

Abstract
Over the last two decades there has been significant progress towards understanding the neural substrates that underlie age-related cognitive decline. Although many of the exact molecular and cellular mechanisms have yet to be fully understood, there is consensus that alterations in neuronal calcium homeostasis contribute to age-related deficits in learning and memory. Furthermore, it is thought that the age-related changes in calcium homeostasis are driven, at least in part, by changes in calcium channel expression. In this review, we focus on the role of a specific class of calcium channels: L-type voltage-gated calcium channels (LVGCCs). We provide the reader with a general introduction to voltage-gated calcium channels, followed by a more detailed description of LVGCCs and how they serve to regulate neuronal excitability via the post burst afterhyperpolarization (AHP). We conclude by reviewing studies that link the slow component of the AHP to learning and memory, and discuss how age-related increases in LVGCC expression may underlie cognitive decline by mediating a decrease in neuronal excitability.
Keywords: age-related cognitive decline, neuronal excitability, L-type voltage-gated calcium channels, CaV1.2, CaV1.3, slow afterhyperpolarization, sAHP
Introduction to Voltage-Gated Calcium Channels
Voltage-gated calcium channels (VGCCs) are multi-subunit membrane complexes that permit calcium to cross the plasma membrane in response to depolarizing shifts in membrane potential. In mammals, VGCCs are widely distributed across multiple organ systems and cell types. As with other ionic species, the direction of flow across the membrane is driven in a concentration-dependent manner, and in the case of calcium, the extracellular concentration far exceeds that measured inside the cell. In neurons, the intracellular Ca2+ concentration is in the tens of nanomolar range while the extracellular concentration is in the millimolar range, a difference of a thousand-fold or greater (Clapham, 2007)! This steep concentration gradient ensures a rapid and robust signaling mechanism for a multiplicity of neuronal functions.
Voltage-gated calcium channels in neurons are typically comprised of a pore forming subunit and several “auxiliary” subunits. The pore forming (α1) subunit proteins are relatively large (~200 kD) and the entire ion channel pore is formed from a single α1 subunit, consisting of four repeated domains each containing six transmembrane domains (for review, see Catterall, 2011). The auxiliary subunits (α2δ and β) are important for α1 subunit trafficking to the plasma membrane with subcellular domain specificity and can modulate α1 subunit voltage dependency and kinetics (Dolphin, 2016). In addition, there exists a γ subunit which appears to be part of the calcium channel complex found in skeletal muscle but is not found in neurons (Dolphin, 2016).
For the uninitiated, the nomenclature surrounding VGCCs can be somewhat confusing. This is in part because calcium channels are still often referred to in terms of the underlying currents, many of which were characterized prior to the identification of the actual channel proteins (and the genes that encode them). Calcium currents were originally classified based upon their voltage of activation (high voltage activated [HVA] vs low voltage activated [LVA]), inactivation kinetics and sensitivity to specific blockers (which were determined empirically). For example, the “L-type” current is classically described as being large and long-lasting while the “T-type” current is tiny and transient, and “N-type” are somewhere in between, being neither L- nor T-type (reviewed by Tsien, Lipscombe, Madison, Bley, and Fox, 1988). In addition, L-type currents are blocked by dihydropyridines such as nimodipine and N-type currents are blocked by the cone snail toxin ω-conotoxin, but T-type currents are not sensitive to either (Tsien et al., 1988). Further classification of calcium currents was greatly facilitated by the use of venom extracted from the funnel web spider (Agelenopsis aperta). Initial experiments used crude fractions (Llinas, Sugimori, and Cherksey, 1989) but before long the active peptide (ω-agatoxin-IVA) was isolated, which effectively blocked high-threshold calcium currents in the rat cerebellum (Mintz, Venema, Swiderek, Lee, Bean, and Adams, 1992). Because these calcium currents were first described in Purkinje neurons (Llinas et al., 1989), they were named P-type currents. While specific channels are today often referred to using nomenclature derived from the underlying currents (e.g. L-type voltage-gated calcium channel), the channels that gate the P current are referred to as P/Q-type. The Q-current was first described in cultures of cerebellar granule cells and could be separated from the P current on the basis of inactivation kinetics and ω-agatoxin-IVA sensitivity (Randall and Tsien, 1995). However, cloning and expression studies have led to the conclusion that both currents are gated by splice variants of the same α-pore forming subunit (Bourinet, Soong, Sutton, Slaymaker, Mathews, Monteil, Zamponi, Nargeot, and Snutch, 1999), giving rise to the P/Q-type moniker. In the same report first describing the Q-current, the authors also described a high threshold current in cerebellar granule cells which was insensitive to antagonism by nimodipine, ω-agatoxin-IVA, or ω-conotoxin, distinguishing it from the other high voltage activated currents (L, P/Q and N). This current was named R-type as it appeared to be resistant to L, P/Q, and N current antagonists (Randall and Tsien, 1995). While there now exists a standard naming system that is similar to that used for voltage-gated K+ channels (see below), the use of L, N, R, P/Q, and T when discussing calcium channels is still common practice, in no small part because using the current-derived nomenclature adds a functional connotation.
With the advent and rapid adoption of molecular cloning methods, researchers in the voltage-gated calcium channel field quickly began to identify genes responsible for encoding the pore-forming subunit and auxiliary subunits. In 1994, the field adopted a unified nomenclature which named the genes that encoded the pore forming subunits as α1A though α1Z. At the time, six distinct pore-forming subunits genes had already been cloned. Starting with α1A, these genes were named in the order in which they were discovered. The exception to this rule was α1S to acknowledge the recently deceased Shosaku Numa who cloned the very first LVGCC gene from skeletal muscle (hence the “S” designation). Therefore, the five remaining genes were designated α1A though α1E (Birnbaumer, Campbell, Catterall, Harpold, Hofmann, Horne, Mori, Schwartz, Snutch, Tanabe, and et al., 1994). However, as more α1 subunits were subsequently cloned, there arose the possibility that the pore-forming subunit α1L would be cloned and it may or may not mediate an L-type current, which might create confusion. Thus, the field adopted a new nomenclature similar to that previously established for voltage-gated K+ channels (Chandy and Gutman, 1993). In this scheme, the charge carrying ion (Ca) is denoted with the biophysical mechanism by which it is regulated (voltage; V) in subscript (CaV). Further, the pore-forming subunits were grouped into three families (CaV1, CaV2 & CaV3) based on protein sequence homology; then family members were numbered in order of discovery (Ertel, Campbell, Harpold, Hofmann, Mori, Perez-Reyes, Schwartz, Snutch, Tanabe, Birnbaumer, Tsien, and Catterall, 2000). Table 1 summaries these different nomenclatures and includes the Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC) approved symbol for the human gene (which interestingly still retains the “alpha” to denote the alpha subunit [eg. CACNA1C] in the gene symbol).
Table 1.
