

Summary:
The ability to transfect synthetic mRNAs into cells to measure processes such as translation efficiency or mRNA decay has been an invaluable tool in cell biology. The use of electroporation over other methods of transfection is an easy, inexpensive, highly efficient and scalable method to introduce synthetic mRNA into a wide range of cell types. More recently, coupling of noncoding RNA sequences or protein coding regions from viral pathogens to fluorescent or bioluminescence proteins in RNA “reporters” has permitted study of host-pathogen interactions. These can range from virus infection of cells to translation of the viral genome, replication and stability of viral RNAs or the efficacy of host antiviral responses. In this chapter, we describe a method for electroporating viral RNA reporters into both fibroblastic and myeloid cells that encode firefly or Renilla luciferase, whose reaction with specific substrates and light emitting activity is a measure of viral RNA translation efficiency. We have used this method to examine host interferon dependent responses that inhibit viral translation along with identifying secondary structures in the 5’ non-translated region and microRNA binding sites in the 3’non-translated region that are responsible for antagonizing the host innate immune responses and restricting viral cell tropism.
Keywords: alphavirus, translation, electroporation, neon transfection, luciferase
1. Introduction:
The introduction of synthetic RNA into cells, especially viral RNA, can be a useful technique in molecular virology to generate recombinant viruses and to also study various aspects of host-pathogen interactions. Alphaviruses are mosquito-borne, positive-strand RNA members of the virus family Togaviridae. The ability to convert virus RNA into an infectious DNA clone has enabled the construction of full-length replication-competent alphaviruses that encode fluorescent or bioluminescent proteins (1–14). These “reporter viruses” aid in studying host-pathogen interactions both in vitro and in live animals by providing a quantifiable readout of either fluorescence or bioluminescence activity.
After infection of a cell, the virus RNA genome, which resembles host cell messenger RNA, is translated by host cell translation complexes producing the virus non-structural proteins and initiating virus replication (15). The host translation machinery is able to efficiently translate alphavirus RNA due to the presence of a type 0 7-methylguanosine cap (m7G) structure at the 5’ end of the virus genome and a 3’ poly(A) tail (16, 17). Virus translation is enhanced by conserved sequence elements (CSE) encoded in the virus’ 5’ non-translated region (NTR) as well as two stem-loop secondary structure elements within the coding region of non-structural protein 1 (nsP1) and a 3’ NTR adjacent to the poly(A) tail (15).
To specifically study translation of the incoming alphavirus RNA, a viral translation reporter is constructed that encodes the 5’ NTR and nsP1 CSE fused in-frame with the firefly luciferase gene followed by the virus 3’ NTR and poly (A) (Figure 1). RNA synthesized from this template contains the translation control structures in the parental virus genome thus mimicking translation of the incoming viral genome (6, 18). Such alphavirus translation reporters are being used to study interferon (IFN) dependent host responses that inhibit translation of the virus genome and host factors that restrict cellular tropism of viruses (1, 6, 18–20). Importantly, the construction of alphavirus translation reporters enables the study of aspects of virus life cycle in a biosafety level (BSL) 2 environment instead of the more restrictive BSL3 biocontainment required for some alphaviruses.
Figure 1: Diagram of the alphavirus RNA genome and translation reporter and the host-mimic.
