

Abstract
The search for effective treatment against novel coronavirus (COVID-19) remains a global challenge due to controversies on available vaccines. In this study, data of SARS coronavirus 3C-like protease (3CLpro) inhibitors; a key drug target in the coronavirus genome was retrieved from CHEMBL database. Quantitative Structure-Activity Relationship (QSAR) studies, Molecular docking, Absorption-Distribution-Metabolism-Excretion-Toxicity (ADMET) and molecular dynamics simulation (MDS) were carried out using these 3CLpro inhibitors. QSAR model constructed using the data had correlation coefficient R2 value of 0.907; cross-validated correlation coefficient Q2 value of 0.866 and test set predicted correlation coefficient R2pred value of 0.517. Variance inflation factor (VIF) values for descriptors contained in the model ranged from 1.352 to 1.68, hence, these descriptors were orthogonal to one another. Therefore, the model was statistically significant and can be used to screen and design new molecules for their inhibitory activity against 3CLpro. Molecular docking showed that seven of the compounds (inhibitors) used in the study had a remarkable binding affinity (−9.2 to −10.3 kcal/mol) for 3CLpro. ADMET study revealed that five (CHEMBL Accession IDs 19438, 196635, 377150, 208763, and 210097) of the seven compounds with good binding ability obeyed Lipinski's rule of five. Hence, they were compounds with drug-like properties. MDS analysis revealed that 3CLpro-compound 21, 3CLpro-compound 22, 3CLpro-compound 40 complexes are very stable as compared to the reference 3CLpro-X77 complex. Therefore, this study identified three potent inhibitors of 3CLpro viz. CHEMBL194398, CHEMBL196635, and CHEMBL210097 that can be further explored for the treatment of COVID-19.
Keywords: COVID-19, SARS Coronavirus 3C-like protease, QSAR, Molecular docking, ADMET, Molecular dynamics simulation
1. Introduction
Coronavirus re-emerged recently in the Wuhan region of China as a novel coronavirus (CoVID-19) causing severe upper respiratory tract infection with symptoms that include; fever, pneumonia, dyspnea, and asthenia reported by people in Wuhan [[1], [2], [3]]. Since then, the virus has spread to almost all countries in the world prompting several lockdown measures by governments to curb the spread of this disease. Even with increasing attention on the development of vaccines to stop the daily mortality recorded, all effort so far has proved abortive with about 98.2 million reported cases and over 2.1 million deaths globally [4].
Coronavirus is a positive-stranded RNA virus with the largest genome of all know RNA viruses with a length of about 26–32 kb [5]. The coronavirus genome encodes 4 crucial structural proteins namely; the spike (S) protein, nucleocapsid (N) protein, membrane (M) protein, and the envelope (E) protein, all of which are essential for the production of a structurally complete viral particle [6,7]. Besides encoding structural proteins, a significant part of the coronavirus genome is transcribed and translated into a polypeptide, which encodes proteins essential for viral replication and gene expression [8]. One of the best-characterized drug targets among coronaviruses is the chymotrypsin-like protease (3CLpro) [9]. Together with the papain-like protease (PLpro), 3CLpro is crucial for processing the translated polyproteins from the viral RNA [10]. The highly conserved 3CLpro consisting of about 306 amino acids, is a key enzyme for coronavirus replication. Consequently, it is a vital target for the development of vaccines against coronavirus.
The design and development of pharmaceutical agents for the control of coronavirus infections is of utmost importance in our world and scientist are leaving no stone unturned in this endeavor. To this effect, scientists are into the traditional methods of obtaining new drugs by screening numerous compounds (either synthesized or extracted phytochemical agents) using non-living systems or simplified living systems such as rats, until a suitable lead is identified. These processes are the arbiter of truth in any scientific stride; however, they are time-consuming and costly. Any procedure that can assist in reducing the cost, time and still maintain scientific integrity is a welcome development. This is where computer-aided drug design methodologies (quantitative structure-activity relationship (QSAR), molecular docking, molecular dynamics simulation, and so on) comes in.
QSAR establishes a mathematical relationship between chemical structures of compounds with defined biological activity and their biological activities. This relationship can be used to screen or design new molecules for better biological activity [11]. QSAR is an effective method for optimizing or correlating specific structural features or molecular descriptors like polarizability, lipophilicity, electronic and steric properties within an analogous series of molecules with their biological activities [12]. Also, molecular docking elucidates the binding of small compounds (drugs or ligands) with a known macro-molecular target (receptor) [13]. Chemical structures with the inhibitory activity against 3C-like protease deposited in the CHEMBL database were used in this study with the aim of developing a QSAR model that will reveal the structural feature of the molecules that relates to their inhibitory activity. Besides, molecular docking was carried out to show the interaction between the compounds and amino acids in the binding pocket of 3CLpro. Compounds with the most negative binding affinity were subjected to ADMET studies followed by a 100 ns molecular dynamics simulation to determine the stability of the lead compounds.
2. Material and methods
2.1. Experimental data
Seventy-three (73) compounds with SARS coronavirus 3C-like protease (3CLpro) inhibitory activity retrieved from the CHEMBL database were used as a dataset in this study. The inhibitory activities of the dataset compounds were presented as IC50 (nM). SDF files of these compounds were retrieved with DataWarrior version 5.2.1 software and their biological activity data (IC50) were converted to PIC50 values presented in Table 1 using equation (1) below:
| PIC50 = (9 – Log IC50) | (1) |
Table 1.
Accession ID, binding affinity, experimental and predicted PIC50 of 3C-like protease inhibitors.
