

Summary
Single-molecule fluorescence recovery after photobleaching (smFRAP) is a newly developed technique that combines single-molecule super-resolution microscopy and traditional FRAP microscopy. smFRAP enables researchers to measure the dynamics, spatial locations, and relative concentrations of proteins. Here, we describe a step-by-step protocol for smFRAP on nuclear envelope transmembrane proteins on the inner nuclear membrane and outer nuclear membrane in live cells.
For complete details on the use and execution of this protocol, please refer to Mudumbi et al. (2016a, 2016b, 2020 .
Subject areas: Atomic Force Microscopy (AFM), Microbiology, Molecular Biology, Biotechnology and bioengineering
Graphical abstract
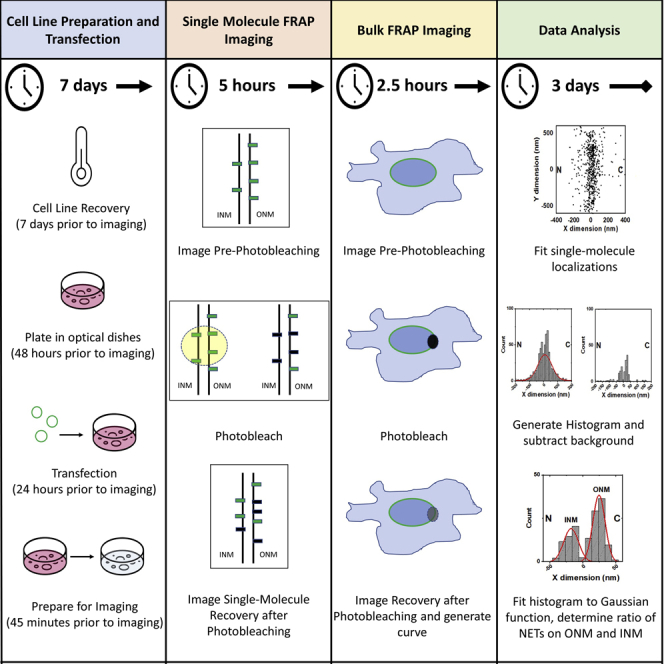
Highlights
-
•
Protocol for smFRAP of nuclear envelope membrane proteins in live cells.
-
•
smFRAP distinguishes the spatial locations of membrane proteins on the nuclear envelope.
-
•
Protocol to measure translocation rates for nuclear envelope transmembrane proteins.
-
•
Steps to quantify diffusion coefficients for membrane proteins on the nuclear envelope.
Single-molecule fluorescence recovery after photobleaching (smFRAP) is a newly developed technique that combines single-molecule super-resolution microscopy and traditional FRAP microscopy. smFRAP enables researchers to measure the dynamics, spatial locations, and relative concentrations of proteins. Here, we describe a step-by-step protocol for smFRAP on nuclear envelope transmembrane proteins on the inner nuclear membrane and outer nuclear membrane in live cells.
Before you begin
Microscopy setup for single molecule fluorescence recovery after photobleaching (smFRAP)
Prior to planning an smFRAP experiment it is critical to identify the cell line in which you plan to perform your experiment. This protocol reports outcomes specifically for HeLa cells, however, this protocol has also been employed to great success in other cell lines including CHO and NIH3T3 cells. Next, it is critical to ensure that the microscope has been setup properly. Ensure that the microscope and laser lines are installed on a research grade optical table to limit vibration that will impact single molecule imaging. Align lasers such that they go through the center of the optical objective and strike the center of the detector. Lastly, ensure that neutral density filters and optical chopper are properly installed in the laser path to regulate laser power and on/off time (Figure 1A).
Figure 1.

A simplified schematic of light microscopy
(A) A schematic of the excitation laser used in smFRAP. A 561 nm laser (green) is directed into the objective. The inset depicts the laser passing through the nuclear envelope while the focal plane is at the nuclear equator, thereby illuminating NETs on both the inner and outer nuclear membrane.
(B) An illustration of the laser power during the different phases of smFRAP. Fluorophores are initially photobleached using high laser power. The chopper is then engaged, allowing fluorescently tagged NETs to diffuse into the detection area.
Mix transport buffer
Timing: 30 min
-
1.
In a beaker with a stir rod, add HEPES, KOAc, NaOAc, MgOAc, and EGTA to 1 liter of distilled water.
-
2.
While mixing, correct the pH to 7.3.
-
3.
Filter contents through 0.22 μm microfilter.
-
4.
Aliquot into 50 mL portions and store at 4°C for no longer than 1 year.
| Reagent | Final concentration (mM or μM) | Amount |
|---|---|---|
| Distilled H2O | n/a | 1 L |
| HEPES | 20 mM | 4.766 g/L |
| Potassium acetate (KOAc) | 110 mM | 10.796 g/L |
| Sodium acetate (NaOAc) | 5 mM | 0.410 g/L |
| Magnesium acetate (MgOAc) | 2 mM | 0.534 g/L |
| EGTA | 1 mM | 0.380 g/L |
Cell line recovery
-
5.
