

Summary:
Adoptive cell transfer (ACT) with tumor-infiltrating lymphocytes (TILs) can generate durable clinical responses in patients with metastatic melanoma and ongoing trials are evaluating efficacy in other advanced solid tumors. The aim of this study was to develop methods for the expansion of tumor-reactive TIL from resected soft tissue sarcoma to a degree required for the ACT. From 2015 to 2018, 70 patients were consented to an institutional review board-approved protocol, and fresh surgical specimens were taken directly from the operating room to the laboratory. Fragments of the tumor (1 mm3) or fresh tumor digest were placed in culture for a period of 4 weeks. Successfully propagated TIL from these cultures were collected and analyzed by flow cytometry. TIL were cocultured with autologous tumor and function was assessed by measurement of interferon-γ in the supernatant by enzyme-linked immunosorbent assay. Initial TIL cultures were further expanded using a rapid expansion protocol. Nearly all specimens generated an initial TIL culture (91% fragment method, 100% digest method). The phenotype of the TIL indicated a predominant CD3+ population after culture (43% fragment, 52% digest) and TIL were responsive to the autologous tumor (56% fragment, 40% digest). The cultured TIL expanded to a degree required for clinical use following rapid expansion protocol (median: 490-fold fragment, 403-fold digest). The data demonstrate the feasibility of TIL culture from fresh soft tissue sarcoma. The derived TIL have tumor-specific reactivity and can be expanded to clinically relevant numbers. An active ACT clinical trial using the methods described in this report is now approved for patients with metastatic soft tissue sarcoma.
Keywords: soft tissue sarcoma, TIL, adoptive cell therapy
Soft tissue sarcoma represents a heterogenous set of malignancies which arise from connective tissue. Despite aggressive multidisciplinary treatment of localized disease, 30% of those with high-grade tumors will develop distant metastatic disease or unresectable recurrent disease, collectively called “advanced disease.” Patients with advanced sarcoma have limited effective systemic treatment options. Options that exist confer a median duration of response of only 9 months and a median survival of only 1 year.1,2 Unfortunately, many patients with sarcoma fall within the pediatric and young adult age group; therefore, death from sarcoma results in a significant loss of productive life-years for malignancies affecting both children and adults.
Recent advances in immunotherapy have shown promise for patients with advanced, widely metastatic malignancies. There are 2 primary strategies to deliver immunotherapy. First, monoclonal antibodies have been developed which abrogate several tumor-specific suppressive pathways. These new medications, collectively called immune checkpoint blockade, have revolutionized cancer therapy in the past decade for patients with melanoma and non–small cell lung cancer, among others.3–9 Second, cellular immunotherapy strategies have been developed which involve infusion of tumor-specific T cells. This strategy has been most recently popularized using gene-modified T cells which are targeted towards a single tumor-specific antigen.10 The chimeric antigen receptor (CAR) T-cell strategy has been very successful in patients with diffuse large B-cell lymphoma, and many trials are underway using this treatment in other hematologic malignancies.11,12 Unfortunately, the same method has not, to date, shown convincing efficacy in solid tumors.13
Reports of immunotherapy-based clinical trials in patients with sarcoma have demonstrated results generally inferior to other solid tumors such as metastatic melanoma and non–small cell lung cancer.14 The seminal trial of checkpoint inhibitor therapy for sarcoma patients utilized anti-programmed death ligand-1 (PD-1) treatment. Patients on SARC028 were enrolled into the separate bone and soft tissue sarcoma cohorts and treated with anti-PD-1 monotherapy. Results demonstrated an objective response only in soft tissue sarcoma patients, all of whom (4/10, 40%) had undifferentiated pleomorphic sarcoma.14 Cellular immunotherapy trials for patients with soft tissue sarcoma have included 2 landmark studies leveraging T cells targeting known tumor antigens. In one trial, patients received a transgenic T-cell receptor (TCR) product targeting the cancer-testis antigen NY-ESO1. Both melanoma and synovial sarcoma patients were enrolled on this protocol and a partial response was demonstrated in 4/6 (67%) patients with synovial sarcoma with a duration of response between 5 and 18 months.15 In another report, a CAR-T–cell product targeting human epidermal growth factor 2 was used in sarcoma patients with tumors expressing the human epidermal growth factor 2 receptor. Of 18 patients treated (16 with osteosarcoma), there were no objective responses noted. The authors noted that this may have been due to the lack of nonmyeloablative lymphodepletion before cell infusion.13
There are several theoretical mechanisms that underlie the lack of efficacy associated with CAR-T treatment of solid malignancies, and the application of cellular therapy to solid malignancies may require a different approach. Adoptive cell immunotherapy using tumor-infiltrating lymphocytes (TILs) for patients with metastatic melanoma is one such approach that has resulted in durable complete responses that are not possible with other conventional or emerging investigational treatments.16–19 This approach at cellular immunotherapy relies on the polyclonal population of T cells that reside within the tumor, suppressed by an array of oncogenic mechanisms. By culturing the tumor-specific TIL ex vivo, away from the suppressive tumor microenvironment, the balance is shifted in favor of the tumor-reactive lymphocytes. Prior work at our institution and others have focused on methods to optimize TIL therapy for solid tumors19–30 and our subsequent clinical trials have shown durable efficacy on par with other reports.17,31 Compared with this prior experience, the primary challenge to the culture of TIL from soft tissue sarcoma is the densely fibrotic stroma, which may inhibit the TIL emigration required by classic methods. The purpose of this work was to develop a reliable method to generate a tumor-reactive TIL product from soft tissue sarcoma in sufficient numbers for clinical use.