| Comp. No | CHEMBL Accession ID | BA (kcal mol−1) | Exp. PIC50 | Pred. PIC50 |
|---|---|---|---|---|
| 1a | CHEMBL45830 | −7.0 | 4.32 | 4.51 |
| 2 | CHEMBL118596 | −7.2 | 4.30 | 4.35 |
| 3 | CHEMBL358279 | −8.5 | 6.43 | 5.96 |
| 4 | CHEMBL348660 | −7.7 | 4.9 | 5.36 |
| 5 | CHEMBL148483 | −6.1 | 4.15 | 4.04 |
| 6 | CHEMBL1518673 | −7.1 | 4.48 | 4.32 |
| 7 | CHEMBL363535 | −8.1 | 4.83 | 5.10 |
| 8 | CHEMBL187266 | −7.5 | 4.44 | 4.62 |
| 9 | CHEMBL188487 | −7.8 | 5.03 | 5.20 |
| 10 | CHEMBL426082 | −8.0 | 4.88 | 5.44 |
| 11 | CHEMBL365134 | −7.9 | 6.01 | 5.54 |
| 12 | CHEMBL187579 | −8.6 | 5.14 | 4.88 |
| 13 | CHEMBL185698 | −7.7 | 4.87 | 5.15 |
| 14 | CHEMBL187598 | −8.0 | 5.32 | 5.76 |
| 15a | CHEMBL188983 | −7.9 | 4.63 | 5.39 |
| 16 | CHEMBL187717 | −8.6 | 5.70 | 5.32 |
| 17a | CHEMBL365469 | −7.9 | 4.95 | 5.43 |
| 18 | CHEMBL190743 | −7.6 | 6.02 | 5.90 |
| 19 | CHEMBL370923 | −8.4 | 4.76 | 4.53 |
| 20 | CHEMBL191575 | −7.9 | 4.90 | 5.19 |
| 21 | CHEMBL194398 | −10.3 | 4.35 | 4.11 |
| 22 | CHEMBL196635 | −9.6 | 4.15 | 4.05 |
| 23a | CHEMBL377150 | −9.8 | 5.00 | 4.73 |
| 24 | CHEMBL210092 | −8.6 | 4.96 | 4.62 |
| 25a | CHEMBL210525 | −6.7 | 4.6 | 4.96 |
| 26 | CHEMBL379727 | −6.8 | 4.72 | 4.97 |
| 27 | CHEMBL209227 | −8.0 | 4.85 | 4.95 |
| 28 | CHEMBL210497 | −8.9 | 4.40 | 4.54 |
| 29 | CHEMBL210632 | −8.5 | 4.22 | 3.96 |
| 30 | CHEMBL207207 | −7.6 | 4.00 | 4.32 |
| 31 | CHEMBL208763 | −9.5 | 4.82 | 4.73 |
| 32a | CHEMBL208584 | −6.7 | 4.52 | 3.87 |
| 33 | CHEMBL208732 | −8.7 | 5.52 | 5.46 |
| 34a | CHEMBL209287 | −5.4 | 4.18 | 3.22 |
| 35a | CHEMBL383725 | −8.0 | 5.96 | 5.33 |
| 36a | CHEMBL210612 | −7.7 | 4.4 | 4.61 |
| 37 | CHEMBL380470 | −8.4 | 4.35 | 4.46 |
| 38a | CHEMBL210487 | −8.9 | 4.22 | 3.83 |
| 39 | CHEMBL378674 | −8.2 | 4.92 | 5.28 |
| 40a | CHEMBL210097 | −9.5 | 4.82 | 5.88 |
| 41 | CHEMBL209667 | −8.9 | 4.82 | 4.69 |
| 42 | CHEMBL212218 | −9.2 | 6.52 | 6.23 |
| 43 | CHEMBL427404 | −7.7 | 5.30 | 5.24 |
| 44 | CHEMBL212190 | −7.6 | 5.00 | 4.84 |
| 45 | CHEMBL211969 | −8.2 | 4.89 | 4.58 |
| 46a | CHEMBL378700 | −7.3 | 4.82 | 3.71 |
| 47a | CHEMBL212019 | −6.6 | 4.80 | 4.09 |
| 48 | CHEMBL212399 | −7.2 | 4.74 | 4.54 |
| 49a | CHEMBL384739 | −7.2 | 4.82 | 5.14 |
| 50 | CHEMBL215732 | −8.2 | 4.8 | 5.06 |
| 51 | CHEMBL215733 | −6.5 | 4.74 | 5.07 |
| 52 | CHEMBL375130 | −6.8 | 4.7 | 4.75 |
| 53 | CHEMBL214372 | −5.3 | 4.4 | 4.44 |
| 54 | CHEMBL212240 | −7.3 | 4.8 | 4.47 |
| 55a | CHEMBL377253 | −7.7 | 4.8 | 4.28 |
| 56 | CHEMBL215397 | −7.3 | 4.6 | 4.59 |
| 57 | CHEMBL378342 | −10.0 | 4.49 | 4.18 |
| 58 | CHEMBL379642 | −6.8 | 5.52 | 5.53 |
| 59 | CHEMBL212454 | −8.4 | 6.05 | 6.50 |
| 60 | CHEMBL213581 | −7.3 | 5.22 | 5.49 |
| 61 | CHEMBL380403 | −8.1 | 4.92 | 5.16 |
| 62a | CHEMBL212504 | −7.8 | 4.89 | 4.32 |
| 63a | CHEMBL215254 | −8.4 | 4.58 | 4.72 |
| 64 | CHEMBL222769 | −8.0 | 7.2 | 6.61 |
| 65a | CHEMBL222840 | −6.1 | 7.22 | 6.04 |
| 66 | CHEMBL426898 | −7.7 | 6.77 | 7.14 |
| 67a | CHEMBL222234 | −6.1 | 7.3 | 5.98 |
| 68a | CHEMBL225515 | −7.3 | 7.19 | 6.79 |
| 69 | CHEMBL222893 | −7.7 | 7.02 | 6.64 |
| 70 | CHEMBL222628 | −6.0 | 6.57 | 6.49 |
| 71a | CHEMBL222735 | −6.2 | 6.47 | 6.35 |
| 72a | CHEMBL1358724 | −7.9 | 5.11 | 4.79 |
| 73 | CHEMBL2146517 | −8.5 | 4.87 | 4.96 |
= Test set compounds; BA - Binding affinity; Exp. PIC50 - Experimental IC50; Pred. PIC50 - Predicted IC50.
2.2. Geometry optimization and molecular descriptor calculation
The geometries of the spatial data (SDF) files of the dataset compounds were optimized in order to make the conformations have the least potential energy using the GROMOS96 force field in Swiss-PDB viewer [14]. The optimized structures were imported into PaDEL-Descriptors [15], which calculated about 1875 molecular descriptors for each molecule. The calculated descriptors and their corresponding activity values for each molecule were arranged in an n × m matrix format (Supplementary Table 1). This constituted the dataset used in the study, where n is the number of molecules and m is the number of descriptors.
2.3. Normalization of descriptor and data division
Descriptor values were normalized to values between 0 and 1 using equation (2) below in order to convert them to values with a similar unit which is needed for regression analysis and to reduce skewness in the measured values [16].
| (2) |
In equation (2), XI is the scaled descriptor value, Xmax and Xmin represent maximum and minimum descriptor values respectively in a column. The normalized descriptors and the activity values were arranged in a matrix as the dataset. The dataset of 73 molecules was divided into 51 training sets and 22 test sets using the Kennard-Stone algorithm available in Dataset Division GUI v1.2 software. The training set was used for model development, while the test set was used for the validation of the developed model.
2.4. Variable selection and model construction
The selection of combinations of important descriptors that best explain the variability in the activity values of the compounds was done using a genetic algorithm (GA) which divides the descriptors into proper subsets from where models can be generated. Multiple linear regression (MLR) method available in MLRplus Validation 1.3 software was used to construct the model.
2.5. Model validation
The quality of the model developed in this study was assessed using validation parameters calculated by the MLRplus Validation 1.3 software. These parameters include determination coefficient R2, adjusted determination coefficient R2 adj, Variance ratio F, Standard errors of estimate SEE, and Golbraikh and Tropsha criteria for an acceptable model [17].
Furthermore, co-linearity between the descriptors contained in the model was checked using the descriptors correlation matrix and their corresponding variance inflation factor. The model variance inflation factor (VIF) also known as inverse of tolerance [18] was calculated using the equation below:
| (3) |
In equation (3), R2 j, is the coefficient of determination of the regression of descriptor j on other descriptors contained in the model.
2.6. Model applicability domain
A model cannot be used to predict the biological activity for the entire chemicals in the universe except for those in its region of reliable/acceptable prediction, which is defined in terms of descriptors contained in the model. This region is known as the applicability domain (AD) of the model. In this study, the AD of the developed model was defined using the extent of the extrapolation method. This method employs leverage h values of dataset molecules and the standardized prediction residual (SDR) of the models to define their AD. The result of this method is often visualized by the plot of h versus SDR (Williams plot).
Leverage h is a special type of distance measures used to show similarity/dissimilarity among objects and it's obtained as the diagonal element of a hat matrix H:
| (4) |
In equation (4), X is the model's descriptor matrix and X T is the transpose of matrix X. Generally, AD of models in the study was defined by a square area with vertical boundary 0 <h i < h* and horizontal boundary −3 < SDR <3, where h i's were molecules leverages values and h* was the models warning leverage expressed as:
| (5) |
In equation (5), k is the number of descriptors in the model, and n is the number of training set molecules. Standardized residual (SDR) was calculated with the equation below:
| (6) |
In equation (6), are observed and predicted response respectively for either training or test set molecules, n is the number of dataset molecule.
2.7. Molecular docking studies
2.7.1. Protein preparation
The crystal structure of SARS coronavirus 3C-like protease with PDB ID 6W63 was retrieved from the protein databank (www.rcsb.org). The structure was prepared by removing existing ligand and water molecules while missing hydrogen atoms were added using Autodock version 4.2 program, Scripps Research Institute [19]. Thereafter, non-polar hydrogens were merged while polar hydrogens were added to the protein and subsequently saved into pdbqt format for molecular docking.
