

Conspectus.
Halogenated natural products number in the thousands, but only in rare cases are the evolutionary advantages conferred by the halogens understood. We set out to investigate the lissoclimide family of cytotoxins, which includes several chlorinated members, because of our longstanding interest in the synthesis of chlorinated secondary metabolites.
Our initial success in this endeavor was a semi-synthesis of chlorolissoclimide (CL) from the commercially available sesquiterpenoid sclareolide. Featuring a highly selective and efficient—and plausibly biomimetic—C–H chlorination, we were able to access enough CL for collaborative studies, including X-ray co-crystallography with the eukaryotic ribosome. Through this experiment, we learned that CL’s chlorine atom engages in a novel halogen-π dispersion interaction with a neighboring nucleobase in the ribosome E-site.
Owing to the limitations of our semi-synthesis approach, we established an analogue-oriented approach to access numerous lissoclimide compounds to both improve our understanding of structure/activity relationships and to learn more about the halogen-π interaction. In the course of these studies, we made over a dozen lissoclimide-like compounds, the most interesting of which contained chlorine-bearing carbons with unnatural configurations. Rationalizing the retained potency of these compounds that appeared to be a poor fit for the lissoclimide binding pocket, we came to realize that the chlorine atoms would engage in these same halogen-π interactions even at the expense of a chair to twist-boat conformational change, which also permitted the compounds to fit in the binding site.
Finally, because neither of the first two approaches could easily access the most potent natural lissoclimides, we designed a synthesis that took advantage of rarely used terminal epoxides to initiate polyene cyclizations. In this case, the chlorine atom was incorporated early, and helped control the stereochemical outcome of the key step.
Over the course of this project, three different synthesis approaches were designed and executed, and our ability to access numerous lissoclimides fueled a range of collaborative biological studies. Further, chlorine played impactful roles throughout various aspects of both synthesis and biology. We remain inspired to learn more about the mechanism of action of these compounds, and to deeply investigate the potentially valuable halogen-π dispersion interaction in the context of small molecule/nucleic acid binding. In that context, our work offers an instance wherein we might have gained a rudimentary understanding of the evolutionary importance of the halogen in a halogenated natural product.
Graphical Abstract

Introduction.
Natural product inhibitors of the ribosome are architecturally complex and demonstrate incredible structural diversity based on the numerous function-altering binding sites available on this enormous and critical biochemical machine.5 Figure 1 shows representative inhibitors of eukaryotic translation, and indicates their binding site. Of course, shutting down translation generally leads to cell death and, as a result, selective inhibition of translation in pathogenic organisms leads to antiinfective activity.6 On the other hand, inhibiting eukaryotic translation is, surprisingly, a viable pathway to antiproliferative agents for human cancer chemotherapy,7 as demonstrated by the FDA approval of homoharringtonine as Synribo for the treatment of chronic myeloid leukemia.8
Figure 1.
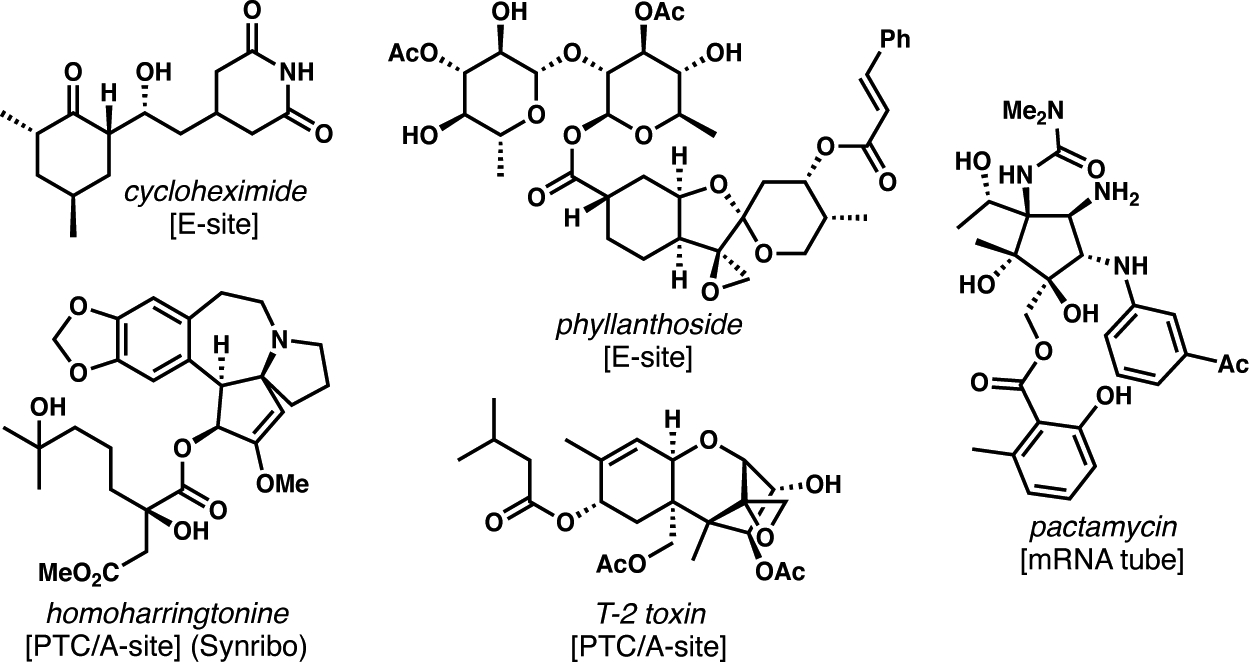
Representative translation inhibitors that exert their effects by binding to the ribosome
Owing to our longstanding interest in the synthesis of polychlorinated compounds (Figure 2),9 we became interested in the synthesis of dichlorolissoclimide (DCL, 1, Figure 3). A labdane diterpenoid with an unusual halogenation pattern and a rare succinimide motif, DCL and its monochlorinated analogue chlorolissoclimide (CL, 2) were reported by Malochet-Grivois and co-workers in the early 1990s.10 These authors also disclosed potent cytotoxicity against several cancer cell lines including, for example for DCL, murine leukemia (P388, IC50 of 2.4 nM), human oral carcinoma (KB, 33 nM) and human non-small-cell lung cancer (NSCLC, 22 nM).
Figure 2.
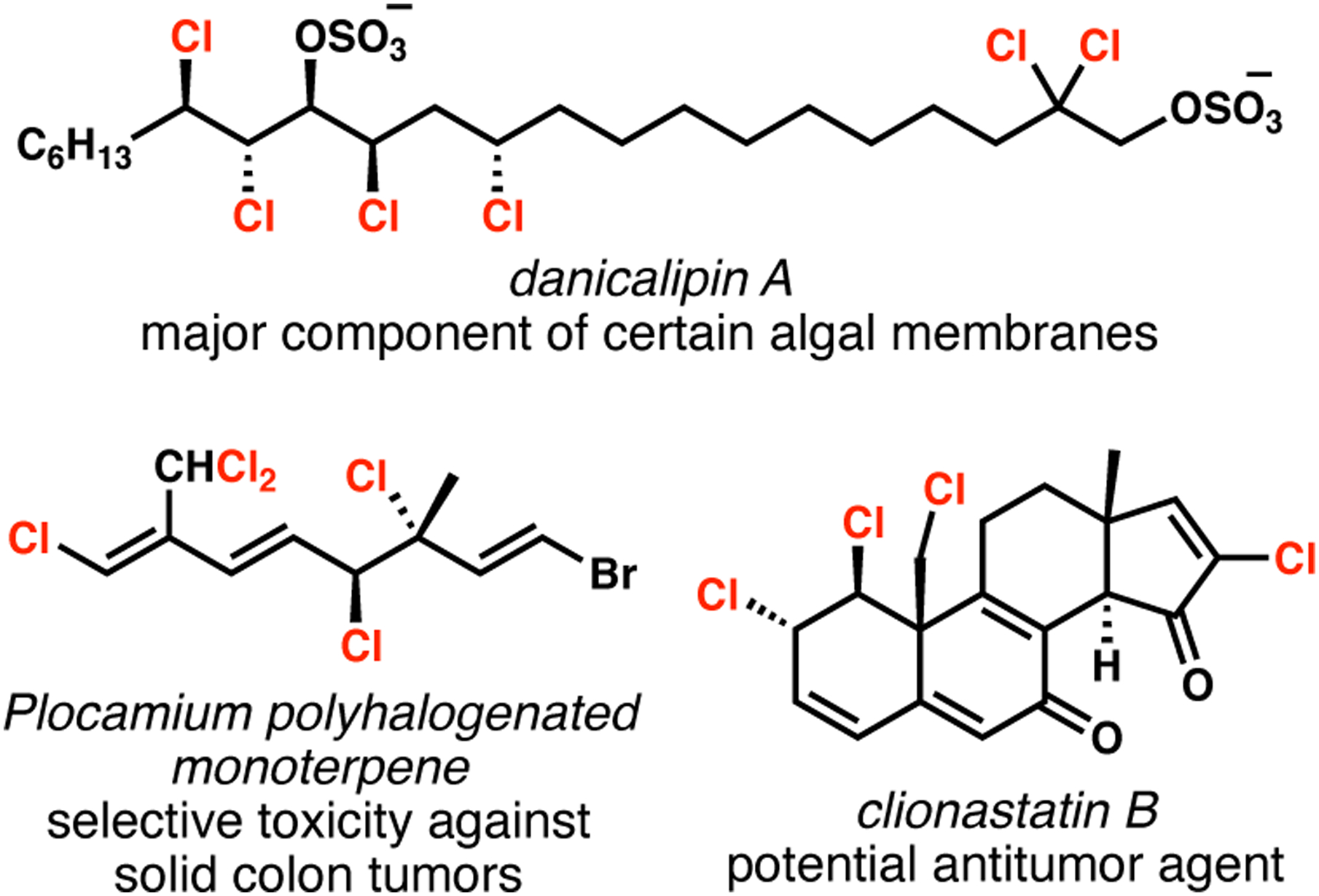
Representative polyhalogenated natural products targeted by the Vanderwal group
Figure 3.

