

Abstract
C3-substituted 1H-indazoles are useful and important substructures in many pharmaceuticals. Methods for direct C3-functionalization of indazoles are relatively rare, compared to reactions developed for the more nucleophilic N1 and N2 positions. Herein, we report a highly C3-selective allylation reaction of 1H--(benzoyloxy)indazoles using CuH catalysis. A variety of C3-allyl 1H-indazoles with quaternary stereocenters were efficiently prepared with high levels of enantioselectivity. Density functional theory (DFT) calculations were performed to explain the reactivity differences between indazole and indole electrophiles, the latter of which was used in our previously reported method. The calculations suggest that the indazole allylation reaction proceeds through an enantioselectivity-determining six-membered Zimmerman-Traxler-type transition state, rather than an oxidative addition/reductive elimination sequence, as we proposed in the case of indole alkylation. The enantioselectivity of the reaction is governed both by the ligand-substrate steric interactions and the steric repulsions involving the pseudoaxial substituent in the six-membered allylation transition state.
INTRODUCTION
The functionalization of nitrogen-containing heterocycles is a key area of research in organic synthesis due to the importance of these molecules in pharmaceutical applications.1 In particular, the preparation of indazole derivatives is of great interest as a result of their versatile pharmacological activities2 and their utility as indole bioisosteres in medicinal chemistry (Figure 1a).3 The direct alkylation of indazoles is one of the most efficient methods to derivatize these molecules for medicinal chemistry studies. Conventionally, indazoles are employed as nucleophiles in these transformations, and either the N1- or N2-isomer is formed, depending on the reaction conditions.4,5 Direct C3-alkylation processes, however, are rare due to the lack of nucleophilicity at the C3-position, even when N1- or N2-position is protected (Figure 1b).6
Figure 1.
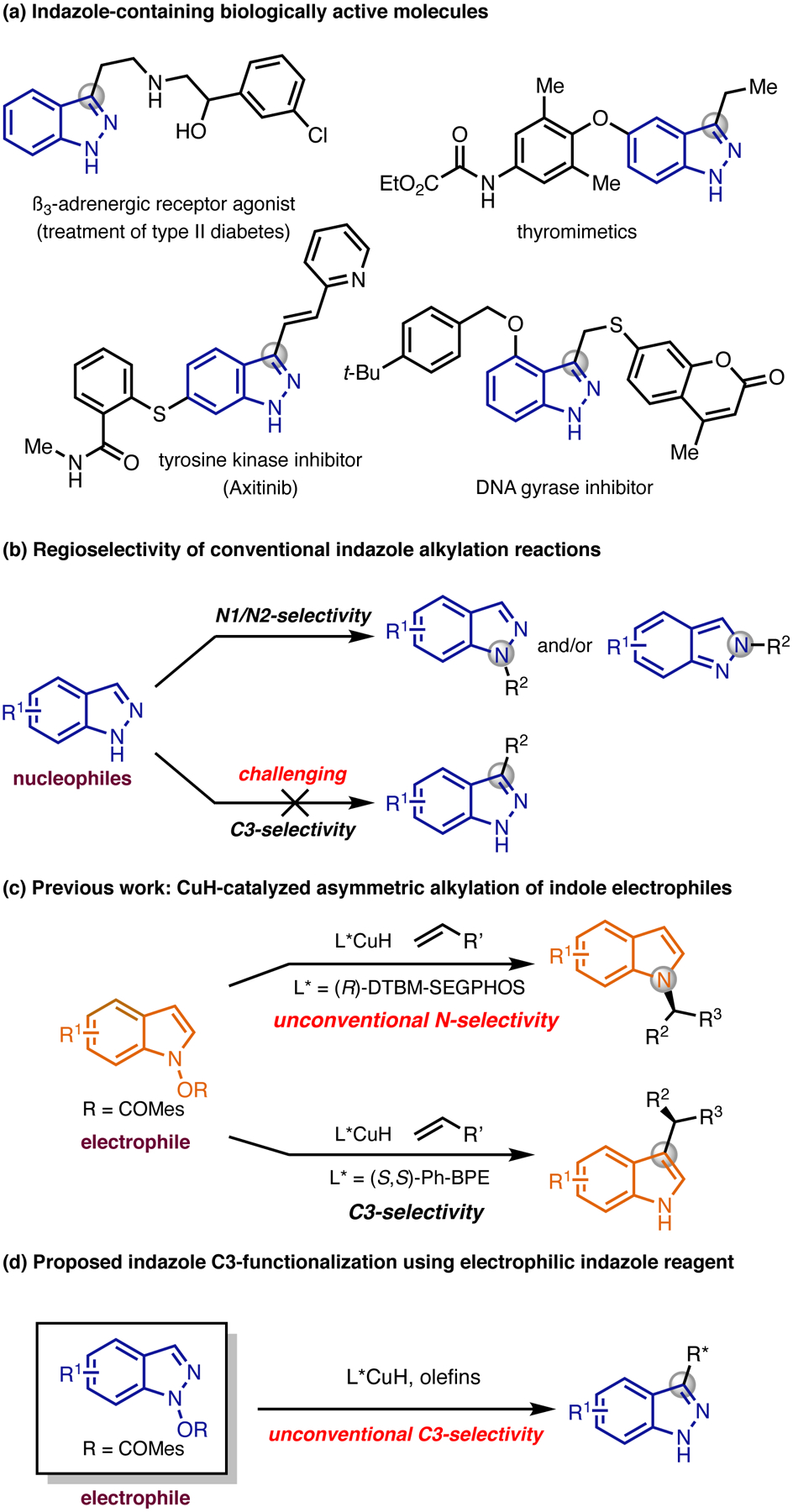
(a) Indazole-containing biologically active molecules. (b) Regioselectivity of conventional indazole alkylation reactions. (c) CuHcatalyzed asymmetric alkylation of indole electrophiles. (d) Proposed CuH-catalyzed asymmetric C3-allylation of indazole electrophiles. Mes: mesityl group.