Resuscitate required cells at least one week before transfection experiment. The frozen stocks (−80°C) of cells are thawed at 37°C and then put into a 25 cm2 culture flask with 4-5 mL prewarmed (37°C) DMEM media (DMEM high glucose GlutaMAX™ Supplement Gibco™ from Thermo Fisher, 10% fetal bovine serum, 1% penicillin-streptomycin).
-
6.
Incubate the culture flask at 37°C with 5% CO2 for 12 to 18 h.
-
7.
Exchange the culture media in culture flask with fresh DMEM media the next day, and incubate at 37°C with 5% CO2 for 24 h.
-
8.
Split the culture cells at least three times over the week in order to guarantee the cells will be at optimal health for transfection and optimal optical conditions for smFRAP experiment. All procedures are finished via aseptic technique. The cell lines used to generate the anticipated data section contain wild-type (WT) HeLa cells, Pom121-GFP stably expressing cells and Pom121-mCherry stably expressing cells.
Plating
-
9.
Two days prior to the smFRAP experiments, split and plate cultured cells onto 2-4 glass bottom optical dishes.
-
10.
Incubate at 37°C with 5% CO2 for 12 to 18 h. The cells plated here should be ~50% confluent in order to achieve optimal transfection results. All procedures are finished via aseptic technique.
Transfection
-
11.
According to the protocol from the manufacturer, we transfect cells on glass bottom optical dishes with plasmids encoding the NETs of interest. In our case, using TransIT-X2 transfection reagent (Mirus). The Opti-MEM™ reduced serum medium (Thermo Fisher), transfection reagent and plasmids are added dropwise to the glass bottom optical dishes and then mixed by gently flicking for getting homogeneous solution.
-
12.
Incubate optical dishes in an incubator at 37°C with 5% CO2 for 12 to 18 h so that fluorescent protein fused NETs will have reasonable expression levels during smFRAP experiments. All procedures are performed following best aseptic technique.
Prepare for imaging
-
13.
The day of the smFRAP experiment, remove the culture media from the glass bottom optical dishes. Use 1 mL of prewarming (37°C) PBS buffer to wash optical dishes twice. Removing DMEM media to reduce fluorescent backgrounds during the smFRAP experiment. This step is performed because the phenol red in DMEM yields too high fluorescent backgrounds for single-molecule experiment. Then, add 1 mL of prewarmed (37°C) transport buffer (20 mM HEPES, 110 mM KOAc, 5 mM NaOAc, 2 mM MgOAc, 1 mM EGTA, pH adjusted to 7.3 with HCl) to the optical dish and incubate transfected cells for 30-45 mins.
Key resources table
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Experimental models: cell lines | ||
| HeLa cells stably expressing mCherry on the N terminus of POM121 | (Li et al., 2021) | RRID: CVCL_A916 |
| Software and algorithms | ||
| SlideBook 6 | 3i | https://www.intelligent-imaging.com/slidebook |
| Fiji ImageJ (Schindelin et al., 2012) | ImageJ | https://imagej.net/Fiji/Downloads |
| GDSC SMLM ImageJ plugin (Herbert, 2013) | University of Sussex | http://www.sussex.ac.uk/gdsc/intranet/microscopy/UserSupport/AnalysisProtocol/imagej/gdsc_plugins/ |
| FRAP profiler ImageJ plugin | ImageJ | https://imagej.net/mbf/intensity_vs_time_ana.htm |
| OriginPro 2019 | OriginLab | https://www.originlab.com/2019 |
| cellSens | Olympus | https://www.olympus-lifescience.com/en/software/cellsens/ |
| Other | ||
| Olympus IX81 equipped with 1.4-NA 100× oil immersion apochromatic objective | Olympus | UPLSAPO 100XO |
| Experimental models: cell lines | ||
| On-chip multiplication gain charge-coupled-device camera | Roper Scientific | Cascade 128+ |
| Confocal laser scanning microscope | Olympus | FV3000 |
| Dichroic filter | Semrock | Di01-R405/488/561/635-25x36 |
| Emission filter | Semrock | NF01- 405/488/561/635-25X5.0 |
| Two circular variable metallic neutral density filters | Newport | Cat. No. 50G02AV.1/50Q04AV.2 |
| Top beam steering accessory SM micrometers | Newport | Cat. No. 670-RCT-M |
| Bottom steering accessory | Newport | Cat. No. 670-RCB |
| Damped optical post, gear rack mounting base 14 in | Newport | Model 75 |
| ObisTM solid state 561-nm LS 50 mW laser | Coherent Inc. | Laser head 1230935 Laser system 1230936 |
| ObisTM solid state 488-nm LX 50 mW laser | Coherent Inc. | Laser head 1185053 Laser system 1178764 |
| Pneumatic isolator pre-mounted to research grade optical table | Newport | Cat. No. RS4000-46-12 |
