

Abstract
Epigenetic mechanisms involve the placing (‘writing’) or removal (‘erasing’) of histone modifications that allows euchromatin to transition to the open, activated chromatin state necessary for transcription. A third, less studied epigenetic pathway involves the ‘reading’ of these specific histone marks once placed. The bromodomain and extra-terminal containing protein family (BETs), which includes BRD2, BRD3, BRD4 and the testis-restricted BRDT, are epigenetic reader proteins that bind to specific acetylated lysine residues on histone tails where they facilitate the assembly of transcription complexes including transcription factors and transcriptional machinery like RNA Polymerase II. As reviewed here, considerable recent data establishes BETs as novel determinants of induced transcriptional programs in vascular cells, like endothelial cells and vascular smooth muscle cells, cardiac myocytes and inflammatory cells, like monocyte/macrophages, cellular settings where these epigenetic reader proteins couple proximal stimuli to chromatin, acting at super-enhancer regulatory regions to direct gene expression. BET inhibition, including the use of specific chemical BET inhibitors like JQ1, has many reported effects in vivo in the cardiovascular setting, like decreasing atherosclerosis, angiogenesis, intimal hyperplasia, pulmonary arterial hypertension and cardiac hypertrophy. At the same time, data in endothelial cells, adipocytes and elsewhere suggests BETs also help regulate gene expression under basal conditions. Studies in the cardiovascular setting have highlighted BET action as a means of controlling gene expression in differentiation, cell identity and cell state transitions, whether physiologic or pathologic, adaptive or maladaptive. While distinct BET inhibitors are being pursued as therapies in oncology, a large prospective clinical cardiovascular outcome study investigating the BET inhibitor RVX-208 (now called apabetalone) has already been completed. Independent of this specific agent and this one trial or the numerous unanswered questions that remain, BETs have emerged as novel epigenetic players involved in the execution of coordinated transcriptional programs in cardiovascular health and disease.
Keywords: Epigenetic Reader, Epigenetics, BETs, Transcriptional programs, Cardiovascular diseases, atherosclerosis, transcriptional regulation, inflammation
Subject Terms: Atherosclerosis, epigenetics, inflammation
BETs and the Reading of the Epigenetic Code
The term “genetic code” has reached common parlance - a broad reference to universal rules that explain how DNA sequence defines protein structure, function and hence, phenotype.1 The genetic code’s power is manifest with disease-causing single DNA nucleotide variants and increasingly revealed, as in large population genome-wide studies linking DNA variants to distinct clinical conditions, including cardiovascular disease.2, 3 Advances in understanding DNA since the double helix structure 66 years ago also enabled the intense, ongoing work into how non-coding DNA sequences, like promoters and enhancers control gene expression.4
The genetic code offers context for the more recent recognition of a distinct, elaborate, highly-regulated system operating independent (‘above’) DNA sequence that governs transcription: the epigenetic code. The massive DNA expanses present in every nucleus creates a space problem, one that is solved by DNA compaction - tightly winding chromosomal material around spool-like histones.5 However, transcription requires histone-wound DNA, or euchromatin, to open up and transition to chromatin, which allows transcription factors (TFs) and transcriptional machinery, like RNA Poylmerase II (RNA RNAPII), to access relevant DNA regulatory regions (Figure 1). Chemical modifications of specific amino acid residues on histone tails can help enable or repress gene expression. In simple but helpful terms, epigenetics refers to enzymatic processes that ‘write’ (place), as achieved through methylation of histidine or acetylation of lysine, or ‘erase’ (remove) as with de-methylation or de-acetylation of these same histone marks. Similarly, epigenetic modifications in the DNA methylation at the 5’ carbon on the pyrimidine ring of cytosine nucleotides can also be observed as an important mechanism that controls gene expression without affecting DNA sequence. The prospects entailed by understanding and controlling transcription through studies on histone and DNA modifications has received extensive attention.6 However, a third critical process exists in epigenetic communication via chromatin remodeling – the reading of histone marks once placed.7 While most attention has focused on enzymatic placement or removal of histone marks, the essential role for epigenetic reader proteins in executing gene expression has often been overlooked, including in the vasculature.8 By binding to modified histone tails, epigenetic reader proteins (Table 1) allow for the engines of transcription, like RNA Polymerase II, and specific transcription drivers, like master proximal TFs - which often control the regulatory activity of other TFs and related genes - to be coupled in the regulatory regions of defined gene cassettes. The bromodomain extra-terminal (BET)-containing family of epigenetic reader proteins, including BRD2, BRD3, BRD4 and the testis-restricted BRDT (referred to as BETs throughout), provides a robust example of how epigenetic reader proteins can orchestrate transcriptional programs, provide new insight into physiologic and pathologic cellular states and offer potentially novel therapeutic strategies. This compendium review considers the recent and rapidly expanding evidence for BETs in vascular biology and atherosclerosis, after beginning with a conceptual framework for epigenetic reader proteins as a prelude to considering basic BET biology.
Figure 1. The BET Bromodomain Family of Epigenetic Reader Proteins In Transcriptional Regulation.
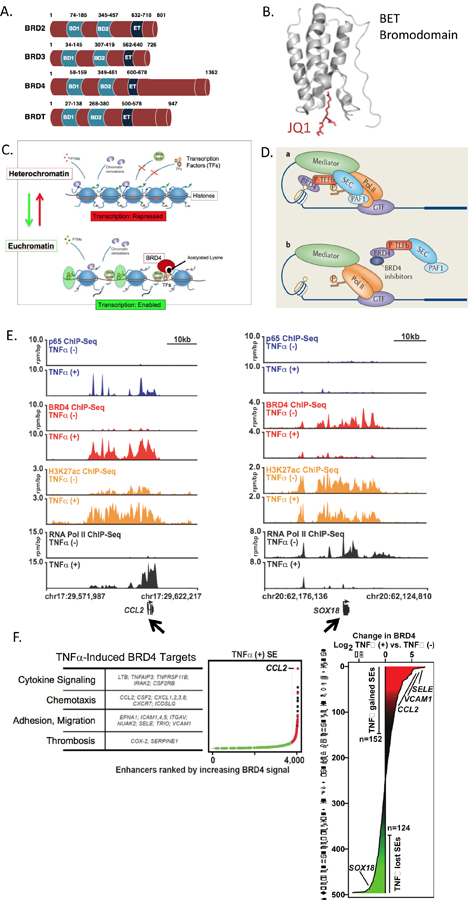
A. The Bromodomain and Extra-Terminal (BET) family members BRD2, BRD3, BRD4 and testes-restricted BRDT, all possess two bromodomains BD1 and BD2, as well as an extra-terminal (ET) domain. BRD4’s unique c-terminal domain (CTD) may facilitate its interaction with P-TEFb and RNA Polymerase II (RNAPII). B. BET bromodomain module predicted ribbon structure includes a hydrophobic pocket that binds to acetylated lysine residues on the 5’ end of histone tails. The pan-BETi JQ1 fits tightly and irreversibly in this pocket, disrupting BET’s association with chromatin and regulation of transcription. C. BET action is coupled to epigenetic mechanisms involved in the transition from heterochromatin to euchromatin. Heterochromatin consists of DNA tightly wound around histone spools, which results in DNA compaction but precludes transcription factor (TF) access to regulatory regions of DNA, repressing gene expression. For transcription to proceed, chromatin transitions to the euchromatin state, which opens DNA regulatory regions, allowing access for transcriptional machinery. BETs, in this case BRD4, binds to acetylated lysines on histone tails and facilitates assembly of TFs and RNAPII. D. Schematic representation of BRD4 binding to a histone tail and allowing for assembly of key players of the transcriptional apparatus, including RNAPII and P-TEFb. Mediator involves 30 protein subunits involved in the “pre-initiation complex”. In the presence of a BETi, BRD4 histone tail binding is disrupted, preventing BRD4 from its function as a transcriptional scaffold and mediator. E. Browser views of ChIP-Seq data in human umbilical vein ECs at baseline and after TNFα stimulation serves as an example of BRD4 regulation of vascular cell gene expression. E Left. Immunoprecipitation of individual proteins amassing at the 5’ end of the chemokine CCL2 gene body (arrow, chr17:29,622,217). Data is shown in pairs, without (upper) or with (lower) TNFα stimulation. At the top, in blue, absent TNFα stimulation, minimal immunoprecipitated p65 is found proximal to the CCL2 start site. As expected, TNFα stimulation recruits p65 to the CCL2 promoter region. In red, BRD4 protein accumulates upstream of the CCL2 start site after TNFα stimulation but was not present under basal conditions. In orange, H3K27Ac indicates an activated promoter after TNFα stimulation. In black, RNAPII is present only after TNFα stimulation and is evident ‘walking down’ the CCL2 gene body, consistent with transcription. JQ1 treatment of ECs blocks BRD4 and blocks these responses (not shown). E. Right. The ChIP-Seq browser view data for Sox18 (arrow) is shown. In contrast to CCL2 and other TNFα-induced, BRD4-regulated genes, Sox18 shows BRD4 and RNAPII association under basal conditions that is “lost” upon TNFα stimulation. F. Left: Multiple TNFα-stimulated pro-atherosclerotic target genes involve BRD4 action at super-enhancer regions. Right: BRD4 accumulation is shown where TNFα either increased (red, “gained”) or decreased (green, “lost”) BRD4 at respective super-enhancers. Each horizontal line corresponds to a distinct gene, including e-selectin (SELE), VCAM1, CCL2 and SOX18. For additional details, see text and associated references.
Table 1. Key epigenetic reader proteins grouped by respective chromatin modification, modification sites and protein binding domain.
| Modification | Modification Site(s) | Binding domain(s) | Reader Protein |
|---|---|---|---|
| Acetylated histone tails | H3: K9, K14, K18, K27, K56, K122 H4: K5, K8, K13, K16 |
BDs | BRD2, BRD3, BRD4, BRDT |
| Methylated CpG | CpG | MBD | MeCP2, MBDs1–6 |
| MBD/ SET | SETDB1, SETDB2 | ||
| MBD/DDT/ PHD/BD |
BAZ2A/B | ||
| Methylated Histones | H3K4 | Chromo | CHD1 |
| PHD | BPTF, TAF3, ING4, CFP1, SPIN1, PHF2/23, PYGO2 | ||
| Tudor | Sgf29 | ||
| H3K9 | Chromo | HP1α/β/γ | |
| Tudor | UHRF1 | ||
| H3K27 | Chromo | CBX7 | |
| WD40 | EED | ||
| BAH | BAHD1 | ||
| PWWP | NSD2 | ||
| H3K36 | PWWP | DNMT3A, LEDGF, ZMYND11 | |
| Chromo | MRG15 | ||
| - | NBS1, Ku70 | ||
| H3K79 | Tudor | TP53BP1 | |
| H4K20 | Tudor | TP53BP1 | |
| MBT | L3MBTL1 | ||
| BAH | ORC1 | ||
| WD repeat | ORCA | ||
| PWWP | PDP1 |
Abbreviations: Histone 3 (H3), Histone 4 (H4), Lysine (K), Methyl-CpG Binding Domain (MBD), Methyl-CpG binding protein 2 (MeCP2), Methyl-CpG Binding Domain Protein 1–6 (MBD1–6), Su(var)3–9, Enhancer-of-zeste and Trithorax (SET), SET Domain Bifurcated Histone Lysine Methyltransferase 1–2 (SETDB1–2), DDT domain, Plant homeodomain (PHD), Bromodomain Adjacent To Zinc Finger Domain 2A/B (BAZ2A/B), Chromodomain Helicase DNA Binding Protein 1 (CHD1), Bromodomain PHD Finger Transcription Factor (BPTF), TATA-Box Binding Protein Associated Factor 3 (TAF3), Inhibitor Of Growth 4 (ING4), CxxC zinc finger protein 1 (CFP1), Spindlin 1 (SPIN1), PHD Finger Protein 2/23 (PHF2/23), Pygopus 2 (PYGO2), SAGA Complex Associated Factor 29 (Sgf29), Heterochromatin Protein 1 α/β/γ (HP1α/β/γ), Ubiquitin Like With PHD And Ring Finger Domains 1 (UHRF1), Chromobox 7 (CBX7), Tryptophan-Aspartic acid 40 (WD40), Embryonic Ectoderm Development (EED), Bromo-Adjacent Homology (BAH), Bromo Adjacent Homology Domain-containing Protein 1 (BAHD1), PWWP domain, Nuclear Receptor Binding SET Domain Protein 2 (NSD2), DNA Methyltransferase 3 Alpha (DNMT3A), Lens Epithelium-Derived Growth Factor (LEDGF), Zinc Finger MYND-Type Containing 11 (ZMYND11), MORF-Related Gene On Chromosome 15 (MRG15), Nijmegen Breakage Syndrome (NBS1), Ku Autoantigen, 70 (Ku70), Tumor Protein P53 Binding Protein 1 (TP53BP1), Malignant Brain Tumor (MBT) domain, Lethal(3) Malignant Brain Tumor-like Protein 1 (L3MBTL1), Origin Recognition Complex Subunit 1 (ORC1), Origin Recognition Complex Associated (ORCA), Pyruvate Dehyrogenase Phosphatase (PDP1).