Recently, we developed a method to prepare chiral alkylated indoles through a CuH-catalyzed nucleophilic alkylation reaction.7 By employing -(benzoyloxy)indoles as electrophiles and with the appropriate choice of ligand, selective N-alkylation or C-alkylation was achieved (Figure 1c). Compared to conventional alkylation reactions, where indoles are used as nucleophiles,8 the regioselectivity of this alkylation protocol was dictated by the catalyst, rather than the intrinsic nucleophilicities of the indole.9 We envisioned that this umpolung strategy10 could be expanded to other nitrogen-containing heterocycles, allowing us to achieve unconventional regioselectivity in the functionalization process of these heterocyclic molecules. Specifically, in the case of indazoles, we were hopeful that by employing N-(benzoyloxy)indazoles as electrophiles, the typically observed N1- or N2-regioselectivity in the nucleophilic substitution reactions could be over-rode, and C3-alkylated indazoles might be accessed (Figure 1d).
RESULTS AND DISCUSSION
Initially, we attempted the coupling of a variety of readily accessible alkenes with indazole 1a under the conditions previously developed for indole alkylation.7 Less than 5% yield of the alkylated indazole products were formed in the cases of styrene (Figure 2a). However, when cyclohexylallene was employed,11 it reacted efficiently with the indazole electrophile 1a, providing the corresponding allyl indazole product (3s) in good yield with a high level of enantioselectivity. Notably, the reaction proceeded with excellent C3-regioselectivity. It is interesting that only the branched allyl indazole was formed, as the same reaction with the indole electrophile 6 produced the corresponding allyl indole product (6a) with exclusive selectivity for the linear isomer (Figure 2a). In addition, unlike the indole alkylation reaction,7 wherein selective N-alkylation could be achieved using DTBM-SEGPHOS ligand, no N-allyl indazole product was observed with all the ligands tested (see the Supporting Information). The intriguing reactivity differences between indazole and indole electrophiles have important implications on the mechanisms of these reactions and were studied in detail with the aid of density functional theory (DFT) calculations (vide infra).
Figure 2:
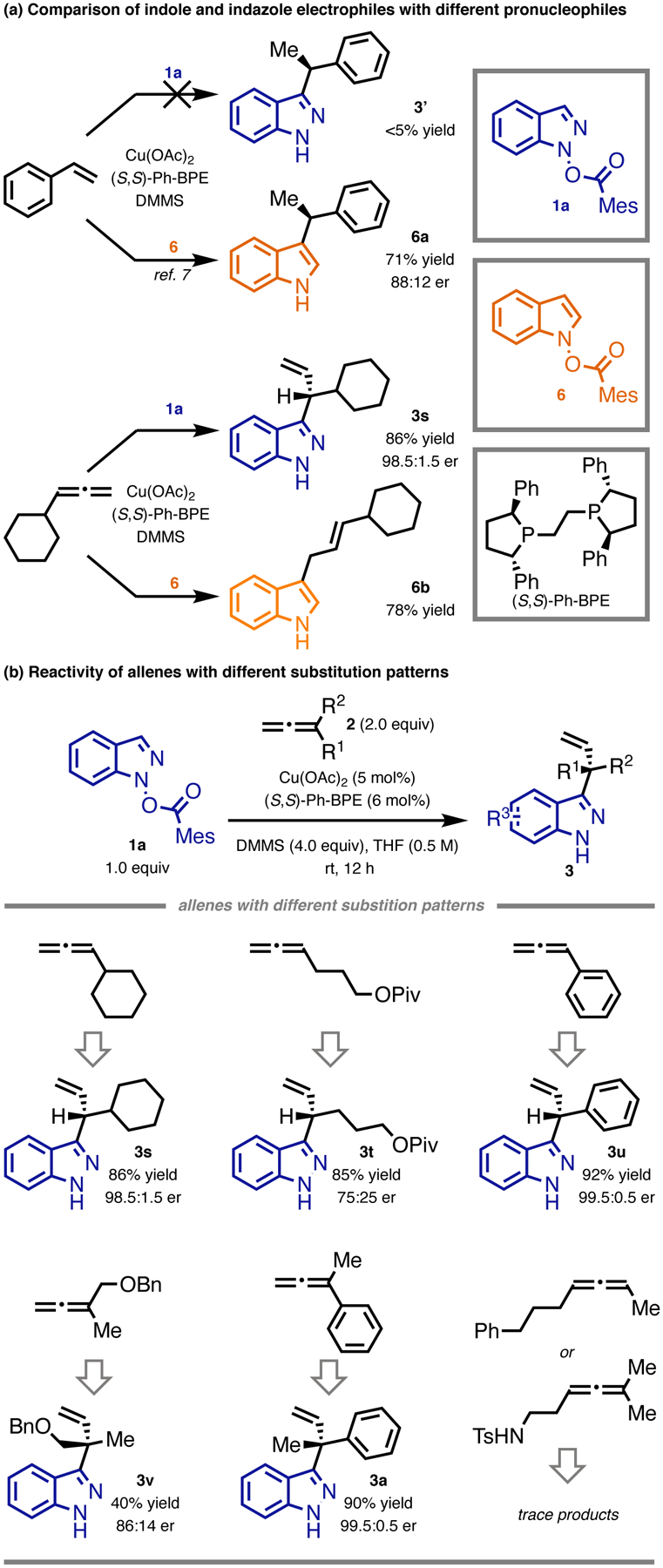
(a) Comparison of indazole and indole electrophiles with styrene and allene pronucleophiles. (b) Test of allenes with different substitution patterns in the indazole allylation reaction. Reactions were conducted on 0.10 mmol scale. Yields were determined by 1 H NMR or GC analysis of the crude reaction mixture. The absolute stereochemistry was signed by analogy to 3a and confirmed by DFT calculations.
We further investigated the reactivity of other types of allenes in this reaction (Figure 2b). A monoalkyl-substituted allene with a substituent sterically smaller than cyclohexyl reacted efficiently albeit with decreased enantioselectivity (3t). A monoaryl-substituted allene could be coupled efficiently (3u). Furthermore, a 1,1-dialkylallene was successfully reacted with 1a in moderate yield and useful enantioselectivity (3v). Finally, 1-aryl-1-alkylallene underwent the reaction with high efficiency, providing C3-allyl indazole (3a) in excellent yield and with a high level of enantioselectivity. However, neither 1,3-disubstituted allenes nor 1,1,3-trisubstituted allenes were suitable substrates.
