

Abstract
A common way to measure the induction of autophagy is to compare the amount of Atg8/LC3-I with that of Atg8-PE/LC3-II using western blot analysis (Kabeya et al. 2000; Mizushima and Yoshimori 2007). This is because changes in the amount of LC3-II are closely associated with changes in the number of autophagosomes present in a cell (Kabeya et al. 2000). Atg8/LC3 is initially synthesized as an unprocessed form, which is proteolytically processed by Atg4 to form Atg8/LC3-I, and then modified into the phosphatidylethanolamine (PE)-conjugated Atg8-PE/LC3-II form (Kabeya et al. 2000; Kirisako et al. 2000). Atg8/LC3-II is the membrane bound form of Atg8/LC3, while Atg8-PE/LC3-I is cytosolic (Kabeya et al. 2000). Atg8-PE/LC3-II associates with both the inner and outer membrane of the autophagosome, thus Atg8-PE/LC3-II is the only autophagy reporter that is reliably associated with completed autophagosomes (Klionsky 2012).
As with mammalian LC3 and yeast Atg8, the C-terminus of LGG-1 appears to be conjugated to PE, with LGG-1-I and LGG-1-II being visible on a western blot as one major band and minor band, respectively (Kang et al. 2007; Alberti et al. 2010; Tian et al. 2010). LGG-2 contains two minor bands as opposed to one; yet it is not clearly defined which minor band represents the lipidated form of LGG-2 (Alberti et al. 2010). Under normal non-stress conditions, the protein levels of LGG-1-I are higher than that of LGG-1-II. Under conditions that induce autophagy, LGG-1-I levels still appear higher than that of LGG-1-II; however, an overall increase in the amount of LGG-1-II is apparent (Kang et al. 2007; Alberti et al. 2010). A similar increase is also found for LGG-2 protein levels (Alberti et al. 2010). Therefore, changes in LGG-1-II and LGG-2 protein levels may be used to monitor autophagy activity in C. elegans.
Although Atg8-PE/LC3-II is reliably associated with the autophagosome, its protein levels may not change in a predictable manner upon autophagy induction (Mizushima and Yoshimori 2007; Klionsky 2012). For example, upon autophagy induction in mammalian cells, the total levels of LC3 may not change; instead an increase in the conversion of LC3-I to LC3-II, or a decrease in the level of LC3-II relative to that of LC3-I, may result. The decrease in LC3-II can be due to the rapid lysosomal degradation of LC3-II (Huang et al. 2000; Klionsky 2012).
Therefore, although changes in the protein levels of LGG-1 can be used to monitor autophagy, caution should be used when evaluating such changes.
Materials:
Reagents:
Bleach Solution <R>:
ECL Detection Kit
M9 Minimal Medium Buffer <R>:
Ponceau S solution <R>:
Anti-GFP and/or anti-LGG-1Primary Antibodies
Secondary Antibody
Protease inhibitor cocktail
1X SDS gel-loading buffer <R>:
10% SDS polyacrylamide gel (20% gel) <R>:
5% Stacking gel <R>:
1X Transfer Buffer <R>:
10X Tris-Buffered Saline (TBS) <R>:
TBST blocking buffer <R>:
Tris-Buffered Saline, 0.1% Tween-20 (TBST) <R>:
1X Tris-glycine electrophoresis buffer (running buffer) <R>:
Equipment:
Fiber pads and Filter paper
Gel electrophoresis cassette and power supply
Gel Transfer cell and sandwich cassette
Nitrocellulose or PVDF membrane
Method:
Note: The following Protocol has been adapted from Molecular Cloning: A Laboratory Manual Sambrook and Russell
Sample Preparation:
-
1
Wash L4 larvae or young adult hermaphrodites from feeding plates using 1 mL of M9 buffer and transfer animals to a 1.5 mL microcentrifuge tube. Rinse worms several times with M9 buffer and centrifuge worms for 1 minute at 2000 rpm in between washes. For embryo preparation, collect gravid adults by washing plates with sterile water. In a sterile 15mL conical centrifuge tube collect worms with bleach solution (should be made fresh every time). Shake well or vortex tube for a few seconds, and repeat a few times, for not longer than 10 min. Adults will dissolve while embryos remain intact. Wash embryos several times with M9 buffer and centrifuge in between washes at 2000 rpm for 1 minute. After the final wash, remove the supernatant leaving only a pellet of embryos and continue with step 2 below.
