

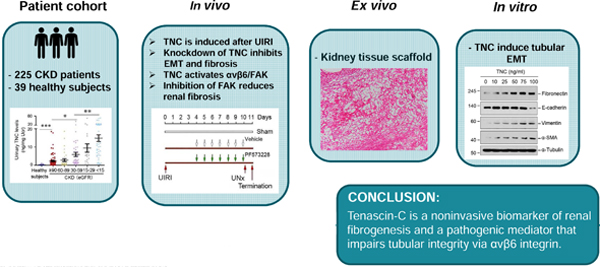
Abstract
Tenascin-C is an extracellular matrix glycoprotein that plays a critical role in kidney fibrosis by orchestrating a fibrogenic niche. Here, we demonstrate that tenascin-C is a biomarker and a mediator of kidney fibrogenesis by impairing tubular integrity. Tenascin-C was found to be increased in kidney biopsies from patients with chronic kidney disease (CKD). In a cohort of 225 patients with CKD, the urinary tenascin-C level was markedly elevated, compared to 39 healthy individuals. Moreover, the level of urinary tenascin-C in CKD was correlated with the severity of kidney dysfunction and fibrosis. In mouse model of acute kidney injury-to-CKD induced by ischemia/reperfusion, depletion of tenascin-C preserved tubular integrity and ameliorated renal fibrotic lesions. In vitro, tenascin-C impaired tubular cell integrity by inducing partial epithelial-mesenchymal transition. Using decellularized kidney tissue scaffolds, we found that tenascin-C-enriched scaffolds facilitated tubular epithelial-mesenchymal transition ex vivo. Mechanistically, tenascin-C specifically induced integrins αvβ6 in tubular cells and activated focal adhesion kinase (FAK). Blocking αvβ6 integrins or inhibition of FAK restored tubular integrity by repressing epithelial-mesenchymal transition and alleviated kidney fibrosis. Thus, our studies underscore that tenascin-C is a noninvasive biomarker of kidney fibrogenesis and a pathogenic mediator that impairs tubular integrity. Hence, blockade of the tenascin-C /αvβ6 integrin/FAK signal cascade may be a novel strategy for therapeutic intervention of kidney fibrosis.
Keywords: Tenascin-C, renal fibrosis, integrin, EMT, microenvironment, chronic kidney disease
Graphical Abstract
INTRODUCTION
Chronic kidney disease (CKD) affects more than 10% of the population worldwide.1, 2 Studies show that patients with CKD are at high risk of progressing into end-stage kidney disease, a devastating condition requiring renal replacement therapies. Irrespective of the etiologies, progression of CKD is characterized by the relentless accumulation of extracellular matrix (ECM) leading to scar formation in renal parenchyma.3–5 In many ways, kidney fibrosis is considered as the common pathway underlying various CKD.6 Current therapeutic options for CKD, however, are limited; and there is no specific treatment for targeting fibrosis per se. This enormous unmet medical need calls for better understanding of the mechanism underlying fibrotic CKD.
The pathophysiology of kidney fibrosis is complex, which involves many types of cells including fibroblasts, pericytes, tubular epithelial cells, endothelial cells and infiltrated inflammatory cells.3, 4, 7 The role of tubular cells is particularly intriguing, because they are often the primary targets of kidney injury in CKD. Increasing evidence shows that tubular cells are not a bystander or a victim, they are dynamically involved in the evolution of CKD as an active player.8, 9 In response to various insults, tubular cells undergo different changes such as partial epithelial-mesenchymal transition (EMT), cell cycle arrest, cellular senescence or metabolic reprogramming.10–14 These alterations render tubular cells a secretory phenotype producing a host of profibrotic factors triggering fibroblast activation.15 The distribution of tubular lesions in CKD is not even throughout renal parenchyma, implying the existence of special microenvironment in controlling local response of renal tubules. However, little is known about the impact of extracellular microenvironment in regulating tubular cell behavior under pathologic conditions.
Tenascin-C (TNC) is a large hexameric ECM glycoprotein and consists of four distinct domains: a cysteine-rich assembly domain, tandem epidermal growth factor (EGF)-like repeats, numerous fibronectin type III-like repeats, and a fibrinogen-like globe.16,17 TNC is barely detectable in normal adult tissues but induced under pathologic conditions.18 As a matricellular protein, TNC modulates cell signaling by binding to cell surface receptors such as integrins or toll-like receptor.16, 19 Whether TNC plays a role in modulating tubular integrity in CKD remains unknown.
In this study, we show that TNC was increased in the kidney and urine of CKD patients. TNC impaired tubular cell integrity by activating integrin αvβ6 and focal adhesion kinase (FAK). Furthermore, pharmacologic inhibition of FAK halted CKD progression after acute kidney injury (AKI). Our studies show that TNC/integrin αvβ6/FAK signal cascade is a therapeutic target of CKD.
RESULTS
Urinary TNC levels are increased in human CKD
We first examined TNC protein in human kidney biopsies by immunohistochemical staining. As shown in Figure 1a, TNC was undetectable in normal human kidneys. However, it was induced in renal biopsies from patients with various CKD including IgA nephropathy (IgAN), membranous nephritis (MN), focal and segmental glomerulosclerosis (FSGS), hypertensive renal disease (HRD) and lupus nephritis (LN). TNC protein was predominantly localized in the tubulointerstitium of diseased kidneys with little staining in the glomeruli. Notably, TNC distribution was not even, rather focal and patchy, throughout renal parenchyma.
Figure 1. TNC expression is associated with the severity of CKD in humans.

(a) Representative micrographs show the abundance and localization of TNC protein in a variety of human CKD. Control, non-tumor kidney tissue from the patients with renal cell carcinoma used as normal controls; IgAN, IgA nephropathy; MN, membranous nephritis; FSGS, focal segmental glomerulosclerosis; HRD, hypertensive renal disease; LN, lupus nephritis. Arrow indicates positive TNC staining. Glo, glomerulus. Scale bar, 50 µm. (b) Western blot analyses show urinary TNC protein in healthy subjects and CKD patients. Representative Western blot and quantitative data are shown. Numbers (1~4) indicate urine samples from each individual subject. **P < 0.01. (c) Graphic presentation shows urinary TNC protein levels in cohorts of CKD patients (n=225) and healthy subjects (n=39). Urinary TNC levels were presented as nanograms per mg urinary creatinine (Ucr). **P < 0.01. (d) Graphic presentation shows urinary TNC protein levels in different stages of CKD. *P < 0.05, **P < 0.01, ***P < 0.001. (e) Linear regression shows a negative correlation between urinary TNC protein and kidney function (eGFR). (f-h) Linear regression shows a significant correlation between urinary TNC levels and serum creatinine (Scr) (f), blood urea nitrogen (BUN) (g) and urinary albumin creatinine ratio (UACR) (h). (i) Representative micrographs show renal collagen deposition in kidney biopsy specimens of CKD patients. Kidney biopsies sections were subjected to Masson’s trichrome staining (MTS). Urinary TNC levels (ng/mg creatinine) were given at the top of micrographs. Arrows indicate positive staining. Scale bar, 50 µm. (j) Linear regression shows urinary TNC protein levels were closely correlated with the severity of kidney fibrosis. Fibrosis score was assessed by two individuals who were blinded to urinary TNC data.