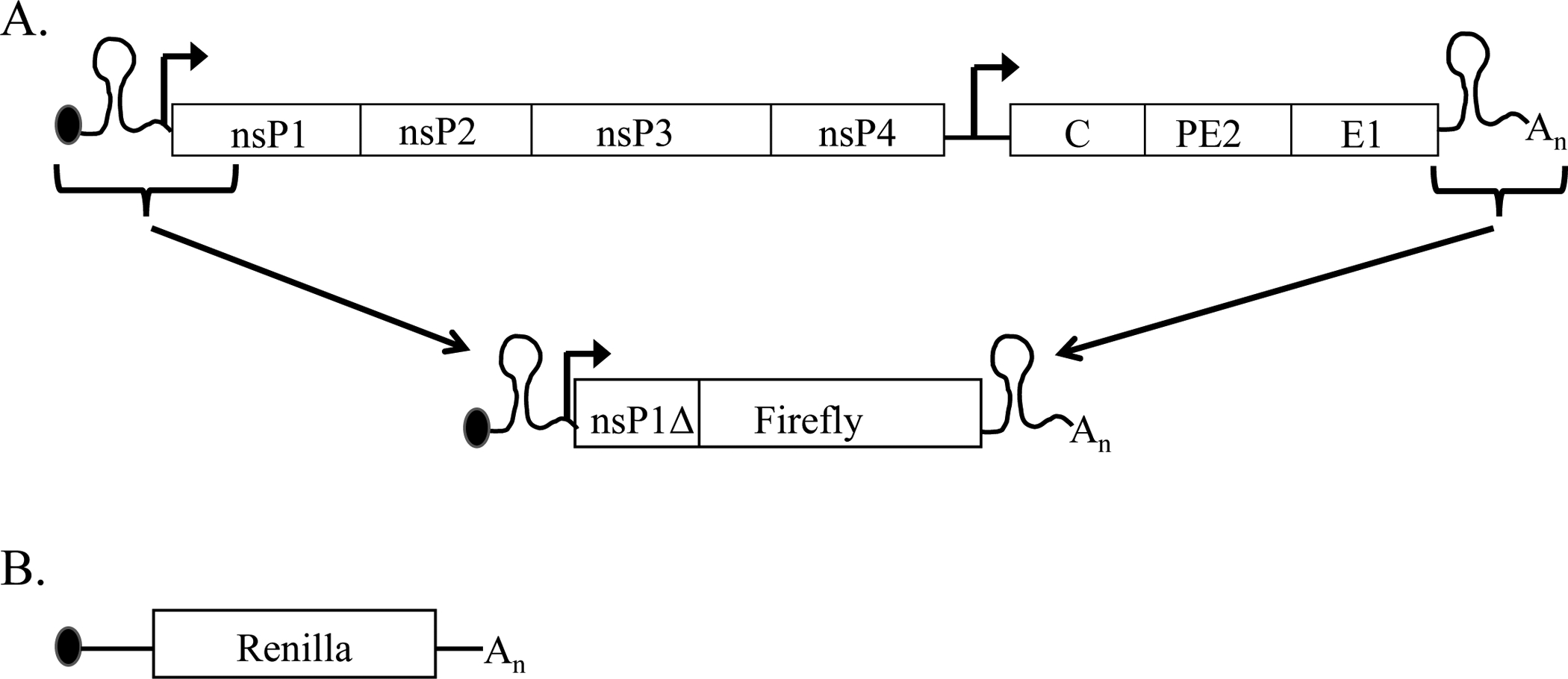
(A) The construction of the translation reporter from the alphavirus genome. The alphavirus RNA translation reporters has the m7G capped alphavirus 5’NTR, nsP1 CSE with an in-frame fusion to firefly luciferase, alphavirus 3’NTR and polyA tail. (B) The host mimic as an m7G capped 5’NTR (random sequence), Renilla luciferase, 3’NTR (random sequence) and polyA tail.
The recent work on the NTRs of alphaviruses has led to the discovery that these genetic regions are important for more than just replication of the virus (Review (21)) and contribute directly to virus pathogenesis. The secondary structures in the 5’NTR of many alphaviruses can antagonize host innate immune responses that inhibit translation of non-self mRNAs in the cytoplasm (20). Additionally, microRNA binding sites in the 3’NTR of some alphaviruses can limit immune sentinel cell tropism greatly suppressing induction of the innate immune response (19). There are currently no antiviral therapies or FDA licensed vaccines for alphaviruses and the use of synthetic mRNAs has been suggested as a possible therapeutic. It can now be understood that the non-translated regions have a tremendous impact on modulating cell tropism, replication efficiency and induction of innate immune responses..
