

Abstract
Potassium ion channels are emerging as promalignant factors involved in cancer progression. In this study, we found that invading human gastric cancer cells express high levels of inwardly rectifying potassium channel 2.1 (Kir2.1). Silencing Kir2.1 markedly reduced the invasive and metastatic capabilities as well as the epithelial–mesenchymal transition (EMT) of gastric cancer cells. The promalignant nature of Kir2.1 in gastric cancer cells was independent of potassium permeation but relied on its interaction with serine/threonine-protein kinase 38 (Stk38) to inhibit ubiquitination and degradation of mitogen-activated protein kinase kinase kinase 2 (MEKK2). Degradation of MEKK2 was mediated by small mothers against decapentaplegic-specific E3 ubiquitin protein ligase 1 (Smurf1), which resulted in activation of the MEK1/2–ERK1/2–Snail pathway in gastric cancer cells. In human gastric cancer tissues, expression was high and positively correlated with invasion depth and metastatic status of the tumors as well as poor overall patient survival. Cox regression analysis identified Kir2.1 as an independent prognostic indicator for patients with gastric cancer. Our results suggest that Kir2.1 is an important regulator of gastric cancer malignancy and acts as a novel prognostic marker and a therapeutic target for gastric cancer.
Significance:
Kir2.1 contributes to invasion and metastasis by a noncanonical ion permeation–independent signaling pathway and may act as a novel prognostic marker and therapeutic target for gastric cancer.
Introduction
Gastric cancer is a major malignant tumor in the digestive system with high mortality worldwide, especially in East Asia (1, 2). Despite the advances in surgical and other supplemental treatments, the prognosis for patients with advanced gastric cancer remains poor (3). Invasion and metastasis are crucial factors in the progression of gastric cancer (4). Therefore, insight into the mechanisms underlying gastric cancer invasion and metastasis should benefit the development of more effective therapies.
Recently, potassium channels are implicated in promoting the malignancy of tumor cells. Potassium channels are pore-forming transmembrane proteins that regulate a multitude of biological processes by selectively transporting potassium ions across the cell membrane. On the basis of structure and activation mechanisms, 78 potassium channels are divided into four main classes: voltage-gated channels (Kv), calcium-activated channels (KCa), inward rectifying channels (Kir), and two-pore domain channels (K2P; ref. 5). Various dysregulated potassium channels that cover all four classes have been found in different types of human cancers. For example, voltage-gated potassium channel Kv10.1 was normally expressed in selected brain areas, but was aberrantly expressed in over 70% human tumors and involved in tumor cell proliferation, survival, angiogenesis, migration, and invasion (6). In triple-negative breast cancer (TNBC), KCa3.1 is implicated in the proliferation, apoptosis, migration, and epithelial–mesenchymal transition (EMT) of tumor cells (7). In highly progressive human astrocytic tumors, the expression of Kir4.1 at both mRNA and protein levels was markedly increased (8). TREK-1, a two-pore domain [K(2P)] potassium channel, was highly expressed in prostate cancer associated with abnormal tumor cell proliferation (9). However, it is unknown whether potassium channels are involved in gastric cancer invasion and metastasis. Hence, we examined the difference in potassium current in invading and noninvading gastric cancer cells by whole-cell patch-clamp. The results showed that potassium current in invading gastric cancer cells was markedly higher than that in noninvading gastric cancer cells. Moreover, the elevated potassium current in invading gastric cancer cells exhibited characteristics of an inwardly rectifying potassium current (IK+), implying that Kir might be associated with the invasive capability of gastric cancer cells. qRT-PCR scanning revealed that within 15 distinct subunits of Kir family, Kir2.1 was most prominently expressed in invading gastric cancer cells. Whole-cell patch-clamp also showed that Kir2.1 was the most important contributor of IK+ in invading gastric cancer cells. We therefore further investigated the role of Kir2.1 in the invasion and metastasis of human gastric cancer and the clinical relevance. Our results indicate that Kir2.1 actively promoted gastric cancer cell invasion and metastasis independent of its potassium transport function, but by interaction with serine/threonine-protein kinase 38 (Stk38) to enhance the signaling of MEKK2–MEK1/2–ERK1/2–Snail pathway in gastric cancer cells. Studies of clinical gastric cancer specimens showed a positive correlation between the levels of Kir2.1 expression and the invasion depth and metastatic status of human gastric cancer in association with poorer overall patient survival. Thus, Kir2.1 plays an important role in promoting gastric cancer malignancy and acts as an independent indicator of gastric cancer prognosis as well as a potential therapeutic target.
Materials and Methods
Patients and specimens
A total of 349 formalin-fixed and paraffin-embedded gastric cancer surgical specimens, in which 131 specimens contained both tumor and matched adjacent normal tissues, were collected from patients enrolled in the Southwest Hospital from 2006 to 2007 without prior radiotherapy or chemotherapy. A separate set of samples, which contain 6 fresh surgical tumor and the adjacent normal tissues, was also collected from the Southwest Hospital. Follow-up information was available for all patients. According to the World Health Organization standard and American Joint Commission on Cancer TNM staging system (10), each specimen was histologically examined and the tumor was graded by two experienced pathologists. Written informed consent was obtained from the patients or their guardians. This study was performed by the principles of the Helsinki Declaration and approved by the Ethical Committee of the Southwest Hospital.
Cells and cell culture
MGC803 human gastric cancer cell line was purchased from the Cell Bank of Shanghai Institute of Cell Biology. Primary gastric cancer cell line XN0422 was generated in our laboratory as described previously (11). All the cells were tested and confirmed negative for Mycoplasma contamination using EZ-PCR Mycoplasma Test Kit (Bio-Ind) and authenticated by short tandem repeat profiling and passaged for less than 6 months according to the manufacturer’s instructions. The cells were cultured in RPMI1640 medium supplemented with 10% FBS (Gibco) at 37°C in 5% CO2.
IHC and immunoreactivity scores
IHC staining was performed on 4-μm tissue sections using an EnVision Kit (DAKO). After deparaffinized, rehydrated in graded ethanol, antigen retrieval and blocking, slides were incubated with an anti-Kir2.1 antibody (1:300, catalog no. ab85492, Abcam) at 4°C overnight. After washing with PBS, a horseradish peroxidase (HRP)-conjugated secondary antibody (DAKO) was added and incubated at 37°C for 30 minutes. Sections were stained by 3,39-diaminobenzidine (DAB, DAKO) and counter-stained with hematoxylin.
IHC scoring method was performed as previously described (12). Briefly, five random IHC images of each slide were captured using an Olympus BX51 microscope. The images were opened by Image-Pro Plus 5.0 software. The area sum and integrated optical density (IOD) sum of the positive site (brown) were measured in pixels using the software. The expression intensity of Kir2.1 was expressed by the mean value of IOD sum/area sum of 5 photographs for each slide. To ensure data comparability, the same parameter settings were kept for all photographs. The best predictive cut-off value of expression intensity was determined to be 0.03438 analyzed with SPSS 20.0. Expression intensity > 0.03438 was defined as Kir2.1high, otherwise was defined as Kir2.1low.
FACS
Single gastric cancer cell suspension was prepared by trypsinization of cultured adherent cells and incubated with antihuman Kir2.1 antibody (catalog no. orb184788, Biorbyt) for 30 minutes at room temperature followed by labeled with APC-conjugated goat anti-rabbit secondary antibody. Then the Kir2.1high and Kir2.1low gastric cancer cells were sorted by the FACSAria II cell sorter (BD Biosciences). Forward side scatter and pulse-width gating were used for excluding the dead cells, debris, and aggregates. Isotype-matched primary antibodies were used as controls.
In vitro gastric cancer cell invasion assay
Matrigel transwell analyses were performed as previously described (13). To acquire invading and noninvading gastric cancer cells, transwell chambers (8-μm pore size, Millipore) were coated with 10-μL mixed Matrigel (Matrigel and RPMI1640, 1:3, v/v). Gastric cancer cells were implanted at 5 × 104 cells/well in 200 μL serum-free RPMI1640. The bottom chambers were filled with 600-μL RPMI1640 medium supplemented with 10% FBS. After incubation for 24 hours at 37°C, the invading and noninvading cells were obtained by trypsin enzyme-digesting technique. To examine the affectation of Kir2.1 agonist and antagonist and signaling inhibitors on the invasiveness of gastric cancer cells, the Matrigel-transwell system were cultured with or without ML133 (20 μmol/L, TOCRIS) or zacopride (10 μmol/L, Sigma-Aldrich) or PD98059 (10 μmol/L, Selleck Chemicals) or LY3009120 (20 μmol/L, Selleck Chemicals). After incubation for 24 hours at 37°C, the invasive cells were stained with 1% crystal violet and then counted from five random visual areas under a microscope.