The presence and clinical significance of TIL in soft tissue sarcoma have been demonstrated previously, though, to our knowledge, there has been no report of successful ex vivo TIL culture.32–35 Here we present preclinical data supporting the use of adoptive cell therapy for patients with sarcoma. First, we show that protocols developed and validated from our significant melanoma experience using tumor fragments can also yield TIL cultures from soft tissue sarcoma. Second, we show that modifications in the manufacturing process, specifically the culture of TIL from fresh tumor digest, enhance the ability to generate a tumor-specific TIL product from resected adult primary sarcoma specimens. Using these methods, the TIL grown from sarcoma specimens were expanded to a clinically meaningful number, and these TIL have tumor-specific activity.
MATERIALS AND METHODS
All patients were enrolled on an institutional review board–approved tissue acquisition protocol and informed consent for the research on tumor tissue was obtained separately from the operative informed consent document. Eligibility criteria included age over 18 years, pathologically confirmed diagnosis of a soft tissue sarcoma, and recommended treatment plan included a curative-intent resection. Patients were enrolled only if, in the surgeon’s discretion, there would be sufficient excess tissue for research purposes outside of the diagnostic pathologic assessment. Patients receiving neoadjuvant chemotherapy and/or radiation (within 42 d of resection) were included and all subtypes of soft tissue sarcoma were considered eligible. Patients with metastatic disease were included if the metastastectomy was performed as a standard of care procedure.
Following tumor resection, a portion of the tumor (≥ 1 g) was prosecuted by the surgeon for research purposes, in a sterile manner after the surgical margin was delineated with ink. The specimen was deidentified and labeled with a unique study ID. One quarter of the specimen was processed for TIL culture, and the remaining three quarters of the specimen were used to generate a tumor line for functional assays. The 2 methods for TIL culture were classified as: (1) the “fragment” method which was identical to those employed on prior metastatic melanoma adoptive cell transfer clinical trials, and (2) the “digest” method which was a second approach developed for soft tissue sarcoma. In addition, the hematoxylin and eosin sections of the tumor were reviewed with a single soft tissue pathologist (M.B.) in an effort to understand the architectural features of the immune infiltration.
The Fragment Method
Tumors were prepared using techniques previously described.17,31 Briefly, tumor fragments were minced into pieces ~1 mm3 in size. Tumor fragments were explanted into at least 12 wells (1 fragment/well) of a 24-well plate in culture media containing interleukin (IL-2) (6000 IU/mL). One half of the media containing IL-2 was replenished every 3 days to maintain the proliferation of the TIL. In some cases, the agonistic anti-41BB antibody was used in the TIL culture at 10 μg/mL initial media concentration and 1 μg/mL for subsequent media exchanges (Bristol-Myers Squibb, New York City, NY). When TIL were 80% confluent within the well, they were expanded to a new well of a 24-well plate.
The Digest Method
The whole tumor from the operating room was processed into a single cell suspension using both mechanical and enzymatic disruption. First, the tumor was mechanically dissociated using a scalpel or rotary tool (Tissue Tearor; Fisher Scientific, Hampton, NH). The tumor slurry was then placed into enzymatic media containing DNase type IV (30,000 U/L), hyaluronidase type V (100 mg/L), collagenase type IV (1000 mg/L), gentamicin (500 mg/L), penicillin-streptomycin (5000 U/mL), L-glutamine (292 mg/L), amphotericin B (62.5 μg/L) and processed on a GentleMACS (Miltenyi Biotec, Bergisch Gladbach, Germany). After filtration through a 100 μm filter, the red blood cells within the sample were then lysed using RBC Lysis Buffer (Biolegend, San Diego, CA). The cell suspension was then assessed for viability using Trypan blue and then resuspended in complete media containing IL-2 (6000 IU/mL) at a concentration of 5×105 live cells/mL. A total of 1×106 live cells were placed into a single well of a 24-well tissue culture plate. One half of the media containing IL-2 (6000 IU/mL) was replenished every 3 days to maintain the proliferation of the TIL. A portion of the single-cell suspension was cryopreserved for downstream analyses.
TIL Phenotype
To determine the phenotype of the immune infiltrate in fresh digest and cultured TIL, flow cytometry was performed on a Celesta flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ). The complete profile of the product included cell surface markers CD3, CD4, CD8, CD56, α/β TCR, γ/δ TCR, PD-1, and CD16. Data were acquired using FACSDiva (Becton-Dickinson and Company) analyzed using FlowJo software (Becton, Dickinson and Company). The phenotype of the TIL product was reported as a percentage of the live, single cell population.