As TNC is a secreted protein, we reasoned whether an increased renal TNC could lead to its elevation in the urine. To test this, we assessed urinary TNC by Western blotting. As shown in Figure 1b, TNC was readily detectable in the urine of CKD patients, but not in healthy subjects. We further quantitatively measured urinary TNC levels in 39 healthy subjects and a cohort of 225 CKD patients using a specific ELISA. The demographic and clinical data of the patients are presented in Supplementary Table S1. As shown in Figure 1c, urinary TNC levels were elevated in CKD patients, compared to healthy subjects. Of note, serum TNC was also increased in CKD patients, but the magnitude of its increase was trivial (Supplementary Figure S1a and b). There was no correlation between urinary and serum TNC levels in CKD patients (Supplementary Figure S1c).
Urinary TNC is associated with kidney dysfunction and fibrosis in humans
We found that urinary TNC levels were correlated with the severity of CKD (Figure 1d). Notably, patients with relatively normal kidney function (eGFR >90 ml/min/1.73 m2) exhibited a significant increase in urinary TNC, compared to healthy subjects (Figure 1d). We next analyzed the relationship between urinary TNC and kidney function. As shown in Figure 1e, TNC levels were inversely associated with eGFR. Similarly, urinary TNC was also correlated with serum creatinine (Figure 1f), blood urea nitrogen (Figure 1g), urinary albumin creatinine ratio (Figure 1h), and urinary total protein (Supplementary Figure S1d).
We further assessed fibrotic lesions in kidney biopsies of CKD patients. As shown in Figure 1i, renal collagen deposition was increased in CKD patients. The severity of renal fibrosis was associated with the levels of urinary TNC (Figure 1i). Of the 30 CKD patients who had biopsy specimens, there was a correlation between urinary TNC and kidney fibrotic score (Figure 1j), suggesting that urinary TNC may be a biomarker of renal fibrogenesis in humans.
Endogenous TNC promotes partial EMT after AKI in vivo
Figure 2, a and b, shows the dynamic of TNC induction in mouse kidney after unilateral ischemia/reperfusion injury (UIRI), a model of AKI to CKD progression.20–23 To elucidate the role of TNC in this model, we carried out in vivo studies by knocking down TNC using short hairpin RNA (shRNA)-mediated approach. Mice were intravenously injected with either control-shRNA or TNC shRNA vectors (Figure 2c). As shown in Figure 2d, renal TNC mRNA was induced at 11 days after UIRI, which was abolished after shRNA-mediated inhibition. Western blotting and immunostaining for TNC protein showed the similar results (Figure 2e–g).
Figure 2. Knockdown of TNC ameliorates kidney injury, dysfunction and fibrosis after IRI.

(a, b) Renal TNC expression is induced at different time points after UIRI. TNC protein levels were assessed by Western blot analyses at 4, 7, and 11 days after UIRI. Representative Western blot (a) and quantitative data (b) are shown. Numbers (1–2) indicate each individual animal in a given group. **P < 0.01, ***P < 0.001 versus sham (n=6). (c) Experimental design. Black arrows indicate the injection of control pLVX-shRNA (Ctrl-shR) or pLVX-shTNC (TNC-shR) plasmids. Red arrows indicate the timing of IRI surgery. UNx, unilateral nephrectomy. (d) Quantitative real-time RT-PCR (qRT-PCR) analyses of renal TNC mRNA expression at 11 days after IRI. Mice were injected with either control shRNA or TNC shRNA at 4 days after unilateral IRI. ***P < 0.001 versus sham; ††P < 0.01 versus control shRNA (n=6). (e, f) Western blot analyses of renal TNC expression at 11 days after IRI in different groups as indicated. Representative Western blot (e) and quantitative data (f) are shown. Numbers (1–3) indicate each individual animal in a given group. ***P < 0.001 versus sham; ††† P < 0.001 versus control shRNA (n=6). (g) Representative immunohistochemical staining shows an effective knockdown of TNC protein in IRI kidney by shRNA-mediated strategy. Arrow indicates positive TNC expression. Scale bar, 50 µm. (h, i) Graphic presentation shows serum creatinine (Scr) and blood urea nitrogen (BUN) levels in different groups as indicated. ***P < 0.001 versus sham; ††P < 0.01, † P < 0.05 versus control shRNA (n=11). (j) Representative micrographs show renal collagen deposition at 11 days after IRI in various groups as indicated. Arrows indicate positive staining. Scale bar, 50 µm. (k) Graphic presentation of renal fibrotic lesions in different groups as indicated. ** P < 0.01 versus sham; ††P < 0.01 versus control shRNA (n=6).
We next examined the effect of TNC depletion on kidney function and fibrosis after UIRI. As illustrated in Figure 2h and i, serum creatinine and BUN were increased at 11 days after UIRI but reduced after TNC depletion. Similarly, knockdown of TNC also ameliorated collagen deposition in the kidney (Figure 2j and k).
We further investigated the effect of TNC on the expression of several fibrosis-related proteins. As shown in Figure 3a–f, knockdown of TNC inhibited renal expression of α-smooth muscle actin (α-SMA), vimentin, fibroblast-specific protein-1 (Fsp-1) and fibronectin in UIRI mice. TNC depletion also restored, at least partially, renal E-cadherin expression (Figure 3a and b). Of note, injection of control shRNA did not affect profibrotic responses of the kidney after UIRI (Supplementary Figure S2a–e). Immunostaining for E-cadherin, α-SMA, vimentin, fibronectin and Fsp1 proteins gave rise to the similar results (Figure 3g). Comparable data were also obtained when renal mRNA expression of α-SMA, vimentin, fibronectin and Fsp-1 was assessed by quantitative real-time PCR (qPCR) (Supplementary Figure S2f–i).
Figure 3. Knockdown of TNC protects tubular cell integrity after IRI.

(a-f) Western blot analyses show renal expression of E-cadherin, α-smooth muscle actin (α-SMA), vimentin, Fsp-1 and fibronectin proteins at 11 days after IRI in different groups as indicated. Representative Western blot (a) and quantitative data (b-f) are shown. Numbers (1–3) indicate each individual animal in a given group. The samples used for detecting α-tubulin in (a) were same as that in Figure 2e. **P < 0.01, ***P < 0.001 versus sham; †P < 0.05, †† P < 0.01, ††† P < 0.001 versus control shRNA (n=6). (g) Representative micrographs show renal E-cadherin, α-SMA, vimentin and fibronectin expression at 11 days after IRI in different groups as indicated. Kidney sections were subjected to immunofluorescence staining for E-cadherin and α-SMA, and immunohistochemical staining for vimentin, fibronectin and Fsp-1, respectively. Arrows indicate positive staining. Scale bar, 50 µm.
TNC induces tubular cells to undergo partial EMT in vitro
To confirm a role for TNC in tubular lesions, we performed in vitro experiments using both stable cell line and primary tubular epithelial cells. As shown in Figure 4a, incubation of human kidney proximal tubular cells (HKC-8) with TNC induced de novo expression of fibronectin, vimentin and α-SMA. High concentration of TNC at 100 ng/ml also suppressed E-cadherin expression in HKC-8 cells (Figure 4a). Of note, the doses of TNC needed to trigger EMT was greater than that to induce fibroblast activation (Supplementary Figure S3).
Figure 4. TNC induces tubular cells to undergo partial epithelial-mesenchymal transition (EMT) in vitro.