Whole-cell patch-clamp recording
Potassium current was detected by whole-cell patch-clamp as described previously (14). Recordings were made at room temperature. Bath (external) solutions were perfused into the chamber using a gravity-driven perfusion system. The standard bath solution consisted of (in μmol/L): 100 d-glucose, 5 KCl, 2 MgCl2, 50 NaOH, and 5 glucose. Recording pipettes were filled with an intracellular solution containing (in μmol/L): 110 KCl, 5 MgATP, 5 NaCl, 2 MgCl2, and 5 μmol/L glucose. Patch pipettes were made from thin-walled borosilicate glass and fire polished with a microforge. Data were acquired using an Axopatch 200B amplifier and filtered at 5 Hz with a Digidata 1322A board. Acquisition and analysis were performed using an EPC-10 software.
RNA extraction and quantitative PCR
Total RNA was isolated using RNAiso TRIzol reagent (Takara), and reverse-transcribed with PrimeScript RT Master Mix (Takara) according to the manufacturer’s instructions. Then a SYBR Fast qPCR Mix (Takara) in a Bio-Rad CFX96 Real-Time PCR Detection System (Bio-Rad) was used for qRT-PCR. qRT-PCR was performed in triplicate and the results were normalized against β-actin. The sequences of all primers for qRT-PCR are listed in Supplementary Table S1.
Western blot analysis
Immunoblot analyses were performed as described previously (13). Gastric cancer cells were washed twice with ice-cold PBS then lysed with protein extraction reagent (Thermo Fisher Scientific) containing 1% protease inhibitors (Thermo Fisher Scientific). The lysate was centrifuged at 4°C at 16,000 × g for 15 minutes and proteins were separated by 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore). After being blocked with 5 % skim milk for 2 hours, the membranes were incubated at 4°C overnight with primary antibodies (detailed information given in the Supplementary Materials and Methods). After washing with PBST, the membranes were incubated with HRP-conjugated secondary antibodies (Beyotime Institute of Biotechnology) for 1 hour. Then proteins were visualized with SuperSignal West Femto Maximum Sensitivity Substrate (ECL, Thermo Fisher Scientific) and detected by a ChemiDocXRS system (Bio-Rad).
Kir2.1 knockdown, overexpression, and site-directed mutagenesis
To knock down Kir2.1 in gastric cancer cells, three targeting shRNAs (named sh-Kir2.1) and a nontargeting scrambled RNA (named mock) were constructed in lentivirus vectors (Supplementary Table S2). For Kir2.1 overexpression in gastric cancer cells, full-length human Kir2.1 (named over-Kir2.1) and empty vector (named ctrl) were constructed with a lentivirus vector. To produce a nonconducting Kir2.1 channel subunit (named mut-Kir2.1), the 144–146 (Gly-Tyr-Gly) signature sequence that serve as the selectivity filter of Kir2.1 was mutated to Ala-Ala-Ala (15) and inserted into a lentivirus vector. All lentivirus particles were packaged by Life Technologies Co. Ltd. and used to infect gastric cancer cells with 2 μg/mL polybrene. Stably transfected cells were selected by 3 μg/mL puromycin in culture.
Peritoneal metastasis in NOD/SCID mice
NOD/SCID mice aging from 4 to 5 weeks and weighing between 17 and 20 grams were obtained from the Experimental Animal Center of Third Military Medical University (Chongqing, China). Gastric cancer cells suspended in PBS were inoculated intraperitoneally into mice at 1 × 104 cells per mouse. The mice were sacrificed at the end of 4 weeks after the implantations. Peritoneal metastases in mesentery were evaluated. Animal experiments were approved by the Third Military Medical University Animal Ethics Committee in accordance with the Guide for the Care and Use of Laboratory Animals.
Human phosphokinase array
Cell lysates were diluted and mixed with a cocktail of biotinylated detection antibodies then added on the Human phosphokinase array (R&D Systems). After overnight incubation at 4°C, Streptavidin-HRP and chemiluminescent detection mix were added, and chemiluminescence was detected using the ChemiDoc MP System (Bio-Rad), Image Lab (Version 5.2 build 14), and automatic exposure settings.
Coimmunoprecipitation
Coimmunoprecipitation (Co-IP) was performed using the Thermo Scientific Pierce Co-IP kit (Thermo Fisher Scientific) following the manufacturer’s protocol. Ten micrograms each of antibody and IgG were immobilized on AminoLink Plus coupling resin for 2 hours, then washed and incubated with 250 μg gastric cancer cell protein lysate. After overnight incubation at 4°C, the resin was washed and eluted using an elution buffer. The eluted proteins were separated by SDS-PAGE and immunoblotted with indicated antibodies as Western blot analysis.
Ubiquitination assay
Ubiquitination assay was performed as previously described (16). Briefly, gastric cancer cells were transfected with plasmids expressing HA-ubiquitin, His-Smurf1, V5-Stk38, and Flag-Kir2.1 alone or in combination as designed protocol. After 48 hours, cells were treated with proteasome inhibitors MG132 (10 μmol/L, Selleck Chemicals) for another 6 hours. Then the ubiquitination degradation was detected by immunoprecipitation.
Statistical analysis
All experiments were performed at least three times and results from representative experiments are presented as the mean ± SD by Student t test using SPSS 20.0 software (IBM) and GraphPad Prism. The cut-off value of Kir2.1 IHC staining scores was analyzed with SPSS. χ2analysis was used to evaluate the relationship between Kir2.1high rate and gastric cancer clinicopathologic features. The overall survival (OS) and progression-free survival (PFS) of patients with gastric cancer were estimated using Kaplan–Meier method. Cox proportional hazard regression model was established for univariate and multivariate analyses of the combined contribution of Kir2.1 and clinicopathologic features to the OS of patients. P < 0.05 was considered as statistically significant.
Results
Kir2.1 is highly expressed in invading gastric cancer cells
We first measured the potassium current by using a whole-cell patch-clamp method in both invading and noninvading primary XN0422 gastric cancer cells. Compared with noninvading gastric cancer cells, invading cells displayed high level of potassium current, which was blocked by barium, a nonspecific potassium current blocker (Fig. 1A). As shown in Fig. 1B, the current density–voltage (pA/pF-V) relation of the potassium current exhibited typical IK+ characteristics in invading gastric cancer cells. The average IK+ at 100 mV in invading cells was markedly higher than that in noninvading cells (−36.0130 ± 4.9826 vs. −3.2529 ± 0.9738, n = 20; Fig. 1C). Similar results were also achieved in gastric cancer cell line MGC803 (Supplementary Fig. S1). These results suggest the presence of functional Kir channels in invading gastric cancer cells. We then scanned gene expression of 15 Kir members by qRT-PCR and found Kir2.1 expression at a higher level in invading gastric cancer cells (Fig. 1D). We subsequently compared Kir2.1 expression between high (> 50 pA) and low (< 5 pA) IK+-possessed gastric cancer cells. As shown in Fig. 1E, markedly increased expression of Kir2.1 was found in high IK+ gastric cancer cells. Increased Kir2.1 protein in invading gastric cancer cells was detected in both gastric cancer cell lines MGC803 and XN0422 (Fig. 1F). These results suggest that aberrant expression of functional Kir2.1 is associated with the invasive and metastatic capabilities of gastric cancer cells.
Figure 1.

Invading gastric cancer cells highly express functional Kir2.1. A, Whole-cell patch-clamp measurement showing invading XN0422 gastric cancer cells that displayed higher potassium current compared with noninvading cells. Potassium current was inhibited by barium (5 μmol/L). B, The current density–voltage (pA/pF-V) curves derived from same data presented in A with a typical characteristic of IK+. C, Higher average IK+ at 100 mV in invading gastric cancer cells than in noninvading gastric cancer cells. Data are shown as mean ± SD (n = 20; *, P < 0.05, Student t-test). D, The mRNA level of Kir family in invading and noninvading gastric cancer cells. Data are shown as mean ± SD (n = 3). E, Increased Kir2.1 mRNA expressed by gastric cancer cells with high current (IK+ > 50 pA) compared with low current (IK+ < 5 pA) gastric cancer cells as detected by qRT-PCR. Data are shown as mean ± SD (n = 6; ***, P < 0.0001, Student t test). F, Higher level of Kir2.1 protein in invading gastric cancer cells compared with noninvading gastric cancer cells as detected by Western blotting.