| Glass bottom culture dishes, 35 mm petri dish, 14 mm Microwell with No. 0 cover glass (0.085-0.13 mm) | MatTek | Cat. No. P35G-0-14-C |
| Corning® 25 cm2 vented cell culture flasks | Sigma-Aldrich | Cat. No. CLS430639 |
| Low autofluorescence immersion oil | Olympus | Cat. No. IMMOIL-F30CC |
| Dulbecco's Modified Eagle Medium (DMEM), high glucose GlutaMAXTM with 10% FBS (vol/vol), 10 mg/mL streptomycin, and 100 U/mL penicillin. | GibcoTM/ Thermo Fisher Scientific | Cat. No. 10566016 |
| Opti-MEMTM Reduced Serum Medium | GibcoTM/ Thermo Fisher Scientific | Cat. No. 31985088 |
| Trypsin-EDTA (0.25%), phenol red | GibcoTM/ Thermo Fisher Scientific | Cat. No. 25200056 |
| TransIT-X2 Transfection Reagent | Mirus | Cat. No. MIR 6000 |
| GibcoTM 1× phosphate buffered solution (PBS), pH 7.4 | GibcoTM/ Thermo Fisher Scientific | Cat. No. 10010023 |
| Transport buffer | This protocol | N/A |
Step-by-step method details
smFRAP experiment: data collection
This major step describes how to align the laser as well as single-molecule imaging parameters to perform smFRAP.
-
1.Ensure that the required laser is aligned to precisely strike the center of the detector in a perpendicular manner and is aligned with the center of the detector’s guides. The detector we used is an on-chip EMCCD camera (Cascade 128+, Roper Scientific Inc.). The guides delineate the center of the detector and can be toggled on or off within the capture program, here we will be using SlideBook 6.1. In our case, the 561-nm lasers are aligned as our NETs of interest are fluorescently tagged with mCherry. This setup may be modified to make use of a laser appropriate to the specific fluorophore (Figure 1A).
-
a.To align the laser to strike the detector in a perpendicular manner begin by adjust the laser pathway to strike the center of the mirror positioned on the micrometer stage.
-
b.At 10× magnification, adjust the micrometer stage so the laser strikes the center of the detector’s guides.
-
c.Increase the magnification to 100× and adjust the micrometer stage so the laser once again strikes the center of the detector’s guides.
-
d.Adjust the focus to evaluate the laser pattern. If the laser is striking the detector at 90°, the laser should appear to be symmetrical as it is adjusted in and out of focus.
-
e.If the laser pattern is not symmetrical, gently adjust the mirrors positioned prior to the iris on the laser line until the laser pattern appears symmetrical.
-
f.After the laser pattern is symmetrical, adjust the focus until the laser is at the smallest point and capture an image. This is to be used as a reference point to determine where the laser is striking in relation to your sample.
-
a.
-
2.
Utilize a 100× oil immersion objective to visualize cells under a mercury lamp with the corresponding filter sets for mCherry.
-
3.
It is critical to target only cells that exhibit a healthy nuclear morphology, defined here as free from nuclear blebbing or other signs of apoptosis (Figures 2B and 2D) as well as sufficient expression of fluorescence fused NETs (Figures 2A and 2C). Sufficient expression of fluorescence fused NETs can be determined by capturing an image of the Nucleus using the fluorescence wavelength associated with your protein of interest. This image should clearly display a nuclear ring (illuminated nuclear envelope with a dark nucleoplasm) as NETs are, by definition, embedded in the nuclear envelope. This means that only cells in interphase should be selected, cells in mitosis should be avoided. Bring the focal plane to equator of the cell and then move the left or right edge of the NE (tangent to the edge of NE) to the center of the camera. This ensures that the center of the laser illumination point spread function (PSF) will strike only the edge of the nuclear envelope (Figure 1A).
-
4.
Take a pre-photobleaching image of the cell with 500-ms exposure time with the mercury lamp. Overexposure and saturation of the image should be avoided.
-
5.
Close the shutter to the mercury lamp and switch to the laser. If the intended photobleaching area is at the center of laser PSF, begin photobleaching the desired area by opening the corresponding shutter to the laser. If the intended photobleaching area is off the center of laser PSF, move the area to the PSF center and then begin photobleaching.
-
5.Photobleach a small area of the NE under high laser power for 10-30 s, depending upon your specific fluorophore. The photobleaching area is around 1 μm in diameter (Figure 1B).