Tumor Reactivity
To directly assess antitumor function in vitro, TIL were cocultured with autologous or HLA-matched sarcoma tumor cells at a 1:1 ratio for 24 hours. The supernatant was collected, and interferon-γ (IFNγ) concentration was measured using an enzyme-linked immunosorbent assay. Comparisons were made between IFNγ secretion in response to autologous/matched tumor and HLA-mismatched tumor to determine tumor-specific reactivity. Furthermore, anti-class I major histocompatibility complex blocking antibody (W6/32 clone; Becton, Dickinson and Company) was used to determine CD8+ T-cell–specific mediated tumor response. TIL cultures were deemed to have tumor-specific reactivity if > 100 pg/mL IFNγ was present in the supernatant and there was a statistically significant difference between the autologous tumor and anti-class I major histocompatibility complex blocking condition.
The IFNγ concentration assay as previously shown was used to understand the tumor-specific function in a manner consistent with other reports of TIL expansion from solid tumors. To better understand a more global reactivity profile, the supernatant derived from the coculture method as previously shown was also assayed on a multiplex cytokine platform (V-PLEX Proinflammatory Panel 1 Human Kit; Meso Scale Discovery, Rockville, MD) and the results for 3 separate cytokines were compared.
Rapid Expansion Protocol (REP)
To demonstrate the feasibility of the complete production process, initial TIL cultures were expanded using a REP developed and employed on prior melanoma clinical trials.17,31 TIL were loaded into GREX-10 flasks (Wilson-Wolf, St. Paul, MN) with media containing IL-2 (3000 IU/mL), OKT-3 (30 ng/mL), and irradiated donor lymphocytes. Media was added on days 4, 7, and 10. Cells were counted on days 7 and 14 using Trypan blue. The phenotype and reactivity of the expanded TIL were assessed by methods as previously shown.
RESULTS
Patient Data
Seventy tumors were obtained from the operating room between March 2015 and December 2018. The median age was 68 years (24–92 y), 10% were in the adolescent-young adult population (age 18–40 y), and 64% were male (Table 1). The sarcoma specimens included: 70% high-grade, 7% intermediate-grade, 23% low-grade tumors. Specimens were obtained from the majority of primary tumors (68%, 21% recurrent, 11% metastatic). Neoadjuvant treatment (within 90 d preceding resection) included chemotherapy (11%), chemoradiation (4%), radiation alone (13%), and no treatment (70%). Soft tissue sarcoma subtype diagnoses included undifferentiated pleomorphic sarcoma (19%), dedifferentiated liposarcoma (29%), well-differentiated liposarcoma (11%), malignant peripheral nerve sheath tumor (4%), myxoid liposarcoma (4%), gastrointestinal stromal tumor (4%), and other (29%).
TABLE 1.
Patient Demographics
| n (%) | |
|---|---|
| Age (y) | |
| Adolescent-young adult (age 18–40) | 7 (10.0) |
| Age > 40 | 63 (90.0) |
| Sex | |
| Male | 45 (64.3) |
| Female | 25 (35.7) |
| Diagnosis | |
| Dedifferentiated liposarcoma | 20 (28.6) |
| Undifferentiated pleomorphic sarcoma | 13 (18.6) |
| Well-differentiated liposarcoma | 8 (11.4) |
| Malignant peripheral nerve sheath tumor | 3 (4.3) |
| Myxoid liposarcoma | 3 (4.3) |
| Gastrointestinal stromal tumor | 3 (4.3) |
| Other | 20 (28.6) |
| Grade | |
| Low | 16 (2.9) |
| Intermediate | 5 (7.1) |
| High | 49 (70.0) |
| Disease state | |
| Primary | 47 (67.1) |
| Recurrent | 15 (21.4) |
| Metastatic | 8 (11.4) |
| Prior treatment | |
| None | 49 (70.0) |
| Chemotherapy | 8 (11.4) |
| Radiation therapy | 9 (12.9) |
| Chemotherapy plus radiation | 3 (4.3) |
| Gene-modified T-cell therapy | 1 (1.4) |
Clinical data regarding patients from which the tissue sample was obtained for tumor-infiltrating lymphocyte culture. A single tumor was obtained from each patient.
Lymphocytic Infiltrate Within Soft Tissue Sarcoma
Resected tumors (n = 4) were stained with hematoxylin and eosin to evaluate patterns of lymphocyte infiltration. All tumors demonstrated a paucicellular background with increased stroma relative to the cellular component of the tumor. Two phenotypes of infiltration were evident: lymphocyte excluded, and lymphocyte infiltrated. In the lymphocyte excluded tumors, the TIL were loculated within the dense fibrous bands of the tumor or the tumor capsule. In lymphocyte infiltrated, the TIL were dispersed evenly throughout the cellular component of the tumor but still bound by the hallmark feature of connective tissue neoplasms—a dense acellular connective tissue stroma (Fig. 1A). Tumors with the lymphocyte infiltrated phenotype all generated an acceptable initial TIL culture (pre-REP > 2×107) while none of those with an immune excluded phenotype generated an acceptable TIL culture.
FIGURE 1.