Kir2.1high gastric cancer cells are highly invasive and metastatic in association with EMT
By FACS, Kir2.1-high (Kir2.1high) and -low gastric cancer cells (Kir2.1low) were isolated, and the percentage of Kir2.1high gastric cancer cells was about 22 % in MGC803 and 14 % in XN0422 (Supplementary Fig. S2). As shown in Fig. 2A and Supplementary Fig. S3, Kir2.1high gastric cancer cells exhibited more invasive capability compared with Kir2.1low cells in vitro. The invading cells in Kir2.1high and Kir2.1low subpopulations were 210.60 ± 33.26 versus 96.80 ± 17.02 for MGC803 and 114.40 ± 18.85 versus 65.80 ± 12.40 for XN0422 cells, respectively. Kir2.1high gastric cancer cells also exhibited increased metastatic capability compared with Kir2.1low cells in a mouse peritoneal metastasis model. The observed metastatic nodules derived from Kir2.1high and Kir2.1low gastric cancer cells were 65.60 ± 5.73 versus 2.60 ± 1.60 for MGC803 and 46.60 ± 4.45 versus 2.40 ± 1.50 for XN0422 cells, respectively (Fig. 2B and C). Knockdown of Kir2.1 (sh-Kir2.1, Supplementary Fig. S4) significantly inhibited the invasion of gastric cancer cells. The invading cells in sh-Kir2.1 and mock cells were 18.60 ± 4.04 versus 83.20 ± 14.99 for MGC803 and 12.80 ± 3.70 versus 46.40 ± 9.96 for XN0422 cells, respectively (Fig. 2D; Supplementary Fig. S5). Compared with mock cells, sh-Kir2.1 MGC803 and XN0422 cells formed markedly reduced number of metastatic nodules in mice (Fig. 2E and F). In contrast, Kir2.1 overexpression (over-Kir2.1, Supplementary Fig. S4) significantly not only elevated the invasive capability (Fig. 2G; Supplementary Fig. S5) but also increased the metastatic nodule formation (Fig. 2H and I) by both MGC803 and XN0422 cells. All metastatic nodules in mice formed by gastric cancer cells were confirmed as human gastric cancer by H&E staining (Supplementary Fig. S6). These results suggest that the presence of Kir2.1 is associated with increased invasion and metastasis of gastric cancer cells.
Figure 2.
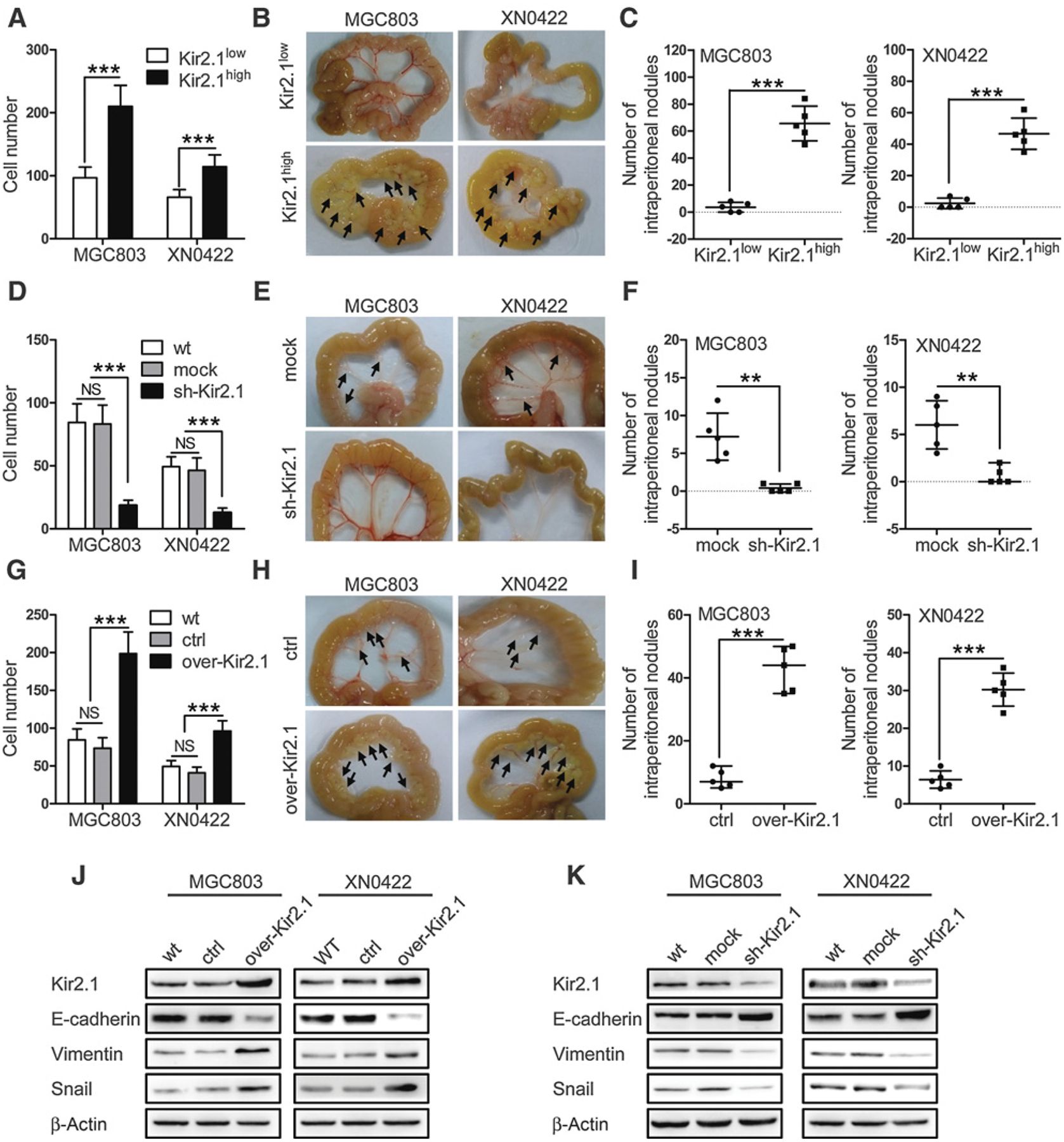
Kir2.1 promotes the invasion and metastasis of gastric cancer cells. A, Matrigel–Transwell invasion assay showing increased invasion capability of Kir2.1high gastric cancer cells compared with Kir2.1low gastric cancer cells. Data are shown as mean ± SD (n = 5; ***, P < 0.0001, Student t test). B, Representative images of intraperitoneal metastasis in NOD/SCID mice showing significantly higher number of metastatic foci formed by Kir2.1high gastric cancer cells than that formed by Kir2.1low gastric cancer cells. C, Quantification of metastatic tumors formed by Kir2.1high and Kir2.1low gastric cancer cells. Data are shown as mean ± SD (n = 5; ***, P < 0.0001, Student t test). D, Reduced invasion capability of sh-Kir2.1 gastric cancer cells compared with mock (scrambled shRNA) gastric cancer cells in vitro. Data are shown as mean ± SD (n = 5; NS, not significant; ***, P < 0.0001, ANOVA test). E and F, Reduced metastasis capability of sh-Kir2.1 gastric cancer cells in vivo. Data are shown as mean ± SD (n = 5; **, P < 0.001, Student t test). G, Increased invasion capability of over-Kir2.1 gastric cancer cells compared with control (empty vector) gastric cancer cells in vitro. Data are shown as mean ± SD (n = 5; NS, not significant; ***, P < 0.0001, ANOVA test). H and I, Increased metastasis capability of over-Kir2.1 gastric cancer cells compared with control (empty vector) gastric cancer cells in vivo. Data are shown as mean ± SD (n = 5; ***, P < 0.0001, Student t test). J, Western blotting showing decreased E-cadherin expression but enhanced vimemtin and Snail expression in over-Kir2.1 gastric cancer cells. K, Western blotting showing upregulated E-cadherin and downregulated vimemtin and Snail in sh-Kir2.1 gastric cancer cells.
EMT is recognized as a pivotal event for tumor cells to enhance their capacity of invasion and metastasis (17, 18). Kir2.1-overexpressing gastric cancer cells contained decreased epithelial marker E-cadherin but increased mesenchymal marker vimentin and the transcription factor Snail (Fig. 2J). In contrast, knockdown of Kir2.1 in gastric cancer cells reversed the EMT phenotype, with upregulated E-cadherin and downregulated vimentin and Snail (Fig. 2K). Although EMT may be regulated by different transcription factors, examination of major EMT-related transcription factors showed that overexpressing Kir2.1 only significantly increased Snail expression in gastric cancer cells (Supplementary Fig. S7). These results suggest that Kir2.1 promotes EMT in gastric cancer cells.
The promalignant effect of Kir2.1 on gastric cancer cells is independent of potassium permeation
Kir2.1 functions either as a potassium transporter (14, 19, 20) or by promoting protein–protein interaction (21–23). We firstly investigated whether the promalignant effect of Kir2.1 was dependent on its channel function in gastric cancer cells. Treatment with ML133, a specific inhibitor of Kir2.1 channel (24), completely interrupted IK+ flux in both over-Kir2.1 and control gastric cancer cells (Supplementary Fig. S8), but did not alter their invasive capability (Fig. 3A; Supplementary Fig. S9) and the levels of EMT-related proteins (Fig. 3B). Also, treatment with zacopride, an IK+ stimulator (25), potently elevated IK+ (Supplementary Fig. S8), but with little effect on the invasion capability (Fig. 3C; Supplementary Fig. S9) and EMT phenotype (Fig. 3D) of both over-Kir2.1 and control gastric cancer cells. As the extracellular pore-forming region of Kir2.1 serves as the “ion-selectivity filter,” in which mutation of GYG into AAA leads to the loss of potassium transport function (15), we introduced a mutant Kir2.1 (GYG to AAA, mut-Kir2.1) into gastric cancer cells. The introduction of mutant Kir2.1 did not increase IK+ (Supplementary Fig. S8), but enhanced the invasive capability (Fig. 3E; Supplementary Fig. S9) of EMT and Snail to the level of over-Kir2.1 in gastric cancer cells (Fig. 3F). These results suggest that the effect of Kir2.1 on increased invasion and metastasis of gastric cancer cells is independent of its ion channel function.