(a) TNC induced tubular cells to undergo partial EMT in a dose-dependent manner. Human kidney proximal tubular epithelial cells (HKC-8) were incubated with different concentrations of recombinant TNC protein as indicated. Cell lysates were subjected to Western blot analyses for fibronectin, E-cadherin, vimentin, α-SMA and α-tubulin. (b, c) TNC promoted HKC-8 cell migration as assessed by Boyden chamber transwell assay. Representative transwell migration assay (b) and quantitative data (c) are presented. Scale bar, 100 µm. *** P < 0.001 versus control. (d) Culture and characterization of mouse primary proximal tubular epithelial cells. Freshly isolated proximal tubules and primary tubular cells (phase contrast) are shown. Primary cells were immunostained with specific antibodies against E-cadherin and vimentin, respectively. Scale bar, 100 µm. (e) Representative Western blot analyses show that TNC promoted mouse primary proximal tubular epithelial cells to undergo partial EMT as well. Primary tubular epithelial cells were incubated with TNC (50 ng/ml) for 2 days. Cell lysates were subjected to Western blot analyses for E-cadherin, α-SMA, vimentin, Fsp-1, fibronectin and GAPDH. (f-j) Graphic presentation shows the relative levels (fold induction over the controls) of E-cadherin (f), α-SMA (g), vimentin (h), Fsp-1 (i) and fibronectin (j) in different groups. *P < 0.05 versus controls (n=3).
We next assessed the ability of TNC to promote HKC-8 cell migration by Boyden chamber assay. As shown in Figure 4b and c, increased migration of HKC-8 cells was observed after incubation with TNC. Similar results were obtained when cell migration was assessed by use of wound healing assay (data not shown).
To corroborate the role of TNC in mediating EMT, we investigated its action in primary tubular epithelial cells. As shown in Figure 4d, primary tubular cells were grown from isolated mouse kidney proximal tubules. Consistently, TNC also inhibited epithelial E-cadherin expression and induced numerous mesenchymal proteins including α-SMA, vimentin, Fsp-1 and fibronectin in primary tubular cells (Figure 4e–j).
TNC-rich tissue scaffold impairs tubular cell integrity ex vivo
To explore whether TNC-rich microenvironment plays a role in modulating tubular integrity, we performed ex vivo studies using decellularized kidney tissue scaffold (KTS).24, 25 As shown in Figure 5a, hematoxylin-eosin staining showed a typical KTS, in which all cells were removed. Western blotting confirmed abundant TNC in the KTS prepared from UIRI kidney, but not from sham controls (Figure 5b). Knockdown of TNC resulted in its deficiency in the KTS of UIRI mice (Figure 5b). As depicted in Figure 5c–f, vimentin, α-SMA and Fsp-1 were induced in HKC-8 cells when cultivated in the TNC-rich KTS from UIRI mice, but not in the TNC-depleted KTS.
Figure 5. TNC-rich kidney tissue scaffold promotes tubular cells to undergo partial EMT.

(a) Representative micrographs show kidney tissue section and decellularized kidney tissue scaffold (KTS) from mice. Sections were stained with hematoxylin-eosin. Scale bar, 100 µm. (b) Knockdown of TNC resulted in its depletion in the KTS. Representative Western blot analyses show TNC protein levels in the KTS prepared from different groups as indicated. (c-f) TNC-rich KTS facilitates tubular cells to undergo partial EMT ex vivo, whereas TNC-deprived KTS inhibited it. Western blots analyses show the levels of vimentin, α-SMA, Fsp-1 in different groups as indicated. Representative Western blot (c) and quantitative data (d-f) are shown. *P < 0.05 versus sham; †P<0.05 versus control shRNA (n=3). (g) Diagram shows the experimental protocol by which HKC-8 cells are cultured on TNC-enriched ECM scaffold. NRK-49F cells were treated with or without Shh to induce TNC expression in the absence or presence of TNC-siRNA. After three days, the ECM scaffold was then prepared after decellularization with EGTA. HKC-8 cells were inoculated on ECM scaffold for 2 days. (h) TNC abundance in the ECM scaffold was assessed by Western blot analyses after various treatments as indicated. (i-l) TNC-enriched ECM scaffold facilitates tubular cells to undergo partial EMT, whereas TNC-depleted ECM scaffold inhibited it. Western blots analyses show the expression of vimentin, α-SMA and Fsp-1 in different groups as indicated. Representative Western blot (i) and quantitative data (j-l) are shown. *P < 0.05 versus controls; †P < 0.05 versus Ctrl-siRNA (n=3).
We aslo prepared TNC-enriched ECM scaffold from sonic hedgehog (Shh)-treated normal rat kidney interstitial fibroblast (NRK-49F) cells using decellularization protocol, as previously reported.24 Figure 5g shows the experimental protocol, in which NRK-49F cells were transfected with control or TNC-specific small interfering RNA (siRNA) in the absence or presence of Shh. The TNC-enriched or TNC-depleted ECM scaffolds were made after decellularization (Figure 5h). When HKC-8 cells were inoculated on these ECM scaffolds, they underwent partial EMT as shown by induction of vimentin, -SMA and Fsp-1. As shown in Figure 5i–l, TNC-enriched ECM scaffold promoted HKC-8 cells to undergo EMT, whereas TNC-depleted one did not. Therefore, the TNC-rich microenvironment provides a unique niche that impairs tubular integrity by inducing EMT.
TNC activates integrin αvβ6/FAK/ERK signal cascade
To investigate the mechanism by which TNC promotes EMT, we studied the integrin signaling in vivo, because TNC binds to and activates integrins.24 As shown in Figure 6a–c, renal expression of integrin αv and β6 was upregulated in the kidneys after UIRI, whereas knockdown of TNC abolished the induction. Although other integrins such as integrin α2, α9 and β3 were also induced in the fibrotic kidney after UIRI, their expression was not substantially affected by TNC depletion (Figure 6d, and Supplementary Figure S4a–d). Consistently, immunostaining revealed a tubule-specific induction of integrin αv and β6 in UIRI kidney, and knockdown of TNC abolished their expression (Figure 6e). Furthermore, co-immunoprecipitation demonstrated that αv and β6 were detectable in the complexes precipitated by anti-TNC antibody from UIRI kidneys (Figure 6f and g). In the reciprocal experiments, TNC was detected in the complexes precipitated by anti-integrin αvβ6 (Figure 6h). As a negative control, the induction of renal integrin α9 after UIRI was neither affected by TNC depletion, nor interacted with TNC (Supplementary Figure S4e and f).
Figure 6. TNC physically interacts with integrin αvβ6.

(a-c) Western blot analyses of renal integrin αv and β6 protein expression at 11 days after IRI in different groups as indicated. Representative Western blot (a) and quantitative data (b and c) are shown. Numbers (1–3) indicate each individual animal in a given group. **P < 0.01, ***P < 0.001 versus sham; †† P < 0.01, ††† P < 0.001 versus control shRNA (n=6). (d) Western blot analyses of renal integrin α2, α9, β1and β3 protein expression at 11 days after IRI in different groups as indicated. Numbers (1–3) indicate each individual animal in a given group. (e) Representative immunohistochemical staining shows renal integrin αv and β6 expression in various groups as indicated. Boxed areas were enlarged. Arrows indicate positive staining. Scale bar, 50 µm. (f-h) Co-immunoprecipitation demonstrates that TNC bound to integrin αvβ6. The lysates from sham and IRI mice injected with control or TNC shRNA were immunoprecipitated (IP) with antibodies against TNC or integrin αvβ6, followed by immunoblotting (IB) with antibodies against TNC, integrin αv, or integrin β6 as indicated.