2.7.2. Ligand preparation
The smiles strings of seventy-three (73) potential inhibitors of 3CLpro were retrieved from the CHEMBL database using DataWarrior software [20]. The structures of this compound were generated with the aid of ChemSketch, (Version 14.01) using the SMILES obtained earlier. The compounds were thereafter converted to PDB chemical format in order to create ligand binding groups using Open babel [21]. Thereafter, the PDB structure of 3CLpro co-crystalized ligand N-(4-tert-butylphenyl)-N-[(1R)-2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl]-1H-imidazole-4-carboxamide (X77) was retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) to serve as the reference compound. Polar hydrogen charges of the Gasteiger-type were assigned and the nonpolar hydrogens were merged with the carbons and the internal degrees of freedom and torsions were set to zero. The compounds were further converted to pdbqt format using Autodocktools [22].
2.7.3. Molecular docking study
Docking of the ligands to 3CLpro and determination of binding affinities was carried out using VINA [23]. Pdbqt format of the receptors and ligands were dragged into their respective columns and the software was run. The grid center for docking was set as X = −4.70, Y = −20.17, Z = 25.47 with the dimensions of the grid box, 67.84 × 46.83 × 72.30 for 3CLpro. A cluster analysis based on Root Mean Square Deviation (RMSD) values, with reference to the starting geometry, was subsequently performed and the lowest energy conformation of the more populated cluster was considered as the most trustable solution. The binding affinities of compounds for 3CLpro were recorded. The compounds were then ranked by their affinity scores and molecular interactions between the receptors and compounds with remarkable binding affinity were viewed with Discovery Studio Visualizer and PoseView [24].
2.7.4ADMET. study
The seven lead compounds identified in molecular docking studies were subjected to Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) studies to determine the drug-likeness of the compounds. ADMET studies were carried out using the Swiss online ADME web tool [[25], [26], [27]] to determine the pharmacokinetic properties of the lead compounds while the US Food and drug administration toxicity risk predictor tool OSIRIS evaluated various toxicity risks properties such as tumorigenicity, mutagenicity, irritation, and reproductive development toxicity.
2.8. Molecular dynamics simulation
Molecular dynamics simulation was employed to validate the docking analysis and quantify the conformational changes of 3CLpro and 3CLpro-screened compound complexes. The dynamics package GROMACS 5.0.7 [28] was used to simulate the system wherein the CHARMM 36 force field was used [29]. Using transferable intermolecular potential water molecules (TIP3Pmodel), the water molecules were added [30], and then neutralization of the complex was achieved by adding Na+ at 310 K temperature. For energy minimization of the complex, the periodic boundary condition was retained where the Particle Mesh Ewald (PME) approach with the steepest descent algorithm was used for the measurement of long-range electrostatic interaction using the Verlet cutoff scheme at 10 kJ mol−1. A dodecahedral simulation box was developed to simulate the complex that was 10 Å greater than the complex. The Berendsen thermostat has been used to monitor the temperature of the simulation system. Initially, the protein-ligand complex and apo-protein structure were cleaned and equilibrated in two stages by the steepest gradient approaches (5000 ps); NVT and NPT ensemble. Lastly, constant temperature and pressure of 300 K and 1 atm were maintained for all the systems subjected to the production MD of 100 ns. The simulation time was maintained using the Parrinello–Rahman with a time step of 2fs for constant pressure simulation. To evaluate the result, the simulation trajectory was saved for every 100 ps.
The simulation results were incorporated with the GROMACS default script. Finally, MD trajectories were evaluated for the measurement of Root-mean-square-deviation (RMSD), Root-mean-square-fluctuation (RMSF), Radius-of-gyration (Rg), Solvent-accessible-surface-area (SASA), Hydrogen bonds (H-bonds), and principal component analysis (PCA). This was worked out to measure the strength of the protein-ligand interaction. In order to get a more accurate MD simulation result, each complex was run three times (n = 3) and the average result was used for analysis.
To calculate the binding free energy, the molecular mechanics Poisson–Boltzmann surface area (MMPBSA) approach was used [31]. The MD trajectories were processed before doing MMPBSA calculations. Binding free energy calculations include free solvation energy (polar + nonpolar solvation energies) and potential energy (electrostatic energies + van der Waals interactions). In the following equation, the whole process of MMPBSA can be summarized:
| ΔGbind = ΔGcomplex(minimized)- [ΔGligand(minimized) + ΔGreceptor(minimized)] |
| ΔGbind = ΔGMM + ΔGPB + ΔGSA-TΔS |
Here, the sum of van der Waals and electrostatic interaction is ΔGMM, the polar and non-polar solving energies are ΔGPB and ΔGSA, and the entropic contribution is TΔS. For average binding energy measurements, the ‘python’ script provided in g_mmpbsa was used. For MM-PBSA measurement, the last one ns MD trajectory files were considered.
3. Results and discussion
3.1. Qualitative structure-activity relationship
The QSAR model produced by the genetic algorithm-multiple linear regression method (GA-MLR) used in this study was represented by equation (7). The model validation parameters were also presented below.
| PIC50 = 5.531(±0.286) + 1.824(±0.214)ALogP2 + 2.209(±0.191)AATS8v – 1.324(±0.206) AATS3i + 1.107(±0.156)MATS6 – 1.227(±0.237)GATS8e – 1.648(±0.227)BCUTp-1h – 1.448(±0.273)ZMIC2 – 1.229(±0.163)VE1_D | (7) |
Internal Validation Parameters: SEE = 0.256; R2 = 0.907; R2 adj = 0.890; F = 51.397; Q2 = 0.866; = 0.817; .
External Validation Parameters: R2 pred. = 0.517; = 0.514; r2 = 0.609; r0 2 = 0.568.
Golbraikh and Tropsha acceptable model criteria's:
-
1.
Q2 = 0.866, Passed (Threshold value: Q2 > 0.5).
-
2.
r2 = 0.609, Passed (Threshold value: r2 > 0.6).
-
3.
|r0 2-r'0 2| = 0.013, Passed (Threshold value |r02-r'02|<0.3).
-
4.
k = 1.045 and [(r2-r0 2)/r2] = 0.067, Passed (Threshold value: [0.85<k < 1.15 and ((r2-r0 2)/r2) < 0.1].
-
5.
OR k ' = 0.94289 and [(r2-r'0 2)/r2] = 0.0873, Passed (Threshold value: 0.85<k'<1.15 and ((r2-r'0 2)/r2) < 0.1))
In equation (7), the alphanumeric terms in the RHS were molecular descriptors (molecular properties encoded in numerical forms) contained in the model which were found to be correlated with the inhibitory activity of 3C-like protease inhibitors (PIC50) used in this study. The details of each descriptor used in this study are presented in Table 2 . The numbers in the parenthesis were the standard deviation of their coefficients. The equation contained 8 descriptors, the maximum ratio of a model descriptors to the number of training set was not violated [32,33]. In addition, the model's determination coefficient R2 was greater than the adjusted R2, therefore, the model was not over-parameterized [34].
Table 2.