Figure 3.
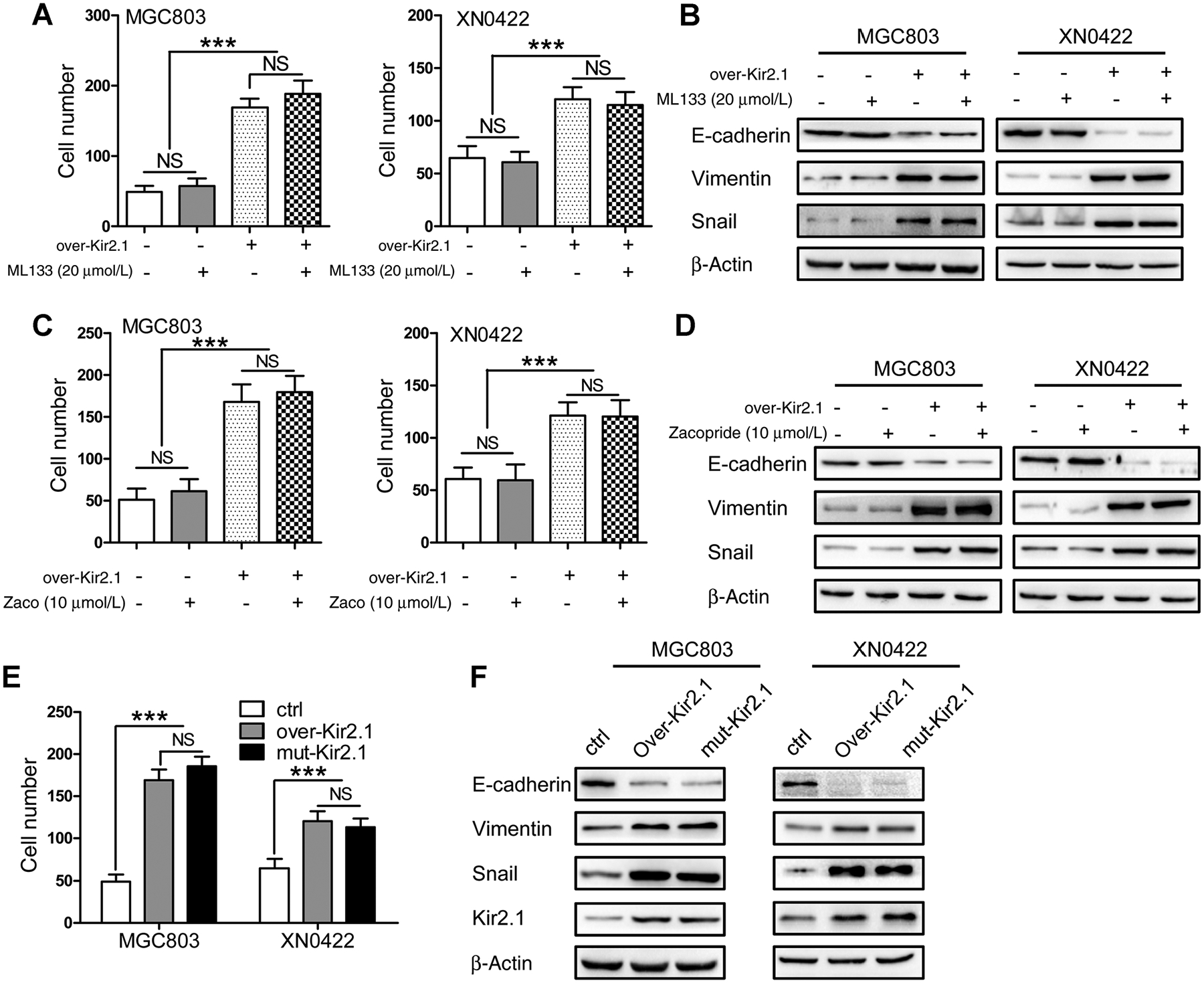
The promalignant effect of Kir2.1 is independent of potassium permeation. A, Similar invasion capability exhibited by gastric cancer cells with or without ML133 (20 μmol/L) treatment. Data are shown as mean ± SD (n = 5; NS, not significant; ***, P < 0.0001, ANOVA test). B, No changes in the expression of E-cadherin, vimemtin, and Snail by gastric cancer cells after treatment with ML133 (20 μmol/L). C, No effect on the invasion capability of gastric cancer cells by zacopride (10 μmol/L) treatment. Data are shown as mean ± SD (n = 5; NS, not significant; ***, P < 0.0001, ANOVA test). D, No changes in the expression of E-cadherin, vimemtin, and Snail by gastric cancer cells after Zacopride (10 μmol/L) treatment. E, Significantly enhanced invasion capability of gastric cancer cells by overexpressing mut-Kir2.1, with similarity to overexpressing wild-type Kir2.1. Data are shown as mean ± SD (n = 5; NS, not significant; ***, P < 0.0001, ANOVA test). F, Mut-Kir2.1 and over-Kir2.1 gastric cancer cells expressing similar levels of E-cadherin, vimemtin, and Snail.
Kir2.1 activates MEK1/2–ERK1/2–Snail–EMT pathway without involvement of Raf
A phosphokinase array was performed to explore the signaling pathways involving Kir2.1 in gastric cancer cells. As shown in Fig. 4A, Kir2.1 overexpression resulted in elevated phosphorylation of 11 kinases in XN0422 cells, among which, the phosphorylation level of ERK1/2 was at the highest level. Markedly enhanced phosphorylation of ERK1/2 by Kir2.1 overexpression but significantly attenuated phosphorylation of ERK1/2 by Kir2.1 knockdown was confirmed by immunoblotting. In contrast to ERK1/2, the phosphorylation of other members of MAPKs, JNK, and p38, was minimally affected in gastric cancer cells by Kir2.1 manipulation (Fig. 4B). We further examined whether Kir2.1-induced ERK1/2 activation involves canonical Ras–Raf–MEK1/2–ERK1/2 pathway. Treatment of over-Kir2.1 gastric cancer cells with PD98059 (26), a MEK1/2 inhibitor, significantly attenuated ERK1/2 phosphorylation (Fig. 4C; Supplementary Fig. S10), accompanied with impaired invasion capability, and downregulated Snail and vimentin as well as upregulated E-cadherin expression (Fig. 4D). LY3009120 (27), an inhibitor of pan-Raf (A-Raf, B-Raf, and C-Raf) only slightly attenuated the invasion capability and phosphorylation of MEK1/2 and ERK1/2 in both control and over-Kir2.1 gastric cancer cells (Fig. 4E and F; Supplementary Fig. S10). Thus, the effect of Kir2.1 on the invasion and EMT of gastric cancer cells is dependent on MEK1/2–ERK1/2–Snail but not Raf.
Figure 4.
