

Abstract
Kynurenine-3-monooxygenase (KMO) is an important therapeutic target for several brain disorders that has been extensively studied in recent years. Potent inhibitors towards KMO have been developed and tested within different disease models, showing great therapeutic potential, especially in models of neurodegenerative disease. The inhibition of KMO reduces the production of downstream toxic kynurenine pathway metabolites and shifts the flux to the formation of the neuroprotectant kynurenic acid. However, the efficacy of KMO inhibitors in neurodegenerative disease has been limited by their poor brain permeability. Combined with virtual screening and prodrug strategies, a novel brain penetrating KMO inhibitor has been developed which dramatically decreases neurotoxic metabolites. This review highlights the importance of KMO as a drug target in neurological disease and the benefits of brain permeable inhibitors in modulating kynurenine pathway metabolites in the central nervous system.
Kynurenine-3-monooxygenase
Kynurenine 3-monooxygenase (KMO, EC 1.14.13.9) is an NADPH-dependent flavin monooxygenase which catalyses the hydroxylation of the L-tryptophan (TRP) metabolite L-kynurenine (L-KYN) into 3-hydroxykynurenine (3-HK). KMO is expressed in microglia and infiltrating macrophages in the brain [1], and at high levels in the liver, kidneys and placenta in the periphery [2]. Specifically, KMO is localised to the outer membrane of mitochondria [3] where it associates with the lipid membrane using C-terminal transmembrane domains, which are also crucial for the catalytic activity of KMO [4]. The cofactor flavin adenine dinucleotide (FAD) binds to KMO at a 1:1 ratio [5]. Following binding of l-KYN to KMO (Figure 1), NADPH acts as an electron donor and reduces FAD, leading to the formation of a L-KYN-FAD-hydroperoxide intermediate. L-KYN is then oxidised, resulting in the release of 3-HK and water. KMO is an important enzyme in the kynurenine pathway (KP, Figure 2), the major catabolic route of TRP, and lies at a critical branchpoint of the KP, which makes it an attractive drug target for several neurodegenerative and neuroinflammatory diseases [6].
Figure 1. The KMO reaction cycle.
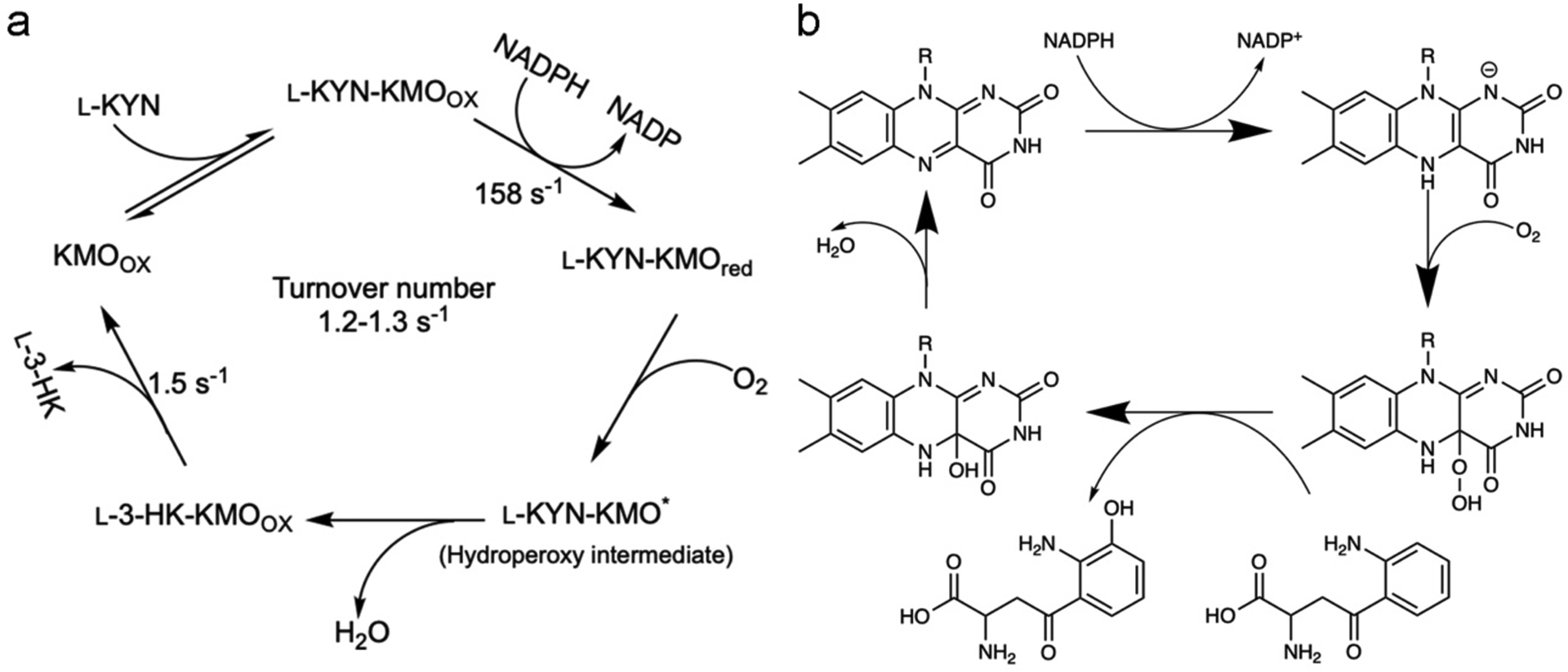
In the KMO reaction, L-KYN binds to the protein first, then NADPH binds and reduces the FAD followed by NADP+ release, allowing for dioxygen binding. A FAD–superoxide radical pair is likely to be formed and rapidly decays to a peroxy-flavin structure, which will act as the electrophile for L-KYN oxidation. C4a-hydroxy flavin is then dehydrated yielding the oxidised flavin. The final step is 3-HK release, which is also the rate limiting step with a slightly faster rate than the overall turnover number [56].
Figure 2. Overview of kynurenine pathway.
