

Abstract
A 42-year-old man with a history of surgery for tongue cancer was referred to our hospital due to an abnormal chest shadow. High-resolution computed tomography showed lower lobe reticulation. A physical examination revealed nail dystrophy, oral leukoplakia, and reticulated hypopigmentation. Lung biopsy revealed subpleural and perilobular fibrosis, suggestive of usual interstitial pneumonia. However, multiple pathological findings, including homogenous fibrosis and cell infiltration in the centrilobular region, which were compatible with nonspecific interstitial pneumonia, and bronchiolitis were also seen. Genetic testing showed a hemizygous missense mutation in the DKC1 gene, and the patient was diagnosed with dyskeratosis congenita. Although anti-fibrotic therapy was initiated, the patient's respiratory function has continued to decrease.
Keywords: dyskeratosis congenita, DKC1 mutation, interstitial pneumonia, nail dystrophy, oral leukoplakia, telomere
Introduction
Dyskeratosis congenita (DC) is a rare genetic disorder with multiple organ involvement and a poor prognosis that is caused by impaired telomere maintenance due to genetic abnormality (1,2). Although DC is characterized by the clinical triad of nail dystrophy, abnormal skin pigmentation, and mucosal leukoplakia, interstitial pneumonia (IP) reportedly develops in 20% of DC patients (3). There are limited reports about pathological findings of lung biopsy in DC patients; most of them are reported as usual interstitial pneumonia (UIP) (3-5). Furthermore, there are few reports on the treatment of interstitial pneumonia in DC patients. We herein report a case of DC with multiple pathological lung findings, in which the patient was treated with anti-fibrotic drugs.
Case Report
A 42-year-old man, with a surgical history of partial resection of tongue carcinoma 15 years previously, was referred to our hospital due to an abnormal chest shadow in January 2018. He was an ex-smoker (1 pack/day for 4 years) and worked on a farm, but had no obvious history of exposure to dust or drugs. Although he did not have a family history of IP, his three maternal uncles each had malignancies, including colorectal cancer and peritonitis carcinomatosa.
A chest radiograph showed a reticular shadow and volume loss in the lower lung field (Fig. 1). High-resolution computed tomography (HRCT) demonstrated reticulation with lower lobe predominance and traction bronchiectasis, which were considered to be a “probable UIP pattern” based on the guidelines for idiopathic interstitial pneumonias (IIPs) (Fig. 2) (6). A physical examination revealed nail dystrophy, oral leukoplakia, and reticulated hypopigmentation (Fig. 3). In addition, mild loss of head hair and dental abnormality were also seen. His skin manifestation and nail dystrophy had started in his early teens and his oral leukoplakia was noticed in his middle 20s. Other physical examinations revealed normal findings.
Figure 1.
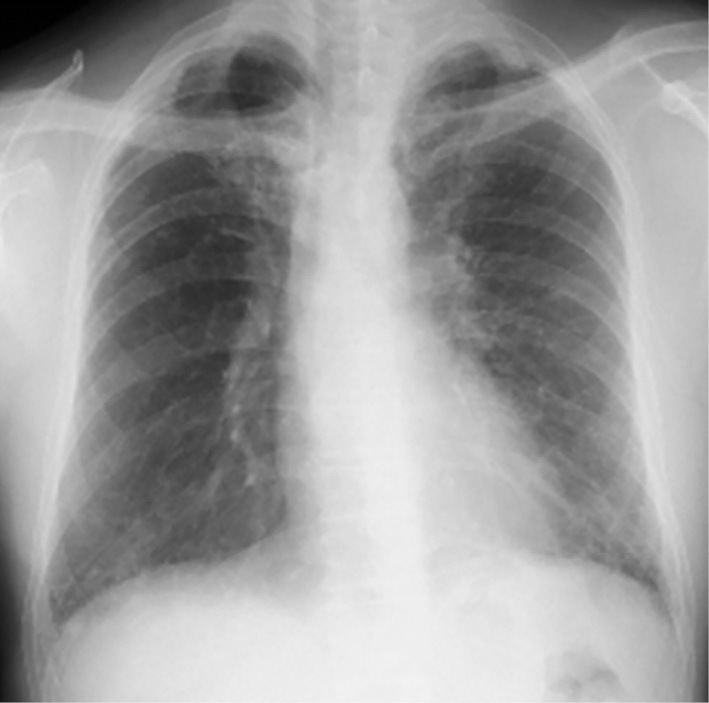
A chest radiograph showed a reticular shadow and volume loss of the lower lung field.
Figure 2.

High-resolution computed tomography showed reticulation with lower lobe predominance and traction bronchiectasis, which were considered a “probable UIP pattern”.
Figure 3.