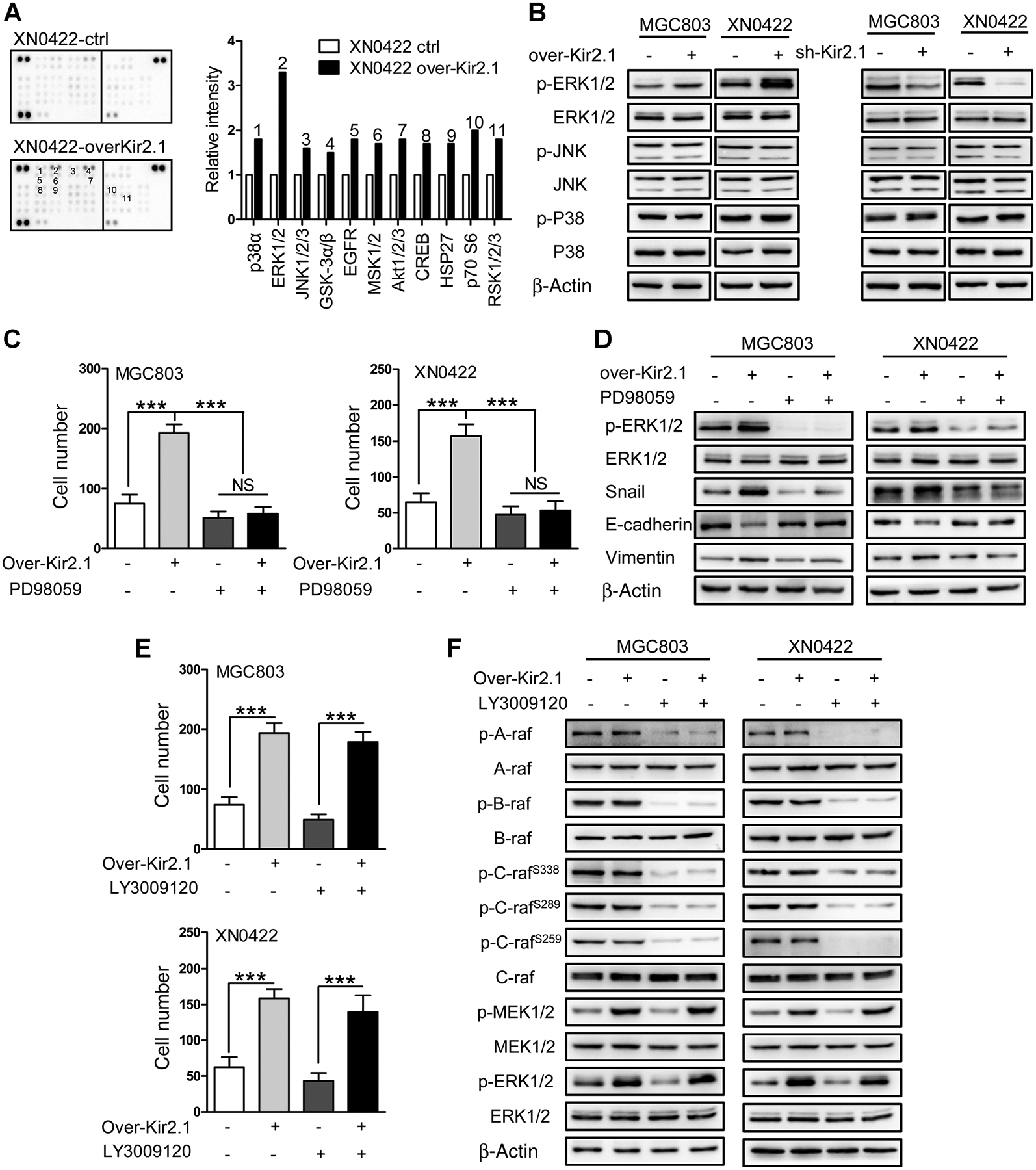
Kir2.1 activates MEK1/2–ERK1/2–Snail–EMT pathway independent of Raf. A, Human phosphokinase antibody array assay showing 11 elevated phosphorylation kinases (left) with ERK1/2 kinases at the highest level (right). Data are shown as mean. B, The over-Kir2.1 gastric cancer cells showing enhanced phosphorylation of ERK1/2 and sh-Kir2.1 gastric cancer cells showing attenuated phosphorylation of ERK1/2. JNK and p38, the other two members of MAPKs in gastric cancer cells, were unaffected by Kir2.1 expression status. C, Decreased invasion capability of gastric cancer cells after treatment with PD98059 (10 μmol/L), a MEK1/2-specific inhibitor. Data are shown as mean ± SD (n = 5; NS, not significant; ***, P < 0.0001, ANOVA test). D, Increased E-cadherin but decreased vimemtin, Snail, and pERK1/2 in PD98059 (10 μmol/L)-treated gastric cancer cells. E, Attenuation of the invasion capability of control gastric cancer cells by treatment with LY3009120 (20 μmol/L), a pan-Raf inhibitor, without effect on over-Kir2.1 gastric cancer cells. Data are shown as mean ± SD (n = 5; ***, P < 0.0001, Student t test). F, Attenuation of the level of pMEK1/2 and pERK1/2 in control gastric cancer cells by LY3009120 (20 μmol/L) treatment, without effect on over-Kir2.1 gastric cancer cells.
Kir2.1–Stk38 interaction upregulates MEKK2 to activate MEK1/2-ERK1/2
As Kir2.1-promoted invasion and metastasis of gastric cancer cells were independent of its potassium channel function, its involvement in protein–protein interaction was hypothesized. With immunoprecipitation–mass spectrometry, proteins potentially interacting with Kir2.1 were identified (Supplementary Table S3), with Stk38 and Rho-associated protein kinase 2 (Rock2), which regulate ERK1/2 pathway (16, 28, 29), were selected for further study. Co-IP confirmed that Stk38, but not Rock2, physically interacted with Kir2.1 in both MGC803 and XN0422 gastric cancer cells (Fig. 5A). Moreover, we further revealed that the interacting site of Kir2.1 with Stk38 was the C-terminal (aa 179–425) region by using truncated mutants (Supplementary Fig. S11). As Stk38 regulates MEK1/2–ERK1/2 pathway mainly by regulating MEKK2, an MAPKKK (30, 31), depletion of MEKK2 with siRNA significantly decreased the phosphorylation of MEK1/2 and ERK1/2 in both control and over-Kir2.1 gastric cancer cells (Fig. 5B).
Figure 5.

Kir2.1–Stk38 interaction upregulates MEKK2 to activate MEK1/2-ERK1/2 in gastric cancer cells. A, Co-IP assay showing interaction of Kir2.1 with Stk38 but not with Rock2. B, Decreased expression of MEKK2, pMEK1/2, and pERK1/2 both in over-Kir2.1 and control gastric cancer cells by siMEKK2. C, Decreased levels of pMEKK2 and non-pMEKK2 accompanied with decreased pMEK1/2 and pERK1/2 both in over-Kir2.1 and control gastric cancer cells by overexpressing Stk38. D, Relative pixel density showing significantly decreased levels of both pMEKK2 and non-pMEKK2 by overexpressing Stk38, without affecting the ratio between pMEKK2 and non-pMEKK2. Data are shown as mean ± SD (n = 3; *, P < 0.05, Student t test). E, Over-Kir2.1 gastric cancer cells showing decreased levels of Smurf1–Stk38 complex compared with paired control gastric cancer cells. F, Overexpressing Kir2.1 blocking ubiquitination of MEKK2 induced by cooverexpressing Stk38 and Smurf1 in cooverexpressing cells.
Stk38 converts MEKK2 from a phosphorylated to a nonphosphorylated form therefore inhibiting MEKK2 (32). As specific anti-pMEKK2 antibody is unavailable, pMEKK2 and non-pMEKK2 may be distinguished by immunoblotting in which the faster migrating band defines nonphosphorylated or hypophosphorylated MEKK2 (non-pMEKK2) while slower migrating band indicates phosphorylated MEKK2 (pMEKK2; refs. 32, 33). We found that overexpressing Stk38 markedly reduced the invasiveness (Supplementary Fig. S12) and the levels of both phosphorylated and nonphosphorylated MEKK2 accompanied by decreased levels of pMEK1/2 and pERK1/2 (Fig. 5C) in gastric cancer cells. However, the relative density of pMEKK2 or non-pMEKK2 remained similar between control and over-Stk38 gastric cancer cells (Fig. 5D), implying that Stk38 regulates MEKK2-MEK1/2-ERK1/2 mainly by changing the level of MEKK2 protein rather than affecting its phosphorylation in gastric cancer cells. As previously reported (16), Stk38 inhibits MEKK2 by interacting with small mothers against decapentaplegic-specific E3 ubiquitin protein ligase 1 (Smurf1) to enhance the ubiquitination and degradation of MEKK2. We thus hypothesized that the interaction between Kir2.1 and Stk38 might attenuate the ubiquitination and degradation of MEKK2, leading to the activation of the MEK1/2–ERK1/2 pathway. With quantitative co-IP and ubiquitination degradation assays, we confirmed that overexpressing Kir2.1 in gastric cancer cells significantly decreased the formation of Smurf1–Stk38 complex (Fig. 5E) and attenuated the ubiquitination degradation of MEKK2 induced by the complex (Fig. 5F). These results suggest that MEKK2–MEK1/2–ERK1/2 pathway is pivotal to the invasion and metastasis of gastric cancer cells, which is enhanced by the interaction of Kir2.1 with Stk38 to inhibit ubiquitinated degradation of MEKK2.