Molecular descriptors used in this study.
| Symbol | Description | Type |
|---|---|---|
| AATS8v | Average Broto-Moreau autocorrelation - lag 8/weighted by van der Waals volumes | 2D autocorrelation |
| AATS3i | Average Broto-Moreau autocorrelation - lag 3/weighted by first ionization potential | 2D autocorrelation |
| MATS6c | Moran autocorrelation - lag 6/weighted by charges | 2D autocorrelation |
| GATS8e | Geary autocorrelation - lag 8/weighted by Sanderson electronegativities | 2D autocorrelation |
| BCUTp-1h | nlow highest polarizability weighted BCUTS | 2D-Matrix based descriptor |
| ZMIC2 | Z-modified information content index (neighborhood symmetry of 2-order) | Information content |
| VE1_D | Coefficient sum of the last eigenvector from Barysz matrix/weighted by atomic number | 2D-Matrix based descriptor |
The model determination coefficient R2 was 0.907 which explained over 90% of the variation in the activity values of dataset compounds. The high Fisher F value of 51.397 indicated that the variation in the activity values explained by the collective descriptors contained in the model was more than could be reasonably attributed to chance. The model cross-validation correlation coefficient Q2 value was 0.866 and its modified correlation coefficient R2 m(loo) was 0.817. These values were greater than 0.5 indicating the model was stable.
The model's descriptors correlation matrix and VIF values presented in Table 3 revealed that inter-correlation between any two descriptors contained in the model was less than 0.5, which implied the descriptors were orthogonal to one another and multi-collinearity problem does not exist in the model. VIF value for each descriptor was less than10 which further confirmed the orthogonal nature of the descriptors and absence of multi-collinearity problem [18,35].
Table 3.
Model's descriptors correlation matrix and variance inflation factors.
| ALogp2 | AATS8v | AATS3i | MATS6c | GATS8e | BCUTp-1h | ZMIC2 | VE1_D | VIF | |
|---|---|---|---|---|---|---|---|---|---|
| ALogp2 | 1 | 1.480 | |||||||
| AATS8v | 0.054 | 1 | 1.647 | ||||||
| AATS3i | 0.025 | −0.176 | 1 | 1.468 | |||||
| MATS6c | 0.078 | −0.019 | −0.159 | 1 | 1.085 | ||||
| GATS8e | −0.056 | −0.341 | −0.246 | 0.142 | 1 | 1.352 | |||
| BCUTp-1h | 0.483 | 0.142 | −0.290 | 0.040 | −0.124 | 1 | 1.681 | ||
| ZMIC2 | 0.311 | 0.454 | −0.070 | −0.054 | −0.135 | 0.330 | 1 | 1.666 | |
| VE1_D | 0.172 | 0.220 | −0.291 | −0.081 | −0.106 | 0.295 | −0.082 | 1 | 1.385 |
The predictive power of the model was further validated by its application to predict the activity values of test set compounds and it was found that its predicted determination coefficient for test set R2 pred was 0.517. This indicated that observed and predicted activity data for the test set by the model were well correlated and the model had good predictive power [16,34] as reflected in Fig. 1 . Y-randomization analysis was performed on training set data to further confirm if the model obtained was not a result of chance correlation. In this analysis, the response values of the dataset were randomly shuffled while the descriptor matrix was untouched, then MLR analysis was performed on the permuted dataset. The result of y-randomization analysis for the model presented in Table 4 showed that the determination coefficient R2, correlation coefficient R and the leave one out determination coefficient Q2 LOO for the model were greater than 5 and that of the randomized models. Furthermore, the model y-randomization parameters cRp 2 were greater than 0.5. These confirmed that the model was not a result of chance correlation [16,36].
Fig. 1.

Predicted inhibitory activity against the experimental inhibitory activity of dataset compounds.
Table 4.
Y-randomization of the model.
| MODEL TYPE | R | R2 | Q2LOO |
|---|---|---|---|
| Original | 0.953 | 0.907 | 0.866 |
| Random 1 | 0.236 | 0.056 | −0.412 |
| Random 2 | 0.397 | 0.157 | −0.256 |
| Random 3 | 0.349 | 0.122 | −0.446 |
| Random 4 | 0.230 | 0.053 | −0.302 |
| Random 5 | 0.332 | 0.110 | −0.437 |
| Random 6 | 0.358 | 0.128 | −0.259 |
| Random 7 | 0.316 | 0.100 | −0.296 |
| Random 8 | 0.430 | 0.185 | −0.314 |
| Random 9 | 0.287 | 0.082 | −0.532 |
| Random 10 | 0.288 | 0.083 | −0.237 |
| Random Models Parameters | |||
| Average R | 0.380 | ||
| Average R2 | 0.180 | ||
| Average Q2(LOO) | −0.239 | ||
| cRp2 | 0.854 | ||
The ability of the model to predict inhibitory activity value for test set data was further confirmed with the Golbraikh and Tropsha criteria for a satisfactorily external predictive model. As presented above, the result showed that the model passed all the criteria. Therefore, the model can be used to predict the inhibitory activity value of external data. In the criteria, r2 represent square correlation coefficients of the plot of observed versus predicted values for the test set with intercept, r2 0 is the square correlation coefficients between observed versus predicted values for the test set without intercept i.e. through the origin, r’2 0 is the reverse of r2 0, k is the slope of the plot of observed versus predicted values for the test set without intercept and k′ is the reverse of k.
3.2. The model applicability domain
The Williams plot showing the applicability domain of the model produced in this study was presented in Fig. 2 . In the figure, AD of the model was defined by a square area bounded by 0 < h* less than 0.52 and −3 < SDR less than 3, where h* is the model's warning leverage. As shown, almost all the data were within the model AD except for training set molecule 59 with a leverage value of 0.54.
Fig. 2.

Williams plot showing the standardized residuals versus leverage values.
The result presented showed that there is no outlier molecule in the dataset because the standardized residual produced by the model for each compound was within the ±3 range. Molecule 59 was not an outlier but an influential molecule with a leverage value greater than the warning leverage h*. Generally, the model reported in this study had a good predictive ability and was well validated. Therefore, it can be used to design and screen new molecules for their inhibitory activity against 3CLpro.
3.3. Molecular docking studies
Of the 73 ligands considered in this study, compounds 21, 22, 23, 31, 40, 42 and 57 were selected based on their remarkable binding affinity of −10.3, −9.6, −9.8, −9.5, −9.7, −9.2 and −10.0 kcal/mol respectively for 3CLpro compared to the reference compound's −10.1 kcal/mol (Table 5 ). Compounds 23, and 42 binds to the catalytic domain of 3CLpro as seen with X77. Alternatively, compounds 21, 22, and 40 occupied a distinct binding site spanning domain 1 and 2 while 57 occupied regions spanning domain 2 and domain 3 but without interaction with any of the catalytic residues in domain 2 (Fig. 3 ). The binding affinities obtained for this compounds were consistent with their predicted activity.
Table 5.
Binding affinity, hydrophobic interactions, hydrogen bonds, and hydrogen bond distance of selected compounds to 3C-like protease.
| Comp No | CHEMBL ID | Binding Affinity (kcal/mol) | Hydrophobic interaction | Hydrogen bonds | Hydrogen bond distance (Å) |
|---|---|---|---|---|---|
| S | X77 | −10.1 | His41, Cys44, Met49, Cys145 |
Gly143 | 2.11 |
| 21 | CHEMBL194398 | −10.3 | Tyr239, Leu287 |
Tyr237 | 3.59 |
| 22 | CHEMBL196635 | −9.6 | Val171, Ala193 |
Lys137, Thr199, Thr196, Asn238 |
3.00, 2.06, 2.11, 2.04 |
| 23 | CHEMBL377150 | −9.8 | His41, Cys44, Met49 Arg118, Asp187 |
Tyr54, Thr24, Met49, Arg188, Asn119, Gly143 |
3.24 2.96 2.40, 2.16 2.19 |
| 31 | CHEMBL208763 | −9.5 | Tyr237, Tyr239, Leu272, Leu287 Lys137 |
Asp197 | 3.33 |
| 40 | CHEMBL210097 | −9.5 | Val171 Ala194, Leu287 |
Gly275, Leu272 |
2.01 2.19 |
| 42 | CHEMBL212218 | −9.2 | Met49, Pro168 |
Thr25, | 1.98 |
| 57 | CHEMBL378342 | −10.0 | Pro108, Ile200, Val202, His246, Ile249 |
Glu240 | 2.32 |
Fig. 3.