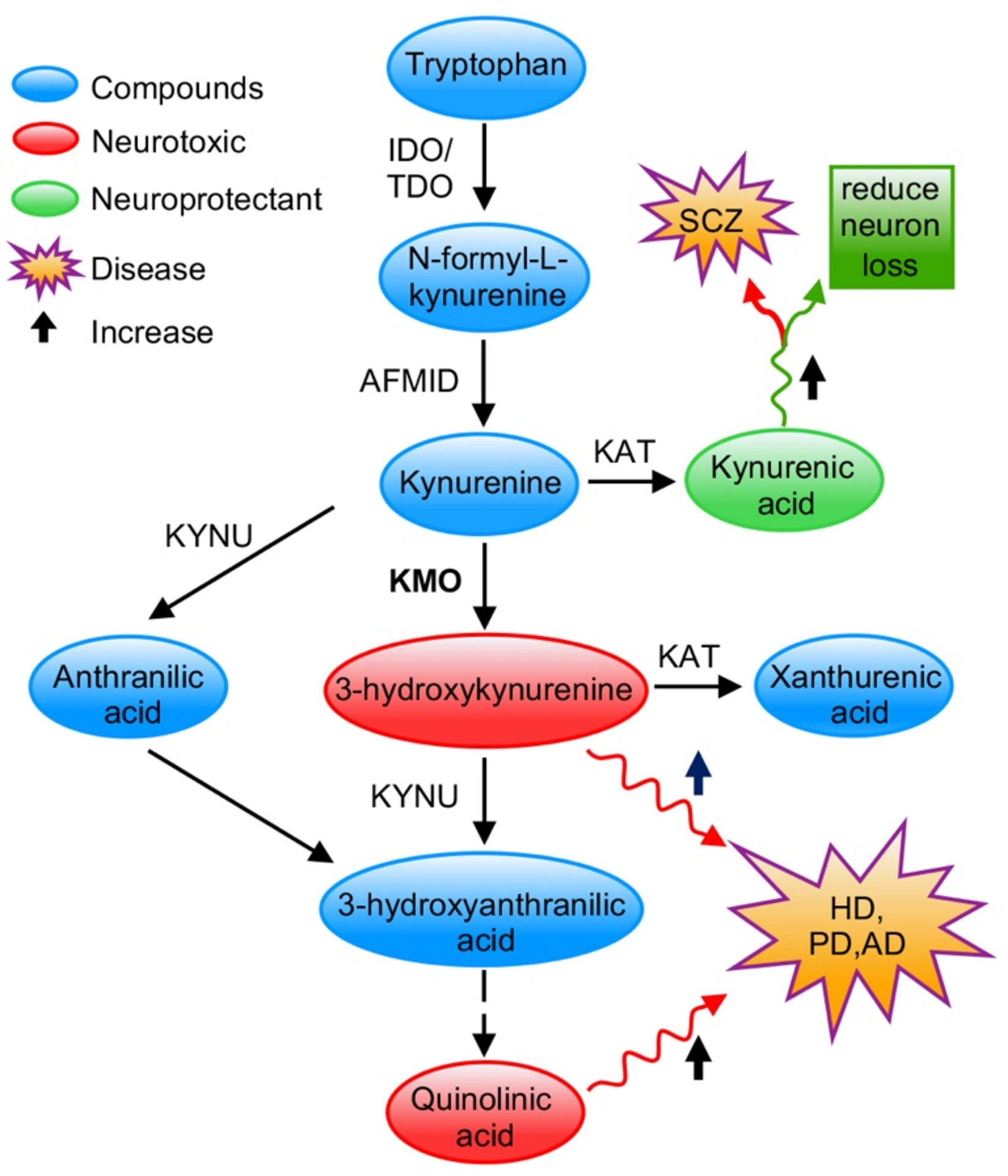
IDO, indole- 2,3-dioxygenase; TDO, tryptophan-2,3- dioxygenase; AFMID, Arylformamidase; KAT, kynurenine aminotransferase; KYNU, kynurenase; HD, Huntington’s disease; PD, Parkinson’s disease; AD, Alzheimer’s disease; SCZ, Schizophrenia. Neurotoxic metabolites 3-hydroxykynurenine and quinolinic acid are highlighted in red. Neuroprotective kynurenic acid is highlighted in green. Their relation with neuro disorders is labelled in the figure.
Overview of the kynurenine pathway
The KP (Figure 2) metabolises more than 95% of TRP [7]. This pathway has been implicated in numerous diseases, including Huntington’s disease, Alzheimer’s disease, Parkinson’s disease, schizophrenia, acute pancreatitis and cancer [8–11]. Hence, there is an increasing interest in identifying new therapeutic strategies by targeting the KP. The KP metabolises TRP into a number of neuroactive metabolites, such as kynurenic acid (KYNA), 3-HK, quinolinic acid (QUIN), xanthurenic and cinnabarinic acids (Figure 2). The role of the KP metabolites in several neurological disorders has been investigated intensively in recent years. Each of the metabolites appears to exert different effects: KYNA is a neuroprotectant [12, 13], 3-HK generates free radicals [14], QUIN is primarily an excitotoxin [15], and xanthurenic and cinnabarinic acids activate metabotropic glutamate receptors [16].
In the KP, the initial step is the conversion of TRP to N-formyl-L-kynurenine through indoleamine 2,3-dioxygenase 1 and 2 (IDO1 and IDO2) or tryptophan 2,3-dioxygenase (TDO) [17]. This results in the synthesis of l-KYN, which can be metabolised by three different enzymes and lies at the key branchpoint of the KP [18]. l-KYN can be metabolised to 3-HK by KMO, or it can form KYNA by a transamination reaction catalysed by kynurenine aminotransferase II (KATII), or alternatively it can be converted to anthranilic acid (AA) by kynureninase, which then feeds back into the 3-HK branch of the KP. Since KMO has the tightest binding affinity for l-KYN under normal conditions, the KMO branch has been considered to be the major metabolic route of the KP [18, 19]. KMO activity plays an essential role in maintaining a balance between the neurotoxic and neuroprotective potential of the pathway. Hence, there has been a focus on KMO inhibition as a potential strategy to treat several neurodegenerative and neuroinflammatory diseases.
KMO inhibition and neurodegenerative diseases
Huntington’s disease
Huntington’s disease is a fatal neurodegenerative disorder which is inherited in an autosomal dominant manner [20]. In the early stages of disease patients often exhibit changes in mood or cognitive ability. As the disorder develops the most characteristic physical symptoms (chorea) will appear. Huntington’s disease is caused by a CAG trinucleotide repeat expansion in the HTT gene, leading to the expansion of a polyglutamine tract in the huntingtin protein (HTT). These mutant forms of HTT can misfold and aggregate, eventually resulting in neuronal cell death [21]. Evidence suggests that the KP may contribute to the neurodegenerative effects observed in patients. Increased levels of the potentially neurotoxic metabolites 3-HK and QUIN have been detected in tissues taken from early stage patients [22–24] and several different studies indicate that there are significantly increased levels of these metabolites in mouse models of Huntington’s disease [25, 26], indicating dysregulation of the KP in Huntington’s disease. Notably, studies using yeast and Drosophila models of Huntington’s disease discovered that genetic inhibition of KMO normalised KP metabolic imbalances and ameliorated disease-related phenotypes [27–29]. Furthermore, pharmacological inhibition of KMO in a Drosophila model of Huntington’s using several KMO inhibitors protected against neuron loss [27, 28, 30]. The KMO inhibitor JM6 has also been shown to reduce loss of synapses and prevent microglial activation in a mouse model of Huntington’s disease [31], while the KMO inhibitor CHDI-340246 was found to ameliorate electrophysiological disturbances [32].
Alzheimer’s disease
Alzheimer’s disease is the most common cause of dementia [33]. It is a chronic neurodegenerative disease that usually develops slowly and causes gradual deterioration over time. The earliest symptom is regularly forgetting recent events and becoming increasingly repetitive. At later stages, patients develop problems with language, thinking and disorientation, as well as a number of behavioural issues. Post-mortem Alzheimer’s brains exhibit extracellular formation and accumulation of senile plaques, primarily comprised of misfolded amyloid-β peptides and also intracellular phosphorylation of tau proteins leading to the formation of neurofibrillary tangles [34]. Alzheimer’s patients tend to exhibit decreased TRP levels, which is possibly due to the increased accumulation of KP metabolites [35]. QUIN has been shown to co-localise with neurofibrillary tangles in cerebral neurons of Alzheimer’s patients, modulate tau phosphorylation [36] and promote tau aggregation[37]. Abnormal IDO1, 3-HK, and QUIN levels have been correlated with specific stages of Alzheimer’s disease [38–40]. It has been demonstrated that treatment with a KMO inhibitor in a mouse model of Alzheimer’s disease significantly prevented synaptic loss and improved spatial memory [31]. Down regulation of KMO gene expression ameliorates several disease-relevant phenotypes in Alzheimer’s model flies, including neurodegeneration, locomotor abnormalities and shortened lifespan [29].