A physical examination revealed nail dystrophy (A), oral leukoplakia (B), and reticulated hypopigmentation (C).
A laboratory examination revealed elevated levels of Krebs von den Lungen-6 (881 U/mL), surfactant protein-D (SP-D) (221 ng/dL), and total immunoglobulin E (9,556 U/mL). In addition, a slight decrease in his platelet count and elevation of his transaminase levels were observed. His white blood cell count, white blood cell fraction and red blood cell count were normal. No autoantibodies were detected. Pulmonary function testing (PFT) showed evidence of mild restrictive ventilation with a forced vital capacity (FVC) of 3.48 L (% predicted, 77.9%) and reduced diffusing capacity of carbon monoxide (DLco) of 16.64 mL/min/Torr (% predicted, 66.1%). Bronchoalveolar lavage fluid from the right middle lobe (B5a) showed a total cell count of 5.1×105 cells/mL (80% macrophages, 14% lymphocytes, 3% neutrophils, and 3% eosinophils) (Table).
Table.
Laboratory Findings at Initial Examination.
| Blood cell couunt | Immunologocal marker | Respiratory function test | |||||||
| WBC | 4,150 | /μL | IgG | 1,196 | mg/dL | VC | 3.34 | L | (73.2%) |
| Neut. | 56.2 | % | IgG4 | 53 | mg/dL | FVC | 3.48 | L | (77.9%) |
| Lymp. | 27.7 | % | IgE | 9,556 | IU/mL | FEV1 | 2.75 | L | (71.1%) |
| Eosi. | 5.3 | % | ANA | 40 | FEV1/FVC | 79.0 | % | ||
| Hb | 14.0 | g/dL | Homo | 40 | %DLCO | 66.1% | |||
| Plt | 14.9 | ×104/μL | RF | 3 | %DLCO/VA | 77.9% | |||
| Biochemistory | Anti CCP | 0.6 | |||||||
| Alb | 4.3 | g/dL | Anti ds-DNA | (-) | Bronchoalveolar lavage | ||||
| AST | 49 | IU/L | Anti SS-A | (-) | Site | Rt B5 | |||
| ALT | 54 | IU/L | Anti SS-B | (-) | Collection rate | 59 | % | ||
| LDH | 289 | IU/L | Anti ARS | (-) | Total cell count | 5.1 | ×105/mL | ||
| CK | 246 | IU/L | Anti Scl-70 | (-) | Macrophages | 80 | % | ||
| Cr | 0.73 | mg/dL | Anti U1-RNP | (-) | Lymphocytes | 14 | % | ||
| CRP | 0.35 | mg/dL | Anti Sm | (-) | Neutrophils | 3 | % | ||
| KL-6 | 881 | U/mL | Anti MPO-ANCA | (-) | Eosinophils | 3 | % | ||
| Sp-D | 221 | ng/dL | Anti PR3-ANCA | (-) | CD4/8 ratio | 2.8 | |||
WBC: white blood cell, Hb: hemoglobin, Plt: platelet, Alb: albumin, AST: aspartate transaminase, ALT: alanine aminotransferase, LDH; lactate dehydrogenase, CK: creatine kinase, Cr: creatinine, CRP: C-reactive protein, KL-6: Krebs von den Lungen-6, Sp-D: surfactant protein-D, IgG: immunoglobulin G, IgE: immunoglobulin E, ANA: antinuclear antibody, CCP: cyclic citrullinated peptide, MPO-ANCA: myeloperoxidase anti-neutrophil cytoplasmic antibody, PR3-ANCA: proteinase-anti-neutrophil cytoplasmic antibody, FVC: forced vital capacity, FEV: forced expiratory volume
Skin biopsy specimens of the pigmented area on his forearm revealed thinning of the epidermis, shedding of basal epithelial cells, and melanin pigmentation (Fig. 4).
Figure 4.

Skin biopsy specimens revealed thinning of the epidermis, loose fibrosis in the dermoepidermal junction (A), shedding of the basal epithelial cells, and melanin pigmentation (B).
Magnetic resonance imaging demonstrated enlargement and blunting of the edge of the hepatic left lobe. The patient was negative for Hepatitis B and C virus antibodies. Liver biopsy prior to the initiation of therapy showed advanced A2/F3 fibrosis (New Inuyama Classification), and pre-liver cirrhosis was diagnosed (7).
To evaluate his IP, a video-assisted thoracoscopic biopsy of the left lingular segment and segment 8 of the lung was performed (Fig. 5). Histologically, the lesion was well defined and showed subpleural and perilobular fibrosis and fibroblastic foci, which was compatible with the histological “UIP pattern” based on the guidelines for IIPs (Fig. 5A, D) (6). However, homogenous fibrosis and cell infiltration in the centrilobular region, bronchiolitis, lymphoid follicles, and the accumulation of macrophages were also observed (Fig. 5B, C). Therefore, the interstitial pneumonia was not considered to be a pure UIP. The multiple pathological findings suggested a differential diagnosis including UIP, fibrotic non-specific interstitial pneumonia (f-NSIP), desquamative interstitial pneumonia (DIP), and bronchiolitis.
Figure 5.