Current voltage-gated calcium channel nomenclature (after Catterall, Lenaeus, and Gamal El-Din, 2020).
| Current | Protein Name | Alpha Subunit | Gene Name (human) | Activation Voltage |
|---|---|---|---|---|
| L-type | CaV1.1 | α1S | CACNA1S | High |
| L-type | CaV1.2 | α1C | CACNA1C | High |
| L-type | CaV1.3 | α1D | CACNA1D | High |
| L-type | CaV1.4 | α1F | CACNA1F | High |
| P/Q-type | CaV2.1 | α1A | CACNA1A | High |
| N-type | CaV2.2 | α1B | CACNA1B | High |
| R-type | CaV2.3 | α1E | CACNA1E | High |
| T-type | CaV3.1 | α1G | CACNA1G | Low |
| T-type | CaV3.2 | α1H | CACNA1H | Low |
| T-type | CaV1.3 | α1I | CACNA1I | Low |
Postsynaptic VGCCs: The L-Type Calcium Channel
If neuroscientists are familiar with voltage-gated calcium channels at all, they are most likely familiar with the P/Q- and N-type channels as these are involved in neurotransmitter release and are often referred to as “presynaptic” channels. On the other hand, L-type VGCCs (referred to hereafter as LVGCCs) are thought of as being “postsynaptic” as they are not involved in neurotransmitter release; instead, in most neurons, LVGCCs gate calcium influx in response to large depolarizing shifts in membrane potential. There are four known LVGCCs but only two, CaV1.2 and CaV1.3, are highly expressed in brain. While CaV1.2 and CaV1.3 share a fair amount of homology both in terms of DNA and protein sequence (Snutch, Leonard, Gilbert, Lester, and Davidson, 1990), they differ significantly in terms of their biophysical properties as well as their distribution throughout the brain and within neuronal compartments. Within the hippocampus, LVGCCs exhibit a differential expression pattern with CaV1.2 being heavily expressed in the pyramidal neuron cell body layer in CA1, in both the cell bodies and dendritic fields of CA2 and CA3, and in the dendrites of dentate gyrus; on the other hand, CaV1.3 is localized to the cell bodies and proximal dendrites throughout the hippocampus (Hell, Westenbroek, Warner, Ahlijanian, Prystay, Gilbert, Snutch, and Catterall, 1993). In addition to having different distributions within the hippocampus, experiments using heterologous expression systems suggest that CaV1.2 and CaV1.3 LVGCCs have different activation thresholds, with CaV1.3 channels opening approximately 20mV more hyperpolarized than CaV1.2 (Helton, Xu, and Lipscombe, 2005; Lipscombe, Helton, and Xu, 2004; Xu and Lipscombe, 2001).
One of the most fascinating aspects of LVGCCs is the diverse roles that they play and the timescales over which they initiate changes in neuronal function. While the bulk of this review will focus primarily on the millisecond to second time range, it should be noted that LVGCCs have been implicated in a wide range of subcellular processes associated with learning and memory that span much longer time frames (hours to weeks). For example, it has been known for some time that the calcium influx gated by LVGCCs plays a key role in adult neurogenesis in the hippocampus. Early work using LVGCC antagonists (such as nifedipine) significantly reduced neurogenesis in rats when administered systemically (Deisseroth, Singla, Toda, Monje, Palmer, and Malenka, 2004). More recent studies using knockout mice have confirmed a role for CaV1.2 and CaV1.3 in adult neurogenesis (Kim, Park, Lee, Choi, Kim, and Kim, 2017; Lee, De Jesus-Cortes, Kabir, Knobbe, Orr, Burgdorf, Huntington, McDaniel, Britt, Hoffmann, Brat, Rajadhyaksha, and Pieper, 2016; Marschallinger, Sah, Schmuckermair, Unger, Rotheneichner, Kharitonova, Waclawiczek, Gerner, Jaksch-Bogensperger, Berger, Striessnig, Singewald, Couillard-Despres, and Aigner, 2015; Temme, Bell, Fisher, and Murphy, 2016; Volkening, Schonig, Kronenberg, Bartsch, and Weber, 2017). Adult neurogenesis has long been implicated in learning and memory, most notably in forms of learning that require “pattern separation/completion” (Tuncdemir, Lacefield, and Hen, 2019). Consistent with this idea, deletion of CaV1.2 disrupts both spatial learning in the water maze and context discrimination during Pavlovian fear conditioning with experimental conditions that are thought to require pattern completion/separation, but spares performance in versions of these tasks that are less reliant upon the dentate gyrus (Temme et al., 2016).
The Post Burst AHP
The post burst AHP (afterhyperpolarization) is, as the name implies, a hyperpolarizing potential that follows a train of action potentials. The post burst AHP is thought to regulate neuronal excitability by holding the cell below the firing threshold for an extended period of time following a burst of action potentials. The post burst AHP is often subdivided by time domains: the fast AHP (fAHP) which immediately follows the burst; the medium AHP (mAHP) which is usually ~200 ms after the burst and often includes the maximum hyperpolarization; and the slow AHP (sAHP) which is often measured at ~1000 ms after the burst and persists for thousands of milliseconds (Faber and Sah, 2002; 2003; Gustafsson and Wigstrom, 1981; Sah, 1996; Sah and Faber, 2002). The ultimate source of each of these three phases of the hyperpolarizing potential is undoubtedly efflux of K+, but the exact identity of the responsible K+ channel(s) and how they regulate neuronal excitability is still somewhat debated. For example, in CA1 pyramidal neurons, a number of reports have suggested that the mAHP (and thus excitability) is regulated by members of the small conductance Ca2+-activated K+ channel (SK, [KCa2.x]) family (Stackman, Hammond, Linardatos, Gerlach, Maylie, Adelman, and Tzounopoulos, 2002; Stocker, 2004; Stocker, Krause, and Pedarzani, 1999) while other reports suggest it is predominated by a combination of voltage-gated K+ channels (KCNQ/M, [KV7.x]) and hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels (Gu, Hu, Vervaeke, and Storm, 2008; Gu, Vervaeke, Hu, and Storm, 2005). More recently, it has been suggested that SK channels play a role in regulating excitability, but only when KCNQ/M channel function has been compromised (Chen, Benninger, and Yaari, 2014). Similarly, there exists some controversy regarding the potassium channels that underlie the sAHP with evidence supporting both a role for (King, Rizwan, Asmara, Heath, Engbers, Dykstra, Bartoletti, Hameed, Zamponi, and Turner, 2015; Turner, Asmara, Engbers, Miclat, Rizwan, Sahu, and Zamponi, 2016) and against (Wang, Mateos-Aparicio, Honigsperger, Raghuram, Wu, Ridder, Sah, Maylie, Storm, and Adelman, 2016) IK channels (intermediate conductance Ca2+-activated K+, [KCa3.1]). Several factors have likely contributed to the field’s inability to settle on a single ground truth regarding the subcellular mechanism(s) that regulate the different phases of the post burst AHP. These include the method of recording (current- vs voltage-clamp; sharp vs whole-cell patch), the organism (mouse, rat, rabbit, cat), the age of the animal, and which cell type is being examined. Even when a number of these factors are made equivalent, details such as the internal recording solution (e.g. Kaczorowski, Disterhoft, and Spruston, 2007; Zhang, Weiner, Valiante, Velumian, Watson, Jahromi, Schertzer, Pennefather, and Carlen, 1994), the genetic background (Moore, Throesch, and Murphy, 2011; Murphy, Fedorov, Giese, Ohno, Friedman, Chen, and Silva, 2004), and recording temperature (Gulledge, Dasari, Onoue, Stephens, Hasse, and Avesar, 2013; Tiwari, Mohan, Biala, and Yaari, 2018) can influence the appearance and plasticity of the AHP. Setting aside the controversies regarding the identity of the K+ channel(s) which underlie the post burst AHP, there is general agreement that the sAHP is dependent upon calcium influx that is gated by LVGCCs (although see Gulledge et al., 2013), which will be the focus of the discussion below.