Parkinson’s disease
Parkinson’s disease is a long-term progressive neurological disorder [41] which mainly affects the motor system in the central nervous system (CNS). Early in the disease, the common symptoms are shaking, rigidity and slowness of movements, with the potential onset of dementia in advanced stages. The cause of Parkinson’s disease is believed to involve both genetic and environmental factors. In addition to more than 20 genes being associated with familial forms of Parkinson’s, meta-analyses of several genome-wide association studies (GWAS) have revealed at least an additional 90 independent genetic risk factors [42, 43]. Notably, the gene encoding the KP enzyme aminocarboxymuconate-semialdehyde-decarboxylase (ACMSD) has been implicated in Parkinson’s via several genetic studies[44]. Similarly to Huntington’s and Alzheimer’s diseases, Parkinson’s disease is a protein misfolding disorder. Indeed, misfolding and aggregation of α-synuclein, which is the major component of Lewy bodies, is associated with Parkinson’s pathogenesis [45, 46]. In a manner similar to tau, QUIN has been found to seed aggregation of α-synuclein[47], which may have ramifications on Parkinson’s pathogenesis. Parkinson’s disease is also associated with oxidative stress, mitochondrial dysfunction, excitotoxicity and inflammation, which again show correlations alterations in KP metabolites [45, 48]. In the basal ganglia of Parkinson’s disease patients, an increased concentration of 3-HK has been found, while l-KYN and KYNA levels are slightly reduced [49, 50]. Increased levels of 3-HK and l-KYN have also been detected in the cerebral spinal fluid of patients with Parkinson’s disease [51]. The inhibition of KMO dramatically decreases the 3-HK/KYNA ratio in Drosophila, ameliorating disease phenotypes in Parkinson’s model flies [29]. Thus, KMO inhibition is a promising therapy for Parkinson’s disease.
Biochemistry and biophysical studies aid the development of brain penetrating inhibitors of KMO
Due to the lack of any structural information at the time, early KMO inhibitors were mainly based on analogues of l-KYN. An array of substrate analogue inhibitors showing therapeutic potential, has been identified, such as m-nitrobenzoylalanine (m-NBA), 3,4-dichlorobenzoylalanine and UPF-648 [52–55]. However, kinetics studies of PfKMO reveal that these substrate analogue inhibitors actually act as effector molecules. Binding of these compounds to KMO stimulates the flavin reduction by NADPH and causes the generation of cytotoxic hydrogen peroxide [56].
More specific and potent KMO inhibitors have been sought from novel and more complex organic compounds. Based on the l-KYN carboxyl group bioisosteres chemical library, a series of N-(4-phenylthiazol-2-yl) benzenesulfonamides KMO inhibitors has been developed [57]. Among these, oral administration of Ro 61–8048, increased the KYNA concentration in the brain by 7.5 fold at a dose of 100 μM/kg [57]. However, Ro 61–8048 shows poor brain permeability in mice, indicating that the protective effects generated by Ro 61–8048 are possibly due to the inhibition of peripheral KMO. The increased L-KYN levels in blood can then be transported across the blood-brain barrier (BBB) into the brain and converted into neuroprotective KYNA. A prodrug (which will be metabolised to the active drug in vivo) derived from Ro 61–8048, named JM6, has also been produced and found to be efficacious [31], although a later study has shown that JM6 is not a prodrug of Ro 61–8048 [58].
Elucidation of the KMO crystal structure and detailed kinetic studies have accelerated the development of KMO inhibitors [8, 56, 59]. A series of novel compounds has been identified that lead to inhibition of KMO activity. KMO inhibitors with IC50 in the nM region have been identified [8, 30, 60, 61]. Nonetheless, poor brain permeability has limited the application of KMO inhibitors within the CNS. KMO inhibitors with brain permeability would be predicted to be more efficacious for treating neurodegenerative diseases than peripheral treatment, as inhibition of KMO in the CNS leads to increased neuroprotective KYNA levels as well as decreased levels of neurotoxic metabolites [30, 62, 63]. To this end, the KMO structure and ligand interaction data were used as a basis for virtual screening and combined with a prodrug strategy to develop brain-permeable KMO inhibitors [30]. Prodrugs are considered as one of the most promising technologies for lead compound optimisation to cross the BBB. For KMO inhibitors, one of the main challenges to cross the BBB is the acidic centre, which mimics the binding of the carboxyl group of l-KYN. In the KMO–inhibitor 1 structure, the carboxylate group of the inhibitor sits close to residues R83/Y97/N368 in the KMO active site (Figure 3). Several interactions between ligand and protein have been identified (Figure 3). This network of polar interactions between protein and ligand makes the compound a potent KMO inhibitor. The complex structure also indicates that the carboxylate region of the inhibitor could be modified as part of a prodrug strategy. By esterification of the carboxyl group, the brain permeability of KMO inhibitor 1 was improved, and a maximal brain:blood ratio of 3.22 at 15 min was achieved[30]. As endothelial cells (ECs) in the CNS are joined together with highly resistant tight junctions, which prevents the passage of polar solutes [64], it is not surprising that esterification of KMO inhibitors could improve its brain permeability. Chemically modifying a drug to become more lipophilic is also a widely used approach to improve BBB penetration of other carboxylic acid containing drugs [65].
Figure 3. Structure basis of brain penetrating KMO inhibitors.