-
a.To determine what timeframe is the most appropriate for your fluorophore, set the photobleaching laser power to 10 times that of the excitation laser power.
-
b.Photobleach a test cell for 10 s. Immediately after photobleaching, capture an image of the photobleached region and use Fiji ImageJ to evaluate for the presence of fluorophores.
-
c.To evaluate for the presence of fluorophores, open the image in Fiji ImageJ. Select two ROIs, one in the center of the photobleached region and one on a dark region, the center of the nucleus is a good option. Use the Measure tool under the Analyze window for each ROI. This tool allows you to gain information regarding the diameter of the region measured, the minimum and maximum pixel intensity within the ROI, and the mean intensity. Compare the mean intensity of the photobleached ROI with that of the background ROI. If the photobleached ROI displays a mean intensity value greater than the background ROI by more than 2, there is likely still fluorophores that were not successfully photobleached within the ROI. If this is the case, select another region and increase the photobleaching time by 10 s and evaluate for fluorescence. Repeat these steps until the appropriate timeframe is identified for your fluorophore.
-
a.
-
6.
Next, use the neutral density filter installed in the laser light path to lower the laser power to excitation level to at ~10% of the photobleaching laser power. The laser power may be adjusted to optimize the signal-to-noise ratio (SNR).
-
7.
Engage the optical chopper at 2-Hz rotation speed with an on time of 1/10 of the total frames recorded (Figure 1B).
-
8.
Record video at a 2 ms per frame for 30 s to produce a video with 15,000 frames. Three total videos are taken consecutively. Here, the SlideBook software is used to record images and videos.
-
9.
After recording videos, take a post-smFRAP image with 500-ms exposure time mercury lamp. The overexposure and saturation of the image should be avoided. Check whether the NE shifted during the experiment by comparing pre-photobleaching image and post-smFRAP image.
Figure 2.

Representative cells
Epi-fluorescent (A) and bright-field (B) microscopy images of healthy cells, representative of the types of cells that should be imaged, exhibiting healthy cell shape when adhering to the bottom of the optic dish. Epi-fluorescent (C) and bright-field (D) microscopy images of unhealthy cells, representative of the types of cells that should not be imaged. Note the shrunken GFP-NE rings as well as being poorly attached to optical dish. Scale bar, 10 μm.
Bulk FRAP experiment (performed with the confocal microscope): data collection
This major step describes how to perform a FRAP experiment at the bulk level.
-
10.
Place the sample on the Olympus FluoView 3000 laser scan confocal microscope.
-
11.
Turn on the required lasers. In our case, both the 488-nm and the 561-nm lasers are turned on.
-
12.
Utilize a 60× oil immersion objective to visualize cells with the laser and the corresponding filter sets for GFP or mCherry.
-
13.
We target only cells with good nuclear morphology and good expression of fluorescence fused NETs (Figure 2). Bring the focal plane to equator of the cell.
-
14.
Select region of interest (ROI). In our case, we select rectangular area including single cell. Then set proper scan size and scan speed for making sure scanning time is short enough. In our case, scan size is 512 × 512 pixel and scan speed is 2 μs/pixel.
-
15.
Take a pre-photobleaching image of the cell by corresponding laser for attaining baseline curve of FRAP curve. If the NET of interest is fused to GFP, a 488-nm laser could be used for imaging. Here, need to set laser intensity, sensitivity, gain and offset in advance to taking an image. In our case, laser intensity is 10%; sensitivity is 700V; gain is 1.0; offset is 0%. The overexposure and saturation of the image should be avoided.
-
16.
Use the sequence manager module on Olympus Fluoview3000 to conduct bulk FRAP experiment.
-
17.
Select region of photobleaching. In our case, we select a circular area covering a small region at the edge of the NE opposite the endoplasmic reticulum (ER). The laser intensity and photobleaching time is dependent on the NETs of interest. In our case, we did 30 s under 100% intensity of 488-nm laser for NET23.
-
18.
Next, take time-lapse images of recovery in 0.5 Hz for 1 min. All of parameter setting should be same as step 6. If 1 min is not sufficient for the fluorescence recovery curve to reach a plateau, the time could be prolonged to 2 mins.
Analysis of bulk FRAP data
This major step describes how to analyze bulk FRAP images captured on the confocal microscope to calculate the proportion of immobile and mobile NETs localized to the Nuclear Envelope.
-
19.
Use ImageJ to open bulk FRAP images in sequencing starting with the pre-photobleaching image, followed by the time-lapse images of recovery (Figure 3).
-
20.
Open the ROI manager (ImageJ/ Analyze/Tools/ROI Manager) and select the two regions of interest. The one ROI is the photobleached region of NE and the second ROI is the edge of NE excluding photobleached area, which must be carefully outlined with the freehand selection tool.