We next explored the downstream signaling of integrin αvβ6 activated by TNC in vivo. As shown in Figure 7a–c, FAK, the principal mediator of integrin signaling,26, 27 was activated in the kidney of UIRI mice, as illustrated by the phosphorylation of FAK at tyrosine 925. Such an activation of renal FAK was abolished after TNC depletion (Figure 7a and b). Accordingly, extracellular signal-regulated kinase-1 and −2 (ERK-1/2), the downstream effector of FAK signaling, were also activated in the kidney of UIRI mice, which was abolished by TNC depletion (Figure 7a and c). Immunostaining exhibited that both activated FAK and ERK-1/2 were localized in renal tubules of UIRI kidney (Figure 7d). Further studies using series sections confirmed that there was spatial association between interstitial TNC and tubular p-FAK and p-ERK expression in the kidney after UIRI (Figure 7e–g).
Figure 7. TNC activates integrin downstream FAK/MAPK signaling in vivo.

(a-c) Western blot analyses of renal p-FAK, FAK, p-ERK1/2 and ERK1/2 protein expression at 11 days after IRI in different groups as indicated. Representative Western blot (a) and quantitative data (b and c) are shown. Numbers (1–3) indicate each individual animal in a given group. **P < 0.01, ***P < 0.001 versus sham; † P < 0.05, ††† P < 0.001 versus control shRNA (Ctrl-shR) (n=6). (d) Representative immunohistochemical staining shows renal p-FAK and p-ERK expression in various groups as indicated. Boxed areas were enlarged. Arrows indicate positive staining. Scale bar, 50 µm. (e) Co-localization of TNC, p-FAK and p-ERK in the ischemic kidney at 11 days after UIRI. Series sections of the kidney were immuostained with antibodies against TNC, p-FAK and p-ERK1/2, respectively. Black arrows indicate the area with interstitial expression of TNC and tubular expression of p-FAK and p-ERK, whereas yellow arrows indicate the area with neither TNC nor p-FAK an p-ERK expression. Scale bar, 50 µm. (f, g) Linear regression shows the correlation between renal TNC protein and p-FAK (f) or p-ERK (g). Three images from each animal were taken (with total 6 animals) and protein abundances of TNC and p-FAK or p-ERK were assessed and expressed as positive cells per high power field and plotted. The correlation coefficient is given in the figures.
Blockade of integrin αvβ6/FAK signaling protects against EMT in vitro
We further delineated the role of integrin αvβ6/FAK/ERK-1/2 signal cascade in mediating TNC action in vitro. As shown in Figure 8a–c, TNC rapidly induced integrin αv and β6 in HKC-8 cells, which led to the activation of FAK and ERK-1/2 (Figure 8d–f). Incubation with a specific integrin αvβ6 blocking antibody (10D5) inhibited FAK and ERK1/2 phosphorylation and activation (Figure 8g–i). 10D5 also abolished TNC-mediated fibronectin, vimentin and α-SMA expression in HKC-8 cells (Figure 8j–m). Furthermore, treatment with PF573228, a specific inhibitor of FAK,28 abolished TNC-triggered FAK and ERK1/2 activation (Figure 8n–p) and fibronectin and α-SMA expression (Figure 8q–s). Notably, TNC did not activate EGF receptor signaling in HKC-8 cells (Supplementary Figure S4g).
Figure 8. TNC activates integrin αvβ6/FAK/MAPK signaling in vitro.

(a-c) TNC induced integrin αvβ6 expression in vitro. HKC-8 cells were treated with TNC (50 ng/ml) for various periods of time as indicated. Representative Western blots (a) and quantitative data (b and c) are presented. *P<0.05, **P<0.01 versus controls (n=3). (d-f) TNC induced a rapid FAK and ERK1/2 phosphorylation in vitro. HKC-8 cells were treated with TNC (50 ng/ml) for various periods of time as indicated. Representative Western blots (d) and quantitative data (e and f) are presented. *P < 0.05, **P < 0.01, ***P < 0.001 versus controls (n=3). (g-i) TNC-mediated FAK and ERK1/2 activation was dependent on αvβ6 integrin signaling. HKC-8 cells were pre-incubated with neutralizing antibody against integrin αvβ6 (10D5; 100 µg/ml) for 1 hour, followed by treatment with TNC (50 ng/ml) for 10 min. Representative Western blots (g) and quantitative data (h and i) are presented. *P < 0.05, **P < 0.01 versus controls; † P < 0.05 versus TNC alone (n=3). (j-m) Blocking of αvβ6 integrin signaling abolished the induction of partial EMT by TNC. HKC-8 cells were pre-incubated with 10D5 neutralizing antibody for 1 hour, followed by treatment with TNC for 2 days. Cell lysates were subjected to Western blot analyses with specific antibodies against fibronectin, vimentin, α-SMA and α-tubulin, respectively. Representative Western blots (j) and quantitative data (k-m) are presented. *P < 0.05 versus controls; † P < 0.05 versus TNC alone (n=3). (n-p) TNC-mediated ERK1/2 activation was dependent on FAK signaling. HKC-8 cells were pre-incubated with FAK inhibitor PF573228 (10 µM) for 1 hour, followed by treatment with TNC for 10 minutes. Representative Western blots (n) and quantitative data (o and p) are presented. *P < 0.05 versus controls; † P < 0.05, †† P < 0.01 versus TNC alone (n=3). (q-s) Blocking of FAK signalling abolished the induction of partial EMT by TNC. HKC-8 cells were pre-incubated with PF573228 for 1 hour, followed by treatment with TNC for 2 days. Representative Western blots (q) and quantitative data (r and s) are presented. *P < 0.05, **P < 0.01 versus controls; † P < 0.05, †† P < 0.01 versus TNC alone (n=3).
Inhibition of FAK preserves tubular integrity and ameliorates renal fibrosis in vivo
To validate the role of TNC/αvβ6/FAK/ERK cascade in mediating AKI-CKD progression, we finally examined whether inhibition of FAK reduces renal fibrosis after UIRI. Mice were injected intraperitoneally with FAK inhibitor PF573228 beginning at 4 days (Figure 9a). As shown in Figure 9b and c, PF573228 repressed renal FAK phosphorylation and activation at 11 days after UIRI, but it did not alter its protein abundance in the kidney. Accordingly, PF573228 also inhibited renal ERK-1/2 activation (Figure 9b, and Supplementary Figure S5a). Immunostaining gave rise to similar results (Figure 9d).
Figure 9. Pharmacologic blockade of FAK signaling preserves tubular cell integrity and attenuates renal fibrosis in vivo.

(a) Experimental design. Green arrows indicate the injection of PF573228. Red arrows indicate the timing of IRI surgery. UNx, unilateral nephrectomy. (b, c) Western blot analyses show inhibition of FAK signaling repressed renal p-FAK and p-ERK1/2 expression. Numbers (1–3) indicate each individual animal in a given group. (c) Graphic presentation demonstrated the relative levels of p-FAK protein expressions in different groups as indicated. ***P < 0.001 versus sham; ††† P < 0.001 versus UIRI controls (n=6). (d) Representative immunohistochemical staining shows renal p-FAK and p-ERK1/2 expression in various groups as indicated. Boxed areas were enlarged. Arrow indicates positive staining. Scale bar, 50 µm. (e, f) Graphic presentation shows serum creatinine (Scr) and BUN levels in different groups as indicated. ***P < 0.001 versus sham; ††P < 0.01, ††† P < 0.001 versus UIRI controls (n=6). (g) Western blot analyses show renal E-cadherin, fibronectin, α-SMA, vimentin and Fsp-1 expression. Numbers (1–3) indicate each individual animal in a given group. (h) Representative micrographs show renal E-cadherin, vimentin protein expression and collagen deposition in various groups as indicated. Kidney sections were subjected to immunohistochemical staining staining for E-cadherin and vimentin, and Masson’s trichrome staining (MTS) for collagen deposition, respectively. Boxed areas were enlarged. Arrows indicate positive staining. Scale bar, 50 µm.