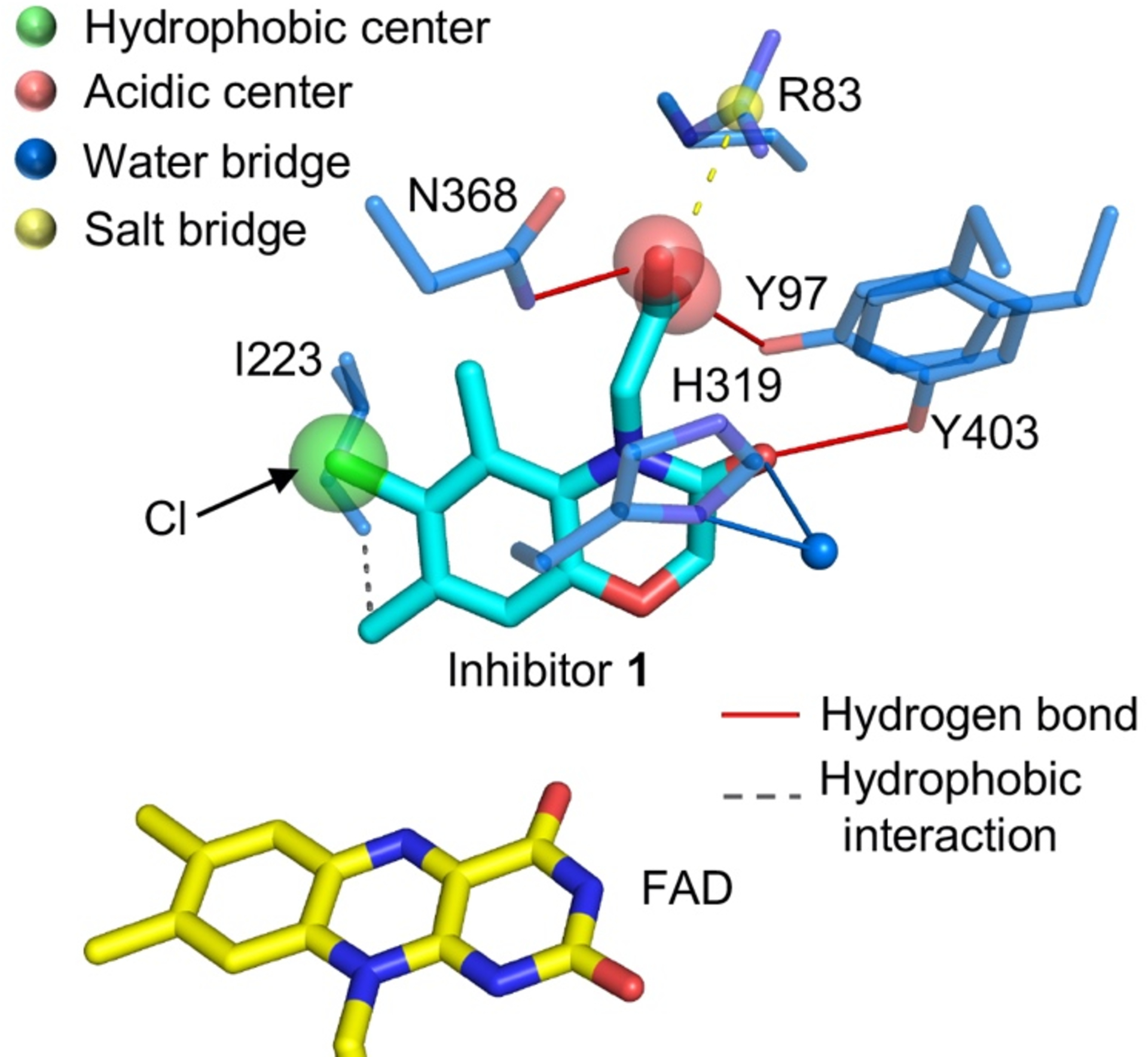
The interaction between compound 1 and Pseudomonas fluorescens KMO [30] (Structure file acquired from Protein Data Bank, 6FP1, https://www.rcsb.org). Key active site residues are shown in atom coloured sticks, carbons of KMO in blue, inhibitor 1 in cyan and FAD in yellow.
As the brain ECs constrain the movement of molecules between the blood and brain, overcoming the BBB remains a challenge for the treatment of CNS disorders. Indeed, the efficacy of traditional oral or systemic administrations of therapeutic drugs is limited by their poor brain permeability. Several strategies have been developed to overcome the issue, with chemical modifications to generate derivatives of a drug proving to be an effective way to improve brain penetration. The novel drug should possess the optimal physicochemical properties to permit passive diffusion, or on the other hand could be recognised as a “substrate” to utilise the endogenous transport systems of the BBB [64]. The previously reported brain penetrating KMO inhibitor (Compound 1) has both characteristics; it is relatively small and lipid-soluble, and the cheminformatics and in vitro cell assays indicate that it can cross the BBB via riboflavin transporters (see below), which are expressed in most of the tissues [30]. For the design of future KMO inhibitors, the polarity and similarity with existing endogenous metabolites could be used as a guide to achieve brain permeability.
The advantages of brain penetrating inhibitors of KMO
The BBB comprises microvascular ECs surrounded by pericytes and the end feet of astrocytes. The apical side of the ECs is in contact with the blood and forms the capillary lumen, whereas the basolateral side is in contact with glial cells of the brain. Several characteristics of the BBB limit the uptake of molecules into the CNS, therefore protecting the brain from toxins and pathogens. Unlike the normal vascular endothelium which allows diffusion of molecules between cells and into tissues, ECs of the BBB are held closely together by tight junctions. These prevent diffusion of most molecules between the ECs across the BBB and into the brain [66, 67]. In addition, low rates of transcytosis by ECs of the BBB reduce vesicle-mediated movement of molecules across the BBB [66]. Although these mechanisms protect the brain, these characteristics of the BBB also prevent the delivery of 98% of small-molecule drugs into the brain [68], limiting the delivery of drugs into the CNS to treat neurological conditions.
Targeting KMO inhibitors to the CNS where they can directly inhibit KMO is preferable to targeting peripheral KMO (Figure 4), as this reduces production of 3-HK and downstream metabolites in the brain and promotes increased levels of neuroprotective KYNA [30]. In general, for drugs to cross the BBB they must have a molecular mass <400 Da and have <8 hydrogen bonds to be lipid soluble [68]. However, efflux pumps expressed on ECs often remove drugs that have transferred from the blood across the plasma membrane of ECs in the BBB [69, 70]. A mechanism used to target drugs into the CNS is to design drugs that can be transported across the BBB by uptake transporters which allow essential nutrients to pass into the brain [71]. The recently developed brain penetrating KMO inhibitor (Compound 1) was shown to cross the BBB using specific uptake transporter proteins expressed in ECs of the BBB known as riboflavin transporters [30]. The SLC52 family of riboflavin transporters are expressed in the brain [72] and transport riboflavin across the plasma membrane [73]. A separate study also identified a riboflavin transporter expressed by microvascular brain ECs that transported riboflavin from the apical to basolateral side of the ECs [74]. The transport of the KMO inhibitor prodrug across the BBB by a riboflavin transporter was supported by the fact that the prodrug out-competed riboflavin uptake in a leukaemia cell line, and the KMO inhibitor prodrug was also shown to release functional KMO inhibitor in the brains of rats shortly after intravenous administration [30]. Crucially, peripheral administration of the prodrug in rats significantly reduced levels of 3-HK in the brain within 1 h, whereas intravenous administration of the KMO inhibitor Ro 61–8048 - which cannot cross the BBB - had no effect on 3-HK levels in the brain [30]. This study shows promising results for the development of a brain permeable KMO inhibitor that could potentially be used to target KMO in the CNS and directly reduce levels of neurotoxic 3-HK and other neurotoxic downstream metabolites of the KP. The development of a brain permeable KMO inhibitor would therefore allow treatment of neurological disorders for which there are currently no available drugs that can enter the CNS.
Figure 4. Advantages of brain penetrating KMO inhibitors.