Cartoon view of the distribution of X77 and seven other ligands in the binding domains of 3C-Like protease.
The reference compound (X77) had formed predominantly hydrophobic interaction with His41, Cys44, Met49, and Cys145 in addition to a single hydrogen bond with Gly143 in the catalytic domain of 3CLpro (Fig. 4 a). Compound 21 had a hydrophobic interaction with Tyr239 and Leu287 along with a donor-donor interaction with Thr199 and a carbon-hydrogen bond with Tyr237 (Fig. 4b). Compound 22 was visualized in mainly hydrogen bond formation with Lys137, Thr196, Thr199 and Asn238 while hydrophobic interactions with Val171 and Ala193 in a similar binding site with compound 21 (Fig. 4c). Hydrophobic interaction was predominant in the binding of 23 to 3CLpro. Catalytic His41, Cys44, and Met49 participated in the hydrophobic interaction in addition to halogen bond formation with Asp187, Arg188, and a hydrogen bond formation with Tyr54, and carbon-hydrogen bond with Tyr54, Thr24, Met49, Asn119, Gly143, and Arg188 (Fig. 4d).
Fig. 4.

Binding of ligands to 3CLpro, the interaction between amino acid in the binding site of 3CLpro and (a) X77 (b) compound 21 (c) compound 22 (d) compound 23. Green dotted line represents hydrogen bond, faint green dotted line represents a carbon-hydrogen bond, the deep pink dotted line represents π-π stacking, the faint pink dotted line represents π-alkyl interaction, the purple dotted line represents π-sigma interaction, the red dotted line represents donor-donor interaction.
The mode of interaction of compound 31 was basically via hydrophobic interaction as seen with Tyr237, Tyr239, Leu272, and Leu287 and a single hydrogen bond with Asp197 (Fig. 5 a). Compound 40 was visualized in a hydrophobic interaction with Val171, Ala194, Leu287, halogen bond formation with Leu271, and a carbon-hydrogen bond with Leu272 and Gly275 (Fig. 5b). The fluoride groups in compound 42 played a significant role in halogen bond formation with catalytic His41, and Cys44 and a hydrogen bond with Thr25 (Fig. 4c). Hydrophobic interaction played a key role in the binding of compound 57 to 3CLpro. The compound interacted with Pro108, Ile200, Val202, His246, and Ile249 via hydrophobic (alkyl) bond formation (Fig. 5d). The residues involved in the binding of the seven selected compounds are listed in Table 5.
Fig. 5.

Binding of ligands to 3CLpro, the interaction between amino acid in the binding sites of 3CLpro and (a) compound 31 (b) compound 40 (c) compound 42 (d) compound 57. Green dotted line represents hydrogen bond, the faint green dotted line represents a carbon-hydrogen bond, the deep pink dotted line represents π-π stacking, the faint pink dotted line represents π-alkyl interaction, cyan dotted line represents π-fluoride interaction, the yellow dotted line represents π-Sulphur interaction, the red dotted line represents donor-donor interaction.
The QSAR model generated in this study provides a valuable approach for ligand base design, while the molecular docking studies provide a valuable approach for structure base design. Previous studies [9,37,38] have reported the presence of three important domains in 3CLpro. The 3CLpro domains 1 and 2 (residues 10–99 and 100–182, respectively) are six-stranded antiparallel β-barrels that contain the substrate-binding site between them. Domain 3 (residues 198–303), a globular cluster of five helices, is involved in regulating dimerization of the 3CLpro mainly through a salt-bridge interaction between Glu290 of one protomer and Arg4 of the other [39]. The binding of compounds identified in this study to domains 1 and 2 of 3CLpro jointly referred to as the N-terminal domain could be exploited in the treatment of coronavirus as the two domains host the complete catalytic machinery of the enzyme. Also, binding of the compounds to domain 3 residues would disrupt the dimerization of the enzyme. Consequently, a promising strategy might be established in which two separate inhibitors, one binding to the active site and another disrupting the dimerization interface on the extra domain (domain 3), are linked together to create a multifunctional inhibitor with significantly enhanced binding affinity and specificity.
3.4. ADMET studies
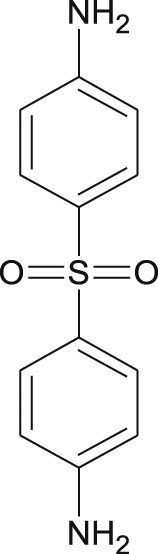
Based on their remarkable binding affinity for 3CLpro, compounds 21, 22, 23, 31, 40, 42, and 57 were subjected to ADMET studies to determine their drug-likeness. Compounds 31 and 42 had all their values within the acceptable range as postulated by the Lipinski rule of 5 (Table 6 ). Also, compounds 21, 22, 23, and 40 had one violation each (Mw > 500 g/mol) which is acceptable. Lipinski's rule states that, generally, an orally active drug has no more than one violation of the following criteria: (1) Not >5 hydrogen bond donors (nitrogen or oxygen atoms with one or more hydrogen atoms). (2) Not >10 hydrogen bond acceptors (nitrogen or oxygen atoms) (3) A molecular mass < 500 g/mol and (4) an octanol-water partition coefficient log P not greater than 5 [40]. Consequently, compound 57 was not considered for further studies due to two Lipinski violations i.e. Mw > 500 g/mol and HBA >10 (Table 6). OSIRIS server identified 2, 4-dichlorotoluene scaffold in compound 42 as a possible mild anti-reproductive unit while the server also highlighted the presence of dapsone constituent in compound 57 that is capable of mild tumorigenic effect (Table 6).
Table 6.
ADMET properties of the seven lead compounds with notable binding affinity for 3C-like protease.
| Properties | Compound 21 | Compound 22 | Compound 23 | Compound 31 | Compound 40 | Compound 42 | Compound 57 |
|---|---|---|---|---|---|---|---|
| Mw (g/mol) | 580.67 | 558.67 | 509.95 | 422.89 | 525.33 | 459.18 | 588.63 |
| LogP | 1.15 | 0.91 | 3.10 | 2.48 | 3.75 | 3.01 | 1.65 |
| HBA | 8 | 8 | 7 | 5 | 8 | 9 | 11 |
| HBD | 3 | 3 | 1 | 1 | 1 | 0 | 2 |
| Solubility (Log S) | −4.33 | −4.19 | −6.04 | −5.35 | −6.79 | −5.75 | −5.80 |
| Lipinski Violation | 1 | 1 | 1 | 0 | 1 | 0 | 2 |
| Mutagenic | No risk | No risk | No risk | No risk | No risk | No risk | No risk |
| Tumorigenic | No risk | No risk | No risk | No risk | No risk | No risk | Medium risk |
| Irritant | No risk | No risk | No risk | No risk | No risk | No risk | No risk |
| Reproductive effect | No risk | No risk | No risk | No risk | No risk | Medium risk | No risk |
| Possible toxic fragment | 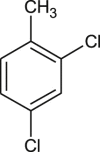 |
 |
Mw - Molecular weight; LogP - Octanol/water partition coefficient; HBA - Hydrogen bond acceptor; HBD – Hydrogen bond donor.
3.5. Molecular dynamics simulation
The docked 3CLpro-compound complexes were subsequently used to study the detailed dynamic, structural, as well as binding behaviors to know how it targets the active site of SARS-CoV2 3CLpro.