We finally examined the consequence of FAK inhibition in UIRI mice. As shown in Figure 9e and f, inhibition of FAK by PF573228 alleviated kidney dysfunction, as assessed by serum creatinine and BUN. Furthermore, PF573228 restored renal E-cadherin and inhibited fibronectin, α-SMA, vimentin and Fsp-1 in the kidney after UIRI (Figure 9g, and Supplementary Figure S5b–f). Similar results were obtained when renal mRNA levels of fibronectin, α-SMA, vimentin and Fsp-1 were assessed by qPCR (Supplementary Figure S5g–j).
Immunostaining revealed that inhibition of FAK largely preserved renal E-cadherin and inhibited the expression of vimentin (Figure 9h), fibronectin, α-SMA and Fsp-1 (Supplementary Figure S5k). PF573228 also inhibited fibroblast proliferation, reduced collagen deposition and mitigated kidney injury in the fibrotic kidney after UIRI (Figure 9h, and Supplementary Figure S5l and S6). Together, these results suggest that FAK could be a novel therapeutic target of fibrotic CKD.
DISCUSSION
Kidney fibrotic lesions are not evenly distributed throughout renal parenchyma, implying that local microenvironment plays a decisive role in eliciting fibrogenic responses. The molecular composition of such microenvironment, as well as the underlying mechanism of action, was poorly characterized. In this study, we show that TNC, an ECM protein identified as a major component of fibrogenic niche,24 is markedly upregulated in renal tubulointerstitium of human CKD and urinary TNC levels are correlated with the severity of renal fibrosis. Furthermore, using in vivo, ex vivo, and in vitro approaches, we have provided unambiguous evidence that TNC is a key pathogenic mediator that impairs tubular integrity by triggering EMT via sequential activation of integrin αvβ6/FAK/ERK-1/2 signal cascade. Finally, we demonstrate that blockade of FAK signaling by a specific inhibitor protects against kidney fibrosis in vivo. These results establish urinary TNC as a noninvasive surrogate that is associated with and may predict renal fibrosis in CKD patients. Our studies also highlight an important role of TNC-rich microenvironment in dictating tubular cell behavior in the evolution of AKI to CKD progression.
Kidney fibrosis is a hallmark of progressive CKD and closely correlated with the decline of renal function. At present, the only method for assessment of fibrotic lesion is through renal biopsy.29 Therefore, there is urgent need to develop a noninvasive biomarker as a surrogate to assess, predict and monitor the development and progression of kidney fibrosis in CKD patients. In this context, the present study identifying urinary TNC as a biomarker for renal fibrogenesis is of importance. One can envision that urinary TNC may be used to monitor CKD progression, predict patient outcomes and guide therapeutic management. Of interest, urinary TNC is already elevated significantly in the patients with early stages of CKD who have normal renal function (eGFR>90 ml/min per 1.73m2) (Figure 1). This suggests that urinary TNC could be an early noninvasive biomarker that illustrates the ongoing fibrogenesis. It should be pointed out that in the advantaged stage of CKD, urinary TNC is not elevated in some patients (Figure 1d). Although not tested, this probably reflects the deficiency of cell activities in failed kidney due to excessive scar formation. Therefore, it is conceivable to speculate that urinary TNC manifests the state of active fibrogenesis, rather than fibrosis per sec. As such, assessing urinary TNC level will afford one to have an ample opportunity for therapeutic intervention.
TNC as a matricellular protein regulates various biologic processes by interacting with growth factors and cell membrane receptors.30–32 Unlike other ECM proteins such as fibronectin, collagen and laminin, TNC is not obligatory for maintaining ECM structural integrity but elicits its biologic actions via regulating cell behaviors.33, 34 TNC has been shown to organize a fibrogenic niche that promotes fibroblast activation and proliferation.24 Furthermore, it recruits and enriches Wnt ligands from surrounding milieu and presents them to the effector cells.32 Interestingly, the present study offers a novel explanation by showing that TNC impairs tubular integrity via inducing partial EMT. Although it remains controversial whether there is a complete EMT in kidney fibrosis,35–37 evidence points to the existence of partial EMT, characterized by tubular cells co-expressing both epithelial and mesenchymal markers but still residing within tubular basement membrane.11, 12, 38 Using a combination of in vitro, ex vivo and in vivo approaches, we show herein that TNC or TNC-rich KTS inhibits epithelial E-cadherin and induces mesenchymal markers including vimentin, Fsp-1, α-SMA and fibronectin (Figures 3–5). Consistently, knockdown of TNC preserves E-cadherin and prevents mesenchymal transition of tubular cells. It is worthwhile to stress that various responses of tubular cells after kidney injury such as partial EMT, cell cycle arrest and cellular senescence might be inter-connected and could represent a different spectrum of tubular response to insults. As partial EMT has been shown to induce subsequent cell cycle arrest,11 the loss of tubular integrity due to partial EMT may be an initial and crucial event in the pathogenesis of kidney fibrosis.
As TNC is produced by activated fibroblasts (Figure 5),24 the findings that TNC impairs tubular integrity highlight an active interplay between fibroblasts and tubular cells in fibrotic CKD. While tubular injury can lead to fibroblast activation by secreting a host of soluble factors such as Shh, Wnts and TGF-β1,39–41 little is known about whether fibroblast activation affects tubular pathology. In this perspective, the present study illustrates for the first time that fibroblasts regulate tubular cell behaviors via orchestrating pro-EMT microenvironment. The mechanism by which TNC impairs tubular integrity remains to be fully delineated, but it is most likely related to the activation of integrin αvβ6/FAK/ERK signal cascade (Figures 6–9). Integrin heterodimers of α2/7/8/9β1 and αvβ1/3/6 are reported to play a role in mediating TNC signaling. Of these integrins, epithelial cells express α2/9β1 and αvβ1/3/6.42 However, because integrins α2, α9, β1 and β3 are not subjected to regulation by TNC (Figure 6), we conclude that αvβ6 are the major integrins mediating TNC action in diseased kidney. In support of this notion, αvβ6 are specifically induced in tubular epithelium and TNC/αvβ6 engagement is evident in vivo (Figure 6). Furthermore, inhibition of αvβ6 signaling with a specific blocking antibody abolishes TNC-mediated FAK/ERK1/2 activation and partial EMT in vitro (Figure 8). Consistently, an earlier report has also shown that TNC induces EMT-like change in breast cancer by binding to αvβ6 integrins.43
It should be noted that integrin αvβ6 as an epithelial-specific integrin is a receptor for numerous ligands including TNC, fibronectin, vitronectin and the latency associated peptide (LAP) of TGF-β.44 The downstream mediator of αvβ6 activation by TNC appears to be FAK, the intermediate effector of integrin signaling,26, 27 as both FAK and its downstream ERK-1/2 are specifically activated in renal tubular epithelial cells (Figure 7). Consistently, TNC induces αvβ6, activates FAK and ERK-1/2, and triggers EMT in cultured tubular cells (Figure 8). Blockade of FAK activation by specific inhibitor abolishes TNC actions in vitro (Figure 8) and renal fibrosis in vivo (Figure 9), underscoring a key role of FAK signaling in mediating TNC-induced tubular injury and kidney fibrosis. Because αvβ6 integrins also play a role in TGF-β activation,45 we cannot exclude the possibility that TNC-mediated αvβ6 activation may upregulate TGF-β signaling as well, thereby promoting tubular EMT and renal fibrosis.
