

Abstract
Coordination compounds of the lanthanide ions (Ln3+) have important applications in medicine due to their photophysical, magnetic, and nuclear properties. To effectively use the Ln3+ ions for these applications, chelators that stably bind them in vivo are required to prevent toxic side effects that arise from localization of these ions in off-target tissue. In this study, two new picolinate-containing chelators, a heptadentate ligand OxyMepa and a nonadentate ligand Oxyaapa, were prepared, and their coordination chemistries with Ln3+ ions were thoroughly investigated to evaluate their suitability for use in medicine. Protonation constants of these chelators and stability constants for their Ln3+ complexes were evaluated. Both ligands exhibit a thermodynamic preference for small Ln3+ ions. The log KLuL = 12.21 and 21.49 for OxyMepa and Oxyaapa, respectively, indicating that the nonadentate Oxyaapa forms complexes of significantly higher stability than the heptadentate OxyMepa. X-ray crystal structures of the Lu3+ complexes were obtained, revealing that Oxyaapa saturates the coordination sphere of Lu3+, whereas OxyMepa leaves an additional open coordination site for a bound water ligand. Solution structural studies carried out with NMR spectroscopy revealed the presence of two possible conformations for these ligands upon Ln3+ binding. Density functional theory (DFT) calculations were applied to probe the geometries and energies of these conformations. Energy differences obtained by DFT are small, but consistent with experimental data. The photophysical properties of the Eu3+ and Tb3+ complexes were characterized, revealing modest photoluminescent quantum yields of < 2%. Luminescence lifetime measurements were carried out in H2O and D2O, showing that the Eu3+ and Tb3+ complexes of OxyMepa have two inner-sphere water ligands, whereas the Eu3+ and Tb3+ complexes of Oxyaapa have zero. Lastly, variable-temperature 17O NMR spectroscopy was performed for the Gd-OxyMepa complex to determine its water exchange rate constant of = (2.8 ± 0.1) × 106 s−1. Collectively, this comprehensive characterization of these Ln3+ chelators provides valuable insight for their potential use in medicine and garners additional understanding on ligand design strategies.
Graphical Abstract
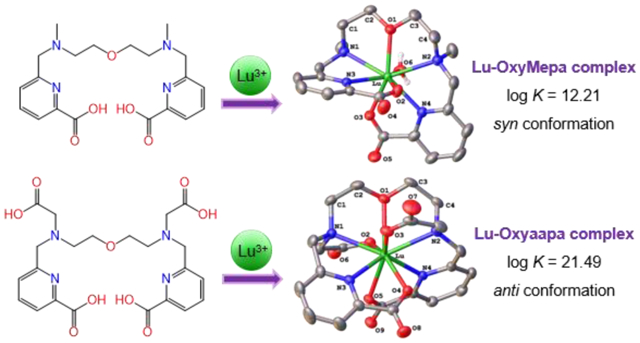
Two new acyclic picolinate-based chelators, OxyMepa and Oxyaapa, are reported. Their coordination chemistries with lanthanide ions (Ln3+) were investigated in detail, revealing that these ligands undergo distinct conformational changes upon binding large and small Ln3+ ions. Furthermore, Oxyaapa forms a Lu3+ complex with high thermodynamic affinity (log KLnL = 21.49) and kinetic inertness, indicating that it would be promising for use in 177Lu3+ radiotherapy.
Introduction
The unique electronic and magnetic properties of the lanthanide ions have driven the use of their coordination compounds in medicine as radiopharmaceutical, photoluminescent imaging, and magnetic resonance imaging (MRI) contrast agents.1–5 To optimally employ the lanthanides for these applications, appropriate chelating agents that stabilize these labile and hydrolysable ions in vivo are needed. Furthermore, special considerations should be made depending on the specific application of these complexes. For example, the hydration number (q) of the central lanthanide ion should be adjusted for MRI applications,6,7 and the ligands must have appropriate electronic excited state energies to achieve useful luminescent applications.3,8
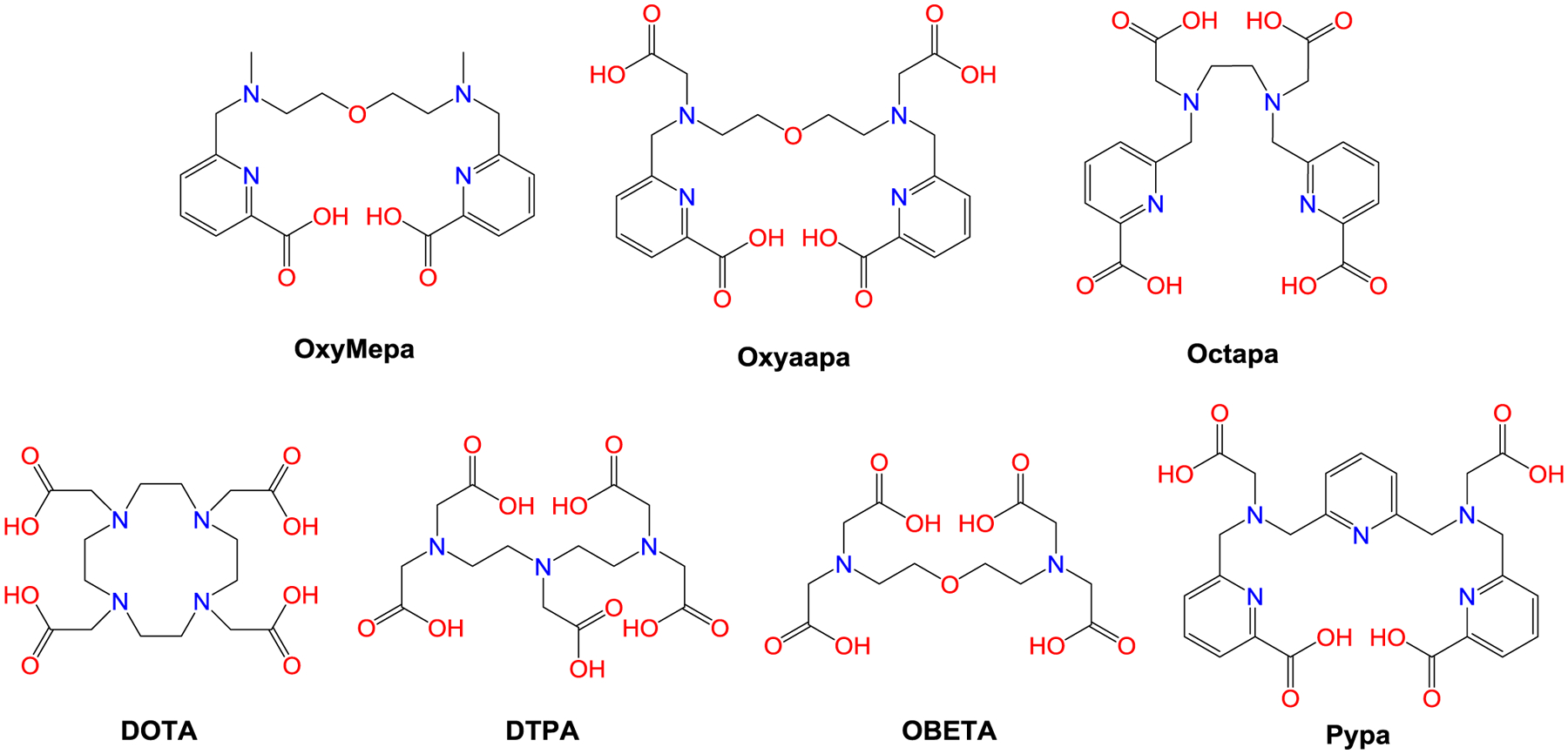
Among the most commonly used chelators for the lanthanide ions (Ln3+) are 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA),9 a macrocycle, and diethylenetriaminepentaacetic acid (DTPA), an acyclic compound (Chart 1). Both chelators form thermodynamically stable complexes with the lanthanides, showing an enhanced affinity for the heavy, smaller ions in the series.10 From a kinetic perspective, macrocyclic ligands such as DOTA generally require significantly longer equilibration times to bind lanthanides but form significantly more inert complexes in comparison to acyclic ligands like DTPA. An extreme example of this situation is given by macrocycles containing a cross-bridged cyclen-based core with pendent picolinate donors for which insertion of lanthanides requires refluxing in n-butanol for 4 d; once inserted, the lanthanides are effectively impossible to displace even with 2 M HCl at 110 °C.11,12 Thus, a potential challenge in the field arises from the fact that desirable enhanced kinetic inertness of macrocycles is often accompanied by slow complexation kinetics in comparison to acyclic chelators.5,13–15 These slow binding kinetics may be especially problematic in the field of nuclear medicine where work with short-lived radionuclides and temperature-sensitive antibodies is often required.
Chart 1.
Chemical structures of the chelators discussed in this work.
Therefore, it is desirable to develop acyclic chelators that retain rapid binding kinetics but exhibit kinetic inertness that is comparable to macrocycles. In this regard, a number of ligands that meet these criteria have been reported within the last 20 years. The ligand Octapa, first reported in 2004,16 is an acyclic octadentate chelator assembled from two picolinate and two acetate donor arms conjugated to an ethylenediamine core (Chart 1). Investigations of this ligand in the context of nuclear medicine revealed it to be an excellent chelator for the rare earth radionuclides 177Lu3+ and 90Y3+ with respect to both its radiolabeling kinetics and stability.17,18 Thus, Octapa is key example of an acyclic ligand that exhibits stability that is comparable to macrocycles. Building upon the success of Octapa, other acyclic chelators bearing picolinate donors have been synthesized and evaluated for use with therapeutically and diagnostically relevant radiolanthanides.19–24 Among these candidates, the nonadentate ligand Pypa (Chart 1) has exhibited some of the most promising properties for radiolabeling 177Lu3+ and 44Sc3+, demonstrating that this structure is highly effective for lanthanide chelation.24,25
The lanthanide ions are hard acids that prefer interacting with anionic bases with high electronegativity such as oxygen donors.26,27 Given the previously reported success with Octapa and Pypa, we hypothesized that the incorporation of another oxygen would give rise to ligands with enhanced lanthanide-binding properties. This strategy, for example, was successful in the design of the ligand OBETA (Chart 1).28,29 In this study, we report our efforts to explore the new acyclic picolinate-containing ligands, OxyMepa and Oxyaapa (Chart 1), which have an additional ethereal oxygen donor to provide enhanced stabilization of the hard lanthanide ions. OxyMepa is heptadentate ligand, which upon the addition of two acetate donors gives Oxyaapa, a nonadentate ligand that we hypothesized would have higher affinity and greater kinetic inertness for the lanthanides. The coordination chemistry of these ligands with the lanthanides was investigated in detail, using analytical titrations, X-ray crystallography, spectroscopy, and computational methods, to reveal the effect of the additional ethereal oxygen donor and the lanthanide-chelation properties of this class of ligands.
Experimental.
Materials and Methods.
Starting material 2,2’-oxybis(N-tosylethan-1-amine) (1) and 6-chloromethylpyridine-2-carboxylic methyl ester were prepared as described in the literature.30,31 Deionized H2O (≥ 18 MΩ·cm) was obtained from an Elga Purelab Flex 2 water purification system. Organic solvents were of ACS grade or higher. All other reagents were purchased from commercial sources and used without further purification.
High-performance liquid chromatography (HPLC) consisted of a CBM-20A communications bus module, an LC-20AP (preparative) or LC-20AT (analytical) pump, and an SPD-20AV UV-Vis detector monitoring at 270 nm (Shimadzu, Japan). Semipreparative purification was performed using an Epic Polar preparative column, 120 Å, 10 μm, 25 cm × 20 mm (ES Industries, West Berlin, NJ) at a flow rate of 14 mL/min. Analytical chromatography was carried out using an Ultra Aqueous C18 column, 100 Å, 5 μm, 250 mm × 4.6 mm (Restek, Bellefonte, PA) at a flow rate of 1.0 mL/min. All HPLC methods employed use a binary mobile phase (H2O and MeOH, 0.1% TFA added for both solvents) gradient. Preparative HPLC runs were carried out with the following methods. Method P1: 0–5 min, 90% H2O/MeOH; 5–25 min, 90% → 0% H2O/MeOH; Method P2: 0–10 min, 90% H2O/MeOH; 10–30 min, 90% → 0% H2O/MeOH. All analytical HPLC runs were carried out with the same method: 0–5 min, 90% H2O/MeOH; 5–25 min, 90% → 0% H2O/MeOH.
All the 1D 1H (except for the La3+ and Lu3+ complexes) and 13C{1H} NMR spectra were acquired on a 500 MHz Bruker AVIII HD spectrometer equipped with a 5 mm, broadband Prodigy cryoprobe operating at 499.76 and 125.68 MHz for 1H and 13C observations, respectively. The 1D 1H and 2D NMR spectra of the La and Lu complexes were acquired on a 600 MHz Varian INOVA spectrometer equipped with a 5-mm HCN inverse probehead operating at 599.50 MHz for 1H observation and 150.76 MHz for 13C decoupling. 2D experiments were performed using the standard Varian pulse sequences HSQCAD, gHMBCAD, gCOSY and ROESYAD as provided in VnmrJ (ver. 3.2). All NMR experiments were carried out at 25 °C unless noted otherwise. 1H NMR spectra of La3+ and Lu3+ complexes were referenced to residual internal HDO solvent peak at 4.77 ppm (25 °C), which was calculated according to the literature equation that accounts for the temperature dependence of the HDO chemical shift.32 The 13C{1H} and 2D NMR spectra were referenced indirectly to the corresponding 1H spectra using the “absolute reference” function provided in MestReNova (Mestrelab Research S.L.). In reporting the NMR peak splitting, the following abbreviations are used: s, singlet; d, doublet; t, triplet; m, multiplet; br, broad; app, apparent. High-resolution mass spectra (HRMS) were acquired on a Thermo Scientific Exactive Orbitrap mass spectrometer with a heated electrospray (HESI) ion source.
Synthesis.
Synthesis of 2,2′-oxybis(N-tosyl-N-methylethan-1-amine) (2).
Methyl p-toluenesulfonate (2.96 g, 15.9 mmol, 4.0 eq) and NaOH (0.80 g, 20.0 mmol, 5.0 eq) were added to a solution of 1 (1.64 g, 3.98 mmol) in MeCN (25 mL). This mixture was heated at 75 °C for 20 h, after which the MeCN was removed under reduced pressure. The residue was washed by CH2Cl2 (3 × 15 mL) followed by filtration to give a light-yellow filtrate. This filtrate was concentrated under reduced pressure to dryness to give a yellow oily residue, which was then purified by column chromatography (silica gel, 1:1 hexane/ethyl acetate) to yield the product as a white solid (1.63 g, 93%). 1H NMR (500 MHz, CDCl3) δ: 7.66 (d, J = 8.1 Hz, 4H, Ph), 7.31 (d, J = 8.1 Hz, 4H, Ph), 3.59 (t, J = 5.6 Hz, 4H, –OCH2–), 3.17 (t, J = 5.6 Hz, 4H, –NCH2–), 2.80 (s, 6H, –NCH3), 2.42 (s, 6H, –PhCH3). 13C{1H} NMR (126 MHz, CDCl3) δ: 143.4, 134.7, 129.7, 127.3, 69.8, 49.8, 36.3, 21.5. ESI-HRMS m/z: 463.1341; calcd for [C20H28N2O5S2 + Na]+: 463.1332.
Synthesis of 2,2’-oxybis(N-methylethan-1-amine) (3).
Compound 2 (1.60 g, 3.63 mmol) and phenol (2.39 g, 25.4 mmol, 7.0 eq) were dissolved in 33% HBr/AcOH (75 mL). The reaction mixture was then heated at 70 °C for 70 h, over which time the solution color gradually turned deep brown. The solvent was then concentrated to a volume of 10 mL under reduced pressure, and 120 mL of acetone was slowly added with stirring, inducing the formation of a precipitate. The precipitate was collected by filtration, washed with acetone, and then dried under vacuum overnight to give the 2HBr salt of 3 as a white solid (0.81 g, 76%). 1H NMR (500 MHz, D2O, pD = 9–10 by NaOD) δ: 3.76 (t, J = 5.0 Hz, 4H, –OCH2–), 3.14 (t, J = 5.0 Hz, 4H, –NCH2–), 2.64 (s, 6H, –CH3). 13C{1H} NMR (126 MHz, D2O, pD = 9–10 by NaOD) δ: 66.9, 49.2, 33.7. ESI-HRMS m/z: 133.1340; calcd for [C6H16N2O + H]+: 133.1335. Elemental analysis: calcd % for C6H16N2O·2HBr: C 24.51, H 6.17, N 9.53; found %: C 24.65, H 5.93, N 9.43.
Synthesis of dimethyl 6,6’-(((oxybis(ethane-2,1-diyl))bis(methylazanediyl))bis(methylene))dipicolinate (4).