The sAHP in Learning and Memory
The sAHP was originally described in 1980 (Alger and Nicoll, 1980; Hotson and Prince, 1980) in the CA1 region of the hippocampus, but it is observed in a variety of brain regions, including the amygdala (Power, Bocklisch, Curby, and Sah, 2011) and the sensorimotor cortex (Schwindt, Spain, and Crill, 1988; Schwindt, Spain, Foehring, Chubb, and Crill, 1988; Schwindt, Spain, Foehring, Stafstrom, Chubb, and Crill, 1988), as well as in interneurons in the striatum (Goldberg and Wilson, 2005). It is well appreciated that the sAHP is modulated by and functions to support several forms of learning (Coulter, Lo Turco, Kubota, Disterhoft, Moore, and Alkon, 1989; Disterhoft, Coulter, and Alkon, 1986; Disterhoft, Golden, Read, Coulter, and Alkon, 1988; Saar, Grossman, and Barkai, 1998; Sehgal, Ehlers, and Moyer, 2014). Many of the early insights into the relationship between the sAHP and learning came from trace eyeblink experiments, initially in rabbits and then in rats (and more recently in mice). In this task, a brief tone (the conditioned stimulus, CS) predicts an air puff delivered to the eye (the unconditioned stimulus, US) after an intervening (trace) interval of several hundred milliseconds. Animals that successfully acquire the task learn this association and close the nictitating membrane covering the eye (conditioned response, CR) to avoid the air puff. Unlike other forms of eyeblink conditioning, trace eyeblink conditioning is dependent upon an intact hippocampus. Therefore, following training in trace eyeblink conditioning, when intracellular recordings are obtained from CA1 pyramidal neurons in ex vivo hippocampal slices, learning performance can be directly correlated with changes in the sAHP. Groundbreaking work from the Disterhoft lab showed that young adult animals that successfully learned the CS-US association exhibited a reduced sAHP (and increased neuronal excitability) when compared to neurons in slices from naïve animals (that had received no training experience), pseudoconditioned animals (that had received unpaired presentations of the CS and US), and, importantly, from “slow-learner” animals that were trained under identical conditions, but did not demonstrate a criterion level of correct CRs and thus were not considered to have learned the task (Disterhoft, Thompson, Moyer, and Mogul, 1996). The reduced sAHP amplitude in the trace eyeblink task appears to be transient in nature: recordings made 7 days after successful learning revealed a sAHP which was indistinguishable from the sAHP recorded in slices from control animals (Moyer, Thompson, and Disterhoft, 1996). This suggests that the reduction in the post-burst AHP may be necessary to permit an animal to learn a task, but likely does not represent the encoded memory itself. While the sAHP has been studied most intensively in the hippocampus, changes in sAHP amplitude have been observed in other brain regions in the context of other learning paradigms, including in the piriform cortex after odor discrimination learning (Saar et al., 1998) and in the amygdala after Pavlovian fear conditioning (Sehgal et al., 2014).
At present it remains somewhat unclear how learning-related changes in the sAHP manifest. Evidence from ex vivo slice preparations suggests that modulation of the sAHP can occur in a cell autonomous fashion because long-lasting reductions in the sAHP have been induced by high frequency stimulation that drives intercellular depolarization (Cohen-Matsliah, Motanis, Rosenblum, and Barkai, 2010; Kaczorowski et al., 2007). Interestingly, both a significantly larger sAHP as well as a significantly smaller sAHP can adversely impact learning and memory. For example, aging has consistently been shown to affect the sAHP. Recordings made in ex vivo slices from aged animals reveal a larger sAHP compared to that recorded in slices from young adult animals (e.g. Landfield and Pitler, 1984; Moyer, Thompson, Black, and Disterhoft, 1992). Further, this increase in magnitude has been associated with poorer performance in behavioral learning and memory tasks (e.g. Kaczorowski and Disterhoft, 2009; Murphy et al., 2004). Perhaps the most convincing evidence linking age-related increases in sAHP amplitude to learning/memory impairments exploits the observation that aged rabbits and rats (and inbred mice under some conditions; see Murphy, Rahnama, and Silva, 2006) often comprise two distinct populations. While not always a 50/50 split, when compared to young animals, approximately one half of the aged animals will exhibit comparable performance (“learners”) while the other half will either take longer to learn the task or fail to learn the task altogether (“slow-learners” or “non-learners”). Following behavioral characterization, animals are sacrificed and ex vivo recordings of the sAHP amplitude are made in acute slices, which can be correlated with the individual animal’s behavioral performance. Using this approach an inverse relationship between sAHP amplitude and memory has been demonstrated in a number of learning paradigms including trace eyeblink conditioning (Matthews, Linardakis, and Disterhoft, 2009; Moyer, Power, Thompson, and Disterhoft, 2000) and the Morris water maze (Tombaugh, Rowe, and Rose, 2005).
Conversely, reduction of the sAHP amplitude has also been shown to lead to learning and memory deficits. For example, genetic deletion of Kcnab2, the gene that encodes KVβ2, which is an auxiliary potassium channel subunit that modulates rates of endogenously inactivating KV1.4 currents (McCormack, McCormack, Tanouye, Rudy, and Stuhmer, 1995), significantly reduces the sAHP in the lateral amygdala. Consistent with the central role that the amygdala plays in Pavlovian fear conditioning, the decreased sAHP observed in the Kcnab2 null mice was associated with disruptions in both contextual and cued (tone CS) fear conditioning (Perkowski and Murphy, 2011). Similarly, deletion of KVβ1.1 significantly reduces the sAHP amplitude and disrupts reversal learning in mice (Giese, Storm, Reuter, Fedorov, Shao, Leicher, Pongs, and Silva, 1998).
Taken together, these data suggest that there may be an effective range for the sAHP (and possibly for the other components as well; see Matthews et al., 2009) in which it is capable of influencing learning and memory. In cases where a genetic or pharmacological intervention has moved the sAHP outside this range (either smaller or larger), training may be unable to sufficiently modulate the sAHP to induce learning and memory. Alternatively, the intracellular mechanisms that are involved in producing a larger or smaller sAHP may occlude the activity-dependent plasticity of the sAHP that is required for learning and memory.