The lissoclimide family of cytotoxins, and two related glutarimide antibiotics
Originally isolated from ascidians (sea squirts), this small family of secondary metabolites was greatly expanded by the discovery of nearly 20 closely related compounds called the haterumaimides by the groups of Ueda and Schmitz (see representatives 3–7, Figure 3), some of which were also obtained from molluscs.11 Some preliminary structure-activity relationships were observed from the collected work of Ueda and co-workers.11e Haterumaimide J (hatJ, 6) emerged as the most potent (sub-nM IC50 against the P388 murine leukemia cell line), while some compounds were completely inactive (not shown but, for example, a C6-ketone abolished all activity, as did changes to the hydroxysuccinimide motif).
CL and DCL were shown by Pelletier and co-workers to be potent inhibitors of eukaryotic translation.12 They each interfere with the elongation step of protein synthesis, resulting in polysomal accumulation and eventual cell death. In this same study, these workers noted the structural homology between CL and the well-studied translation inhibitor CHX (8). Clearly, the glutarimide of CHX and the succinimide of the lissoclimides are structurally similar. CHX and the glutarimide lactimidomycin (9)13 have each been shown to stop the translocation process of protein synthesis by inhibiting the binding of the CCA- end of tRNA to the large ribosomal subunit E-site. Structural studies of Yusupov and co-workers5 and functional studies of Blanchard and co-workers14 are consistent with this understanding. When we began our work in the area, the molecular basis for DCL and CL’s interaction with the ribosome remained unknown, and no successful syntheses of any lissoclimide or haterumaimide had been recorded, though some interesting studies had been reported by the groups of Jung,15 Gonzales,16 and Chai.17
Early approach to dichlorolissoclimide.
At the outset of our work, we aimed to take a “chlorophilic” approach to DCL, as we had with the chlorosulfolipids such as danicalipin A (Figure 2).9a–e That is, we sought to have the chlorine atoms play a key role in the synthesis, rather than simply introduce them at the end via chlorodeoxygenation reactions of hydroxy groups or epoxides. It is critical to note that the trans-diequatorial disposition of the vicinal dichloride motif would appear to preclude its installation via cyclohexene dichlorination, which should favor the formation of the trans-diaxial stereoisomer. Indeed, Jung and Gomez showed exactly this problem in a model study toward the lissoclimides, in which the major product of dichlorination of cyclohexene 10 was trans-diaxial dichloride 11 (Scheme 1). A thermal dyotropic rearrangement of derivative 13 cleanly generated the trans-diequatorial isomer 14 under rather forcing conditions.15 Given the trans-decalin architecture that clearly arose biogenetically from a cationic bicyclization, we wondered if a related cyclization of an α-chlorochloronium ion (16) or equivalent might serve to build the decalin scaffold under the stereochemical control of two chlorine-bearing stereogenic centers, which could be installed stereoselectively in an acyclic setting. To test this idea, we expended great efforts to access compounds of type 15.
Scheme 1.
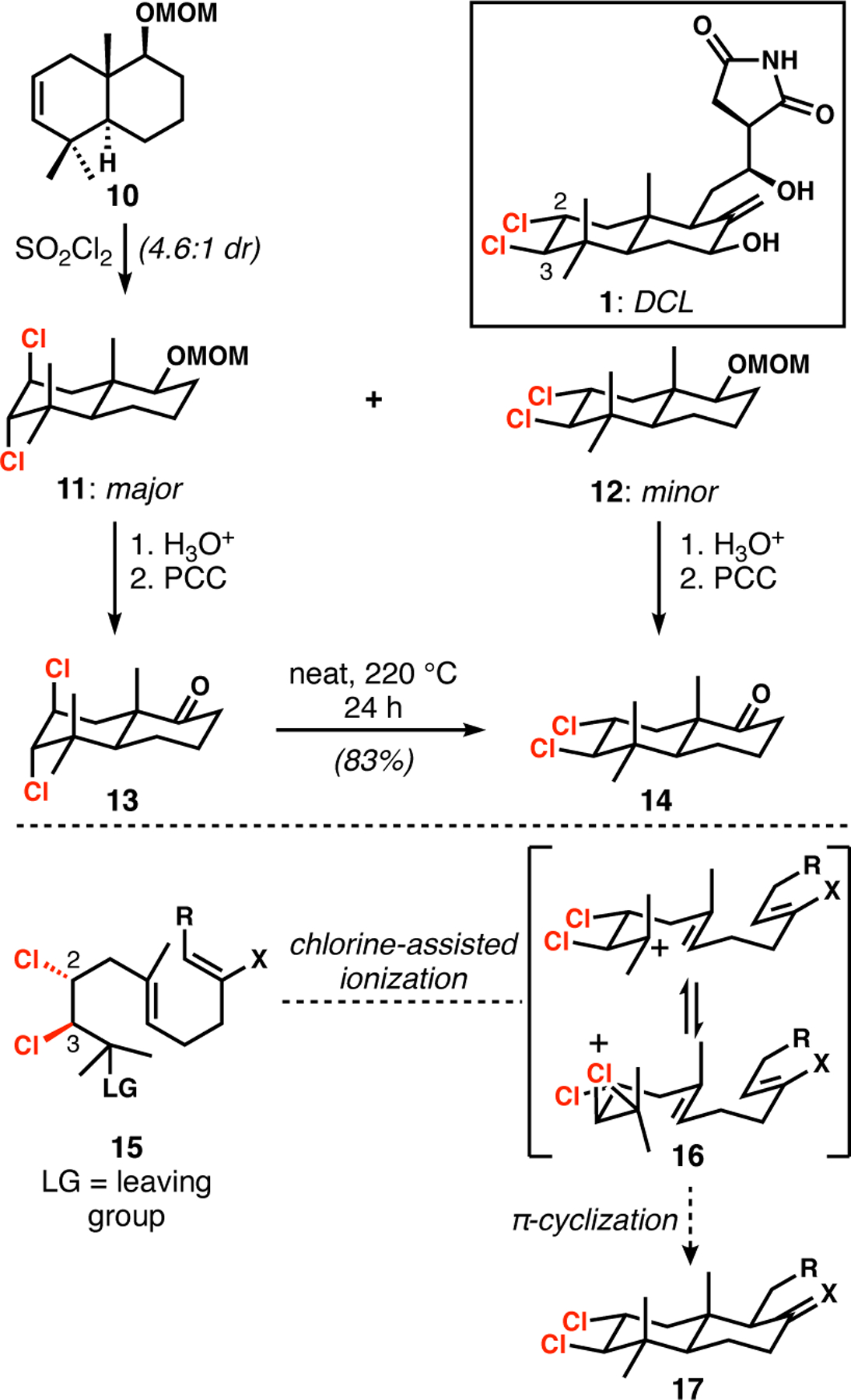
Considerations for the synthesis of DCL
At the time, we considered that a furan would make an attractive terminating group; the work of Tanis documents their utility in this regard,18 and available options for ring-opening of the furan would permit a number of pathways to complete the synthesis. Therefore, we targeted 18 (Scheme 2a), which we aimed to make via allylic/pseudobenzylic cross-coupling19 of reactants 19 and 20. Unfortunately, the synthesis of dichloride 22 proved an enormous challenge, as efforts to introduce the chlorine atoms via either alkene dichlorination or epoxide deoxydichlorination were unsuccessful in the presence of the adjacent fully substituted carbon atom (Scheme 2b). After much work, we were able to procure a potential bicyclization precursor 30 by changing the order of events: we first installed the vicinal dichloride by stereospecific epoxide deoxychlorination of 25,20 followed by coupling to the furan to give 28, and ending with the introduction of the offending geminal methyl groups (Scheme 2c). In this way, we obtained hundred milligram quantities of 30 for testing the key step, which unfortunately was never successful. Dozens of attempts to activate the tertiary alcohol via sulfonation, acylation, protonation, and Lewis acid complexation were either ineffective at inducing any reaction, or led to degradation under more forcing conditions. Still, the idea of a furan-terminated π-cyclization, wherein a chlorine-bearing stereogenic center plays a key role, played a major role in our later experiments, as will be described below.
Scheme 2.
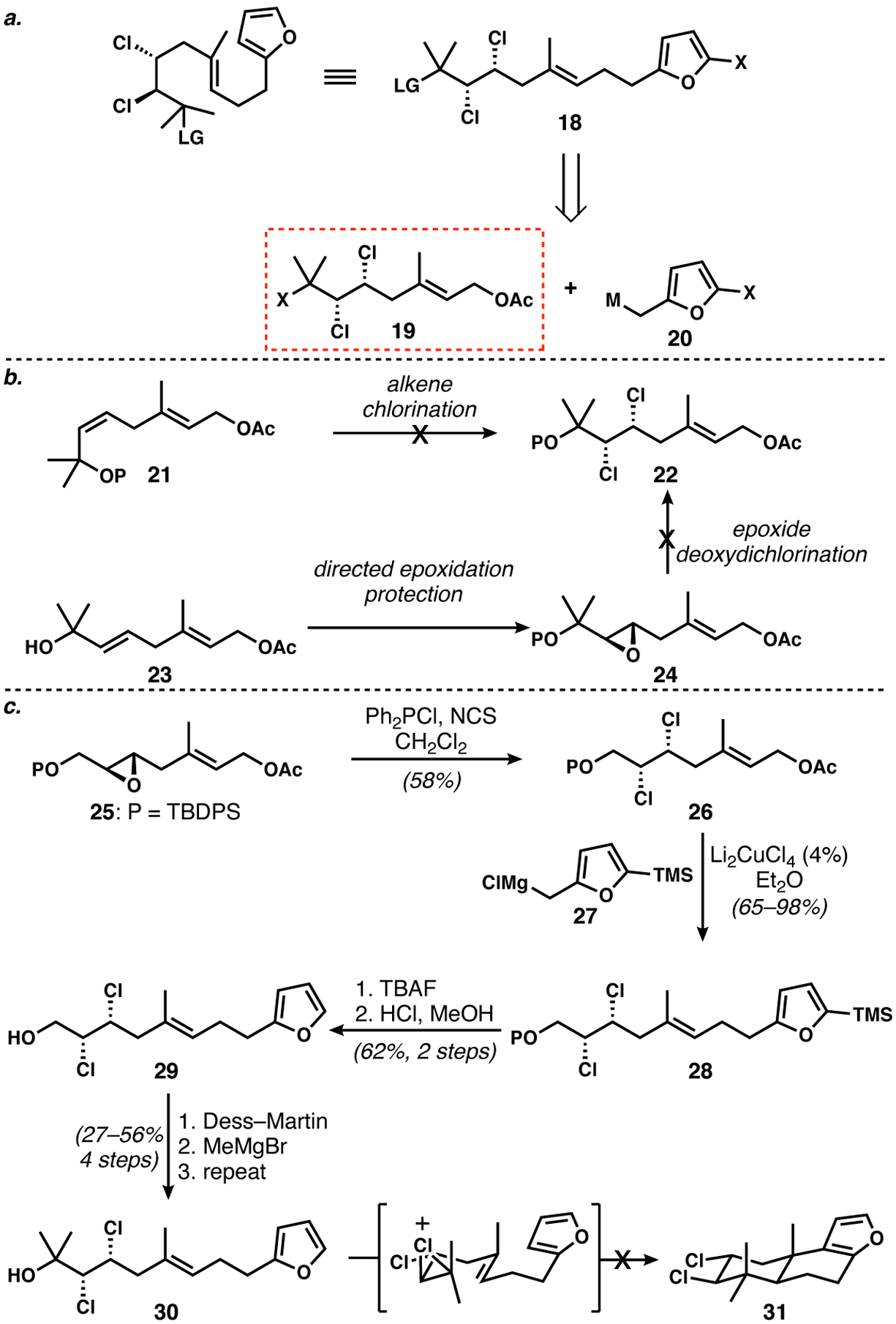
a. Planned π-cyclization precursor. b. Attempted synthesis of dichloride 22. c. Synthesis of potential cyclization precursor 30 and failed trans-decalin formation.