The expression of Kir2.1 in gastric cancer tissues is correlated with clinicopathologic features and the prognosis of patients with gastric cancer
We further investigated the clinical relevance of Kir2.1 expression in gastric cancer. In 349 gastric cancer specimens and 131 paired adjacent normal tissues, Kir2.1 protein was mainly detected in the cytoplasm and membrane of gastric cancer cells (Fig. 6A). Kir2.1 was weakly or not expressed in normal gastric mucosa (Fig. 6Aa). The density of Kir2.1 protein in gastric cancer specimens increased with the invasion depth of tumor (Fig. 6Ab–e). Kir2.1 was also highly expressed in lymph node metastasis nodules of gastric cancer (Fig. 6Af). ROC curve showed a significant cut-off value of 0.03438 for Kir2.1 as assessed by SPSS 20.0 (Supplementary Fig. S13). The IHC scores of Kir2.1 were significantly higher in 131 cancer tissues than in paired adjacent normal tissues (Fig. 6B). In a separate set of specimens, quantitative analysis of Kir2.1 in 6 freshly resected tumor samples and adjacent normal tissues indicated high levels of Kir2.1 expression in tumor tissues than in adjacent normal tissues (Fig. 6C; Supplementary Table S4). Higher expression of Kir2.1 in gastric cancer tumors was also found in GEO GSE13911 and GES27342 (Fig. 6D; Supplementary Table S5). Analysis of clinicopathologic features showed that high expression of Kir2.1 in gastric cancer was positively correlated with increased tumor invasion depth, lymph node metastasis (Fig. 6E), and TNM stage (Table 1). Kaplan–Meier analyses showed significant correlation between higher expression of Kir2.1 and poorer OS and PFS in patients with gastric cancer (Fig. 6F). KMPLOT database also revealed negative correlation between increased Kir2.1 expression and poorer prognosis of patients with gastric cancer (Supplementary Fig. S14). Both univariate and multivariate analyses suggested that Kir2.1 was an independent prognostic indicator for patients with gastric cancer (Supplementary Table S6). Thus, elevated Kir2.1 in gastric cancer is associated with increased invasion and metastasis with poorer prognosis of patients.
Figure 6.
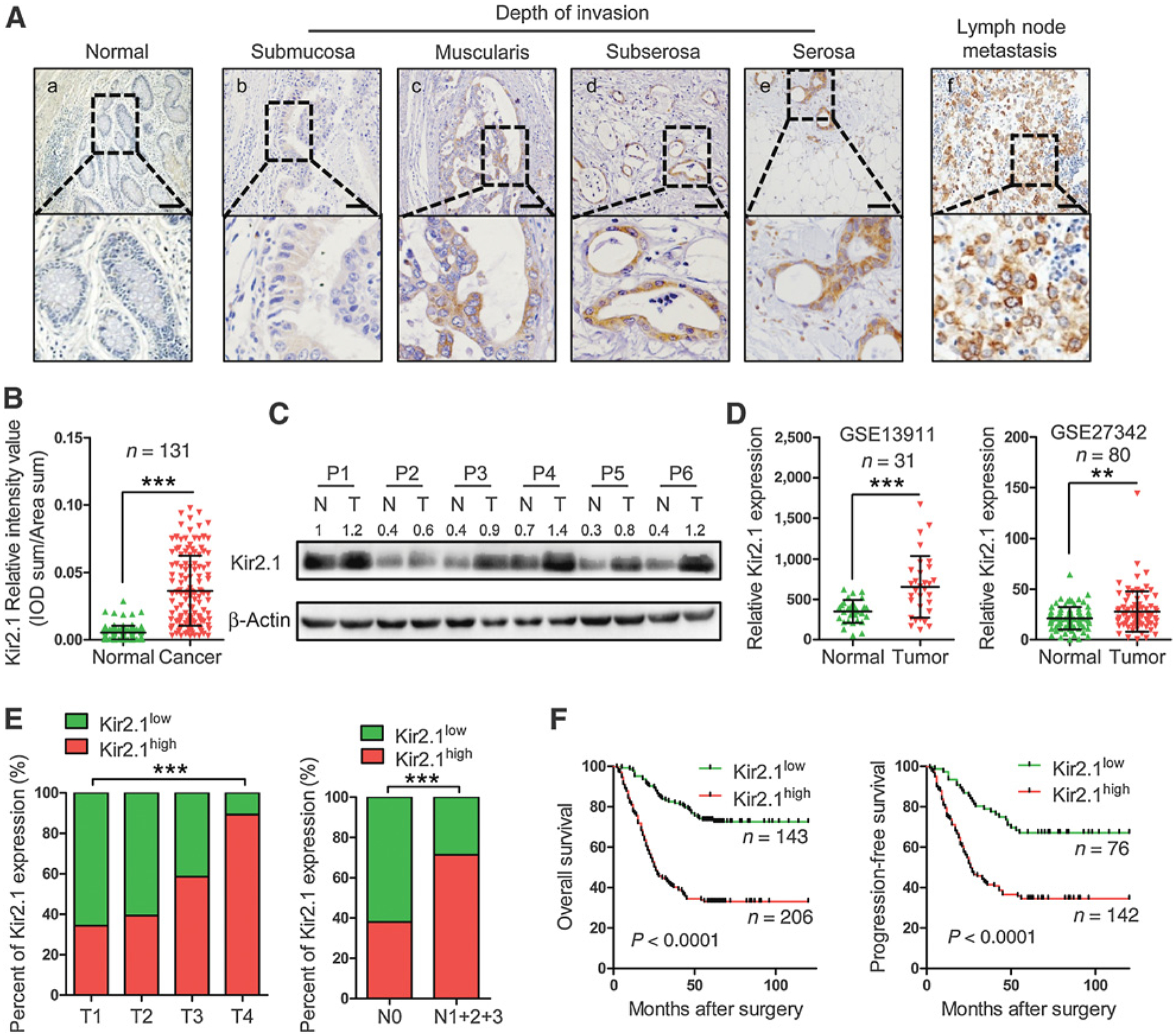
Upregulation of Kir2.1 in human gastric cancer and the correlation with cancer invasion, lymph node metastasis, and outcome of patients with gastric cancer. A, Representative IHC images of Kir2.1 in gastric cancer specimens. Scale bar, 100 μm. a, Absence of Kir2.1 staining in normal gastric mucosa. b–e, Increased Kir2.1 staining intensity with invasion depth. f, High Kir2.1 level in both primary tumor and corresponding metastatic lymph node. B, Higher IHC scores of Kir2.1 in 131 gastric cancer tissues compared with paired adjacent normal tissues. Data are shown as mean ± SD (n = 131; ***, P < 0.0001, paired t test). C, Kir2.1 protein in 6 paired surgically removed gastric tumor tissues (T) and the adjacent normal tissues (N) showing highly expressed Kir2.1 in cancerous tissues. D, Higher mRNA level of Kir2.1 in cancerous tissues than in adjacent normal tissues from GEO GES13911 and GES27342. Data are shown as mean ± SD (n = 31, GSE13911; n = 80, GSE27342; ***, P < 0.0001; **, P < 0.001, paired t test). E, The relationship between Kir2.1 expression and tumor invasion depth and lymph node metastasis of gastric cancer. n = 349; ***, P < 0.0001, χ2 test. F, Kaplan–Meier curves showing the correlation between the levels of Kir2.1 and the overall survival and progression-free survival of patients with gastric cancer [n = 349 (OS), n = 218 (PFS), log-rank test].
Table 1.
The correlation between Kir2.1 expression in gastric cancer tissues and clinicopathologic features of patients with gastric cancer
| Clinicopathologic parameter | Total no. | Kir2.1 | P | |
|---|---|---|---|---|
| Low (%) | High (%) | |||
| Age, y | 0.316 | |||
| ≤60 | 177 | 72 (40.7) | 105 (59.3) | |
| >60 | 172 | 61 (35.5) | 111 (64.5) | |
| Sex | 0.103 | |||
| Female | 89 | 43 (48.3) | 46 (51.7) | |
| Male | 260 | 100 (38.5) | 160 (61.5) | |
| Differentiation | 0.039 | |||
| Well | 31 | 12 (38.7) | 19 (61.3) | |
| Moderate | 115 | 50 (43.5) | 65 (56.5) | |
| Poor | 203 | 60 (29.6) | 143 (70.4) | |
| T stage | 0.000 | |||
| T1 | 32 | 21 (65.6) | 11 (34.4) | |
| T2 | 66 | 40 (60.6) | 26 (39.4) | |
| T3 | 147 | 61 (41.5) | 86 (58.5) | |
| T4 | 103 | 11 (10.7) | 92 (89.3) | |
| Lymph node metastasis | 0.000 | |||
| Absent | 129 | 80 (62.0) | 49 (38.0) | |
| Present | 220 | 63 (28.6) | 157 (71.4) | |
| TNM stage | 0.000 | |||
| I | 68 | 48 (70.6) | 20 (29.4) | |
| II | 117 | 64 (54.7) | 53 (45.3) | |
| III | 160 | 32 (20.0) | 128 (80.0) | |
| IV | 4 | 0 (0) | 4 (100.0) | |
Discussion
It is well known that aberrant functions of potassium channels result in many types of channelopathies, such as epilepsy (34), cardiac arrhythmia (34), and neuromuscular symptoms (35). Emerging evidence demonstrates that potassium channels are involved in carcinogenesis and progression of many cancers, including gastric cancer (36–38). Liu and colleagues (39) found that KCNQ1 polymorphisms appear to be independent predictors of chemotherapeutic response in gastric cancer. Ding and colleagues (40) showed that hERG1 expression was an independent prognostic factor in gastric cancer. In this study, we demonstrated, for the first time, that Kir2.1 was an important player in the invasion and metastasis capacity of gastric cancer cells. Elevated Kir2.1 expression in gastric cancer tissues is associated with cancer invasion depth, lymph node metastasis, and poor outcome of the patients.