Age-related Changes in LVGCC and the sAHP
From the early 1990s, evidence has accumulated suggesting that there is an age-related alteration in calcium homeostasis. One of the first experiments to support this hypothesis found an age-related increase in calcium spike duration, which is a measure of calcium influx via voltage-gated calcium channels (Pitler and Landfield, 1990). Later, in an elegant study using on-cell voltage clamp recordings, Philip Landfield’s group (Thibault and Landfield, 1996) demonstrated that CA1 pyramidal neurons from aged animals had enhanced LVGCC density when compared to young and middle-aged rats. Notably, this increase in current density was correlated with a decrease in performance in the Morris water maze, suggesting that the increase in channel density contributed significantly to the age-related decline in hippocampal-dependent learning (Thibault and Landfield, 1996). These studies demonstrating an age-related dysregulation of intracellular calcium have been corroborated by calcium imaging (e.g. Hemond and Jaffe, 2005; Thibault, Hadley, and Landfield, 2001), which showed an increase in [Ca2+]I in response to repetitive spiking (Hemond and Jaffe, 2005; Thibault et al., 2001) in neurons from aged animals. Importantly, the intracellular calcium concentration [Ca2+]i at rest in these neurons was similar to that observed in young animals. These results suggest that the high voltageactivated, postsynaptically localized LVGCCs represent the source of increased [Ca2+]I in aged animals. This idea is supported by experiments in which LVGCCs were targeted pharmacologically in aged animals. For example, nimodipine (a LVGCC blocker) has been demonstrated to modulate the AHP preferentially in ex vivo slices prepared from aged rabbits (Moyer et al., 1992). Similarly, nimodipine reduced the IsAHP (the current that underlies the sAHP) by a greater magnitude in aged neurons as compared to young neurons from rats and rabbits (Lima and Marrion, 2007; Power, Wu, Sametsky, Oh, and Disterhoft, 2002). Taken together, the data show that expression of LVGCCs is upregulated during aging and that this increase is correlated with cognitive impairments; further, the sAHP, which is Ca2+-dependent, is also increased with age and correlated with cognitive impairments, Therefore, a model has emerged whereby age-related upregulation of LVGCCs leads to an increase in the magnitude of the sAHP, which in turn results in learning and memory deficits in aged animals. However, determining the exact identity of the LVGCC subunit(s) that is upregulated during aging has been somewhat challenging. Early work correlating LVGCC current density with mRNA levels (using RT-PCR) at the level of individual neurons suggested that CaV1.3 expression was upregulated during aging (Chen, Blalock, Thibault, Kaminker, and Landfield, 2000). Similarly, studies using LVGCC antibodies and immunoblotting suggested that CaV1.3 protein levels in CA1 hippocampus were increased in aged rats (Veng and Browning, 2002) which correlated with age-related impairments in working memory (Veng, Mesches, and Browning, 2003). However, it also appears that phosphorylation of CaV1.2 by cAMP-dependent protein kinase A (PKA) is significantly increased in the hippocampus of aged rats (Davare and Hell, 2003), although not in aged mice (Murphy, Shah, Hell, and Silva, 2005). While none of these reports directly conflict with one another, a complete picture has yet to emerge regarding age-related changes in LVGCC protein levels. This is likely due in part to the lack of specificity of the antibodies and the technical and methodological challenges associated with their use in quantitative studies. Voltage-gated ion channels, in general, are known to be difficult to target immunohistochemically (Rhodes and Trimmer, 2006) and LVGCCs appear to be especially sensitive to a variety of experimental conditions including acrylamide concentration (e.g. Buonarati, Henderson, Murphy, Horne, and Hell, 2017). In addition, when LVGCC protein levels are quantified using bulk homogenates, there appears to be an age-related decrease in both CaV1.2 and CaV1.3, but when examination is restricted to protein in/on the cell surface, a region-specific, age-related increase in LVGCC expression is revealed (Nunez-Santana, Oh, Antion, Lee, Hell, and Disterhoft, 2014). While there has yet to be a definitive study which ascribes the relative contributions of CaV1.2 or CaV1.3 to the age-related increase in activity dependent [Ca2+]i, there are several lines of evidence to suggest that calcium influx via CaV1.3 is a major source of calcium associated with the sAHP under normal conditions. For example, in mice genetic ablation of Cacna1d but not Cacna1c resulted in a significant reduction in the sAHP in CA1 hippocampus (Gamelli, McKinney, White, and Murphy, 2011). More recent studies utilizing direct stochastic optical reconstruction microscopy (dSTORM) suggest that CaV1.3 clusters into a complex with KCa3.1 on the plasma membrane and that this complex is anchored to ryanodine receptors localized on the endoplasmic reticulum (Sahu, Wazen, Colarusso, Chen, Zamponi, and Turner, 2019). Importantly, disruption of this complex reduces the IsAHP. Finally, we have recently generated a transgenic mouse line which over-expresses an epitope tagged form of CaV1.3 (Krueger, Moore, Parent, McKinney, Lee, and Murphy, 2017). The expression levels of CaV1.3 in CA1 hippocampus of these mice are similar to what we have previously observed in aged mice and the sAHP is similarly increased (Figure 1).
Figure 1.