Proximal master TFs induce or repress expression of multiple genes, helping explain coordinated transcriptional programs, as seen with nuclear factor kappa B (NF-kB) activation directing complex inflammatory programs or peroxisome proliferator-activated receptors (PPARs) directing energy balance. Epigenetic reader proteins provide another distinct way of controlling integrated transcriptional programs, coupling proximal signals to TFs and chromatin marks. The placement of histone marks, and their subsequent reading, can account for biologic memory - cellular recording of stimuli and exposures as a way of facilitating key transcriptional responses, as might be expected in host defenses9, energy storage10 or coagulation11, all being examples of programs essential to survival. Biologic memory may also direct maladaptive responses, as in chronic disease states or understanding the long-term impact of prior cigarette use12, genetic hypercholesterolemia13 or gestational diabetes.14 Epigenetic reader proteins can also help explain gene expression as a means of defining cell state transitions as required in response to dynamic environments involving multiple stimuli. One fundamental cell state change is stem cell differentiation, as master TFs stimulate pluripotent stem cells to acquire a specific biologic identity and function, as typically defined by marker expression. 15 Mature cells must also mount functional responses to stimuli, whether mechanical, like hemodynamic pressure, or humoral, as with inflammatory cytokines. Data already implicates epigenetic reader proteins in both stem cell differentiation and mature cell function. Although the focus here is on BET regulation of transcription, which has received more attention in the CV setting in this nascent area, it is worth noting that other epigenetic reader proteins exist (Table 1), with no doubt data to come regarding their part in vascular responses. Understanding basic aspects of BET biology will facilitate consideration of the evidence for how this specific epigenetic reader protein family directs gene expression in multiple CV settings and may emerge as a therapeutic target, as has already been explored in clinical cardiovascular trials.
BET Biology: Directing Transcriptional Choreography
A bromodomain is a conserved ~110 amino acid module found in some 60 different proteins in the human genome, including the BET subfamily. The bromodomain structure, characterized by four alpha helices (A, B, C and Z) connected by two loops between their helices BC and ZA, is the only protein domain capable of binding to acetylated lysine residues.16 The bromodomain module forms a deep hydrophobic pocket that recognizes and binds to ε-N-acetylated lysines on the 5’ end of histone tails. While most bromodomain-containing proteins have only one such domain, all four BET family members have an extra-terminal (ET) domain, resulting in two tandem N-terminal bromodomains (BD, specifically BD1 and BD2). Differential BD affinity among BET inhibitors may determine their functional effects. By binding to acetylated lysine marks, BETs serve as a scaffold for assembling TFs and essential transcriptional machinery, including RNA Polymerase II (RNAPII), which occurs at active promoter sites or cis regulatory elements that can facilitate transcription from a distance. Similar BET actions also produce non-coding RNAs as another means of altering functional responses. High-resolution protein crystallography reveals that the affinity for acetylated lysines among BET family BDs is modest for monoacetylated lysine but increases significantly when multiple acetylated sites exist within a span of one to five amino acids 17, 18. The accumulation of BRD4 at enhancer elements can account for the dominant transcriptional activity in specific cell states.19, 20 BRD2 and BRD3 have also been identified in genomic enhancer regions in cell lines. 21–23 The terms super-enhancer (SE) or stretch enhancer has been used to refer to this concentrated transcriptional action at discrete cis-regulatory domains that involves BETs, as potential pioneer signals, acetylated histones, DNA-bound TFs and co-activators like Mediator 1.19, 20 BET localization is also linked to formation of enhancer transcripts, which can use bromodomains cooperatively as docking sites to increase BRD4 affinity for acetylated lysine residues while augmenting other cofactor recruitment and increasing transcriptional activity.24 BET proteins can also interact with acetylated TFs, such as ETS Related Gene (ERG) and Nuclear Factor kappa-light-chain-enhancer of activated B (NF-κB), through BD-dependent and independent mechanisms, a potential contributor to context-specific effects of BET inhibition. Indeed, suppressing BETs action represses downstream targets genes while DNA-bound TFs remain unaffected.25 Although beyond the focus of this review, BETs may also exert non-transcriptional effects, for example in DNA stress responses. 26, 27
BET action in controlling gene expression depends on their function as both scaffolds for localizing transcriptional machinery and as players that actively help initiate transcription and RNA production. Both BRD4 and BRDT include a c-terminal domain (CTD), which facilitates transcription activation and elongation by recruiting and interacting with the positive-transcriptional elongation factor b (P-TEFb), a complex composed of the cyclin-dependent kinase 9 (CDK9) and a regulatory subunit Cyclin T1 or T2. 28, 29. In this regard, BETs, and their role in cardiovascular gene expression, can be understood as integral to complex, fundamental transcriptional mechanisms that continue to be uncovered. After RNAPII promoter recruitment, the multi-protein pre-initiation complex (PIC) forms, which includes the General Transcriptional Factors (GTF) and transcriptional factor human II (TFII) - composed of 10 subunits.30 This complex not only allows RNAPII to initiate transcription but also establishes the pause state, in which recruited, promoter-associated RNAPII halts transcription and RNA elongation when phosphorylated on Serine5 by the cyclin dependent kinase 7 (CDK7) subunit.31 RNAPII pausing is regulated mainly by two complexes: DRB sensitivity-inducing factor (DSIF) and Negative elongation factor (NELF).32 RNAPII release depends on DSIF, NELF and P-TEFb-induced RNAPII phosphorylation on Serine2, which in turn activates transcriptional elongation at the promoter-proximal region33, a process referred to as pause-release.34 BRD4’s association with P-TEFb is essential for RNAPII activation since this interaction stimulates P-TEFb kinase activity (Itzen F, 2014), and prevents P-TEFb interaction with 7SK/HEXIM, a ribonucleoprotein that segregates P-TEFb into its kinase-inactive form.35 BRD4 may also promote elongation in a P-TEFb-independent manner, since acute BRD4 degradation abrogates RNAPII Serine 2 phosphorylation and subsequent elongation, without altering genomic occupancy of P-TEFb’s catalytic subunit CDK9.36 BRD4 may also phosphorylate RNAPII at Serine 2, through an atypical kinase activity, further contributing to transcription initiation.35 Thus, BRD4 is critically involved in several steps that rapidly increases transcription after signal-induced activation is triggered, as with activation, recruitment and execution of TF programs.
More recent studies have extended mechanisms through which BETs can regulate transcription. The ET domain in BRD4 may also promote gene expression by independently recruiting histone modifiers that increase transcriptional activity, including the lysine methyltransferase nuclear receptor binding set domain protein 3 (NSD3) and the arginine demethylase jmjC domain-containing protein 6 (JMJD6)37,38. Furthermore, the ET domain can also interact with ATP-dependent chromatin modifiers like SWI-SNF and CHD2.37 These interactions reveal that BRD4 controls gene expression not only by regulating transcription initiation and elongation through RNAPII and P-TEFb, as outlined above, but also by directing these other chomatin modifiers, which can drive chromatin de-compaction when BRD4 occupies active transcription sites. BETs can also regulate gene expression by associating with the Mediator complex39, a group of ~30 subunits that interact with TFs and helps recruit RNAPII.40 Although the precise interaction site between BETs and the Mediator complex is still unclear, several studies indicate that BRD4 and the Mediator complex co-localize at several binding sites, including super-enhancers, with this BRD4-Mediator interaction stabilizing these proteins at specific genomic locations.41, 42
Recent reports also indicate that BET family proteins, and in particular BRD4, may exhibit intrinsic histone acetyltransferase (HAT) activity, providing another means for BETs to direct gene expression. Devaiah and colleagues reported that histone H3 and H4 in different lysine residues can be directly acetylated by BRD4 through an activity distinct from classical HAT proteins.24 Furthermore, these authors identified that BRD4-mediated acetylation on H3K122 leads to nucleosome eviction and chromatin de-compaction, thus increasing gene transcription.24 Tian and colleagues also found intrinsic BRD4 HAT activity and its induction of transcription, observing that pharmacological BRD4 inhibition reduced acetylation of H3K122 and decreased viral-induced airway inflammation.43 In addition, BRD4 HAT activity and its ET domains have been reported to regulate RNA splicing by interacting with HnRNPM, a key alternative splicing regulatory factor, with BRD4 interacting with alterative exons independent of acetylated lysine binding.44 Although this rapidly expanding data continues to unfold, the evidence establishes BETs as potent determinants of transcription, by bringing master TFs and essential transcriptional machinery like RNAPII and P-TEFb to specific histone sites with acetylated lysine residues. Moreover, BET action in the choreography of transcription is multifaceted - coordinating localization, allowing interaction among the assembled players and exerting activity that all help promote gene expression at these epigenetically determined sites.
BET Inhibitors (BETi): Chemical Tools, Clinical Promise?
Insight into the role of BETs on gene expression and subsequent cellular effects has been greatly advanced by the discovery and development of small-molecule BETi (Table 2).45, 46 Although these compounds have reached clinical development for therapeutic use in various settings, including cardiovascular disease, as further discussed below, they served initially as valuable chemical tools for probing BET function, as evident in studies in models of atherosclerosis and other cardiovascular diseases. BETi were first discovered employing chemical shift mapping and nuclear magnetic resonance from the BD acetyl-binding pocket structure in complex with putative ligands.47 Although the first molecules exhibited low affinity for the BD pocket, they provided proof-of-principle that BDs from the BET family could be inhibited.48 Subsequently, several small-molecules with higher BETs BD affinity were identified, in part through further chemical design based on earlier BET ligand structures.49 The thienotriazolodiazepine JQ1 exhibits a shape that is highly complementary to the tandem BDs in BRD2, BRD3 and BRD4, binding to the conserved asparagine residue present in their hydrophobic pocket. As such, JQ1 mimics the interaction between BETs and the acetyl lysine interaction on histones.50 By irreversibly binding at this location, JQ1 displaces BETs from its location on histone tails, disrupting the assembled transcriptional scaffolding and blocking expression of BET-controlled target genes. Other chemical probes that inhibit BETs, like I-BET762 were also developed but identified through other means, as discussed below. Unlike JQ1, I-BET762 has a dimethylisoxazole ring scaffold that serves as an acetyl lysine mimic.51 Further optimization of i-BET762 yielded I-BET151, which showed enhanced pharmacokinetics and a longer terminal half-life for use in in vivo studies.52 RVX-208, now called apabetalone, is a BETi in clinical trials for cardiovascular benefit, as discussed below.53 Finally, several different distinct BETi structures have been identified that are either in preclinical or clinical development for studying and/or treating cancer.54
Table 2: Summy of BET Targeting Agents in Development.
| Class | Compound | Selectivity | Stage | Action | Ref |
|---|---|---|---|---|---|
| BET inhibitors | JQ1 | BD1 + BD2, Pan-BET | Preclinical | Hematologic malignancies | (132) |
| Lung cancer | (133) | ||||
| Breast cancer | (134) | ||||
| Prostate cancer | (135, 136) | ||||
| Colon cancer | (137) | ||||
| Hepatocellular cancer | (138) | ||||
| RVX-208 (RVX000222, apabetalone) | BD2 > BD1; BRD2/3 > BRD4 | Phase II/III | Dyslipidemia, CAD/Diabetes |
NCT01423188 NCT01067820 NCT02586155 (127) |
|
| Phase I/II | Fabry disease | NCT03228940 | |||
| Phase II | PAH | NCT03655704 | |||
| IBET 762 (GSK525762) | BD1 + BD2; Pan-BET | Phase I | Neoplasm | NCT01943851 | |
| Phase I | Carcinoma, Midline | NCT01587703 | |||
| IBET 151 (GSK1210151 A) | BD1 + BD2; Pan-BET | Preclinical | Mixed Lineage Leukemia | (46) | |
| Melanoma | (139) | ||||
| Myeloma | (140) | ||||
| MK-8628 | BRD2/3/4, No BRDT data | Phase I | NUT Midline Carcinoma | NCT02259114 | |
| Triple Negative Breast Cancer | |||||
| Castration-resistant Prostate Cancer | |||||
| Lung Cancer | |||||
| FT-1101 | Pan-BET | Phase I | Acute Myeloid Leukemia | NCT02543879 | |
| Non-Hodgkin Lymploma | |||||
| CPI-0610 | BD1; BRD2/4/BRDT | Phase I | Multiple Myeloma | NCT02157636 | |
| ABBV-075 | BRD2/3/4 | Phase I | Breast Cancer | NCT02391480 | |
| Prostate Cancer | |||||
| Non-Hodgkin Lymploma | |||||
| Acute Myeloid Leukemia | |||||
| Multiple Myeloma | |||||
| BMS-986158 | Undisclosed | Phase I/II | Avanced Tumors | NCT02419417 | |
| BET degraders | BET-PROTAC (AV-771) | BRD4 | Preclinical | Prostate cancer | (141) |
| Non-Hodgkin Lymploma | (50) | ||||
| BET-PROTAC (AV-824) | BRD2/3/4 | Preclinical | Non-Hodgkin Lymploma | (50) |
Agents are listed (column headings left to right) according to their (1) mechanism of action in blocking BET function; (2) Relative selectivity among bromodomains (BD) and BET family members; (3) Stage of development; (4) Condition or diseases for which they are being studied; (5) Reference or assigned U.S. National Library of Medicine clinical trial number (https://clinicaltrials.gov).
