

Abstract
Maintaining health and well-being of the population is a universal priority. Governments around the globe are therefore seeking greater efficiency and better outcomes from researches being held. Although large randomized trials or systematic review of several large trials provides the highest level of evidence, the intricate cost, time, and difficulties of conventional trials have led to questions about their sustainability commanding search for alternative approaches. Demands for improved competences in medical research have led to mounting interest in newer clinical trial designs. This article provides an insight into newer clinical trial designs, including cluster trials, adaptive designs, the master protocols along with their strengths, weaknesses, and which trials design should be opted for in different clinical scenarios.
Keywords: Adaptive design, new clinical trial designs, pragmatic clinical trials, the master protocol
INTRODUCTION
Evidence-based medicine depends on the systematic accumulation of information about how different treatments affect patients. The process of experimental design translates the research question about a population of interest into a formal experiment or clinical trial protocol which incorporates patient eligibility requirements, detailed descriptions of the experimental and control interventions, as well as definition of objective, measurable outcomes.
Classical clinical trial design, i.e., randomized, double-blind, parallel groups are considered as the gold standard for ascertaining the efficacy of new treatments and are robust, easy to perform, and easy to interpret.[1] Nevertheless, several shortcomings have been noted, including higher cost, need for a large sample size and long study duration, lack of feasibility when sample size requirement far exceeds the available population (in rare diseases), lack of power to evaluate efficacy overall or in important subgroups. It also becomes ethically questionable if already available information is ignored while study is ongoing.
These limitations along with advances in statistical software and computing power have enabled the researchers to pursue more complex study designs and analytical techniques. Efficient alternatives that preserve protections against bias and confounding are thus of considerable interest. Among the most useful are the pragmatic trials conducted in “real-world” settings sometimes referred to as effectiveness trials. Researches using this method enroll a large number of participants with few exclusions usually resulting in patient centred outcomes with practical results applicable to clinical practice [Figure 1]. However, these novel trial designs should be adopted only when there are compelling reasons.[2]
Figure 1.
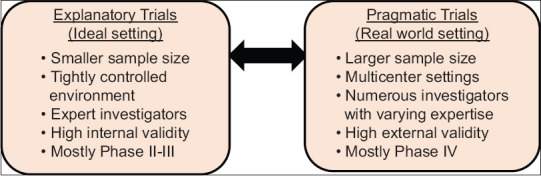
Comparison between explanatory trials and pragmatic trials
NEWER CLINICAL TRIAL DESIGNS
This entity encompasses various study designs which are summarized in the following text.
Cluster trials[2]
These are defined by the exposure being allocated to a “group” of subjects which may include entire hospitals, units within hospitals or even patients under care of a single physician. In each case, as all patients in a particular group are given similar treatment, the need for individual consent is usually waived off. Sample size estimation is complicated but depends essentially on the number of clusters. This design is especially useful for systematic changes such as employment of rapid response teams that cannot be allocated individually or easily reversed. There are numerous variations of cluster design [Figure 2].
Figure 2.
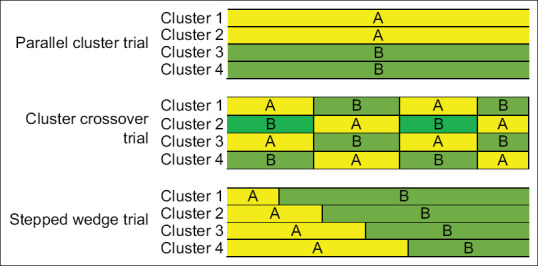
Cluster designs comparing treatment A (yellow) and treatment B (green)
Parallel cluster trials
In this, groups of patients are simultaneously randomly assigned to one of two or more exposures. It resembles conventional parallel-group randomized trials, except here randomization is on the basis of units rather than individuals. The advantages include reduction in the Hawthorne effect, i.e., improvements that results from focussing attention on a particular outcome and principal elimination of learned behavior as clinicians get little opportunity to acquire information from one treatment and apply that to other because each site or clinician is exposed to only one treatment at a time.
Cluster crossover trials (alternating or randomized)
Here, interventions are applied for a limited period after which they get removed from the trial for a comparable amount of time.