Synthetic RNAs can be designed that target virus sequences known to efficiently suppress virus replication in vivo and that RNA drug resistance mutations will specifically reduce the virulence of resultant viruses. Furthermore, knowledge gained regarding virulence loci in coding and non-coding regions can be used for rational design of live-attenuated vaccines (LAV). A crippling problem with current LAV candidates for the alphaviruses that are given as investigational new drugs (INDs) is the high rate of adverse symptoms that resemble infection by the virulent parental virus, presumably due to reversion of the attenuating mutation(s) (Review (22)). The current IND LAVs have only one or two attenuating mutations leading to increase likelihood of the attenuated vaccine reverting to virulence (Review (22)). Currently, there are several LAV candidates that are being tested that have mutations at multiple loci, including the 5’NTR and 3’NTR (22). These mutations have known mechanisms of action and, thus, can be engineered (e.g., by deletion or multiple site mutation) to resist reversion.
An important factor when using the alphavirus translation reporters is determining the optimal electroporation system and electroporation conditions. We utilize the Dual-Luciferase Assay system (Promega), which requires the electroporation of the test luciferase reporter along with a host-mimic reporter that expresses Renilla luciferase. The host-mimic Renilla reporter serves as an internal control for transection efficiency between experiments and is used to normalize the test luciferase reporter results. Two electroporation systems are utilized in this protocol: the Bio-Rad Gene Pulser Xcell™ system and the Invitrogen Neon® transfection system. The Neon® transfection system is reported to be more efficient for primary cells and other hard to transfect cells lines due to a pipette chamber that helps generates a more uniform electric field (23). We have achieved higher transfection efficiencies of myeloid-lineage cells with the Neon® transfection system compared to the Bio-Rad system (1, 19). Both systems allow for manipulation of the electroporation conditions for an optimal balance of transfection efficiency and cell viability. However, the Neon® transfection system is limited by the volume of cells per electroporation cuvette.
This chapter describes a detailed step-by-step protocol for electroporating both fibroblastic and myeloid lineage cells. The Promega Dual Luciferase assay system is used to quantify changes in the abundance of luciferase protein produced by translation of in vitro synthesized alphavirus reporter and control RNAs in the different cell types. Additionally, this chapter describes electroporation conditions for both the Bio-Rad Gene Pulser Xcell system and the Invitrogen Neon® transfection system. For easy-to-transfect fibroblastic cells, we recommend using the Bio-Rad Gene Pulser Xcell system. However, for more difficult cells such as myeloid lineage cells and primary cells, we recommend using the Neon® transfection system with the understanding that this system is limited in scale but offers more consistent results compared to the Bio-Rad Gene Pulser Xcell system.
2. Materials:
2.1. Media/Components for cell harvest
Dulbeccos’ Phosphate-Buffered Saline (DPBS), 1X without calcium and magnesium
Opti-MEM® Reduced Serum media
0.05% trypsin with 0.53 mM EDTA, 1X [−] sodium bicarbonate
Growth media for BHK-21 cells: RPMI with 10% heat-inactivated donor calf serum, 10% tryptose phosphate broth (TPB; Moltox), 1% L-glutamine and 100 U penicillin/streptomycin (per 500 mL of media).
Growth media for RAW264.7 cells: DMEM with 10% heat-inactivated fetal bovine serum, 1% L-glutamine and 100 U penicillin/streptomycin (per 500 mL of media).
Cell scrapers, 25cm
Trypan blue
Hemocytometer
1.5 mL Eppendorf tube
2.2. Translation reporter RNA
Translation reporter RNA encoding firefly luciferase: In vitro transcribed 5’-capped RNA encoding the firefly luciferase gene fused to the alphavirus translational control sequences as described by Ryman et al. and Tesfay et al. (Figure 1) (6, 18) (Note 1).
Translational reporter RNA encoding Renilla luciferase. Host mRNA mimic encoding the Renilla luciferase gene fused to short 5’ and 3’ NTRs (Figure 1B)(8). The Renilla reporter will serve as an internal control to normalize luciferase relative light units to Renilla relative light units in each cell.