Relevant to BETi, the BET bromodomains BD1 and BD2 may exhibit differential functions, including distinct cellular responses to agents with BD-selective versus equal BD targeting as well as individual preferential BET inhibition. Most BETis, including JQ1, ABBV-075 and OTX015, target both BDs equally, as JQ1 while RVX-208 is reportedly BD2-selective.55 Deeper understanding about BD selectivity might improve clinical efficacy and tolerability. 56 Increased anti-tumor activity and reduced gastrointestinal toxicity was reported with the more BRD4-specific BD2-selective inhibitor ABBV-774 as compared to equal BD inhibition.57 For, RVX-208, whose BD2-selectivity may be greater for BRD2 and BRD3 than BRD4, decreased potency was seen in cancer cells lines compared to JQ1.55 As discussed subsequently, BD selectivity is implicated in vascular cell biology58 and may be relevant to RVX-208 clinical effects. Further investigation is needed to clarify specific BD function, distinct BETs family member roles, and specific inhibitor pharmacology in order to optimize BETi approaches.
More recently, the system of PROteolysis Targeting Chimerics (PROTACs) have been applied to BETs to develop a completely novel class of BETi known as BET-PROTACs – ligand-activated chimeric proteins that target specific BETs for proteolytic degradation.59 These heterobifunctional proteins possess two binding regions connected by a linker. One ligand binds specifically to the target protein of interest, as with JQ1 interacting with BETs while the other binding region recruits a modifier protein like the E3-ubiquitin ligase, which causes polyubiquitination of the target protein and proteasomal degradation. PROTACs can be designed for BET protein specificity, for example degrading all BETs, as with BET-PROTAC ARV-771 and ARV-825 or targeting mainly BRD4, as with BD2-selective MZ1 and AT159 or BD1-selective dBET6 and dBET23.60 Recent studies indicate that lower doses of BET degraders might improve therapeutic responses compared to BET inhibitors. The BET-PROTACs ARV-825 and ARV-771 showed enhanced antitumoral activity in vitro and in vivo, respectively as compared to BET inhibitors, which paralleled changes in global transcription.61 Completely distinct responses between BET degraders versus BET inhibitors establishes the need for further study.62 Despite impressive preclinical tumor model effects, no BET-PROTACs have reached clinical trials although their potential use continues to be pursued and potentially relevant for cardiovascular disease. The interest in harnessing BET action for therapeutic use in the CV setting derives from preclinical data seen with BET action and inhibition in endothelial, vascular smooth muscle cells, leukocytes and myocardial settings.
BETs in the Endothelium
BET family members vary in their relative endothelial expression in mouse and human ECs. We reported siRNA knock down of BRD2, BRD3 or BRD4 expression in human umbilical vein endothelial cells (ECs) decreased tumor necrosis factor alpha (TNFα)-induced gene expression, with BRD4 as the key player in this response. Given embryonic lethality of BET deficiency, JQ1 was used to investigate BET effects in vitro and in vivo, with this chemical strategy also offering the advantage of avoiding effects that might occur with genetic BRD4 deficiency from birth.63 JQ1 selectivity and specificity for BRD2, 3 and 4 among other bromodomain-containing proteins has been established.50 Treatment of ECs with either JQ1 or the I-BET BETi blocked TNFα induction of multiple pro-inflammatory and pro-atherosclerotic endothelial genes like vascular cell adhesion molecule 1 (VCAM-1) and functional EC responses such as vascular adhesion and trans-endothelial migration of leukocytes in vitro and ex vivo.63 As these findings would predict, administering BETi in vivo decreased atherosclerosis in the LDL-receptor deficient mouse model.63 Similar effects were seen with RVX-208 treatment of ApoE-deficient mice.64 BET regulation of endothelial responses prompted consideration of global gene expression as well as genome-wide analysis of BET association and action in the endothelial transcriptome using chromatin immunoprecipitation and high throughput sequencing (ChIP-Seq).65 ChIP-Seq involves stimulating cells prior to immunoprecipitating relevant proteins, like BRD4 and RNAPII, and the DNA fragments attached to these proteins; subsequent massive parallel sequencing and alignment of the DNA co-immunoprecipitated with these proteins fixes where that protein, like BRD4, was located across the entire genome under defined conditions.
Applying global ChIP-Seq approaches to BRD4 in human ECs yielded insights relevant to BET action in vascular biology, atherosclerosis and other cardiovascular settings. First, after cytokine stimulation, BRD4 facilitated expression of multiple pro-inflammatory and pro-atherosclerotic targets involved in pathways like thrombosis, leukocyte adhesion and endothelial barrier function, thus identifying BRD4 helps orchestrate a coordinated transcriptional program relevant to atherosclerosis. Second, BRD4 activity was concentrated at specific regulatory super enhancer regions. This BET-restricted pattern of gene expression suggests the complex state like atherosclerosis may be reduced to a more tractable gene set, as proposed for BRD4 in multiple myeloma.41 Of note, relatively little overlap existed between BRD4-associated super enhancers in macrophages and ECs.63 It is worth noting that the gene identified as regulated by a given BRD4-associated enhancer is a predictive exercise, typically based on the nearest gene’s start site. Given transcriptional regulation over significant distances, the predicted transcriptional regulation of any specific gene requires more direct confirmation. In response to TNFα, BRD4’s location aligned closely with p65 across the genome, strongly supporting BRD4 as coupling this key NFκB component to chromatin and the execution of the TNFα transcriptional program. ChIP-Seq data also revealed that while TNFα stimulation induced BRD4 association and the induced expression of canonical pro-inflammatory, pro-atherosclerotic endothelial gene targets, e.g. VCAM-1, BRD4 association and expression of another distinct set of endothelial genes was lost in response to this inflammatory cytokine, e.g. Sox18 (Figure 1) This finding implicates BRD4 in basal EC gene expression and that TNFα stimulation prompts BRD4 redeployment to newly formed enhancers that defines a new inflammatory cell state through this induced transcriptional program (Figure 2). Similar basal versus stimulated regulation by BETs have been seen elsewhere, including adipogenesis.66
Figure 2. BET Epigenetic Reader Proteins In Cell State Transitions and Cellular Identity.
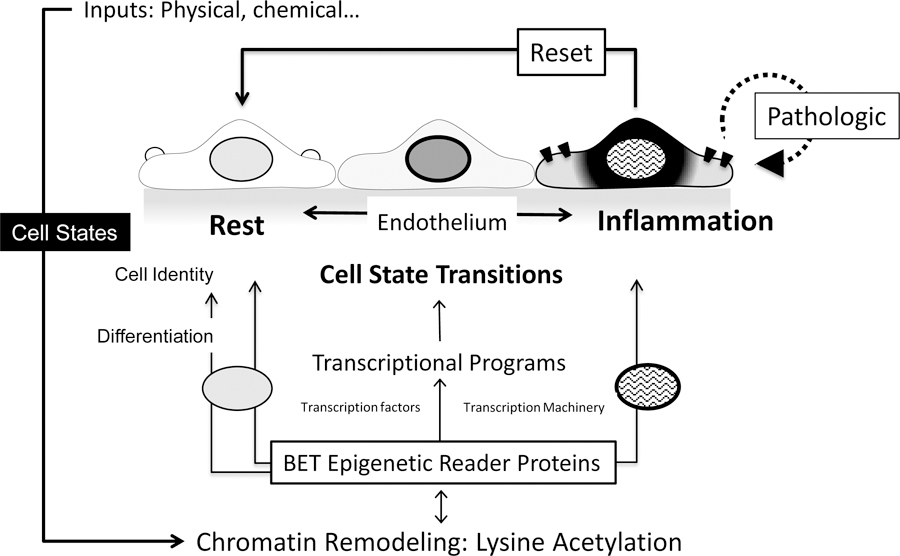
BETs are known to direct stem cell differentiation, cellular identity and transitions in mature cell states, as relevant in the CV system. EC changes are used here to illustrate concepts applicable to BETs in other vascular and inflammatory cells. Multiple inputs including physical forces like pressure, wall stress, fluid dynamics and chemical stimuli, e.g. inflammatory cytokines, stimulate cellular responses that include chromatin remodeling. BETs link these inputs to coordinated transcriptional programs by serving as a recruited scaffold for transcriptional mediators, e.g. RNAPII. BET involvement in stem cell differentiation may influence mature vascular and inflammatory cells characteristics, function and identity. In response to inputs, mature ECs undergo dynamic cell state transitions as defined by distinct expression patterns, as with resting versus inflammatory stimulated endothelium. BETs govern transcriptional programs at rest and after stimulation, as shown in response to cytokines, VEGF and conditions like hypoxia. Persistent or recorded, via histone marks, exposures may direct pathologic, maladaptive BET-regulated transcriptional programs, promoting chronic conditions like atherosclerosis. BETs are also involved in changes in mature cell identity, as shown for endothelial mesenchymal transition. Although BET mechanisms must be able to be terminated or reset, including removal of histone lysine acetylation marks, much less is known about these potentially protective processes. Similar cell identity and state issues apply to BET action in smooth muscle cells, leukocytes and cardiomyocytes, each with their own distinctive stimuli and cell states, as well as other settings, including adipogenesis. References as per text.
These BRD4 actions relate to other studies identifying BET involvement in endothelial biology. BETs help coordinate vascular endothelial growth factor (VEGF) responses in angiogenesis and hypoxia, effects first identified in oncology. For example, BET inhibition was found to decrease hypoxia’s potent induction of carbonic anhydrase (CA) IX expression, which may promote tumor heterogeneity and predict worse therapeutic responses67 and to limit vascularization and tumor growth in childhood sarcoma models.68 BET inhibition has also been shown to regulate transcriptional responses to hypoxia, including CA expression, can decrease tumor growth in breast cancer.69 BET inhibition in HUVECs through JQ1 treatment or shRNA to BRD2 or BRD4 repressed VEGF-mediated angiogenesis and vascular permeability, decreasing endothelial nitric oxide activation and phosphorylation of VEGF receptor 2 (VEGFR2) and PAK1 but without altering VEGFR2 expression while in vivo, JQ1 decreased Matrigel angiogenesis and retinal neovascularization.70 In detailed studies, Pu and colleagues identified that VEGF stimulation increased ETS1 acetylation, prompting its association with BRD4 and the induction of various genes that promote angiogenesis.71
ECs may be a unique setting that offers insight into BET action. The endothelium must rapidly and effectively transduce multiple stimuli, whether sensing mechanical forces, detecting a loss of vascular integrity to initiate coagulation, marshalling inflammatory responses or any one of the other known roles for this dynamic organ. The endothelium is essential for initiating and often propagating such responses in both health and disease. The EC response to these stimuli represent cell state transitions. Cell state changes can also be understood as contributing to pathogenesis, as with chronic inflammation, hypertension and many other scenarios. BETs facilitate the rapid execution of transcriptional programs that define these cell states, including their part in reading epigenetic marks recorded from prior exposures. From this perspective, the failure to reset BET action can help explain chronic disease states and highlights the importance of missing insight into how BET signals are terminated.