Compound 3·2HBr (0.47 g, 1.60 mmol) was suspended in a solution of dry MeCN (30 mL) containing 6-chloromethylpyridine-2-carboxylic methyl ester (0.65 g, 3.50 mmol, 2.2 eq). N,N-Diisopropylethylamine (1250 μL, 7.18 mmol, 4.5 eq) was slowly added with stirring, and the suspension dissolved within 5 min. This solution was then heated at 35 °C with a drying tube equipped on the reaction flask. After 48 h, the mixture was concentrated to dryness under reduced pressure to afford a pale-yellow solid. The crude material was then dissolved in H2O containing 10% MeOH and 0.1% TFA (2.5 mL). Following filtration, this solution was injected into the preparative HPLC system and subjected to the gradient elution Method P1. Pure fractions were combined, concentrated under reduced pressure, and lyophilized to yield 4 as a colorless oil (0.65 g). Based on the peak integrations in the 1H NMR and 19F NMR spectra of this compound relative to an internal standard of fluorobenzene, 3 was estimated to be a 3TFA salt. The entirety of this material was used in the next step. 1H NMR (500 MHz, CD3OD) δ: 8.14 (d, J = 7.7 Hz, 2H, py), 8.09 (t, J = 7.7 Hz, 2H, py), 7.72 (d, J = 7.7 Hz, 2H, py), 4.70 (br s, 4H, –NCH2py–), 4.01 (t, 4H, J = 5.0 Hz, –NCH2CH2O–), 3.97 (s, 6H, –COOCH3), 3.61 (app s, 4H, –NCH2CH2O–), 3.02 (s, 6H, –NCH3). 13C{1H} NMR (126 MHz, CD3OD) δ: 166.6, 152.2, 148.7, 140.8, 128.8, 126.6, 66.1, 60.6, 57.1, 53.7, 42.4. ESI-HRMS m/z: 431.2298; calcd for [C22H30N4O5 + H]+: 431.2289. Analytical HPLC: tR = 16.98 min.
Synthesis of 6,6’-(((oxybis(ethane-2,1-diyl))bis(methylazanediyl))bis(methylene))dipicolinic acid (OxyMepa).
Compound 4 (0.65 g) was dissolved in 4 M HCl (7.5 mL) and heated at 80 °C for 18 h. The solution was then concentrated under reduced pressure to obtain a yellow oily residue. To ensure the complete removal of the TFA coming from 3, 4 M HCl (6 mL) was added to the oily residue, and the resulting solution was concentrated to dryness under reduced pressure. The residue was then dissolved in H2O (3 mL) and lyophilized to yield the 4HCl·3.5H2O salt of OxyMepa as a pale-yellow solid (0.46 g, 47% from 3). 1H NMR (500 MHz, D2O, pD ≈ 8 by NaOD) δ: 7.90 (t, J = 7.7 Hz, 2H, py), 7.84 (d, J = 7.7 Hz, 2H, py), 7.47 (d, J = 7.7 Hz, 2H, py), 4.46 (s, 4H, –NCH2py–), 3.80 (app s, 4H, –NCH2CH2O–), 3.41 (app s, 4H, –NCH2CH2O–), 2.84 (s, 6H, –CH3). 13C{1H} NMR (126 MHz, D2O, pD ≈ 8 by NaOD) δ: 172.5, 153.4, 150.2, 139.6, 126.1, 124.3, 65.2, 60.6, 56.3, 42.3. ESI-HRMS m/z: 403.1984; calcd for [C20H26N4O5 + H]+: 403.1976. Analytical HPLC: tR = 14.34 min. Elemental analysis: calcd % for C20H26N4O5·4HCl·3.5H2O: C 39.29, H 6.10, N 9.16; found %: C 39.27, H 6.05, N 9.12.
Synthesis of dimethyl 6,6’-(((oxybis(ethane-2,1-diyl))bis(tosylazanediyl))bis(methylene))dipicolinate (5).
Starting material 1 (1.00 g, 2.42 mmol) and 6-chloromethylpyridine-2-carboxylic methyl ester (0.99 g, 5.33 mmol, 2.2 eq) were dissolved in MeCN (30 mL), and K2CO3 (1.00 g, 7.24 mmol, 3.0 eq) was added to the resulting solution. This mixture was heated at 75 °C, while being monitored by TLC (1:2 hexane/ethyl acetate), and the reaction was found to have reached completion within 6 d. The mixture was then allowed to cool to rt, transferred into a centrifugation tube, and centrifuged. The supernatant was collected by decantation. The remaining pellet, presumed to contain mostly inorganic salts, was rinsed with 10 mL of MeCN and centrifuged again. This supernatant was combined with the previous, and they were concentrated under reduced pressure to obtain an orange oil. This oil was purified by column chromatography (silica gel, 1:2 n-hexane/ethyl acetate) to yield the product as a white solid (1.48 g, 86%). 1H NMR (500 MHz, CDCl3) δ: 7.99 (d, J = 7.8 Hz, 2H, py), 7.84 (t, J = 7.8 Hz, 2H, py), 7.75 (d, J = 7.8 Hz, 2H, py), 7.68 (m, 4H, Ph), 7.29 (d, J = 8.0 Hz, 4H, Ph), 4.50 (s, 4H, –NCH2py–), 3.97 (s, 6H, –COOCH3), 3.19 (app s, 8H, –NCH2CH2O–), 2.42 (s, 6H, –PhCH3). 13C{1H} NMR (126 MHz, CDCl3) δ: 165.5, 158.3, 147.1, 143.7, 137.8, 135.9, 129.8, 127.2, 125.5, 124.0, 68.9, 55.0, 52.9, 49.0, 21.5. ESI-HRMS m/z: 733.1951; calcd for [C34H38N4O9S2 + Na]+: 733.1972.
Synthesis of 6,6’-(((oxybis(ethane-2,1-diyl))bis(azanediyl))bis(methylene))dipicolinic acid (6).
Compound 5 (0.71 g, 1.00 mmol) and phenol (0.66 g, 7.01 mmol, 7.0 eq) were dissolved in 33% HBr/AcOH (10 mL). This mixture was heated at 70 °C for 72 h, over which time the solution color turned deep brown and a precipitate gradually formed. The reaction mixture was then allowed to cool to rt and transferred into a centrifuge tube. After centrifugation, the supernatant was removed by decantation. To the remaining solid pellet was added H2O (7 mL), giving an emulsion that was filtered through a 0.2-μm polytetrafluoroethylene (PTFE) syringe membrane to obtain a light-yellow filtrate. The majority of the species within this filtrate is the desired product 6, but unhydrolyzed methyl esters of 3 and other impurities were also present. Concentrated HCl (7 mL) was added to this filtrate, giving a final HCl concentration of 6 M, and the solution was heated at 90 °C for 2.5 h. The solution was then concentrated to 2 mL under reduced pressure, and 40 mL of acetone was slowly added with stirring, causing precipitation. The precipitate was collected by filtration, washed with acetone, and dried under vacuum overnight to give the 3HCl salt of 6 as a white solid (0.31 g, 64%). 1H NMR (500 MHz, D2O, pD ≈ 9 by NaOD) δ: 7.82 (t, J = 7.7 Hz, 2H, py), 7.73 (d, J = 7.7 Hz, 2H, py), 7.41 (d, J = 7.7 Hz, 2H, py), 4.01 (s, 4H, –NCH2py–), 3.71 (t, J = 5.2 Hz, 4H, –NCH2CH2O–), 2.98 (t, J = 5.2 Hz, 4H, –NCH2CH2O–). 13C{1H} NMR (126 MHz, D2O, pD ≈ 9 by NaOD) δ: 173.4, 156.2, 153.5, 139.1, 125.1, 123.2, 68.8, 53.1, 47.7. ESI-HRMS m/z: 375.1654; calcd for [C18H22N4O5 + H]+: 375.1663. Analytical HPLC: tR = 13.06 min. Elemental analysis: calcd % for C18H22N4O5·3HCl: C 44.69, H 5.21, N 11.58; found %: C 44.33, H 4.92, N 11.38.
Synthesis of dimethyl 6,6’-(((oxybis(ethane-2,1-diyl))bis(azanediyl))bis(methylene))dipicolinate (7).
Compound 6·3HCl (0.41 g, 0.85 mmol) was suspended in MeOH (15 mL), and SOCl2 (200 μL) was added with vigorous stirring. This mixture was heated at 50 °C for 2.5 h, over which period dissolution of the suspension occurred. The MeOH was then removed under reduced pressure. Saturated NaHCO3 (8 mL) was added dropwise, giving a weakly basic solution. This aqueous solution was subsequently extracted with 8 × 20 mL of CHCl3, and the combined organics were dried over Na2SO4. The CHCl3 was then removed under reduced pressure to give 7 as a pale-yellow oil (0.32 g, 94%). 1H NMR (500 MHz, CDCl3) δ: 7.99 (dd, J = 7.7 Hz, 2H, py), 7.79 (t, J = 7.7 Hz, 2H, py), 7.62 (d, J = 7.7 Hz, 2H, py), 4.07 (s, 4H, –NCH2py–), 3.99 (s, 6H, –CH3), 3.63 (t, J = 5.2 Hz, 4H, –NCH2CH2O–), 2.88 (t, J = 5.2 Hz, 4H, –NCH2CH2O–), 2.59 (br s, –NH– and HDO). 13C{1H} NMR (126 MHz, CDCl3) δ: 165.8, 160.4, 147.5, 137.5, 125.7, 123.6, 70.2, 54.9, 52.9, 48.9. ESI-HRMS m/z: 403.1962; calcd for [C20H26N4O5 + H]+: 403.1976. Analytical HPLC: tR = 16.07 min.
Synthesis of 6,6’-(((oxybis(ethane-2,1-diyl))bis((methylcarboxymethyl)azanediyl))bis(methylene))dipicolinate (8).
Compound 7 (0.35 g, 0.87 mmol) and methyl bromoacetate (173 μL, 1.83 mmol, 2.1 eq) were dissolved in dry MeCN (20 mL). N,N-Diisopropylethylamine (380 μL, 2.18 mmol) was slowly added to this solution with stirring. This solution was then heated at 40 °C with a drying tube equipped on the reaction flask. After 40 h, the mixture was concentrated to dryness under reduced pressure to give a pale-orange solid. H2O containing 10% MeOH and 0.1% TFA (2.5 mL) was added to dissolve this solid. The resulting solution was filtered and then injected onto a preparative HPLC system with a gradient elution following Method P2. Pure fractions were combined, concentrated under reduced pressure, and lyophilized to yield 8 as a colorless oil (0.48 g). Based on the peak integrations in the 1H NMR and 19F NMR spectra of this compound relative to an internal standard of fluorobenzene, 8 was estimated to be a 4TFA salt. The entirety of this material was used in the next step. 1H NMR (500 MHz, CD3OD) δ: 8.15 (d, J = 7.8 Hz, 2H, py), 8.08 (t, J = 7.8 Hz, 2H, py), 7.73 (d, J = 7.8 Hz, 2H, py), 4.81 (s, 4H, –NCH2py–), 4.33 (s, 4H, –NCH2COO–), 3.99 (s, 6H, –pyCOOCH3), 3.94 (t, 4H, J = 4.8 Hz, –NCH2CH2O–), 3.80 (s, 6H, –CH2COOCH3), 3.68 (t, 4H, J = 4.8 Hz, –NCH2CH2O–). 13C{1H} NMR (126 MHz, CD3OD) δ: 168.4, 166.5, 152.8, 148.5, 140.7, 128.7, 126.4, 66.8, 59.5, 56.3, 55.6, 53.6. ESI-HRMS m/z: 547.2383; calcd for [C26H34N4O9 + H]+: 547.2399. Analytical HPLC: tR = 18.20 min.
Synthesis of 6,6’-(((oxybis(ethane-2,1-diyl))bis((carboxymethyl)azanediyl))bis(methylene))dipicolinic acid (Oxyaapa).
Compound 8 (0.48 g) was dissolved in 4 M HCl (7.5 mL) and heated at 80 °C. After 24 h, the solution was then concentrated under reduced pressure to obtain colorless oily residue. To ensure the complete removal of the TFA counterion coming from 8, 4 M HCl (6 mL) was added to dissolve the residue, and the resulting solution was concentrated under reduced pressure. The residue was dissolved in H2O (3 mL) and lyophilized to yield the 3HCl·H2O salt of Oxyaapa as a white solid (0.32 g, 60% from 7). 1H NMR (500 MHz, D2O, pD ≈ 9 by NaOD) δ: 7.83 (m, 4H, py), 7.47 (dd, J = 7.2, 1.6 Hz, 2H, py), 4.05 (s, 4H, –NCH2py–), 3.46 (t, J = 5.3 Hz, 4H, –NCH2CH2O–), 3.37 (s, 4H, –NCH2COO−), 2.94 (app s, 4H, –NCH2CH2O–). 13C{1H} NMR (126 MHz, D2O, pD ≈ 9 by NaOD) δ: 177.1, 173.3, 156.1, 153.4, 139.1, 126.1, 123.4, 67.6, 60.2, 58.8, 54.3. ESI-HRMS m/z: 491.1759; calcd for [C22H26N4O9 + H]+: 491.1773. Analytical HPLC: tR = 13.39 min. Elemental analysis: calcd % for C22H26N4O9·3HCl·H2O: C 42.77, H 5.06, N 9.07; found %: C 43.09, H 5.09, N 9.03.
Potentiometric Titrations.
Potentiometric titrations were carried out using a Metrohm Titrando 888 titrator equipped with a Ross Orion combination electrode (8103BN, ThermoFisher Scientific) and a Metrohm 806 exchange unit with an automatic burette (10 mL). This titration system was controlled by Tiamo (ver. 2.5) software. The titration vessel was fitted into a removable glass cell (≈ 70 mL) and thermostated at 25.0 °C (pKw = 13.78)33 using a Thermomix 1442D circulating water bath. CO2 was excluded from the vessel prior to and during the titrations by an argon flow, which was passed through an aqueous 30 wt% KOH solution. Carbonate-free KOH (0.1 M) was prepared by dissolving KOH pellets (semiconductor grade, 99.99% trace metals basis, Sigma-Aldrich) in freshly boiled water and was standardized by potentiometric titration against potassium hydrogen phthalate. Hydrochloric acid (0.1 M, J.T. Baker) was standardized against Tris (base form). Titration solutions were maintained at a constant ionic strength of 0.1 M with KCl (BioUltra, ≥ 99.5%, Sigma-Aldrich) and were equilibrated for 15 minutes prior to the addition of titrant. The electrode was calibrated before each titration by titrating a solution of standardized HCl with standardized KOH, and the data were analyzed using the program Glee34 (ver. 3.0.21) to obtain the standard electrode potential and slope factor.
Ligand stock solutions were made by dissolving the solid ligand in H2O, and their exact concentrations were determined based on the endpoints of the potentiometric titration curves obtained during the protonation constant determinations. The concentrations determined from titration curves matched the concentrations calculated from the ligand masses, where the MWs of the ligands were estimated from the elemental analysis results. Ln3+ stock solutions were made by dissolving the corresponding LnCl3 hydrate salts (99.9% purity or higher) in standardized HCl (0.1 M). The exact concentrations were determined by complexometric titrations35 with a standardized Na2H2EDTA solution (Alfa Aesar). The complexometric titrations were performed at pH = 5.4 maintained by a HOAc-NaOAc buffer, and the endpoint was indicated by xylenol orange.
Protonation constant (OxyMepa and Oxyaapa) or stability constant (Ln-OxyMepa) determinations were carried out by titrating an acidic solution containing free ligand or both ligand and metal with standardized KOH. For protonation constant determinations, the ligand concentration was 1 mM. For stability constant determinations, both the ligand and metal ion concentrations were 1 mM. The total analyte volumes were 15–20 mL. Equilibration times of 3 min (protonation constant determinations) or 5 min (stability constant determinations) were used before recording the solution pH after the addition of an aliquot of base. The solutions were inspected throughout the titrations for signs of Ln(OH)3 precipitation. Data points of the titration curves were excluded from analysis if any precipitate was observed. These titration data were refined with Hyperquad 201336 software to determine protonation and stability constants. The data points within the pH range of 2.5–11.3 for OxyMepa and 2.0–11.3 for Oxyaapa were used for analysis. At least 6 independent titrations for protonation constant measurements, and 3 titrations for stability constant measurements were carried out for each L and LnL system.
UV-Vis Spectroscopic Titrations.
