingdom, direct transmission events between pairs of matched STs could only be ruled out by WGS. The range of SNPs similarly varied between different STs, with one pair of ST-44 isolates differing by more than 4,000 SNPs. Direct transmission between unmatched ST types was not investigated (24).
WGS of C. difficile has been compared to other conventional typing methods for investigation of transmission events. Eyer et al. compared WGS to MLVA (25). In that study, the authors showed that while WGS had more discriminatory power overall than MLVA, the two methods were highly concordant in their ability to identify related isolates during an outbreak investigation. With the TATs, costs, and levels of data analysis expertise needed for both methods being relatively similar, either method could be used as part of a two-step algorithm. In fact, a recent ESCMID study group suggested an approach similar to what we propose in our study for reasons we presented, including TAT, cost, and accessibility to WGS tools. The authors recommended that outbreak investigation could be performed using a two-step algorithm, starting with capillary electrophoresis PCR ribotyping followed by further typing of epidemiologically linked strains of common ribotypes by either WGS or MLVA (28). Gateau and colleagues recently investigated a C. difficile outbreak in two geriatric inpatient units at a French hospital using multiple typing methods, both conventional and WGS-based methods (29). All C. difficile strains recovered during the outbreak period were typed by PCR ribotyping and MLST first, followed by further analysis using more discriminatory methods, including MLVA and WGS, the latter two performed only on concordant STs or ribotypes, an approach similar to what we proposed in our study. This approach confirmed that WGS provided better resolution in differentiating between epidemic and nonepidemic RT-18 strains.
Our study has some limitations. The first is its retrospective design. Isolates from hospital A were selected to represent the most common ST strains, as determined from historical data. While a few less common STs were included in the analysis, it is possible that more pronounced differences in clustering could be observed with a collection of STs not included in the study. However, similar conclusions were drawn from the hospital B isolates, which were collected and typed by MLST prospectively. Second, not all C. difficile isolates recovered were successfully sequenced and analyzed, which could have an impact on the concordance results established between MLST and WGS. Third, the cost and turnaround time analyses are based on local information and may differ between institutions or laboratories.
In conclusion, we show in this study that compared to WGS, a two-tiered approach starting with MLST and followed by wgSNP analysis was a cost-saving, potentially more rapid approach than direct wgSNP for routine surveillance of C. difficile.
Supplementary Material
ACKNOWLEDGMENTS
Nur Hassan, Brian Fanelli, and Manoj Dadlani are employed by CosmosID.
Research reported in this publication was supported by the National Center For Advancing Translational Sciences of the National Institutes of Health under award number UL1TR000457. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also funded in part through the National Institutes of Heath/National Cancer Institute Cancer Center Support (grant P30 CA008748).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. 2015. Burden of Clostridium difficile infection in the United States. N Engl J Med 372:825–834. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuijper EJ, Coignard B, Tull P, Difficile E, States EUM, European Centre for Disease Prevention and Control. 2006. Emergence of Clostridium difficile-associated disease in North America and Europe. Clin Microbiol Infect 12(Suppl 6):2–18. doi: 10.1111/j.1469-0691.2006.01580.x. [DOI] [PubMed] [Google Scholar]
- 3.Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault AM, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson TJ, Horn R, Rene P, Monczak Y, Dascal A. 2005. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med 353:2442–2449. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 4.Knetsch CW, Lawley TD, Hensgens MP, Corver J, Wilcox MW, Kuijper EJ. 2013. Current application and future perspectives of molecular typing methods to study Clostridium difficile infections. Euro Surveill 18:20381. doi: 10.2807/ese.18.04.20381-en. [DOI] [PubMed] [Google Scholar]
- 5.Griffiths D, Fawley W, Kachrimanidou M, Bowden R, Crook DW, Fung R, Golubchik T, Harding RM, Jeffery KJ, Jolley KA, Kirton R, Peto TE, Rees G, Stoesser N, Vaughan A, Walker AS, Young BC, Wilcox M, Dingle KE. 2010. Multilocus sequence typing of Clostridium difficile. J Clin Microbiol 48:770–778. doi: 10.1128/JCM.01796-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker AS, Eyre DW, Wyllie DH, Dingle KE, Harding RM, O'Connor L, Griffiths D, Vaughan A, Finney J, Wilcox MH, Crook DW, Peto TE. 2012. Characterisation of Clostridium difficile hospital ward-based transmission using extensive epidemiological data and molecular typing. PLoS Med 9:e1001172. doi: 10.1371/journal.pmed.1001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durovic A, Widmer AF, Tschudin-Sutter S. 2018. New insights into transmission of Clostridium difficile infection-narrative review. Clin Microbiol Infect 24:483–492. doi: 10.1016/j.cmi.2018.01.027. [DOI] [PubMed] [Google Scholar]