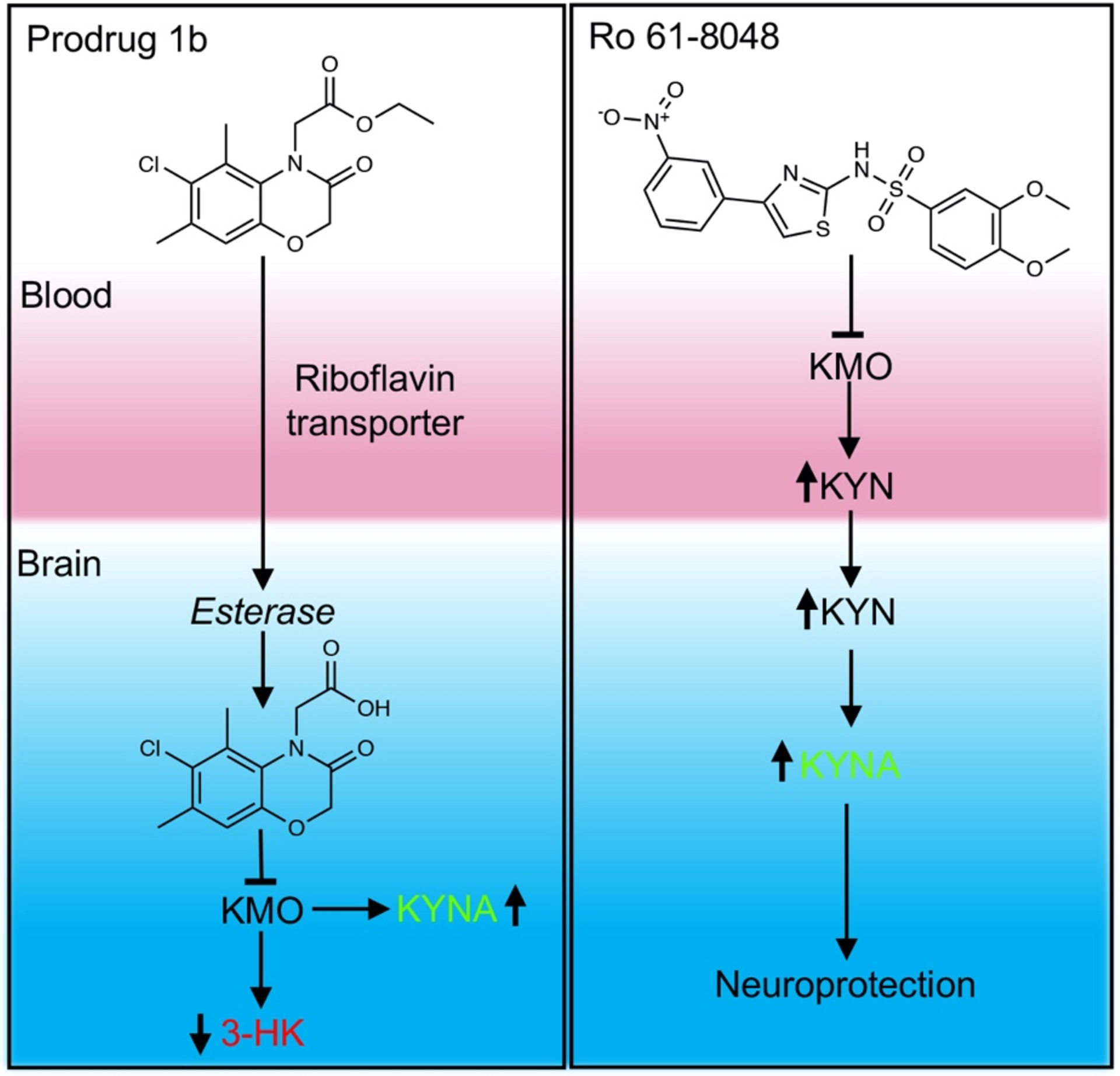
Brain penetrating KMO inhibitor transferred into brain and activated by esterase. The activated KMO inhibitor suppresses the activity of KMO in brain, leading to the decrease of neurotoxic 3-HK and increase of neuroprotective KYNA. While the non-brain penetrating inhibitors act in the periphery, raising levels of L-KYN in the blood. The L-KYN will be transported to brain and converted to neuroprotective KYNA.
Future perspectives
The KP regulates many important physiological processes and plays an adverse role in several pathological states, such as neurodegenerative and neuroinflammatory diseases, cancer and chronic infection [8–11]. The inhibition of KMO has been found to normalise pathological KP imbalances in several disease models, ameliorating disease phenotypes [31, 75–78]. The development of potent KMO inhibitors has become straightforward due to the availability of the protein structure and the ease of functional studies. The next challenge is how to improve its clinical behaviour to avoid side effects and have a better efficacy. A brain permeable inhibitor would be highly desirable as it can prevent the accumulation of neurotoxic metabolites in the brain, while increasing levels of neuroprotective KYNA. In addition, several KMO inhibitors are based on L-KYN analogues, and most of these work as effectors rather than a true inhibitor, which can stimulate the reduction of flavin and produce cytotoxic hydrogen peroxide [30, 56, 79]. Thus, in future KMO inhibitor development, these effector type inhibitors should be avoided and for CNS disorders brain permeability should also be enhanced. Regarding the brain permeable KMO inhibitor (Compound 1) [30], thorough testing in different disease models is now required to validate its therapeutic potential. Modification of the inhibitor to improve the efficacy should also be considered.
Acknowledgements
MC and FG are supported by funding from the National Institute of Mental Health (Silvio O. Conte Center for Translational Mental Health Research - MH-103222). SZ, DJH and NSS are supported by the UK Engineering and Physical Sciences Research Council (awards EP/S01778X/1 and EP/S030336/1).
Footnotes
Competing Interests statement
A patent (new compounds and uses) from the University of Leicester and University of Manchester by N.S.S. and F.G is pending, application number 1719327.7. The authors declare there are no other competing financial interests.