2.3. Electroporation Components
Bio-Rad Gene Pulser II with capacitance extender or Bio-Rad Gene Pulser Xcell
Pre-warmed complete media
0.4 cm gap sterile electroporation cuvette
1.5 mL Eppendorf tubes or 96-well U-bottom tissue culture treated plate
2.4. Luciferase Assay Components
Orion or similar microplate luminometer with injectors (sensitivity range 300–630nm)
Dual-Luciferase® Reporter Assay System (Promega)
HyClone biology grade water
Passive lysis buffer (Promega): Dilute the 5X passive lysis buffer included in the Dual-Luciferase kit to 1X with biology grade water
Luciferase assay substrate: Resuspend the lyophilized luciferase assay substrate with 10 mL of the luciferase assay buffer II. Luciferase assay substrate can be stored at −20°C for up to 1 month or −70°C for 1 year (Note 2).
1X Stop & Glo® substrate (50X): Dilute the Stop & Glo substrate 1:50 in the Stop & Glo buffer. Stop & Glo reagent should be prepared prior to use each time.
White polystyrene, flat bottom, non-treated 96-well plates
2.5. Alternate Neon Electroporation Components:
Neon transfection system (Invitrogen)
Neon 100 μL kit (Invitrogen)
Pre-warmed complete cell media without antibiotics
3. Methods
3.1. Harvesting Cells
Grow Baby hamster kidney cells (BHK-21; ATCC# CCL-10), or the monocyte/macrophage cell line, RAW264.7 (ATCC #TIB-71), to 80–90% confluency in a T-175 cm2 flask or 150 mm dish (Note 3).
Remove the media from the cells.
Wash cells with DPBS to remove remaining traces of media.
For BHK cells, add 5 mL of trypsin to the dish. Incubate at 37°C until cells become nonadherent (~2–5 min). For RAW 264.7 cells, use a cell scraper to gently remove the cells from the flask or dish. For RAW 264.7 cells or other primary cells and myeloid cells, proceed to Section 3.3.
Add 10 mL of complete media to the flask or dish to transfer the cells to a 50-mL conical. (Note 4).
Count the cells using hemocytometer. Add 10 μL of cells to 90 μL of Trypan blue in a 1.5 mL Eppendorf tube. Transfer 10 μL of cells to hemocytometer to count the cells.
Transfer ~1.2 × 107 cells to a 1.5 mL Eppendorf tube.
Centrifuge the cells at 200 g for 5 minutes at 4°C to gently pellet.
Decant the supernatant and disrupt the cell pellet by gently flicking the tube.
Add 1 mL of Opti-MEM® and centrifuge at 200 g for 5 min at 4°C. Repeat 2 times.
Decant the supernatant and resuspend the cells in 0.5 mL of Opti-MEM® for each electroporation.
3.2. Electroporation of Cells using a Bio-Rad Electroporator
Add 7.5 μg of the viral RNA firefly luciferase translation reporter and 0.75 μg of the host mimic Renilla translation reporter to the BHK cells in the Eppendorf tube and mix well.
Transfer the cells and RNA to a 0.4 cm gap sterile electroporation cuvette.
For BHK cells, electroporate the cells and RNA at 220V, 1000 μF capacitance. Repeat electroporation for a total of two pulses (Note 5) (Table 1).
Transfer the cells to 15-mL conical tube containing 13 mL of pre-warmed complete media Rinse the cuvette with complete media to recover remaining cells.
Invert the conical tube twice to gently mix the cells and then place 1 mL of cells per 1.5 mL Eppendorf tube (Note 6) and place at 37°C.
Table 1:
Electroporation Conditions for Various Cell Types
| Electroporator | Cell Type | Voltage1 (V) | Capacitance (uF) | Pulse Width (ms) | Pulses | Alphavirus Translation Reporter Concentration (μg) | Host Translation Reporter Concentration (μg) |
|---|---|---|---|---|---|---|---|
| BioRad Electroportor | BHK | 220 | 1000 | N/A2 | 2 | 7.5 | 0.75 |
| MEF | 220 | 1000 | N/A | 2 | 7.5 | 7.5 | |
| C7/10 | 360 | 1000 | N/A | 1 | ND3 | ND | |
| Neon® Electroporator | BHK | 1200 | N/A | 30 | 1 | ND | ND |
| RAW264.7 | 1750 | N/A | 25 | 1 | 7.5 | 0.75 | |
| RAJI | 1375 | N/A | 30 | 1 | ND | ND |
3.3. Alternate Cell Harvesting Protocol for using Neon® Transfection System
The optimal electroporation conditions and amount of RNA will need to be determined prior to the experiment (Table 1).