We considered 1-aryl-1-alkylallenes attractive coupling partners because the corresponding C3-allyl indazole products with an acyclic quaternary stereocenter are potentially valuable molecules in medicinal chemistry, and challenging to access using existing methods.12
Therefore a variety of 1-alkyl-1-arylallenes are found to couple efficiently to N-(benzoyloxy)indazole electrophiles (Table 1). Substituents including 4-methoxy (3b), 2-fluoro (3c), 4-bromo (3g), 3-chloro (3i), 4-trifluoromethyl (3j), and 2-methyl (3k) on the phenyl ring of the allenes are well tolerated. Allenes containing heterocycles, such as furan (3d), 2-methoxypyridine (3e), and -Ts-pyrrole (3f), are also compatible in the reaction. We observed a slight decrease in both efficiency and enantioselectivity with the increasing steric hindrance of alkyl substituents on the allenes. Methyl- (3a), ethyl- (3g), isobutyl- (3l), and cyclopropyl- (3h) substituted indazoles were prepared from the corresponding allenes with decreasing enantiomeric ratios (99.5:0.5 to 92:8). In addition, an indazole derivative with a fused dihydropyran (3i) was prepared from a chromane-derived allene efficiently.
Table 1.
Substrate Scope for the C3-Allylation of Indazole Electrophiles.a
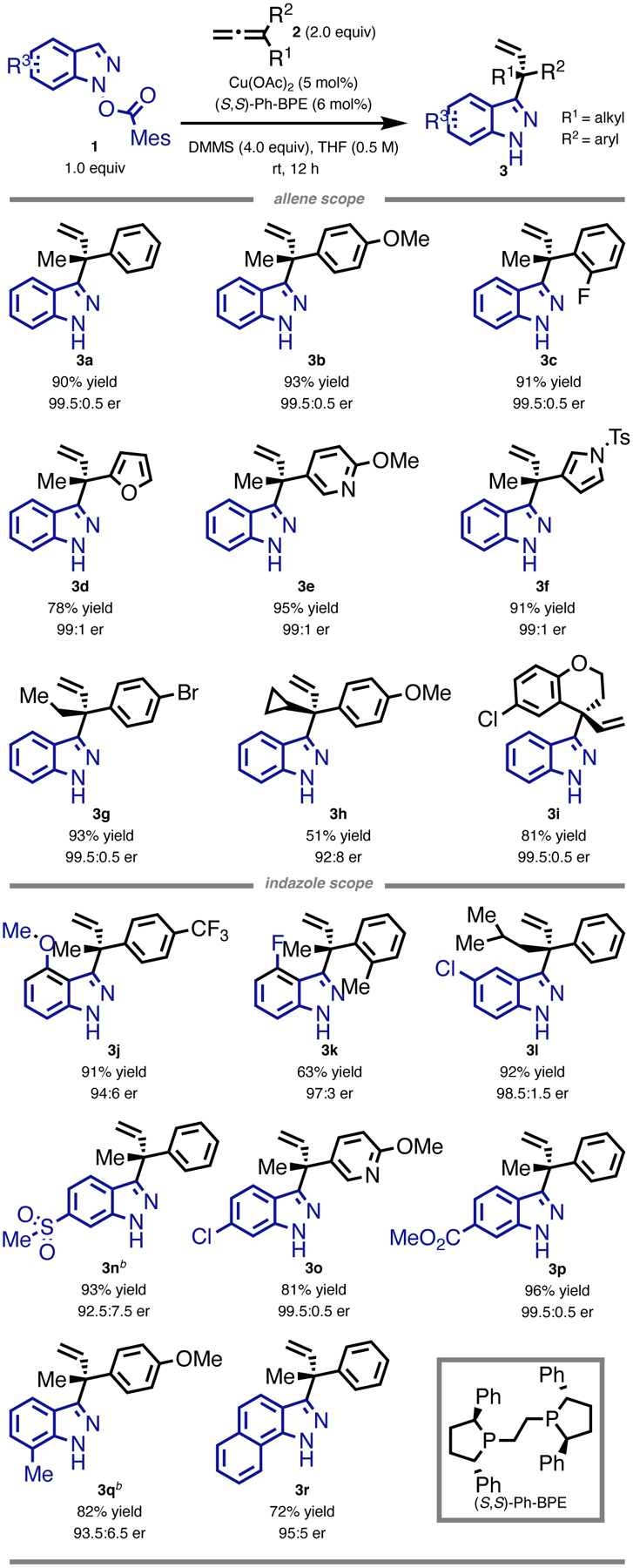
|
All yields represent average isolated yields of two runs, performed with 0.5 mmol of indazole electrophile.
60 °C.
The reaction is broadly tolerant of substituents appended to the indazole electrophiles, including 4-methoxy (3j), 4-fluoro (3k), 5-chloro (3l), 6-methylsulfonyl (3n), 6-chloro (3o), 6-carbomethoxy (3p), and 7-methyl (3q). Moreover, a benzoindazole electrophile was also found to be a competent coupling partner in this transformation (3r).
To briefly demonstrate the potential synthetic utility of these coupling products, indazole 3b was converted to a primary alcohol in good yield under hydroboration-oxidation process (Scheme 1, a). Furthermore, the terminal double bond of 3b could be easily reduced to an ethyl group, generating a stereocenter containing both methyl and ethyl substituents with excellent enantioselectivity (Scheme 1, b). An Ullmann coupling reaction of 3a with an aryl iodide was performed successfully as well. Although transition-metal-catalyzed N-arylation of indazoles usually generates a mixture of N1- and N2-arylated products,12 high N1-selectivity (N1:N2>20:1) of 3b was observed in this case, presumably due to the steric hindrance of the C3-substituent (Scheme 1, c).
Scheme 1. Further Functionalizations of Allylated Indazoles.a.