UV-Vis spectra were recorded on a Cary 8454 UV-Vis spectrometer (Agilent Technologies, Santa Clara, CA) with a 1-cm quartz cuvette. The stability constants of Ln-Oxyaapa were determined by UV-Vis spectroscopic titrations with a batch method. A set of solutions (15–20 samples) containing the same concentration of Ln3+ and Oxyaapa (1:1 or 1.5:1 metal to ligand molar ratio) adjusted to different pH values were prepared. All samples were maintained at low pH (0.7 ≤ pH ≤ 2.4), and the pH was adjusted with standardized HCl (0.1 M or 1 M). Under these conditions, pH cannot be accurately measured with the glass electrode; pH values were calculated directly from the concentration of HCl used in each sample. When 1.0 ≤ pH ≤ 2.4, the ionic strength was fixed at 0.1 M with KCl. When 0.7 ≤ pH < 1.0, the contribution from HCl to the ionic strength was more than 0.1 M, and the ionic strength was dictated by the HCl concentration. The solutions were incubated at 25 °C overnight after which all samples had reached equilibrium. The pH-dependent spectroscopic data were analyzed with HypSpec 201437 software. The absorption band from 240–300 nm was used for analysis and the molar extinction coefficients for all HiL species (i = 0–5) were included in the fitting. Three independent titrations were performed for each LnL system.
DTPA Challenges.
UV–Vis spectra were recorded on a Cary 8454 UV-Vis spectrometer (Agilent Technologies, Santa Clara, CA) with a 1-cm quartz cuvette. A stock solution of DTPA (200 mM) was prepared in H2O, and its pH was adjusted to 7.4 with NMe4OH. The concentrations of OxyMepa, Oxyaapa, and metal stock solutions were determined as described above. The ligand (0.3 μmol) and metal (0.3 μmol) were mixed in a cuvette and diluted to 2850 μL with 0.1 M MOPS buffer (pH = 7.4) to form the complex in situ. After allowing these solutions to equilibrate for 5 min, 150 μL of the DTPA stock solution was added, giving a final DTPA concentration of 10 mM. Upon addition of DTPA, the reaction was monitored immediately by UV–Vis spectroscopy at 22 °C. The absorbance at wavelengths of the most significant spectral changes (282 nm for OxyMepa and 279 for Oxyaapa) were plotted as a function of time. The half-lives for these first order processes were calculated from the slopes of ln (At – A∞) vs t plots, where the slope was obtained by linear regression, t is the reaction time, At and A∞ are the absorbances at time t and infinite time respectively. Three independent replicates were performed for each LnL system.
X-Ray Crystallography.
Single crystals of the Lu3+ complex of OxyMepa were grown via the following procedure. OxyMepa·4HCl·3.5H2O (24.4 mg, 0.040 mmol) and LuCl3·6H2O (17.9 mg, 0.046 mmol) were dissolved in 800 μL of H2O. The pH of this solution was adjusted to 5–6 by the addition of 1 M NaOH (≈ 240 μL) with stirring. After 10 min, solid KPF6 (22.1 mg, 0.120 mmol) was added to this mixture, causing the formation of a white precipitate within several minutes. This suspension was allowed to stir for another 30 min and then transferred to a 2-mL centrifuge tube. It was centrifuged, and the supernatant was removed. The pellet was resuspended in H2O and isolated by centrifugation twice, to wash away water-soluble impurities. The remaining pellet was transferred into a scintillation vial as a suspension in H2O, and then lyophilized to give a white solid (24.6 mg). Slow evaporation of an acetone-methanol-water solution of this material afforded X-ray quality crystals of the Lu-OxyMepa complex. Single crystals of the Lu3+ complex of Oxyaapa were grown via the following procedure. Oxyaapa·3HCl·H2O (34.7 mg, 0.056 mmol) and LuCl3·6H2O (29.6 mg, 0.076 mmol) were dissolved in 200 μL of H2O. The pH of this solution was adjusted to ≈ 4 with 1 M NaOH (≈ 360 μL), added while stirring. It was allowed to stir for 15 min and then filtered. Slow evaporation of the filtrate at rt afforded X-ray quality crystals of the Lu-Oxyaapa complex.
Low-temperature X-ray diffraction data for single crystals of Lu-OxyMepa and Lu-Oxyaapa were collected on a Rigaku XtaLAB Synergy diffractometer coupled to a Rigaku Hypix detector with Mo Kα radiation (λ = 0.71073 Å), from a PhotonJet micro-focus X-ray source at 253 K. The diffraction images were processed and scaled using the CrysAlisPro38 software. The structures were solved through intrinsic phasing using SHELXT39 and refined against F2 on all data by full-matrix least squares with SHELXL40 following established refinement strategies.41 All non-hydrogen atoms were refined anisotropically. All hydrogen atoms bound to carbon were included in the model at geometrically calculated positions and refined using a riding model. Hydrogen atoms bound to oxygen were located in the difference Fourier synthesis and subsequently refined semi-freely with the help of distance restraints. In the structure of Lu-Oxyaapa, the hydrogen atoms on 1.5 oxygen atoms, presumed to be outer-sphere water molecules, were not located and therefore not included in the model. The isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the U(eq) value of the atoms they are linked to (1.5 times for methyl groups). Details of the data quality and a summary of the residual values of the refinements are listed in Supporting Information, SI.
Photophysical Studies.
Photoluminescence quantum yield measurements were carried out using a Varian Eclipse Fluorometer and a Beckman Coulter DU800 UV-Vis instrument. These measurements employed a relative method, in which the quantum yield is compared to a known standard with optically dilute samples (Abs < 0.1), following established protocols.42 [Ru(bpy)3]Cl2 was used as the reference (Φ = 4.0% in air-saturated H2O)43, which was cross-calibrated with 9,10-diphenylanthracene (Φ = 97% in cyclohexane)43 in our laboratory with a < 10% error.
Luminescence lifetime measurements was carried out with the system described below. Excitation of the complexes was provided by pulsing a 265 nm UV LED (M265L3, Thorlabs, Newton, NJ) using a lab-built nanosecond pulsed current source. The LED pulses were 100 ns in duration. The LED was mounted 1 mm from the side of the sample-filled cuvette and phosphorescence was collected at 90 degrees by a large area (25 mm diameter photocathode) Bialkali photomultiplier tube (HC125, Hamamatsu, Bridgewater, NJ) mounted 1 cm from the cuvette. The luminescence of Tb complexes was filtered through a 525 bandpass filter (525/50, Chroma Technology, Bellows Falls, VT) before detection, while for Eu complexes a 600 nm longpass filter was used (HQ600 M, Chroma Technology, Bellows Falls, VT). The time-resolved photon counts were collected using a SR430 multi-channel scaler (Stanford Research, Sunnyvale, CA). Data was transferred to a PC via the SR430 GPIB bus and fit to the standard exponential decay model with MagicPlot Pro (ver. 2.7).
DFT Calculations.
DFT calculations were executed using Gaussian 09.44 The geometries of complexes were optimized using the B3LYP exchange-correlation functional.45,46 The 6–31G(d,p) basis set47,48 was used for light atoms (C, H, N, O), whereas the large-core relativistic effective core potential (LCRECP) and the associated (7s6p5d)/[5s4p3d] basis set49 was assigned to the lanthanide atoms. The LCRECP calculation includes (46 + 4fn) electrons in the core of the lanthanide, leaving the outermost 5s, 5p, 5d, and 6s electrons explicitly dealt with. The use of the this LCRECP for other Ln3+ complexes has been validated by previous studies50–52 and is justified by the fact that 4f orbitals do not have a significant contribution to bonding. The effect of the aqueous environment was considered during the geometry optimizations using the polarizable continuum model (PCM). In particular, the SMD solvation model53 and the parameterized PCM radii of Ln3+ ions52 were applied in our computations. In addition, frequency calculations were carried out on optimized geometries to confirm, via the absence of imaginary frequencies, that these structures are local minima on the potential energy surface. Zero-point energies were also afforded by the frequency calculations.
17O NMR Measurements.
Variable-temperature 17O NMR experiments were carried out on a 600 MHz Varian INOVA spectrometer (14.1 T, 81.4 MHz for 17O) using a broadband 10-mm probe. The thermocouple used for measuring temperature was calibrated using neat ethylene glycol54 with an error of ± 0.2 °C. The transverse relaxation rate (1/T2) was measured by the Carr-Purcell-Meiboom-Gill spin echo technique55 using the standard Varian pulse sequence CPMG as provided in VnmrJ (ver. 3.2). A 10-mm spherical NMR tube was used to contain the solutions. Acidified water (pH = 3.4 HClO4 solution) was used as the reference solution, and 3.0 mM Gd-OxyMepa solution (5% ligand excess, pH = 7.4 maintained by 0.1M MOPS buffer) was taken as the sample solution. Both solutions were enriched to ≈ 1% 17O abundance by 10% H217O (Cambridge Isotope Laboratories). The 1/T2 measurements were carried out over a temperature range of 5–90 °C. Solutions were allowed to thermally equilibrate for at least 15 min prior to data acquisition at each temperature.
Details regarding the equations used for data analysis are given in the SI. The experimental data were fitted globally to obtain all unknown parameters in a single fit. The data fitting procedure was performed using Mathematica (ver. 11.1)56 with the built-in NMinimize function in conjugation with both the Random Search57 (RS) and Nelder-Mead58 (simplex) optimization methods, which are well-known optimization methods that do not require gradient calculations. To increase the robustness of the data fitting process, the Leave-One-Out cross-validation (LOOCV) procedure59 was employed to minimize overfitting. With experimental data measured at 18 different temperatures, the LOOCV procedure ended up with 18 different predictions for each parameter, from which the population means were taken as the final results and the corresponding standard deviations were used to quantify the uncertainties in the parameter values. Because both optimization methods yielded consistent results, only the parameter values obtained using the Nelder-Mead method were presented herein.
Result and Discussion
Synthesis.
The synthetic routes followed to obtain OxyMepa and Oxyaapa are shown in Schemes 1 and 2. The syntheses of both ligands commence with the same starting material 1, tosyl-protected 2,2’-oxybis(ethan-1-amine). The general synthetic strategy for both ligands is similar; the first set of donors was attached to this ether core, followed by deprotection of the tosyl group, and then installation of the second set of donor groups.
Scheme 1.
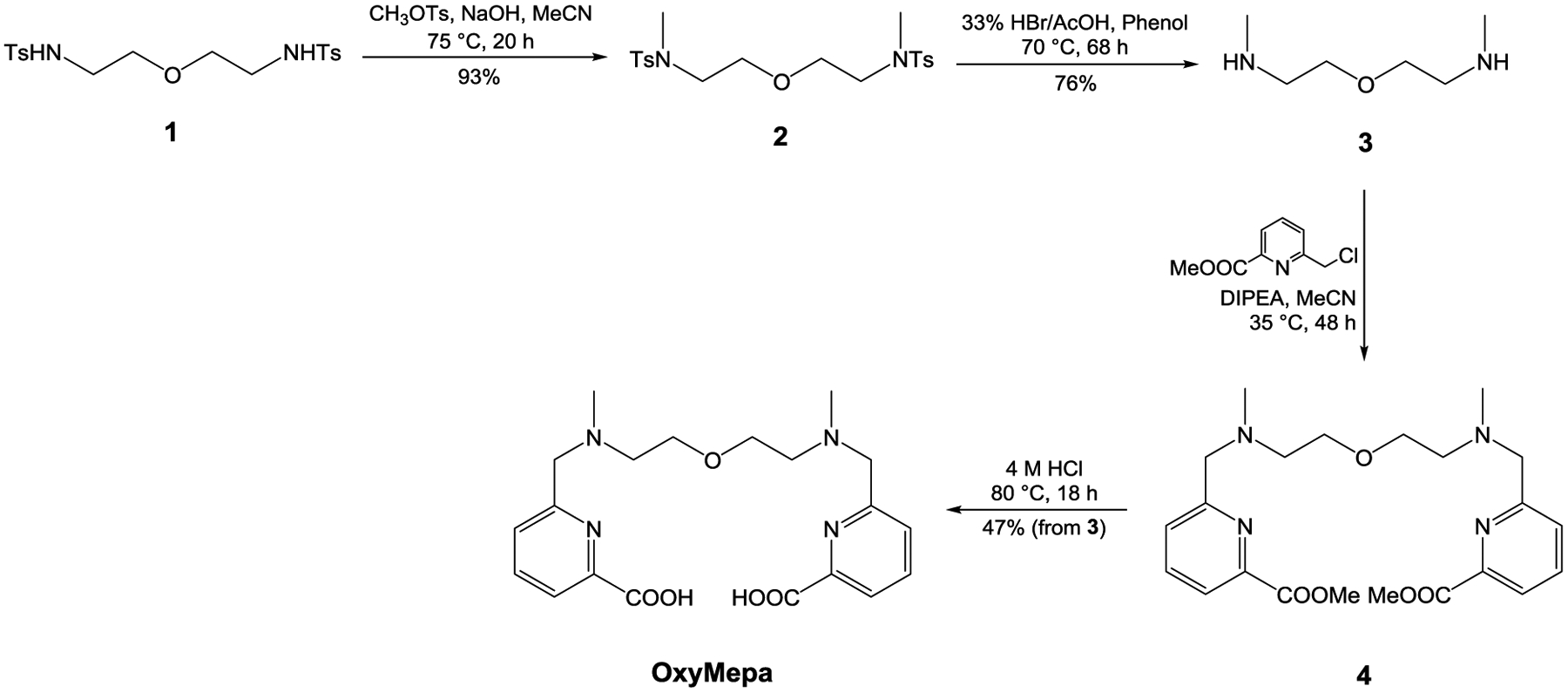
Synthetic route to OxyMepa.
Scheme 2.
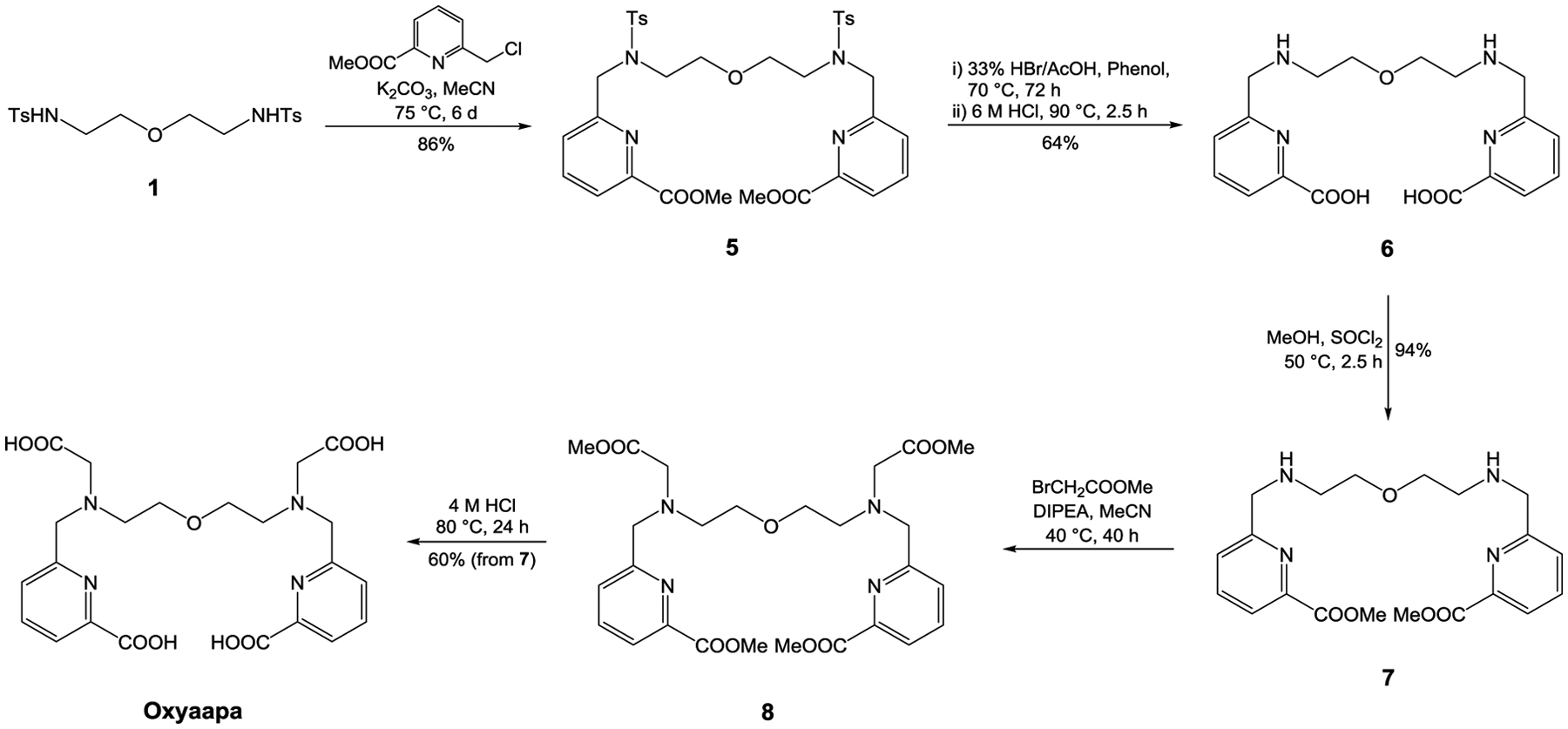
Synthetic route to Oxyaapa.