References
- [1].Alberati-Giani D, Ricciardi-Castagnoli P, Kohler C, Cesura AM, Regulation of the kynurenine pathway by IFN-gamma in murine cloned macrophages and microglial cells, Adv Exp Med Biol 398 (1996) 171–5. [DOI] [PubMed] [Google Scholar]
- [2].AlberatiGiani D, Cesura AM, Broger C, Warren WD, Rover S, Malherbe P, Cloning and functional expression of human kynurenine 3-monooxygenase, FEBS Lett 410 (1997) 407–412. [DOI] [PubMed] [Google Scholar]
- [3].Okamoto H, Yamamoto S, Nozaki M, Hayaishi O, On the submitochondrial localization of l-kynurenine-3-hydroxylase, Biochem Biophys Res Commun 26 (1967) 309–14. [DOI] [PubMed] [Google Scholar]
- [4].Hirai K, Kuroyanagi H, Tatebayashi Y, Hayashi Y, Hirabayashi-Takahashi K, Saito K, Haga S, Uemura T, Izumi S, Dual role of the carboxyl-terminal region of pig liver l-kynurenine 3-monooxygenase: mitochondrial-targeting signal and enzymatic activity*, J Biochem 148 (2010) 639–650. [DOI] [PubMed] [Google Scholar]
- [5].Uemura T, Hirai K, L-kynurenine 3-monooxygenase from mitochondrial outer membrane of pig liver: Purification, some properties, and monoclonal antibodies directed to the enzyme, J Biochem 123 (1998) 253–262. [DOI] [PubMed] [Google Scholar]
- [6].Amaral M, Outeiro TF, Scrutton NS, Giorgini F, The causative role and therapeutic potential of the kynurenine pathway in neurodegenerative disease, J Mol Med 91 (2013) 705–13. [DOI] [PubMed] [Google Scholar]
- [7].Stone TW, Darlington LG, Endogenous kynurenines as targets for drug discovery and development, Nat Rev Drug Discov 1 (2002) 609–620. [DOI] [PubMed] [Google Scholar]
- [8].Mole DJ, Webster SP, Uings I, Zheng X, Binnie M, Wilson K, Hutchinson JP, Mirguet O, Walker A, Beaufils B, Ancellin N, Trottet L, Beneton V, Mowat CG, Wilkinson M, Rowland P, Haslam C, McBride A, Homer NZ, Baily JE, Sharp MG, Garden OJ, Hughes J, Howie SE, Holmes DS, Liddle J, Iredale JP, Kynurenine-3-monooxygenase inhibition prevents multiple organ failure in rodent models of acute pancreatitis, Nat Med 22 (2016) 202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vecsei L, Szalardy L, Fulop F, Toldi J, Kynurenines in the CNS: recent advances and new questions, Nat Rev Drug Discov 12 (2013) 64–82. [DOI] [PubMed] [Google Scholar]
- [10].Stone TW, Darlington LG, The kynurenine pathway as a therapeutic target in cognitive and neurodegenerative disorders, Brit J Pharmacol 169 (2013) 1211–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ, Kynurenines in the mammalian brain: when physiology meets pathology, Nat Rev Neurosci 13 (2012) 465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fazio F, Lionetto L, Molinaro G, Bertrand HO, Acher F, Ngomba RT, Notartomaso S, Curini M, Rosati O, Scarselli P, Di Marco R, Battaglia G, Bruno V, Simmaco M, Pin JP, Nicoletti F, Goudet C, Cinnabarinic acid, an endogenous metabolite of the kynurenine pathway, activates type 4 metabotropic glutamate receptors, Mol Pharmacol 81 (2012) 643–56. [DOI] [PubMed] [Google Scholar]
- [13].Andine P, Lehmann A, Ellren K, Wennberg E, Kjellmer I, Nielsen T, Hagberg H, The excitatory amino acid antagonist kynurenic acid administered after hypoxic-ischemia in neonatal rats offers neuroprotection, Neurosci Lett 90 (1988) 208–12. [DOI] [PubMed] [Google Scholar]
- [14].Okuda S, Nishiyama N, Saito H, Katsuki H, Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine, Proc Natl Acad Sci USA 93 (1996) 12553–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schwarcz R, Whetsell WO, Mangano RM, Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain, Science 219 (1983) 316–318. [DOI] [PubMed] [Google Scholar]
- [16].Copeland CS, Neale SA, Salt TE, Actions of Xanthurenic acid, a putative endogenous Group II metabotropic glutamate receptor agonist, on sensory transmission in the thalamus, Neuropharmacology 66 (2013) 133–42. [DOI] [PubMed] [Google Scholar]
- [17].Ball HJ, Sanchez-Perez A, Weiser S, Austin CJ, Astelbauer F, Miu J, McQuillan JA, Stocker R, Jermiin LS, Hunt NH, Characterization of an indoleamine 2, 3-dioxygenase-like protein found in humans and mice, Gene 396 (2007) 203–213. [DOI] [PubMed] [Google Scholar]
- [18].Fujigaki H, Yamamoto Y, Saito K, L-Tryptophan-kynurenine pathway enzymes are therapeutic target for neuropsychiatric diseases: Focus on cell type differences, Neuropharmacology 112 (2017) 264–274. [DOI] [PubMed] [Google Scholar]
- [19].Lugo-Huitron R, Ugalde Muniz P, Pineda B, Pedraza-Chaverri J, Rios C, Perez-de la Cruz V, Quinolinic acid: an endogenous neurotoxin with multiple targets, Oxid Med Cell Longev 2013 (2013) 104024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, Wild EJ, Tabrizi SJ, Huntington disease, Nat Rev Dis Primers 1 (2015) 15005. [DOI] [PubMed] [Google Scholar]
- [21].Ross CA, Tabrizi SJ, Huntington’s disease: from molecular pathogenesis to clinical treatment, The Lancet Neurol 10 (2011) 83–98. [DOI] [PubMed] [Google Scholar]
- [22].Guidetti P, Luthi-Carter RE, Augood SJ, Schwarcz R, Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease, Neurobiol Dis 17 (2004) 455–61. [DOI] [PubMed] [Google Scholar]
- [23].Beal MF, Matson WR, Storey E, Milbury P, Ryan EA, Ogawa T, Bird ED, Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex, J Neurol Sci 108 (1992) 80–7. [DOI] [PubMed] [Google Scholar]
- [24].Pearson SJ, Reynolds GP, Increased brain concentrations of a neurotoxin, 3-hydroxykynurenine, in Huntington’s disease, Neurosci Lett 144 (1992) 199–201. [DOI] [PubMed] [Google Scholar]
- [25].Guidetti P, Bates GP, Graham RK, Hayden MR, Leavitt BR, MacDonald ME, Slow EJ, Wheeler VC, Woodman B, Schwarcz R, Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice, Neurobiol Dis 23 (2006) 190–7. [DOI] [PubMed] [Google Scholar]