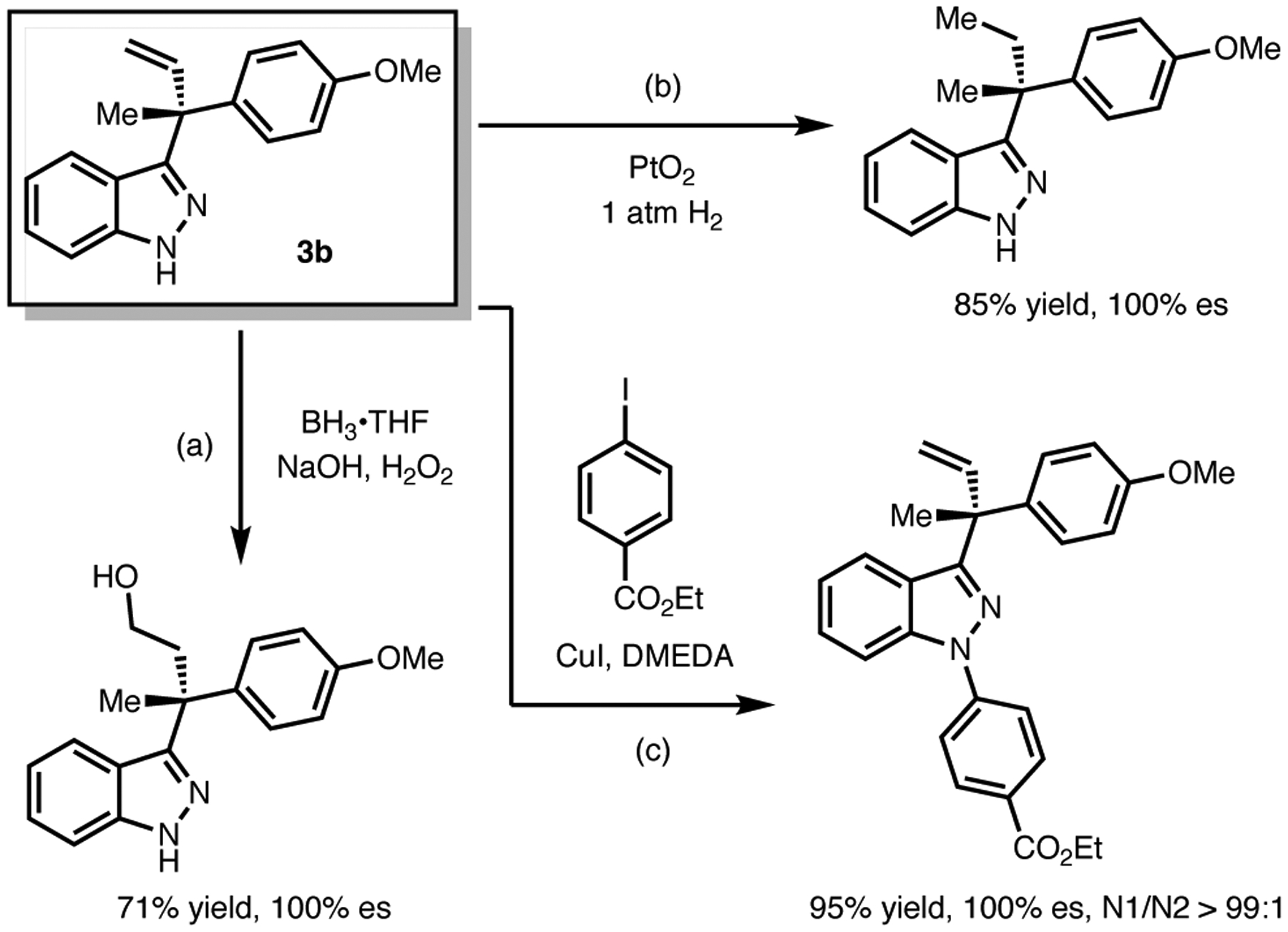
aReactions were conducted on 0.2 mmol scale. See the Supporting Information for detailed conditions.
As mentioned earlier, we noted prominent differences in reaction outcomes between indazole and indole electrophiles under copper hydride catalysis. To obtain more mechanistic understanding of the origin of reactivity differences, we computed the energy profiles of allylation reactions of 1a and 6 with 1-phenyl-1-methylallene 2a using density functional theory (DFT) calculations (Figure 3). Both reactions initiate via the hydrocupration of allene 2a with copper hydride 7 with a 15.9 kcal/mol activation energy (TS1a, see the Supporting Information, Figure S1, for other less favorable hydrocupration pathways). This step leads to the irreversible formation of the -isomer of the terminal allylic copper species (8). Complex 8 can rapidly isomerize to form either diastereomers of the tertiary benzylic copper intermediate (9), which undergoes subsequent isomerization to afford the thermodynamically more stable E-isomer of the terminal allylic copper (10). 14 In the presence of the indole electrophile 6, the most favorable reaction pathway proceeds through the SN2′ type oxidative addition (TS3′, ΔG‡ = 23.3 kcal/mol with respect to 10), leading to the formation of C3-allyl indole product with linear selectivity, which is consistent with the experimental results (Figure 2b). The competing SN2 type oxidative addition TS4′ leading to the N-allyl indole product is disfavored by 5.9 kcal/ mol. These results are consistent with the previously studied ligand effects, where Ph-BPE ligand promoted the formation of C3-alkylated product.7
Figure 3.