A semi-synthesis of chlorolissoclimide.
At the time of our struggles with the π-cyclization approach to DCL, we questioned whether or not our laser-focus on one member of the lissoclimides was sensible. With around 20 members and interesting biological activity that might be better understood with collaborative studies using synthetic material, we broadened our program, and began with a semi-synthesis effort toward the monochlorinated congener, CL. This approach was predicated on the knowledge that selective C–H functionalizations of the inexpensive sesquiterpenoid sclareolide (32) at C2 were known,21 and the other required manipulations (Figure 4) should be straightforward.
Figure 4.

A plan for a semi-synthesis of CL from sclareolide
At the time, C–H amination,21a oxidation,21b chlorination,21c and fluorination21d,e reactions at this position of sclareolide were already known; however, the efficiencies were rather low in many cases. We had already attempted the Groves chlorination21c (manganese porphyrin catalyst, bleach), which worked but would not provide sufficient material throughput. However, during a seminar from Professor Erik Alexanian (UN Chapel Hill) at UCI, our group learned of their development of N-bromoamide reagents—essentially sterically and electronically tuned variants of NBS—for selective radical C–H bromination reactions of aliphatic compounds, including sclareolide.22 A brief discussion revealed that they were also developing chlorinating agents, and a collaboration was born. Together, members of each group optimized the chlorination of sclareolide to the point where it reliably produced gram quantities of 2-chlorosclareolide in single reaction vessels. The efficiency of this sclareolide functionalization is unmatched, with yields of 2-chlorosclareolide routinely above 80% (32, Scheme 3) and complete regio- and stereochemical selectivity1 It is worth pointing out that the selectivity of C–H abstraction on sclareolide has been nicely explained by Chen, Eschenmoser, and Baran,21a with additional insights provided by White and co-workers.21b The result of both the inherent selectivity and the optimization of reagent and conditions was a robust chlorination reaction that served admirably as the first step in the semi-synthesis of CL.
Scheme 3.

Semi-synthesis of CL from sclareolide
Both the Gonzales16 and Chai17 groups had used sclareolide to advance to complex, lissoclimide-like compounds, taking advantage of the procedure of Boukouvalas23 to turn sclareolide into an aldehyde related to 37 but without the chlorine atom or the C7-oxygenation. Therefore, ring-opening to the Weinreb amide and dehydration gave 35, which we then oxidized at the now allylic C7 position to afford the α-hydroxy group; oxidation provided the ketone 36. Double reduction and protection set up for an Evans-aldol-based introduction of the succiminide to aldehyde 37, patterned after the work of Chai, whose group struggled with the formation of the γ-lactone from aldol addition products of type 39.17 We circumvented this problem by not decomposing the boron aldolate 39, and rather exposing it directly to methanolic ammonia; a solvent switch and addition of sodium hydride pushed the material safely in the direction of the imide. The 9-step synthesis clearly relies much on the prior work of others, but proved an excellent showcase of the Alexanian chlorination chemistry and was efficient enough to procure CL for our collaborative biological studies (see below).1
It is worth pointing out that the key chlorination reaction is likely biomimetic. The trans-decalin of the lissoclimides is most likely biosynthesized by cation-induced polyene cyclization of an unhalogenated substrate (many lissoclimides and haterumaimides lack chlorine).11 Further tailoring of the decalin by halogenases and oxygenases leads to the diversity of structures that have been isolated so far. An interesting corollary of this hypothesis is that the enzyme(s) responsible for halogenation might not have been evolved so much for selectivity, necessarily, but more for reactivity. While we have tried to take this chemistry a step further, by chlorinating hatQ (5) to directly generate CL, this reaction did not work, likely owing to the presence of allylic hydrogen atoms. Of course, we do not know at what stage in the post-cyclization biosynthesis that the halogenation reaction takes place, or if the enzyme is indeed able to steer halogenation away from allylic positions.
An analogue-oriented synthesis of lissoclimides
Although our sclareolide-based semi-synthesis approach permitted the synthesis of CL, hatQ, and two other simpler analogues,1 it did not provide ready access to much structural diversity in the synthesis of natural and non-natural lissoclimide compounds. Therefore, we aimed to develop a synthesis that could address compounds with varied substitution on the A and B rings. We targeted an intermediate of type 45 (Scheme 4), which we believed should be readily available by an epoxide-initiated, enoxysilane-terminated π-cyclization, followed by elimination of the C3 alcohol. Two series were envisioned, one with C18 oxygenation (like hatJ, 6) and one without, dictated by the choice of starting material.
Scheme 4.

Proof-of-principle synthesis of hatQ via route designed for procurement of lissoclimide analogues
We made use of Corey’s clever convergent enoxysilane synthesis,24 in which acylsilane 40 (Scheme 4) is first made by alkylation of the metalloenamine derived from acetyltrimethylsilane with 2,3-epoxy geranyl bromide (not shown). Addition of organolithium reagent 41 to the acylsilane triggers a Brook rearrangement and subsequent sulfinate elimination, thus generating enoxysilane 42 exclusively as the E-stereoisomer. Lewis-acid-catalyzed polyene cyclization provided B-ring ketone 43 after epimerization of C9; the identity of the Lewis acid dictated whether or not silyl transfer to the C3 hydroxy group took place. Elimination of this hydroxy group provided a versatile C2–C3 alkene 45 that could be functionalized in a variety of ways, and in some cases was dichlorinated, with the expected high diaxial diastereoselectivity. As shown in the scheme, it could also be hydrogenated, prior to conversion of the B-ring ketone into the useful allylsilane of 46. The selectivity for SE’ reaction with electrophiles always favored α-attack, and in the case at hand, bromination cleanly gave the axial allylic bromide, which was surprisingly robust to the next sequence of events that included dioxolane hydrolysis and the aldol-based three-stage succinimide introduction. The final step was an invertive hydrolysis of the C–Br bond of 48 using potassium superoxide, delivering hatQ (5). Admittedly, we could make hatQ from sclareolide in 8 steps; however, this was proof-of-principle of our approach that proceeded via intermediates that provided opportunities for the synthesis of diverse lissoclimide analogues, including the important compound C45 (see below).2
The same overall strategy was adapted to the synthesis of C18-oxygenated lissoclimide compounds, in the expectation that they would be particularly active, given the potency of hatJ (6). We had to “preoxygenate” C18 in the earliest stage by allylic oxidation of geranyl acetate, and thus made acylsilane 49 (Scheme 5), which was subjected to similar operations as just described to obtain A-ring alkene/B-ring ketone 50. While dichlorination of 45 (Scheme 4, reaction not shown) and homoallylic alcohol 50 each gave exclusively the expected diaxial product, the temporary conversion of the C18-alcohol to its trifluoroacetate provided a molecular situation in which dichlorination yielded an equimolar mixture of diaxial and diequatorial products (51). We do not understand this outcome, but were inspired to try the use of electron-deficient esters because we previously observed surprising changes in stereochemical control in the dichlorination of allylic and homoallylic alcohols/trihaloacetates during our efforts toward the chlorosulfolipids,9a–e,25 the Plocamium monoterpenoids,9f and the clionastatins.9g After three-step conversion of the diastereomeric mixture to the aldehyde substrates for succinimide introduction, the isomers 52 and 53 could be separated and each processed to the final lissoclimide analogues 54 and 55. In the end, nearly 20 analogues were made via the combination of the semi-synthesis and the analogue-oriented approaches, essentially doubling the number of lissoclimide-type compounds.2
Scheme 5.