- [26].Sathyasaikumar KV, Stachowski EK, Amori L, Guidetti P, Muchowski PJ, Schwarcz R, Dysfunctional kynurenine pathway metabolism in the R6/2 mouse model of Huntington’s disease, J Neurochem 113 (2010) 1416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ, A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease, Nat Genet 37 (2005) 526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Campesan S, Green EW, Breda C, Sathyasaikumar KV, Muchowski PJ, Schwarcz R, Kyriacou CP, Giorgini F, The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s Disease, Curr Biol 21 (2011) 961–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Breda C, Sathyasaikumar KV, Idrissi SS, Notarangelo FM, Estranero JG, Moore GGL, Green EW, Kyriacou CP, Schwarcz R, Giorgini F, Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites, P Natl Acad Sci USA 113 (2016) 5435–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang S, Sakuma M, Deora GS, Levy CW, Klausing A, Breda C, Read KD, Edlin CD, Ross BP, Wright Muelas M, Day PJ, O’Hagan S, Kell DB, Schwarcz R, Leys D, Heyes DJ, Giorgini F, Scrutton NS, A brain-permeable inhibitor of the neurodegenerative disease target kynurenine 3-monooxygenase prevents accumulation of neurotoxic metabolites, Commun Biol 2 (2019) 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zwilling D, Huang SY, Sathyasaikumar KV, Notarangelo FM, Guidetti P, Wu HQ, Lee J, Truong J, Andrews-Zwilling Y, Hsieh EW, Louie JY, Wu T, Scearce-Levie K, Patrick C, Adame A, Giorgini F, Moussaoui S, Laue G, Rassoulpour A, Flik G, Huang Y, Muchowski JM, Masliah E, Schwarcz R, Muchowski PJ, Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration, Cell 145 (2011) 863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Beaumont V, Mrzljak L, Dijkman U, Freije R, Heins M, Rassoulpour A, Tombaugh G, Gelman S, Bradaia A, Steidl E, Gleyzes M, Heikkinen T, Lehtimaki K, Puolivali J, Kontkanen O, Javier RM, Neagoe I, Deisemann H, Winkler D, Ebneth A, Khetarpal V, Toledo-Sherman L, Dominguez C, Park LC, Munoz-Sanjuan I, The novel KMO inhibitor CHDI-340246 leads to a restoration of electrophysiological alterations in mouse models of Huntington’s disease, Exp Neurol 282 (2016) 99–118. [DOI] [PubMed] [Google Scholar]
- [33].Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM, Alzheimer’s disease, Lancet 388 (2016) 505–17. [DOI] [PubMed] [Google Scholar]
- [34].Citron M, Alzheimer’s disease: strategies for disease modification, Nat Rev Drug Discov 9 (2010) 387–98. [DOI] [PubMed] [Google Scholar]
- [35].Widner B, Leblhuber F, Walli J, Tilz GP, Demel U, Fuchs D, Tryptophan degradation and immune activation in Alzheimer’s disease, J Neural Transm 107 (2000) 343–53. [DOI] [PubMed] [Google Scholar]
- [36].Rahman A, Ting K, Cullen KM, Braidy N, Brew BJ, Guillemin GJ, The excitotoxin quinolinic acid induces tau phosphorylation in human neurons, PloS one 4 (2009) e6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Esmaeili S, Ghobadi N, Akbari V, Moradi S, Shahlaie M, Ghobadi S, Jalalvand AR, Amani M, Khodarahmi R, Pyridine-2,3-dicarboxylate, quinolinic acid, induces 1N4R Tau amyloid aggregation in vitro: Another evidence for the detrimental effect of the inescapable endogenous neurotoxin, Chem Biol Interact 315 (2020) 108884. [DOI] [PubMed] [Google Scholar]
- [38].Bonda DJ, Mailankot M, Stone JG, Garrett MR, Staniszewska M, Castellani RJ, Siedlak SL, Zhu X, H.-g. Lee, G. Perry, Indoleamine 2, 3-dioxygenase and 3-hydroxykynurenine modifications are found in the neuropathology of Alzheimer’s disease, Redox Report 15 (2010) 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Guillemin GJ, Brew BJ, Noonan CE, Takikawa O, Cullen KM, Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer’s disease hippocampus, Neuropath Appl Neurobiol 31 (2005) 395–404. [DOI] [PubMed] [Google Scholar]
- [40].Guillemin GJ, Williams KR, Smith DG, Smythe GA, Croitoru-Lamoury J, Brew BJ, Quinolinic acid in the pathogenesis of Alzheimer’s disease, Adv Exp Med Biol 527 (2003) 167–76. [DOI] [PubMed] [Google Scholar]
- [41].Kalia LV, Lang AE, Parkinson’s disease, Lancet 386 (2015) 896–912. [DOI] [PubMed] [Google Scholar]
- [42].Blauwendraat C, Nalls MA, Singleton AB, The genetic architecture of Parkinson’s disease, Lancet Neurol 19 (2020) 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, Bras J, Young E, von Coelln R, Simon-Sanchez J, Schulte C, Sharma M, Krohn L, Pihlstrom L, Siitonen A, Iwaki H, Leonard H, Faghri F, Gibbs JR, Hernandez DG, Scholz SW, Botia JA, Martinez M, Corvol JC, Lesage S, Jankovic J, Shulman LM, Sutherland M, Tienari P, Majamaa K, Toft M, Andreassen OA, Bangale T, Brice A, Yang J, Gan-Or Z, Gasser T, Heutink P, Shulman JM, Wood NW, Hinds DA, Hardy JA, Morris HR, Gratten J, Visscher PM, Graham RR, Singleton AB, andMe Research T, System C Genomics of Parkinson’s Disease, C. International Parkinson’s Disease Genomics, Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies, Lancet Neurol 18 (2019) 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Thirtamara-Rajamani K, Li P, Escobar Galvis ML, Labrie V, Brundin P, Brundin L, Is the Enzyme ACMSD a Novel Therapeutic Target in Parkinson’s Disease?, J Parkinsons Dis 7 (2017) 577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lim CK, Fernandez-Gomez FJ, Braidy N, Estrada C, Costa C, Costa S, Bessede A, Fernandez-Villalba E, Zinger A, Herrero MT, Guillemin GJ, Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease, Prog Neurobiol 155 (2017) 76–95. [DOI] [PubMed] [Google Scholar]
- [46].Lee JM, Tan V, Lovejoy D, Braidy N, Rowe DB, Brew BJ, Guillemin GJ, Involvement of quinolinic acid in the neuropathogenesis of amyotrophic lateral sclerosis, Neuropharmacology 112 (2017) 346–364. [DOI] [PubMed] [Google Scholar]
- [47].Tavassoly O, Sade D, Bera S, Shaham-Niv S, Vocadlo DJ, Gazit E, Quinolinic Acid Amyloid-like Fibrillar Assemblies Seed alpha-Synuclein Aggregation, J Mol Biol 430 (2018) 3847–3862. [DOI] [PubMed] [Google Scholar]
- [48].Smith JR, Jamie JF, Guillemin GJ, Kynurenine-3-monooxygenase: a review of structure, mechanism, and inhibitors, Drug Discov Today 21.2 (2015) 315–324.. [DOI] [PubMed] [Google Scholar]
- [49].Knyihar-Csillik E, Chadaide Z, Mihaly A, Krisztin-Peva B, Fenyo R, Vecsei L, Effect of 6-hydroxydopamine treatment on kynurenine aminotransferase-I (KAT-I) immunoreactivity of neurons and glial cells in the rat substantia nigra, Acta Neuropathol 112 (2006) 127–37. [DOI] [PubMed] [Google Scholar]
- [50].Ogawa T, Matson WR, Beal MF, Myers RH, Bird ED, Milbury P, Saso S, Kynurenine pathway abnormalities in Parkinson’s disease, Neurology 42 (1992) 1702–6. [DOI] [PubMed] [Google Scholar]
- [51].Iwaoka K, Otsuka C, Maeda T, Yamahara K, Kato K, Takahashi K, Takahashi K, Terayama Y, Impaired metabolism of kynurenine and its metabolites in CSF of parkinson’s disease, Neurosci Lett 714 (2020) 134576. [DOI] [PubMed] [Google Scholar]
- [52].Pellicciari R, Natalini B, Costantino G, Mahmoud MR, Mattoli L, Sadeghpour BM, Moroni F, Chiarugi A, Carpenedo R, Modulation of the Kynurenine Pathway in Search for New Neuroprotective Agents - Synthesis and Preliminary Evaluation of (M-Nitrobenzoyl)Alanine, a Potent Inhibitor of Kynurenine-3-Hydroxylase, J Med Chem 37 (1994) 647–655. [DOI] [PubMed] [Google Scholar]
- [53].Carpenedo R, Chiarugi A, Russi P, Lombardi G, Carla V, Pellicciari R, Moroni F, Mattoli L, Inhibitors of Kynurenine Hydroxylase and Kynureninase Increase Cerebral Formation of Kynurenate and Have Sedative and Anticonvulsant Activities, Neuroscience 61 (1994) 237–244. [DOI] [PubMed] [Google Scholar]
- [54].Speciale C, Wu HQ, Cini M, Marconi M, Varasi M, Schwarcz R, (R,S)-3,4-dichlorobenzoylalanine (FCE 28833A) causes a large and persistent increase in brain kynurenic acid levels in rats, Eur J Pharmacol 315 (1996) 263–7. [DOI] [PubMed] [Google Scholar]