-
2
After washing, centrifuge animals for 1 minute at 2000 rpm and remove supernatant leaving a pellet of animals. “Snap freeze” the animals in liquid nitrogen and add an equal volume of 1X SDS gel-loading buffer containing protease inhibitors to the sample and boil at 100°C for 3–10 minutes. It is important to also prepare marker proteins of known molecular weight for control purposes.
-
3
After boiling, spin down samples and cool on ice for 5 minutes. Load the samples onto the SDS polyacrylamide gel.
SDS-PAGE Preparation (modified from Sambrook and Russel, 2001):
-
4
Assemble the polyacrylamide gel apparatus by inserting two glass plates into the gel caster as described by the manufacturer.
-
5
Using an Erlenmeyer flask, prepare a 12%−20% polyacrylamide resolving gel. Swirl the mixture rapidly and pour it into the gap between the glass plates. Be sure to leave enough space for the stacking gel. Overlay the polyacrylamide gel with isopropanol. Allow the gel to polymerize for ~30 minutes. Pour the resolving gel immediately after adding TEMED.
-
6
Once polymerization is complete, pour off the isopropanol and rinse the top of the resolving gel a few times with deionized water. Remove all residual isopropanol and water.
-
7
Prepare a 5% stacking gel and pour the stacking gel solution onto the polymerized resolving gel. Carefully insert a clean gel comb into the stacking gel solution by avoiding air bubbles. Allow the stacking gel to polymerize at room temperature. Polymerization of the stacking gel will begin once TEMED is added, so the pouring step has to be done quickly.
-
8
After polymerization is complete, remove the gel comb and wash all wells with running buffer to remove any unpolymerized acrylamide. Mount the gel in the electrophoresis apparatus and add running buffer to the top and bottom reservoirs of the apparatus. Load ~15 uL of sample into each well, using gel-loading tips. One can add a higher volume on to the wells using a pipette with longer tips.
-
9
Attach the electrophoresis apparatus to the power supply as described by the manufacturer’s instructions. Run the gel at ~180 to 200 volts or until the bromophenol blue dye in the sample buffer reaches the bottom of the gel.
-
10
Once electrophoresis is complete, gently remove the glass plates from the gel apparatus and carefully release the polyacrylamide gel.
Membrane Transfer (Modified from Bio-Rad Laboratories Mini-Trans-Blot instruction manual):
-
11
Cool 1X transfer buffer at 4°C. Cut the nitrocellulose membrane (or PVDF membrane) and filter paper to the dimensions of the polyacrylamide gel. Soak the membrane, filter paper, and fiber pad in transfer buffer until the Western blotting preparation is ready to be made.
-
12
Prepare the Western blot sandwich in the following order: Start with the clear side of the case, followed by the sponge pad, Whatman filter paper, membrane, gel, Whatman filter paper, sponge pad, and the black side of the cassette. Close the cassette firmly and ensure that all bubbles are removed from the sandwich.
-
13
Place the transfer cassette in the transfer cell with the black side facing black, and fill the cell with cooled 1X transfer buffer. Transfer for 1h, at 4°C and ~90 volts. Make sure that the cassette is positioned in the correct direction so that the proteins in the gel transfer to the membrane. The voltage required may vary according to the manufacturer’s instructions.
Immunoblotting (Modified from (You et al. 2006)):
-
14
Once transfer is complete, remove the membrane from the transfer cassette and incubate the membrane in TBST blocking buffer (which contains 5% non-fat dry milk) for ~1 hour at room temperature. This can be done in a plastic bag or a Tupperware dish that can be placed on a shaker. Ponceau S stain can be administered to the membrane to check for successful transfer of proteins before blocking. Prepare the 1X Ponceau S solution as described by the manufacturer and incubate the blot for 1 to 5 minutes in the solution on a rocker. Rinse with distilled water to rid the blot of stain, until protein bands are clearly visible. Wrap blot with plastic wrap and take a picture or a Xerox. Keep the membrane from drying. After Ponceau S staining is completed, continue with the blocking step 14.
-
15
Dilute the primary antibody, (i.e. anti-LGG-1 (Tian et al. 2010) or anti-GFP (Kang et al. 2007; Alberti et al. 2010), in 2ml TBST blocking buffer (which contains 5% non-fat dry milk) at the appropriate concentration and incubate the membrane in a Ziploc bag overnight at 4°C, placed on a rocker. For primary antibodies, use the dilution factor as suggested by the manufacturer; anti-GFP from Roche has been commonly used at a 1:500 dilution ((Alberti et al. 2010; Djeddi et al. 2012). As antibodies vary, and the protocols may vary, determine the concentration of the primary antibody empirically before the start of the experiment.