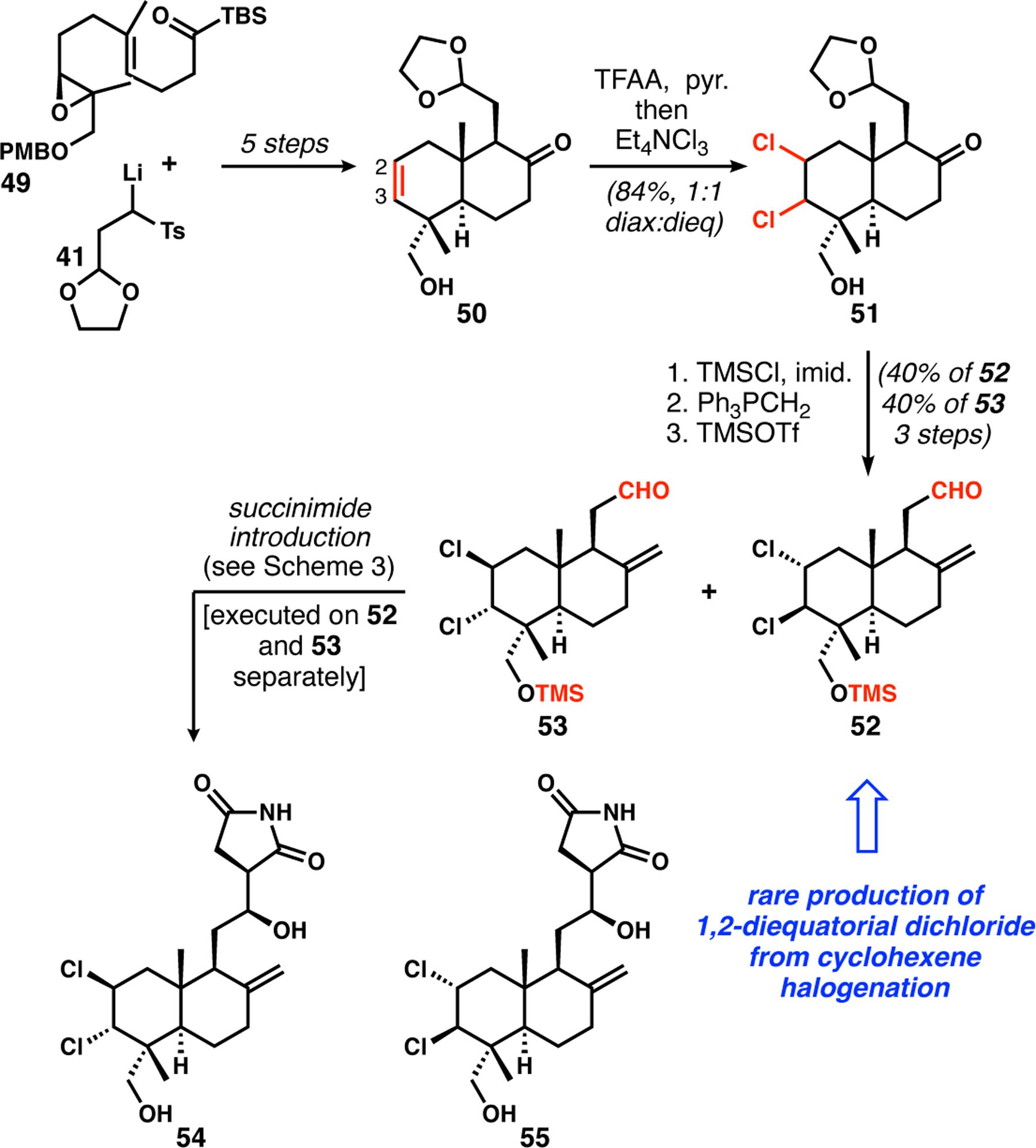
Synthesis of lissoclimide analogues with C18 oxygenation
Biological investigations of synthetic lissoclimides
Pelletier had shown biochemically that CL and DCL inhibit translation and had made the critical structural connection between these succinimide-containing compounds with the glutarimide antibiotics,12 the flagship of which is cycloheximide (CHX). At about the time when we had first made CL via semi-synthesis, we learned of the beautiful results of the Yusupov group on the molecular basis for transcription inhibition by over a dozen ribosome binders.5 These co-crystallization studies were an exceptional follow-up to their groundbreaking first X-ray crystallographic structure determination of the eukaryotic ribosome.26 The list of inhibitors included the glutarimide E-site binders CHX and LTM, which prompted us to contact them about the possibility of collaboration. Some months later, the Yusupov group obtained and refined a beautiful X-ray co-crystal structure of CL with the 80S ribosome (Figure 5), showing how the succinimide of CL binds in a nearly identical way to the glutarimides of CHX and LTM.2 More importantly, they identified a novel interaction of the chloride residue with the six-membered ring of the nucleobse of G-2794. The chlorine atom was located nearly directly above the centroid of this ring, and in close (3.2 Å) proximity, suggesting a dispersive stabilizing interaction. Such halogen–π dispersive interactions were unknown at the time in nucleic acid solid-state structures, though similar phenomena were certainly known in protein-ligand complexes.27 Shortly after we reported this structure, the groups of Yusupov, Romo, Green, Liu and co-workers reported a similar arrangement of a heterocyclic bromide from agelastatin with a ribosomal uracil.28 Previously, the fluorine atom of an aminoglycoside analogue had arranged itself in a related manner with the six-membered ring of a guanine in a bacterial ribosome, as shown by Hanessian and co-workers; however, the fluorine was not quite oriented over the centroid of the ring.29
Figure 5.
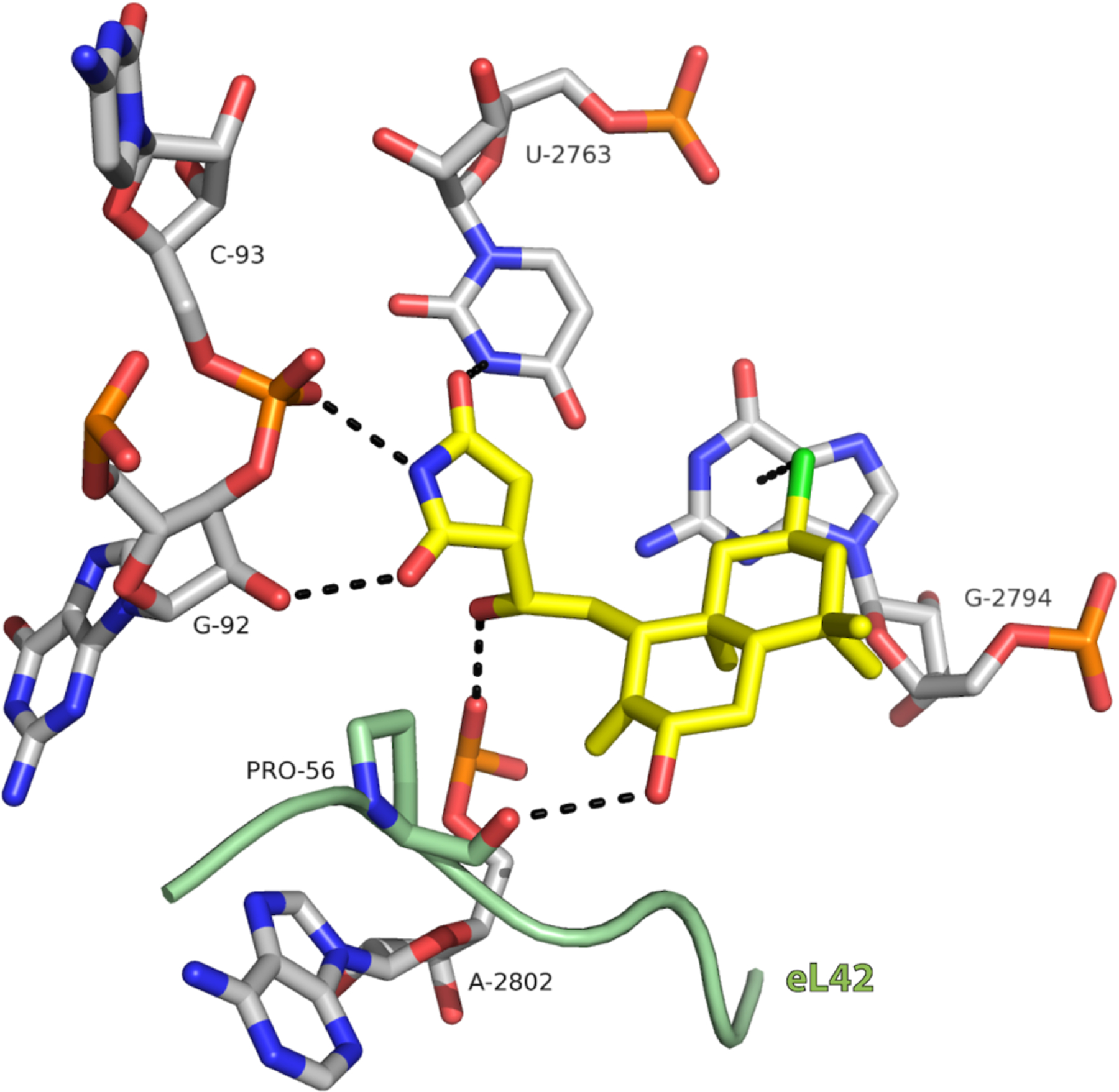
Co-crystal structure of a portion of the Saccharomyces 80S ribosome E-site bound with synthetic CL, showing the chlorine–π interaction with G-2794. Reproduced with permission from ref. 2. Copyright 2017 Springer Nature.
In collaboration with the Furche group at UCI, we studied the expected strength of this and related interactions. Using the PBE-D3 method, which is designed to take into account dispersion interactions via semi-empirical correction, they found that the arrangement shown in the co-crystal structure likely benefits from a stabilizing interaction of approximately 1.1 kcal/mol.2 Based on preliminary calculations, we expect that this type of interaction will increase in the series F>Cl>Br. That does not consider the full structure of the lissoclimide analogue or the ribosome, including the flexibility of the binding site and its ability to accommodate larger atoms. Nonetheless, this work has spurred on our ongoing efforts to make fluorolissoclimide and bromolissoclimide, as well as methyllissoclimide, so that we can study them through structural analysis and biochemical and biological assays.
During our forays into lissoclimide synthesis, we started collaborative investigations into their biological activities with the Horne lab at City of Hope. Because all natural lissoclimides had been tested against the murine leukemia P388 cell line,11 they used that, along with invasive prostate (DU145) and melanoma (A2058) cell lines, to learn about the antiproliferative activities of our synthetic compounds. To supplement this information, the Pelletier lab contributed translation inhibition assays, because that provided a set of data that could connect structural biology and cytotoxicity. With knowledge of the way in which CL binds in the ribosome E-site, we could hypothesize which compounds might be reasonable E-site binders and thus likely strong inhibitors of translation and ultimately cytotoxicity. To further refine our approach, and given the complexity of obtaining X-ray co-crystal structures of many compounds with the ribosome, we worked with the Mobley lab on computational docking. Collectively, these studies showed that cytotoxicity tracked reasonably well with translation inhibition activity, as expected, and yielded some SAR results that included some surprises. A few key data are shown in Figure 6.1,2
Figure 6.