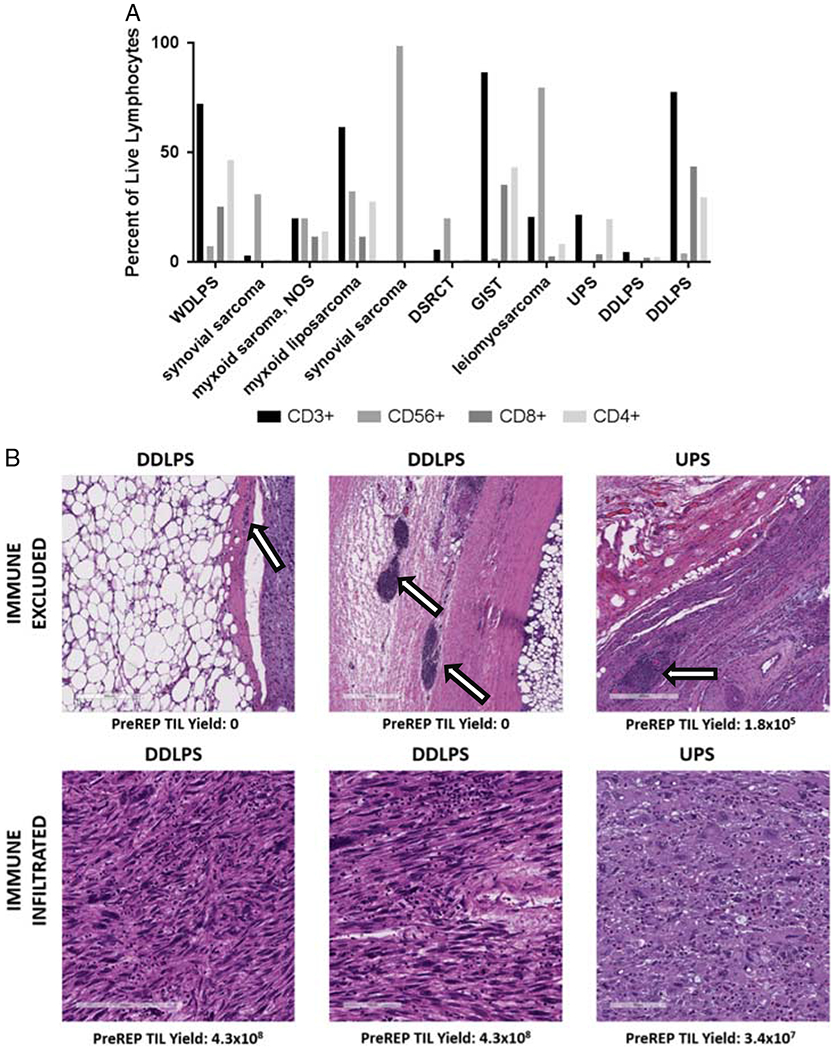
De novo immune infiltrate in soft tissue sarcoma. A, Flow cytometry of fresh tumor digest reveals infiltration of CD8+, CD4+ T cells, and CD3-CD56+ natural killer cells with significant heterogeneity of the immune infiltrate across sarcoma subtype histologies. B, Hematoxylin and eosin (stain of resected specimens reveals 2 patterns of infiltration: immune excluded and immune infiltrated. Nests of lymphocytes are trapped within the fibrous capsule of tumors (arrow) in tumors that are immune excluded. DDLPS indicates dedifferentiated liposarcoma; DSRCT, desmoplastic small round cell tumor; GIST, gastrointestinal stromal tumor; NOS, not otherwise specified; REP, rapid expansion protocol; TIL, tumor-infiltrating lymphocyte; UPS, undifferentiated pleomorphic sarcoma; WDLPS, well-differentiated liposarcoma.
Fresh tumor digest was analyzed by flow cytometry for lymphocyte populations in 11 patients from 10 different subtype diagnoses. Lymphocyte subpopulations included CD3+ (median: 20.6%, range: 0.4%–86.7%), CD3−CD56+ (20.1%, 0%–98.6%), CD3+CD4+ (13.9%, 0.37%–46.6%), and CD3+CD8+ (3.63%, 0.11%–43.8%). There was significant heterogeneity between samples but gastrointestinal stromal tumor and liposarcoma (both well-differentiated and dedifferentiated) contained the highest percentage of CD3+ lymphocytes (86.7% and 51.6%, respectively) (Fig. 1B).
Primary TIL Culture From Tumor Fragments
Primary cultures were initiated from fragments of 64 tumors. Lymphocytes were grown from nearly all tumors (58/64, 91%). Five tumors (3 dedifferentiated liposarcoma, 1 undifferentiated pleomorphic sarcoma, and 1 malignant peripheral nerve sheath tumor) did not generate a primary TIL culture, and 1 tumor was contaminated early in the culture. The median number of TIL grown from 12 tumor fragments was 4.5×106 live lymphocytes (range: 1.2×104−4.4×108) and 16 tumors (25%) generated a primary TIL culture with ≥ 2×107 live lymphocytes, the threshold required for subsequent expansion to a clinically significant infusion product (Fig. 2A). Tumor fragments cultured in media containing both IL-2 and agonistic anti-41BB antibody generated more TIL than those culture in IL-2 alone (average: 4.55×107 vs. 1.33×106, P=0.0007). The lymphocyte phenotypes of the primary TIL cultures derived from fragments included CD3+ (median: 64.6%, range: 3.2%–98.7%), CD3+CD8+ (54.2%, 3%–95.4%), CD3+CD4+ (2.5%, 0.03%–44.73%), and CD3−CD56+ (19.5%, 0.05%–93.43%) (Fig. 2B).
FIGURE 2.