-
16
Remove the membrane from the Ziploc bag and wash it three times with ~50–100 mL of TBST for 5 minutes each, in a small Tupperware on a shaker.
-
17
Incubate the membrane with the secondary antibody at the correct concentration (as directed by the antibody manufacturer, or previously described (You et al. 2006; Alberti et al. 2010), for 1 hour at room temperature, in a Ziploc bag, on a rocker.
-
18
Rinse the membrane three times with ~50–100 mL of TBST blocking buffer in 10 minute intervals. Then rinse with distilled water once. After the last wash, use an ECL detection kit as instructed by the manufacturer to visualize the protein bands.
Data Analysis:
Western blot analysis provides a convenient way to measure any changes in the levels of the different forms of LGG-1; however, caution is advised when interpreting the quantity of LGG-1-II using western blot analysis.
A direct way to measure changes in overall LGG-1-II levels is through quantification, by comparing the protein levels of LGG-1-II with the protein levels of a housekeeping gene (i.e. tubulin) or with LGG-1-I (Kang et al. 2007; Michelet et al. 2009; Alberti et al. 2010; Barth et al. 2010). In addition, it is important to use appropriate standardization controls to ensure equal loading between samples, as this may change the amount of LGG-1-I and LGG-1-II protein between samples (Klionsky 2012). Furthermore, the stress condition of the animals prior to experimental manipulation should be at a minimum, to ensure unaltered levels of LGG-1-I and LGG-1-II protein at the start of an experiment.
Increased levels of LGG-1-II relative to LGG-1-I can reflect autophagosome accumulation, due to increased autophagy, or an accumulation of autophagosomes, as a result of defective lysosomal degradation (Michelet et al. 2009; Alberti et al. 2010; Barth et al. 2010; Lu et al. 2011; Klionsky 2012). Alternatively, based on mammalian studies, lower levels of LGG-1-II compared to LGG-1-I can represent defective autophagy, as a result of poor LGG-1-I to LGG-1-II conversion, or increased autophagic flux, resulting in the rapid degradation of LGG-1-II (Mizushima and Yoshimori 2007). The use of lysosomal inhibitors can be one way to distinguish between all these possibilities (Oka and Futai 2000; Ji et al. 2006; Mizushima and Yoshimori 2007; Pivtoraiko et al. 2010).
Alternatively, protein extracts isolated from mutant animals previously shown to alter the lipidation of LGG-1 can also be useful as positive and/or negative controls in blots that measure changes in LGG-1 protein levels. gpb-2 is a G-protein β subunit involved in the muscarinic signaling pathway, and gpb-2 mutants, following starvation, have elevated levels of autophagy in pharyngeal muscles, visualized by the expression of GFP::LGG-1, and also have a higher ratio of lipidated LGG-1 to non-lipidated LGG-1, compared to wild-type controls (Kang et al. 2007). lgg-2 mutants, which have defects in the acidification and degradation of autophagosomes, result in elevated levels of both lipidated and non-lipidated forms of LGG-1 (Manil-Segalen et al. 2014b). In addition, protein extracts isolated from animals fed dsRNA against the C. elegans ortholog of TOR, let-363 (Long et al. 2002), and rab-7, the small GTPase involved in endosome/ lysosomal fusion events (Bucci et al. 2000), can also be used as controls to monitor changes in the levels of the different forms of LGG-1 protein (Alberti et al. 2010). RNAi against let-363 induces autophagy, observed by elevated levels of GFP::LGG-1 in the hypodermis and intestine, and was reported to display a decrease in the levels of non-lipidated LGG-1, but an increase in the levels of lipidated LGG-1, compared to empty vector controls. On the other hand, RNAi against rab-7, which leads to increased levels of GFP::LGG-1 as a result of defective lysosomal fusion, resulted in elevated levels of both lipidated and non-lipidated LGG-1, compared to controls (Alberti et al. 2010).
It is important to note that the loss of certain autophagy genes can inhibit autophagy without affecting LC3-II/Atg8-PE formation (Klionsky 2012), which appears to be also true for LGG-1-II in C. elegans (Tian et al. 2010; Lu et al. 2011; Liang et al. 2012). Therefore, additional methods may be required to determine if autophagy is functional when observing changes in the protein levels of LGG-1.