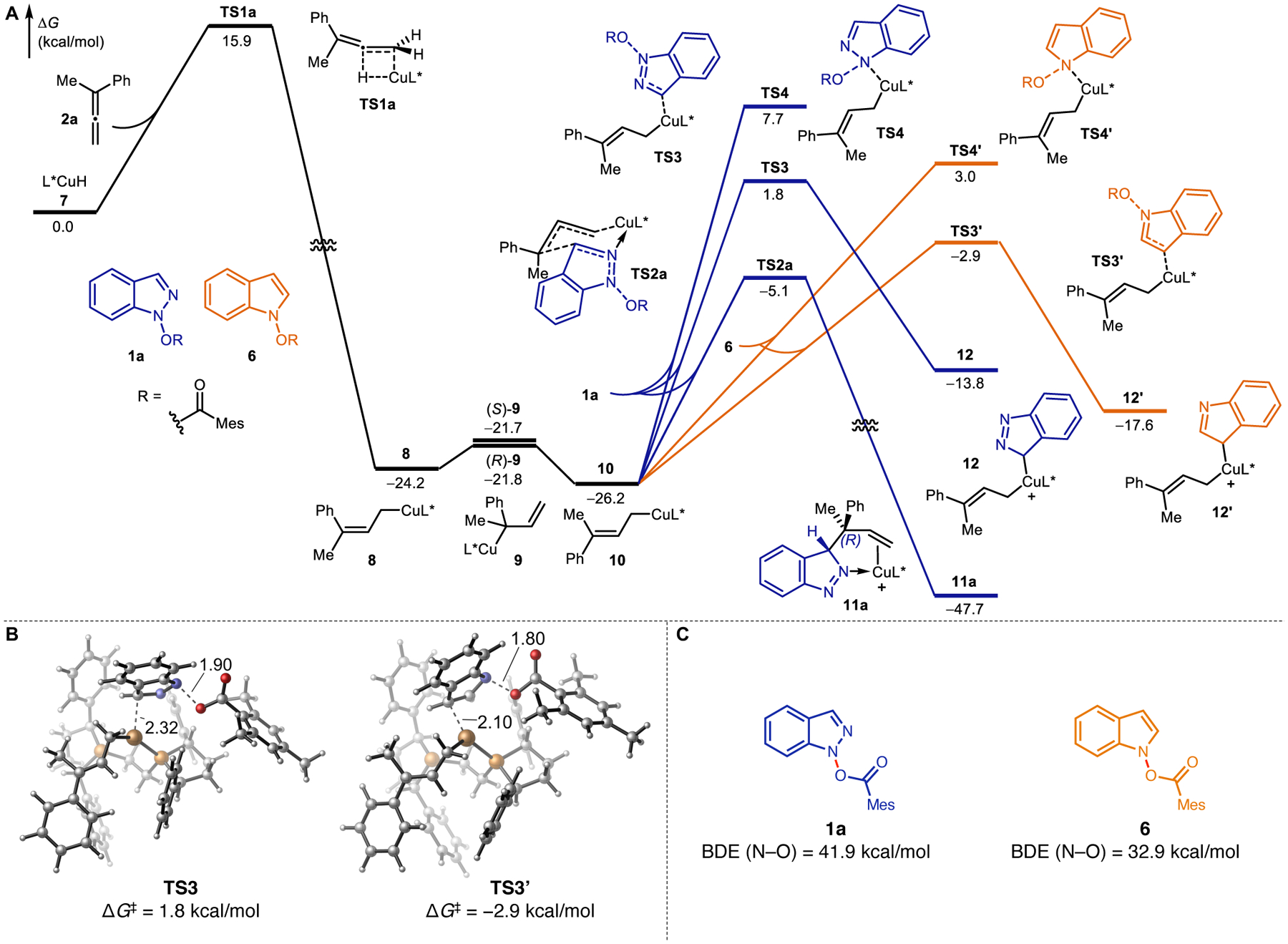
(A) Energy profiles of the allylation of indazole (1a) and indole (6) electrophiles. (B) Optimized structures of the C3-oxidative addition transition states with indazole (TS3) and indole (TS3′) substrates. (C) Calculated N−O bond dissociation enthalpies (BDEs) of 1a and 6.
In the reaction with the indazole electrophile (1a), we found that the SN2′ oxidative addition (TS3) requires a higher activation barrier of 28.0 kcal/mol with respect to 10, when compared to the reaction with indole (TS3′), and the product is thermodynamically destabilized by 3.6 kcal/mol (12 versus 12′). Since the C3 oxidative additions are endergonic, the transition states are more product-like and exhibit significant N−O bond elongations (Figure 3B). The computed kinetic and thermodynamic trends can therefore be attributed to the cleavage of a stronger N−O bond in the indazole electrophile, which is supported by calculated BDEs where the cleavage of the N−O bond in 1a requires 9.0 kcal/mol higher energy than the corresponding bond cleavage in 6 (Figure 3C). In addition to the relatively high calculated energy barrier, this oxidative addition pathway would lead to the linear allylation products, which are inconsistent with the branched selectivity observed in experiment.
Our DFT calculations revealed a more feasible mechanism with indazole 1a via a Zimmerman-Traxler type six-member transition state (TS2a).15 This mechanism is favored because it forgoes the generation of the less stable Cu(III) intermediate. Furthermore, this transition state is stabilized by the presence of dative Cu−N2 bond, which is not available with the indole substrate. This model is consistent with the branched regioselectivity as well as the observed enantioselectivity in the reaction (Figure 4). The indazole electrophile 1a can add at either face of the C=C bond of 8 or 10 (TSa-d). Here, the C−C bond formation and the dissociation of 2,4,6-trimethylbenzoate anion are concerted processes, leading directly to 3H-indazole complexes (11a–d), which form the 1H-indazole product upon tautomerization. The enantioselectivity of the C3-allylation product is determined in the indazole addition step (TS2). Among the four competing transition states, TS2c and TS2d originating from the -allyl complex 8 are both disfavored (3.3 and 8.0 kcal/mol higher than TS2a, respectively) due to the pseudoaxial placement of the bulky phenyl group, which leads to increased repulsions with the indazole ring (Figure 4B). In TS2a and TS2b, the smaller methyl group is placed at the pseudoaxial position and thus the steric repulsions about the forming C−C bond are decreased. From intermediate 10, the addition of the indazole to form product (S)-3a through TS2b is 5.4 kcal/mol less favorable than the addition to form (R)-3a through TS2a. The relative instability of TS2b arises from unfavorable steric repulsions between the (S,S)-Ph-BPE ligand and the 2,4,6-trimethylbenzoate leaving group. In TS2b, the bulky leaving group is placed in the quadrant occupied by a “proximal” phenyl group on the ligand (Figure 4C). By contrast, in TS2a, the leaving group is in a less occupied quadrant with a “distal” phenyl group. The increased ligand-substrate steric repulsions in TS2b are evidenced by the more significant distortion of the Ph-BPE ligand in TS2b than in TS2a (ΔΔEdist-Ph-BPE = 3.7 kcal/mol, see Figure S2).
Figure 4.