In summary, we have shown herein that TNC is markedly induced in wide variety of CKD in humans and its levels in the urine of CKD patients could be used as a noninvasive biomarker for assessing the state of kidney fibrogenesis. We show that TNC-enriched microenvironment impairs tubular cell integrity by triggering EMT via αvβ6 integrins/FAK/ERK-1/2 pathway. Therefore, targeting this signal cascade could be a novel strategy for therapeutic intervention of renal fibrosis.
CONCISE METHODS
Human urine and kidney biopsies samples
Human urine, serum and kidney specimens were obtained from diagnostic renal biopsies performed at the Nanfang Hospital, Southern Medical University. Some urine and serum samples were also collected from healthy volunteers. All the studies involving human samples were approved by the Ethic Committee on Human Subjects of the Nanfang Hospital, Sothern Medical University.
TNC ELISA
Human Tenascin-C Assay Kit was purchased from the Immuno-Biological Laboratories (IBL) (#27767; IBL Company, Gunma, Japan). Human urinary and serum TNC were measured according to the assay procedures specified by the manufacturer.
Animal models
Male BALB/c mice were obtained from the Southern Medical University Animal Center (Guangzhou, China). UIRI was performed as described previously.40, 46 At day 10 after UIRI, the contralateral intact kidney was removed. Mice were sacrificed at 11 days post-UIRI, and serum and kidney tissues collected for various analyses. All animal studies were approved by the Experimental Animal Committee at the Nanfang Hospital, Southern Medical University.
Knockdown of TNC in vivo
Knockdown of TNC expression in vivo was performed using shRNA-mediated approach, as recently reported.32 Male BALB/c mice were divided into three groups (n=6 in each group): (i) sham-operated mice, (ii) UIRI mice injected with control shRNA, and (iii) UIRI mice injected with TNC-shRNA. Four days after UIRI, mice were injected with either pLVX-shTNC or control (pLVX-control) plasmids via tail-vein injection.
Inhibition of FAK in vivo
Mice were divided into three groups (n=6 in each group): (i) sham-operated mice, (ii) UIRI injected with vehicle, and (iii) UIRI injected with FAK inhibitor. Mice were daily injected intraperitoneally with FAK inhibitor PF573228 at 5 mg/kg body wt beginning from day 4 after surgery. Mice were sacrificed at 11 days post-IRI, and serum and kidney tissues were analyzed.
Cell culture and treatment
Human kidney proximal tubular cell line (HKC-8) was described previously.47 HKC-8 cells were treated by human TNC at different dosages for various periods of time as indicated. For some experiments, HKC-8 cells were pretreated with integrin αvβ6 blocking antibody (10D5) or PF573228 for 1 hour, followed by incubating with vehicle or TNC (50 ng/ml).
Mouse primary proximal tubular epithelial cells
Mouse primary proximal tubular cells were isolated and cultured as previously described.48 Briefly, the cortical part of mouse kidneys were minced, then digested in pre-warmed 0.75 mg/ml collagenase 4 for 40 min at 37°C, after which the mashed tissue was sieved in DMEM/F-12. The tubular tissues were centrifuged using 31% Percoll gradients for 10 minutes, resuspended and washed twice with DMEM/F-12. Cells were cultivated for 4–8 days until they reached 60%–80% confluency and characterized by staining for epithelial E-cadherin and mesenchymal vimentin.
Western blot analyses
Protein expression was analyzed by Western blot analysis as described previously.49 The antibodies used are summarized in Supplemental Table S2.
Quantitative real-time RT-PCR
Total RNA was isolated, and qPCR was performed on an ABI PRISM 7000 Sequence Detection System as described previously.50 The mRNA levels of different genes were calculated after normalization with β-actin. The sequences of primer pairs are presented as Supplemental Table S3.
Histology, immunohistochemical and immunofluorescence staining
Paraffin-embedded mouse kidney sections were prepared by a routine procedure. The sections were stained with Masson’s trichrome staining (MTS) reagents. Immunohistochemical staining was performed as described previously.24 Antibodies used are summarized in Supplemental Table S2.
Preparation of fibroblast-derived ECM scaffold
Serum-starved NRK-49F cells were stimulated by Shh at 50 ng/ml for 3 days. Decellularization was carried out with EGTA in calcium-free PBS.
Preparation of kidney tissue scaffold
KTS was prepared according to an established protocol.24 Briefly, kidney was cut into 3–4 slices of same thickness along the sagittal plane. The kidney slices were then subjected to multiple steps of decellularization procedures as described.24
Co-immunoprecipitation
The interaction of TNC with integrin αvβ6 in mice was determined by co-immunoprecipitation. Briefly, kidney homogenates were immunoprecipitated with anti-TNC antibody and protein A/G plus agarose. The precipitated complexes were immunoblotted with anti-integrin αv, anti-integrin β6, anti-integrin α9 or anti-TNC antibodies, respectively. In the reciprocal experiments, kidney homogenates were immunoprecipitated with anti-integrin αvβ6 antibody, followed by immunoblotting with anti-TNC antibody.
Statistical analyses
All data examined were expressed as mean ± SEM. Statistical analyses of the data were performed using SPSS 13.0 (SPSS Inc, Chicago, IL). Comparisons between groups were made by t test, or using one-way ANOVA followed by the Student-Newman-Kuels test or Dunnett’s T3 test. Spearman (nonparametric) correlation analysis was used to assess the relationship between urinary TNC and other variables. P<0.05 was considered significant.
Supplementary Material
Figure S1. Serum TNC levels are slightly increased in human CKD. (a) Serum TNC levels in CKD patients (n=126) and healthy subjects (n=26). Graphic presentation shows serum TNC levels (µg/ml) in individual patients with CKD and healthy subjects. **P < 0.01. (b) Serum TNC levels in CKD patients with eGFR > 90 or < 90 ml/min per 1.73 m2. **P < 0.01 versus healthy subjects. (c) Linear regression showed no correlation between urinary and serum TNC levels in CKD patients. (d) There was a correlation between urinary total protein and urinary TNC levels in CKD patients. Correlation coefficient is given in the figure.
Figure S2. Knockdown of TNC prevents the mRNA induction of mesenchymal cell markers in the kidney. (a-e) Control scrambled siRNA did not affect renal expression of profibrotic proteins after UIRI. Representative Western blots (a) and quantitative data on fibronectin (b), E-cadherin (c), vimentin (d) and a-SMA (e) are shown. (f-i) Knockdown of TNC inhibited renal mRNA induction of mesenchymal markers after UIRI. Quantitative real-time RT-PCR analyses of renal α-SMA (f), fibronectin (g), vimentin (h) and Fsp-1 (i) mRNA levels in different groups as indicated. ***P < 0.001, **P < 0.01 versus sham; †P < 0.05 versus control shRNA (n=6).
Figure S3. High concentration of TNC is required for triggering partial EMT. (a) Rat renal interstitial fibroblast (NRK-49F) cells were incubated with different concentrations of recombinant TNC protein as indicated. Cell lysates were subjected to Western blot analyses for fibronectin, α-SMA, c-fos, PCNA, cyclin D1 and α-tubulin. (b) Human kidney proximal tubular (HKC-8) cells were incubated with different concentrations of recombinant TNC protein as indicated. Cell lysates were subjected to Western blot analyses for fibronectin, vimentin, α-SMA and α-tubulin, respectively.