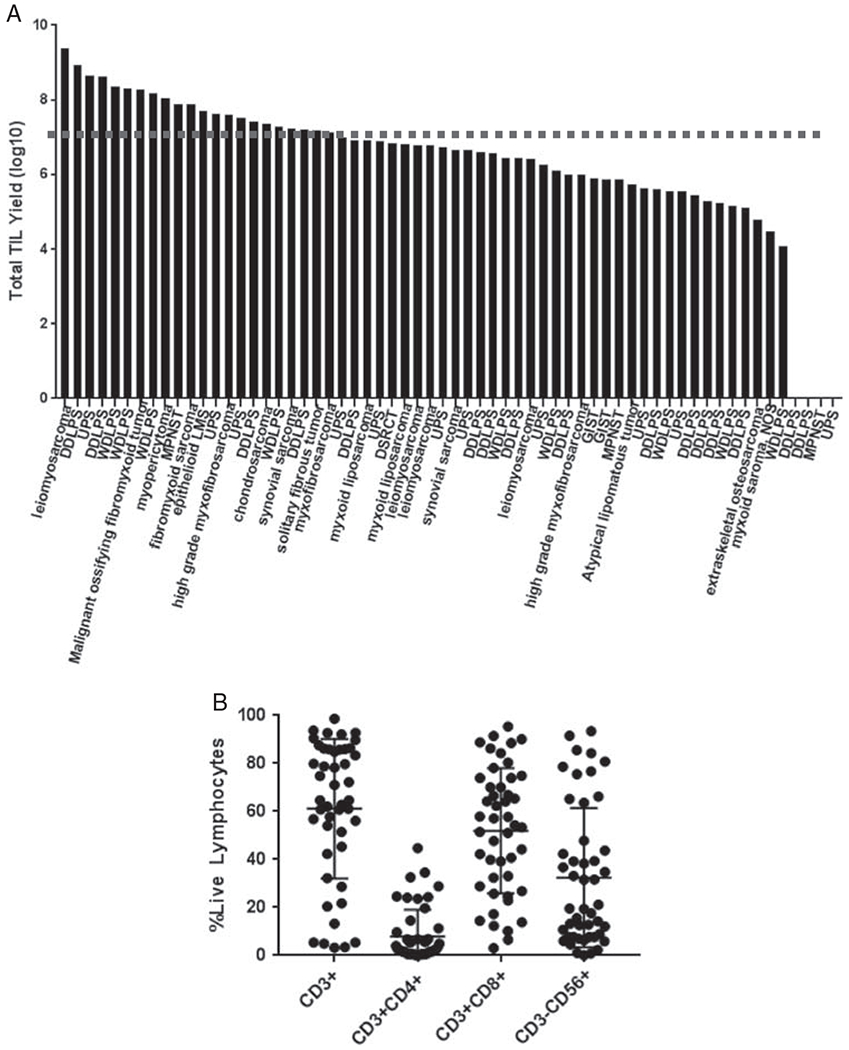
Cultured TIL (pre-rapid expansion protocol) from fresh soft tissue sarcoma specimens. A, Expansion of CD3+ TIL at a threshold for clinical grade expansion (2×107, dashed line) is possible at 4 weeks for several different histologic subtypes. B, Distribution of TIL culture phenotype from multiple tumor fragments reveals significant heterogeneity. DDLPS indicates dedifferentiated liposarcoma; DSRCT, desmoplastic small round cell tumor; GIST, gastrointestinal stromal tumor; LMS, leiomyosarcoma; MPNST, malignant peripheral nerve sheath tumor; NOS, not otherwise specified; TIL, tumor-infiltrating lymphocyte; UPS, undifferentiated pleomorphic sarcoma; WDLPS, well-differentiated liposarcoma.
Primary TIL Culture From Fresh Tumor Digest
To overcome the densely fibrotic stroma associated with soft tissue sarcoma, an alternative method for the initial culture of TIL from soft tissue sarcoma was undertaken in 22 patients. As previously shown, the culture conditions were the same as the fragment method except for the starting tumor material in a culture where a tumor digest was used. Using fold expansion as a primary endpoint, 15 (68%) tumors generated TIL that expanded > 2-fold from the number of total cells at initiation (median: 2.96-fold, range: 0.11- to 143.35-fold) (Fig. 3A). When normalized to the CD3+ fraction of the primary tumor, the TIL expansion was greater (median: 23.04, range: 0.26–663.67). The phenotype of the TIL grown using the digest method compared with the fragment method was comparable for individual populations including CD3+ (P=0.39), CD3−CD56+ (P=0.23), CD3+CD4+ (P = 0.32), and CD3+CD8+ (P = 0.05) (Fig. 3B).
FIGURE 3.