BETs in Vascular Smooth Muscle Cells (SMCs)
Despite SMCs constituting the integrity, strength and contractility of the artery wall, even highly differentiated SMCs demonstrate remarkable plasticity, reactivity and contributions to vascular pathology, in keeping with BET-governed cell state transitions.72 Indeed, such shifts in cellular phenotype in cells with a set genome, are by definition epigenetic in nature.73 Well-established SMC changes, whether in response to mechanical forces, injury and cytokine stimulation, which can induce micro-proliferation of intimal hyperplasia (IH) found in atherosclerosis, (re)stenosis and other vascular pathologies, have all been suggested to involve BETs, and in particular BRD4, as a key determinant of SMC phenotype and pathologic states.74, 75
Wang et al observed pronounced increases in BRD4 expression in the neointima of human artery and vein samples, and in the rat carotid artery IH model of balloon angioplasty injury.76 In cultured rat primary SMCs, the BETi JQ1(+), but not its inactive enantiomer, abrogated cell proliferation and migration; siRNA to BRD4 but not siBRD2 or siBRD3 as well as direct JQ1-containing perivascular hydrogel recapitulated these SMC effects.76 Further studies suggest BET inhibition can have unique effects in vascular cells. For example, in balloon-injured rat carotid arteries, JQ1 delivered intravenously via biomimetic, platelet membrane-coated nanoclusters decreased IH without impairing re-endothelialization of the injury site while similarly delivered rapamycin also reduced IH but decreased re-endothelialization,77 consistent with BET inhibition protecting against impaired growth, inflammation and apoptosis.63 The effects seen with BET inhibition in SMCs effects suggests this as a means of circumventing the offsetting pro-thrombotic EC effects of current rapamycin- or paclitaxel-eluting stents.78
BETs as epigenetic reader proteins orchestrating transcriptional programs may involve another dimension especially relevant to the vasculature: intra-cellular communications. Endothelial function and dysfunction can direct SMC responses in a paracrine and/or endocrine manner, producing biologically-active mediators like nitric oxide, controlling the uptake of metabolites and nutrients like fatty acids and glucose, responding to and releasing inflammatory mediators, all of which can contribute to changes in SMC phenotype. As such, BET effects in either ECs or SMCs may modulate SMC responses. BETs are also involved in another mechanism that spans EC and SMC biology in terms endothelial-to-mesenchymal transition (EndMT), a variation of epithelial to mesenchymal transition (EMT) discussed in oncology and other settings.79 In EndMT, ECs begin expressing mesenchymal markers like Taglin, while concurrently losing expression of EC-restricted proteins like CD31 - a definitive example of cell state transition. EndMT contributes directly to IH in atherosclerosis80 and vein graft loss81 in humans. BRD4 has been shown to govern EndMT in human and rodent ECs. 58, 82 Interestingly, in a deeper analysis of bromodomain function, the decrease in IH seen with BET inhibition appeared to depend on the BD2 but not BD1 domain of BRD4.58 Although it remains unclear if this effect derived from inhibiting BRD4 in ECs or SMCs could not be established in vivo, these findings provide strong evidence for BRD4’s role in SMC phenotypic transitions.83
Pulmonary arterial hypertension (PAH) is a distinct vascular disease state that has been reported to involve disruption of normal EC and SMC biology, EndMT and BET involvement.84 Bonnet and colleagues have reported increased BRD4 levels in lungs and specifically pulmonary artery SMCs (PASMCs) of PAH patients, with evidence that BET inhibition decreases PASMC proliferation and enhances apoptosis in a BRD4-dependent manner. 84, 85 Mechanisms proposed for these BET-mediated effects involved decreased expression of three major PAH-associated oncogenes that occurred with BET inhibition or specific siBRD4 knockdown: nuclear factor of activated T cells (Nfatc2), B-cell lymphoma 2 (Bcl-2) and survivin. The increase in BRD4 in PAH PASMCs was found to depend on microRNA-204, which was a previously reported contributor to PAH. In the established Sugen hypoxia rat model, JQ1 treatment reversed PAH severity.84 Intriguingly, these same investigators reported that interleukin 6 (IL-6) increased BRD4 expression, which might then promote both PAH and the association between PAH and increased coronary artery disease (CAD). Higher BRD4 levels were found in SMCs from both pulmonary and coronary PAH patient arteries while in rat PAH models, IL-6 stimulation increased both BRD4 levels in SMCs and BRD4-dependent proliferation.85 In vivo experiments with JQ1 or siBRD4 produced similar responses in the rat Sugen hypoxia PAH model. This data prompted the hypothesis that systemic inflammation increases BRD4, propelling SMC proliferation in both pulmonary and coronary arteries and helping explain the increased CAD observed in PAH.86 BET inhibition as a means of improving PAH is supported by the proposed BETi apabetalone (RVX-208) effects in microvascular ECs and SMCs from PAH patients and in other preclinical studies and well as pooled data in human studies.87
While the studies noted support a key role for BRD4 in SMC proliferation/migration and vascular wall thickening, the underlying molecular mechanisms remained unclear. Natarajan and colleagues investigated BRD4 in mediating SMC changes in response to angiotensin (AngII).87 AngII was found to induce activity at enhancers/super-enhancer associated with key cell signaling TFs including AP-1, ETS and specific kinases like Jun. This coordinated AngII-activated SMC transcriptional program was abolished by either specific enhancer deletion or JQ1 treatment. Long noncoding RNAs (lncRNAs) that overlapped with the relevant identified enhancer regions, namely lnc-Ang184 and lnc-Ang383, were also found to be involved in AngII stimulation, an example of the novel concept that lncRNAs may contribute to BET action at specific enhancers. Finally, intraperitoneal JQ1 treatment of mice ameliorated AngII-induced hypertension, arterial wall medial hypertrophy and inflammation. These findings further extend BETs in integrating complex SMC transcriptional programs, linking these epigenetic readers to AngII-induced gene expression, lncRNAs and hypertension.
Other reports identify BET involvement in PAH and in other pulmonary settings like airway SMCs indirectly. Chemical library screening for modulators of PASMC proliferation identified emetine, a principal alkaloid extracted from the Brazilian ipecac root in use as an emetic and antiprotozoal inhibited PASMC proliferation, ultimately finding that this occurred through decreased expression of BRD4 and survivin.88 Separate studies report JQ1 can inhibit airway SMC proliferation89, inflammation90 and enhance antioxidant gene expression.91 Whether these effects derived from BRD2, BRD3 or BRD4 remains unclear. While airway SMCs differ considerably from vascular SMCs, these reports may help further identify BET-directed effects and functional responses in vascular SMCs and elsewhere. Early evidence that BET inhibition may limit aneurysm formation and/or expansion may also involve SMC effects.92
BETs in Leukocyte Biology: Monocytes, Macrophages, T cells
The evidence for BETs in directing pathologic responses has included a common theme of BET inhibition decreasing transcriptional programs in inflammation. Such findings build upon earlier studies demonstrating BET involvement in responses to extracellular pathogens and sepsis.51 The evidence for BET coupling TNFα signaling to NF-κB activation in ECs aligns with and would predict effects in other settings regarding BET inhibition decreasing p65 signaling in other settings, in keeping with the overlap between acute inflammatory responses essential for host defenses and the low-grade, chronic inflammation involved in atherosclerosis.93 Many prior reports established a pro-inflammatory role for BETs in various in vitro and in vivo pathogenic conditions, as considered in recent reviews.94–96 Herein we focus mainly on new progress regarding BETs in leukocyte-related function.
Inflammatory cell modulation by BET inhibition was among the first reports implicating this epigenetic reader protein family outside of oncology. In genome-wide analyses in bone marrow-derived macrophages (BMDM), the BETi I-BET suppressed lipopolysaacharide (LPS)-induced inflammatory gene expression, including IL6, IL1-beta and chemokines like CXC9 and CCL12, all of which are mediators also involved in atherosclerosis.51 Particularly impressive was the finding that I-BET administration to mice tempered sepsis and improved animal survival. Subsequent reports using BETi or individual BET knockdowns extended these findings to pro-inflammatory activation or differentiation of monocytes97, 98, macrophages99,100, 101, T cells102, 103, natural killer cells104, dendritic cells105, 106, B cells106 and microglia.107, 108. Issues remain however, including some divergence among studies and individual roles among BRD2, BRD3 or BRD4.
While the BMDM isolated from hypomorphic heterozygous BRD2-deficient mice (Brd2lo), which have lower BRD2 expression versus wild type) were found to produce less inflammatory cytokines, despite having intact and unchanged BRD3 and BRD4, silencing each BET in vitro in wild type BMDM reduced inflammatory cytokine expression to a similar extent.109 In another study, use of a novel proteolysis-targeting chimera (PROTAC) to delete BRD2 and BRD4 proteins, LPS-stimulated microglia activation was dampened110, suggesting a limited role for BRD3 in LPS-induced inflammation. In the N9 microglia cell line, siRNA to BRD2 blocked LPS-induced inflammatory cytokine gene expression most potently, with siBRD4 and siBRD3 showing less and no effect, respectively111. Despite this, BRD3 deficiency in mouse RAW246.7 macrophages inhibited virus-triggered IFN-β production. 112 Bao et al demonstrated that myeloid-specific Brd4 deficiency resulted in resistance to LPS-induced sepsis, with much more marked diminution of inflammatory gene expression in Brd4-deficient BMDMs.113 Interestingly, more recently, using cell type-specific BRD4 deficiency and subsequent ChIP-Seq/RNAseq analysis, Dey et al found a limited role for BRD4 in BMDM development and LPS-induced inflammation.114 These investigators reported that Il1β, TNFα, and Ccl5 were actually BRD4-independent and p65 binding was enhanced rather than decreased after BRD4 depletion. The divergence between these studies and reports in other settings suggest BET action may have cell context-dependency in inflammation, sensitivity to potential variables among specific models and/or compensatory changes. Indeed, increased BRD2 and BRD3 levels were observed in BRD4-deficient BMDMs.114 The nature of specific inhibitors may also be a factor, given differences among inhibitors regarding preferential BD1 versus BD2 targeting. While blocking BD2 (in all BETs) more effectively repressed LPS-induced inflammation in mouse N9 cells111, BD1 was found to exert a more dominant role in the murine oligodendrocyte115 and Th17 cell116 differentiation.
To date, BET-directed control over pro-inflammatory responses in leukocytes has been most often attributed to BRD4 associating with the master TF NFκB.63, 114 and the P-TEFb transcription elongation complex, which then activates RNAPII106, 114. Brd4-deficient mouse BMDM also showed increased IκBα expression, which reduced NF-κB binding to inflammatory gene promoters.113 Such counter-regulatory responses have been seen in other BET settings, for example with TNFα stimulation of ECs resulting in BRD4-mediated increases in inflammatory target gene expression while also inducing targets that limit NFκB activation.63 Such findings support BETs as coordinating balanced gene expression to maintain biologic equipoise. In another study, glucocorticoid-receptor activation inhibited BMDM responses by countering p300 and BRD4 recruitment.117 Interactions have also been reported between BRD4 and the TF IRF1 114 and between BRD3 and IRF3/p300.112 While BRD4 is considered a prominent super-enhancer mediator, BRD4 has been reported to be unnecessary for some LPS-induced super-enhancer function in BMDMs.114 In contrast, others have identified separate yet inter-dependent BRD2 and BRD4 function in Th17 cell development and adaptive immunity.21 While BRD2 was involved in localizing STAT3 to active enhancers occupied by TFs IRF4 and BATF, it was BRD4 that controlled RNAPII pause release, doing so in a temporal manner.21 More recently, BRD4 in thymocytes and T cells was reported to interact with RNA splicing machinery, regulating alternative splicing.44 Using pan-BETi, additional BET regulated responses have been identified in distinct inflammatory cell responses - increasing autophagy in human macrophages118, augmenting anti-oxidant gene expression a human monocyte cell line91 and inhibiting PD-1/PD-L1 immune-checkpoint responses in human and mouse B cells. 119 Although many aspects of these and other BET actions require further delineation, like how specific cis and trans elements carry out BET responses and how individual BETs are involved in leukocyte different subtypes, differentiation pathways and in response to different stimuli, the importance of BETs in leukocyte biology and its relevance to atherosclerosis and other cardiovascular pathologies is already apparent, and provides a basis for pursuing such studies.