Harvest cells using steps 1–6 from Method 3.1.
Transfer 6×106 cells to a 1.5 mL Eppendorf tube (Note 7).
Centrifuge the cells at 200 g for 5 minutes at 4°C to pellet.
Decant the supernatant and disrupt the cell pellet by gently flicking the tube.
Add 1 mL of DPBS and centrifuge at 200 g for 5 min at 4°C. Repeat 2 times.
Resupend the cells in 110 μL of Buffer R per electroporation (Note 8).
3.4. Electroporation of Cells using Neon® Transfection System
In the electroporation cuvette, add 3 mL of Buffer E2 and insert the cuvette into the electroporation station (Note 9).
Load the 100 μL tip on the pipette by pressing the plunger to the second stop and add the tip to the pipette according to the manufactures guidelines (Note 10).
In the Eppendorf tube that has either BHK or RAW264.7 cells resuspended in Buffer R, add 7.5 μg of the viral RNA translation reporter and 0.75 μg of the host mRNA translation reporter. Volume of RNA should not exceed 10% of the total Buffer R volume.
Add the cells and RNA into the 100 μL tip, making sure to eliminate all air bubble from the tip (Note 11).
Enter the appropriate protocol on the Neon® transfection system (Table 1) and then press Start.
When the electroporation is complete, a message will appear on the Neon® screen, then eject the cells by depressing the plunger to the second stop into 13 mL of pre-warmed antibiotic free complete media (Note 6).
3.3. Harvesting cells for Dual-luciferase Assay
At predetermined times post electroporation, harvest the cells for the luciferase assay. For translation reporters, time points vary from 30 minutes to 4 hours post electroporation.
Centrifuge the cells at 200 × g for 5 min at room temperature to pellet the cells.
Decant the supernatant and add 1 mL of DPBS to wash the cells, making sure to resuspend the pellet. Repeat 2 additional times.
After the last wash, decant the supernatant and resuspend the cells in 100 μL of 1X passive lysis buffer (Note 12).
Store the lysates at −80°C and allow to freeze for 1 hour.
Freeze-thaw the samples an additional time to completely lyse the cells (Note 13).
3.4. Dual-Luciferase Assay:
Add 25 μL of the cell lysate into a white polystyrene 96-well plate (Note 14).
Calculate the volume of the luciferase assay substrate and the Stop & Glo substrate you will need for the luciferase assay. Use 25 μL of each substrate per sample and then add 10% to total volume to account for priming the injectors with the substrates.
To measure firefly luciferase activity, prime luminometer injector 1 with the luciferase assay II substrate. To measure Renilla luciferase activity, prime injector 2 with the Stop & Glo substrate. The Stop & Glo substrate will quench the firefly luciferase substrate and subsequently react with the Renilla luciferase protein (Note 15).
Adjust the luminometer to inject 25 μL of the luciferase assay substrate per well with a 10 sec delay before measuring luciferase activity (Note 16).
After measuring firefly luciferase activity, inject 25 μL of the Stop & Glo substrate per well with a 10 second delay before measuring the Renilla luciferase activity (Note 16).
The data can be analyzed with two different methods. First, relative light units (RLUs) of firefly luciferase can be normalized to the Renilla luciferase RLUs in each sample to generate a ratio of firefly RLUs to Renilla RLUs that can be used to compare samples between independent experiments. A second method is to calculate the change in the RLU ratio between the initial and final time point for each reporter (Note 17).
4. Notes
The translation reporters encode either a T7 or SP6 promoter driving in vitro transcription. The in vitro transcription kit (Ambion) must contain a type 0 cap analog to generate 5’ capped RNA for efficient translation of the alphavirus reporters.
The luciferase assay substrate can be freeze-thawed up to 6 times before losing luciferase activity. The resuspended luciferase assay substrate can be stored at −20°C for 1 month or −80°C for up to 1 year.