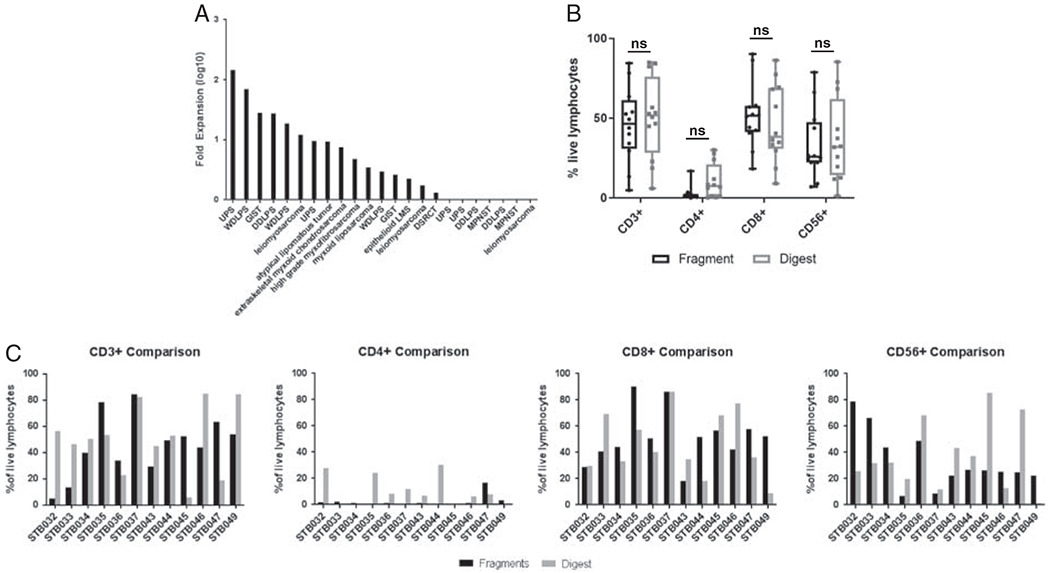
Cultured tumor-infiltrating lymphocyte (TIL) (pre-rapid expansion protocol) from soft tissue sarcoma using digest method. A, TIL can be expanded from soft tissue sarcoma using the digest method to a clinically meaningful degree. B, The phenotype of primary TIL cultures in matched pairs derived the from digest method is similar to the fragment method. C, Data regarding the expansion of lymphocytes from the fresh tumor are presented for individual samples to demonstrate linked differences between digest and fragment method of TIL culture. DDLPS indicates dedifferentiated liposarcoma; DSRCT, desmoplastic small round cell tumor; GIST, gastrointestinal stromal tumor;LMS, leiomyosarcoma; MPNST, malignant peripheral nerve sheath tumor; NS, nonsignificant; UPS, undifferentiated pleomorphic sarcoma; WDLPS, well-differentiated liposarcoma.
Expansion of Primary TIL Culture
A REP was used to expand the primary TIL culture for 10 samples to demonstrate feasibility in the process of product manufacturing. This subset was chosen to include a representative sample of each histology with adequate primary TIL growth. This method resulted in a median expansion of 443-fold (range: 16- to 2541-fold). The expansion of TIL derived from tumor fragments (median: 476-fold, n = 7) was similar to TIL derived from tumor digest (median: 410-fold, n = 3). While all primary TIL cultures demonstrated some degree of expansion, there was one TIL culture derived from leiomyosarcoma which demonstrated expansion < 100-fold (Fig. 4A).
FIGURE 4.
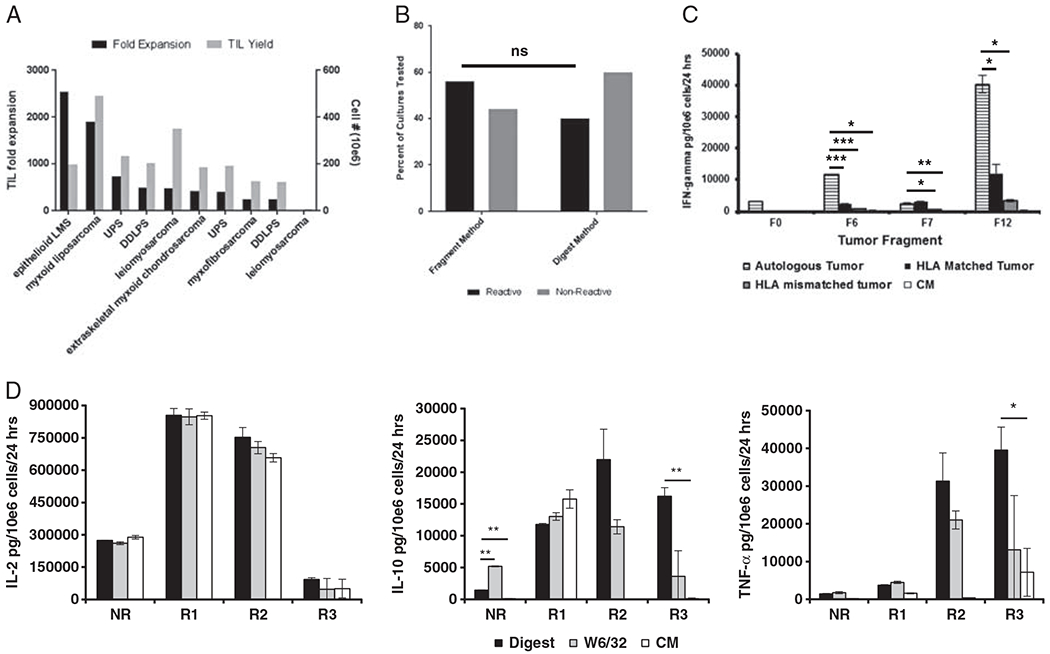
TIL from soft tissue sarcoma can be expanded and have tumor-specific activity. A, Cultured TIL can be expanded through rapid expansion protocol to achieve quantities sufficient for infusion. B, There is no difference in the likelihood of tumor-specific reactivity in TIL cultures derived from fragment compared with those derived from the fresh digest. C, Individual patient reactivity profile demonstrating 2 fragment TIL cultures (F6, F12) with tumor-specific activity. D, Multiplex cytokine assay results following coculture of TIL with tumor digest, class I major histocompatibility complex blocking antibody (W6/32) and control media (CM). Four separate tumor specimens are represented; 1 nonreactive (NR) and 3 reactive samples (R1, R2, R3). *P = 0.05; **P = 0.01; ***P = 0.001. DDLPS indicates dedifferentiated liposarcoma; IFN, interferon; IL, interleukin; LMS, leiomyosarcoma; NS, nonsignificant; TIL, tumor-infiltrating lymphocyte; TNF, tumor necrosis factor; UPS, undifferentiated pleomorphic sarcoma.