-
21.
Generate FRAP curve by running FRAP Profiler plugin (https://imagej.net/mbf/intensity_vs_time_ana.htm) and determine the immobilized fraction for overall NE by using the normalized output.
-
22.
Next, calculate the immobilized fraction of NETs on the INM. This is done by evaluating the FRAP curve generated by FRAP profiler. The fraction recovery at the point the FRAP curve plateaus is the fraction of mobile NETs whereas the difference between the prephotobleached and the plateaued recovery curve represents the fraction of non-mobile NETs (Figure 7D).
-
23.
Determine the final concentration ratio of NETs on the ONM and INM by using the diffusion-based concentration ratio and the immobilized fraction on the INM. For example, if the diffusion-based concentration ratio (ONM: INM) is 1:0.30 and the immobilized fraction on the INM is 0.72. Then, the final corrected concentration ratio 1:1.1 (ONM: INM) is obtained by 1:(0.30 /(1-0.72)).
Figure 3.

Bulk FRAP analysis flowchart
Shown here is a step by step flowchart demonstrating the individual tools and methods to utilize FRAP Profiler in order to determine the proportion of mobile fluorescent proteins on the NE.
Figure 7.

Expected results of smFRAP
(A) Two-dimension (2d) super-resolution image of LBR on the NE, in which the INM shown in red and the ONM shown in purple.
(B) Two-peak Gaussian fittings of the points collected from the NE showing the distribution of LBR along the NE. The shaded regions represent the width of the INM and ONM as determined by the full width at half maximum (FWHM) as determined by the fitting.
(C) Approximate concentration ratios of LBR’s distribution along the INM (red) and ONM (purple). The corrected ratios can be found in Table 1.
(D) The FRAP curve for LBR. The mobile fraction of whole NE is ~0.39. INM: inner nuclear membrane; ONM: inner nuclear membrane.
Analysis of smFRAP data
This major step describes how to analyze single-molecule FRAP data, including how to fit the INM and the ONM to determine the proportion of fluorophore tagged NETs localized to each discrete membrane.
-
24.
Convert videos generated with Slidebook into image sequence .tiff files.
-
25.
Use the peak fit (series) function of the ImageJ plugin GDSC SMLM (http://www.sussex.ac.uk/gdsc/intranet/microscopy/UserSupport/AnalysisProtocol/imagej/gdsc_plugins/) to localize each data point from the .tiff files (Figure 4).
-
26.
Filter the raw data from GDSC SMLM by using the signal to noise ratio (SNR), localization precision and the width of the Gaussian fitting (wi). In our case, the threshold for SNR is > 10; the localization precision is <20 nm; and the wi is 0.5~2.0. These filters are selected as this method allows for the detection between INM and ONM, which are separated by a perinuclear space of ~40 nm (7). The localization precision of the detected NETs must be, at most, half that of the perinuclear space to ensure that the detected particle is assigned to the appropriate membrane.
-
27.
Calculate the diffusion coefficient of the filtered points using GDSC SMLM with the built-in trace diffusion tool (Figure 4). Ensure that the MSD correction feature is selected as this will account for distances between discrete steps of the trace, and not simply average the distance between the locations of the beginning and end point. It is important to gather sufficient traces to ensure the quality of the calculated diffusion coefficient. Samples with a more consecutive frames are desirable. For our purposes, traces consisting of seven or more consecutive frames are considered separately from traces consisting of two to six consecutive frames. Approximately 50 single-molecule traces of the seven or more class or approximately 500 of the two to six class are required. Finally, an averaged diffusion coefficient is then calculated from the observed traces.
-
28.
Use OriginPro 2019 to plot all filtered data points localized via GDSC SMLM. This enables the user to visualize the distribution of data points, and will be plotted in the rough shape of the right or left edge of NE (Figure 5A).
-
29.
From here, plot the frequency distribution histogram by using the x coordinates of these data points (Figure 5B). In our case, the line perpendicular to the right or left NE edge is parallel to the x axis. Therefore, we use the x coordinates. Use y coordinates to plot frequency histogram if targeting up or down edge of the NE.
-
30.
Still using OriginPro 2019, select and mask the binned localizations ±40nm from the two highest peaks of the frequency distribution histogram of x coordinates. This will exclude these data points from the subsequent Gaussian fitting.
-
31.
Fit the histogram with a single Gaussian function (Figure 3B). The area under the curve is representative of background noise. For a complete explanation, please consult Mudumbi et. al. 2016 (Mudumbi et al., 2016a).
-
32.
Using the built in subtract function of OriginPro 2019, subtract the area under the curve from the entirety of the x-coordinate histogram (Figure 5C).
-
33.Further fit the background reduced histogram with a two-peak Gaussian, yielding distribution of NETs on inner and outer NE membranes (Figure 5D).