Additionally, LGG-1-II levels can be influenced by the type of anti-LGG-1 antibodies used, as well as the type of membrane used during protein transfer (Barth et al. 2010; Klionsky 2012). In C. elegans, experiments that visualize the lipidated and non-lipidated forms of LGG-1 by western blotting may utilize anti-GFP or anti-LGG-1 primary antibodies, nitrocellulose membranes, as well as protein samples from larvae and embryos (Kang et al. 2007; Alberti et al. 2010; Tian et al. 2010).
In mammalian cells, during the initial periods of starvation, the amount of LC3-I may be inversely proportional to that of LC3-II; however, as the starvation period is prolonged, the levels of both LC3-I and LC3-II have been shown to decrease (Mizushima and Yoshimori 2007). Although this has not been fully examined for LGG-1 in C. elegans, one should consider the appropriate length of time that animals are exposed to starvation. Lastly, LGG-1 has been shown to localize to phagosomes in cells that engulf and degrade apoptotic cells in C. elegans (Li et al. 2012). Thus, conditions that induce apoptosis should be at a minimum when evaluating levels of LGG-1-II during autophagy
Overall, a way to measure autophagy induction is to analyze the levels of LGG-1-II and compare them to the levels of LGG-1-I; however, several considerations should be made to ensure the proper interpretation of results.
Troubleshooting (Modified from Abcam site (url: http://www.abcam.com/index.html?pageconfig=resource&rid=11352):
Problem: No signal detected on membrane (Step 17)
Solution: Consider the following:
Lysate preparation was not carried out correctly.
Incorrect primary or secondary antibody was used or not enough primary or secondary antibodies were used
ECL detection kit was not used properly.
Poor transfer to membrane. See solution below.
Problem: High background intensity on membrane (Step 17)
Solution: Consider the following:
Concentration of primary and secondary antibodies is too high. Dilute and repeat.
Not enough washes were done to remove unbound antibodies in step 17. Include more washes and repeat.
Cross-reactivity between blocking buffer and primary and/or secondary antibodies. Substituting non-fat dry milk with BSA may circumvent this issue.
Problem: Poor transfer of protein to membrane (Step 11–13)
Solution: Consider the following:
Transfer buffer was prepared incorrectly.
Transfer Cassette and sandwich were not setup properly according to the manufacturer’s instruction
If a PDVF membrane was used, pre-soak in methanol before use
Discussion:
Western blot analysis may be used to monitor autophagy by evaluating the levels of lipidated and non-lipidated Atg8/LC3/LGG-1. However, an increase in the lipidated form of Atg8/LC3/LGG-1-II can reflect the induction of autophagy and/or inhibition of autophagy, and is therefore not a direct measure of autophagic flux without the use of additional methods (Klionsky 2012). In mammalian cells, the accumulation of LC3-II can result from an increase in autophagy activity or defective lysosomal degradation (Mizushima and Yoshimori 2007). The addition of lysosomal protease inhibitors, the introduction of a mutation, or RNAi treatment that results in defective lysosomal degradation, may differentiate between an increase in autophagy or the reduction of autophagic function. As described by Mizushima and Yoshimori, an additional increase in LGG-1-II levels, observed under conditions that block the fusion between autophagosomes and lysosomes, would be indicative of autophagy induction (Mizushima and Yoshimori 2007). In contrast, no change in LGG-1-II levels after treatment with agents or RNAi that decrease lysosomal degradation would indicate a block in the autophagic pathway. In conclusion, with careful interpretation of results, changes in the protein levels of LGG-1 can provide a way to measure changes in autophagy activity.
Recipes:
Bleach Solution (should be made fresh each time):
3.5 mL sterile H2O
0.5 mL 5N NaOH
1 mL of household bleach (sodium hypochlorite)
M9 Minimal Medium Buffer:
22mM KH2PO4
22mM Na2HPO4
85mM NaCl
1mM MgSO4
Autoclave for 15 minutes on liquid cycle
Ponceau S solution:
2g Ponceau S
30g trichloracetic acid
30g sulfosalicyclic acid
H2O to 100 ml
1X SDS gel-loading buffer:
50mM Tris-Cl (pH 6.8)
100mM dithiothreitol
2% (w/v) SDS
0.1% bromophenol blue
10% (v/v) glycerol
10% SDS polyacrylamide gel (20% gel):
30% acrylamide mix
1.5M Tris (pH 8.8)
10% SDS
10% ammonium sulfate
TEMED
5% Stacking gel:
30% acrylamide mix
1.5M Tris (pH 8.8)
10% SDS
10% ammonium sulfate
TEMED
1X Transfer Buffer:
25mM Tris
192mM glycine
20% (v/v) methanol (or ethanol)
pH 8.3
10X Tris-Buffered Saline (TBS):
24.2g Tris Base
80g NaCl
pH 7.6
Add distilled water to final volume of 1L
TBST blocking buffer:
1X TBS
0.1% Tween-20
5% w/v non-fat dry milk
Tris-Buffered Saline, 0.1% Tween-20 (TBST):
1X TBS
0.1% Tween-20
1X Tris-glycine electrophoresis buffer (running buffer):
25mM Tris Base
192mM glycine
0.1% SDS
pH 8.8
FIGURE 1.