3.5.1. RMSD, RMSF, and Rg of 3CLpro-compound complexes during MDS
The MDS trajectories of 100 ns simulations were examined to study the detailed structural and dynamic mechanisms of the 3CLpro protein and 3CLpro-compound complexes. The RMSD, RMSF, Rg fluctuations profile of all systems during the period of 100 ns simulation are presented in Fig. 6 . The RMSD of the backbone atoms computed over 100 ns revealed that the 3CLpro protein reached stability after approximately 50 ns, whereas all the 3CLpro-compound complexes took only 5–10 ns to become stable (Fig. 6A) 3CLpro-X77 complex (reference) as well as all the 3CLpro-compound complexes were stabilized until the end of the MD production run and converged overall except 3CLpro-compound 21 complex which is stable up to 90 ns and after that, it showed a little fluctuation of about 0.15–0.2 ns and become stable at the end. The average RMSD values of 3CLpro, 3CLpro-X77 complex, 3CLpro-compound 21 complex, 3CLpro-compound 22 complex, 3CLpro-compound 23 complex, 3CLpro-compound 31 complex, and 3CLpro-compound 40 complex were found to be 0.18 nm, 0.17 nm, 0.20 nm, 0.17 nm, 0.20 nm, 0.20 nm, and 0.15 nm, respectively (Table 7 ). Interestingly, the RMSD values of all the systems are very similar and do not exceed 0.2 nm, which denotes the structural integrity of the 3CLpro protein. The RMSD profile suggested that upon compound binding no significant variation or conformational changes were taking place in the 3CLpro structure.
Fig. 6.

Simulation result showing RMSD (A), RMSF (B), and Rg (C) plots of 3CLpro and 3CLpro-ligand complexes during the period of simulation.
Table 7.
The average values of different parameters, RMSD, RMSF, RG, SASA, Number of H-bond, and Gibbs Energy.
| S No. | Protein/Protein-ligand complex | Average RMSD (nm) | Average RMSF (nm) | Average RG (nm) | Average SASA (nm2) | Number of H-bond | Gibbs Energy (kJ mol−1) |
|---|---|---|---|---|---|---|---|
| 1 | 3CLpro | 0.18 ± 0.03 | 0.09 ± 0.05 | 1.92 ± 0.13 | – | ||
| 2 | 3CLpro-X77 (Reference) complex | 0.17 ± 0.02 | 0.13 ± 0.07 | 1.88 ± 0.26 | 149.29 ± 2.77 | 4 | 12.5 |
| 3 | 3CLpro-compound 21 complex | 0.20 ± 0.06 | 0.12 ± 0.08 | 1.67 ± 0.27 | 151.33 ± 2.75 | 4 | 14.8 |
| 4 | 3CLpro-compound 22 complex | 0.17 ± 0.02 | 0.10 ± 0.05 | 1.82 ± 0.21 | 158.04 ± 3.05 | 3 | 12.9 |
| 5 | 3CLpro-compound 23 complex | 0.20 ± 0.04 | 0.11 ± 0.08 | 1.90 ± 0.18 | 152.58 ± 4.85 | 3 | 13.5 |
| 6 | 3CLpro-compound 31 complex | 0.20 ± 0.03 | 0.10 ± 0.05 | 1.79 ± 0.21 | 153.63 ± 3.49 | 4 | 12.4 |
| 7 | 3CLpro-compound 40 complex | 0.15 ± 0.01 | 0.09 ± 0.04 | 1.72 ± 0.25 | 150.21 ± 2.55 | 4 | 12.9 |
Structural flexibility was evaluated by the residue-wise RMSF in 3CLpro protein and 3CLpro-compound complexes. RMSF mainly specifies the flexible region of the protein and analyzes the portion that diverges from the overall structure. A higher RMSF value indicates greater flexibility (less stability) during the MD simulation while the lower value of RMSF reveals less flexibility (good stability) of the system. The 3CLpro, 3CLpro-X77 complex, 3CLpro-compound 21 complex, 3CLpro-compound 22 complex, 3CLpro-compound 23 complex, 3CLpro-compound 31 complex, and 3CLpro-compound 40 complex showed an average RMSF value of 0.09 nm, 0.13 nm, 0.12 nm, 0.10 nm, 0.11 nm, 0.10 nm, and 0.09 nm, respectively (Table 7). All the 3CLpro-compound complexes exhibited overall lower RMSF than the 3CLpro-X77 complex during the simulation (Fig. 6B). The RMSF results predicted that all the 3CLpro-compound complexes were stable and can act as potential drug candidates against SARS-CoV2.
The Rg of the protein and protein-ligand complex indicates the degree of compactness and rigidity of the protein. Therefore, we investigated the Rg of 3CLpro and 3CLpro-compound complexes to know how they show their compactness during the simulation run. For this, we have calculated the Rg of 3CLpro and 3CLpro-compound complexes during the 100 ns simulation time. Fig. 6C showed that all the 3CLpro-compound complexes have almost similar stability as the 3CLpro protein and 3CLpro-X77 complex. The average Rg values of the 3CLpro and 3CLpro-X77 complex were found to be 1.92 nm and 1.88 nm respectively. Similarly, Rg values were found to be 1.67 nm, 1.82 nm, 1.90 nm, 1.79 nm, and 1.72 nm for the 3CLpro-compound 21 complex, 3CLpro-compound 22 complex, 3CLpro-compound 23 complex, 3CLpro-compound 31 complex, and 3CLpro-compound 40 complex, respectively (Table 7). From Rg profiles, it has been observed that the 3CLpro-compound complexes exhibited a more compact behavior than the 3CLpro protein without ligand. The lower RMSD, reduced residue-wise fluctuation, and higher compact nature in the 3CLpro-compound complexes indicate their overall stability as well as convergence.
3.5.2. H-bonds, solvent accessible surface area, and PCA analyses of 3CLpro-compound complexes
The H-bonds are essential for drug specificity, metabolization, and stability. Therefore, the H-bonding pattern was evaluated to understand the H-bond and its contributions to the overall stability of the systems. From Fig. 7 A, it can be observed that the maximum numbers of intermolecular hydrogen bond interactions were found to be 4 for, 3CLpro-X77 complex, 3CLpro-compound 21 complex, 3CLpro-compound 31 complex, and 3CLpro-compound 40 complex respectively, while, the 3CLpro-compound 22 complex, 3CLpro-compound 23 complex formed 3 hydrogen bond interactions during the 100 ns simulation period. By analyzing the result, it was found that all 3CLpro-compound complexes did not deviate and almost similar numbers of hydrogen bonds were formed between the 3CLpro-compound complexes and 3CLpro-X77 complex which indicates that all the compounds were bound to the 3CLpro as tightly and effectively as its standard inhibitor X77. This result reflects that the H-bonds probably played a crucial role in the stability of the 3CLpro-X77 complex during the simulation and also indicates stability to the 3CLpro-compound complexes. Fig. 7B showed that the SASA of 3CLpro-X77 complex and 3CLpro-compound complexes. The average SASA values were found to be 158.04 nm2 for 3CLpro-compound 22 complex, 153.63 nm2 for 3CLpro-compound 31 complex respectively. 3CLpro-compound 21 complex (151.33 nm2), 3CLpro-compound 23 complex (152.58 nm2), and 3CLpro-compound 40 complex (150.21 nm2) was found to have a slightly similar SASA value compared to the 3CLpro-X77 complex which showed the average SASA value of 149.29 nm2. However, after 40 ns 3CLpro-X77 complex as well as all the 3CLpro-compound complexes showed almost similar surface area (Fig. 7B). The results showed a similar assessable surface area of compounds to the reference X77 in the aqueous system which indicates equivalent stability of compounds with 3CLpro as X77.
Fig. 7.

Simulation result showing the number of H-bond (A), fluctuations in the solvent accessibility surface area (B), and Principal component analysis showing eigenvalues vs. first 40 eigenvectors (C) during the simulation period.