-
a.This information enables you to calculate the ratiosmFRAP of NETs on outer nuclear membrane (ONM) to inner nuclear membrane (INM). This is accomplished by dividing the area under the ONM curve by the area under the INM curve.
-
a.
-
34.The concentration ratio of NETs on the ONM and INM must now be corrected. This is accomplished by factoring in the impact of the diffusion coefficient of NETs protein on both the INM and ONM. The formulas were originally published in our previous works (Mudumbi and Yang, 2017, Mudumbi et al., 2016b, Mudumbi, 2020, Mudumbi et al., 2016a).
-
a.The equation G(i,D,t) represents the probability of finding a randomly diffusing particle at location i following diffusion at a diffusion constant of D within the time t (Figure 6A).
(Equation 1) -
b.The equation f(D,t) refers to observing a molecule diffusing into the detection area from any locations within the entire area (Rmax) (Figures 6B and 6C), in which 2R is an approximation for the length of the arc from of a circle of radius i that falls within the gray circle of radius R.
(Equation 2) -
c.The equation V(D) denotes the total area that a particle travels from t1 to t1 + 30 s.
(Equation 3) -
d.From this series of equations, the corrected concentrations of the ONM and INM can be derived using the following relationship:
where NONM and NINM are the originally derived values from step 10(a), VONM and VINM is calculated using equation 3. CONM and CINM represent the actual concentrations of NETs, and can be derived by solving this relationship.(Equation 4)
-
a.
-
35.
Next, calculate the fraction of mobile NETs on the INM. Here the actual proportion of the NETs on the ONM is denoted as NONM and the INM is NINM. The immobile fraction of INM is defined as x. Fm denotes the mobile fraction as determined by FRAP in step 3 of bulk FRAP analysis. R represents the corrected concentration ratio after considering the effects of diffusion coefficients on the NETs. These formulas were originally published previously in earlier work (Mudumbi and Yang, 2017, Mudumbi et al., 2016b, Mudumbi et al., 2016a, Mudumbi et al., 2020).
| (Equation 5) |
| (Equation 6) |
Figure 4.

GDSC single-molecule localization flowchart
Shown here is a flowchart depicting individual settings, tool locations, and parameters for localizing and calculating the diffusion coefficient of Single-Molecules using GDSC. Note the settings are correct only for the smFRAP microscope setup described in this protocol.
Figure 5.

Analysis of smFRAP data
(a) Two-dimension (2D) localizations of a NET protein of interest on the NE.
(b) Frequency distribution histogram for the locations of NETs protein along the X dimension. The background noise (without the data from the two peaks) is fit with a single Gaussian function (red curve). Previous experiments verified that the background-noise distribution was generated by randomly diffusing fluorophores around the NE, rather than the membrane proteins located at the NE (3).
(c) Background signal is subtracted to generate a histogram with two clear peaks. The peak positions for membrane proteins on the NE can only be precisely determined after subtracting the background-noise distribution.
(d) The resultant data are fit with a two-peak Gaussian function (red curves) to determine the localization of NETs on the NE. N: nucleus; C: cytoplasm; INM: inner nuclear membrane; ONM: inner nuclear membrane.
Figure 6.

Correction of ONM:INM ratios by factoring in the impact of the molecular diffusion coefficient derived from single-molecule trajectories
This calculation accounts for discrepancies in diffusion coefficients of transmembrane proteins on the nuclear envelope as they enter the detection area (depicted as a gray circle). The distribution ratio is also corrected to reflect the actual transmembrane protein concentrations along the nuclear envelope. The outer ring (Rmax) is representative of the nuclear envelope circumference. The detected molecule (green) may diffuse into the detection area from any direction within the distance x from the center of the photobleached area (depicted by the inner ring).
(A) In this equation G(i,D,t) denotes the probability of finding a randomly diffusing molecule at location i with a diffusion constant of D within time t.
(B) The integrated probability that a molecule with a beginning location at i (green) will diffuse into the detection area (gray).
(C) f(D,t) refers to the summed probability of observing a molecule diffusing into the detection area from any locations within the entire area (Rmax). In the equation, 2R is an approximation for the length of the arc from of a circle of radius i that falls within the gray circle of radius R. This figure was originally published in Nature Communications (Mudumbi et al., 2016a) and is redrawn here with permission.
Expected outcomes
A successfully completed smFRAP experiment, and corresponding data analysis, yields 2D super-resolution images of the distribution of the NETs of interest (Figure 7A). Subsequently, the 2D distribution of the NETs of interest are utilized to quantitatively measure the relative concentration ratio of NETs along the INM and ONM (Figures 5, 7B, and 7C). For example, we first determined the concentration ratio of Lamin B receptor (LBR) along the INM and ONM within live HeLa cells (Mudumbi et al., 2016a). As shown in the Figure 3, the relative concentration ratiosmFRAP of LBR is 0.53:1 (ONM: INM), and the distance between LBR on INM and ONM is ~41 nm.