- [55].Pellicciari R, Amori L, Costantino G, Giordani A, Macchiarulo A, Mattoli L, Pevarello P, Speciale C, Varasi M, Modulation of the kynurine pathway of tryptophan metabolism in search for neuroprotective agents. Focus on kynurenine-3-hydroxylase, Adv Exp Med Biol 527 (2003) 621–8. [DOI] [PubMed] [Google Scholar]
- [56].Crozier-Reabe KR, Phillips RS, Moran GR, Kynurenine 3-Monooxygenase from Pseudomonas fluorescens: Substrate-like Inhibitors both Stimulate Flavin Reduction and Stabilize the Flavin-Peroxo Intermediate yet Result in the Production of Hydrogen Peroxide, Biochemistry 47 (2008) 12420–12433. [DOI] [PubMed] [Google Scholar]
- [57].Rover S, Cesura AM, Huguenin P, Kettler R, Szente A, Synthesis and biochemical evaluation of N-(4-phenylthiazol-2-yl)benzenesulfonamides as high-affinity inhibitors of kynurenine 3-hydroxylase, J Med Chem 40 (1997) 4378–4385. [DOI] [PubMed] [Google Scholar]
- [58].Beconi MG, Yates D, Lyons K, Matthews K, Clifton S, Mead T, Prime M, Winkler D, O’Connell C, Walter D, Toledo-Sherman L, Munoz-Sanjuan I, Dominguez C, Metabolism and pharmacokinetics of JM6 in mice: JM6 is not a prodrug for Ro-61–8048, Drug Metab Dispos 40 (2012) 2297–306. [DOI] [PubMed] [Google Scholar]
- [59].Amaral M, Levy C, Heyes DJ, Lafite P, Outeiro TF, Giorgini F, Leys D, Scrutton NS, Structural basis of kynurenine 3-monooxygenase inhibition, Nature 496 (2013) 382–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Toledo-Sherman LM, Prime ME, Mrzljak L, Beconi MG, Beresford A, Brookfield FA, Brown CJ, Cardaun I, Courtney SM, Dijkman U, Hamelin-Flegg E, Johnson PD, Kempf V, Lyons K, Matthews K, Mitchell WL, O’Connell C, Pena P, Powell K, Rassoulpour A, Reed L, Reindl W, Selvaratnam S, Friley WW, Weddell DA, Went NE, Wheelan P, Winkler C, Winkler D, Wityak J, Yarnold CJ, Yates D, Munoz-Sanjuan I, Dominguez C, Development of a series of aryl pyrimidine kynurenine monooxygenase inhibitors as potential therapeutic agents for the treatment of Huntington’s disease, J Med Chem 58 (2015) 1159–83. [DOI] [PubMed] [Google Scholar]
- [61].Hutchinson JP, Rowland P, Taylor MRD, Christodoulou EM, Haslam C, Hobbs CI, Holmes DS, Homes P, Liddle J, Mole DJ, Uings I, Walker AL, Webster SP, Mowat CG, Chung CW, Structural and mechanistic basis of differentiated inhibitors of the acute pancreatitis target kynurenine-3-monooxygenase, Nat Commun 8 (2017) 15827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Amori L, Guidetti P, Pellicciari R, Kajii Y, Schwarcz R, On the relationship between the two branches of the kynurenine pathway in the rat brain in vivo, J Neurochem 109 (2009) 316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Giorgini F, Huang SY, Sathyasaikumar KV, Notarangelo FM, Thomas MA, Tararina M, Wu HQ, Schwarcz R, Muchowski PJ, Targeted deletion of kynurenine 3-monooxygenase in mice: a new tool for studying kynurenine pathway metabolism in periphery and brain, J Biol Chem 288 (2013) 36554–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pavan B, Dalpiaz A, Ciliberti N, Biondi C, Manfredini S, Vertuani S, Progress in drug delivery to the central nervous system by the prodrug approach, Molecules 13 (2008) 1035–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zawilska JB, Wojcieszak J, Olejniczak AB, Prodrugs: a challenge for the drug development, Pharmacol Rep 65 (2013) 1–14. [DOI] [PubMed] [Google Scholar]
- [66].Coomber BL, Stewart PA, Morphometric analysis of CNS microvascular endothelium, Microvasc Res 30 (1985) 99–115. [DOI] [PubMed] [Google Scholar]
- [67].Brightman MW, Reese TS, Junctions between intimately apposed cell membranes in the vertebrate brain, J Cell Biol 40 (1969) 648–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pardridge WM, The blood-brain barrier: bottleneck in brain drug development, NeuroRx 2 (2005) 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Schinkel AH, Wagenaar E, Mol CA, van Deemter L, P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs, J Clin Invest 97 (1996) 2517–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CA, van der Valk MA, Robanus-Maandag EC, te Riele HP, et al. , Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs, Cell 77 (1994) 491–502. [DOI] [PubMed] [Google Scholar]
- [71].Hu C, Tao L, Cao X, Chen L, The solute carrier transporters and the brain: Physiological and pharmacological implications, Asian J Pharm Sci 15 (2020) 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Yao Y, Yonezawa A, Yoshimatsu H, Masuda S, Katsura T, Inui K, Identification and comparative functional characterization of a new human riboflavin transporter hRFT3 expressed in the brain, J Nutr 140 (2010) 1220–6. [DOI] [PubMed] [Google Scholar]
- [73].Jin C, Yao Y, Yonezawa A, Imai S, Yoshimatsu H, Otani Y, Omura T, Nakagawa S, Nakagawa T, Matsubara K, Riboflavin Transporters RFVT/SLC52A Mediate Translocation of Riboflavin, Rather than FMN or FAD, across Plasma Membrane, Biol Pharm Bull 40 (2017) 1990–1995. [DOI] [PubMed] [Google Scholar]
- [74].Patel M, Vadlapatla RK, Pal D, Mitra AK, Molecular and functional characterization of riboflavin specific transport system in rat brain capillary endothelial cells, Brain Res 1468 (2012) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zakhary G, Sherchan P, Li Q, Tang J, Zhang JH, Modification of kynurenine pathway via inhibition of kynurenine hydroxylase attenuates surgical brain injury complications in a male rat model, J Neurosci Res 98 (2020) 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ray A, Song Y, Du T, Tai YT, Chauhan D, Anderson KC, Targeting tryptophan catabolic kynurenine pathway enhances antitumor immunity and cytotoxicity in multiple myeloma, Leukemia 34 (2020) 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sundaram G, Lim CK, Brew BJ, Guillemin GJ, Kynurenine pathway modulation reverses the experimental autoimmune encephalomyelitis mouse disease progression, J Neuroinflammation 17 (2020) 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Swainson LA, Ahn H, Pajanirassa P, Khetarpal V, Deleage C, Estes JD, Hunt PW, Munoz-Sanjuan I, McCune JM, Kynurenine 3-Monooxygenase Inhibition during Acute Simian Immunodeficiency Virus Infection Lowers PD-1 Expression and Improves Post-Combination Antiretroviral Therapy CD4+ T Cell Counts and Body Weight, J Immunol 203 (2019) 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kim HT, Na BK, Chung J, Kim S, Kwon SK, Cha H, Son J, Cho JM, Hwang KY, Structural Basis for Inhibitor-Induced Hydrogen Peroxide Production by Kynurenine 3-Monooxygenase, Cell Chem Biol 25 (2018) 426–438. [DOI] [PubMed] [Google Scholar]