- 9.Maiden MC, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, McCarthy ND. 2013. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol 11:728–736. doi: 10.1038/nrmicro3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Fernandez S, Frentrup M, Steglich M, Gonzaga A, Cobo M, Lopez-Fresnena N, Cobo J, Morosini MI, Canton R, Del Campo R, Nubel U. 2019. Whole-genome sequencing reveals nosocomial Clostridioides difficile transmission and a previously unsuspected epidemic scenario. Sci Rep 9:6959. doi: 10.1038/s41598-019-43464-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janezic S, Rupnik M. 2019. Development and implementation of whole genome sequencing-based typing schemes for Clostridioides difficile. Front Public Health 7:309. doi: 10.3389/fpubh.2019.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McMillen T, Kamboj M, Babady NE. 2016. Comparison of multilocus sequence typing and the Xpert C. difficile/Epi assay for identification of Clostridium difficile 027/NAP1/BI. J Clin Microbiol 54:775–778. doi: 10.1128/JCM.03075-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Babady NE, Stiles J, Ruggiero P, Khosa P, Huang D, Shuptar S, Kamboj M, Kiehn TE. 2010. Evaluation of the Cepheid Xpert Clostridium difficile Epi assay for diagnosis of Clostridium difficile infection and typing of the NAP1 strain at a cancer hospital. J Clin Microbiol 48:4519–4524. doi: 10.1128/JCM.01648-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sim JH, Anikst V, Lohith A, Pourmand N, Banaei N. 2015. Optimized protocol for simple extraction of high-quality genomic DNA from Clostridium difficile for whole-genome sequencing. J Clin Microbiol 53:2329–2331. doi: 10.1128/JCM.00956-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wust J, Sullivan NM, Hardegger U, Wilkins TD. 1982. Investigation of an outbreak of antibiotic-associated colitis by various typing methods. J Clin Microbiol 16:1096–1101. doi: 10.1128/JCM.16.6.1096-1101.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeno-Tarraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet 38:779–786. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 17.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Hebshi NN, Baraniya D, Chen T, Hill J, Puri S, Tellez M, Hasan NA, Colwell RR, Ismail A. 2019. Metagenome sequencing-based strain-level and functional characterization of supragingival microbiome associated with dental caries in children. J Oral Microbiol 11:1557986. doi: 10.1080/20002297.2018.1557986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Treangen TJ, Ondov BD, Koren S, Phillippy AM. 2014. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol 15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunter PR. 1990. Reproducibility and indices of discriminatory power of microbial typing methods. J Clin Microbiol 28:1903–1905. doi: 10.1128/JCM.28.9.1903-1905.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunter PR, Fraser CA. 1989. Application of a numerical index of discriminatory power to a comparison of four physiochemical typing methods for Candida albicans. J Clin Microbiol 27:2156–2160. doi: 10.1128/JCM.27.10.2156-2160.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Y, Burnham CA, Hink T, Chen L, Shaikh N, Wollam A, Sodergren E, Weinstock GM, Tarr PI, Dubberke ER. 2014. Phenotypic and genotypic analysis of Clostridium difficile isolates: a single-center study. J Clin Microbiol 52:4260–4266. doi: 10.1128/JCM.02115-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Didelot X, Eyre DW, Cule M, Ip CL, Ansari MA, Griffiths D, Vaughan A, O'Connor L, Golubchik T, Batty EM, Piazza P, Wilson DJ, Bowden R, Donnelly PJ, Dingle KE, Wilcox M, Walker AS, Crook DW, Peto TE, Harding RM. 2012. Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol 13:R118. doi: 10.1186/gb-2012-13-12-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eyre DW, Fawley WN, Best EL, Griffiths D, Stoesser NE, Crook DW, Peto TE, Walker AS, Wilcox MH. 2013. Comparison of multilocus variable-number tandem-repeat analysis and whole-genome sequencing for investigation of Clostridium difficile transmission. J Clin Microbiol 51:4141–4149. doi: 10.1128/JCM.01095-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson ER, Walker TM, Pallen MJ. 2013. Genomics and outbreak investigation: from sequence to consequence. Genome Med 5:36. doi: 10.1186/gm440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jagadeesan B, Gerner-Smidt P, Allard MW, Leuillet S, Winkler A, Xiao Y, Chaffron S, Van Der Vossen J, Tang S, Katase M, McClure P, Kimura B, Ching Chai L, Chapman J, Grant K. 2019. The use of next generation sequencing for improving food safety: translation into practice. Food Microbiol 79:96–115. doi: 10.1016/j.fm.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krutova M, Wilcox MH, Kuijper EJ. 2019. A two-step approach for the investigation of a Clostridium difficile outbreak by molecular methods. Clin Microbiol Infect 25:1300–1301. doi: 10.1016/j.cmi.2019.07.022. [DOI] [PubMed] [Google Scholar]
- 29.Gateau C, Deboscker S, Couturier J, Vogel T, Schmitt E, Muller J, Menard C, Turcan B, Zaidi RS, Youssouf A, Lavigne T, Barbut F. 2019. Local outbreak of Clostridioides difficile PCR-Ribotype 018 investigated by multi locus variable number tandem repeat analysis, whole genome multi locus sequence typing and core genome single nucleotide polymorphism typing. Anaerobe 60:102087. doi: 10.1016/j.anaerobe.2019.102087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All C. difficile raw sequence reads generated in this study have been deposited in the Sequence Read Archive under BioProject no. PRJNA672122.