OxyMepa was prepared over four steps with a cumulative yield of 33%. The two methyl groups were first attached to the tosyl amide nitrogen atoms of 1 via the SN2 reaction with CH3OTs in the presence of NaOH to give 2. Deprotection of the tosyl groups was achieved with a mixture of HBr, phenol, and AcOH to obtain 3. Installation of the picolinate arms was accomplished by alkylating 3 with 6-chloromethylpyridine-2-carboxylic methyl ester and then hydrolyzing the esters under acidic conditions to afford OxyMepa.
Oxyaapa was synthesized over a five-step sequence with a cumulative yield of 31%. In contrast to the synthesis of OxyMepa, the picolinate arms were attached to 1 in the first step to give 5. Acidic deprotection of the tosyl groups and the methyl esters afforded 6. Compound 6 was then reesterified in MeOH with SOCl2 to afford 7, which was subsequently alkylated with methyl bromoacetate to yield 8. Oxyaapa was obtained after acidic ester hydrolysis of 8.
Both ligands and all intermediates were characterized by 1H and 13C{1H} NMR spectroscopy and HRMS. Analytical HPLC was used to further ascertain purity for samples with sufficient aqueous solubility. Notably, elemental analysis was performed to help identify the protonation states of compounds isolated as HCl or HBr salts. Full characterization data are included in the SI (Figures S1−S24).
Ligand Protonation Constants and Lanthanide Complex Stability Constants.
Metal complex stability constants provide a quantitative measure of the thermodynamic affinity of ligands for metal ions. The magnitudes of these stability constants for different ligands have been useful for assessing their value in different metal chelation applications.15,62 In this context, we evaluated the stability constants of OxyMepa and Oxyaapa with the lanthanide series by potentiometric and UV-Vis spectroscopic titrations. Ligand protonation constants and metal complex stability constants are summarized in Table 1. Data for the structurally relevant Octapa and OBETA are also collected for comparison. These ligand protonation constants (Ki) and metal complex stability constants (KLnL) are defined as (Equations 1) and (2), respectively.
| (1) |
| (2) |
Table 1.
Protonation Constants of OxyMepa, Oxyaapa, Octapa, OBETA and Stability Constants of their Lanthanide Complexes.a
| OxyMepa | Oxyaapa | Octapa | OBETA | |
|---|---|---|---|---|
| log K1 | 8.72(1) | 8.47(4) | 8.52b, 8.58c | 9.34e |
| log K2 | 7.81(2) | 7.63(2) | 5.40b, 5.43c | 8.62e |
| log K3 | 3.09(2) | 3.66(4) | 3.65b, 3.75c | 3.19e |
| log K4 | 2.35(4) | 2.90(1) | 2.97b, 3.08c | 2.19e |
| log K5 | 1.77(12) | 1.66b, 2.21c | 1.77e | |
| log KLaL | 9.93(3) | 19.06(5) | 20.13b | 16.89e |
| log KCeL | 10.74(1) | 17.34f | ||
| log KPrL | 11.25(3) | |||
| log KNdL | 11.49(1) | 19.77(7) | 18.39e | |
| log KSmL | 12.13(2) | 20.20c | 19.02f | |
| log KEuL | 12.15(2) | 19.13f | ||
| log KGdL | 12.02(1) | 20.50(2) | 20.23b | 19.37f |
| log KTbL | 12.17(3) | |||
| log KDyL | 12.18(1) | 20.35c | 18.87f | |
| log KHoL | 12.10(4) | 18.93e | ||
| log KErL | 12.00(3) | 21.33(7) | 18.46f | |
| log KTmL | 12.05(1) | 18.31f | ||
| log KYbL | 12.14(2) | 20.10c | ||
| log KLuL | 12.21(1) | 21.49(4) | 20.49b, 20.08d | 17.93e |
A commonly-used measure of the conditional thermodynamic affinity of a metal-ligand pair at physiological pH is provided by the pM value, which is defined as the −log [M]free when [M]total = 10−6 M and [L]total = 10−5 M at pH = 7.4.63 Calculated pLn values for OxyMepa and Oxyaapa are listed in Table S1 (SI).
The protonation constants of OxyMepa and Oxyaapa were determined by potentiometric titrations (Figures S25−S26, SI). The first four log Ki values for OxyMepa and five log Ki values for Oxyaapa were successfully refined using data within the pH range of the potentiometry experiments. For both ligands, we assign K1 and K2 to protonation events on the two aliphatic tertiary nitrogen atoms. Because pyridinium-2-carboxylic acid has a larger pKa (5.39) than acetic acid (4.76),64 we tentatively assign K3 and K4 to the protonation of two picolinate groups. For Oxyaapa, K5 most likely reflects the protonation equilibrium of one of the pendent acetate donors. We caution, however, that additional experimental data would be needed to verify this assignment with certainty. As a comparison, the log Ki values of the related ligand Octapa are comparable to those for Oxyaapa and OxyMepa with the exception of log K2, which is somewhat larger for the latter ligands. The greater basicity of the second tertiary nitrogen in Oxyaapa and OxyMepa can be explained by the larger separation between the two nitrogen atoms afforded by the central ether linkage. Additionally, similar log Ki values are observed for Oxyaapa and OBETA. The tertiary nitrogen atoms of Oxyaapa are slightly less basic, as reflected by the log K1 and log K2 values, than those of OBETA. The decreased basicity of Oxyaapa is most likely a consequence of the presence of the nearby electron-withdrawing picolinate units, which are absent in OBETA. With two acetate donor groups replaced by picolinates, the log K3 and log K4 values are slightly larger than those of OBETA.
Following measurement of the ligand protonation constants, the stability constants of the Ln-OxyMepa complexes were evaluated by carrying out potentiometric titrations (Figures S27–S40, SI). For Oxyaapa, lanthanide complexation was found to occur even at very low pH values due to its high affinity. Because of challenges associated with pH electrode calibration at these low pH values, potentiometry was found to be unsuitable to determine the stability constants of this ligand with lanthanides. Instead, UV-Vis spectroscopic titrations were carried out at low pH, where pH can be directly calculated from the HCl concentration (Figures S41–S45, SI). The stability constants for Ln-Oxyaapa complexes are given in Table 1. Because these UV-Vis spectroscopic titrations are significantly more labor-intensive than potentiometric titrations, the stability constants were only determined for a representative subset of the entire lanthanide series.
In general, both OxyMepa and Oxyaapa exhibit a thermodynamic preference for late lanthanides, where the log KLnL values increase as the series is traversed from La3+ to Lu3+. This thermodynamic preference for the late lanthanides is a general feature observed for most chelators,10 due to an increasing charge density on the Ln3+ ion across the series that enhances M–L electrostatic interaction.65 With the two extra carboxylate donors, Oxyaapa forms significantly more stable complexes than OxyMepa, as reflected by stability constants that are approximately nine orders of magnitude larger. OBETA, which contains the same 2,2’-oxybis(ethan-1-amine) core, is worth comparing in this context as well. In contrast to OxyMepa and Oxyaapa, the tertiary nitrogen donors of OBETA are functionalized with a total of four acetate groups. OBETA forms significantly more stable Ln3+ complexes than OxyMepa, despite the fact that both ligands provide the same number of donor atoms. This result would seem to indicate that for this class of ligands the binding properties of a single picolinate donor do not match those of two acetate donors. By contrast, Oxyaapa forms consistently more stable Ln3+ complexes than OBETA. In this case, the additional two acetate donors of Oxyaapa provide additional stabilization. In comparison to the ligands Octapa and Pypa, Oxyaapa exhibits similar thermodynamic affinity for the lanthanides. Oxyaapa forms a complex with La3+ with slightly lower stability (log KLaL = 19.06) than those of Octapa (log KLaL = 20.13)60 and Pypa (KLaL = 19.74).24 With the smallest lanthanide Lu3+, the stability of the Oxyaapa complex (log KLuL = 21.49) sits between those of Octapa (log KLuL = 20.08)60 and Pypa (log KLuL = 22.02).24 Oxyaapa does not exhibit a significant enhancement of thermodynamic affinity for the lanthanides in comparison to Octapa and Pypa. Their similar affinities for lanthanides are also indicated by the comparable pLn values of Oxyaapa (Table S1, SI, pLa = 18.5, pLu = 21.1), Octapa (pLa = 19.7, pLu = 19.8), and Pypa (pLa =19.9, pLu = 22.6).24 This result is somewhat surprising, given that Oxyaapa provides an additional oxygen donor atom compared to Octapa and Pypa, which should stabilize the chemically hard Ln3+ ions. Although the ethereal oxygen atom of Oxyaapa is a hard Lewis base,66 it is a weaker donor compared to the central pyridine of Pypa, which is often regarded as a borderline Lewis base.66 This difference in donor strength is reflected in part by the pKa values of the corresponding conjugate acids of these donors; for Et2OH+, the pKa value is −3.6,67 whereas the 2,6-dimethylpyridinium cation has a pKa value of 6.7,68 indicating that the pyridyl nitrogen donor is significantly more basic and thus likely serves as a stronger donor to metal ions.
Transchelation Challenge with DTPA.
When administered in vivo, coordination complexes are often under kinetic rather than thermodynamic control. As such, the kinetic inertness of these lanthanide complexes is an important aspect to evaluate to gauge their potential use in biological applications, because uncomplexed lanthanide ions accumulate in the body and cause various toxicological effects.69 Furthermore, thermodynamic stabilities do not necessarily correlate directly with kinetic inertness. For example, Ln-DOTA complexes are typically significantly more inert than those of Ln-DTPA, despite their comparable thermodynamic stabilities.70–73 For this reason, experiments that explicitly probe kinetic inertness are also necessary. We therefore challenged the La3+, Gd3+, and Lu3+ complexes of OxyMepa and Oxyaapa with a 1000-fold excess of DTPA as a competing chelator and monitored the kinetics of transchelation by UV-Vis spectroscopy. Based on the higher stability constants of DTPA for these metal ions (log KLaDTPA = 19.48, log KGdDTPA = 22.46, log KLuDTPA = 22.44),74 these conditions will thermodynamically favor loss of the Ln3+ from OxyMepa and Oxyaapa, and therefore this transchelation process is dictated by kinetics. We have used this challenge study previously to probe kinetic inertness of related Ln3+ complexes.75–77
Under these conditions, the absorbance change follows pseudo-first-order kinetics, allowing facile determination of the complex half-lives (t1/2) to compare kinetic inertness. The t1/2 values are given in Table 2, and the UV-Vis spectral traces of these processes are shown in Figures S46–S50 (SI). Moving from large La3+, intermediate Gd3+, and small Lu3+, the kinetic inertness of the complexes increases, showing a correlation with the trend in thermodynamic stability constants discussed above. Likewise, complexes of the nonadentate Oxyaapa are significantly more inert than those of the heptadentate OxyMepa. In particular, the kinetic inertness of Oxyaapa with Lu3+ is remarkable; its t1/2 under these conditions is estimated to be around 6 weeks based on extrapolation of the kinetic data (Figure S50, SI). Thus, Oxyaapa is a promising candidate for chelation of the therapeutic radiometal 177Lu3+.
Table 2.
Half-lives of Lanthanide Complexes of OxyMepa and Oxyaapa when Challenged with 100 Equivalents of DTPA ([LnL] = 100 μM, pH = 7.4 in 0.1 M MOPS, 22 °C).
The half-life is too short or too long to measure accurately.
X-Ray Crystallography.
To correlate structural features with thermodynamic and kinetic properties, we determined the crystal structures of the Lu3+ complexes of OxyMepa and Oxyaapa. X-ray quality crystals of [Lu(OxyMepa)(OH2)][PF6] were obtained by slow evaporation of an acetone-methanol-water solution. For the Oxyaapa complex, X-ray quality crystals were obtained by the slow evaporation of a 1.4:1 mixture of LuCl3 and Oxyaapa in aqueous solution with the pH adjusted to 4 by using NaOH. The formula of the complex obtained is [(OH2)3Na(μ-OH2)2Na(μ-OH2)2Na(OH2)3][Lu(Oxyaapa)]2Cl·H2O. The crystal structures and selected interatomic distances of these complexes are shown in Figure 1 and Table 3, and their crystallographic data collection and refinement parameters are listed in Table S2 (SI).
Figure 1.
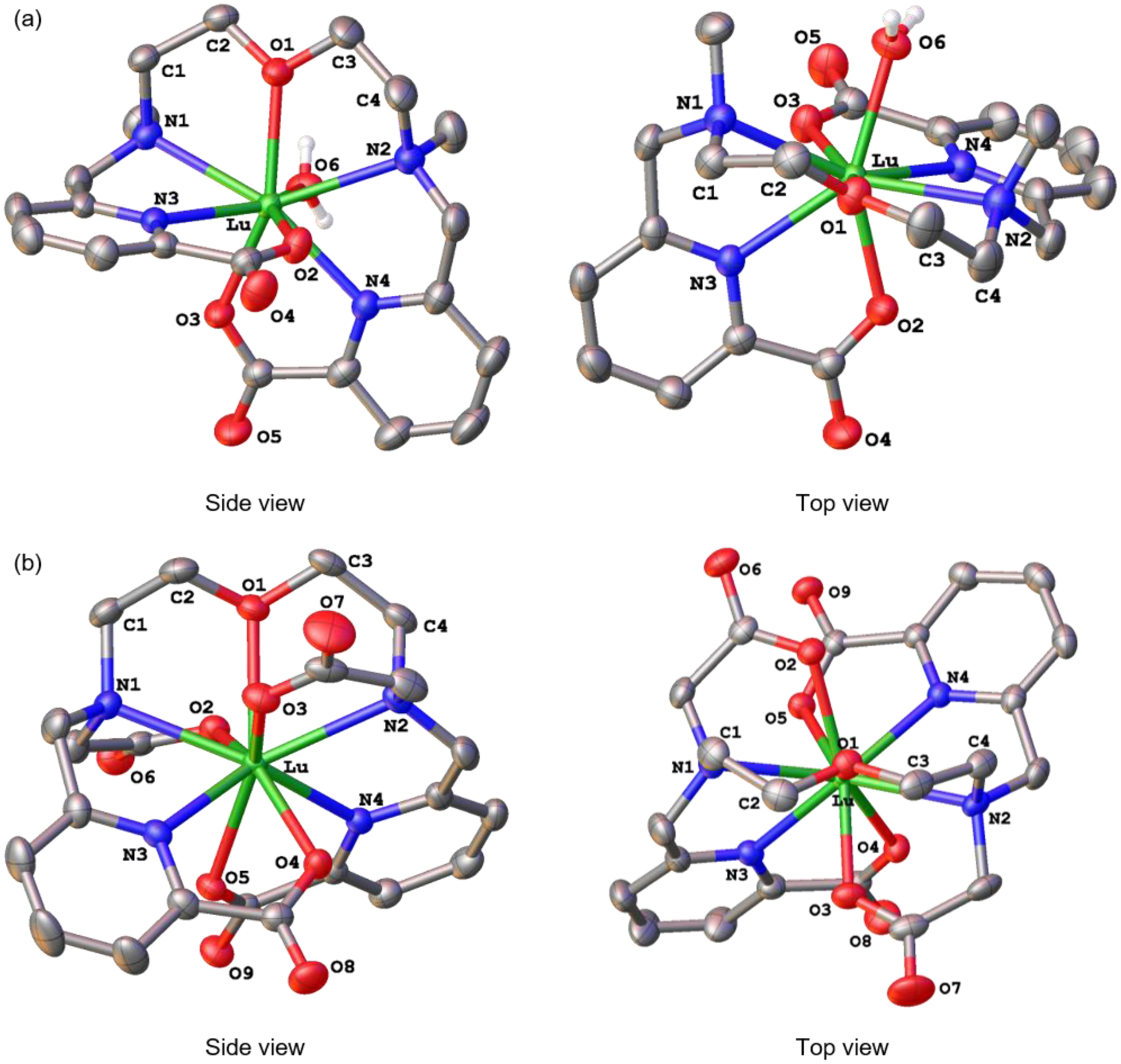
Crystal structures of (a) Lu-OxyMepa and (b) Lu-Oxyaapa. Thermal ellipsoids are drawn at the 50% probability level. Solvent, counterions, and non-acidic hydrogen atoms are omitted for clarity.
Table 3.
Selected Interatomic distances (Å) within the Lu-OxyMepa and Lu-Oxyaapa Crystal Structures.a
| Lu-OxyMepa | Lu-Oxyaapa | ||
|---|---|---|---|
| Lu–N1 | 2.591(3) | Lu–N1 | 2.542(2) |
| Lu–N2 | 2.564(3) | Lu–N2 | 2.540(2) |
| Lu–N3 | 2.420(3) | Lu–N3 | 2.383(2) |
| Lu–N4 | 2.407(3) | Lu–N4 | 2.404(2) |
| Lu–O1 | 2.312(3) | Lu–O1 | 2.504(2) |
| Lu–O2 | 2.274(3) | Lu–O2 | 2.358(2) |
| Lu–O3 | 2.236(2) | Lu–O3 | 2.290(2) |
| Lu–O6 | 2.282(3) | Lu–O4 | 2.287(2) |
| Lu–O5 | 2.401(2) | ||
Atoms are labeled as shown in Figure 1. The values in the parentheses are one standard deviation of the last significant figure.