Figure S4. Protein levels of different integrins in the kidney after various treatments. (a-d) Graphic presentation of the quantitative data shows the relative levels of integrin α2 (a), α9 (b), β1 (c) and β3 (d) proteins in different groups as indicated. *P < 0.05 versus sham; †P < 0.05 versus control shRNA (n=6). (e) Knockdown of TNC did not affect integrin α9 expression in the kidney after UIRI. (f) Co-immunoprecipitation showed no interaction between TNC and integrin α9 in the kidney after UIRI. The lysates from sham and UIRI kidneys injected with Ctrl-shRNA or TNC shRNA were immunoprecipitated (IP) with antibodies against TNC, followed by immunoblotting (IB) with antibodies against TNC or integrin α9 as indicated. (g) TNC (50 ng/ml) did not induce EGFR activation in renal tubular HKC-8 cells. As a positive control, EGF markedly stimulated EGFR phosphorylation at tyrosine-845.
Figure S5. Pharmacologic inhibition of FAK signaling preserves tubular cell integrity and attenuates renal fibrosis. (a-f) Graphic presentation shows the relative levels of p-ERK1/2 (a), E-cadherin (b), fibronectin (c), α-SMA (d), vimentin (e) and Fsp-1 (f) proteins in different groups as indicated. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham; ††P < 0.01, ††† P < 0.001 versus UIRI controls (n=6). (g-j) Quantitative real-time RT-PCR analyses of renal mRNA levels of fibronectin (g), α-SMA (h), vimentin (i) and Fsp-1 (j) in different groups as indicated. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham; †P < 0.05, †† P < 0.01 versus UIRI controls (n=6). (k) Representative immunohistochemical staining shows renal expression of fibronectin, α-SMA and Fsp-1 proteins in different groups as indicated. Boxed areas were enlarged. Arrows indicate positive staining. Scale bar, 50 µm. (l) Graphical representation shows the extent of fibrotic lesions in different groups as indicated. ** P < 0.01 versus sham; ††P < 0.01 versus UIRI controls (n=6).
Figure S6. Preservation of tubular integrity by blocking FAK signaling reduces fibroblast proliferation and kidney injury after UIRI. (a) Representative micrographs show renal PCNA expression and kidney morphology in various groups as indicated. Kidney sections were subjected to immunohistochemical staining for PCNA or PAS staining for kidney morphology, respectively. Yellow arrows indicate PCNA-positive cells in the interstitium, whereas black arrows show PCNA-positive cells in renal tubules. Scale bar, 50 µm. (b) Quantitative determination of the PCNA-positive cells in renal tubular and interstitial compartments. *P < 0.05, ***P < 0.001 versus sham; ††P < 0.01, ††† P < 0.001 versus UIRI 4d; ### P < 0.001 versus UIRI 11d. HPF, high power field.
Table S1. Demographic and clinical data of healthy subjects and CKD patients.
Table S2. The sources of antibodies used in this study.
Table S3. Nucleotide sequences of the primers used for qPCR
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China grants 81521003, 81770715 and 81770737; National Institutes of Health grant DK064005; Guangzhou Regenerative Medicine and Health Guangdong Laboratory grants 2018GZR110104001 and 2018GZR110102004.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
All the authors declared no competing interests.
Supplementary material is linked to the online version of the paper at www.kidney-international.org.
REFERENCES
- 1.Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038–2047. [DOI] [PubMed] [Google Scholar]
- 2.Meguid El Nahas A and Bello AK. Chronic kidney disease: the global challenge. Lancet. 2005;365:331–340. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Djudjaj S and Boor P. Cellular and molecular mechanisms of kidney fibrosis. Mol Aspects Med. 2019;65:16–36. [DOI] [PubMed] [Google Scholar]
- 5.Campanholle G, Ligresti G, Gharib SA, et al. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am J Physiol Cell Physiol. 2013;304:C591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mutsaers HA and Olinga P. Editorial: Organ fibrosis: Triggers, pathways, and cellular plasticity. Front Med. 2016;3:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeisberg M and Neilson EG. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010;21:1819–1834. [DOI] [PubMed] [Google Scholar]
- 8.Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest. 2014;124:2299–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu BC, Tang TT, Lv LL, et al. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 2018;93:568–579. [DOI] [PubMed] [Google Scholar]
- 10.Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lovisa S, LeBleu VS, Tampe B, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21:998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grande MT, Sanchez-Laorden B, Lopez-Blau C, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 2015;21:989–997. [DOI] [PubMed] [Google Scholar]
- 13.Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo C, Zhou S, Zhou Z, et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J Am Soc Nephrol. 2018;29:1238–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou D and Liu Y. Renal fibrosis in 2015: Understanding the mechanisms of kidney fibrosis. Nat Rev Nephrol. 2016;12:68–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tucker RP and Chiquet-Ehrismann R. Tenascin-C: Its functions as an integrin ligand. Int J Biochem Cell Biol. 2015;65:165–168. [DOI] [PubMed] [Google Scholar]
- 17.Midwood KS, Hussenet T, Langlois B, et al. Advances in tenascin-C biology. Cell Mol Life Sci. 2011;68:3175–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhattacharyya S, Wang W, Morales-Nebreda L, et al. Tenascin-C drives persistence of organ fibrosis. Nat Commun. 2016;7:11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Midwood K, Sacre S, Piccinini AM, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–780. [DOI] [PubMed] [Google Scholar]
- 20.Fu Y, Tang C, Cai J, et al. Rodent models of AKI-CKD transition. Am J Physiol Renal Physiol. 2018;315:F1098–F1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Clef N, Verhulst A, D’Haese PC, et al. Unilateral renal ischemia-reperfusion as a robust model for acute to chronic kidney injury in mice. PLoS One. 2016;11:e0152153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao L, Zhou D, Tan RJ, et al. Sustained activation of Wnt/beta-catenin signaling drives AKI to CKD progression. J Am Soc Nephrol. 2016;27:1727–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei J, Zhang J, Wang L, et al. New mouse model of chronic kidney disease transitioned from ischemic acute kidney injury. Am J Physiol Renal Physiol. 2019;317:F286–F295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu H, Tian Y, Zhou L, et al. Tenascin-C is a major component of the fibrogenic niche in kidney fibrosis. J Am Soc Nephrol. 2017;28:785–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross EA, Williams MJ, Hamazaki T, et al. Embryonic stem cells proliferate and differentiate when seeded into kidney scaffolds. J Am Soc Nephrol. 2009;20:2338–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee BY, Timpson P, Horvath LG, et al. FAK signaling in human cancer as a target for therapeutics. Pharmacol Ther. 2015;146:132–149. [DOI] [PubMed] [Google Scholar]
- 27.Lechertier T and Hodivala-Dilke K. Focal adhesion kinase and tumour angiogenesis. J Pathol. 2012;226:404–412. [DOI] [PubMed] [Google Scholar]
- 28.Slack-Davis JK, Martin KH, Tilghman RW, et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–14852. [DOI] [PubMed] [Google Scholar]