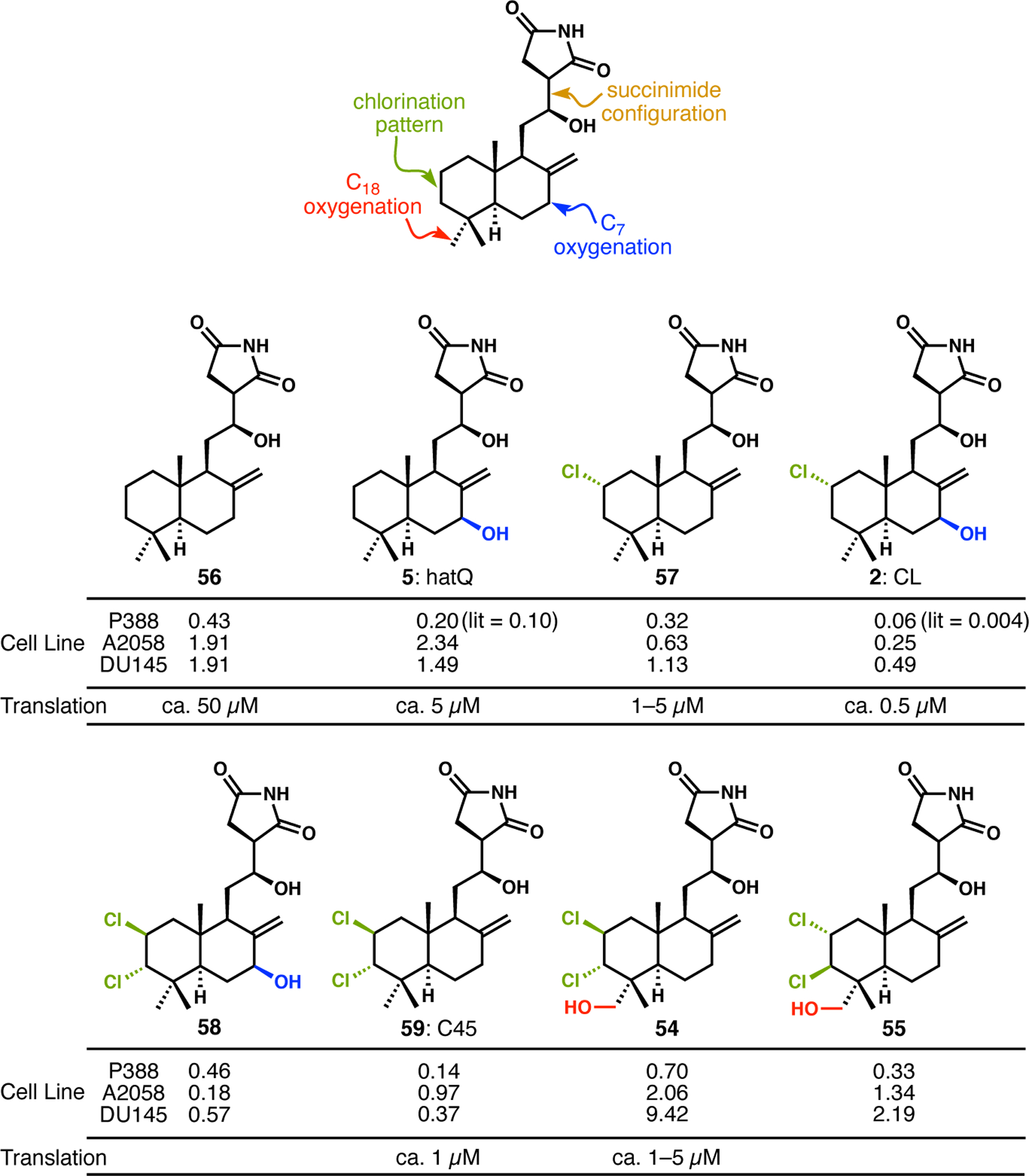
Biological activities of representative lissoclimides
We quickly learned that the succinimide configuration was critical to activity (not shown). This observation was not surprising given that semi-synthetic N-methylated or dehydrated analogues were shown to be inactive by Ueda and coworkers,11e and because this portion of the molecule “anchors” the lissoclimide molecules into the binding site. Other alterations of the A-ring, including a C2–C3 alkene and a C3 hydroxyl group, led to abolishment of cytotoxicity and translation inhibition activity. The most interesting results came from the interplay of A-ring chlorination, B-ring (C7) oxygenation, and C18 oxygenation. We used the “unfunctionalized” and previously unknown lissoclimide analogue 56 as a benchmark. It was a poor inhibitor of translation and a micromolar cytotoxin toward the three cell lines tested. Introduction of the C7 hydroxyl group in hatQ (5) had little effect on cytotoxicity, and the same was true with C2-chlorination (see 57); however, each of these compounds was a significantly better translation inhibitor than 56. Combining the C2-chlorine with the C7-alcohol in CL (2) led to a significantly more potent compound. While we have not yet made DCL (known to be more potent against P388 than CL), we did make four A-ring dichlorinated lissoclimide analogues. Because they were made from A-ring alkene dichlorination, it was easier to obtain the C2/C3-diepi compounds (chlorines nominally axial, see below); the one exception was 55, which combined the diequatorial dichloride of DCL with the C18 hydroxy group of hatJ (6). This compound was surprisingly lackluster, despite having these combined elements that we had believed were important for activity in these two most potent natural lissoclimides. The significant surprise came from the maintained activity of compounds 54, 58, and 59, with the unnaturally configured chlorine-bearing centers. Compound 59, which became known as “C45”, was second only to CL in translation inhibition and cytotoxic activities.2,3
Given what we knew about the E-site binding pocket, and the interaction of the chloride in CL with G2793—with the equatorial chloride residue nestled tightly between it and G2794—we anticipated that C45 and other diepi dichlorides would be completely inactive. To try to understand the failure of this structure-based hypothesis and the relatively strong activity of C45, we engaged in docking studies with the Mobley group at UCI. The results were surprising, but rationalizable. The docking experiment provided a binding pose in which the hydroxysuccinimide and B-ring were relatively unperturbed relative to that of CL; however, it had enforced an A-ring conformational change from a chair to a twist-boat.2 This conformation resulted in the nominally axial chloride residues residing in pseudoequatorial dispositions and, curiously, positioned each chlorine atom above a different guanine residue in range of a potential dispersive attractive interaction of the type that we first observed with CL (Figure 7a).2 Interestingly, the docking experiment is not sophisticated enough to “understand” the attractive dispersive interaction. We calculated the energy difference between the chair conformation (with four axial substituents, including three on one face) to twist-boat conformation, and found that the latter was only 1.5 kcal/mol higher in energy. Therefore, we became comfortable explaining that the relatively low penalty for this conformational change might be offset by the two stabilizing halogen-π interactions, and would also permit a decent fit of C45 into the binding site, thus potentially accounting for its reasonable activity. However, at this stage, we were rationalizing a docking experiment that might well have been in error.2 We were understandably pleased when the Yusupov group provided an X-ray co-crystal structure of the eukaryotic ribosome with C45 that revealed exactly this structural relationship (Figure 7b).3 This second example of an apparent alkyl chloride/guanine interaction gave us increased confidence that this phenomenon might be important, and was worthy of broader, ongoing studies.
Figure 7.

a. Docking experiment of C45 (59) with ribosomal E-site. Reproduced with permission from ref. 2. Copyright 2017 Springer Nature. b. X-ray co-crystal structure of C45 with ribosomal E-site. Reproduced with permission from ref. 3. Copyright 2019 Oxford University Press, distributed under the terms of the Creative Commons C BY license.
Haterumaimide J
Although we had already developed both a semi-synthesis and an analogue-oriented total synthesis approach to the lissoclimides, we had not yet accessed the most potent known natural product in the series, hatJ (6).11c,e While it was not inconceivable that the latter, π-cyclization approach could reach that goal, the presence of the C18 hydroxyl group motivated our investigations into an approach that would interrogate some interesting chemical questions, as well as potentially provide an efficient synthesis. The chlorine atom was again going to play a key role in these studies.
Our π-cyclization studies described above were typical of epoxide-initiated polycyclizations in the use of the C2–C3 epoxide that comes naturally from oxidation of the geraniol-type alkene, and that is also the substrate type for myriad biosynthetic and synthetic cyclizations.30 The exocyclic, C18-oxygenation of hatJ suggested the possibility of using a different epoxide regioisomer to trigger π-cyclization and thus directly install the required hydroxy group (Figure 8). While this approach—making use of rarely studied terminal-epoxide-initiatied π-cyclizations—was clearly bioinspired in the decalin-forming reaction, it was not strictly speaking biomimetic. To our knowledge, nature does not use terminal epoxides in these cyclizations, and the oxidation of the C18 (or C19) methyl groups occurs by post-cyclization P450-mediated reactions.
Figure 8.

Potential application of a terminal-epoxide-initiated π-cyclization toward a synthesis of hatJ
Typical, internal-epoxide-initiated cyclizations (Scheme 6a for an example from Loh and co-workers31) generally provide single stereoisomers of product because of the resident epoxide stereogenic center. Terminal-epoxide-initiated π-cyclizations have only been investigated a few times, with the initial report by Goldsmith and Phillips shown in Scheme 6b;32 some more complex examples in synthesis were later reported by the van Tamelen32 and Corey34 labs. Terminal epoxides such as 62 do not have a resident stereogenic center to enforce the formation of a single diastereomer of product. While the tertiary epoxide C–O bond that is undergoing stereoinvertive attack by the π-bond will lead to stereospecific generation of the C4 stereogenic center, there are two different π-facial arrangements for cyclization, which would lead to two different diastereomeric products (63 and 64). The E-alkene in the substrate enforces trans-decalin formation, so the diastereomers are most easily recognized by the equatorial (C18) or axial (C19) location of the oxidized carbon that derived from the epoxide, and from the C4/C5 relative configuration. The studies of Goldsmith and Phillips implied that there was some selectivity for products with equatorial (C18) oxygenation, as 63 was the only product that they could characterize; however, many other uncharacterized products might well have been formed by a different facial approach of the activated epoxide to the alkene.32 Our driving hypothesis was that the C3 equatorial chlorine atom in hatJ, if present in the cyclization precursor (65, Scheme 6c), would ensure cyclization through the desired conformation owing to the preference to keep the chlorine equatorial. Even with a small A-value of ca. 0.5 kcal/mol, we believed that the combination of the potential inherent stereoselectivity implied by Goldsmith and Phillips with the avoidance of the chlorine being coaxial with two methyl groups would result in high selectivity for 66, a compound with the desired stereochemical arrangement for hatJ. As discussed below, this was borne out by the key reaction, which was a modified example of what we had initially tried to do: a π-cyclization with stereochemical control arising (in part) from a chlorine-bearing stereogenic center and terminated by a furan.18
Scheme 6.