As mentioned above, the relative concentration ratiosmFRAP still must be corrected by considering the effects caused by the diffusion coefficient of NETs on the INM and ONM. The diffusion based corrected concentration ratio is calculated using the formula published in our previous works (Mudumbi and Yang, 2017, Mudumbi et al., 2016b). As a result, the corrected concentration ratio of LBR is 0.58:1 (ONM: INM) (Table 1).
Table 1.
Corrected ratios of NETs on the INM:ONM
| NETs protein | concentration ratiosmFRAP (INM:ONM) | Diffusion coefficient on ONM (μm2 s−1) | Diffusion coefficient on INM (μm2 s−1) | Diffusion based corrected concentration ratio (INM:ONM) | immobilized fraction | Final Concentration ratio (INM:ONM) |
|---|---|---|---|---|---|---|
| LBR | 0.53 : 1 | 2.6 ± 0.8 | 1.9 ± 0.6 | 0.58 : 1 | 61 ± 6% (overall) | 3.1 : 1 |
| 81 ± 6% (INM) |
The final concentration ratio of NETs on the ONM and INM is further attained by correcting the diffusion-based concentration ratio according to the immobilized fraction on the INM. The bulk FRAP and corresponding data analysis provides the FRAP curve for NETs of interest which could be used to calculate the immobilized fraction on the INM. Shown in Figure 2D and Table 1, the immobilized fraction of LBR makes up ~61% of the total fluorescently tagged NETs on the NE and ~81% for INM specifically. Hence, the final concentration ratio of LBR is 3.1:1 (ONM: INM).
Successful completion of this technique will enable researchers to observe the diffusivity of molecules on the nuclear membrane as well as the fraction of immobile molecules. Of particular interest is the ability of this technique to differentiate the relative fractions of NETs located on the discrete inner and outer nuclear membranes, including the mobile and immobile fractions (Tingey et al., 2019). This will allow researchers to draw key inferences into the function and possible protein-protein interactions between NETs of interest and the attendant proteins on the INM and ONM respectively.
Limitations
While this technique does allow for the relative concentration and abundance of NETs on the discreet membranes of the NE, it does not allow a researcher to quantify the exact number of NETs present on the inner and outer membranes.
Although smFRAP employs single-molecule microscopy, bulk FRAP remains diffraction limited and unable to distinguish between the discreet membranes of the NE as they are only separated by ~40 nm. Therefore, any information regarding the diffusion of NETs or the immobile fractions is an aggregate average of the NETs behavior on both the INM and ONM and is not specific to any single membrane.
Lastly, photodamage could be potentially problematic in a smFRAP experiment. This is due to the comparatively higher laser power for pre-photobleaching. Therefore, we choose to utilize the photodamage-resistant cell line, HeLa cells, in our experiment (Wäldchen et al., 2015). To further ameliorate potential photodamage, we employ an optical chopper to break up the laser. To ensure the health of the cell being imaged, we test the potential photodamage and check viability via cell and NE morphology. To accomplish this, we snap a bright-field and fluorescent image of cells before and after laser illumination. These images are then compared to evaluate the cell for distortion of the NE, detachment of adherent cells from the culturing flask, plasma membrane blebbing, the occurrence of large vacuoles, or the aggregation of fluorescent proteins. Typically, after the imaging experiments, we continue culturing the irradiated cells 12 to 18 h under their corresponding culture condition. The cell and NE morphology of these cells are evaluated once more to confirm the health of the cells.
Troubleshooting
Problem 1
Failure of the photobleached area to recover.
Potential solution
The failure of a photobleached region to recover may be due to the excitation laser power being too high. The solution to this problem is to identify the optimal laser power for excitation without photobleaching too quickly. Generally, the concentration gradients of laser power after passing through the neutral density filters are 0.1 mW, 0.2 mW, 0.5 mW, 0.7 mW, 1 mW, 1.5 mW, and 2 mW. If the selected laser power is photobleaching the sample too quickly, half the laser power and test again. Adjust as many times as required until the sample begins to recover. Be aware that lowering laser power too far will result in a poor signal/noise ratio. It is therefore advisable that the optimal excitation power be identified for your specific molecule.
Problem 2
Inability to thoroughly photobleach the sample within 30 s.
Potential solution
There are two potential causes for this issue. First, the laser power used is too weak. To resolve this issue, double the laser power. Second, the diffusion speed of the NETs in question is very high. This causes the protein to move quickly into, or out of, the photobleaching area preventing photobleaching from occurring. To resolve this problem, enlarge the photobleaching area or double the laser power.