BETs in Myocardial Function
The myocardium is another setting in which cell state changes, shifting cell identities and even stem cell differentiation have all been invoked, as with cardiomyocyte changes and cardiac remodeling from either pressure or volume overload, responses to exercise or injury and myocardial fibrosis among others. We identified BRD4 as regulating key TFs involved in heart failure, as seen in response to phenylephrine stimulation; in vivo, BET inhibition altered cardiomyocyte responses to ventricular pressure overload caused by trans-aortic constriction (TAC) and prevented cardiac hypertrophy.120 BETs were found to act as pause-release factors for these master myocardial TFs. In response to TAC, BRD4 was involved in TFs controlling expression of myocardial hypertrophy genes with active gene body elongation via RNAPII while in the setting of BET inhibition, these TFs were more often found at transcription start sites but without movement down the gene body, consistent with RNAPII pausing. These BET-mediated shifts controlled cardiac gene expression involved in cytoskeletal reorganization, extracellular matrix production, cell-cycling, cell growth through paracrine and autocrine stimuli and inflammation, all of which are relevant forces in heart failure. Despite BET control of MYC signaling in cancer and MYC’s established myocardial role, BET action in cardiac myocytes was MYC-independent, further supporting distinct BET effects in different cellular settings. Subsequently, BRD4 was reported to control a fibrotic and inflammatory transcriptional network in different murine heart failure models and in agonist-induced hypertrophic changes in human iPSC-derived cardiomyocytes.121 BRD4 may control cardiac fibrosis and heart failure by targeting genes that induce quiescent cardiac fibroblasts to convert into cells producing extracellular matrix components through tumor growth factor-beta (TGFβ) and p38 Mitogen-activated protein kinase expression.122 In subsequent work consistent with BET-mediated regulation of EndMT reviewed earlier, BET inhibition was reported to limit cardiac fibrosis induced by TAC or TGFβ, with effects on EndMT-associated transcription factors, the SMAD pathway and TGFβ receptor I.82 Controversy exists regarding TAC, EndMT and differences among murine models of cardiac fibrosis123 but simply overexpressing BRD4 increased EndMT responses in the absence of TGFβ stimulation, suggesting a fundamental shift towards a pro-fibrotic cell state.
BETs as a Therapeutic Target For CV Disease
The prospect of interrupting the execution of a coordinated transcriptional program that underlies complex patterns in diseases like atherosclerosis, pulmonary arterial hypertension or heart failure is appealing (Figure 3). The relationship between BETs and clinical phenotypes is evident in the finding that NUT midline carcinoma (NMC), a poorly differentiated squamous cell carcinoma (SCC) with multiple abnormalities, derives from a fusion protein involving the BRD4 and NUT genes.124 While most attention in epigenetic therapeutics has focused on blocking histone mark placement or directing their removal, the prospect of keeping previously placed marks that promote atherosclerosis or other CV disorders left unread is also compelling. Such an approach may be even more appealing given that histone marks may be placed early in life, even in utero, be long-lived, involve maladaptive responses and not required for normal biologic function. Based in part on the evidence reviewed here, clinical therapeutic strategies targeting BETs have been pursued, even reaching largescale prospective clinical cardiovascular trials.
Figure 3. BRD4 Controls Transcriptional Responses In Vascular and Inflammatory Cells.
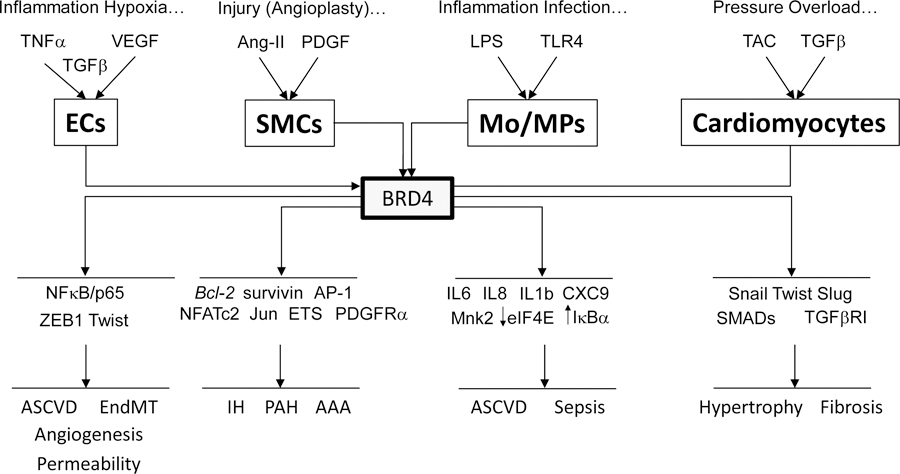
Examples of stimuli, regulated gene targets and pathologic responses involving specifically BRD4 in ECs, SMCs, monocyte/macrophages (Mo/MPs) and cardiomyocytes as reviewed here are shown. These BRD4 effects have been demonstrated through the use of gene expression, BETi responses and global profiling tools of RNA-Seq and/or ChIP-Seq. This data reveals BRD4 exerts broad, integrated transcriptional effects executed in large through its association with super enhancer regions to determine functional cellular responses and associated disease states. An example that supports BET action as directing transcriptional programs to achieve a coordinated output is evident in BET inhibition repressing NFκB-directed gene expression while concurrently increasing translation of IκBα, which also decreases NFκB activity. BRD2 and BRD3 have also been implicated in similar although often distinct responses relevant to the vasculature. Abbreviations, references as per text.
Therapeutic BET inhibition has been first explored in cancer, given the BRD4-NUT oncogene and BET involvement in MYC oncogene effects in hematologic malignancies like mixed-lineage leukemia, acute myeloid leukemia, Burkitt’s and Burkitt-like lymphoma.125 Distinct BETi have been developed and are under study in clinical trials for various cancers (Table 2), based on encouraging preclinical and sometimes translational data.126 Some common adverse events have been observed among BETi, including thrombocytopenia, anemia, neutropenia, gastrointestinal issues, increased bilirubin and fatigue.127 Preclinical studies suggest that combining BETi with other epigenetic modulators may have a synergistic anti-tumor activity, allowing for lower doses and perhaps fewer adverse events, while other strategies involve combining BETi with immune-modulators and hormone therapy for greater efficacy.128 In terms of cardiovascular responses, BETi trials in cancer will provide insight into the safety and tolerability of specific BET agents and perhaps indirect evidence on cardiovascular responses. One emerging issue is BETi resistance - the induction or existence of counter-regulatory responses that may limit longer term effectiveness of BET inhibition in cancer treatment.129 Examples include specific AML cell subsets that are resistant to BETi-induced apoptosis, compensatory induction of MYC through WNT signaling and increased post-translational BRD4 modifications like phosphorylation.130–132 Whether resistance to BET inhibition might influence cardiovascular responses is unknown but BET involvement in basal cellular function, their wide expression and their regulation of genes that offset stimulated TF responses may limit BET efficacy.
Another intriguing clinical angle has been the discovery that BET inhibition can re-activate latent HIV infection. A significant hurdle in HIV eradication is HIV latency - that virus’ ability to maintain itself in cellular reservoirs, like T cells, which avoids viral exposure to therapies.133 Although the mechanism(s) for how BET inhibition activates HIV remains unclear, the hypotheses under study adds insight into how BETs and their inhibitors operate.134,135,136, 137 HIV replication depends on RNAPII activation, which is induced by the viral Tat protein. Related to earlier mechanistic discussions, after HIV initiates transcription, RNAPII pauses, a consequence of two negative elongation factors that inhibit RNAPII activity. For elongation to begin, the P-TEFb complex, consisting of CDK9 and the regulatory subunit Cyclin T1, is recruited, which eliminates the negative elongation factors; the HIV Tat protein recruits P-TEFb. Hypotheses for why BETi may cause HIV re-activation include increases in P-TEFb levels and/or activity, alterations in T cell gene expression and/or removal of BRD4 from its position on the HIV promoter that keeps Tat from allowing transcription to proceed.
The BETi farthest along as a cardiovascular therapeutic agent is RVX-208 (previously RVX000222) and as noted, now called apabetalone.53 Initially this quinazolone compound with BD2-selectivity, was identified during small molecule screening for apolipoprotein A-1 (ApoA1) expression inducers.55, 138, 139 A similar ApoA1 transcription assay screen identified the I-BET inhibitors 140. Interestingly, neither of these screens sought out BETi compounds and it remains unclear if increased ApoA1 contributes to RVX-208 or other BETi effects.
Early studies supported RVX-208 effects on HDL-C levels and particle size as well as cholesterol efflux, including in primates.138 RVX-208 decreased atherosclerosis in ApoE-deficient model,64 similar to JQ1 effects in LDLR-deficient mice.63 RVX-208 decreased atherosclerosis without major lipid level changes, suggesting possible anti-inflammatory effects. The 26 week Phase II ASSURE (Apo A-I Synthesis Stimulation and Intravascular Ultrasound for Coronary Atheroma Regression Evaluation) showed RVX-208 failed to significantly decrease the primary endpoint of percent change in atheroma volume versus placebo although total atheroma volume from baseline declined.141 RVX-208 treated patients had lower inflammatory markers but also elevated liver function tests (7.1% vs 0%, P=.009). The Phase II ASSERT trial studied 299 statin-treated patients with stable CAD receiving placebo or one of four RVX-208 doses.142 A dose-dependent but not statistically significant increase in ApoA1 levels occurred; HDL particle number was significantly increased (~1%). RVX-208 was also associated with transient, reversible increases in alanine transaminase and aspartate transaminase, as previously observed, without bilirubin or creatinine changes; adverse effects seen with other BETi were not evaluated. Pooling Phase 2 trial data suggested stronger evidence for RVX-208 clinical benefits on death, myocardial infarction (MI), coronary revascularization or cardiovascular hospitalization (5.9% vs 10.4%, p = .02), especially in higher risk patients with elevated high sensitivity C-reactive protein levels (hs-CRP, > 2 mg/dL, 5.4 vs 14.2%; P = .02), lower baseline HDL (5.5% vs 12.8%, p = .01) and type 2 diabetes (5.4% vs 12.7%, p = .02). Given the prior negative IVUS data, plaque stabilization could be postulated as a contributing mechanism. A Phase 3 trial was undertaken (BETonMACE) testing apabetalone (RVX-208) versus placebo on first major adverse cardiovascular events in high risk patients with diabetes, low HDL-C and a recent (7–90 days) acute coronary syndrome (ACS) event.143 Carried out at 195 sites in 13 countries, BETonMACE data is not yet published but has been presented. 144 with these presented results considered here given relevance to BET mechanisms and further BETi development and clinical trials.
In BETonMACE, apabetalone showed no significant difference versus placebo on the primary endpoint (combined CV death, MI, or stroke) in 2,425 subjects with T2D, recent ACS and low HDL-C (10.3% vs. 12.4%, hazard ratio of 0.82, p = 0.11) over the average 26 month follow-up.144, 145 Participants (mean age 62 years, 26% female) excluded those with a prior or current diagnosis of heart failure, recent coronary bypass (90 days), planned coronary revascularization, advanced kidney disease and all underwent mandatory high intensity statin run-in (atorvastatin 40 or 80 mg or rosuvastatin 20 or 40 mg). At study initiation, subjects had a median LDL-C 65 mg/dL, HDL-C 33 mg/dL and an A1C of 7.3%. A primary endpoint MACE sensitivity analysis showed a hazard ratio of 0.79 (p=0.06). Nominal but insignificant decreases were observed with apabetalone on CV death and MI (9.2% vs.11.5%, p > 0.05) and all-cause mortality (5.0% vs. 5.7%, p > 0.05). While HDL-C increased significantly from baseline (16.2% vs. 10.4% (p = 0.001) there was no change in hs-CRP. Although involving smaller numbers, prespecified subgroup analyses posted by the sponsor suggests possible greater benefit on heart failure hospitalizations and in those with decreased kidney function and lower LDL-C levels. Adverse events were seen more common in patients receiving apabetalone including discontinuations (114, 9.4% vs 69, 5.7%) and increased liver function tests (78, 6.4% vs 18, 1.5%), all versus placebo. Analysis of BETonMACE and insight into the trial’s implications on BET inhibition as a strategy for atherosclerotic complications awaits full publication. The FDA recently granted Breakthrough Therapy Designation for apabetalone, a status designed for expediting drugs that have preliminary clinical evidence indicating a potential substantial improvement over available therapy on clinically-significant endpoint.146
More germane to this discussion are the scientific implications from BETonMACE. While clinical trial data analysis and interpretation requires defined rigor, mechanistically, the BETonMACE trends suggests BET inhibition may limit CV events, aligning with preclinical data. Apabetalone is but one chemical entity aimed at BET inhibition; the prospects for improving CV outcomes by disrupting BET action as an epigenetic reader cannot be reduced to a single agent or trial. Indeed, in oncology, multiple distinct BETi are being pursued in different cancer types. Apabetalone is a less potent and BD2-selective BETi that may target BRD2 and BRD3 more than BRD4.55 How BD selectivity, potency of BET inhibition or selectivity/potency against individual BET family members translates into clinical cardiovascular efficacy remains unknown. Of particular interest will be how BETonMACE data aligns with the reported basic science effects of BET inhibition, for example in terms of inflammatory markers/mediators, cardiac hypertrophy/heart failure and/or hypertension. Analysis of global gene expression patterns, mediator levels or ChIP-Seq in patient samples, like circulating leukocytes, is possible but unfortunately not done in BETonMACE. Such deeper analyses could shed light on responders and non-responders, especially given the obvious complexity of BET biology.