If more than one T175 cm2 flask or 150 mm dish is being used for the electroporation at one time just combine the flask or dishes into the same 50-ml conical with up to 3 flask/dishes into the same conical (i.e. electroporating BHK cells with multiple mRNA translation reporters, when harvesting the cells all the cells can be combined together to ensure each electroporation receives the same number of cells).
Adding complete media to the BHK cells after typsinizing will inactivate the trypsin. Inactivation of trypsin is important to ensure that the cells maintain optimal viability.
Electroporation conditions must be optimized for each Bio-Rad electroporator because the actual electroporation voltage may change during the electroporation resulting in suboptimal conditions.
A smaller volume can be used if a 96-well U-bottom plate will be used instead of 1.5 mL Eppendorf tubes.
Each of the 100 μL electroporation tips can hold up to 6×106 cells, and each tip can be used for two electroporations for a total of 12×106 cells.
For two electroporations, double the volume of Buffer R to 220 μL. The extra volume helps in removing air bubbles from the tip.
Replace the E2 buffer in the cuvette or replace the entire cuvette between electroporations with different translation reporters.
If the tip is properly loaded onto the pipette, the gold electrode will move to allow for the cell-RNA mixture to enter the electroporation chamber. Remember that the pipette has two stops when discharging liquid and the second stop ejects the tip.
If air bubbles remain in the tip, a spark will occur during electroporation and cells can be ejected from the tip. Additional Buffer R can be added to help in eliminating air bubbles.
To ensure better lysis of the cells, pipet up and down several times.
During the thaw, place the tubes or plates on a shaker to further facilitate the lysis of the cells.
Contamination from neighboring wells can occur if a plate other than the white polystyrene 96-well plates is used to measure luciferase activity.
In the injectable luminometer, rinse out the injectors with water before priming the injectors with the substrates.
The luminometer will report the luciferase activity as relative light units (RLU).
Different cell types will have different levels of translation and/or electroporation efficiency resulting in variable luciferase assay results (e.g., BHK cells will give higher values than RAW264.7 cells) and generally the host mRNA translation reporter will have higher translation compared to the viral RNA translation reporter and thus, less of the host mimic reporter is used compared to the viral reporter.
Acknowledgements
This work was supported by 1R01AI095436 (WBK) and 1R01AI081886 (KDR).
References:
- 1.Gardner CL, Burke CW, Tesfay MZ, Glass PJ, Klimstra WB, Ryman KD. 2008. Eastern and Venezuelan equine encephalitis viruses differ in their ability to infect dendritic cells and macrophages: impact of altered cell tropism on pathogenesis. Journal of virology 82:1063410646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryman KD, Gardner CL, Burke CW, Meier KC, Thompson JM, Klimstra WB. 2007. Heparan sulfate binding can contribute to the neurovirulence of neuroadapted and nonneuroadapted Sindbis viruses. Journal of virology 81:3563–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryman KD, Gardner CL, Meier KC, Biron CA, Johnston RE, Klimstra WB. 2007. Early restriction of alphavirus replication and dissemination contributes to age-dependent attenuation of systemic hyperinflammatory disease. The Journal of general virology 88:518–529. [DOI] [PubMed] [Google Scholar]
- 4.Ryman KD, White LJ, Johnston RE, Klimstra WB. 2002. Effects of PKR/RNase L-dependent and alternative antiviral pathways on alphavirus replication and pathogenesis. Viral immunology 15:53–76. [DOI] [PubMed] [Google Scholar]