Problem 3
Movement of the NE during imaging
Potential solution
The movement of the NE over the course of recording may be a result of the video being too long. It is also possible that the membrane shift is due to unacceptable vibration of the microscope. First, ensure that the microscope is not being manipulated during imaging. It is also advisable to ensure that the pneumatic isolators at the base of the microscope are functioning correctly. Moreover, we generally finished the photobleaching step within ~30 s, and excitation and video recording within ~60 s, because our control experiment showed that the shifting of NE of live HeLa cells is ~6 nm within 2 mins. Finally, we imaged and localized the position of the NPC or NE before and after video recording, and then compared the pre-photobleaching video and post-photobleaching video for the locations of the NPC or NE. If the shift observed is larger than 10 nm, which is our standard localization precision for a moving particle, then the video files are discarded.
Problem 4
The Gaussian fitted peak of one membrane is dramatically larger than the other, or there is only a single peak during analysis
Potential solution
There are two potential causes for this eventuality. The first is that the protein of interest is only located on one membrane and is not present on the other. However, to confirm this, one must first rule out the possibility that the laser has shifted. If the laser has moved during imaging, it may strike one membrane unequally causing much higher fluorescence and resulting in a single peak or a disproportionately large peak during data analysis. To check for this, first refer to the reference image taken in step 1F of the smFRAP experiment. Determine that the laser was striking the center of the membrane. If the original alignment is found to be striking the center of the alignment, take an image of the current laser strike location on the detector. Using GDSC, localize the position of the laser. Compare the original alignment location with the current location. If it is found that the laser has shifted and is no longer striking the center of the detectors, the solution is to re-align the laser so that it is striking the center of the detector, as delineated by the guides, in a 90° fashion.
Problem 5
Unable to fit the data by Gaussian functions.
Potential solution
If, during data analysis, it is found that you are unable to fit Gaussian functions to the data, it is most probably due to the bin size being too large or too small. To identify the best bin size, begin with a bin size equal to the localization precision of the fittings. Then adjust the bin size by up in a stepwise fashion until an optimal Gaussian fitting is realized.
Resource availability
Lead contact
Further questions regarding this protocol and the materials used should be directed to the lead contact, Dr. Weidong Yang (weidong.yang@temple.edu).
Material availability
This study did not generate new or unique reagents.
Data and code availability
This study did not generate new or unique code.
Acknowledgments
The project was supported by grants from the US National Institutes of Health (NIH GM116204 and GM22552 to W.Y.). The authors would like to acknowledge Dr. Krishna C. Mudumbi (Yale Cancer Biology Institute) for his assistance in assembling this manuscript.
Author contributions
The manuscript and figures were written and generated by Y.L and M.T. The manuscript was edited by Y.L., M.T., and W.Y.
Declaration of interests
The authors declare no competing interests.
Reference
- Herbert A. GDSC Single Molecule Light Microscopy (SMLM) ImageJ Plugins. 2013. http://www.sussex.ac.uk/gdsc/intranet/microscopy/UserSupport/AnalysisProtocol/imagej/gdsc_plugins/ University of Sussex.
- Li Y., Tingey M., Ruba A., Yang W. High-speed super-resolution imaging of rotationally symmetric structures using SPEED microscopy and 2D-to-3D transformation. Nat. Protocol. 2021;16:532–560. doi: 10.1038/s41596-020-00440-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudumbi K.C. Nucleoplasmic signals promote directed transmembrane protein import simultaneously via multiple channels of nuclear pores. Nature Communications. 2020;11:2184. doi: 10.1038/s41467-020-16033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudumbi K.C., Schirmer E.C., Yang W. Single-point single-molecule FRAP distinguishes inner and outer nuclear membrane protein distribution. Nat. Commun. 2016;7:12562. doi: 10.1038/ncomms12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudumbi K.C., Yang W. Determination of Membrane Protein Distribution on the Nuclear Envelope by Single-Point Single-Molecule FRAP. Curr. Protoc. Cell Biol. 2017;76:21 11 1–21 11 13. doi: 10.1002/cpcb.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudumbi K.C., Yang W., Ma J., Schirmer E.C. Single-Point Frap Distinguishes Inner and Outer Nuclear Membrane Protein Distribution. Biophys. J. 2016;110:596a. doi: 10.1038/ncomms12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingey M., Mudumbi K.C., Schirmer E.C., Yang W. Casting a wider net: differentiating between inner nuclear envelope and outer nuclear envelope transmembrane proteins. Int. J. Mol. Sci. 2019;20:5248. doi: 10.3390/ijms20215248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wäldchen S., Lehmann J., Klein T., Van De Linde S., Sauer M. Light-induced cell damage in live-cell super-resolution microscopy. Sci. Rep. 2015;5:15348. doi: 10.1038/srep15348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate new or unique code.