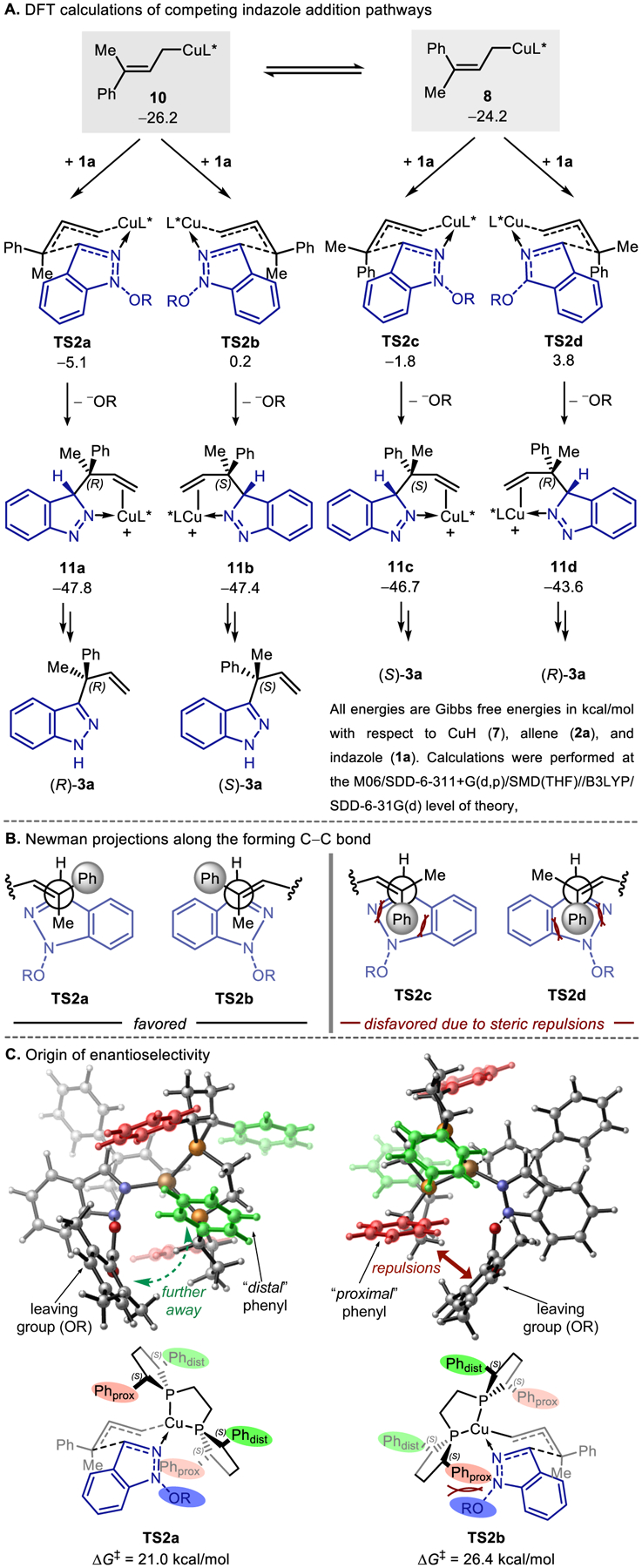
Origin of enantioselectivity in the C3-allylation with the indazole electrophile 1a.
To further verify the mechanistic model, we calculated the enantioselectivities of the allylation reaction with allenes containing substituents of varying degrees of steric hindrance. The enantioselectivities were computed from transition states TSa and TSc arising from the same facial addition of 1a to the E- and Z-isomers of the corresponding allylic copper species (Figure 5). The calculated enantioselectivity trend is in a good qualitative agreement with the experimental data (Figure 2). While reactions with allenes 2a and 2s are both highly enantioselective, using a less bulky primary alkyl allene (2t) almost completely diminishes the predicted er. Although this computed value is underestimated when compared to the observed er, both computational and experimental results demonstrated the role of steric effects of allene substituents on the er of the allylation product.
Figure 5.
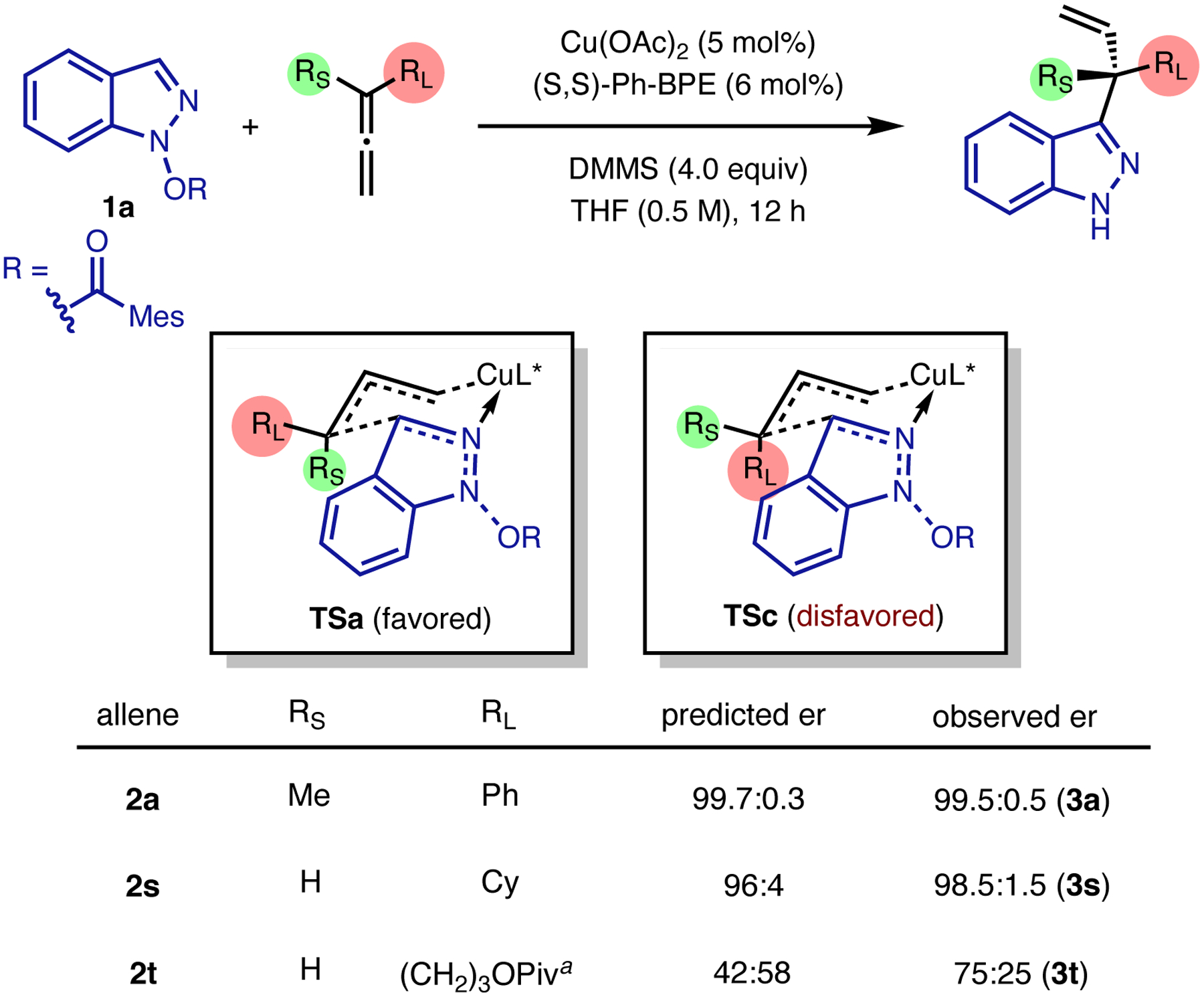
RL= n-Pr was used in calculations as a model of the 3- pivaloyloxypropyl group in 2t. Computed enantioselectivities with different allene substrates.