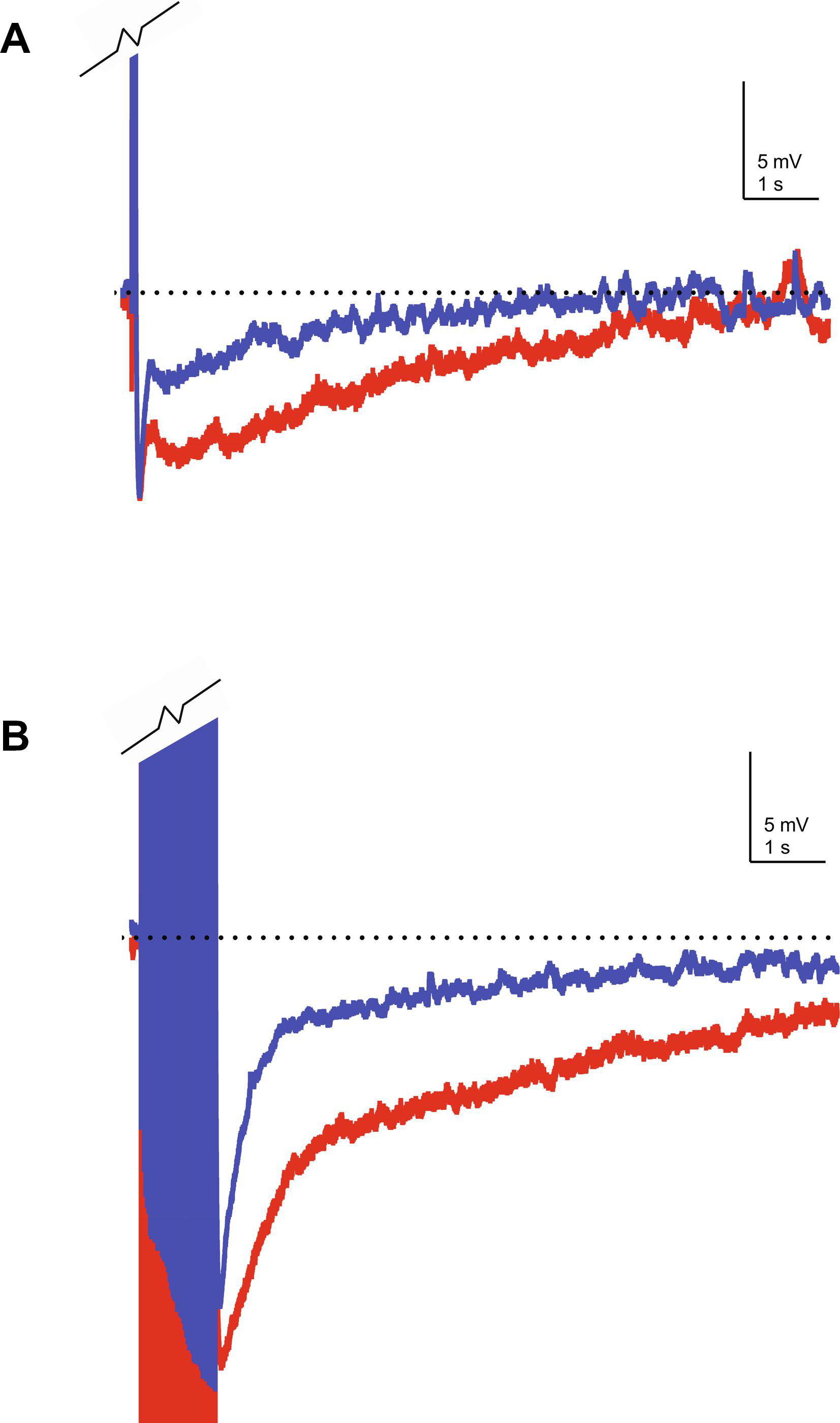
Representative sAHP recordings from CaV1.3 over-expressing mice. Whole-cell current-clamp recordings were made in CA1 hippocampus pyramidal neurons in ex vivo slices prepared from young (4 mo) wild-type (blue) and CaV1.3 over-expressing (red) mice. A) The sAHP elicited by a 50 ms depolarizing step that is sufficient to elicit five action potentials. B) The sAHP recorded in response to a 1 s train of action potentials at 50 Hz. In both cases, the sAHP was increased in the CaV1.3 over-expressing mice compared to that observed in wild-type mice. Action potentials have been truncated to emphasize the sAHP. Dotted line approximates the resting membrane potential.
Interestingly, young mice over-expressing Cav1.3 also show impairments in learning and memory tasks, similar to those observed in aged wild-type mice (Moore, S. personal observation; see also Moore, Slater, and Murphy, 2012). Taken collectively, these data suggest that an age-related increase in CaV1.3 results in enhanced activity-dependent [Ca2+]I, mediates the enhanced sAHP, and contributes to learning and memory deficits in aged animals.
Additional Mechanisms Contributing to Age-Related Changes in Ca2+ Homeostasis
The focus of this review is to highlight the role that LVGCCs play in age-related changes in neuronal excitability and cognition. However, it should be noted that the alterations in LVGCC expression/function occur concurrently with other age-related changes in calcium homeostasis. For example, several studies have suggested that intracellular stores provide the source of calcium for activating the Ca2+-activated K+ channel(s) that mediate the sAHP. Both depletion of intracellular stores or blockage of ryanodine receptors, which mediate release of calcium from intracellular stores, reduced the sAHP in aged animals (Bodhinathan, Kumar, and Foster, 2010; Gant, Sama, Landfield, and Thibault, 2006; Kumar and Foster, 2004). Interestingly, LVGCCs are known to be one source of calcium that can trigger Ca2+-induced Ca2+ release from intracellular stores and the effect of depletion of intracellular stores on the sAHP can be reversed using a LVGCC agonist (Kumar and Foster, 2004). It is likely that this and other processes could contribute to the age-related dysregulation of Ca2+ homeostasis, and may be differentially recruited in certain cell types or regions, and/or may act in synergistically in concert to disrupt calcium signaling in aged animals.
Concluding Thoughts
Although the evidence supporting the idea that aging is accompanied by an increase in LVGCC expression and a subsequent increase in the sAHP is substantial, many questions remain unanswered. One obvious question is: why? Is the age-related upregulation of LVGCCs a consequence of some other, unknown neurosenescence? Or perhaps the upregulation of LVGCC expression is a compensatory response. Given that even modest prolonged elevations of [Ca2+]i can be neurotoxic and the fact that neurons expend a significant amount of energy (in the form of ATP) to maintain minimal levels of [Ca2+]i,, this seems unlikely. An equally important and related question is: how? If one accepts the premise that the end result of the age-related upregulation of LVGCCs is impaired cognitive function, this proves to be a critical question. At present, there are no subunit specific LVGCC blockers. Because CaV1.2 is abundantly expressed in cardiac myocytes and vascular tissue, off target effects of the existing non-specific antagonists somewhat limits their use. In this regard the results from the recent phase III NILVAD trial are informative (Lawlor, Segurado, Kennelly, Olde Rikkert, Howard, Pasquier, Borjesson-Hanson, Tsolaki, Lucca, Molloy, Coen, Riepe, Kalman, Kenny, Cregg, O’Dwyer, Walsh, Adams, Banzi, Breuilh, Daly, Hendrix, Aisen, Gaynor, Sheikhi, Taekema, Verhey, Nemni, Nobili, Franceschi, Frisoni, Zanetti, Konsta, Anastasios, Nenopoulou, Tsolaki-Tagaraki, Pakaski, Dereeper, de la Sayette, Senechal, Lavenu, Devendeville, Calais, Crawford, Mullan, and Group, 2018). In this randomized, placebo-controlled, double-blind trial, patients with mild- and moderate-stage Alzheimer’s disease where treated with nilvadipine (a LVGCC blocker similar to nimodipine) or a placebo. Unfortunately, nilvadipine did not alter the trajectory of cognitive decline in this study at the selected dose (8 mg/day). This dose was selected in part because it was thought that a higher dosing regimen might reduce blood pressure to an unsafe level. Although treated patients on average did have a small reduction in blood pressure at the end of the trial, the authors concluded that “the effect on blood pressure quite modest, so it would probably have been safe to give a higher dose.” (Lawlor et al., 2018). These results highlight the difficulty in even designing a trial in the absence of subunit specific pharmacology and emphasize the need for a renewed effort toward drug discovery in this area. Finally, it is worth noting that, in addition to cognitive aging, dysregulation of voltage-gated calcium channels has been implicated in a number of other conditions. For example, LVGCCs have been implicated in Parkinson’s disease (Liss and Striessnig, 2019) and substance abuse (Burgdorf, Schierberl, Lee, Fischer, Van Kempen, Mudragel, Huganir, Milner, Glass, and Rajadhyaksha, 2017) and variants in CACNA1C have been implicated in a wide array of psychiatric disorders (Cross-Disorder Group of the Psychiatric Genomics, 2013). In this regard, future advances made in better understanding the role of LVGCCs in aging and cognition will likely have considerable impacts on a broad range of neurological and psychiatric disease states.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alger BE, & Nicoll RA (1980). Epileptiform burst afterhyperpolarization: calcium-dependent potassium potential in hippocampal CA1 pyramidal cells. Science, 210, 1122–1124. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L, Campbell KP, Catterall WA, Harpold MM, Hofmann F, Horne WA, Mori Y, Schwartz A, Snutch TP, Tanabe T, & et al. (1994). The naming of voltage-gated calcium channels. Neuron, 13, 505–506. [DOI] [PubMed] [Google Scholar]
- Bodhinathan K, Kumar A, & Foster TC (2010). Redox sensitive calcium stores underlie enhanced after hyperpolarization of aged neurons: role for ryanodine receptor mediated calcium signaling. J Neurophysiol, 104, 2586–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, & Snutch TP (1999). Splicing of alpha 1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci, 2, 407–415. [DOI] [PubMed] [Google Scholar]