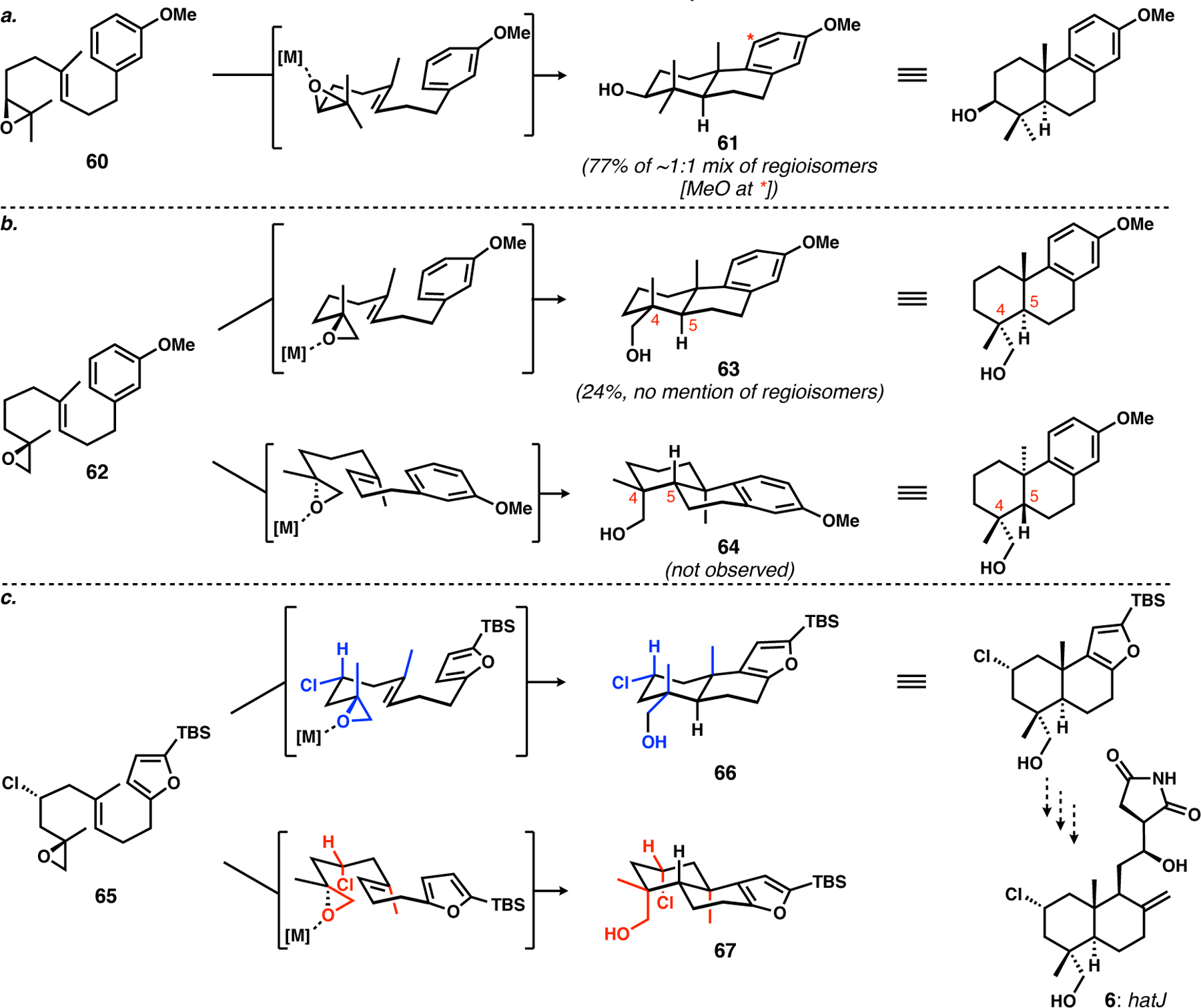
a. Typical stereocontrolled bicyclization initiated by Lewis-acid-activation of internal epoxides by Loh and co-workers31. b. The analogous terminal-epoxide-initiated reaction by Goldsmith and Phillips32. c. Our plan to use the C2-chlorine atom to bias the cyclization of 65 toward the diastereomer needed for a synthesis of hatJ.
The synthesis was reduced to practice4 by a stereocontrolled synthesis of vinyl iodide 68 with the specific relative configuration shown of chlorine bearing C3-stereogenic center and epoxide (Scheme 7a). It was coupled with the alkyl iodide 69 under modified Suzuki–Miyaura conditions35 to give cyclization precursor 65. Under optimized conditions, this substrate behaved exactly as expected, affording a single stereoisomer of trans-decalin 66, whose silylfuran was transformed into alkene/aldehyde 70 for the final succinimide introduction to deliver hatJ. As satisfying as the stereochemical outcome was, the control experiment lacking the chlorine led to a 4.5:1 selectivity for the stereoisomer with C18 oxygenation (not shown); in other words, the chlorine atom played only a supporting role in the stereochemical control. However, we were pleased to see that diastereomer 71, when subjected to the same cyclization conditions that were not reoptimized for this substrate, afforded an 11:1 ratio of diastereomers favoring C19 oxygenation; the strength of the chlorine directing group36 is revealed here, in the mismatched case.37
Scheme 7.
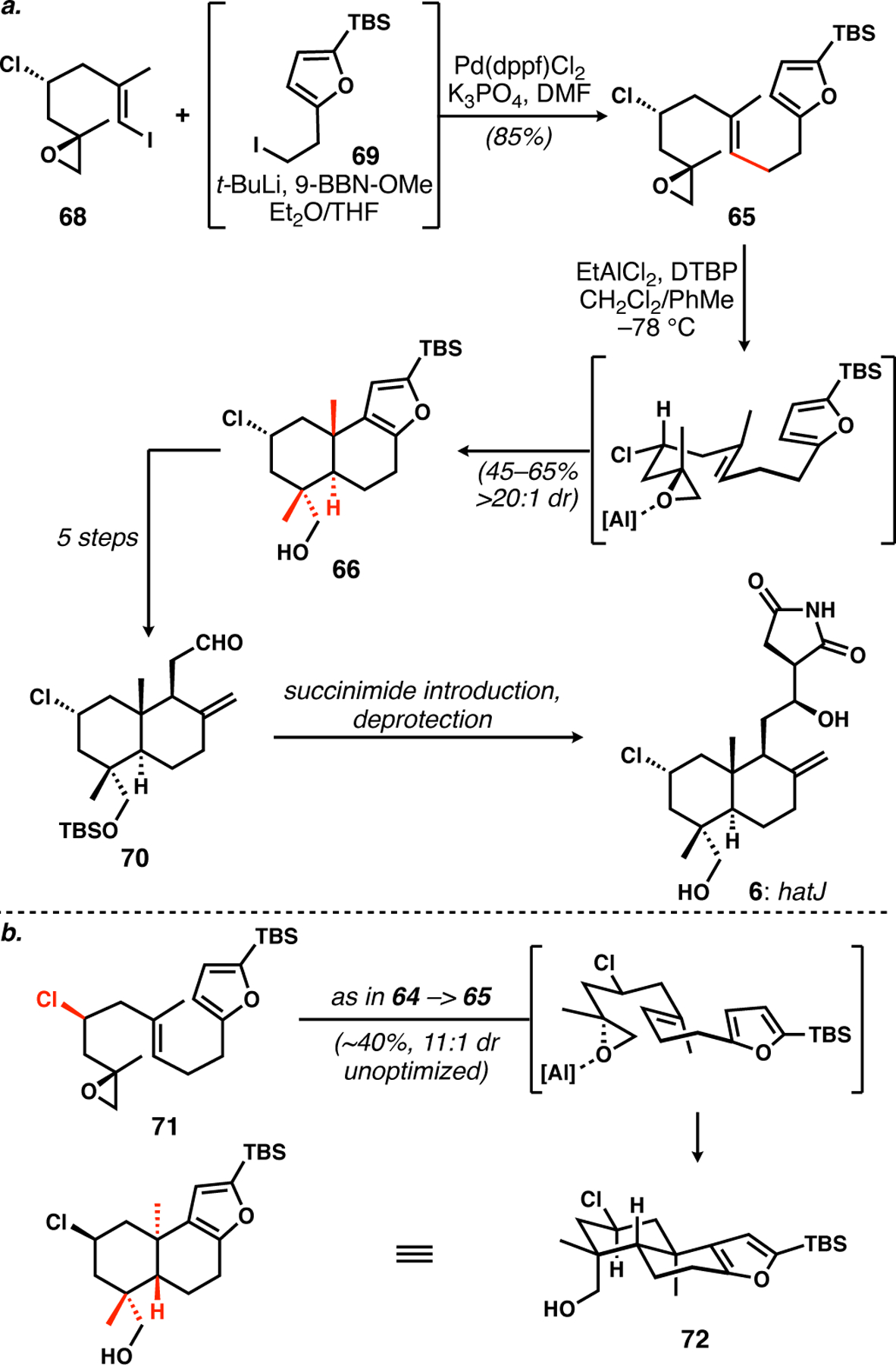
a. Stereocontrolled synthesis of hatJ via terminal-epoxide-initiated bicyclization. b. Chlorine-bearing stereogenic center can control the stereochemical outcome of these cyclizations.