Alternating controlled cluster trials
Defining aspect is studying of entire hospital units and applying intervention of interest alternatingly to clusters within given units during the designated periods. Trial is called controlled because exposure allocation is not determined by the patient or the physician preference. It operates best under the circumstance where exposure is tolerated by nearly all the patients. The benefits include low cost, reduced difficulty, excellent generalizability, and limited Hawthorne effect as observation period include times with and without intervention rather than just after intervention. The limitations involve lack of blinding and allocation concealment and clinician learning during intervention periods that increases care during nonintervention periods.
Randomized crossover cluster trials
Here, clusters are allocated treatment randomly rather than alternately which makes it the preferred approach due to the advantage of elimination of selection and measurement biases through allocation blinding.
Stepped wedge trial
In this, at the beginning of trial, experimental treatment is used at none of the study sites. Subsequently, one site is randomly selected where experimental treatment is implemented followed by another randomly selected site after a suitable interval (may be months). This process is continued till all the study sites become active, thus producing a growing “wedge” of active sites. The merit of this design is conversion of all the trial units eventually to experimental treatment in contrast to only half the units with parallel-group cluster approach. The demerits include longer study duration which, in turn, leads to increased dropout rates and risk of attrition bias, lack of blinding, difficult to plan and undertake interim analyses, and more complex analysis of results. For example, TRACE trial (routine posTsuRgical Anesthesia visit to improve patient outComE), a prospective, multicentre trial evaluating diminution in risk of postoperative complications by routine postoperative anesthesiologist visits. In this, all hospitals start simultaneously with a control phase providing standard care and crossing over sequentially in a randomized order to the intervention phase (routine visits).[3]
Real-time automated enrolment and randomization[2]
This design is applicable in conditions where immediate treatment is warranted and it is not feasible or practical to obtain the consent. It is useful for appraising responses to relatively uncommon intraoperative events such as anaphylaxis, severe airway problems, or serious hypotension. It may use a Deferred consent model (e.g., critical care trials) whereby next-of-kin is informed of the research and is given an opportunity to opt-out and included surviving patients are later asked to provide consent for inclusion of data in the trial; Outright waiver when the test interventions are of low risk or likely to be helpful compared with routine care; Modified consent might be requested by the institutional review board whereby information is provided in advance enabling opt out or requesting posteriori consent from qualifying subjects with data inclusion only after approval.
Practice preference randomization[2]
This design harnesses the natural clinical variation present across the countries and favors allocation of subjects according to the existing clinical practice. It involves three steps: (i) Trial sites or individual physicians are clustered into groups according to their current practice routine; (ii) Eligible patients are then randomly assigned in unequal ratio (say 2:1) favouring current practice for each site; and (iii) Patients are enrolled with consent being obtained only for nonstandard arm [Figure 3]. The advantages include increased trial engagement because clinician preference is respected and increased enrolment as only a fraction of patients are approached for consent at each site. Drawbacks involve increase in baseline imbalance, confounding, and selection bias.
Figure 3.

Patient preference randomization
Adaptive designs
Conventional clinical trials often include prespecified thresholds that govern whether a trial should be stopped or continued, whereas this design systematically reviews the accruing data and alters the protocol as warranted. It may include pre-planned decision rules that permit changes to study population, assignment ratio, sample size or study drug administration, or dose [Figure 4]. The advantages of this design are shorter timeframe, decreased number of patients, reduced costs, lessened Type I error, and enhanced likelihood of finding the true benefit, if any, of the treatment being studied. Complexities involve difficult statistical evaluation as it needs to account for multiple testing because of frequent interim analyses, confounding caused by baseline imbalance, and difficult safety profile due to reduced number of participants in the trial.[4]
Figure 4.

Adaptive trial design. All eligible patients are randomized to Placebo (p) or treatment Groups A, B, C or D. Interim analysis is run at defined time points and any treatment arm with low probability of benefit or potential harm is subjected to dose modification accordingly. A newly discovered treatment (e) can be added at later stage if required
TYPES OF ADAPTIVE DESIGN
Adaptive dose response
In early phase, adaptive designs allow researchers to learn and optimize based on amassing information related to dosing, exposure, differential participant response, or biomarker responses. This design seeks to determine maximum tolerated dose, i.e., highest dose for some percent of treated participants (e.g., 33 or 50%) having dose-related toxicities using Bayesian adaptive model-based approach called the continual reassessment method [Figure 5].