Tumor-specific Reactivity
The cultured TIL product was assessed for tumor-specific reactivity by IFNγ release assay in 51 samples. The tumor-specific activity was noted in 56.3% of patients (9/16) using the fragment method and in 40% (14/35) using the digest method (P = 0.37 comparing fragment vs. digest methods) (Fig. 4B). Individual patient TIL cultures often demonstrated notable heterogeneity in tumor-specific reactivity between those derived from separate fragments of the same tumor (Fig. 4C). This heterogeneity in response was mirrored by a diversity of lymphocyte subpopulations within the separate TIL cultures. A more comprehensive assessment of cytokine production after coculture with autologous tumor demonstrated increased secretion of tumor necrosis factor-α and IL-10 in one sample that also demonstrated reactivity as measured by conventional IFNγ release assay. There was no decrease in IL-2 after coculture with autologous tumor noted in the reactive samples (Fig. 4D).
DISCUSSION
The treatment options available for patients with advanced soft tissue sarcoma are limited, and current regimens produce a median overall survival of 12 months. Despite recent notable advancements in therapy for other solid tumors, patients with soft tissue sarcoma have not seen a commensurate increase in the number of effective treatment options. This is particularly true for treatments focused on enhancing the immune system response to malignancy. Immunotherapy options for metastatic melanoma, non–small cell lung cancer, and genitourinary malignancies have revolutionized care and outcomes for these patients.
The 2 most promising immunotherapy approaches have been monoclonal antibody therapy designed to eliminate tumor-specific immune suppressive mechanisms and cellular immunotherapy designed to deliver tumor-specific lymphocytes capable of inducing direct tumor cell death. The former option has not produced the degree of efficacy in soft tissue sarcoma patients derived from other solid tumor malignancies. Similarly, approaches using cellular immunotherapy have generally not been successful in treating patients with solid tumors, which is in stark contrast to the success of this approach for hematologic malignancies. One such reason for this difference is the clonal nature of the cellular product, which is not sufficient to address the intratumoral antigenic heterogeneity of solid tumors. Adoptive cell therapy with TILs addresses this heterogeneity as it is fundamentally based on a polyclonal lymphocyte product.
To our knowledge, we present the first data on the expansion of a tumor-specific lymphocyte product from soft tissue sarcoma. The methods described in this work were undertaken relative to the fundamental thresholds that are required to justify the expansion of adoptive cell therapy to solid tumors, namely the degree of TIL expansion and tumor-specific activity. The data demonstrate the feasibility and success rate of this process in sarcoma patients with a wide array of sarcoma histologic subtypes. To demonstrate the feasibility of this process in support of an adoptive cell therapy clinical trial, all aspects of the manufacturing process were completed in a serial fashion (ie, pre-REP culture through full-scale REP) with a success rate of 90% (9/10) for those tumors that were used to test the entire manufacturing process.
Expansion of a TIL product was not unique to single subtype histology, though the interpatient variability was large. In contrast to prior methods of TIL expansion from solid tumors, it should be noted that the samples in our work were derived from primary tumors rather than tumor-bearing lymph nodes as has been done most often in prior reports. This difference is not subtle, as the immune landscape within the primary tumor is notably different from that within a tumor-bearing lymph node. As soft tissue sarcoma metastasizes to lymph nodes only in very rare cases, we believe the methods reported here for the culture of TIL from primary tumors are significant to the application of adoptive cell therapy in patients with soft tissue sarcoma.
The primary objective of this work was to establish a method to derive a tumor-specific TIL product for infusion in patients with advanced soft tissue sarcoma. The initial work was largely based on prior methods using tumor fragments for the initiation of the lymphocyte cultures. While early data suggested that the addition of agonistic anti-41BB antibody improved the yield of initial TIL culture, the commercial source for this product discontinued production and thus another method was needed to reliably generate an initial TIL culture. This new method involved using tumor digest rather than fragments and was developed with 2 primary aims. First, this method offers a way to scale the initial growth process, thereby allowing a starting cell number from which a target number of lymphocytes are generated after expansion. Second, the digest method overcomes the physical limitations of the fibrous, dense, and paucicellular nature of the primary sarcoma tumors. Through the elimination of the acellular, connective tissue stroma (which is quite significant in most soft tissue sarcomas), the T cells are physically free to expand and tumor-specific lymphocyte clones are liberated to interact directly with the tumor cells.