Independent of this one trial, targeting BETs for CV benefit entails both great promise and inherent hurdles. The powerful nature of BET action in integrating expression of multiple targets in response to distinct proximal stimuli via an epigenetic mechanism identifies BETs as warranting further study in the CV system. At the same time, the extent of BET control of transcriptional programs raises cautionary notes. Systemic BET inhibition may cause loss of necessary adaptive responses while evidence for BET activity under basal conditions and their wide expression patterns suggest their involvement in transcriptional programs directing normal physiology. Perhaps more selective BET interventions, like delivery to specific cells or clinical circumstances, or agents with less potent BET inhibition may be necessary. No doubt such paths forward are ultimately dependent on better understanding BET biology in vascular and inflammatory cells.
Future BETs: Directions and Conclusions
The elucidation of epigenetic mechanisms, which operate ‘above’ the genome, has revealed a completely new dimension to how control over gene expression can dictate phenotypes in health and disease (Figure 3). Chromatin remodeling adds a dynamic component to fixed genotypes, with processes that can record prior exposures, circumstances or stimuli, with functional effects. While the writing and erasing of histone marks requires attention in CV diseases, we now know that the BET bromodomain-containing epigenetic reader proteins are critical determinants of executing this epigenetic code. As evident in the data reviewed here, BETs help orchestrate transcriptional programs involved in the endothelium, smooth muscle cells, inflammatory cells and the myocardium, directing differentiation, cellular identity and cell state transitions in response to forces commonly encountered in the heart, lungs and vasculature.
Despite exciting progress regarding BET action, essential information is missing about how these epigenetic reader proteins determine transcription in CV settings. What are the distinct roles of individual BETs, how is that selectivity determined and how does this or other mechanisms account for gene selectivity for BET regulation as well as BET inhibitors? Do changes in expression of BETs themselves impact transcriptional output, as suggested in some vascular settings, or do other forces modulate BET activity or enhancer association? What common features define those genes that do, or do not, involve BET transcriptional control? How do BETs drive basal cellular function in vascular and inflammatory cells? Although BETs bind to acetylated lysines on histone tails, what determines which acetylated lysines interact with BETs and how is this integrated into the three-dimensional space of chromatin? Perhaps particularly important given the chronicity of CV diseases, what determines temporal aspects of BET action, including signal termination? These and many other questions are prompted by the startling and relatively recent recognition of BETs as epigenetic reader proteins in coordinating gene expression in the CV setting. Despite rapid advances in this area, even reaching clinical CV BETi trials, continued progress will require answers to these and other questions, especially if the predictive and therapeutic potential of BET biology is to be fully realized.
Funding:
Lemann Foundation (PDB), R01HL133665 (LQ - PI), R56HL125894, R01DK107239, R01HL133665, EuroPro Foundation, Neissa Foundation, Linda Joy Pollin Research Fund (JP)
Footnotes
Disclosures: None
References
- 1.Marshall J The genetic code. Proc Natl Acad Sci U S A. 2014;111:5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Georgakis MK, Gill D, Rannikmae K, Traylor M, Anderson CD, Lee JM, Kamatani Y, Hopewell JC, Worrall BB, Bernhagen J, Sudlow CLM, Malik R and Dichgans M. Genetically Determined Levels of Circulating Cytokines and Risk of Stroke. Circulation. 2019;139:256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leopold JA, Maron BA and Loscalzo J. The application of big data to cardiovascular disease: paths to precision medicine. J Clin Invest. 2020;130:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robson MI, Ringel AR and Mundlos S. Regulatory Landscaping: How Enhancer-Promoter Communication Is Sculpted in 3D. Mol Cell. 2019;74:1110–1122. [DOI] [PubMed] [Google Scholar]
- 5.Liu J, Ali M and Zhou Q. Establishment and evolution of heterochromatin. Ann N Y Acad Sci. 2020. [DOI] [PMC free article] [PubMed]
- 6.Feinberg AP. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N Engl J Med. 2018;378:1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keating ST, Plutzky J and El-Osta A. Epigenetic Changes in Diabetes and Cardiovascular Risk. Circ Res. 2016;118:1706–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rizzacasa B, Amati F, Romeo F, Novelli G and Mehta JL. Epigenetic Modification in Coronary Atherosclerosis: JACC Review Topic of the Week. J Am Coll Cardiol. 2019;74:1352–1365. [DOI] [PubMed] [Google Scholar]
- 9.Obata Y, Furusawa Y and Hase K. Epigenetic modifications of the immune system in health and disease. Immunol Cell Biol. 2015;93:226–32. [DOI] [PubMed] [Google Scholar]
- 10.Ling C and Ronn T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019;29:1028–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patsouras MD and Vlachoyiannopoulos PG. Evidence of epigenetic alterations in thrombosis and coagulation: A systematic review. J Autoimmun. 2019;104:102347. [DOI] [PubMed] [Google Scholar]
- 12.Kaur G, Begum R, Thota S and Batra S. A systematic review of smoking-related epigenetic alterations. Arch Toxicol. 2019;93:2715–2740. [DOI] [PubMed] [Google Scholar]
- 13.Bogsrud MP, Ulven SM and Holven KB. Does intrauterine exposure to hypercholesterolemia adversely affect familial hypercholesterolemia phenotype? Curr Opin Lipidol. 2016;27:382–7. [DOI] [PubMed] [Google Scholar]
- 14.Elliott HR, Sharp GC, Relton CL and Lawlor DA. Epigenetics and gestational diabetes: a review of epigenetic epidemiology studies and their use to explore epigenetic mediation and improve prediction. Diabetologia. 2019;62:2171–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atlasi Y and Stunnenberg HG. The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet. 2017;18:643–658. [DOI] [PubMed] [Google Scholar]
- 16.Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK and Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–6. [DOI] [PubMed] [Google Scholar]
- 17.Moriniere J, Rousseaux S, Steuerwald U, Soler-Lopez M, Curtet S, Vitte AL, Govin J, Gaucher J, Sadoul K, Hart DJ, Krijgsveld J, Khochbin S, Muller CW and Petosa C. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature. 2009;461:664–8. [DOI] [PubMed] [Google Scholar]
- 18.Filippakopoulos P and Knapp S. The bromodomain interaction module. FEBS letters. 2012;586:2692–704. [DOI] [PubMed] [Google Scholar]
- 19.Pott S and Lieb JD. What are super-enhancers? Nat Genet. 2015;47:8–12. [DOI] [PubMed] [Google Scholar]
- 20.Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, Program NCS, Black BL, Visel A, Pennacchio LA, Collins FS, National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program A and Authors NCSP. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. 2013;110:17921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheung KL, Zhang F, Jaganathan A, Sharma R, Zhang Q, Konuma T, Shen T, Lee JY, Ren C, Chen CH, Lu G, Olson MR, Zhang W, Kaplan MH, Littman DR, Walsh MJ, Xiong H, Zeng L and Zhou MM. Distinct Roles of Brd2 and Brd4 in Potentiating the Transcriptional Program for Th17 Cell Differentiation. Mol Cell. 2017;65:1068–1080 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinz S, Unser S and Rascle A. Signal transducer and activator of transcription STAT5 is recruited to c-Myc super-enhancer. BMC molecular biology. 2016;17:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murakami S, Li R, Nagari A, Chae M, Camacho CV and Kraus WL. Distinct Roles for BET Family Members in Estrogen Receptor alpha Enhancer Function and Gene Regulation in Breast Cancer Cells. Molecular cancer research : MCR. 2019;17:2356–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devaiah BN, Case-Borden C, Gegonne A, Hsu CH, Chen Q, Meerzaman D, Dey A, Ozato K and Singer DS. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nature structural & molecular biology. 2016;23:540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roe JS, Mercan F, Rivera K, Pappin DJ and Vakoc CR. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol Cell. 2015;58:1028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donati B, Lorenzini E and Ciarrocchi A. BRD4 and Cancer: going beyond transcriptional regulation. Mol Cancer. 2018;17:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Dulak AM, Hattersley MM, Willis BS, Nikkila J, Wang A, Lau A, Reimer C, Zinda M, Fawell SE, Mills GB and Chen H. BRD4 facilitates replication stress-induced DNA damage response. Oncogene. 2018;37:3763–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bisgrove DA, Mahmoudi T, Henklein P and Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci U S A. 2007;104:13690–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroder S, Cho S, Zeng L, Zhang Q, Kaehlcke K, Mak L, Lau J, Bisgrove D, Schnolzer M, Verdin E, Zhou MM and Ott M. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. The Journal of biological chemistry. 2012;287:1090–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murakami K, Tsai KL, Kalisman N, Bushnell DA, Asturias FJ and Kornberg RD. Structure of an RNA polymerase II preinitiation complex. Proc Natl Acad Sci U S A. 2015;112:13543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Core L and Adelman K. Promoter-proximal pausing of RNA polymerase II: a nexus of gene regulation. Genes & development. 2019;33:960–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, Hasegawa J and Handa H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell. 1999;97:41–51. [DOI] [PubMed] [Google Scholar]
- 33.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN and Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–34. [DOI] [PubMed] [Google Scholar]
- 34.Chen FX, Smith ER and Shilatifard A. Born to run: control of transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. 2018;19:464–478. [DOI] [PubMed] [Google Scholar]
- 35.Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, Ozato K, Sims RJ 3rd and Singer DS. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci U S A. 2012;109:6927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, Roberts JM, Zeid R, Scott TG, Paulk J, Lachance K, Olson CM, Dastjerdi S, Bauer S, Lin CY, Gray NS, Kelliher MA, Churchman LS and Bradner JE. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell. 2017;67:5–18 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW and Howley PM. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Molecular and cellular biology. 2011;31:2641–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang J, Aggarwal A and Rosenfeld MG. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell. 2013;155:1581–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Donner AJ, Ebmeier CC, Taatjes DJ and Espinosa JM. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nature structural & molecular biology. 2010;17:194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang YW, Veschambre P, Erdjument-Bromage H, Tempst P, Conaway JW, Conaway RC and Kornberg RD. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc Natl Acad Sci U S A. 1998;95:8538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI and Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Micco R, Fontanals-Cirera B, Low V, Ntziachristos P, Yuen SK, Lovell CD, Dolgalev I, Yonekubo Y, Zhang G, Rusinova E, Gerona-Navarro G, Canamero M, Ohlmeyer M, Aifantis I, Zhou MM, Tsirigos A and Hernando E. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell reports. 2014;9:234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian B, Liu Z, Yang J, Sun H, Zhao Y, Wakamiya M, Chen H, Rytting E, Zhou J and Brasier AR. Selective Antagonists of the Bronchiolar Epithelial NF-kappaB-Bromodomain-Containing Protein 4 Pathway in Viral-Induced Airway Inflammation. Cell reports. 2018;23:1138–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uppal S, Gegonne A, Chen Q, Thompson PS, Cheng D, Mu J, Meerzaman D, Misra HS and Singer DS. The Bromodomain Protein 4 Contributes to the Regulation of Alternative Splicing. Cell reports. 2019;29:2450–2460 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu L, Wang Z, Zhang Z, Ren X, Lu X and Ding K. Small-molecule BET inhibitors in clinical and preclinical development and their therapeutic potential. Curr Top Med Chem. 2015;15:776–94. [DOI] [PubMed] [Google Scholar]