The mechanisms by which potassium channels regulate the malignancy of cancer vary among different potassium channels and in different cancers. The mechanisms may be grouped into two types: canonical ion permeation-dependent and noncanonical ion permeation-independent. As the basic function of potassium channels in excitable and nonexcitable cells is to conduct potassium across the membrane to dominate the resting membrane potential, aberrant potassium channel expression may contribute to the progression of cancer by affecting the resting membrane potential. It is well known that rapidly proliferating embryonic cells, stem cells, and cancer cells in general are more depolarized, with resting membrane potential at −20 to −40 mV, whereas differentiated cell types such as neurons or cardiomyocytes are hyperpolarized, with resting membrane potential at −60 to −80 mV (41). A change in resting membrane potential directly or indirectly regulates cancer cell behaviors, such as proliferation and migration (42–44). Potassium channels also regulate cancer cell behaviors through noncanonical ion permeation-independent mechanisms, in which potassium channels utilize their cytoplasmic domains for protein–protein interactions (17, 39). For example, transfecting either wild-type or nonconducting Drosophila melanogaster eag channels into NIH3T3 cells induced comparable levels of cell proliferation (40). Downie and colleagues (45) found that EAG1 enhanced HIF-1 activity and VEGF secretion, hence tumor vascularization independently of its ion channel function in NIH3T3 cells. Kir2.2 promoted prostate cancer cell growth via protein–protein interaction with RelA (42). We found that Stk38 was critical to mediating a noncanonical ion permeation-independent signaling for Kir2.1 to enhance gastric cancer cell invasion and metastasis.
ERK1/2 signaling pathway is one of the key regulators that drive EMT switch-dependent metastasis of cancer cells (46–48). We showed that Kir2.1 potently activated ERK1/2 pathway in gastric cancer cells and inhibition of ERK1/2 blocked the effect of Kir2.1 on the capability of gastric cancer cell invasion and metastasis and reversed Kir2.1-induced EMT in gastric cancer cells, indicating that ERK1/2 pathway exerts a pivotal role in the process of Kir2.1-driven gastric cancer cell malignancy.
Classic cascade of ERK1/2 activation includes Ras–Raf–MEK–ERK sequentially, that is (i) Ras recruits and activates Raf (MAP3K) and (ii) Raf phosphorylates MEK1/2 (MAP2K) then activates ERK1/2 (49, 50). The Ras–Raf–MEK1/2–ERK1/2 signaling (canonical ERK1/2 pathway) is one of the most well-characterized cascades in cancer cells (51–53). In invading gastric cancer cells, however, elevated Kir2.1 activates ERK1/2 in a MEK1/2-dependent manner, but independent of Raf, implying that other MAP3Ks may be involved. In fact, there are over 20 MAP3Ks that selectively phosphorylate and activate different combinations of seven MAP2Ks (54) including MEKK2. Previous studies have demonstrated that MEKK2 transduces mitogenic signals emanating from EGFR and FGF2R to JNK and ERK5 cascades (55, 56). In recent years, MEKK2 was also found to exert important activities in ERK pathway (16, 30, 57). In this study, we demonstrated that MEKK2 was the necessary MAP3K for Kir2.1 to activate ERK1/2 pathway. MEKK2 is regulated by Stk38, a binding partner of Kir2.1 in gastric cancer cells. Stk38 is a serine–threonine protein kinase that belongs to a subfamily of AGC family of kinases (58). Studies have provided compelling evidence for the important roles of Stk38 in the regulation of MEKK2 signaling. Stk38 converts MEKK2 from a phosphorylated to a nonphosphorylated form and inhibits MEKK2 (32). Stk38 also promotes Smurf1-mediated polyubiquitination of MEKK2 by interacting with Smurf1 (16). In our study, Stk38 was found to inhibit the function of MEKK2 by Smurf1-mediated polyubiquitination and degradation of MEKK2. The binding between Kir2.1 and Stk38 interrupted the function of Stk38, resulting in MEKK2 enrichment and activation of MEKK2–MEK1/2–ERK1/2 signaling. Activated ERK1/2 triggers EMT of gastric cancer cells with increased invasion and metastasis potential. A working model was presented in Supplementary Fig. S15. The function of Kir2.1 in controlling other malignant behaviors of gastric cancer cells, such as proliferation, drug resistance and apoptosis, requires further investigation.
In conclusion, we report that Kir2.1 control gastric cancer cell invasion and metastasis with a noncanonical ion permeation–independent signaling pathway. Thus, Kir2.1 acts not only as an additional diagnostic marker, but also as a novel therapeutic target for gastric cancer.
Supplementary Material
Acknowledgments
This research was supported by the Ministry of Science and Technology of the People’s Republic of China (China National Science and Technology Major Project, 2016YFA0101203 to X.-W. Bian); the National Natural Science Foundation of China (81372555 to Y.-H. Cui); the National Natural Science Foundation of China (81402080 to Y.-X. Wang); and the Chongqing Science and Technology Commission (cstc2015jcyjA10114 to D.-F. Xiang).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- 2.Balakrishnan M, George R, Sharma A, Graham DY. Changing trends in stomach cancer throughout the world. Curr Gastroenterol Rep 2017;19:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu N, Wang X. [Current status and research progress of perioperative chemotherapy in advanced gastric cancer]. Zhonghua Wei Chang Wai Ke Za Zhi 2015;18:983–5. [PubMed] [Google Scholar]
- 4.Wadhwa R, Song S, Lee JS, Yao Y, Wei Q, Ajani JA. Gastric cancer-molecular and clinical dimensions. Nat Rev Clin Oncol 2013;10:643–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang X, Jan LY. Targeting potassium channels in cancer. J Cell Biol 2014;206:151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ouadid-Ahidouch H, Ahidouch A, Pardo LA. Kv10.1 K(+) channel: from physiology to cancer. Pflugers Arch 2016;468:751–62. [DOI] [PubMed] [Google Scholar]
- 7.Zhang P, Yang X, Yin Q, Yi J, Shen W, Zhao L, et al. Inhibition of SK4 potassium channels suppresses cell proliferation, migration and the epithelial–mesenchymal transition in triple-negative breast cancer cells. PLoS One 2016;11:e0154471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan G, Sun SQ, Yuan DL. Expression of Kir 4.1 in human astrocytic tumors: correlation with pathologic grade. Biochem Biophys Res Commun 2008;367:743–7. [DOI] [PubMed] [Google Scholar]
- 9.Voloshyna I, Besana A, Castillo M, Matos T, Weinstein IB, Mansukhani M, et al. TREK-1 is a novel molecular target in prostate cancer. Cancer Res 2008;68:1197–203. [DOI] [PubMed] [Google Scholar]
- 10.Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 2010;17:1471–4. [DOI] [PubMed] [Google Scholar]
- 11.Wang B, Liu J, Ma LN, Xiao HL, Wang YZ, Li Y, et al. Chimeric 5/35 adenovirus-mediated Dickkopf-1 overexpression suppressed tumorigenicity of CD44(+) gastric cancer cells via attenuating Wnt signaling. J Gastroenterol 2013;48:798–808. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Z, Ji Z, Wang Y, Li J, Cao H, Zhu HH, et al. TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology 2014;147:1043–54. [DOI] [PubMed] [Google Scholar]
- 13.Liu JJ, Liu JY, Chen J, Wu YX, Yan P, Ji CD, et al. Scinderin promotes the invasion and metastasis of gastric cancer cells and predicts the outcome of patients. Cancer Lett 2016;376:110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lam D, Schlichter LC. Expression and contributions of the Kir2.1 inward-rectifier K(+) channel to proliferation, migration and chemotaxis of microglia in unstimulated and anti-inflammatory states. Front Cell Neurosci 2015;9:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLerie M, Lopatin AN. Dominant-negative suppression of I(K1) in the mouse heart leads to altered cardiac excitability. J Mol Cell Cardiol 2003;35:367–78. [DOI] [PubMed] [Google Scholar]
- 16.Wen M, Ma X, Cheng H, Jiang W, Xu X, Zhang Y, et al. Stk38 protein kinase preferentially inhibits TLR9-activated inflammatory responses by promoting MEKK2 ubiquitination in macrophages. Nat Commun 2015;6:7167. [DOI] [PubMed] [Google Scholar]
- 17.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial–mesenchymal transitions in development and disease. Cell 2009;139:871–90. [DOI] [PubMed] [Google Scholar]