The synthesis of hatJ has facilitated ongoing studies into its biology, as the most potent member of the lissoclimides isolated or made to date. As we can see from this successful endeavor, our original idea of a chlorine-directed, furan-terminated polyene cyclization toward lissoclimide-type compounds has come full circle, and led us into a relatively underexplored area of terminal-epoxide-initiated π-cyclizations. Moreover, it provides another interesting example of the potential power of halogens as single-atom auxiliaries to control the stereochemical outcomes of C–C bond-forming (architecture-building) events.
Conclusions and outlook.
Starting from an interest in the synthesis of polyhalogenated natural products, our work on the lissoclimides evolved with the invention of three separate synthesis designs and with multi-faceted collaborative biological investigations. The first approach made use of sclareolide as a precursor in a semi-synthesis of CL and simple congeners; the second was an analogue-oriented strategy that permitted access to numerous unnatural lissoclimides, and the third focused on the most potent lissoclimides using a terminal-epoxide-initiated bicyclization. Throughout all of our studies, chlorine played a central role: (1) a potentially biomimetic chlorination of sclareolide permitted our semi-synthesis of CL; (2) the chlorine atom in CL engaged in a novel halogen-π dispersion interaction in its X-ray co-crystal structure with the ribosome; and (3) a chlorine atom played a stereocontrolling role in the epoxide-initiated cyclization chemistry en route to hatJ. Studies with our collaborators defined structure-activity relationships and established the molecular basis for translation inhibition by the lissoclimides. Our ongoing work in this area seeks to gain an improved understanding of the biology of the lissoclimides and the halogen-π interaction that was uncovered by our work in this area.
Acknowledgments
The work described herein was accomplished by virtue of the outstanding commitment of my previous co-workers, as well as the co-authors of this account. Drs. Anne Szklarski, Yvonne Schmidt, Zef Könst, and Sharon Michalak were instrumental in our past achievements on the lissoclimide project. Our extensive network of collaborators helped turn a chemical synthesis project into an interdisciplinary science project, and we are indebted to them for their enthusiastic and critical contributions: Yusupov group (IGBMC, Strasbourg), Mobley and Furche groups (UCI), Horne lab (City of Hope), Pelletier lab (McGill), and Blanchard group (St Jude).
Bonnie S. Pak was raised in Palm Springs, CA, and obtained her B.S. degree in chemistry in 2017 from University of California, San Diego. She conducted undergraduate research with Professor Michael Burkart and interned in the medicinal chemistry department at Takeda Pharmaceuticals. She subsequently joined Professor Christopher Vanderwal’s research group in 2018 as a graduate student at University of California, Irvine. Her graduate research currently focuses on the synthesis of lissoclimide analogues to evaluate the halogen-π interaction and gain a deeper understanding of the lissoclimides’ biological activities.
Nantamon Supantanapong earned her B.Sc. in chemistry from Prince of Songkla University in Thailand (2016). She carried out her M.Sc. thesis work on one-pot synthesis of 3-carbonyl-4-arylbenzo[f]indoles at the Chulabhorn Graduate Institute in Bangkok (2019), under the supervision of Prof. Jumreang Tummatorn. In the fall of 2018, Nantamon worked in the group of Prof. Daniel Seidel at University of Florida. Since then, she has been pursuing her Ph.D. at University of California, Irvine working on synthesis of lissoclimide natural product derivatives under Christopher Vanderwal.
Christopher D. Vanderwal grew up in Ottawa, Canada, where he earned BSc (Biochemistry) and MSc (Chemistry) degrees from the University of Ottawa. He obtained his PhD from the Scripps Research Institute, where he worked with Professor Erik Sorensen on the synthesis and biological evaluation of the cytotoxin FR182877. After a postdoctoral stay in asymmetric catalysis working with Professor Eric Jacobsen at Harvard, he began his independent academic career in the Department of Chemistry at UC Irvine in 2005. He is currently Professor of Chemistry and of Pharmaceutical Sciences, and his research program continues to focus on the synthesis of complex, bioactive natural products and collaborative biological investigations thereof.
References
- 1.Quinn RK; Könst ZA; Michalak SE; Schmidt Y; Szklarski AR; Flores AR; Nam S; Horne DA; Vanderwal CD; Alexanian EJ Site-Selective Aliphatic C–H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc 2016, 138, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]; A new method for selective C–H chlorination developed by the Alexanian group was leveraged in a semi-synthesis of chlorolissoclimide and analogues.
- 2.Könst ZA; Szklarski AR; Michalak SE; Pellegrino S; Meyer M; Zanette C; Cencic R; Nam S; Voora V; Horne DA; Pelletier J; Mobley DL; Yusupov M; Vanderwal CD Synthesis Facilitates an Understanding of the Structural Basis for Translation Inhibition by the Lissoclimides. Nature Chem. 2017, 9, 1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]; An X-ray co-crystal structure of chlorolissoclimide with the eukaryotic ribosome revealed how it inhibits protein synthesis, and prompted an analogue-oriented synthesis to learn about structure-activity relationships in the lissoclimide series.
- 3.Pellegrino S; Meyer M; Könst ZA; Holm M; Voora VK; Kashinskaya D; Zanette C; Mobley DL; Yusupova G; Vanderwal CD; Blanchard SC; Yusupov M Understanding the Role of Intermolecular Interactions between Lissoclimides and the Eukaryotic Ribosome. Nucl. Acids. Res 2019, 47, 3223–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]; A combination of X-ray co-crystallography, computation, and single-molecule FRET studies provided a deeper understanding of the interactions between chlorolissoclimide and analogue “C45” with the ribosome.
- 4.Michalak SE; Nam S; Kwon DM; Horne DA; Vanderwal CD A Chlorine-Atom-Controlled Terminal-Epoxide-Initiated Bicyclization Cascade Enables a Synthesis of the Potent Cytotoxins Haterumaimides J and K. J. Am. Chem. Soc 2019, 141, 9202–9206. [DOI] [PMC free article] [PubMed] [Google Scholar]; A synthesis of the most potent lissoclimide was executed by an uncommon polyene cascade initiated by a terminal epoxide.
- 5.Garreau de Loubresse N; Prokhorova I; Holtkamp W; Rodnina MV; Yusupova G; Yusupov M Structural Basis for the Inhibition of the Eukaryotic Ribosome. Nature 2014, 513, 517–522. [DOI] [PubMed] [Google Scholar]
- 6.Wilson DN The A–Z of Bacterial Translation Inhibitors. Critical Rev. Biochem. Mol. Biol 2009, 44, 393–433. [DOI] [PubMed] [Google Scholar]
- 7.Malina A; Mills JR; Pelletier J Emerging Therapeutics Targeting mRNA Translation. Cold Spring Harb. Perspect. Biol 2012, doi: 10.1101/cshperspect.a012377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gandhi V; Plunkett W; Cortes JE Omacetaxine: A Protein Translation Inhibitor for Treatment of Chronic Myelogenous Leukemia. Clin. Cancer Res 2014, 20, 1735–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Bedke DK; Shibuya GM; Pereira A; Gerwick WH; Haines TH; Vanderwal CD Relative Stereochemistry Determination and Synthesis of the Major Chlorosulfolipid from Ochromonas danica. J. Am. Chem. Soc 2009, 131, 7570–7572. [DOI] [PubMed] [Google Scholar]; (b) Bedke DK; Shibuya GM; Pereira A; Gerwick WH; Vanderwal CD A Concise Enantioselective Synthesis of the Chlorosulfolipid Malhamensilipin A. J. Am. Chem. Soc 2010, 132, 2542–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chung W.-j.; Carlson JS; Bedke DK; Vanderwal CD A Synthesis of the Chlorosulfolipid Mytipilin A via a Longest Linear Sequence of Seven Steps. Angew. Chem. Int. Ed 2013, 52, 10052–10055. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chung W.-j.; Carlson JS; Vanderwal CD A General Approach to the Synthesis of the Chlorosulfolipids Danicalipin A, Mytilipin A, and Malhamensilipin A in Enantioenriched Form. J. Org. Chem 2014, 79, 2226–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chung W.-j.; Vanderwal CD Approaches to the Chemical Synthesis of the Chlorosulfolipids. Acc. Chem. Res 2014, 47, 718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Vogel CV; Pietraszkiewicz H; Sabry OM; Gerwick WH; Valeriote FA; Vanderwal CD Enantioselective, Divergent Syntheses of Several Polyhalogenated Plocamium Monoterpenes, and Evaluation of their Activity and Selectivity toward Solid Tumors. Angew. Chem. Int. Ed 2014, 53, 12205–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Tartakoff SS; Vanderwal CD A Synthesis of the ABC Tricyclic Core of the Clionastatins, Highly Chlorinated Androstane Steroids, Serves to Corroborate their Proposed Structures. Org. Lett 2014, 16, 1458–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Malochet-Grivois C; Cotelle P; Biard JF; Hénichart JP; Debitus C; Roussakis C; Verbist J-F Dichlorolissoclimide, a New Cytotoxic Labdane Derivative from Lissoclinum voeltzkowi Michaelson (Urochordata). Tetrahedron Lett. 1991, 32, 6701–6702. [Google Scholar]; (b) Malochet-Grivois C; Roussakis C; Robillard N; Biard JF; Riou D; Debitus C; Verbist J-F Effects in vitro of Two Marine Substances, Chlorolissoclimide and Dichlorolissoclimide, on a Non-small-cell Bronchopulmonary Carcinoma Line (NSCLC-N6). Anti-Cancer Drug Design 1992, 7, 493–502. [PubMed] [Google Scholar]; (c) Roussakis C; Charrier J; Riou D; Biard JF; Malochet C; Meflah K; Verbist JF Chemotherapeutic Inhibition of erb-B2 Oncogene Expression on a Non-small-cell Cancer Line (NSCLC-N6) by Marine Substances. Anti-Cancer Drug Design 1994, 9, 119–128. [PubMed] [Google Scholar]