The structure of the Lu-OxyMepa complex reveals that the coordination sphere of the Lu3+ center comprises eight donor atoms, with seven of these arising from the OxyMepa ligand and the eighth from a coordinated H2O molecule. The overall symmetry of this complex is C1, and the coordination geometry of the Lu3+ ion most closely matches that of a biaugmented trigonal prism, as indicated by SHAPE78 program (ver. 2.1). The relative orientation of the two picolinate donors is approximately perpendicular, and the two N–CH3 groups are pointing in the same direction (syn).
The structure of the Lu3+ complex of Oxyaapa shows that all nine donor atoms of this ligand coordinate directly to the lanthanide center, thus preventing the coordination of solvent molecules. The complex attains a distorted C2 symmetry, and the coordination polyhedron about the Lu3+, afforded by the nine donor atoms, most closely matches a spherical capped square antiprism, as determined by SHAPE.78 The two acetate donors on the tertiary nitrogen bind the metal center from opposite faces. Thus, in contrast to the OxyMepa structure where the N–CH3 groups attain a syn configuration, the N–CH2CO2– donors in this structure assume an anti configuration. This structure is comparable to that of the Lu3+ complex of Pypa.24 In this case, the central pyridine N donor of Pypa is replaced by the central ether oxygen donor (O1) of Oxyaapa, but the relative orientation of the other donor atoms is similar. Another noteworthy feature of this structure is the somewhat unusual counterion, [(OH2)3Na(μ-OH2)2Na(μ-OH2)2Na(OH2)3]3+ (Figure S51, SI). This trisodium cluster has been observed previously in several other crystal structures.79,80
Both OxyMepa and Oxyaapa can attain several different diastereomeric conformations upon binding to metal ions. These conformations will differ based on the absolute chiralities (S or R) at the tertiary nitrogen donors (N1 and N2), N1SN2S, N1SN2R, N1RN2S, or N1RN2R, and the chiralities (δ or λ)81 of the two five-membered chelate rings (Ln–N1–C1–C2–O1–Ln and Ln–O1–C3–C4–N2–Ln), δδ, δλ, λδ, or λλ (Chart 2). Thus, 16 stereoisomers (8 enantiomeric pairs) are possible upon complexation of these ligands. If the absolute configurations of N1 and N2 are the same (N1SN2S or N1RN2R), two methyl groups of OxyMepa or two acetate groups of Oxyaapa point to opposite directions, thus attaining what we described above as the anti conformation. If the nitrogen configurations are different (N1SN2R or N1RN2S) then these groups will be oriented in the same direction, giving rise to the syn conformer. The crystal structure of the Lu3+ complex of OxyMepa shows that it attains the N1SN2R(δλ) / N1RN2S(λδ) configurations as an enantiomeric pair, whereas the structure of the Oxyaapa complex contains the enantiomeric N1SN2S(λδ) / N1RN2R(δλ) configurations. The relative energies of these conformations will be discussed further in the following sections.
Chart 2.
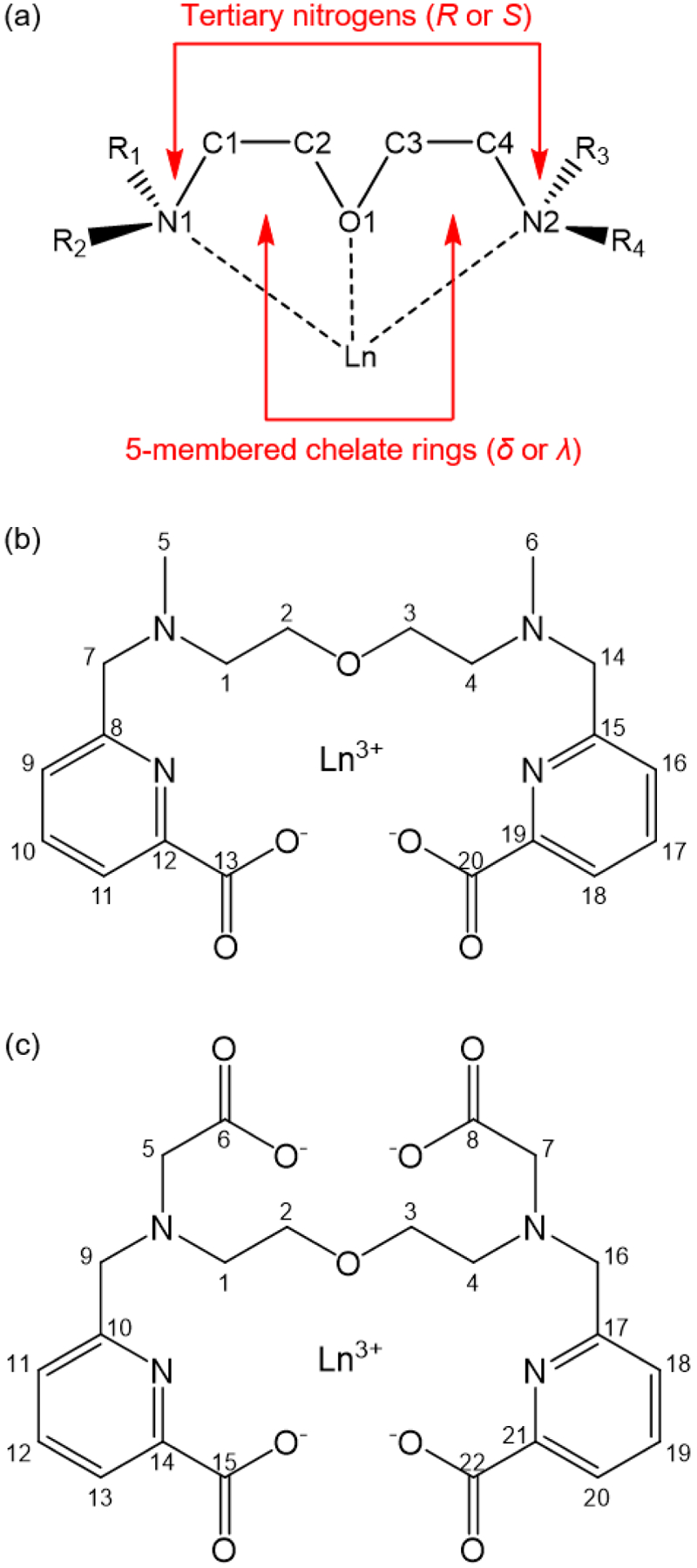
(a) Depiction of the sources of chirality in Ln-OxyMepa or Ln-Oxyaapa complex; (b) Numbering scheme of Ln-OxyMepa complex; (c) Numbering scheme of Ln-Oxyaapa complex.
Solution Structures of Complexes.
The 1H and 13C{1H} NMR spectra of the diamagnetic La3+ and Lu3+ complexes of OxyMepa and Oxyaapa were acquired in D2O at pD = 7 to assess their solution structures and dynamics. A comprehensive set of 2D NMR experiments were also carried out on all four complexes to fully assign the 1H and 13C chemical shifts and obtain a more thorough understanding of their solution structures. The 1H NMR spectra of all four complexes are shown in Figure 2, and the atoms are numbered as shown in Chart 2. In these spectra, the hydrogen resonances on the methylene groups linking the picolinate donor arms (H-7 and H-14 for OxyMepa, H-9 and H-16 for Oxyaapa; Chart 2) of OxyMepa and Oxyaapa resolve into two well-separated doublets. The loss of equivalency of the two methylenic hydrogens is consistent with the generation of chirality in the complex upon metal binding. Because these doublets are easily identifiable in the spectra, they serve as good indicators of the solution structures of the complexes. Additionally, the N–CH3 peaks of OxyMepa (H-5 and H-6), and the methylene groups linking the acetate donor arms of Oxyaapa (H-5 and H-7), can act in a similar capacity. Full spectral-width 1H NMR spectra with peak integrations and complete assignments, as well as the corresponding 13C and 2D NMR spectra are given in the SI (Figures S52–S87).
Figure 2.
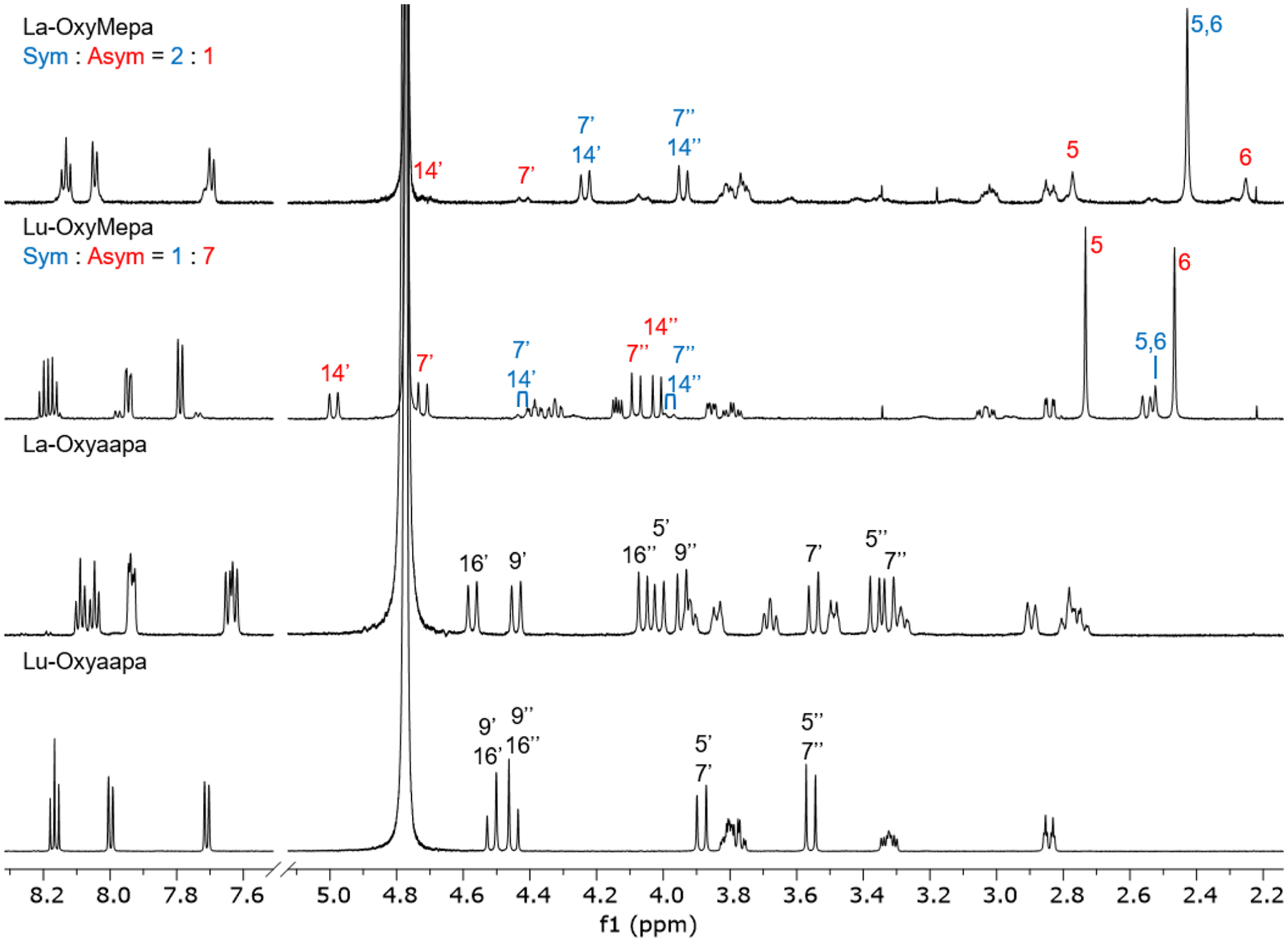
1H NMR spectra of OxyMepa and Oxyaapa complexes formed with La3+ and Lu3+ (600 MHz, D2O, pD = 7, 25 °C). Diagnostic peaks are labeled and the numbering scheme is shown in Chart 2. The high-intensity peak near 4.8 ppm arises from residual HDO in the NMR solvent.
The 1H NMR spectra of the La- and Lu-OxyMepa complexes show that there are two different species in solution, which we hypothesize to be conformational isomers. One of these conformations is symmetric (Sym) on the NMR timescale, showing only one N–CH3 resonance (H-5 and H-6 in blue), whereas the other conformation is asymmetric (Asym), as reflected by the presence of two distinct N–CH3 resonances (H-5 and H-6 in red). The methylene resonances (H-7, H-14) similarly exist only as one set of doublets in the symmetric conformer, but are separated into two sets of doublets in the asymmetric conformer. For the La3+ complex, the symmetric conformer is the predominant species in solution. Based on total peak integrations of the two isomers, the ratio of symmetric to asymmetric conformer is approximately 2:1. Because this ratio does not change over time, we assumed that this ratio represents that system at equilibrium, thus allowing us to calculate a small standard free energy difference of 1.7 kJ/mol between these conformers by using the established relationship between K and ΔG° (ΔG° = –RT ln K, where R and T are the ideal gas constant and temperature, respectively). By contrast, the major conformer for the Lu3+ complex is the asymmetric species. The ratio of symmetric to asymmetric conformers for Lu3+ is approximately 1:7, reflecting a larger free energy difference of 4.8 kJ/mol between these conformers. 2D ROESY spectra were useful for further probing these complexes. For the La3+ complex (Figures S58–S59, SI), the symmetric and asymmetric species are correlated by chemical-exchange (EXSY) crosspeaks with the same phase as the diagonal, verifying the two different species arise from the same compound and indicating they are interconverting within the 200-ms mixing time of the NMR experiment. For the Lu3+ complex (Figures S66–S67, SI), these EXSY crosspeaks were not observed using the same mixing time. Thus, the Lu3+ complex is more structurally rigid in solution, a feature that may give rise to its greater kinetic inertness. Moreover, analysis of the ROESY crosspeaks with the opposite phase to the diagonal, which arise from through-space interactions, was used as a further confirmation of the assignment of the major isomer in solution. The intensities of the crosspeaks of the major asymmetric conformer are consistent with the interatomic distances obtained from the crystal structure of this complex (Figure 1), in which the N–CH3 groups are arranged in a syn orientation. As shown in Figure S67 and Table S5 (SI), through-space correlations are present for all hydrogens that are less than 2.7 Å apart in the crystal structure (Figure 1) with relative intensities directly related to the distance of their separation. With the major species assigned, the minor symmetric species can then be deduced to be the conformer in which the N–CH3 resonance are arranged in an anti configuration.
In contrast to OxyMepa, the 1H NMR spectra of both the La3+ and Lu3+ complexes of Oxyaapa reveal the presence of only a single conformer in solution (Figure 2). The symmetries of the La3+ and Lu3+ complexes, however, are different from each other. The La3+ complex is asymmetric, as reflected by the observation of two distinct sets of resonances for the picolinate arms and the methylenic hydrogens (H-5, H-7, H-9, H-16). The Lu3+ complex is symmetric; only a single environment for the picolinate arms is observed and two sets of doublets from the methlyenic hydrogens are present. The ROESY spectra of La- and Lu-Oxyaapa complexes show differing degrees of conformational flexibility. No EXSY crosspeaks are observed for the Lu3+ complex (Figures S86–S87, SI), indicating its high stereochemical rigidity. By contrast, the ROESY spectrum of the La3+ complex shows clear intramolecular EXSY crosspeaks (Figures S76–S77, SI), which indicate, for example, that H-2′ and H-3″ are exchanging as well as H-2″ and H-3′. Similar correlations are observed for H-1 and H-4, H-5 and H-7, H-9 and H-16. Thus, the La3+ complex is stereochemically less rigid compared to the Lu3+ complex, which may contribute to its lower kinetic inertness. Drawing analogies to OxyMepa, the symmetric conformer corresponds to an anti arrangement of the acetate pendent donors, whereas the asymmetric conformer arises from a syn arrangement. The crystal structure of the Lu-Oxyaapa complex, shown in Figure 1, clearly displays the anti arrangement of the pendent acetate donors and contains a pseudo-C2 axis. Thus, the solid-state conformation of Lu-Oxyaapa complex matches its conformation and symmetry in the solution state, as deduced via NMR spectroscopy.
Photophysical Properties.