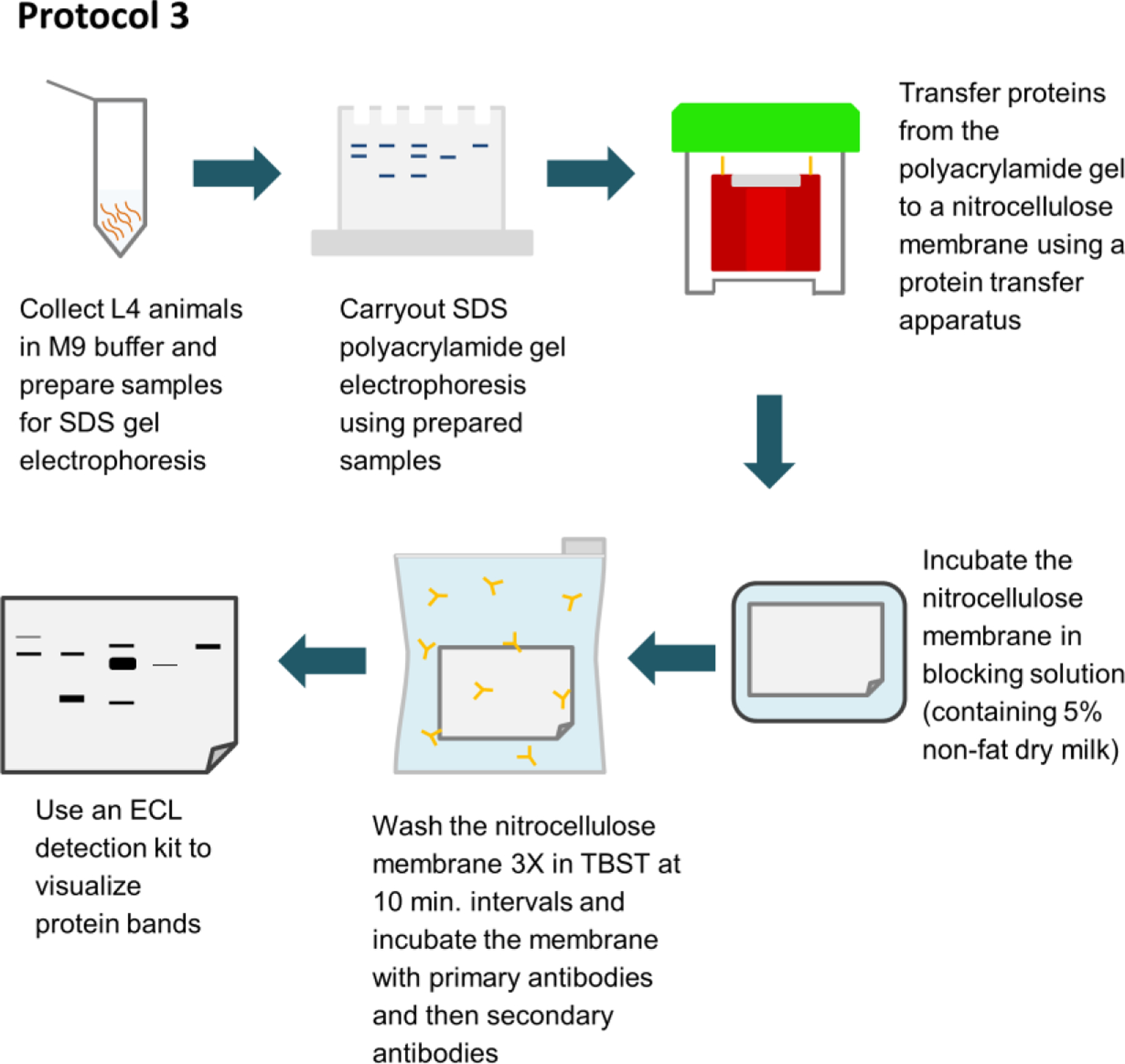
Flowchart summarizing a protocol for quantitating the levels of autophagy marker proteins by western blotting in the nematode Caenorhabditis elegans. ECL, enhanced chemiluminescence; L4, larval stage 4; TBST, Tris-buffered saline containing 0.1% Tween-20.