- Buonarati OR, Henderson PB, Murphy GG, Horne MC, & Hell JW (2017). Proteolytic processing of the L-type Ca (2+) channel alpha 11.2 subunit in neurons. F1000Res, 6, 1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorf CE, Schierberl KC, Lee AS, Fischer DK, Van Kempen TA, Mudragel V, Huganir RL, Milner TA, Glass MJ, & Rajadhyaksha AM (2017). Extinction of Contextual Cocaine Memories Requires Cav1.2 within D1R-Expressing Cells and Recruits Hippocampal Cav1.2-Dependent Signaling Mechanisms. J Neurosci, 37, 11894–11911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (2011). Voltage-Gated Calcium Channels. Cold Spring Harbor Perspectives in Biology, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Lenaeus MJ, & Gamal El-Din TM (2020). Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu Rev Pharmacol Toxicol, 60, 133–154. [DOI] [PubMed] [Google Scholar]
- Chandy KG, & Gutman GA (1993). Nomenclature for mammalian potassium channel genes. Trends Pharmacol Sci, 14, 434. [DOI] [PubMed] [Google Scholar]
- Chen KC, Blalock EM, Thibault O, Kaminker P, & Landfield PW (2000). Expression of α1D subunit mRNA is correlated with L-type Ca2+ channel activity in single neurons of hippocampal “zipper” slices. Proc Natl Acad Sci U S A, 97, 4357–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Benninger F, & Yaari Y (2014). Role of small conductance Ca(2)(+)-activated K(+) channels in controlling CA1 pyramidal cell excitability. J Neurosci, 34, 8219–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE (2007). Calcium signaling. Cell, 131, 1047–1058. [DOI] [PubMed] [Google Scholar]
- Cohen-Matsliah SI, Motanis H, Rosenblum K, & Barkai E (2010). A novel role for protein synthesis in long-term neuronal plasticity: maintaining reduced postburst afterhyperpolarization. J Neurosci, 30, 4338–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Lo Turco JJ, Kubota M, Disterhoft JF, Moore JW, & Alkon DL (1989). Classical conditioning reduces amplitude and duration of calcium-dependent afterhyperpolarization in rabbit hippocampal pyramidal cells. J Neurophysiol, 61, 971–981. [DOI] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics C (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet, 381, 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, & Hell JW (2003). Increased phosphorylation of the neuronal L-type Ca2+ channel CaV1.2 during aging. Proc Natl Acad Sci U S A, 100, 16018–16023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Singla S, Toda H, Monje M, Palmer TD, & Malenka RC (2004). Excitation-Neurogenesis Coupling in Adult Neural Stem/Progenitor Cells. Neuron, 42, 535–552. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Coulter DA, & Alkon DL (1986). Conditioning-specific membrane changes of rabbit hippocampal neurons measured in vitro. Proc Natl Acad Sci U S A, 83, 2733–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disterhoft JF, Golden DT, Read HL, Coulter DA, & Alkon DL (1988). AHP reductions in rabbit hippocampal neurons during conditioning correlate with acquisition of the learned response. Brain Res, 462, 118–125. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Thompson LT, Moyer JR Jr., & Mogul DJ (1996). Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus. Life Sciences, 59, 413–420. [DOI] [PubMed] [Google Scholar]
- Dolphin AC (2016). Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol, 594, 5369–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, & Catterall WA (2000). Nomenclature of Voltage-Gated Calcium Channels. Neuron, 25, 533–535. [DOI] [PubMed] [Google Scholar]
- Faber ES, & Sah P (2002). Physiological role of calcium-activated potassium currents in the rat lateral amygdala. Journal of Neuroscience, 22, 1618–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, & Sah P (2003). Calcium-activated potassium channels: multiple contributions to neuronal function. Neuroscientist, 9, 181–194. [DOI] [PubMed] [Google Scholar]
- Gamelli AE, McKinney BC, White JA, & Murphy GG (2011). Deletion of the L-type calcium channel CaV1.3 but not CaV1.2 results in a diminished sAHP in mouse CA1 pyramidal neurons. Hippocampus, 21, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Sama MM, Landfield PW, & Thibault O (2006). Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci, 26, 3482–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Storm JF, Reuter D, Fedorov NB, Shao L-R, Leicher T, Pongs O, & Silva AJ (1998). Reduced K+ channel inactivation, spike broadening, and after-hyperpolarization in Kvß1.1-deficient mice with impaired learning. Learning and Memory, 5, 257–273. [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, & Wilson CJ (2005). Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium-activated potassium currents in striatal cholinergic interneurons. J Neurosci, 25, 10230–10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Hu H, Vervaeke K, & Storm JF (2008). SK (KCa2) channels do not control somatic excitability in CA1 pyramidal neurons but can be activated by dendritic excitatory synapses and regulate their impact. J Neurophysiol, 100, 2589–2604. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, & Storm JF (2005). Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol (Lond), 566, 689–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Dasari S, Onoue K, Stephens EK, Hasse JM, & Avesar D (2013). A sodium-pump-mediated afterhyperpolarization in pyramidal neurons. J Neurosci, 33, 13025–13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson B, & Wigstrom H (1981). Evidence for two types of afterhyperpolarization in CA1 pyramidal cells in the hippocampus. Brain Res, 206, 462–468. [DOI] [PubMed] [Google Scholar]
- Hell J, Westenbroek R, Warner C, Ahlijanian M, Prystay W, Gilbert M, Snutch T, & Catterall W (1993). Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. Journal of Cell Biology, 123, 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton TD, Xu W, & Lipscombe D (2005). Neuronal L-type calcium channels open quickly and are inhibited slowly. Journal of Neuroscience, 25, 10247–10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemond P, & Jaffe DB (2005). Caloric restriction prevents aging-associated changes in spike-mediated Ca2+ accumulation and the slow afterhyperpolarization in hippocampal CA1 pyramidal neurons. Neuroscience, 135, 413–420. [DOI] [PubMed] [Google Scholar]
- Hotson JR, & Prince DA (1980). A calcium-activated hyperpolarization follows repetitive firing in hippocampal neurons. Journal of Neurophysiology, 43, 409–419. [DOI] [PubMed] [Google Scholar]