Figure 5.
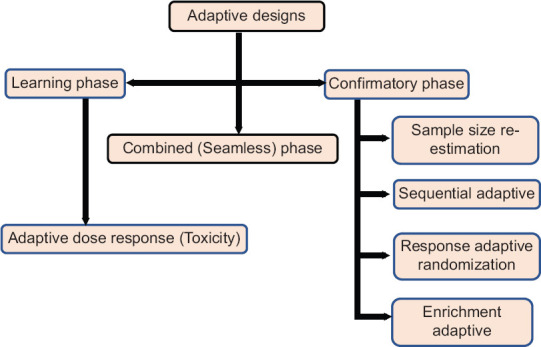
Types of adaptive design
Seamless design
In this, researchers combine the learning stage of Phase II and the confirmatory stage of Phase III trials. In the beginning of Phase II, subjects are randomized into treatment arms of A, B, combined therapy of A and B or control. Interim analysis is then performed to determine which active arm should be dropped. In the confirmatory stage of Phase III, treatment groups with residual effective active arm and control arm are investigated further. The end point of both the phases is mostly similar but may be different in some cases (Eg. Biomarker vs regular clinical endpoint).[5] E.g., INHANCE trial, a two stage, randomized clinical trial which evaluated efficacy of inhaled indacaterol, a long-acting bronchodilator for the treatment of chronic obstructive pulmonary disease (COPD).[6] In Stage 1, patients with COPD were assigned randomly to receive one of the seven regimens: Four doses of indacaterol, placebo, formoterol, or tiotropium and latter two were considered as comparators of standard care. In Stage 2, two out of the four indacaterol doses were selected for further testing together with placebo and tiotropium. Final analysis was based on combined data from two stages. This design has two variants:
Inference seamless: Here, subjects carry the treatment arm from Phase 2 to Phase 3 and data from both the phases is combined together for final analyses
Operational seamless: Phase 2 subjects are not continued in Phase 3 and data in both phases is analysed separately with final analysis based only on data from Phase 3.
Sample size re-estimation
Choosing a fixed sample size for the study is complicated by the need to choose a clinically meaningful treatment effect and to stipulate values for nuisance parameters (such as variance or overall event rate) in turn causing underpowered or overpowered study if estimates are inaccurate. This design allows parameter estimates to be updated during the ongoing trial and adjust the sample size accordingly. A variant of it is known as Internal pilot in which for 1st phase (Pilot phase), sample size is estimated based on previous studies and study is continued till this sample size is achieved (definitive phase). For trial continuation, an interim analysis is performed and sample size is readjusted accordingly. E.g., CHAMPION PHOENIX trial designed to evaluate whether infusion of antiplatelet agent cangrelor reduces the ischemic complications of percutaneous coronary intervention as compared to oral clopidogrel.[7] In this, sample size was re-estimated by interim analysis after enrolment of 70% patients based on observed percentage lowering in relative risk. No change in sample size was made in unfavourable zone (<13.6%) and favourable zone (>21.2%). Sample size was increased in promising zone (≥13.6% to ≤ 21.2) to increase the probability of achieving statistical significance.
Sequential adaptive design
It allows for repeated interim analysis and stoppage once the end point of efficacy, safety or futility is achieved. Analysis is performed after each patient (continuous sequential) or after fixed or variable number of patients (group sequential). The final number of participants required is unknown at the time of initiation and trial ends immediately at first interim analysis meeting the preset stopping criteria. This design is applicable when enrolment in study is expected to be prolonged and treatment outcomes occur relatively soon after recruitment. Complexities involve power calculation, appropriate selection of timing and number of interim analyses.
Response adaptive (play the winner, drop the loser) design
Here, the probability of being randomized to one of the groups is modified according to results being obtained with previous patients. In “play the winner” design, more study subjects are randomized to effective intervention. In “drop the loser” approach, study subjects are removed from the ineffective intervention arm. Merits include enhanced exposure of the subjects to effective intervention hence increased chances of recruitment. Demerits involve unequal group sizes adversely affecting the statistical power and complex sample size calculations.