CONCLUSIONS
In summary, we developed a method for the preparation of C3-allyl indazoles bearing quaternary stereocenters in high yield with excellent levels of enantioselectivity using CuH catalysis. Key to the success of this unique C3-selectivity in indazole alkylation reaction is the use of an umpolung strategy: in contrast to the conventional use of indazoles as nucleophiles, electrophilic indazoles (-(benzoyloxy)indazoles) are employed as electrophiles in the reaction. With the aid of DFT calculations, we discussed the fundamental reactivity differences between the indazole and the previously reported indole electrophiles. In addition, a mechanistic model was developed to account for the branched selectivity of the allyl indazole products and explain the observed enantioselectivity in the reaction. Expanding this a polarity reversal strategy to achieve novel reactivities in other nitrogen-containing heterocycle functionalization reactions is currently underway.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Solvias AG for donations of ligands used in this project. We are grateful to the National Institutes of Health (R35-GM122483, R35-GM128779). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health. We thank Yujing Zhou, Drs. Richard Liu, Scott McCann, and Christine Nguyen for their advice on the preparation of this manuscript. We acknowledge Drs. Peter Müller (MIT) and Charlene Tsay (MIT) for X-ray crystallographic analysis. We thank the National Institutes of Health for a supplemental grant for the purchase of supercritical fluid chromatography (SFC) equipment (GM058160-17S1). I.K. is grateful to the University of Pittsburgh and the Pittsburgh Quantum Institute for fellowships. DFT calculations were performed at the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the NSF.
Footnotes
The authors declare no competing financial interest.
Supporting Information.
The supporting Information is available free of charge on the ACS Publications website.
Experimental details and computational data (PDF)
Spectroscopic data (PDF)
REFERENCES
- (1).(a) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem, 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (b) Lamberth C; Dinges J Bioactive Heterocyclic Compound Classes: Pharmaceuticals and Agrochemicals; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- (2).Cerecetto H; Gerpe A; González M; Arán VJ; de Ocáriz CO Pharmacological Properties of Indazole Derivatives: Recent Developments. Mini-Reviews in Medicinal Chemistry 2005, 5, 869–878. [DOI] [PubMed] [Google Scholar]
- (3).(a) Brown N Bioisosterism in Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]; (b) Ainsworth C Indazole Analog of Tryptamine: A New Synthesis of Indazoles. J. Am. Chem. Soc 1957, 79, 5242–5245. [Google Scholar]; (c) Ainsworth C The Indazole Analog of Serotonin. J. Am. Chem. Soc 1957, 79, 5245–5247. [Google Scholar]
- (4).Reviews on indazole synthesis:; (a) Schmidt A; Beutler A; Snovydovych B Recent Advances in the Chemistry of Indazoles. Eur. J. Org. Chem 2008, 4073–4095. [Google Scholar]; (b) Zhang S-G; Liang C-G; Zhang W-H Recent Advances in Indazole-Containing Derivatives: Synthesis and Biological Perspective. Molecules 2018, 23, 2783–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Examples of indazoles alkylation with N1/N2 regioselectivity:; (a) Hunt KW; Moreno DA; Suiter N; Clark CT; Kim G Selective Synthesis of 1-Functionalized-Alkyl-1H-Indazoles. Org. Lett 2009, 11, 5054–5057. [DOI] [PubMed] [Google Scholar]; (b) Lin M-H; Liu H-J; Lin W-C; Kuo C-K; Chuang T-H Regioselective Synthesis of 2H-Indazoles through Ga/Al- and Al-Mediated Direct Alkylation Reactions of Indazoles. Org. Biomol. Chem 2015, 13, 11376–11381. [DOI] [PubMed] [Google Scholar]; (c) Haydl AM; Breit B Regio-and Enantioselective Synthesis of N-Substituted Pyrazoles by Rho-dium-Catalyzed Asymmetric Addition to Allenes. Angew. Chem., Int. Ed 2015, 54, 7149–7153. [DOI] [PubMed] [Google Scholar]; (d) Slade DJ; Pelz NF; Bodnar W; Lampe JW; Watson PS Indazoles: Regioselective Protection and Subsequent Amine Coupling Reactions. J. Org. Chem 2009, 74, 6331–6334. [DOI] [PubMed] [Google Scholar]; (e) Luo G; Chen L; Dubowchik G Regioselective Protection at N-2 and Derivatization at C-3 of Indazoles. J. Org. Chem 2006, 71, 5392–5395. [DOI] [PubMed] [Google Scholar]
- (6).A rare example of direct C3-alkylation using indazoles as nucleophiles:; Campetalla S; Palmieri A; Petrini M Synthesis of 3-(Tosyl-alkyl)indazoles and their Desulfonylation Reactions-A New Entry to 3-Substituted Indazoles by an Unprecedented Friedel-Crafts Process. Eur. J. Org. Chem 2009, 3184–3188. [Google Scholar]
- (7).Ye Y; Kim S-T; Jeong J; Baik M-H; Buchwald SL CuH-Catalyzed Enantioselective Alkylation of Indole Derivatives with Lig-and-Controlled Regiodivergence. J. Am. Chem. Soc 2019, 141, 3901–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Chen JB; Jia YX Recent Progress in Transition-Metal-Catalyzed Enantioselective Indole Functionalizations. Org. Biomol. Chem 2017, 15, 3550–3567. [DOI] [PubMed] [Google Scholar]; (b) Bandini M; Eichholzer A Catalytic Functionalization of Indoles in a New Dimension. Angew. Chem., Int. Ed 2009, 48, 9608–9644. [DOI] [PubMed] [Google Scholar]
- (9).(a) Lakhdar S; Westermaier M; Terrier F; Goumont R; Boubaker T; Ofial AR; Mayr H Nucleophilic Reactivities of Indoles. J. Org. Chem 2006, 71, 9088–9095. [DOI] [PubMed] [Google Scholar]; (b) Otero N; Mandado M; Mosquera RA Nucleophilicity of Indole Derivatives: Activating and Deactivating Effects Based on Proton Affinities and Electron Density Properties. J. Phys. Chem. A 2007, 111, 5557–5562. [DOI] [PubMed] [Google Scholar]