PCA analysis was performed to get insights into the correlation of atomic motions in the protein-ligand interaction, which was obtained from the significant motion of atoms regulated by the secondary structure of a protein. Typically, the overall motion of the protein subspace where the majority of the protein dynamics occur is defined by only the first few eigenvectors. For this study, the stable PCA clusters for the 3CLpro-X77 complex and 3CLpro-compound complexes were visualized and analyzed (Fig. 7C). The figure shows the eigenvalues from the diagonalization of the covariance matrix of atomic motions and the corresponding eigenvector for 3CLpro-X77 and 3CLpro-compound complexes. Here, the first 40 eigenvectors were selected to calculate collective motions. It was analyzed that out of the selected eigenvectors, the first ten eigenvectors accounted for 74.59% in 3CLpro-X77 complex, 81.35% in 3CLpro-compound 21 complex, 71.10% in 3CLpro-compound 22 complex, 77.46% in 3CLpro-compound 23 complex, 68.68% in 3CLpro-compound 31 complex, and 63.44% in 3CLpro-compound 40 complex of overall collective motions, respectively. All the studied 3CLpro-compound complexes showed very similar dynamic motions as reference complex. So we can conclude that all compounds showed fewer motions and form very stable complexes with 3CLpro.
3.5.3. Gibbs free energy analyses of 3CLpro-compound complexes
The Gibbs energy plots were generated from the PC1 and PC2 coordinates and are shown in Fig. 8 . In these plots, ΔG values ranging from 0 to 12.5 kJ mol−1, 0–14.8 kJ mol−1, 0–12.9 kJ mol−1, 0–13.5 kJ mol−1, 0–12.4 kJ mol−1, and 0–12.9 kJ mol−1 for 3CLpro-X77 complex, 3CLpro-compound 21 complex, 3CLpro-compound 22 complex, 3CLpro-compound 23 complex, 3CLpro-compound 31 complex, and 3CLpro-compound 40 complex, respectively. All the 3CLpro-compound complexes represent significantly similar energy as the 3CLpro-X77 complex, which indicates that these compounds follow the energetically favorable transitions during the MDS.
Fig. 8.

Gibbs energy plot of (A) 3CLpro-X77 complex, (B) 3CLpro-Compound 21 complex, (C) 3CLpro-Compound 22 complex, (D) 3CLpro-Compound 23 complex, (E) 3CLpro-Compound 31 complex, and (F) 3CLpro-Compound 40 complex.
3.5.4. Binding free energy calculations in 3CLpro-compound complexes
To determine how strongly compounds bind to 3CLpro and their respective associated binding modes, the binding free energies were calculated using the MM-PBSA approach. The MD trajectories were analyzed through MM-PBSA to know the binding free energy values and their energy component. For this purpose, the last ns trajectory was investigated to calculate binding energies and insights into the binding modes of compounds with 3CLpro. The reference molecule X77 was found to display binding energy of −57.380 kJ mol−1 for 3CLpro (Table 8 ). Computation of the binding energy of compounds for the 3CLpro revealed that compound 40, 21, and 22 exhibited a higher affinity −70.419 kJ mol−1, -53.415 kJ mol−1, and -47.490 kJ mol−1 respectively, while compound 23 and 31 displayed a lower affinity −28.003 kJ mol−1and 8.739 kJ mol−1 respectively for the 3CLpro. The detailed study of the individual energy components revealed that all components including the van der Waals energy, Electrostatic Energy, and SASA energy, except the polar solvation energy contributed to the efficient binding of compounds with 3CLpro.
Table 8.
Table displaying binding energy of 3CLpro-compound complexes obtained by MM–PBSA.
| S No. | Protein/Protein-ligand complex | Van der Waal Energy (KJmol−1) |
Electrostatic Energy (KJ mol−1) |
Polar salvation energy (KJ mol−1) | SASA energy (KJ mol−1) | Binding Energy (KJ mol−1) |
|---|---|---|---|---|---|---|
| R | 3CLpro -X77 complex | −114.85 ± 8.88 | −8.42 ± 6.92 | 81.55 ± 11.92 | −15.657 ± 1.025 | −57.380 ± 9.773 |
| 1 | 3CLpro-compound 21 complex | −112.59 ± 8.72 | −13.90 ± 8.01 | 88.28 ± 13.74 | −15.199 ± 1.494 | −53.415 ± 10.654 |
| 2 | 3CLpro-compound 22 complex | −93.1 ± 18.14 | −20.05 ± 1.03 | 77.97 ± 40.20 | −12.302 ± 2.176 | −47.490 ± 34.959 |
| 3 | 3CLpro-compound 23 complex | −27.24 ± 8.26 | −2.62 ± 11.15 | 6.07 ± 47.33 | −4.210 ± 1.751 | −28.003 ± 48.454 |
| 4 | 3CLpro-compound 31 complex | −0.23 ± 0.22 | 0.10 ± 1.43 | 8.86 ± 60.08 | 0.010 ± 1.502 | 8.739 ± 60.651 |
| 5 | 3CLpro-compound 40 complex | −111.82 ± 17.44 | −14.84 ± 9.42 | 71.04 ± 20.59 | −14.801 ± 2.107 | −70.419 ± 11.211 |
The comprehensive study shows that all three compounds have very good binding efficiency against the 3CLpro. From the overall RMSD, RMSF, Rg, SASA, hydrogen bonds, PCA, and binding free energy analysis results, we conclude that 3CLpro-compound 21, 3CLpro-compound 22, 3CLpro-compound 40 complexes are very stable as compared to reference 3CLpro-X77 complex. It means that these compounds may potent inhibitors of SARS-CoV-2 3CLpro and could be used as potential drug candidates against SARS-CoV-2. However, further studies are necessary to reveal the action of these compounds. This study provides an insight into the structure of new compounds that have not been previously reported. With an effective binding to SARS-COV-2 3CLpro, these compounds can regulate their role in replicase polyprotein processing and the release of functional proteins during virus maturation.
For the last 1 ns of MD simulation trajectories, a per residue interaction energy profile was also developed using the MM-PBSA approach to identify the essential residues involved in ligand binding toward 3CLpro protein. Fig. 9 shows a per-residue decomposition plot of the total binding energy of the 3CLpro-ligand complexes. Only residues that contribute most to overall binding energy are illustrated in the figure for a better representation of the results. The plot showed that the strongly involved amino acids in all complexes were Thr25, Leu27, His41, Cys44, Met49, Gly143, Cys145, Met165, Pro168, and Asp187. The per-residue interaction plot revealed that the majority of residues had negative binding energy, while only a few had positive binding energy. The residues with a negative binding affinity were important in maintaining the stable protein-ligand complex. Fig. 10 depicts the per-residue decomposition plot of active site residues of 3CLpro in various 3CLpro-ligand complexes. When compared to other active site residues, Thr25, Leu27, Met49, Gly143, Cys145, Met165, and Pro168 showed higher binding affinity. The overall results revealed that Met49, Cys145, and Met165 play the most significant roles in 3CLpro-ligand stabilization, which is consistent with previous research [41].
Fig. 9.

The contributions of individual amino acid residues of 3CLpro to the total binding.
Fig. 10.

The per-residue decomposition plot of active site residues of 3CLpro in studied 3CLpro-ligand complexes.
4. Conclusion
The present study aimed to identify novel inhibitors against the SARS-CoV-2 3CLpro. In this study, quantitative structure-activity relationship and molecular docking were used to evaluate the relationship between molecular properties and inhibitory activity of selected compounds against SARS coronavirus 3C-like protease from the CHEMBL database. The model obtained in the study was robust and statistically significant. This study revealed five compounds with a remarkable binding affinity for 3CLpro, good ADMET properties, and important pharmacophore features. Finally, the relative stability of these compounds was validated by 100 ns MD simulation. The MD trajectories analysis showed that three compounds viz. compound 21, 22, and 40 reflect good structural stability with 3CLpro. Hence from this study, three hits i.e. compound 21 (CHEMBL ID 19438), 22 (CHEMBL ID 196635), 40 (CHEMBL ID 210097) were identified against SARS-CoV-2 3CLpro and these can be considered as a possible treatment for COVID-19. However, further in-vitro and in-vivo studies are necessary to investigate the efficacy of these potential compounds against SARS-CoV-2.