Enrichment adaptive design
It accomplishes the desire to target therapies to those who can benefit the most from the treatment. Study is conducted in two phases with first period revealing participant groups most likely to benefit from the test agent (discovery phase) with subsequent randomization of subgroup members to receive either active agent or control (validation phase). Power for the chosen subgroups is increased due to increase in sample size as nonpromising groups are discarded. Most appealing area for this design is pharmacogenetic research where it may allow isolation of one or two genetic marker subgroups predictive of treatment response.
THE MASTER PROTOCOLS
Conduct of a series of clinical trials each investigating one or two interventions in a single disease has become more expensive and cumbersome. The onset of “precision medicine” trials to evaluate targeted therapies creates complexities in recruitment of patients with rare genetic subtypes of disease.[8] Methodologic innovation responsive to the need involves coordinated efforts to evaluate more than one treatment in more than one patient type or disease within the same trial frame. Here comes the Master protocols defined as one overarching protocol designed to answer multiple questions. Main objective of these trials is to deliver new therapies to patients safely, more quickly and competently than in the past. Challenges include cost in time and resources to establish needed infrastructure, up-front planning and coordination to meet the objectives of different stakeholders, more complex trial designs and real-time decision making.[9] Arrival of new drugs in the market changing the standard care may in turn jeopardize the long running master protocol. Included in it are three distinct entities: Umbrella, basket and platform trials[10] [Figures 6 and 7].
Figure 6.

(a) Umbrella trial and (b) basket trial
Figure 7.
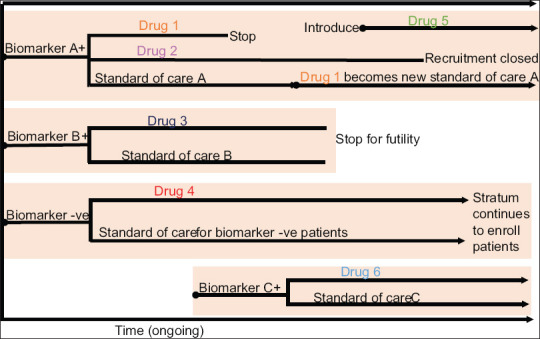
Platform trial. Patients are screened for known biomarkers A and B. Biomarker A + subjects are randomized into drug 1 and 2 versus standard of care. When drug 1 meets criteria for success, it replaces previous standard of care as control. If new drug 5 becomes available, the patients are randomized to this group. Similarly, if drug 3 shows no benefit, the stratum is stopped and biomarker B+ subjects are assigned into biomarker negative stratum. If at some point a new biomarker C and drug 6 becomes available, a new stratum is opened with subsequent screening of patients and assignment to Group A and C
Umbrella trial
Involves study of multiple targeted therapies in the context of a single disease. Eg. BATTLE-1 trial, a Phase 2, single centre trial comparing three monotherapies (erlotinib, vandetanib, and sorafenib) and one combination therapy (erlotinib plus bexarotene) in refractory non-small cell lung cancer.[11]
Basket trial
Involves study of a single targeted therapy in the context of multiple disease or disease subtypes. Eg. B2225 trial, a Phase 2, multicenter trial to determine response of 40 solid tumours and hematologic cancers to single imatinib therapy (400 or 800 mg/day).[12]
Platform trial
Involves study of multiple targeted therapies in the context of a single disease in a perpetual manner with therapies and specific standards of care allowed to enter or leave the platform on the basis of a decision algorithm. This design is different from multi-Arm multi-Stage design which uses pre specified treatment selection rules and allows for comparison of multiple experimental treatments against a single control group.[13] E.g., I-SPY 2 trial, a Phase 2, multicenter trial comparing standard chemotherapy to neoadjuvant therapy for locally advanced breast cancer treatment on the basis of three biomarkers (hormone-receptor status, HER2 status, and MammaPrint risk score).[14] This trial uses response adaptive randomization to assign patients to most promising treatment regimens or their combinations in respective genetic breast cancer sub groups.