- 29.Farris AB, Adams CD, Brousaides N, et al. Morphometric and visual evaluation of fibrosis in renal biopsies. J Am Soc Nephrol. 2011;22:176–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sawyer AJ and Kyriakides TR. Matricellular proteins in drug delivery: Therapeutic targets, active agents, and therapeutic localization. Adv Drug Deliv Rev. 2016;97:56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen S, Fu H, Wu S, et al. Tenascin-C protects against acute kidney injury by recruiting Wnt ligands. Kidney Int. 2019;95:62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida T, Akatsuka T and Imanaka-Yoshida K. Tenascin-C and integrins in cancer. Cell Adh Migr. 2015;9:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paron I, Berchtold S, Voros J, et al. Tenascin-C enhances pancreatic cancer cell growth and motility and affects cell adhesion through activation of the integrin pathway. PLoS One. 2011;6:e21684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quaggin SE and Kapus A. Scar wars: mapping the fate of epithelial-mesenchymal-myofibroblast transition. Kidney Int. 2011;80:41–50. [DOI] [PubMed] [Google Scholar]
- 36.Kriz W, Kaissling B and Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest. 2011;121:468–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.LeBleu VS, Taduri G, O’Connell J, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou D, Li Y, Zhou L, et al. Sonic hedgehog is a novel tubule-derived growth factor for interstitial fibroblasts after kidney injury. J Am Soc Nephrol. 2014;25:2187–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou D, Fu H, Zhang L, et al. Tubule-derived Wnts are required for fibroblast activation and kidney fibrosis. J Am Soc Nephrol. 2017;28:2322–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan RJ, Zhou D and Liu Y. Signaling crosstalk between tubular epithelial cells and interstitial fibroblasts after kidney injury. Kidney Dis. 2016;2:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Humphries JD, Byron A and Humphries MJ. Integrin ligands at a glance. J Cell Sci. 2006;119:3901–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katoh D, Nagaharu K, Shimojo N, et al. Binding of alphavbeta1 and alphavbeta6 integrins to tenascin-C induces epithelial-mesenchymal transition-like change of breast cancer cells. Oncogenesis. 2013;2:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bandyopadhyay A and Raghavan S. Defining the role of integrin alphavbeta6 in cancer. Curr Drug Targets. 2009;10:645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mamuya FA and Duncan MK. aV integrins and TGF-beta-induced EMT: a circle of regulation. J Cell Mol Med. 2012;16:445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skrypnyk NI, Harris RC and de Caestecker MP. Ischemia-reperfusion model of acute kidney injury and post injury fibrosis in mice. J Vis Exp. 2013; (78): doi: 10.3791/50495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou D, Tian Y, Sun L, et al. Matrix metalloproteinase-7 is a urinary biomarker and pathogenic mediator of kidney fibrosis. J Am Soc Nephrol. 2017;28:598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernhardt A, Fehr A, Brandt S, et al. Inflammatory cell infiltration and resolution of kidney inflammation is orchestrated by the cold-shock protein Y-box binding protein-1. Kidney Int. 2017;92:1157–1177. [DOI] [PubMed] [Google Scholar]
- 49.Zhou L, Chen X, Lu M, et al. Wnt/beta-catenin links oxidative stress to podocyte injury and proteinuria. Kidney Int. 2019;95:830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Y, Wang C, Hong X, et al. Wnt/beta-catenin signaling mediates both heart and kidney injury in type 2 cardiorenal syndrome. Kidney Int. 2019;95:815–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Serum TNC levels are slightly increased in human CKD. (a) Serum TNC levels in CKD patients (n=126) and healthy subjects (n=26). Graphic presentation shows serum TNC levels (µg/ml) in individual patients with CKD and healthy subjects. **P < 0.01. (b) Serum TNC levels in CKD patients with eGFR > 90 or < 90 ml/min per 1.73 m2. **P < 0.01 versus healthy subjects. (c) Linear regression showed no correlation between urinary and serum TNC levels in CKD patients. (d) There was a correlation between urinary total protein and urinary TNC levels in CKD patients. Correlation coefficient is given in the figure.
Figure S2. Knockdown of TNC prevents the mRNA induction of mesenchymal cell markers in the kidney. (a-e) Control scrambled siRNA did not affect renal expression of profibrotic proteins after UIRI. Representative Western blots (a) and quantitative data on fibronectin (b), E-cadherin (c), vimentin (d) and a-SMA (e) are shown. (f-i) Knockdown of TNC inhibited renal mRNA induction of mesenchymal markers after UIRI. Quantitative real-time RT-PCR analyses of renal α-SMA (f), fibronectin (g), vimentin (h) and Fsp-1 (i) mRNA levels in different groups as indicated. ***P < 0.001, **P < 0.01 versus sham; †P < 0.05 versus control shRNA (n=6).
Figure S3. High concentration of TNC is required for triggering partial EMT. (a) Rat renal interstitial fibroblast (NRK-49F) cells were incubated with different concentrations of recombinant TNC protein as indicated. Cell lysates were subjected to Western blot analyses for fibronectin, α-SMA, c-fos, PCNA, cyclin D1 and α-tubulin. (b) Human kidney proximal tubular (HKC-8) cells were incubated with different concentrations of recombinant TNC protein as indicated. Cell lysates were subjected to Western blot analyses for fibronectin, vimentin, α-SMA and α-tubulin, respectively.
Figure S4. Protein levels of different integrins in the kidney after various treatments. (a-d) Graphic presentation of the quantitative data shows the relative levels of integrin α2 (a), α9 (b), β1 (c) and β3 (d) proteins in different groups as indicated. *P < 0.05 versus sham; †P < 0.05 versus control shRNA (n=6). (e) Knockdown of TNC did not affect integrin α9 expression in the kidney after UIRI. (f) Co-immunoprecipitation showed no interaction between TNC and integrin α9 in the kidney after UIRI. The lysates from sham and UIRI kidneys injected with Ctrl-shRNA or TNC shRNA were immunoprecipitated (IP) with antibodies against TNC, followed by immunoblotting (IB) with antibodies against TNC or integrin α9 as indicated. (g) TNC (50 ng/ml) did not induce EGFR activation in renal tubular HKC-8 cells. As a positive control, EGF markedly stimulated EGFR phosphorylation at tyrosine-845.
Figure S5. Pharmacologic inhibition of FAK signaling preserves tubular cell integrity and attenuates renal fibrosis. (a-f) Graphic presentation shows the relative levels of p-ERK1/2 (a), E-cadherin (b), fibronectin (c), α-SMA (d), vimentin (e) and Fsp-1 (f) proteins in different groups as indicated. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham; ††P < 0.01, ††† P < 0.001 versus UIRI controls (n=6). (g-j) Quantitative real-time RT-PCR analyses of renal mRNA levels of fibronectin (g), α-SMA (h), vimentin (i) and Fsp-1 (j) in different groups as indicated. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham; †P < 0.05, †† P < 0.01 versus UIRI controls (n=6). (k) Representative immunohistochemical staining shows renal expression of fibronectin, α-SMA and Fsp-1 proteins in different groups as indicated. Boxed areas were enlarged. Arrows indicate positive staining. Scale bar, 50 µm. (l) Graphical representation shows the extent of fibrotic lesions in different groups as indicated. ** P < 0.01 versus sham; ††P < 0.01 versus UIRI controls (n=6).
Figure S6. Preservation of tubular integrity by blocking FAK signaling reduces fibroblast proliferation and kidney injury after UIRI. (a) Representative micrographs show renal PCNA expression and kidney morphology in various groups as indicated. Kidney sections were subjected to immunohistochemical staining for PCNA or PAS staining for kidney morphology, respectively. Yellow arrows indicate PCNA-positive cells in the interstitium, whereas black arrows show PCNA-positive cells in renal tubules. Scale bar, 50 µm. (b) Quantitative determination of the PCNA-positive cells in renal tubular and interstitial compartments. *P < 0.05, ***P < 0.001 versus sham; ††P < 0.01, ††† P < 0.001 versus UIRI 4d; ### P < 0.001 versus UIRI 11d. HPF, high power field.
Table S1. Demographic and clinical data of healthy subjects and CKD patients.
Table S2. The sources of antibodies used in this study.
Table S3. Nucleotide sequences of the primers used for qPCR