The rich photoluminescent properties of Eu3+ and Tb3+ have enabled their use for biological imaging applications. To assess the suitability of OxyMepa and Oxyaapa for these applications, we investigated the photophysical properties of their Eu3+ and Tb3+ complexes. The UV–Vis absorption and emission spectra of these complexes in 0.1 M MOPS (pH = 7.4) at room temperature are shown in Figure 3, and a summary of their photophysical properties are collected in Table 4. The absorption spectra of these complexes exhibit an absorption band centered at 273 nm, which can be attributed to the π → π* transition of the picolinate arms.82 Upon excitation of this ligand absorption band, all complexes exhibit characteristic lanthanide-based f-f emission spectra. Emission from these f-f excited states occurs via the well-established antenna effect.3,83
Figure 3.
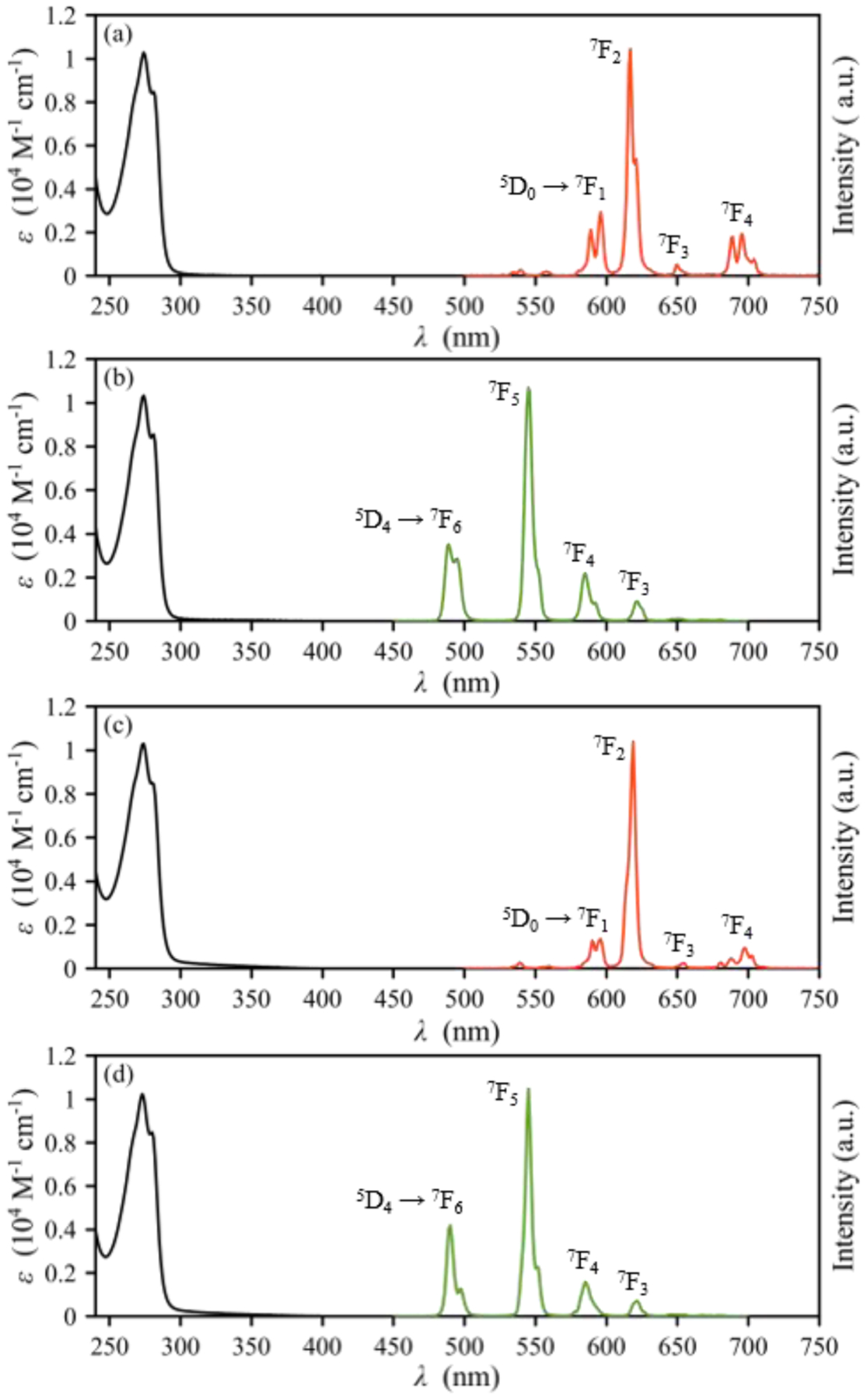
UV–Vis absorption (black) and emission (red, Eu3+; green, Tb3+) spectra of (a) Eu-OxyMepa, (b) Tb-OxyMepa, (c) Eu-Oxyaapa and (d) Tb-Oxyaapa complexes (pH = 7.4 in 0.1 M MOPS, 22 °C). Major transitions are labeled accordingly.
Table 4.
Photophysical Properties of the Eu3+ and Tb3+ Complexes Formed with OxyMepa and Oxyaapa (pH = 7.4 in 0.1 M MOPS, 22 °C)a
| λmax/nm | ε(λmax)/(M−1 cm−1) | QY/% | τ(H2O)/ms | τ(D2O)/ms | q | |
|---|---|---|---|---|---|---|
| Eu-OxyMepa | 273 | 10300 | 0.36(1) | 0.390(1) | 1.671(4) | 1.8 |
| Tb-OxyMepa | 273 | 10300 | 1.4(1) | 1.231(4) | 2.558(6) | 1.8 |
| Eu-Oxyaapa | 273 | 10300 | 0.86(4) | 1.145(4) | 1.635(3) | −0.1 |
| Tb-Oxyaapa | 273 | 10200 | 2.1(2) | 1.818(3) | 1.905(4) | −0.2 |
The values in the parentheses are one standard deviation of the last significant figure.
The emission spectra of the Eu3+ complexes of both OxyMepa and Oxyaapa reveal the presence of the characteristic Eu-based 5D0 → 7FJ transitions,8 where J = 1–4. The 5D0 → 7F0 transition only has appreciable intensity when the Eu3+ center possesses a Cnv, Cn or Cs symmetry.84 The absence of this transition for both Eu3+ complexes indicates that they are of low symmetry in solution on the timescale of this experiment. The low-intensity transitions observed between 530 and 570 nm for both complexes are attributed to emission from higher excited states such as 5D1 → 7FJ.84 For the Tb3+ complexes, the emission peaks corresponding to 5D4 → 7FJ transitions,8 where J = 6–3, are observed clearly. The transitions for the relaxation of the 5D4 state to the 7F2, 7F1, and 7F0 states are not readily detected.
The photoluminescent quantum yields of these complexes in aqueous solution were measured relative to a reference compound [Ru(bpy)3]Cl2. These complexes are weakly emissive, as reflected by their low quantum yields that are less than 2%. Notably, the Tb3+ complexes have a higher quantum yields than the Eu3+ complexes. This observation is typical for Ln3+ complexes with picolinate-based ligands82,85–87 because the triplet state of picolinate donors are closer in energy to the emissive f-f excited state of Tb3+ than that of Eu3+, a property that leads to more efficient antenna effect energy transfer to Tb3+. In comparing these two ligands, Oxyaapa gives rise to Eu3+ and Tb3+ complexes with higher quantum yields than OxyMepa. As evident from the crystal structure of the Lu3+ complexes of these ligands, Oxyaapa saturates the metal coordination sphere, whereas OxyMepa allows for the direct coordination of a water molecule. Thus, the lower quantum yields of the OxyMepa complexes most likely arise from the presence of the O–H oscillator from the coordinated water, which is an effective quencher of lanthanide luminescence.3,8,83,88
In order to probe the hydration states of these Eu3+ and Tb3+ complexes in solution, we determined their luminescence lifetimes in both H2O and D2O (Figures S88–S89, SI). A comparison of lifetimes in these two solvents can be used to afford the number of inner-sphere water molecules, or q. (Equations 3) and (4), are used for Eu3+ and Tb3+, respectively:89,90
| (3) |
| (4) |
In both equations, Δkobs = 1/τ(H2O) – 1/τ(D2O) in ms−1. Based on the lifetimes we measured (Table 4), we were able to calculate the hydration numbers for all four complexes. For the Eu3+ and Tb3+ complexes of OxyMepa, a q of approximately 2 was obtained. The crystal structure of the Lu3+ complex of OxyMepa (Figure 1) clearly shows that only 1 water molecule is directly coordinated to this central lanthanide. The higher hydration numbers of the Eu3+ and Tb3+ complexes are most likely a consequence of the larger ionic radii of these lanthanides, which can accommodate higher coordination numbers in comparison to Lu3+. By contrast, the Eu3+ and Tb3+ complexes of Oxyaapa were found by this method to contain no inner-sphere water molecules. This result is consistent with the crystal structure of the Lu3+ complex of this ligand, which likewise shows that no bound water molecule is present. The higher denticity of Oxyaapa in comparison to OxyMepa is thus able to effectively saturate the coordination spheres of the lanthanide ions. The previously reported octadentate ligand Octapa forms Eu3+ and Tb3+ complexes with a single bound water molecule;16 thus the nine donors afforded by Oxyaapa is sufficient to coordinately saturate these ions, but the eight donors of Octapa is not.
DFT Calculations.
DFT calculations were carried out to understand the geometries and relative energies of the different conformations of lanthanide complexes with OxyMepa and Oxyaapa. For these calculations, we explored complexes of the largest lanthanide La3+ and the smallest lanthanide Lu3+ to understand how ligand conformation changes in a manner that is dependent on central metal ion size. The extensively used hybrid B3LYP functional and LCRECP for the Ln3+ center were applied in our calculations. Once we obtained optimized geometries, frequency calculations were carried out to confirm that they corresponded to local minima on the potential energy surface and to obtain zero-point energies. To more effectively capture the solution dynamics of these complexes, an implicit SMD solvation model was employed within these calculations as well. Complete computational details are described in the Experimental Section. Depictions of all the optimized structures (Figures S90–S127, SI), their coordinates, and their output energies are included in the SI.
In carrying out the DFT structure optimizations, an important point to consider is the number of inner-sphere water molecules, which could influence the energies of the different conformations. Based on the crystal structures, 1 and 0 water molecules are present within the inner-sphere of the Lu3+ complexes of OxyMepa and Oxyaapa, respectively. Thus, we used these hydration numbers for the DFT calculations of the Lu3+ complexes. For La3+, no inner-sphere water molecules were included in our DFT calculations of the Oxyaapa complex. The use of 0 inner-sphere water molecules is supported by our luminescence lifetime measurements of the Eu3+ and Tb3+ complexes, which suggest that the coordination spheres of these large ions is saturated by the ligand. For the OxyMepa complex, we applied 1 inner-sphere water molecule in our calculations to match what was observed experimentally in the crystal structure of the Lu-OxyMepa complex. Although luminescence lifetime measurements for Eu3+ and Tb3+ suggested that 2 water molecules were coordinated to these larger Ln3+ ions, efforts to optimize La3+ complexes of OxyMepa in this way were not successful. During some of these geometry optimizations, only one water molecule would stay directly bound to the metal center, while the second moved to the outer-sphere and remained bound only via hydrogen-bonding interactions. We have therefore carried out two sets of calculations for La-OxyMepa. In one set, we only include a single bound water molecule; in the other set, we used two bound waters, but omitted the optimizations for which the second water moved to the outer-sphere.
Using these hydration numbers, the geometries of the La3+ and Lu3+ complexes were optimized, varying the ligand conformation to be one of the eight possible diastereomers. As noted above in our discussion on the X-ray crystal structures and shown in Chart 2, both the absolute chirality of the tertiary nitrogen donors and the tilt of the ether-based five-membered chelate rings bring about these different conformations. The optimization of all eight possibilities provided distinct minima on the potential energy surface, and the relative free energies of these conformation are given in Table 5. For the Lu3+ complexes, the lowest-energy conformers were calculated to be N1SN2R(δλ) / N1RN2S(λδ) and N1SN2S(λδ) / N1RN2R(δλ) for OxyMepa and Oxyaapa, respectively. These are the conformations that are observed experimentally by X-ray crystallography. Additionally, these results are consistent with 1H NMR spectroscopy, which shows the complex to exist predominately as the syn conformer for OxyMepa and anti conformer for Oxyaapa. Notably, the calculation captured the relative energies of these conformers. For example, by 1H NMR spectroscopy we see that the ratio of the syn to anti conformer for Lu-OxyMepa is 7:1, corresponding to a free energy difference of 4.8 kJ/mol. This energy difference obtained from DFT calculations is 6.2 kJ/mol, a value that is very close to the experimental result. For Lu-Oxyaapa, the calculated energy difference between the anti and syn conformers is slightly larger, at 9.1 kJ/mol, with the anti conformer being more stable. This larger energy difference is also supported by the 1H NMR spectrum of this complex, where only one conformer is detected. Calculations of the La-Oxyaapa complexes predict that the syn configuration is more stable than the anti configuration by 10.0 kJ/mol, a result that is consistent with our NMR experiments. As noted above, for the La-OxyMepa we carried out two sets of calculations employing both q = 1 and q = 2. For the q = 1 set of calculations, the experimental observation was matched as the anti configuration was found to be more stable by 5.5 kJ/mol. For the q = 2 calculations, however, we found the opposite result in contradiction with experiment; the syn configuration was determined to be more stable than the anti configuration by 7.8 kJ/mol. Upon inspection of the most stable DFT geometry of the syn configuration, it became apparent that the two bound water molecules are interacting with each other via hydrogen bonding interactions (Figure S106, SI). We believe that our model calculations overestimate the energetic importance of these hydrogen bonding interactions because there would be an array of water molecules available in aqueous solution that could act as hydrogen bonding partners for both the syn and anti configurations of the complex. Thus, these calculations highlight some of the potential pitfalls and challenges in probing the aqueous coordination chemistry of the lanthanides. The inclusion of a larger quantity of water molecules within the secondary coordination sphere, a strategy that has previously been applied successfully in modeling Gd-based MRI contrast agents,91 could also be used to address these issues.
Table 5.
The Relative Free Energies (kJ·mol−1) at 298 K of DFT-Optimized Conformations for Lu3+ and La3+ Complexes Formed with OxyMepa and Oxyaapa. The Enantiomeric Pair with the Lowest Calculated Energy of Each Complex is Referenced as Zero.
| ΔG(Lu-OxyMepa) | ΔG(Lu-Oxyaapa) | ΔG(La-OxyMepa, q = 1) | ΔG(La-OxyMepa, q = 2) | ΔG(La-Oxyaapa) | ||
|---|---|---|---|---|---|---|
| anti conformations | N1SN2S(λλ) / N1RN2R(δδ) | +34.7 | +17.4 | +17.0 | --b | +21.2 |
| N1SN2S(λδ) / N1RN2R(δλ) | +6.2 | 0a | +0.5 | +11.3 | +19.8 | |
| N1SN2S(δλ) / N1RN2R(λδ) | +40.9 | +7.0 | +15.8 | +7.8 | +10.0 | |
| N1SN2S(δδ) / N1RN2R(λλ) | +27.3 | +7.2 | 0 | +12.0 | +21.6 | |
| syn confromations | N1SN2R(λλ) / N1RN2S(δδ) | +19.8 | +20.9 | +14.3 | +8.8 | +8.0 |
| N1SN2R(λδ) / N1RN2S(δλ) | +35.0 | +23.0 | +14.8 | +16.3 | +12.5 | |
| N1SN2R(δλ) / N1RN2S(λδ) | 0a | +9.1 | +5.5 | --b | 0 | |
| N1SN2R(δδ) / N1RN2S(λλ) | +22.5 | +22.0 | +10.4 | 0 | +13.9 |
The conformation found in the crystal structure.
One inner-sphere water molecule moved to the outer-sphere during the optimization.
Variable-Temperature 17O NMR Measurements.
The water exchange rate of lanthanide coordination complexes can span several orders of magnitude.6,92 These relative exchange rates are often related to structural features of the coordination complexes and can therefore provide additional characterization data of such complexes in aqueous solutions. To gain further insight on this dynamic behavior, we probed the water exchange kinetics of the Gd3+ complex of OxyMepa using variable-temperature 17O NMR spectroscopy. The temperature dependence of the 17O NMR transverse relaxation rate of bulk water with and without the paramagnetic complex, or the reduced transverse rate, 1/T2r, can afford the water exchange rate upon carrying out the proper analysis.92 This method, which has a well-established theoretical background, has been used extensively for Gd3+ complexes due to their relevance as MRI contrast agents.6,7 Hence, Gd3+ was chosen as a representative lanthanide to study the water exchange kinetics afforded by complexes of OxyMepa. Because complexes of Oxyaapa contain no coordinated exchangeable water ligands, we did not explore this chelator by 17O NMR spectroscopy.