- (10).Seebach D Methods of Reactivity Umpolung. Angew. Chem. Int. Ed. Engl 1979, 18, 239–258. [Google Scholar]
- (11).(a) Kieslich K; Koch H-J; Kosmol H; Rufer C; Schröder E; Vössing R Totalsynthese von Natürlichem Östradiolmethyläther. Tetrahedron Lett. 1966, 7, 2321–2330. [Google Scholar]; (b) Fuji K Asymmetric Creation of Quaternary Carbon Centers. Chem. Rev 1993, 93, 2037–2066. [Google Scholar]; (c) Corey EJ; Guzman-Perez A The Catalytic Enantioselective Construction of Molecules with Quaternary Carbon Stereocenters. Angew. Chem. Int. Ed 1998, 37, 388–401. [DOI] [PubMed] [Google Scholar]; (d) Christoffers J; Mann A Enantioselective Construction of Quaternary Stereocenters. Angew. Chem. Int. Ed 2001, 40, 4591–4597. [DOI] [PubMed] [Google Scholar]; (e) Denissova I; Barriault L Stereoselective Formation of Quaternary Carbon Centers and Related Functions. Tetrahedron 2003, 59, 10105–10146. [Google Scholar]; (f) Douglas CJ; Overman LE Catalytic Asymmetric Synthesis of All-Carbon Quaternary Stereocenters. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 5363–5367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Trost BM ; Jiang C Catalytic Enantioselective Construction of All-Carbon Quaternary Stereocenters. Synthesis 2006, 369–396. [Google Scholar]; (h) Das J; Marek I Enantioselective Synthesis of All-Carbon Quaternary Stereogenic Centers in Acyclic Systems. Chem. Commun 2011, 47, 4593–4623. [DOI] [PubMed] [Google Scholar]; (i) Quasdorf KW; Overman LE Catalytic Enantioselective Synthesis of Quaternary Carbon Stereocentres. Nature 2014, 516, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Gibian H; Marek I; Minko Y; Pasco M; Mejuch T; Gilboa N; Chechik H; Das JP All-Carbon Quaternary Stereogenic Centers in Acyclic Systems through the Creation of Several C–C Bonds per Chemical Step. J. Am. Chem. Soc 2014, 136, 2682–2694. [DOI] [PubMed] [Google Scholar]; (k) Liang T; Rivas F All-Carbon Quaternary Centers in Natural Products and Medicinal Chemistry: Recent Advances. Tetrahedron 2016, 72, 6729–6777. [Google Scholar]; (l) Feng J; Holmes M; Krische MJ Acyclic Quaternary Carbon Stereocenters via Enantioselective Transition Metal Catalysis. Chem. Rev 2017, 117, 12564–12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Kazzouli SE; Guillaumet G Functionalization of Indazoles by Means of Transition Metal-Catalyzed Cross-Coupling Reactions. Tetrahedron 2016, 72, 6711–6727. [Google Scholar]; (b) Antilla JC; Baskin JM; Barder TE; Buchwald SL Copper-Diamine-Catalyzed N-Arylation of Pyrroles, Pyrazoles, Indazoles, Imidazoles, and Triazoles. J. Org. Chem 2004, 69, 5578–5587. [DOI] [PubMed] [Google Scholar]
- (13).For asymmetric allylation using allyl copper species, see:; (a) Holmes M; Schwartz LA; Krische MJ Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev 2018, 118, 6026–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shao X; Li K; Malcolmson SJ Enantioselective Synthesis of Anti-1,2-Diamines by Cu-Catalyzed Reductive Coupling of Azadienes with Aldimines and Ketimines. J. Am. Chem. Soc 2018, 140, 7083–7087. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li M; Wang J; Meng F Cu-Catalyzed Enantioselective Reductive Coupling of 1,3-Dienes and Aldimines. Org. Lett 2018, 20, 7288–7292. [DOI] [PubMed] [Google Scholar]; (d) Liu RY; Yang Y; Buchwald SL Regiodivergent and Diastere-oselective CuH-Catalyzed Allylation of Imines with Terminal Allenes. Angew. Chem. Int. Ed 2016, 55, 14077–14080. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Li C; Liu RY; Jesikiewicz LT; Yang Y; Liu P; Buchwald SL CuH-Catalyzed Enantioselectvie Ketone Allylation with 1,3-Dienes: Scope, Mechanism, and Application. J. Am. Chem. Soc 2019, 141, 5062–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Grayson MN; Krische MJ; Houk KN Ruthenium-Catalyzed Asymmetric Hydrohydroxyalkylation of Butadiene: The Role of the Formyl Hydrogen Bond in Stereochemical Control. J. Am. Chem. Soc 2015, 137, 8838–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mejuch T; Gilboa N; Gayon E; Wang H; Houk KN; Marek I Axial Preferences in Allylation Reactions via the Zimmerman–Traxler Transition State. Acc. Chem. Res 2013, 46, 1659–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim SW; Meyer CC; Mai BK; Liu P ; Krische MJ Inversion of Enantioselectivity in Allene Gas versus Allyl Acetate Reductive Aldehyde Allylation Guided by Metal-Centered Stereogenicity: An Experimental and Computational Study. ACS Catal. 2019, 9, 9158–9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Grayson MN; Krische MJ; Houk KN Ruthenium-Catalyzed Asymmetric Hydrohydroxyalkylation of Butadiene: The Role of the Formyl Hydrogen Bond in Stereochemical Control. J. Am. Chem. Soc 2015, 137, 8838–8850, DOI: 10.1021/jacs.5b04844 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mejuch T; Gilboa N; Gayon E; Wang H; Houk KN; Marek I Axial Preferences in Allylation Reactions via the Zimmerman–Traxler Transition State. Acc. Chem. Res 2013, 46, 1659–1669, DOI: 10.1021/ar4000532 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim SW; Meyer CC; Mai BK; Liu P; Krische MJ Inversion of Enantioselectivity in Allene Gas versus Allyl Acetate Reductive Aldehyde Allylation Guided by Metal-Centered Stereogenicity: An Experimental and Computational Study. ACS Catal. 2019, 9, 9158–9163, DOI: 10.1021/acscatal.9b03695 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.