CONCLUSION
Although conventional trials remain a priority for establishing the efficacy of new treatments, however, their inadequacies remain the limiting factors in medical innovation. The newer clinical trial designs are gaining popularity among clinicians because they introduce flexibility and competence by speeding enrolment, addressing heterogeneous populations in real world scenarios, accommodating bundled interventions and lowering the cost of research. Recent advances in precision medicine represent an unprecedented opportunity for development of designs tailored for individual patients and translating laboratory innovations quickly to clinical evaluation. However, these alternative approaches also raise new challenges in terms of planning, organization, ethical surveillance and statistical analysis.[15] Thus, study “a priorism” and functional virtue should be warranted, especially when inspections are done on an interim basis and decisions are taken at the earliest convenience.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Pontes C. New methods and designs of clinical trials. Clin Ther. 2016;38:e2–e3. doi: 10.1016/j.clinthera.2016.07.018. [DOI] [PubMed] [Google Scholar]
- 2.Sessler DI, Myles PS. Novel clinical trial designs to improve the efficiency of research. Anesthesiology. 2020;132:69–81. doi: 10.1097/ALN.0000000000002989. [DOI] [PubMed] [Google Scholar]
- 3.Smit-Fun VM, de Korte-de Boer D, Posthuma LM, Stolze A, Dirksen CD, Hollmann MW, et al. TRACE (Routine posTsuRgical Anesthesia visit to improve patient outComE): A prospective, multicenter, stepped-wedge, cluster-randomized interventional study. Trials. 2018;19:586. doi: 10.1186/s13063-018-2952-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhatt DL, Mehta C. Adaptive designs for clinical trials. N Engl J Med. 2016;375:65–74. doi: 10.1056/NEJMra1510061. [DOI] [PubMed] [Google Scholar]
- 5.Chow SC, Tu YH. On two-stage seamless adaptive design in clinical trials. J Formos Med Assoc. 2008;107:52–60. doi: 10.1016/s0929-6646(09)60009-7. [DOI] [PubMed] [Google Scholar]
- 6.Donohue JF, Fogarty C, Lötvall J, Mahler DA, Worth H, Yorgancioglu A, et al. Once-daily bronchodilators for chronic obstructive pulmonary disease: Indacaterol versus tiotropium. Am J Respir Crit Care Med. 2010;182:155–62. doi: 10.1164/rccm.200910-1500OC. [DOI] [PubMed] [Google Scholar]
- 7.Bhatt DL, Stone GW, Mahaffey KW, Gibson CM, Steg PG, Hamm CW, et al. Effect of platelet inhibition with cangrelor during PCI on ischemic events. N Engl J Med. 2013;368:1303–13. doi: 10.1056/NEJMoa1300815. [DOI] [PubMed] [Google Scholar]
- 8.Janiaud P, Serghiou S, Ioannidis JPA. New clinical trial designs in the era of precision medicine: An overview of definitions, strengths, weaknesses, and current use in oncology. Cancer Treat Rev. 2019;73:20–30. doi: 10.1016/j.ctrv.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Cecchini M, Rubin EH, Blumenthal GM, Ayalew K, Burris HA, Russell-Einhorn M, et al. Challenges with novel clinical trial designs: Master protocols. Clin Cancer Res. 2019;25:2049–57. doi: 10.1158/1078-0432.CCR-18-3544. [DOI] [PubMed] [Google Scholar]
- 10.Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377:62–70. doi: 10.1056/NEJMra1510062. [DOI] [PubMed] [Google Scholar]
- 11.Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR, Jr, Tsao A, et al. The BATTLE trial: Personalizing therapy for lung cancer. Cancer Discov. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinrich MC, Joensuu H, Demetri GD, Corless CL, Apperley J, Fletcher JA, et al. Phase II, open-label study evaluating the activity of imatinib in treating life-threatening malignancies known to be associated with imatinib-sensitive tyrosine kinases. Clin Cancer Res. 2008;14:2717–25. doi: 10.1158/1078-0432.CCR-07-4575. [DOI] [PubMed] [Google Scholar]
- 13.Lin J, Bunn V. Comparison of multi-arm multi-stage design and adaptive randomization in platform clinical trials. Contemp Clin Trials. 2017;54:48–59. doi: 10.1016/j.cct.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Barker AD, Sigman CC, Kelloff GJ, Hylton NM, Berry DA, Esserman LJ. I-SPY 2: An adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin Pharmacol Ther. 2009;86:97–100. doi: 10.1038/clpt.2009.68. [DOI] [PubMed] [Google Scholar]
- 15.Bowalekar S. Adaptive designs in clinical trials. Perspect Clin Res. 2011;2:23–7. doi: 10.4103/2229-3485.76286. [DOI] [PMC free article] [PubMed] [Google Scholar]