References
- Alberti A, Michelet X, Djeddi A, Legouis R. 2010. The autophagosomal protein LGG-2 acts synergistically with LGG-1 in dauer formation and longevity in C. elegans. Autophagy 6: 622–633. [DOI] [PubMed] [Google Scholar]
- Barth S, Glick D, Macleod KF. 2010. Autophagy: Assays and artifacts. J Pathol 221: 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bio-Rad Laboratories. Mini-Trans Blot Electrophoretic Transfer Cell Instruction Manual, Rev K. Bio-Rad Laboratories, Hercules, CA. [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. 2000. Rab7: A key to lysosome biogenesis. Mol Biol Cell 11: 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djeddi A, Michelet X, Culetto E, Alberti A, Barois N, Legouis R. 2012. Induction of autophagy in ESCRT mutants is an adaptive response for cell survival in C. elegans. J Cell Sci 125: 685–694. [DOI] [PubMed] [Google Scholar]
- Huang WP, Scott SV, Kim J, Klionsky DJ. 2000. The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J Biol Chem 275: 5845–5851. [DOI] [PubMed] [Google Scholar]
- Ji YJ, Choi KY, Song HO, Park BJ, Yu JR, Kagawa H, Song WK, Ahnn J. 2006. VHA-8, the E subunit of V-ATPase, is essential for pH homeostasis and larval development in C. elegans. FEBS Lett 580: 3161–3166. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, You YJ, Avery L. 2007. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev 21: 2161–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M, Takao T, Noda T, Ohsumi Y. 2000. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol 151: 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ. 2012. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8: 1–100.22082964 [Google Scholar]
- Liang Q, Yang P, Tian E, Han J, Zhang H. 2012. The C. elegans ATG101 homolog EPG-9 directly interacts with EPG-1/Atg13 and is essential for autophagy. Autophagy 8: 1426–1433. [DOI] [PubMed] [Google Scholar]
- Long X, Spycher C, Han ZS, Rose AM, Muller F, Avruch J. 2002. TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr Biol 12: 1448–1461. [DOI] [PubMed] [Google Scholar]
- Lu Q, Yang P, Huang X, Hu W, Guo B, Wu F, Lin L, Kovacs AL, Yu L, Zhang H. 2011. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell 21: 343–357. [DOI] [PubMed] [Google Scholar]
- Manil-Segalen M, Lefebvre C, Jenzer C, Trichet M, Boulogne C, Satiat-Jeunemaitre B, Legouis R. 2014. The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Dev Cell 28: 43–55. [DOI] [PubMed] [Google Scholar]
- Michelet X, Alberti A, Benkemoun L, Roudier N, Lefebvre C, Legouis R. 2009. The ESCRT-III protein CeVPS-32 is enriched in domains distinct from CeVPS-27 and CeVPS-23 at the endosomal membrane of epithelial cells. Biol Cell 101: 599–615. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. 2007. How to interpret LC3 immunoblotting. Autophagy 3: 542–545. [DOI] [PubMed] [Google Scholar]
- Oka T, Futai M. 2000. Requirement of V-ATPase for ovulation and embryogenesis in Caenorhabditis elegans. J Biol Chem 275: 29556–29561. [DOI] [PubMed] [Google Scholar]
- Pivtoraiko VN, Harrington AJ, Mader BJ, Luker AM, Caldwell GA, Caldwell KA, Roth KA, Shacka JJ. 2010. Low-dose bafilomycin attenuates neuronal cell death associated with autophagy-lysosome pathway dysfunction. J Neurochem 114: 1193–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. 2001. Molecular cloning: A laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, et al. 2010. C elegans screen identifies autophagy genes specific to multicellular organisms. Cell 141: 1042–1055. [DOI] [PubMed] [Google Scholar]
- You YJ, Kim J, Cobb M, Avery L. 2006. Starvation activates MAP kinase through the muscarinic acetylcholine pathway in Caenorhabditis elegans pharynx. Cell Metab 3: 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]