The reduced transverse rate (1/T2r) at different temperatures (T) were measured for the Gd3+ complex of OxyMepa by 17O NMR spectroscopy at pH = 7.4 (Table S8, SI) and then analyzed based on the theory developed by Swift and Connick.93 Relevant theoretical background, including a summary of the equations applied,94–97 are included in the SI. The 1/T2r data were fit to a four-parameter model97 to obtain the water exchange rate constant at 298.15 K , the enthalpy of activation for this process (ΔH‡), the electronic relaxation rate at 298.15 K , and the activation energy for electronic relaxation (ΔET1e). The hydration number of the Eu3+ and Tb3+ complexes of OxyMepa, q = 1.8, was used for the data fitting of the Gd-OxyMepa complex. Additionally, we fixed the hyperfine coupling constant (A/ħ) at –3.8 × 106 rad·s−1 because this value does not vary significantly among different complexes of Gd3+.95,98 Data fitting at different A/ħ values verifies that the value of this parameter does not significantly affect the final values of the parameters above (Table S9, SI).
The 1/T2r data along with the best-fit curve are shown in Figure 4. The key parameters obtained from the fit are = (2.8 ± 0.1) × 106 s−1 and ΔH‡ = 39.8 ± 1.1 kJ·mol−1 (Table S9, SI). From these two parameters, the entropy of activation (ΔS‡) can be calculated using the Eyring equation to be +12.2 ± 3.9 J·mol−1·K−1. The positive ΔS‡ for water exchange on Gd-OxyMepa may reflect a dissociative or interchange dissociative mechanism. Although more data, such as the volume of activation ΔV‡, would be needed to provide more support for this mechanism, it is reasonable given that the formation of an associative ten-coordinate complex from the nine-coordinate Gd-OxyMepa complex is unexpected due to steric reasons. Furthermore, positive ΔS‡ values were generally found for Gd3+ complexes that undergo dissociative exchange.7,92
Figure 4.
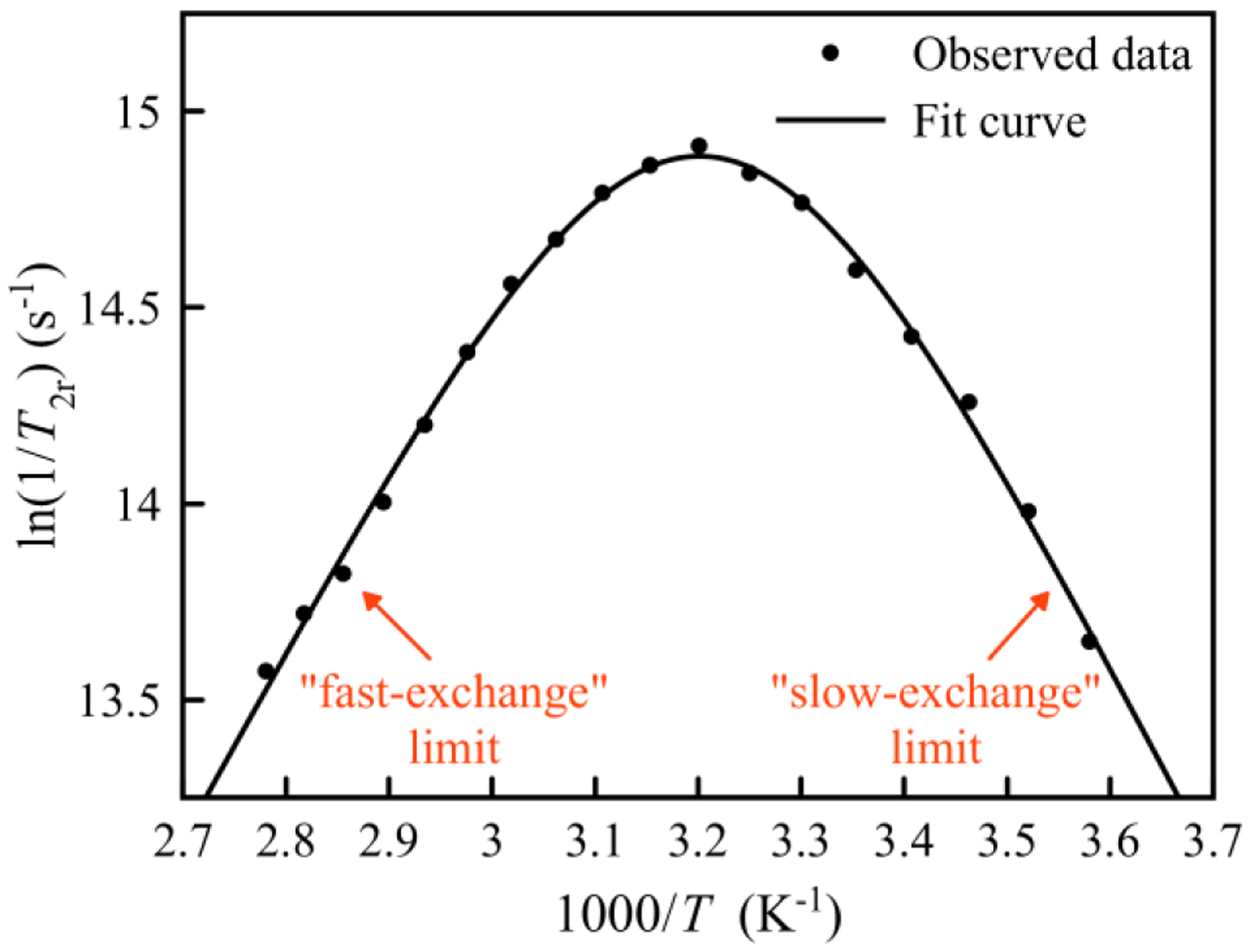
Temperature dependence of the reduced transverse 17O relaxation rate of 3.0 mM Gd-OxyMepa complex (pH = 7.4 in 0.1 M MOPS).
A qualitative visual of the 1/T2r data in Figure 4 shows that this quantity reaches a maximum when the temperature is approximately 40 °C, showing both a “slow-exchange” regime below this temperature and a “fast-exchange” regime above this temperature. This slow-to-fast changeover was also observed for Gd-DOTA ( = 4.1 × 106 s−1) and Gd-DTPA ( = 3.3 × 106 s−1),94,95 both of which have comparable water exchange rates. The related Gd-Octapa complex has a slightly faster water-exchange rate ( = 5.0 × 106 s−1)16 than Gd-OxyMepa. The ΔH‡ for this process in the Gd-Octapa complex (ΔH‡ = 40.1 kJ·mol−1)16 is effectively the same as that measured for Gd-OxyMepa. Thus, the greater of Gd-Octapa is a consequence of its larger ΔS‡.
Conclusion.
OxyMepa and Oxyaapa, two open-chain picolinate-containing chelators with a central ethereal oxygen donor, were synthesized, and their coordination chemistry with lanthanide ions was investigated in detail. Not surprisingly, the heptadentate chelator OxyMepa formed complexes with much lower thermodynamic affinities and kinetic inertnesses in comparison to the nonadentate chelator Oxyaapa. An interesting feature within these two ligand systems was the large conformational shifts that they underwent upon binding lanthanide ions of different sizes. A ligand design strategy suggested by this observation would be to reduce conformational freedom of such systems in order to enhance lanthanide ion selectivity. Although we hypothesized that the presence of an additional hard oxygen donor from the ether backbone would give rise to enhanced stability, our measurement of the thermodynamic stability of showed no significant increase in affinity in comparison to related chelators like Pypa and Octapa that contain fewer oxygen donors. This lack of significant increase in affinity can be accounted for by the fact that ethereal oxygen atoms are weaker donors in a general sense than pyridine, regardless of hard-soft acid-base considerations. In any case, the kinetic inertness of Lu-Oxyaapa was found to be excellent, and thus additional studies probing the use of this ligand in 177Lu3+ radiopharmaceutical agents are warranted.
Supplementary Material
Acknowledgement.
This research was supported in part by seed funding from the Academic Integration grant program at Cornell University and from the College of Arts and Sciences at Cornell University. This research made use of the NMR Facility at Cornell University, which is supported, in part, by the NSF under Award CHE-1531632. Prof. P. Chen is thanked for providing access to a fluorimeter.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c00372.
Full characterization of compounds, representative titration data, crystallographic details, coordinates and energies for DFT-optimized structures, details on the variable-temperature 17O NMR measurements (PDF).
Accession Codes
CCDC 1982239–1982240 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif,orbyemailingdata_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
References.
- (1).Parker D; Dickins RS; Puschmann H; Crossland C; Howard JAK Being Excited by Lanthanide Coordination Complexes: Aqua Species, Chirality, Excited-State Chemistry, and Exchange Dynamics. Chem. Rev 2002, 102, 1977–2010. [DOI] [PubMed] [Google Scholar]
- (2).Bottrill M; Kwok L; Long NJ Lanthanides in Magnetic Resonance Imaging. Chem. Soc. Rev 2006, 35, 557–571. [DOI] [PubMed] [Google Scholar]
- (3).Heffern MC; Matosziuk LM; Meade TJ Lanthanide Probes for Bioresponsive Imaging. Chem. Rev 2014, 114, 4496–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Teo RD; Termini J; Gray HB Lanthanides: Applications in Cancer Diagnosis and Therapy. J. Med. Chem 2016, 59, 6012–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kostelnik TI; Orvig C Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem. Rev 2019, 119, 902–956. [DOI] [PubMed] [Google Scholar]
- (6).Wahsner J; Gale EM; Rodríguez-Rodríguez A; Caravan P Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers. Chem. Rev 2019, 119, 957–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Caravan P; Esteban-Gómez D; Rodríguez-Rodríguez A; Platas-Iglesias C Water Exchange in Lanthanide Complexes for MRI Applications. Lessons Learned over the Last 25 Years. Dalton Trans 2019, 48, 11161–11180. [DOI] [PubMed] [Google Scholar]
- (8).Bünzli J-CG Lanthanide Luminescence for Biomedical Analyses and Imaging. Chem. Rev 2010, 110, 2729–2755. [DOI] [PubMed] [Google Scholar]
- (9).Stasiuk GJ; Long NJ The Ubiquitous DOTA and its Derivatives: the Impact of 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic Acid on Biomedical Imaging. Chem. Commun 2013, 49, 2732–2746. [DOI] [PubMed] [Google Scholar]
- (10).Cacheris WP; Nickle SK; Sherry AD Thermodynamic study of Lanthanide Complexes of 1,4,7-Triazacyclononane-N,N′,N″-triacetic Acid and 1,4,7,10-Tetraazacyclododecane-N,N′,N″,N‴-tetraacetic Acid. Inorg. Chem 1987, 26, 958–960. [Google Scholar]
- (11).Rodríguez-Rodríguez A; Esteban-Gómez D; Tripier R; Tircsó G; Garda Z; Tóth I; de Blas A; Rodríguez-Blas T; Platas-Iglesias C Lanthanide(III) Complexes with a Reinforced Cyclam Ligand Show Unprecedented Kinetic Inertness. J. Am. Chem. Soc 2014, 136, 17954–17957. [DOI] [PubMed] [Google Scholar]
- (12).Rodríguez-Rodríguez A; Regueiro-Figueroa M; Esteban-Gómez D; Tripier R; Tircsó G; Kálmán FK; Bényei AC; Tóth I; de Blas A; Rodríguez-Blas T; Platas-Iglesias C Complexation of Ln3+ Ions with Cyclam Dipicolinates: A Small Bridge that Makes Huge Differences in Structure, Equilibrium, and Kinetic Properties. Inorg. Chem 2016, 55, 2227–2239. [DOI] [PubMed] [Google Scholar]
- (13).Hancock RD Chelate Ring Size and Metal Ion Selection. The Basis of Selectivity for Metal Ions in Open-Chain Ligands and Macrocycles. J. Chem. Educ 1992, 69, 615–621. [Google Scholar]
- (14).Wängler B; Schirrmacher R; Bartenstein P; Wängler C Chelating Agents and Their Use in Radiopharmaceutical Sciences. Mini-Rev. Med. Chem 2011, 11, 968–983. [DOI] [PubMed] [Google Scholar]
- (15).Price EW; Orvig C Matching Chelators to Radiometals for Radiopharmaceuticals. Chem. Soc. Rev 2014, 43, 260–290. [DOI] [PubMed] [Google Scholar]
- (16).Platas-Iglesias C; Mato-Iglesias M; Djanashvili K; Muller RN; Vander Elst L; Peters JA; de Blas A; Rodríguez-Blas T Lanthanide Chelates Containing Pyridine Units with Potential Application as Contrast Agents in Magnetic Resonance Imaging. Chem. - Eur. J 2004, 10, 3579–3590. [DOI] [PubMed] [Google Scholar]
- (17).Price EW; Zeglis BM; Cawthray JF; Ramogida CF; Ramos N; Lewis JS; Adam MJ; Orvig C H4octapa-Trastuzumab: Versatile Acyclic Chelate System for 111In and 177Lu Imaging and Therapy. J. Am. Chem. Soc 2013, 135, 12707–12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Price EW; Edwards KJ; Carnazza KE; Carlin SD; Zeglis BM; Adam MJ; Orvig C; Lewis JS A Comparative Evaluation of the Chelators H4octapa and CHX-A″-DTPA with the Therapeutic Radiometal 90Y. Nucl. Med. Biol 2016, 43, 566–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ramogida CF; Cawthray JF; Boros E; Ferreira CL; Patrick BO; Adam MJ; Orvig C H2CHXdedpa and H4CHXoctapa—Chiral Acyclic Chelating Ligands for 67/68Ga and 111In Radiopharmaceuticals. Inorg. Chem 2015, 54, 2017–2031. [DOI] [PubMed] [Google Scholar]
- (20).Price EW; Zeglis BM; Cawthray JF; Lewis JS; Adam MJ; Orvig C What a Difference a Carbon Makes: H4octapa vs H4C3octapa, Ligands for In-111 and Lu-177 Radiochemistry. Inorg. Chem 2014, 53, 10412–10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Price EW; Cawthray JF; Bailey GA; Ferreira CL; Boros E; Adam MJ; Orvig C H4octapa: An Acyclic Chelator for 111In Radiopharmaceuticals. J. Am. Chem. Soc 2012, 134, 8670–8683. [DOI] [PubMed] [Google Scholar]
- (22).Price EW; Zeglis BM; Lewis JS; Adam MJ; Orvig C H6phospa-trastuzumab: Bifunctional Methylenephosphonate-Based Chelator with 89Zr, 111In and 177Lu. Dalton Trans. 2014, 43, 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bailey GA; Price EW; Zeglis BM; Ferreira CL; Boros E; Lacasse MJ; Patrick BO; Lewis JS; Adam MJ; Orvig C H2azapa: a Versatile Acyclic Multifunctional Chelator for 67Ga, 64Cu, 111In, and 177Lu. Inorg. Chem 2012, 51, 12575–12589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li L; Jaraquemada-Peláez M de G.; Kuo, H.-T.; Merkens, H.; Choudhary, N.; Gitschtaler, K.; Jermilova, U.; Colpo, N.; Uribe-Munoz, C.; Radchenko, V.; Schaffer, P.; Lin, K.-S.; Bénard, F.; Orvig, C. Functionally Versatile and Highly Stable Chelator for 111In and 177Lu: Proof-of-Principle Prostate-Specific Membrane Antigen Targeting. Bioconjugate Chem. 2019, 30, 1539–1553. [DOI] [PubMed] [Google Scholar]
- (25).Li L; Jaraquemada-Peláez M de G; Aluicio-Sarduy E; Wang X; Jiang D; Sakheie M; Kuo H-T; Barnhart TE; Cai W; Radchenko V; Schaffer P; Lin K-S; Engle JW; Bénard F; Orvig C [nat/44Sc(pypa)]−: Thermodynamic Stability, Radiolabeling, and Biodistribution of a Prostate-Specific-Membrane-Antigen-Targeting Conjugate. Inorg. Chem 2020, 59, 1985–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cotton SA Scandium, Yttrium & the Lanthanides: Inorganic & Coordination Chemistry. In Encyclopedia of Inorganic Chemistry. John Wiley & Sons, Ltd.: Chichester, U.K., 2006. [Google Scholar]
- (27).Huang C; Bian Z Introduction. In Rare Earth Coordination Chemistry: Fundamentals and Applications; Huang C, Eds.; John Wiley & Sons (Asia) Pte Ltd: Singapore, 2010; pp 1–39. [Google Scholar]
- (28).Baranyai Z; Botta M; Fekete M; Giovenzana GB; Negri R; Tei L; Platas-Iglesias C Lower Ligand Denticity Leading to Improved Thermodynamic and Kinetic Stability of the Gd3+ Complex: The Strange Case of OBETA. Chem. - Eur. J 2012, 18, 7680–7685. [DOI] [PubMed] [Google Scholar]
- (29).Negri R; Baranyai Z; Tei L; Giovenzana GB; Platas-Iglesias C; Bényei AC; Bodnár J; Vágner A; Botta M Lower Denticity Leading to Higher Stability: Structural and Solution Studies of Ln(III)–OBETA Complexes. Inorg. Chem 2014, 53, 12499–12511. [DOI] [PubMed] [Google Scholar]
- (30).Griffin JLW; Coveney P V; Whiting, A.; Davey, R. Design and Synthesis of Macrocyclic Ligands for Specific Interaction with Crystalline Ettringite and Demonstration of a Viable Mechanism for the Setting of Cement. J. Chem. Soc., Perkin Trans 2 1999, 1973–1981. [Google Scholar]
- (31).Mato-Iglesias M; Roca-Sabio A; Pálinkás Z; Esteban-Gómez D; Platas-Iglesias C; Tóth É; de Blas A; Rodríguez-Blas T Lanthanide Complexes Based on a 1,7-Diaza-12-crown-4 Platform Containing Picolinate Pendants: A New Structural Entry for the Design of Magnetic Resonance Imaging Contrast Agents. Inorg. Chem 2008, 47, 7840–7851. [DOI] [PubMed] [Google Scholar]
- (32).Gottlieb HE; Kotlyar V; Nudelman A NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem 1997, 62, 7512–7515. [DOI] [PubMed] [Google Scholar]