- 18.Tiwari N, Gheldof A, Tatari M, Christofori G. EMT as the ultimate survival mechanism of cancer cells. Semin Cancer Biol 2012;22:194–207. [DOI] [PubMed] [Google Scholar]
- 19.Zhang YY, Li G, Che H, Sun HY, Xiao GS, Wang Y, et al. Effects of BKCa and Kir2.1 channels on cell cycling progression and migration in human cardiac c-kit+ progenitor cells. PloS One 2015;10:e0138581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao Z, Feng Z, Long C. Agonist of inward rectifier K+ channels enhances the protection of ischemic postconditioning in isolated rat hearts. Perfusion 2014;29:321–6. [DOI] [PubMed] [Google Scholar]
- 21.Leonoudakis D, Conti LR, Anderson S, Radeke CM, McGuire LM, Adams ME, et al. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J Biol Chem 2004;279:22331–46. [DOI] [PubMed] [Google Scholar]
- 22.Dart C, Leyland ML. Targeting of an A kinase-anchoring protein, AKAP79, to an inwardly rectifying potassium channel, Kir2.1. J Biol Chem 2001;276:20499–505. [DOI] [PubMed] [Google Scholar]
- 23.Sampson LJ, Leyland ML, Dart C. Direct interaction between the actin-binding protein filamin-A and the inwardly rectifying potassium channel, Kir2.1. J Biol Chem 2003;278:41988–97. [DOI] [PubMed] [Google Scholar]
- 24.Wang HR, Wu M, Yu H, Long S, Stevens A, Engers DW, et al. Selective inhibition of the K(ir)2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem Biol 2011;6:845–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu QH, Li XL, Xu YW, Lin YY, Cao JM, Wu BW. A novel discovery of IK1 channel agonist: zacopride selectively enhances IK1 current and suppresses triggered arrhythmias in the rat. J Cardiovasc Pharmacol 2012;59:37–48. [DOI] [PubMed] [Google Scholar]
- 26.Arana-Argaez VE, Delgado-Rizo V, Pizano-Martinez OE, Martinez-Garcia EA, Martin-Marquez BT, Munoz-Gomez A, et al. Inhibitors of MAPK pathway ERK1/2 or p38 prevent the IL-1{beta}-induced up-regulation of SRP72 autoantigen in Jurkat cells. J Biol Chem 2010;285:32824–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henry JR, Kaufman MD, Peng SB, Ahn YM, Caldwell TM, Vogeti L, et al. Discovery of 1-(3,3-dimethylbutyl)-3-(2-fluoro-4-methyl-5-(7-methyl-2-(methylamino)pyrido[2,3-d]pyrimidin-6-yl)phenyl)urea (LY3009120) as a pan-RAF inhibitor with minimal paradoxical activation and activity against BRAF or RAS mutant tumor cells. J Med Chem 2015;58:4165–79. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Suzuki YJ, Day RM, Fanburg BL. Rho kinase-induced nuclear translocation of ERK1/ERK2 in smooth muscle cell mitogenesis caused by serotonin. Circ Res 2004;95:579–86. [DOI] [PubMed] [Google Scholar]
- 29.Li F, Jiang Q, Shi KJ, Luo H, Yang Y, Xu CM. RhoA modulates functional and physical interaction between ROCK1 and Erk1/2 in selenite-induced apoptosis of leukaemia cells. Cell Death Dis 2013;4:e708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maruyama T, Kadowaki H, Okamoto N, Nagai A, Naguro I, Matsuzawa A, et al. CHIP-dependent termination of MEKK2 regulates temporal ERK activation required for proper hyperosmotic response. EMBO J 2010;29: 2501–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Widmann C, Sather S, Oyer R, Johnson GL, Dreskin SC. In vitro activity of MEKK2 and MEKK3 in detergents is a function of a valine to serine difference in the catalytic domain. Biochim Biophys Acta 2001;1547: 167–73. [DOI] [PubMed] [Google Scholar]
- 32.Enomoto A, Kido N, Ito M, Morita A, Matsumoto Y, Takamatsu N, et al. Negative regulation of MEKK1/2 signaling by serine-threonine kinase 38 (STK38). Oncogene 2008;27:1930–8. [DOI] [PubMed] [Google Scholar]
- 33.Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX, et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 2005;121:101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohling R, Wolfart J. Potassium channels in epilepsy. Cold Spring Harb Perspect Med 2016;6:pii: a022871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fontaine B Muscle channelopathies and related diseases. Handb Clin Neurol 2013;113:1433–6. [DOI] [PubMed] [Google Scholar]
- 36.Stringer BK, Cooper AG, Shepard SB. Overexpression of the G-protein inwardly rectifying potassium channel 1 (GIRK1) in primary breast carcinomas correlates with axillary lymph node metastasis. Cancer Res 2001; 61:582–8. [PubMed] [Google Scholar]
- 37.Hemmerlein B, Weseloh RM, Mello de Queiroz F, Knotgen H, Sanchez A, Rubio ME, et al. Overexpression of Eag1 potassium channels in clinical tumours. Mol Cancer 2006;5:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pillozzi S, Brizzi MF, Balzi M, Crociani O, Cherubini A, Guasti L, et al. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia 2002;16:1791–8. [DOI] [PubMed] [Google Scholar]
- 39.Liu X, Chen Z, Zhao X, Huang M, Wang C, Peng W, et al. Effects of IGF2BP2, KCNQ1 and GCKR polymorphisms on clinical outcome in metastatic gastric cancer treated with EOF regimen. Pharmacogenomics 2015;16: 959–70. [DOI] [PubMed] [Google Scholar]
- 40.Ding XW, Yang WB, Gao S, Wang W, Li Z, Hu WM, et al. Prognostic significance of hERG1 expression in gastric cancer. Dig Dis Sci 2010;55: 1004–10. [DOI] [PubMed] [Google Scholar]
- 41.Yang M, Brackenbury WJ. Membrane potential and cancer progression. Front Physiol 2013;4:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang X, He Y, Dubuc AM, Hashizume R, Zhang W, Reimand J, et al. EAG2 potassium channel with evolutionarily conserved function as a brain tumor target. Nat Neurosci 2015;18:1236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang X, Dubuc AM, Hashizume R, Berg J, He Y, Wang J, et al. Voltage-gated potassium channel EAG2 controls mitotic entry and tumor growth in medulloblastoma via regulating cell volume dynamics. Genes Dev 2012; 26:1780–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hammadi M, Chopin V, Matifat F, Dhennin-Duthille I, Chasseraud M, Sevestre H, et al. Human ether a-gogo K(+) channel 1 (hEag1) regulates MDA-MB-231 breast cancer cell migration through Orai1-dependent calcium entry. J Cell Physiol 2012;227:3837–46. [DOI] [PubMed] [Google Scholar]
- 45.Downie BR, Sanchez A, Knotgen H, Contreras-Jurado C, Gymnopoulos M, Weber C, et al. Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J Biol Chem 2008;283:36234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol 2013;15:677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang L, Yan Q, Fang S, Liu M, Li Y, Yuan YF, et al. Calcium binding protein 39 promotes hepatocellular carcinoma growth and metastasis by activating ERK signaling pathway. Hepatology 2017;66:1529–45. [DOI] [PubMed] [Google Scholar]
- 48.Ichikawa K, Kubota Y, Nakamura T, Weng JS, Tomida T, Saito H, et al. MCRIP1, an ERK substrate, mediates ERK-induced gene silencing during epithelial–mesenchymal transition by regulating the co-repressor CtBP. Mol Cell 2015;58:35–46. [DOI] [PubMed] [Google Scholar]
- 49.Caunt CJ, Finch AR, Sedgley KR, McArdle CA. Seven-transmembrane receptor signalling and ERK compartmentalization. Trends Endocrinol Metab 2006;17:276–83. [DOI] [PubMed] [Google Scholar]
- 50.Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene 2007;26:3122–42. [DOI] [PubMed] [Google Scholar]
- 51.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 2010;1802:396–405.20079433 [Google Scholar]
- 52.De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets 2012;16:S17–27. [DOI] [PubMed] [Google Scholar]
- 53.Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 2009;19:230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cuevas BD, Abell AN, Johnson GL. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene 2007;26: 3159–71. [DOI] [PubMed] [Google Scholar]
- 55.Sun W, Wei X, Kesavan K, Garrington TP, Fan R, Mei J, et al. MEK kinase 2 and the adaptor protein Lad regulate extracellular signal-regulated kinase 5 activation by epidermal growth factor via Src. Mol Cell Biol 2003;23: 2298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kesavan K, Lobel-Rice K, Sun W, Lapadat R, Webb S, Johnson GL, et al. MEKK2 regulates the coordinate activation of ERK5 and JNK in response to FGF-2 in fibroblasts. J Cell Physiol 2004;199:140–8. [DOI] [PubMed] [Google Scholar]
- 57.Li Y, Zhang Z, Zhou X, Li L, Liu Q, Wang Z, et al. The oncoprotein HBXIP enhances migration of breast cancer cells through increasing filopodia formation involving MEKK2/ERK1/2/Capn4 signaling. Cancer Lett 2014;355:288–96. [DOI] [PubMed] [Google Scholar]
- 58.Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci 2002;27:514–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.