- 46.Zhang F and Ma S. Disrupting Acetyl-lysine Interactions: Recent Advance in the Development of BET Inhibitors. Curr Drug Targets. 2018;19:1148–1165. [DOI] [PubMed] [Google Scholar]
- 47.Cochran AG, Conery AR and Sims RJ 3rd. Bromodomains: a new target class for drug development. Nat Rev Drug Discov. 2019;18:609–628. [DOI] [PubMed] [Google Scholar]
- 48.Sachchidanand, Resnick-Silverman L, Yan S, Mutjaba S, Liu WJ, Zeng L, Manfredi JJ and Zhou MM. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chemistry & biology. 2006;13:81–90. [DOI] [PubMed] [Google Scholar]
- 49.Lu T, Lu W and Luo C. A patent review of BRD4 inhibitors (2013–2019). Expert Opin Ther Pat. 2020;30:57–81. [DOI] [PubMed] [Google Scholar]
- 50.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S and Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K and Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJ and Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schooling CM and Zhao JV. How Might Bromodomain and Extra-Terminal (BET) Inhibitors Operate in Cardiovascular Disease? Am J Cardiovasc Drugs. 2019;19:107–111. [DOI] [PubMed] [Google Scholar]
- 54.Pervaiz M, Mishra P and Gunther S. Bromodomain Drug Discovery - the Past, the Present, and the Future. Chem Rec. 2018;18:1808–1817. [DOI] [PubMed] [Google Scholar]
- 55.Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, Diez-Dacal B, Philpott M, Bountra C, Lingard H, Fedorov O, Muller S, Brennan PE, Knapp S and Filippakopoulos P. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc Natl Acad Sci U S A. 2013;110:19754–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolden JE, Tasdemir N, Dow LE, van Es JH, Wilkinson JE, Zhao Z, Clevers H and Lowe SW. Inducible in vivo silencing of Brd4 identifies potential toxicities of sustained BET protein inhibition. Cell reports. 2014;8:1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, Zhang L, Bui MH, Sheppard GS, Wang L, Sehgal V, Lin X, Huang X, Lu X, Uziel T, Hessler P, Lam LT, Bellin RJ, Mehta G, Fidanze S, Pratt JK, Liu D, Hasvold LA, Sun C, Panchal SC, Nicolette JJ, Fossey SL, Park CH, Longenecker K, Bigelow L, Torrent M, Rosenberg SH, Kati WM and Shen Y. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature. 2020;578:306–310. [DOI] [PubMed] [Google Scholar]
- 58.Zhang M, Wang B, Urabe G, Huang Y, Plutzky J, Kent KC and Guo LW. The BD2 domain of BRD4 is a determinant in EndoMT and vein graft neointima formation. Cell Signal. 2019;61:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zengerle M, Chan KH and Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS chemical biology. 2015;10:1770–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun B, Fiskus W, Qian Y, Rajapakshe K, Raina K, Coleman KG, Crew AP, Shen A, Saenz DT, Mill CP, Nowak AJ, Jain N, Zhang L, Wang M, Khoury JD, Coarfa C, Crews CM and Bhalla KN. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia. 2018;32:343–352. [DOI] [PubMed] [Google Scholar]
- 61.Saenz DT, Fiskus W, Qian Y, Manshouri T, Rajapakshe K, Raina K, Coleman KG, Crew AP, Shen A, Mill CP, Sun B, Qiu P, Kadia TM, Pemmaraju N, DiNardo C, Kim MS, Nowak AJ, Coarfa C, Crews CM, Verstovsek S and Bhalla KN. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia. 2017;31:1951–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang CY, Qin C, Bai L and Wang S. Small-molecule PROTAC degraders of the Bromodomain and Extra Terminal (BET) proteins - A review. Drug discovery today Technologies. 2019;31:43–51. [DOI] [PubMed] [Google Scholar]
- 63.Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, Bair S, Newton G, Lichtman A, Kung A, Yang T, Wang H, Luscinskas FW, Croce K, Bradner JE and Plutzky J. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jahagirdar R, Zhang H, Azhar S, Tobin J, Attwell S, Yu R, Wu J, McLure KG, Hansen HC, Wagner GS, Young PR, Srivastava RA, Wong NC and Johansson J. A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis. 2014;236:91–100. [DOI] [PubMed] [Google Scholar]
- 65.Wade JT. Mapping Transcription Regulatory Networks with ChIP-seq and RNA-seq. Adv Exp Med Biol. 2015;883:119–34. [DOI] [PubMed] [Google Scholar]
- 66.Brown JD, Feldman ZB, Doherty SP, Reyes JM, Rahl PB, Lin CY, Sheng Q, Duan Q, Federation AJ, Kung AL, Haldar SM, Young RA, Plutzky J and Bradner JE. BET bromodomain proteins regulate enhancer function during adipogenesis. Proc Natl Acad Sci U S A. 2018;115:2144–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ledaki I, McIntyre A, Wigfield S, Buffa F, McGowan S, Baban D, Li JL and Harris AL. Carbonic anhydrase IX induction defines a heterogeneous cancer cell response to hypoxia and mediates stem cell-like properties and sensitivity to HDAC inhibition. Oncotarget. 2015;6:19413–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bid HK and Houghton PJ. Targeting angiogenesis in childhood sarcomas. Sarcoma. 2011;2011:601514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.da Motta LL, Ledaki I, Purshouse K, Haider S, De Bastiani MA, Baban D, Morotti M, Steers G, Wigfield S, Bridges E, Li JL, Knapp S, Ebner D, Klamt F, Harris AL and McIntyre A. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene. 2017;36:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang M, Qiu Q, Xiao Y, Zeng S, Zhan M, Shi M, Zou Y, Ye Y, Liang L, Yang X and Xu H. BET Bromodomain Suppression Inhibits VEGF-induced Angiogenesis and Vascular Permeability by Blocking VEGFR2-mediated Activation of PAK1 and eNOS. Sci Rep. 2016;6:23770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen J, Fu Y, Day DS, Sun Y, Wang S, Liang X, Gu F, Zhang F, Stevens SM, Zhou P, Li K, Zhang Y, Lin RZ, Smith LEH, Zhang J, Sun K, Melero-Martin JM, Han Z, Park PJ, Zhang B and Pu WT. VEGF amplifies transcription through ETS1 acetylation to enable angiogenesis. Nat Commun. 2017;8:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bennett MR, Sinha S and Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gomez D, Swiatlowska P and Owens GK. Epigenetic Control of Smooth Muscle Cell Identity and Lineage Memory. Arterioscler Thromb Vasc Biol. 2015;35:2508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bradner JE, Hnisz D and Young RA. Transcriptional Addiction in Cancer. Cell. 2017;168:629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA and Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang B, Zhang M, Takayama T, Shi X, Roenneburg DA, Kent KC and Guo LW. BET Bromodomain Blockade Mitigates Intimal Hyperplasia in Rat Carotid Arteries. EBioMedicine. 2015;2:1650–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang B, Chen G, Urabe G, Xie R, Wang Y, Shi X, Guo LW, Gong S and Kent KC. A paradigm of endothelium-protective and stent-free anti-restenotic therapy using biomimetic nanoclusters. Biomaterials. 2018;178:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Inoue T, Croce K, Morooka T, Sakuma M, Node K and Simon DI. Vascular inflammation and repair: implications for re-endothelialization, restenosis, and stent thrombosis. JACC Cardiovascular interventions. 2011;4:1057–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G and Baker AH. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73:190–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA and Simons M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. 2015;125:4514–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cooley BC, Nevado J, Mellad J, Yang D, Hilaire CS, Negro A, Fang F, Chen G, San H, Walts AD, Schwartzbeck RL, Taylor B, Lanzer JD, Wragg A, Elagha A, Beltran LE, Berry C, Feil R, Virmani R, Ladich E, Kovacic JC and Boehm M. TGF-beta signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med. 2014;6:227ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Song S, Liu L, Yu Y, Zhang R, Li Y, Cao W, Xiao Y, Fang G, Li Z, Wang X, Wang Q, Zhao X, Chen L, Wang Y and Wang Q. Inhibition of BRD4 attenuates transverse aortic constriction- and TGF-beta-induced endothelial-mesenchymal transition and cardiac fibrosis. J Mol Cell Cardiol. 2019;127:83–96. [DOI] [PubMed] [Google Scholar]
- 83.Ostriker AC and Martin KA. Bromodomain Blockade for Intimal Hyperplasia - A Good BET? EBioMedicine. 2015;2:1574–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meloche J, Potus F, Vaillancourt M, Bourgeois A, Johnson I, Deschamps L, Chabot S, Ruffenach G, Henry S, Breuils-Bonnet S, Tremblay E, Nadeau V, Lambert C, Paradis R, Provencher S and Bonnet S. Bromodomain-Containing Protein 4: The Epigenetic Origin of Pulmonary Arterial Hypertension. Circ Res. 2015;117:525–35. [DOI] [PubMed] [Google Scholar]
- 85.Meloche J, Lampron MC, Nadeau V, Maltais M, Potus F, Lambert C, Tremblay E, Vitry G, Breuils-Bonnet S, Boucherat O, Charbonneau E, Provencher S, Paulin R and Bonnet S. Implication of Inflammation and Epigenetic Readers in Coronary Artery Remodeling in Patients With Pulmonary Arterial Hypertension. Arterioscler Thromb Vasc Biol. 2017;37:1513–1523. [DOI] [PubMed] [Google Scholar]
- 86.Dutta P, Gomez D and Gladwin MT. Do BRD(4)S of a Feather Flock Together? How an Inflammation-Driven Epigenetic Regulator May Link Pulmonary Hypertension and Coronary Artery Disease. Arterioscler Thromb Vasc Biol. 2017;37:1428–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dai Z and Zhao YY. BET in Pulmonary Arterial Hypertension: Exploration of BET Inhibitors to Reverse Vascular Remodeling. Am J Respir Crit Care Med. 2019;200:806–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Siddique MAH, Satoh K, Kurosawa R, Kikuchi N, Elias-Al-Mamun M, Omura J, Satoh T, Nogi M, Sunamura S, Miyata S, Ueda H, Tokuyama H and Shimokawa H. Identification of Emetine as a Therapeutic Agent for Pulmonary Arterial Hypertension: Novel Effects of an Old Drug. Arterioscler Thromb Vasc Biol. 2019;39:2367–2385. [DOI] [PubMed] [Google Scholar]
- 89.Perry MM, Durham AL, Austin PJ, Adcock IM and Chung KF. BET bromodomains regulate transforming growth factor-beta-induced proliferation and cytokine release in asthmatic airway smooth muscle. The Journal of biological chemistry. 2015;290:9111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Clifford RL, Patel JK, John AE, Tatler AL, Mazengarb L, Brightling CE and Knox AJ. CXCL8 histone H3 acetylation is dysfunctional in airway smooth muscle in asthma: regulation by BET. Am J Physiol Lung Cell Mol Physiol. 2015;308:L962–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Michaeloudes C, Mercado N, Clarke C, Bhavsar PK, Adcock IM, Barnes PJ and Chung KF. Bromodomain and extraterminal proteins suppress NF-E2-related factor 2-mediated antioxidant gene expression. J Immunol. 2014;192:4913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Duan Q, Mao X, Liao C, Zhou H, Sun Z, Deng X, Hu Q, Qi J, Zhang G, Huang H, Plutzky J and Yang T. Inhibition of BET bromodomain attenuates angiotensin II induced abdominal aortic aneurysm in ApoE(−/−) mice. Int J Cardiol. 2016;223:428–432. [DOI] [PubMed] [Google Scholar]
- 93.Libby P, Ridker PM and Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–25. [DOI] [PubMed] [Google Scholar]
- 94.Andrieu G, Belkina AC and Denis GV. Clinical trials for BET inhibitors run ahead of the science. Drug discovery today Technologies. 2016;19:45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Felisbino MB and McKinsey TA. Epigenetics in Cardiac Fibrosis: Emphasis on Inflammation and Fibroblast Activation. JACC Basic Transl Sci. 2018;3:704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klein K Bromodomain protein inhibition: a novel therapeutic strategy in rheumatic diseases. RMD Open. 2018;4:e000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hishiki K, Akiyama M, Kanegae Y, Ozaki K, Ohta M, Tsuchitani E, Kaito K and Yamada H. NF-kappaB signaling activation via increases in BRD2 and BRD4 confers resistance to the bromodomain inhibitor I-BET151 in U937 cells. Leuk Res. 2018;74:57–63. [DOI] [PubMed] [Google Scholar]