- (33).Sweeton FH; Mesmer RE; Baes CF Jr. Acidity Measurements at Elevated Temperatures. VII. Dissociation of Water. J. Solution Chem 1974, 3, 191–214. [Google Scholar]
- (34).Gans P; O’Sullivan B GLEE, a New Computer Program for Glass Electrode Calibration. Talanta 2000, 51, 33–37. [DOI] [PubMed] [Google Scholar]
- (35).Wuhan University. Analytical Chemistry I, 5th ed.; Higher Education Press: Beijing, 2006. [Google Scholar]
- (36).Gans P; Sabatini A; Vacca A Investigation of Equilibria in Solution. Determination of Equilibrium Constants with the HYPERQUAD Suite of Programs. Talanta 1996, 43, 1739–1753. [DOI] [PubMed] [Google Scholar]
- (37).Gans P; Sabatini A; Vacca A Determination of Equilibrium Constants from Spectrophometric Data Obtained from Solutions of Known pH: the Program pHab. Ann. Chim 1999, 89, 45–49. [Google Scholar]
- (38).CrysAlisPro. Rigaku OD: The Woodlands, TX; 2015. [Google Scholar]
- (39).Sheldrick GM SHELXT – Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr., Sect. A: Found. Adv 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sheldrick GM A Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]
- (41).Müller P Practical Suggestions for Better Crystal Structures. Crystallogr. Rev 2009, 15, 57–83. [Google Scholar]
- (42).Brouwer AM Standards for Photoluminescence Quantum Yield Measurements in Solution (IUPAC Technical Report). Pure Appl. Chem 2011, 83, 2213–2228. [Google Scholar]
- (43).Suzuki K; Kobayashi A; Kaneko S; Takehira K; Yoshihara T; Ishida H; Shiina Y; Oishi S; Tobita S Reevaluation of Absolute Luminescence Quantum Yields of Standard Solutions Using a Spectrometer with an Integrating Sphere and a Back-Thinned CCD Detector. Phys. Chem. Chem. Phys 2009, 11, 9850–9860. [DOI] [PubMed] [Google Scholar]
- (44).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr., Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT: 2009. [Google Scholar]
- (45).Lee C; Yang W; Parr RG Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (46).Becke AD Density‐Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- (47).Hehre WJ; Ditchfield R; Pople JA Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys 1972, 56, 2257–2261. [Google Scholar]
- (48).Hariharan PC; Pople JA The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- (49).Dolg M; Stoll H; Savin A; Preuss H Energy-Adjusted Pseudopotentials for the Rare Earth Elements. Theor. Chim. Acta 1989, 75, 173–194. [Google Scholar]
- (50).Maron L; Eisenstein O Do f Electrons Play a Role in the Lanthanide-Ligand Bonds? A DFT Study of Ln(NR2)3; R = H, SiH3. J. Phys. Chem. A 2000, 104, 7140–7143. [Google Scholar]
- (51).Eisenstein O; Maron L DFT Studies of Some Structures and Reactions of Lanthanides Complexes. J. Organomet. Chem 2002, 647, 190–197. [Google Scholar]
- (52).Regueiro-Figueroa M; Esteban-Gómez D; de Blas A; Rodríguez-Blas T; Platas-Iglesias C Understanding Stability Trends along the Lanthanide Series. Chem. - Eur. J 2014, 20, 3974–3981. [DOI] [PubMed] [Google Scholar]
- (53).Marenich A V; Cramer, C. J.; Truhlar, D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- (54).Ammann C; Meier P; Merbach A A Simple Multinuclear NMR Thermometer. J. Magn. Reson 1982, 46, 319–321. [Google Scholar]
- (55).Meiboom S; Gill D Modified Spin-Echo Method for Measuring Nuclear Relaxation Times. Rev. Sci. Instrum 1958, 29, 688–691. [Google Scholar]
- (56).Mathematica, version 11.1; Wolfram Research, Inc.: Champaign, IL: 2017. [Google Scholar]
- (57).Bekey GA; Masri SF Random Search Techniques for Optimization of Nonlinear Systems with Many Parameters. Math. Comput. Simul 1983, 25, 210–213. [Google Scholar]
- (58).Nelder JA; Mead R A Simplex Method for Function Minimization. Comput. J 1965, 7, 308–313. [Google Scholar]
- (59).Hastie T; Tibshirani R; Friedman J Model Assessment and Selection. In The Elements of Statistical Learning: Data Mining, Inference, and Prediction; Springer Science+Business Media: New York, 2009; pp 219–259. [Google Scholar]
- (60).Kálmán FK; Végh A; Regueiro-Figueroa M; Tóth É; Platas-Iglesias C; Tircsó G H4octapa: Highly Stable Complexation of Lanthanide(III) Ions and Copper(II). Inorg. Chem 2015, 54, 2345–2356. [DOI] [PubMed] [Google Scholar]
- (61).Jaraquemada-Peláez M de G; Wang X; Clough TJ; Cao Y; Choudhary N; Emler K; Patrick BO; Orvig C. H4octapa: Synthesis, Solution Equilibria and Complexes with Useful Radiopharmaceutical Metal Ions. Dalton Trans. 2017, 46, 14647–14658. [DOI] [PubMed] [Google Scholar]
- (62).Martell AE; Hancock RD Metal Complexes in Aqueous Solutions; Plenum Press: New York, 1996. [Google Scholar]
- (63).Harris WR; Carrano CJ; Raymond KN Coordination Chemistry of Microbial Iron Transport Compounds. 16. Isolation, Characterization, and Formation Constants of Ferric Aerobactin. J. Am. Chem. Soc 1979, 101, 2722–2727. [Google Scholar]
- (64).Harris DC Quantitative Analytical Chemistry, 8th ed.; W. H. Freeman and Company: New York, 2010. [Google Scholar]
- (65).Piguet C; Bünzli J-CG Mono- and Polymetallic Lanthanide-Containing Functional Assemblies: A Field between Tradition and Novelty. Chem. Soc. Rev 1999, 28, 347–358. [Google Scholar]
- (66).Pearson RG Chemical Hardness: Applications from Molecules to Solids; Wiley-VCH: Weinheim, 1997. [Google Scholar]
- (67).Arnett EM; Wu CY Base Strengths of Some Saturated Cyclic Ethers in Aqueous Sulfuric Acid. J. Am. Chem. Soc 1962, 84, 1684–1688. [Google Scholar]
- (68).Andon RJL; Cox JD; Herington EFG The Ultra-Violet Absorption Spectra and Dissociation Constants of Certain Pyridine Bases in Aqueous Solution. Trans. Faraday Soc 1954, 50, 918–927. [Google Scholar]
- (69).Rim KT; Koo KH; Park JS Toxicological Evaluations of Rare Earths and Their Health Impacts to Workers: A Literature Review. Saf. Health Work 2013, 4, 12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Wang X; Jin T; Comblin V; Lopez-Mut A; Merciny E; Desreux JF A Kinetic Investigation of the Lanthanide DOTA Chelates. Stability and Rates of Formation and of Dissociation of a Macrocyclic Gadolinium(III) Polyaza Polycarboxylic MRI Contrast Agent. Inorg. Chem 1992, 31, 1095–1099. [Google Scholar]
- (71).Stimmel JB; Kull FC Jr. Samarium-153 and Lutetium-177 Chelation Properties of Selected Macrocyclic and Acyclic Ligands. Nucl. Med. Biol 1998, 25, 117–125. [DOI] [PubMed] [Google Scholar]
- (72).Byegård J; Skarnemark G; Skålberg M The Stability of Some Metal EDTA, DTPA and DOTA Complexes: Application as Tracers in Groundwater Studies. J. Radioanal. Nucl. Chem 1999, 241, 281–290. [Google Scholar]
- (73).Port M; Idée J-M; Medina C; Robic C; Sabatou M; Corot C Efficiency, Thermodynamic and Kinetic Stability of Marketed Gadolinium Chelates and Their Possible Clinical Consequences: A Critical Review. BioMetals 2008, 21, 469–490. [DOI] [PubMed] [Google Scholar]
- (74).Moeller T; Thompson LC Observations on the Rare Earths—LXXV: The Stabilities of Diethylenetriaminepentaacetic Acid Chelates. J. Inorg. Nucl. Chem 1962, 24, 499–510. [Google Scholar]
- (75).Thiele NA; Brown V; Kelly JM; Amor-Coarasa A; Jermilova U; MacMillan SN; Nikolopoulou A; Ponnala S; Ramogida CF; Robertson AKH; Rodríguez-Rodríguez C; Schaffer P; Williams C Jr.; Babich JW; Radchenko V; Wilson JJ An Eighteen-Membered Macrocyclic Ligand for Actinium-225 Targeted Alpha Therapy. Angew. Chem. Int. Ed 2017, 56, 14712–14717. [DOI] [PubMed] [Google Scholar]
- (76).Thiele NA; Woods JJ; Wilson JJ Implementing f-Block Metal Ions in Medicine: Tuning the Size Selectivity of Expanded Macrocycles. Inorg. Chem 2019, 58, 10483–10500. [DOI] [PubMed] [Google Scholar]
- (77).Aluicio-Sarduy E; Thiele NA; Martin KE; Vaughn BA; Devaraj J; Olson AP; Barnhart TE; Wilson JJ; Boros E; Engle JW Establishing Radiolanthanum Chemistry for Targeted Nuclear Medicine Applications. Chem.- Eur. J 2020, 26, 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Alvarez S; Alemany P; Casanova D; Cirera J; Llunell M; Avnir D Shape Maps and Polyhedral Interconversion Paths in Transition Metal Chemistry. Coord. Chem. Rev 2005, 249, 1693–1708. [Google Scholar]
- (79).Abrahams BF; Abrahams CT; Haywood MG; Hudson TA; Moubaraki B; Murray KS; Robson R 3d-Metal Derivatives of the [CuI(SO3)4]7− Ion: Structure and Magnetism. Dalton Trans. 2012, 41, 4091–4099. [DOI] [PubMed] [Google Scholar]
- (80).Adonin SA; Peresypkina EV; Sokolov MN; Fedin VP. Trinuclear Iodobismuthate Complex [Na3(Me2CO)12][Bi3I12]: Synthesis and Crystal Structure. Russ. J. Coord. Chem 2014, 40, 867–870. [Google Scholar]
- (81).Wilson JJ; Birnbaum ER; Batista ER; Martin RL; John KD Synthesis and Characterization of Nitrogen-Rich Macrocyclic Ligands and an Investigation of Their Coordination Chemistry with Lanthanum(III). Inorg. Chem 2015, 54, 97–109. [DOI] [PubMed] [Google Scholar]
- (82).Chatterton N; Bretonnière Y; Pécaut J; Mazzanti M An Efficient Design for the Rigid Assembly of Four Bidentate Chromophores in Water-Stable Highly Luminescent Lanthanide Complexes. Angew. Chem. Int. Ed 2005, 44, 7595–7598. [DOI] [PubMed] [Google Scholar]
- (83).Moore EG; Samuel APS; Raymond KN From Antenna to Assay: Lessons Learned in Lanthanide Luminescence. Acc. Chem. Res 2009, 42, 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Binnemans K Interpretation of Europium(III) Spectra. Coord. Chem. Rev 2015, 295, 1–45. [Google Scholar]
- (85).Nonat A; Gateau C; Fries PH; Mazzanti M Lanthanide Complexes of a Picolinate Ligand Derived from 1,4,7-Triazacyclononane with Potential Application in Magnetic Resonance Imaging and Time-Resolved Luminescence Imaging. Chem. - Eur. J 2006, 12, 7133–7150. [DOI] [PubMed] [Google Scholar]
- (86).Nocton G; Nonat A; Gateau C; Mazzanti M Water Stability and Luminescence of Lanthanide Complexes of Tripodal Ligands Derived from 1,4,7-Triazacyclononane: Pyridinecarboxamide versus Pyridinecarboxylate Donors. Helv. Chim. Acta 2009, 92, 2257–2273. [Google Scholar]
- (87).Roca-Sabio A; Bonnet CS; Mato-Iglesias M; Esteban-Gómez D; Tóth É; de Blas A; Rodríguez-Blas T; Platas-Iglesias C Lanthanide Complexes Based on a Diazapyridinophane Platform Containing Picolinate Pendants. Inorg. Chem 2012, 51, 10893–10903. [DOI] [PubMed] [Google Scholar]
- (88).Thibon A; Pierre VC Principles of Responsive Lanthanide-Based Luminescent Probes for Cellular Imaging. Anal. Bioanal. Chem 2009, 394, 107–120. [DOI] [PubMed] [Google Scholar]
- (89).Supkowski RM; Horrocks WD Jr. On the Determination of the Number of Water Molecules, q, Coordinated to Europium(III) Ions in Solution from Luminescence Decay Lifetimes. Inorg. Chim. Acta 2002, 340, 44–48. [Google Scholar]
- (90).Beeby A; Clarkson IM; Dickins RS; Faulkner S; Parker D; Royle L; de Sousa AS; Williams JAG; Woods M Non-Radiative Deactivation of the Excited States of Europium, Terbium and Ytterbium Complexes by Proximate Energy-Matched OH, NH and CH Oscillators: An Improved Luminescence Method for Establishing Solution Hydration States. J. Chem. Soc., Perkin Trans 2 1999, 493–503. [Google Scholar]
- (91).Esteban-Gómez D; de Blas A; Rodríguez-Blas T; Helm L; Platas-Iglesias C Hyperfine Coupling Constants on Inner-Sphere Water Molecules of GdIII-Based MRI Contrast Agents. ChemPhysChem 2012, 13, 3640–3650. [DOI] [PubMed] [Google Scholar]
- (92).Helm L; Merbach AE Inorganic and Bioinorganic Solvent Exchange Mechanisms. Chem. Rev 2005, 105, 1923–1960. [DOI] [PubMed] [Google Scholar]
- (93).Swift TJ; Connick RE NMR‐Relaxation Mechanisms of O17 in Aqueous Solutions of Paramagnetic Cations and the Lifetime of Water Molecules in the First Coordination Sphere. J. Chem. Phys 1962, 37, 307–320. [Google Scholar]
- (94).Micskei K; Helm L; Brücher E; Merbach AE 17O NMR Study of Water Exchange on [Gd(DTPA)(H2O)]2– and [Gd(DOTA)(H2O)]– Related to NMR Imaging. Inorg. Chem 1993, 32, 3844–3850. [Google Scholar]
- (95).Powell DH; Dhubhghaill OMN; Pubanz D; Helm L; Lebedev YS; Schlaepfer W; Merbach AE Structural and Dynamic Parameters Obtained from 17O NMR, EPR, and NMRD Studies of Monomeric and Dimeric Gd3+ Complexes of Interest in Magnetic Resonance Imaging: An Integrated and Theoretically Self-Consistent Approach. J. Am. Chem. Soc 1996, 118, 9333–9346. [Google Scholar]
- (96).Burai L; Tóth É; Sour A; Merbach AE Separation and Characterization of the Two Diastereomers for [Gd(DTPA-bz-NH2)(H2O)]2–, a Common Synthon in Macromolecular MRI Contrast Agents: Their Water Exchange and Isomerization Kinetics. Inorg. Chem 2005, 44, 3561–3568. [DOI] [PubMed] [Google Scholar]
- (97).Caravan P; Parigi G; Chasse JM; Cloutier NJ; Ellison JJ; Lauffer RB; Luchinat C; McDermid SA; Spiller M; McMurry TJ Albumin Binding, Relaxivity, and Water Exchange Kinetics of the Diastereoisomers of MS-325, a Gadolinium(III)-Based Magnetic Resonance Angiography Contrast Agent. Inorg. Chem 2007, 46, 6632–6639. [DOI] [PubMed] [Google Scholar]
- (98).Peters JA; Huskens J; Raber DJ Lanthanide Induced Shifts and Relaxation Rate Enhancements. Prog. Nucl. Magn. Reson. Spectrosc 1996, 28, 283–350. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.