- Kaczorowski CC, Disterhoft J, & Spruston N (2007). Stability and plasticity of intrinsic membrane properties in hippocampal CA1 pyramidal neurons: effects of internal anions. Journal of Physiology, 578, 799–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski CC, & Disterhoft JF (2009). Memory deficits are associated with impaired ability to modulate neuronal excitability in middle-aged mice. Learn Mem, 16, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Park YR, Lee B, Choi B, Kim H, & Kim CH (2017). Reduction of Cav1.3 channels in dorsal hippocampus impairs the development of dentate gyrus newborn neurons and hippocampal-dependent memory tasks. PLoS One, 12, e0181138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B, Rizwan AP, Asmara H, Heath NC, Engbers JD, Dykstra S, Bartoletti TM, Hameed S, Zamponi GW, & Turner RW (2015). IKCa channels are a critical determinant of the slow AHP in CA1 pyramidal neurons. Cell Rep, 11, 175–182. [DOI] [PubMed] [Google Scholar]
- Krueger JN, Moore SJ, Parent R, McKinney BC, Lee A, & Murphy GG (2017). A novel mouse model of the aged brain: Over-expression of the L-type voltage-gated calcium channel CaV1.3. Behavioural brain research, 322, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, & Foster TC (2004). Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. J Neurophysiol, 91, 2437–2444. [DOI] [PubMed] [Google Scholar]
- Landfield PW, & Pitler TA (1984). Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science, 226, 1089–1092. [DOI] [PubMed] [Google Scholar]
- Lawlor B, Segurado R, Kennelly S, Olde Rikkert MGM, Howard R, Pasquier F, Borjesson-Hanson A, Tsolaki M, Lucca U, Molloy DW, Coen R, Riepe MW, Kalman J, Kenny RA, Cregg F, O’Dwyer S, Walsh C, Adams J, Banzi R, Breuilh L, Daly L, Hendrix S, Aisen P, Gaynor S, Sheikhi A, Taekema DG, Verhey FR, Nemni R, Nobili F, Franceschi M, Frisoni G, Zanetti O, Konsta A, Anastasios O, Nenopoulou S, Tsolaki-Tagaraki F, Pakaski M, Dereeper O, de la Sayette V, Senechal O, Lavenu I, Devendeville A, Calais G, Crawford F, Mullan M, & Group NS (2018). Nilvadipine in mild to moderate Alzheimer disease: A randomised controlled trial. PLoS Med, 15, e1002660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, De Jesus-Cortes H, Kabir ZD, Knobbe W, Orr M, Burgdorf C, Huntington P, McDaniel L, Britt JK, Hoffmann F, Brat DJ, Rajadhyaksha AM, & Pieper AA (2016). The Neuropsychiatric Disease-Associated Gene cacna1c Mediates Survival of Young Hippocampal Neurons. eNeuro, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima PA, & Marrion NV (2007). Mechanisms underlying activation of the slow AHP in rat hippocampal neurons. Brain Research. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Helton TD, & Xu W (2004). L-Type Calcium Channels: The Low Down. J Neurophysiol, 92, 2633–2641. [DOI] [PubMed] [Google Scholar]
- Liss B, & Striessnig J (2019). The Potential of L-Type Calcium Channels as a Drug Target for Neuroprotective Therapy in Parkinson’s Disease. Annu Rev Pharmacol Toxicol, 59, 263–289. [DOI] [PubMed] [Google Scholar]
- Llinas RR, Sugimori M, & Cherksey B (1989). Voltage-dependent calcium conductances in mammalian neurons. The P channel. Ann N Y Acad Sci, 560, 103–111. [DOI] [PubMed] [Google Scholar]
- Marschallinger J, Sah A, Schmuckermair C, Unger M, Rotheneichner P, Kharitonova M, Waclawiczek A, Gerner P, Jaksch-Bogensperger H, Berger S, Striessnig J, Singewald N, Couillard-Despres S, & Aigner L (2015). The L-type calcium channel Cav1.3 is required for proper hippocampal neurogenesis and cognitive functions. Cell Calcium, 58, 606–616. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Linardakis JM, & Disterhoft JF (2009). The fast and slow afterhyperpolarizations are differentially modulated in hippocampal neurons by aging and learning. J Neurosci, 29, 4750–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack K, McCormack T, Tanouye M, Rudy B, & Stuhmer W (1995). Alternative splicing of the human Shaker K+ channel beta 1 gene and functional expression of the beta 2 gene product. FEBS Lett, 370, 32–36. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, & Adams ME (1992). P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature, 355, 827–829. [DOI] [PubMed] [Google Scholar]
- Moore S, Slater N, & Murphy G The role of L-type voltage-gated calcium channels in age-related cognitive decline. Program No. 919.01. 2012 Neuroscience Meeting Planner. New Orleans, LA: Society for Neuroscience, 2012. Online. [Google Scholar]
- Moore SJ, Throesch BT, & Murphy GG (2011). Of mice and intrinsic excitability: genetic background affects the size of the postburst afterhyperpolarization in CA1 pyramidal neurons. Journal of Neurophysiology, 106, 1570–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR Jr., Power JM, Thompson LT, & Disterhoft JF (2000). Increased excitability of aged rabbit CA1 neurons after trace eyeblink conditioning. J Neurosci, 20, 5476–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR Jr., Thompson LT, Black JP, & Disterhoft JF (1992). Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. Journal of Neurophysiology, 68, 2100–2109. [DOI] [PubMed] [Google Scholar]
- Moyer JR Jr., Thompson LT, & Disterhoft JF (1996). Trace eyeblink conditioning increases CA1 excitability in a transient and learning-specific manner. J Neurosci, 16, 5536–5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy GG, Fedorov NB, Giese KP, Ohno M, Friedman E, Chen R, & Silva AJ (2004). Increased neuronal excitability, synaptic plasticity, and learning in aged Kvβ1.1 knockout mice. Current Biology, 14, 1907–1915. [DOI] [PubMed] [Google Scholar]
- Murphy GG, Rahnama NP, & Silva AJ (2006). Investigation of Age-Related Cognitive Decline Using Mice as a Model System: Behavioral Correlates. The American Journal of Geriatric Psychiatry, 14, 1004–1011. [DOI] [PubMed] [Google Scholar]
- Murphy GG, Shah V, Hell JW, & Silva AJ (2005). Investigation of age-related cognitive decline using mice as a model system: neurophysiological correlates. Am J Geriatr Psychiatry. [DOI] [PubMed] [Google Scholar]
- Nunez-Santana FL, Oh MM, Antion MD, Lee A, Hell JW, & Disterhoft JF (2014). Surface L-type Ca2+ channel expression levels are increased in aged hippocampus. Aging Cell, 13, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkowski JJ, & Murphy GG (2011). Deletion of the mouse homolog of KCNAB2, a gene linked to monosomy 1p36, results in associative memory impairments and amygdala hyperexcitability. Journal of Neuroscience, 31, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, & Landfield PW (1990). Aging-related prolongation of calcium spike duration in rat hippocampal slice neurons. Brain Research, 508, 1–6. [DOI] [PubMed] [Google Scholar]