- 98.Chan CH, Fang C, Yarilina A, Prinjha RK, Qiao Y and Ivashkiv LB. BET bromodomain inhibition suppresses transcriptional responses to cytokine-Jak-STAT signaling in a gene-specific manner in human monocytes. Eur J Immunol. 2015;45:287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Joshi S, Singh AR, Liu KX, Pham TV, Zulcic M, Skola D, Chun HB, Glass CK, Morales GA, Garlich JR and Durden DL. SF2523: Dual PI3K/BRD4 Inhibitor Blocks Tumor Immunosuppression and Promotes Adaptive Immune Responses in Cancer. Mol Cancer Ther. 2019;18:1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ha SD, Cho W, DeKoter RP and Kim SO. The transcription factor PU.1 mediates enhancer-promoter looping that is required for IL-1beta eRNA and mRNA transcription in mouse melanoma and macrophage cell lines. The Journal of biological chemistry. 2019;294:17487–17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Das A, Chai JC, Yang CS, Lee YS, Das ND, Jung KH and Chai YG. Dual transcriptome sequencing reveals resistance of TLR4 ligand-activated bone marrow-derived macrophages to inflammation mediated by the BET inhibitor JQ1. Sci Rep. 2015;5:16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mele DA, Salmeron A, Ghosh S, Huang HR, Bryant BM and Lora JM. BET bromodomain inhibition suppresses TH17-mediated pathology. J Exp Med. 2013;210:2181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gegonne A, Chen QR, Dey A, Etzensperger R, Tai X, Singer A, Meerzaman D, Ozato K and Singer DS. Immature CD8 Single-Positive Thymocytes Are a Molecularly Distinct Subpopulation, Selectively Dependent on BRD4 for Their Differentiation. Cell reports. 2018;24:117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gibbons HR, Mi DJ, Farley VM, Esmond T, Kaood MB and Aune TM. Bromodomain inhibitor JQ1 reversibly blocks IFN-gamma production. Sci Rep. 2019;9:10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ceribelli M, Hou ZE, Kelly PN, Huang DW, Wright G, Ganapathi K, Evbuomwan MO, Pittaluga S, Shaffer AL, Marcucci G, Forman SJ, Xiao W, Guha R, Zhang X, Ferrer M, Chaperot L, Plumas J, Jaffe ES, Thomas CJ, Reizis B and Staudt LM. A Druggable TCF4- and BRD4-Dependent Transcriptional Network Sustains Malignancy in Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancer Cell. 2016;30:764–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sun Y, Wang Y, Toubai T, Oravecz-Wilson K, Liu C, Mathewson N, Wu J, Rossi C, Cummings E, Wu D, Wang S and Reddy P. BET bromodomain inhibition suppresses graft-versus-host disease after allogeneic bone marrow transplantation in mice. Blood. 2015;125:2724–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang J, Chen J, Jin H, Lin D, Chen Y, Chen X, Wang B, Hu S, Wu Y, Wu Y, Zhou Y, Tian N, Gao W, Wang X and Zhang X. BRD4 inhibition attenuates inflammatory response in microglia and facilitates recovery after spinal cord injury in rats. J Cell Mol Med. 2019;23:3214–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sanchez-Ventura J, Amo-Aparicio J, Navarro X and Penas C. BET protein inhibition regulates cytokine production and promotes neuroprotection after spinal cord injury. J Neuroinflammation. 2019;16:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Belkina AC, Nikolajczyk BS and Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol. 2013;190:3670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.DeMars KM, Yang C, Castro-Rivera CI and Candelario-Jalil E. Selective degradation of BET proteins with dBET1, a proteolysis-targeting chimera, potently reduces pro-inflammatory responses in lipopolysaccharide-activated microglia. Biochem Biophys Res Commun. 2018;497:410–415. [DOI] [PubMed] [Google Scholar]
- 111.Zhao L, Li J, Fu Y, Zhang M, Wang B, Ouellette J, Shahi PK, Pattnaik BR, Watters JJ, Wong WT and Guo LW. Photoreceptor protection via blockade of BET epigenetic readers in a murine model of inherited retinal degeneration. J Neuroinflammation. 2017;14:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ren W, Wang C, Wang Q, Zhao D, Zhao K, Sun D, Liu X, Han C, Hou J, Li X, Zhang Q, Cao X and Li N. Bromodomain protein Brd3 promotes Ifnb1 transcription via enhancing IRF3/p300 complex formation and recruitment to Ifnb1 promoter in macrophages. Sci Rep. 2017;7:39986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bao Y, Wu X, Chen J, Hu X, Zeng F, Cheng J, Jin H, Lin X and Chen LF. Brd4 modulates the innate immune response through Mnk2-eIF4E pathway-dependent translational control of IkappaBalpha. Proc Natl Acad Sci U S A. 2017;114:E3993–E4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dey A, Yang W, Gegonne A, Nishiyama A, Pan R, Yagi R, Grinberg A, Finkelman FD, Pfeifer K, Zhu J, Singer D, Zhu J and Ozato K. BRD4 directs hematopoietic stem cell development and modulates macrophage inflammatory responses. EMBO J. 2019;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gacias M, Gerona-Navarro G, Plotnikov AN, Zhang G, Zeng L, Kaur J, Moy G, Rusinova E, Rodriguez Y, Matikainen B, Vincek A, Joshua J, Casaccia P and Zhou MM. Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chemistry & biology. 2014;21:841–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cheung K, Lu G, Sharma R, Vincek A, Zhang R, Plotnikov AN, Zhang F, Zhang Q, Ju Y, Hu Y, Zhao L, Han X, Meslamani J, Xu F, Jaganathan A, Shen T, Zhu H, Rusinova E, Zeng L, Zhou J, Yang J, Peng L, Ohlmeyer M, Walsh MJ, Zhang DY, Xiong H and Zhou MM. BET N-terminal bromodomain inhibition selectively blocks Th17 cell differentiation and ameliorates colitis in mice. Proc Natl Acad Sci U S A. 2017;114:2952–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sacta MA, Tharmalingam B, Coppo M, Rollins DA, Deochand DK, Benjamin B, Yu L, Zhang B, Hu X, Li R, Chinenov Y and Rogatsky I. Gene-specific mechanisms direct glucocorticoid-receptor-driven repression of inflammatory response genes in macrophages. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Campbell GR, Bruckman RS, Herns SD, Joshi S, Durden DL and Spector SA. Induction of autophagy by PI3K/MTOR and PI3K/MTOR/BRD4 inhibitors suppresses HIV-1 replication. The Journal of biological chemistry. 2018;293:5808–5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, Beavis PA, Darcy PK, Martin BP, Spencer A, Traunbauer AK, Sadovnik I, Bauer K, Valent P, Bradner JE, Zuber J, Shortt J and Johnstone RW. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell reports. 2017;18:2162–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, Cappola TP, Lemieux M, Plutzky J, Bradner JE and Haldar SM. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154:569–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, Huang Y, Zhang R, Sahadevan A, Lemieux ME, Brown JD, Srivastava D, Bradner JE, McKinsey TA and Haldar SM. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stratton MS, Farina FM and Elia L. Epigenetics and vascular diseases. J Mol Cell Cardiol. 2019;133:148–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bacmeister L, Schwarzl M, Warnke S, Stoffers B, Blankenberg S, Westermann D and Lindner D. Inflammation and fibrosis in murine models of heart failure. Basic Res Cardiol. 2019;114:19. [DOI] [PubMed] [Google Scholar]
- 124.French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, Kutok JL, Toretsky JA, Tadavarthy AK, Kees UR, Fletcher JA and Aster JC. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008;27:2237–42. [DOI] [PubMed] [Google Scholar]
- 125.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L and Sims RJ 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Alqahtani A, Choucair K, Ashraf M, Hammouda DM, Alloghbi A, Khan T, Senzer N and Nemunaitis J. Bromodomain and extra-terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future Sci OA. 2019;5:FSO372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stathis A and Bertoni F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018;8:24–36. [DOI] [PubMed] [Google Scholar]
- 128.Xu Y and Vakoc CR. Targeting Cancer Cells with BET Bromodomain Inhibitors. Cold Spring Harb Perspect Med. 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bechter O and Schoffski P. Make your best BET: The emerging role of BET inhibitor treatment in malignant tumors. Pharmacol Ther. 2020:107479. [DOI] [PubMed]
- 130.Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, Hoffmann T, Boryn LM, Axelsson E, Schweifer N, Tontsch-Grunt U, Dow LE, Gianni D, Pearson M, Valent P, Stark A, Kraut N, Vakoc CR and Zuber J. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525:543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, Giotopoulos G, Lugo D, Jeffrey P, Lee SC, Carpenter C, Gregory R, Ramsay RG, Lane SW, Abdel-Wahab O, Kouzarides T, Johnstone RW, Dawson SJ, Huntly BJ, Prinjha RK, Papenfuss AT and Dawson MA. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525:538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Togel L, Nightingale R, Chueh AC, Jayachandran A, Tran H, Phesse T, Wu R, Sieber OM, Arango D, Dhillon AS, Dawson MA, Diez-Dacal B, Gahman TC, Filippakopoulos P, Shiau AK and Mariadason JM. Dual Targeting of Bromodomain and Extraterminal Domain Proteins, and WNT or MAPK Signaling, Inhibits c-MYC Expression and Proliferation of Colorectal Cancer Cells. Mol Cancer Ther. 2016;15:1217–26. [DOI] [PubMed] [Google Scholar]
- 133.Cary DC, Fujinaga K and Peterlin BM. Molecular mechanisms of HIV latency. J Clin Invest. 2016;126:448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bartholomeeusen K, Xiang Y, Fujinaga K and Peterlin BM. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. The Journal of biological chemistry. 2012;287:36609–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li PC, Planelles V, Bradner JE, Zhou MM, Siliciano RF, Weinberger L, Verdin E and Ott M. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle. 2013;12:452–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li Z, Guo J, Wu Y and Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic acids research. 2013;41:277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Abner E, Stoszko M, Zeng L, Chen HC, Izquierdo-Bouldstridge A, Konuma T, Zorita E, Fanunza E, Zhang Q, Mahmoudi T, Zhou MM, Filion GJ and Jordan A. A New Quinoline BRD4 Inhibitor Targets a Distinct Latent HIV-1 Reservoir for Reactivation from Other “Shock” Drugs. J Virol. 2018;92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bailey D, Jahagirdar R, Gordon A, Hafiane A, Campbell S, Chatur S, Wagner GS, Hansen HC, Chiacchia FS, Johansson J, Krimbou L, Wong NC and Genest J. RVX-208: a small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J Am Coll Cardiol. 2010;55:2580–9. [DOI] [PubMed] [Google Scholar]
- 139.Tsujikawa LM, Fu L, Das S, Halliday C, Rakai BD, Stotz SC, Sarsons CD, Gilham D, Daze E, Wasiak S, Studer D, Rinker KD, Sweeney M, Johansson JO, Wong NCW and Kulikowski E. Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism. Clinical epigenetics. 2019;11:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gosmini R, Nguyen VL, Toum J, Simon C, Brusq JM, Krysa G, Mirguet O, Riou-Eymard AM, Boursier EV, Trottet L, Bamborough P, Clark H, Chung CW, Cutler L, Demont EH, Kaur R, Lewis AJ, Schilling MB, Soden PE, Taylor S, Walker AL, Walker MD, Prinjha RK and Nicodeme E. The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J Med Chem. 2014;57:8111–31. [DOI] [PubMed] [Google Scholar]
- 141.Nicholls SJ, Puri R, Wolski K, Ballantyne CM, Barter PJ, Brewer HB, Kastelein JJ, Hu B, Uno K, Kataoka Y, Herrman JP, Merkely B, Borgman M and Nissen SE. Effect of the BET Protein Inhibitor, RVX-208, on Progression of Coronary Atherosclerosis: Results of the Phase 2b, Randomized, Double-Blind, Multicenter, ASSURE Trial. Am J Cardiovasc Drugs. 2016;16:55–65. [DOI] [PubMed] [Google Scholar]
- 142.Nicholls SJ, Gordon A, Johansson J, Wolski K, Ballantyne CM, Kastelein JJ, Taylor A, Borgman M and Nissen SE. Efficacy and safety of a novel oral inducer of apolipoprotein a-I synthesis in statin-treated patients with stable coronary artery disease a randomized controlled trial. J Am Coll Cardiol. 2011;57:1111–9. [DOI] [PubMed] [Google Scholar]
- 143.Ray KK, Nicholls SJ, Ginsberg HD, Johansson JO, Kalantar-Zadeh K, Kulikowski E, Toth PP, Wong N, Cummings JL, Sweeney M and Schwartz GG. Effect of selective BET protein inhibitor apabetalone on cardiovascular outcomes in patients with acute coronary syndrome and diabetes: Rationale, design, and baseline characteristics of the BETonMACE trial. Am Heart J. 2019;217:72–83. [DOI] [PubMed] [Google Scholar]
- 144.Ray KK. Effect of BET Protein Inhibition With Apabetalone on Cardiovascular Outcomes in Patients With Acute Coronary Syndrome and Diabetes - BETonMACE. 2019. [DOI] [PubMed]
- 145.Dharam J Kumbhani M. Effect of BET Protein Inhibition With Apabetalone on Cardiovascular Outcomes in Patients With Acute Coronary Syndrome and Diabetes - BETonMACE. American College of Cardiology. 2019;2020. [Google Scholar]
- 146.Benz EJ Jr. Breakthrough Therapy Designation for New Drugs. JAMA. 2018;320:2042. [DOI] [PubMed] [Google Scholar]