- 5.Sun C, Gardner CL, Watson AM, Ryman KD, Klimstra WB. 2014. Stable, high-level expression of reporter proteins from improved alphavirus expression vectors to track replication and dissemination during encephalitic and arthritogenic disease. Journal of virology 88:2035–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tesfay MZ, Yin J, Gardner CL, Khoretonenko MV, Korneeva NL, Rhoads RE, Ryman KD, Klimstra WB. 2008. Alpha/beta interferon inhibits cap-dependent translation of viral but not cellular mRNA by a PKR-independent mechanism. Journal of virology 82:2620–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas JM, Klimstra WB, Ryman KD, Heidner HW. 2003. Sindbis virus vectors designed to express a foreign protein as a cleavable component of the viral structural polyprotein. Journal of virology 77:5598–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bick MJ, Carroll JW, Gao G, Goff SP, Rice CM, MacDonald MR. 2003. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. Journal of virology 77:11555–11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donnelly ML, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, Ryan MD. 2001. The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. The Journal of general virology 82:1027–1041. [DOI] [PubMed] [Google Scholar]
- 10.Brault AC, Foy BD, Myles KM, Kelly CL, Higgs S, Weaver SC, Olson KE, Miller BR, Powers AM. 2004. Infection patterns of o’nyong nyong virus in the malaria-transmitting mosquito, Anopheles gambiae. Insect molecular biology 13:625–635. [DOI] [PubMed] [Google Scholar]
- 11.Huang HV, Rice CM, Xiong C, Schlesinger S. 1989. RNA viruses as gene expression vectors. Virus genes 3:85–91. [DOI] [PubMed] [Google Scholar]
- 12.Pugachev KV, Mason PW, Shope RE, Frey TK. 1995. Double-subgenomic Sindbis virus recombinants expressing immunogenic proteins of Japanese encephalitis virus induce significant protection in mice against lethal JEV infection. Virology 212:587–594. [DOI] [PubMed] [Google Scholar]
- 13.Tsetsarkin K, Higgs S, McGee CE, De Lamballerie X, Charrel RN, Vanlandingham DL. 2006. Infectious clones of Chikungunya virus (La Reunion isolate) for vector competence studies. Vector borne and zoonotic diseases 6:325–337. [DOI] [PubMed] [Google Scholar]
- 14.Xiong C, Levis R, Shen P, Schlesinger S, Rice CM, Huang HV. 1989. Sindbis virus: an efficient, broad host range vector for gene expression in animal cells. Science 243:1188–1191. [DOI] [PubMed] [Google Scholar]
- 15.Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiological reviews 58:491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castello A, Sanz MA, Molina S, Carrasco L. 2006. Translation of Sindbis virus 26S mRNA does not require intact eukariotic initiation factor 4G. Journal of molecular biology 355:942–956. [DOI] [PubMed] [Google Scholar]
- 17.Berben-Bloemheuvel G, Kasperaitis MA, van Heugten H, Thomas AA, van Steeg H, Voorma HO. 1992. Interaction of initiation factors with the cap structure of chimaeric mRNA containing the 5’-untranslated regions of Semliki Forest virus RNA is related to translational efficiency. European journal of biochemistry / FEBS 208:581–587. [DOI] [PubMed] [Google Scholar]
- 18.Ryman KD, Meier KC, Nangle EM, Ragsdale SL, Korneeva NL, Rhoads RE, MacDonald MR, Klimstra WB. 2005. Sindbis virus translation is inhibited by a PKR/RNase L-independent effector induced by alpha/beta interferon priming of dendritic cells. Journal of virology 79:1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trobaugh DW, Gardner CL, Sun C, Haddow AD, Wang E, Chapnik E, Mildner A, Weaver SC, Ryman KD, Klimstra WB. 2014. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 506:245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hyde JL, Gardner CL, Kimura T, White JP, Liu G, Trobaugh DW, Huang C, Tonelli M, Paessler S, Takeda K, Klimstra WB, Amarasinghe GK, Diamond MS. 2014. A viral RNA structural element alters host recognition of nonself RNA. Science 343:783–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyde JL, Chen R, Trobaugh DW, Diamond MS, Weaver SC, Klimstra WB, Wilusz J. 2015. The 5’ and 3’ ends of alphavirus RNAs - Non-coding is not non-functional. Virus research 206:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trobaugh DW, Ryman KD, Klimstra WB. 2014. Can understanding the virulence mechanisms of RNA viruses lead us to a vaccine against eastern equine encephalitis virus and other alphaviruses? Expert review of vaccines 13:1423–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JA, Cho K, Shin MS, Lee WG, Jung N, Chung C, Chang JK. 2008. A novel electroporation method using a capillary and wire-type electrode. Biosensors & bioelectronics 23:1353–1360. [DOI] [PubMed] [Google Scholar]