Declaration of competing interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Acknowledgments
The authors are thankful to the Head Department of Botany, Kumaun University, Nainital for providing the facility, space, and resources for this work. The authors also acknowledge Kumaun University, Nainital for providing high-speed internet facilities. We also extend our acknowledge to Rashtriya Uchchattar Shiksha Abhiyan (RUSA), Ministry of Human Resource Development, Government of India to provide Computational infrastructure for the establishment of Bioinformatics Centre in Kumaun University, S.S.J Campus, Almora.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2021.104483.
Funding
There was no funding source to carry out this research work.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Hui D.S., I Azhar E., Madani T.A., Ntoumi F., Kock R., Dar O., Ippolito G., Mchugh T.D., Memish Z.A., Drosten C., Zumla A., Petersen E. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health — the latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020;91:264–266. doi: 10.1016/j.ijid.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu H., Stratton C.W., Tang Y.W. Outbreak of pneumonia of unknown etiology in Wuhan, China: the mystery and the miracle. J. Med. Virol. 2020;92:401–402. doi: 10.1002/jmv.25678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W. A novel coronavirus from patients with pneumonia in China. N. Engl. J. Med. 2019:2020. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization Coronavirus disease (COVID-19) weekly epidemiological update, geneva. 2021. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports
- 5.Schoeman D., Fielding B.C. Coronavirus envelope protein: current knowledge. Virol. J. 2019;16:1–22. doi: 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mortola E., Roy P. Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett. 2004;576:174–178. doi: 10.1016/j.febslet.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang C., Zheng X., Gai W., Zhao Y., Wang H., Wang H., Feng N., Chi H., Qiu B., Li N., Wang T., Gao Y., Yang S., Xia X. Oncotarget; 2017. MERS-CoV Virus-like Particles Produced in Insect Cells Induce Specific Humoural and Cellular Imminity in Rhesus Macaques. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X., Wang X.J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genomics. 2020;47:119. doi: 10.1016/j.jgg.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) Structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. 80- [DOI] [PubMed] [Google Scholar]
- 10.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nantasenamat C., Isarankura-Na-Ayudhya C., Naenna T., Prachayasittikul V. A practical overview of quantitative structure-activity relationship. EXCLI J. 2009;8:74–88. [Google Scholar]
- 12.Abdel-Hamid M.K., Abdel-Hafez A.A., El-Koussi N.A., Mahfouz N.M. Quantitative structureactivity relationship (QSAR) studies on a series of 1,3,4-thiadiazole-2-thione derivatives as tumor-associated carbonic anhydrase IX inhibitors. J. Enzym. Inhib. Med. Chem. 2009;24:722–729. doi: 10.1080/14756360802361514. [DOI] [PubMed] [Google Scholar]
- 13.Huang S.Y., Zou X. Advances and challenges in Protein-ligand docking. Int. J. Mol. Sci. 2010;11:3016–3034. doi: 10.3390/ijms11083016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Gunsteren W.F., Billeter S.R., Eising A.A., Hunenberger P.H., Krüger P., Mark A.E., Scott W.R.P., Tironi I.G., Hünenberger P.H., Huenenberger P.H. 1996. The GROMOS96 Manual and User Guide. [Google Scholar]
- 15.Yap C.W. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011;32:1466–1474. doi: 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- 16.Adedirin O., Uzairu A., Shallangwa G.A., Abechi S.E. Computational studies on α-aminoacetamide derivatives with anticonvulsant activities. Beni-Suef Univ. J. Basic Appl. Sci. 2018;7:709–718. doi: 10.1016/j.bjbas.2018.08.005. [DOI] [Google Scholar]
- 17.Golbraikh A., Tropsha A. Beware of q2! J. Mol. Graph. Model. 2002;20:269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- 18.Sivakumar P.M., Babu S.K.G., Mukesh D. QSAR studies on chalcones and flavonoids as anti-tuberculosis agents using genetic function approximation (GFA) method. Chem. Pharm. Bull. 2007;55:44–49. doi: 10.1248/cpb.55.44. [DOI] [PubMed] [Google Scholar]
- 19.Goodsell D.S., Morris G.M., Olson A.J. Automated docking of flexible ligands: applications of AutoDock. J. Mol. Recogn. 1996;9:1–5. doi: 10.1002/(SICI)1099-1352(199601)9:1<1::AID-JMR241>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 20.Sander T., Freyss J., Von Korff M., Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 21.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open babel: an open chemical toolbox. J. Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morris G.M., Goodsell D.S., Pique M.E., Lindy Lindstrom W., Huey R., Forli S., Hart W.E., Halliday S., Belew R., Olson A.J. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trott O., Olson A.J., Vina AutoDock. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stierand K., Maaß P.C., Rarey M. Molecular complexes at a glance: automated generation of two-dimensional complex diagrams. Bioinformatics. 2006;22:1710–1716. doi: 10.1093/bioinformatics/btl150. [DOI] [PubMed] [Google Scholar]
- 25.Daina A., Michielin O., Zoete V. ILOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014;54:3284–3301. doi: 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- 26.Daina A., Zoete V. A BOILED‐egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11:1117–1121. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M.R., Smith J.C., Kasson P.M., Van Der Spoel D., Hess B., Lindahl E. Gromacs 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A.D., Jr. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Izadi S., Onufriev A.V. Accuracy limit of rigid 3-point water models. J. Chem. Phys. 2016;145 doi: 10.1063/1.4960175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumari R., Kumar R., Consortium O.S.D.D., Lynn A. g _ immpress - a GROMACS tool for MM-PBSA and its optimization for high-throughput binding energy calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 32.Oluwaseye A., Uzairu A., Adamu S.G., Eyije A.S. Quantitative structure activity relationship studies on some N-benzylacetamide and 3-(phenylamino) propanamide derivatives with anticonvulsant properties. Int. J. Geol. Agric. Environ. Sci. 2017;5:5–22. [Google Scholar]
- 33.Topliss J.G., Costello R.J. Chance correlations in structure-activity studies using multiple regression analysis. J. Med. Chem. 1972;15:1066–1068. doi: 10.1021/jm00280a017. [DOI] [PubMed] [Google Scholar]
- 34.Tropsha A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010;29:476–488. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 35.Beheshti A., Pourbasheer E., Nekoei M., Vahdani S. QSAR modeling of antimalarial activity of urea derivatives using genetic algorithm–multiple linear regressions. J. Saudi Chem. Soc. 2016;20:282–290. [Google Scholar]
- 36.Roy K. On some aspects of validation of predictive quantitative structure-activity relationship models. Expet Opin. Drug Discov. 2007;2:1567–1577. doi: 10.1517/17460441.2.12.1567. [DOI] [PubMed] [Google Scholar]
- 37.Shi J., Wei Z., Song J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme. Defining the extra domain as a new target for design of highly specific protease inhibitors. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi J., Song J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006;273:1035–1045. doi: 10.1111/j.1742-4658.2006.05130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lipinski C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 41.Joshi T., Sharma P., Joshi T., Pundir H., Mathpal S., Chandra S. Structure-based screening of novel lichen compounds against SARS Coronavirus main protease (Mpro) as potentials inhibitors of COVID-19. Mol. Divers. 2020:1–13. doi: 10.1007/s11030-020-10118-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.