- 11.(a) Uddin MJ; Kokubo S; Suenaga K; Ueda K; Uemura D Haterumaimides A–E, Five New Dichlorolissoclimide-Type Diterpenoids from an Ascidian Lissoclinum Species. Heterocycles 2001, 54, 1039–1047. [Google Scholar]; (b) Uddin MJ; Kokubo S; Ueda K; Suenaga K; Uemura D Haterumaimides F–I, Four New Cytotoxic Diterpene Alkaloids from an Ascidian Lissoclinum Species. J. Nat. Prod 2001, 64, 1169–1173. [DOI] [PubMed] [Google Scholar]; (c) Uddin MJ; Kokubo S; Ueda K; Suenaga K; Uemura D Haterumaimides J and K, Potent Cytotoxic Diterpene Alkaloids from the Ascidian Lissoclinum Species. Chem. Lett 2002, 10, 1028–1029. [Google Scholar]; (d) Fu X; Palomar AJ; Hong EP; Schmitz FJ; Valeriote FA Cytotoxic Lissoclimide-type Diterpenes from the Molluscs Pleurobranchus albiguttatus and Pleurobranchus forskalii. J. Nat. Prod 2004, 67, 1415–1418. [DOI] [PubMed] [Google Scholar]; (e) Uddin J; Ueda K; Siwu ERO; Kita M; Uemura D Cytotoxic Labdane Alkaloids from an Ascidian Lissoclinum sp.: Isolation, Structure Elucidation, and Structure-activity Relationship. Bioorg. Med. Chem 2006, 14, 6954–6961. [DOI] [PubMed] [Google Scholar]
- 12.Robert F; Hao HQ; Donia M; Merrick WC; Hamann MT; Pelletier J Chlorolissoclimides: New Inhibitors of Eukaryotic Protein Synthesis. RNA 2006, 12, 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Sugawara K; Nishiyama Y; Toda S; Komiyama N; Hatori M; Moriyama T; Sawada Y; Kamei H; Konishi M; Oki T Lactimidomycin, a New Glutarimide Group Antibiotic. Production, Isolation, Structure and Biological Activity. J. Antibiot 1992, 45, 1433–1441. [DOI] [PubMed] [Google Scholar]; (b) Ju J; Rajski SR; Lim SK; Seo JW; Peters NR; Hoffmann FM; Shen B Lactimidomycin, iso-Migrastatin and Related Glutarimide-containing 12-Membered Macrolides Are Extremely Potent Inhibitors of Cell Migration. J. Am. Chem. Soc 2009, 131, 1370–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schneider-Poetsch T; Ju J; Eyler DE; Dang Y; Bhat S; Merrick WC; Green R; Shen B; Liu JO Inhibition of Eukaryotic Translation Elongation by Cycloheximide and Lactimidomycin. Nat. Chem. Biol 2010, 6, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferguson A; Wang L; Altman RB; Terry DS; Juette MF; Burnett BJ; Alejo JL; Dass RA; Parks MM; Vincent CT; Blanchard SC Functional Dynamics within the Human Ribosome Regulate the Rate of Active Protein Synthesis. Mol. Cell 2015, 60, 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung ME; Gomez AV Efficient Method for the Preparation of 2α,3β-Dichloro-4,4,10-trimethyl-decalin Systems as a Route for the Synthesis of Dichlorolissoclimide. Tetrahedron Lett. 1993, 34, 2891–2894. [Google Scholar]
- 16.González MA; Romero D; Zapata B; Betancur-Galvis L First Synthesis of Lissoclimide-type Alkaloids. Lett. Org. Chem 2009, 6, 289–292. [Google Scholar]
- 17.Nguyen TM; Nguyen QV; Youte J-J; Lau J; Cheong A; Ho YS; Tan BSW; Yogonathan K; Butler MS; Chai CLL, A Fast and Straightforward Route Towards the Synthesis of the Lissoclimide Class of Anti-Tumour Agents. Tetrahedron 2010, 66, 9270–9276. [Google Scholar]
- 18.(a) Tanis SP; Herrinton PM Furans in Synthesis. 3. Furans as Terminators in Cationic Cyclization. J. Org. Chem 1983, 48, 4572–4580. [Google Scholar]; (b) Tanis SP; Chuang Y-H; Head DB Furans in Synthesis. 8. Formal Total Syntheses of (±)- and (+)-Aphidicolin. J. Org. Chem 1988, 53, 4929–4938. [Google Scholar]
- 19.Gansäuer A; Justicia J; Rosales A; Worgull D; Rinker B; Cuerva JM; Oltra JE Transition-Metal-Catalyzed Allylic Substitution and Titanocene-Catalyzed Epoxypolyene Cyclization as a Powerful Tool for the Preparation of Terpenoids. Eur. J. Org. Chem 2006, 4115–4127. [Google Scholar]
- 20.Yoshimitsu T; Fukumoto N; Tanaka T Enantiocontrolled Synthesis of Polychlorinated Hydrocarbon Motifs: A Nucleophilic Multiple Chlorination Process Revisited. J. Org. Chem 2009, 74, 696–702. [DOI] [PubMed] [Google Scholar]
- 21.(a) Chen K; Eschenmoser A; Baran PS Strain Release in C–H Bond Activation? Angew. Chem. Int. Ed 2009, 48, 9705–9708. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen MS; White MC Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. [DOI] [PubMed] [Google Scholar]; (c) Liu W; Groves JT Manganese Porphyrins Catalyze Selective C-H Bond Halogenations. J. Am. Chem. Soc 2010, 132, 12847–12849. [DOI] [PubMed] [Google Scholar]; (d) Liu W; Huang X; Cheng M-J; Nielsen RJ; Goddard III WA; Groves JT Oxidative Aliphatic C–H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin. Science 2012, 337, 1322–1325. [DOI] [PubMed] [Google Scholar]; (e) Halperin SD; Fan H; Chang S; Martin RE; Britton R A Convenient Photocatalytic Fluorination of Unactivated C–H Bonds. Angew. Chem. Int. Ed 2014, 53, 4690–4693. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt VA; Quinn RK; Brusoe AT; Alexanian EJ Site-selective Aliphatic C–H Bromination Using N-Bromoamides and Visible Light. J. Am. Chem. Soc 2014, 136, 14389–14392. [DOI] [PubMed] [Google Scholar]
- 23.Boukouvalas J; Wang J-X; Marion O; Ndzi B Synthesis and Stereochemistry of the Antitumor Diterpenoid (+)-Zerumin B. J. Org. Chem 2006, 71, 6670–6673. [DOI] [PubMed] [Google Scholar]
- 24.Corey EJ; Luo G; Lin LS A Simple Enantioselective Synthesis of the Biologically Active Tetracyclic Marine Sesterterpene Scalarenedial. J. Am. Chem. Soc 1997, 119, 9927–9928. [Google Scholar]
- 25.Shibuya GM; Kanady JS; Vanderwal CD Stereoselective Dichlorination of Allylic Alcohol Derivatives to Access Key Stereochemical Arrays of the Chlorosulfolipids. J. Am. Chem. Soc 2008, 130, 12514–12518. [DOI] [PubMed] [Google Scholar]
- 26.Ben-Shem A; Garreau de Loubresse N; Melnikov S; Jenner L; Yusupova G; Yusupov M The Structure of the Eukaryotic Ribosome at 3.0 Å Resolution. Science 2011, 334, 1524–1529. [DOI] [PubMed] [Google Scholar]
- 27.Imai YN; Inoue Y; Nakanishi I; Kitaura K Cl–π Interactions in Protein–ligand Complexes. Protein Sci. 2008, 17, 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McClary B; Zinshteyn B; Meyer M; Jouanneau M; Pellegrino S; Yusupova G; Schuller A; Reyes JCP; Lu J; Guo Z; Ayinde S; Luo C; Dang Y; Romo D; Yusupov M; Green R; Liu JO Inhibition of Eukaryotic Translation by the Antitumor Natural Product Agelastatin A. Cell Chem. Biol 2017, 24, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanessian S; Saavedra OM; Vilchis-Reyes MA; Maianti JP; Kanazawa H; Dozzo P; Matias RD; Serio A; Kondo J Synthesis, Broad Spectrum Antibacterial Activity, and X-ray Co-crystal Structure of the Decoding Bacterial Ribosomal A-site with 4ʹ-Deoxy-4ʹ-fluoro Neomycin Analogs. Chem. Sci 2014, 5, 4621–4632. [Google Scholar]
- 30.Yoder RA; Johnston JN A Case Study in Biomimetic Total Synthesis: Polyolefin Carbocyclizations to Terpenes and Steroids. Chem. Rev 2005, 105, 4730–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao J-F; Zhao Y-J; Loh T-P Indium Tribromide-Promoted Arene-terminated Epoxy Olefin Cyclization. Chem. Commun 2008, 1353–1355. [DOI] [PubMed] [Google Scholar]
- 32.Goldsmith DJ; Phillips CF The Structural and Stereochemical Course of in Vitro Epoxy Olefin Cyclization. Diterpenoid Intermediates. J. Am. Chem. Soc 1969, 91, 5862–5870. [Google Scholar]
- 33.van Tamelen EE; Zawacky SR; Russell RK; Carlson JG Biogenetic-Type Total Synthesis of (±)-Aphidicolin. J. Am. Chem. Soc 1983, 105, 142–143. [Google Scholar]
- 34.Corey EJ; Liu K Enantioselective Total Synthesis of the Potent anti-HIV Agent Neotripterifordin. Reassignment of Stereochemistry at C(16). J. Am. Chem. Soc 1997, 119, 9929–9930. [Google Scholar]
- 35.Marshall JA; Schaaf GM Total Synthesis and Structure Confirmation of Leptofuranin D. J. Org. Chem 2003, 68, 7428–7432. [DOI] [PubMed] [Google Scholar]
- 36.Halperin S; Britton R Chlorine, an Atom Economical Auxiliary for Asymmetric Aldol Reactions. Org. Biomol. Chem 2013, 11, 1702–1705. [DOI] [PubMed] [Google Scholar]
- 37.Masamune S; Choy W; Petersen JS; Sita LR Double Asymmetric Synthesis and a New Strategy for Stereochemical Control in Organic Synthesis. Angew. Chem. Int. Ed 1985, 24, 1–30. [Google Scholar]