- Power JM, Bocklisch C, Curby P, & Sah P (2011). Location and function of the slow afterhyperpolarization channels in the basolateral amygdala. J Neurosci, 31, 526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Wu WW, Sametsky E, Oh MM, & Disterhoft JF (2002). Age-related enhancement of the slow outward calcium-activated potassium current in hippocampal CA1 pyramidal neurons in vitro. Journal of Neuroscience, 22, 7234–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall A, & Tsien RW (1995). Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci, 15, 2995–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes KJ, & Trimmer JS (2006). Antibodies as valuable neuroscience research tools versus reagents of mass distraction. J Neurosci, 26, 8017–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saar D, Grossman Y, & Barkai E (1998). Reduced after-hyperpolarization in rat piriform cortex pyramidal neurons is associated with increased learning capability during operant conditioning. European Journal of Neuroscience, 10, 1518–1523. [DOI] [PubMed] [Google Scholar]
- Sah P (1996). Ca2+-activated K+ currents in neurones: types, physiological roles and modulation. Trends in Neurosciences, 19, 150–154. [DOI] [PubMed] [Google Scholar]
- Sah P, & Faber ES (2002). Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol, 66, 345–353. [DOI] [PubMed] [Google Scholar]
- Sahu G, Wazen RM, Colarusso P, Chen SRW, Zamponi GW, & Turner RW (2019). Junctophilin Proteins Tether a Cav1-RyR2-KCa3.1 Tripartite Complex to Regulate Neuronal Excitability. Cell Rep, 28, 2427–2442 e2426. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, & Crill WE (1988). Influence of anomalous rectifier activation on afterhyperpolarizations of neurons from cat sensorimotor cortex in vitro. J Neurophysiol, 59, 468–481. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Foehring RC, Chubb MC, & Crill WE (1988). Slow conductances in neurons from cat sensorimotor cortex in vitro and their role in slow excitability changes. J Neurophysiol, 59, 450–467. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Foehring RC, Stafstrom CE, Chubb MC, & Crill WE (1988). Multiple potassium conductances and their functions in neurons from cat sensorimotor cortex in vitro. J Neurophysiol, 59, 424–449. [DOI] [PubMed] [Google Scholar]
- Sehgal M, Ehlers VL, & Moyer JR Jr. (2014). Learning enhances intrinsic excitability in a subset of lateral amygdala neurons. Learn Mem, 21, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snutch TP, Leonard JP, Gilbert MM, Lester HA, & Davidson N (1990). Rat brain expresses a heterogeneous family of calcium channels. Proc Natl Acad Sci U S A, 87, 3391–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, & Tzounopoulos T (2002). Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. Journal of Neuroscience, 22, 10163–10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M (2004). Ca2+-Activated K+ channels:Molecular determinants and fuction of the SK family. Nat Rev Neurosci, 5, 758–770. [DOI] [PubMed] [Google Scholar]
- Stocker M, Krause M, & Pedarzani P (1999). An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proceedings of the National Academy of Sciences of the United States of America, 96, 4662–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme SJ, Bell RZ, Fisher GL, & Murphy GG (2016). Deletion of the Mouse Homolog of CACNA1C Disrupts Discrete Forms of Hippocampal-Dependent Memory and Neurogenesis within the Dentate Gyrus. eNeuro, 3, ENEURO.0118–0116.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Hadley R, & Landfield PW (2001). Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci, 21, 9744–9756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, & Landfield PW (1996). Increase in single L-type calcium channels in hippocampal neurons during aging. Science, 272, 1017–1020. [DOI] [PubMed] [Google Scholar]
- Tiwari MN, Mohan S, Biala Y, & Yaari Y (2018). Differential contributions of Ca(2+) - activated K(+) channels and Na(+) /K(+) -ATPases to the generation of the slow afterhyperpolarization in CA1 pyramidal cells. Hippocampus, 28, 338–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, & Rose GM (2005). The slow afterhyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. J. Neurosci, 25, 2609–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RW, Lipscombe D, Madison DV, Bley KR, & Fox AP (1988). Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci, 11, 431–438. [DOI] [PubMed] [Google Scholar]
- Tuncdemir SN, Lacefield CO, & Hen R (2019). Contributions of adult neurogenesis to dentate gyrus network activity and computations. Behav Brain Res, 374, 112112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RW, Asmara H, Engbers JD, Miclat J, Rizwan AP, Sahu G, & Zamponi GW (2016). Assessing the role of IKCa channels in generating the sAHP of CA1 hippocampal pyramidal cells. Channels (Austin), 10, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veng LM, & Browning MD (2002). Regionally selective alterations in expression of the α1D subunit (CaV1.3) of L-type calcium channels in the hippocampus of aged rats. Brain Res Mol Brain Res, 107, 120–127. [DOI] [PubMed] [Google Scholar]
- Veng LM, Mesches MH, & Browning MD (2003). Age-related working memory impairment is correlated with increases in the L-type calcium channel protein α1D (CaV1.3) in area CA1 of the hippocampus and both are ameliorated by chronic nimodipine treatment. Brain Res Mol Brain Res, 110, 193–202. [DOI] [PubMed] [Google Scholar]
- Volkening B, Schonig K, Kronenberg G, Bartsch D, & Weber T (2017). Deletion of psychiatric risk gene Cacna1c impairs hippocampal neurogenesis in cell-autonomous fashion. Glia, 65, 817–827. [DOI] [PubMed] [Google Scholar]
- Wang K, Mateos-Aparicio P, Honigsperger C, Raghuram V, Wu WW, Ridder MC, Sah P, Maylie J, Storm JF, & Adelman JP (2016). IK1 channels do not contribute to the slow afterhyperpolarization in pyramidal neurons. Elife, 5, e11206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, & Lipscombe D (2001). Neuronal CaV1.3 α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. Journal of Neuroscience, 21, 5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Weiner JL, Valiante TA, Velumian AA, Watson PL, Jahromi SS, Schertzer S, Pennefather P, & Carlen PL (1994). Whole-cell recording of the Ca(2+)-dependent slow afterhyperpolarization in hippocampal neurones: effects of internally applied anions. Pflugers Arch, 426, 247–253. [DOI] [PubMed] [Google Scholar]