The results presented are not without limitations. First, the proportion of patients that reliably generate a TIL product of clinically meaningful number is lower (30%) than that which has been reported with malignant melanoma (95%), the solid tumor on which this treatment strategy has been based for 3 decades.17,31 Second, the resultant TIL product does not consistently contain tumor-specific reactivity as defined by prior reports for patients with melanoma (40%–56% vs. 74%–77%).17,31 Despite this difference, the success rate we report does offer a novel treatment option for many more patients who otherwise have no alternative treatment. If the clinical responses in these patients with TIL adoptive cell transfer prove positive in patients with advanced soft tissue sarcoma, this approach might be offered as an alternative to current second-line or third-line systemic treatments.
In summary, this report demonstrates, for the first time, that it is feasible to generate a clinically significant number of tumor-reactive TIL from resected sarcomas. The cell production methods from a primary tumor have not been previously in this select group of patients and now form the basis of manufacturing an expanded TIL product on a pilot clinical trial. There are opportunities demonstrated for improvement on the methods described herein with the goal of further increasing the probability of a successful tumor-specific TIL expansion from soft tissue sarcoma patients, and these potential modifications will be explored in parallel to our current trial.
Acknowledgments
CONFLICTS OF INTEREST/FINANCIAL DISCLOSURES
Supported in part by the Chotiner Foundation and the Ocala Royal Dames Foundation. This work was also supported in part by the Cell Therapy Facility, Tissue Core Facility, and Flow Cytometry Core Facility at the Moffitt Cancer Center, and in part by the Cancer Center Support Grant P30 CA076292 from the National Cancer Institute.
All authors have declared that there are no financial conflicts of interest with regard to this work.
REFERENCES
- 1.Martin-Broto J, Pousa AL, de Las Penas R, et al. Randomized phase II study of trabectedin and doxorubicin compared with doxorubicin alone as first-line treatment in patients with advanced soft tissue sarcomas: a Spanish Group for Research on Sarcoma Study. J Clin Oncol. 2016;34:2294–2302. [DOI] [PubMed] [Google Scholar]
- 2.Judson I, Verweij J, Gelderblom H, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014;15:415–423. [DOI] [PubMed] [Google Scholar]
- 3.Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber J, Mandala M, Del Vecchio M, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med. 2017;377:1824–1835. [DOI] [PubMed] [Google Scholar]
- 5.Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375:1845–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–330. [DOI] [PubMed] [Google Scholar]
- 7.Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017;18:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robbins PF, Kassim SH, Tran TL, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21:1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmed N, Brawley VS, Hegde M, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T Cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33:1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tawbi HA, Burgess M, Bolejack V, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017;18:1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Besser MJ, Shapira-Frommer R, Itzhaki O, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res. 2013;19:4792–4800. [DOI] [PubMed] [Google Scholar]
- 17.Pilon-Thomas S, Kuhn L, Ellwanger S, et al. Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J Immunother. 2012;35:615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenberg SA, Yannelli J, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and IL-2. J Natl Cancer Inst. 1994;86:1159–1166. [DOI] [PubMed] [Google Scholar]
- 19.Goff SL, Dudley ME, Citrin DE, et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol. 2016;34:2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chacon JA, Wu RC, Sukhumalchandra P, et al. Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS One. 2013;8:e60031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chacon JA, Pilon-Thomas S, Sarnaik AA, et al. Continuous 4-1BB co-stimulatory signals for the optimal expansion of tumor-infiltrating lymphocytes for adoptive T-cell therapy. Oncoimmunology. 2013;2:e25581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chacon JA, Sarnaik AA, Chen JQ, et al. Manipulating the tumor microenvironment ex vivo for enhanced expansion of tumor-infiltrating lymphocytes for adoptive cell therapy. Clin Cancer Res. 2015;21:611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radvanyi LG, Bernatchez C, Zhang M, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2012;18:6758–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dudley ME, Wunderlich J, Nishimura MI, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–373. [DOI] [PubMed] [Google Scholar]
- 25.Dudley ME, Wunderlich JR, Yang JC, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunol. 2002;25:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dudley ME, Gross CA, Langhan MM, et al. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res. 2010;16:6122–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deniger DC, Kwong ML, Pasetto A, et al. Apilot trial of the combination of vemurafenib with adoptive cell therapy in patients with metastatic melanoma. Clin Cancer Res. 2017; 23:351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mullinax JE, Hall M, Prabhakaran S, et al. Combination of ipilimumab and adoptive cell therapy with tumor-infiltrating lymphocytes for patients with metastatic melanoma. Front Oncol. 2018;8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tseng WW, Malu S, Zhang M, et al. Analysis of the intratumoral adaptive immune response in well differentiated and dedifferentiated retroperitoneal liposarcoma. Sarcoma. 2015;2015:547460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Angelo SP, Shoushtari AN, Agaram NP, et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum Pathol. 2014; 46:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sorbye S, Kilvaer T, Valkov A, et al. Prognostic impact of lymphocytes in soft tissue sarcomas. PLoS One. 2011;6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sorbye SW, Kilvaer TK, Valkov A, et al. Prognostic impact of peritumoral lymphocyte infiltration in soft tissue sarcomas. BMC Clin Pathol. 2012;12:5. [DOI] [PMC free article] [PubMed] [Google Scholar]