(A) Surgical lung biopsy from the lingular segment showed subpleural and perilobular fibrosis, which was compatible with the histological “UIP pattern”. (B, C) Lung tissue from segment 8 showed homogenous fibrosis and cell infiltration in the centrilobular region, bronchiolitis, lymphoid follicle, and accumulation of macrophages. (D) Alcian blue staining showed patchy fibroblastic foci.
Based on these results, genetic disease was suspected. After genetic counseling, whole exome sequencing of the patient and his parents was performed and a variant of the DKC1 gene (p.Try416Asn) was detected (Fig. 6A, B). This variant was not present in database of single nucleotide variations including the Genome Aggregation Database and our in-house Japanese exome controls. In addition, p.Tyr416Asn predicted to be deleterious by four [SIFT, protein variation effect analyzer (PROVEAN), Mutation Taster and CADD] bioinformatic tools. Based on these results and the clinical findings, the patient was diagnosed with DC. Furthermore, no rare variants were found in any gene for which a pathogenic variant has been reported to cause dyskeratosis congenita, with the exception of the DKC1 variant. Additionally, quantitative polymerase chain reaction to measure the telomere length was performed using a peripheral blood sample, using the CFX384 Touch Real-Time polymerase chain reaction (PCR) Detection System (Bio-Rad, Hercules, USA). This showed that he had a very short telomere length for his age (3.4 kilo base pairs, age-adjusted median ±2SD =7.99±3.42), which was consistent with a diagnosis of DC.
Figure 6.

(A) The patient did not have a family history of IP or DC. His 3 maternal uncles were affected with malignancy. (B) The candidate variant was extracted by trio whole-exome sequencing (III-2, II-1 and II-9). By sanger confirmation, the affected male and his mother were hemizygous and heterozygous, respectively, for a missense variant of the DKC1 gene (c.1246T>A: p.Tyr416Asn in NM_001363.3). This variant was predicted to be pathogenic.
IP was considered as a complication of DC, and the patient was carefully followed up; however, his IP had gradually progressed with a decrease in his respiratory function (FVC 2.98 L, %FVC 65.8%, %DLco 46%, in August 2019). Therefore, treatment with pirfenidone was initiated in September 2019, but his respiratory function has continued to decrease (Fig. 7).
Figure 7.

IP had gradually progressed with a decrease in the respiratory function despite the initiation of treatment with pirfenidone.
Discussion
DC is a rare heritable disorder of telomere maintenance, with a prevalence of approximately 1 per 1,000,000 (1,2). In addition to the clinical triad of nail dystrophy, abnormal skin pigmentation, and mucosal leukoplakia, DC may present with multiple organ involvement, including bone marrow failure, secondary malignancy, interstitial lung disease, and liver disorder, as was seen in our case (2,8). In general, skin manifestations and bone marrow failure appear early in many cases of DC (9). However, in some cases, such as the present case, pulmonary lesions and liver cirrhosis may precede bone marrow failure, making the diagnosis difficult (4). Thus, in cases with physical findings characteristic of DC, as in our case, blood cell telomere measurements should be performed first, and if telomere length is short, a genetic analysis should be added.
Because DC may develop due to an abnormality of any gene that encodes the telomerase complex, such as DKC1, TERC, TERT, and NHP2, DC can exhibit all three patterns of inheritance, including autosomal dominant, autosomal recessive, and X-linked recessive (10,11). Because DKC1 mutations, [the most common mutation in DC (30% of cases)], are inherited in an X-linked recessive pattern, our male patient would be the only one to develop DC in his family (11). Additionally, malignancy and premature death in his family may have been caused by an atypical type of DC.
Our results could provide the following two clinical implications. First, pathological findings of IP in a patient with DC would be diverse. Although there are only limited case reports of pathological lung findings due to the rarity of DC, most case reports described UIP as a common pattern of IP in DC patients (3,7,12). The first pathological case of IP in a DC patient reported by Imokawa et al. in 1994 was described as UIP (4). Similarly, Utz et al. reported the cases of 2 DC patients with the UIP pattern (3). However, the photomicrographs in his report demonstrated subpleural fibrosis with cellular infiltration and lymphoid follicles, which appeared somewhat atypical for a classic UIP pattern of idiopathic pulmonary fibrosis (IPF). Additionally, a few reports have demonstrated substantial bronchiolitis of IP in DC patients, as was noted in our case (13,14). Although the pathology of IP in DC patients is generally similar to the UIP pattern, according to the guidelines for IIPs, accurate pathological findings could be diverse and a detailed assessment may include UIP, f-NSIP, DIP, and bronchiolitis. The cause of IP in DC patients remains unknown. However, recent investigations have suggested that the loss of function of the telomerase complex may impair the turnover and healing of alveolar epithelial cells following a damaging stimulus that causes IP (15). Based on these findings and our case, it was suggested that a disorder of alveolar epithelial cells, and cell infiltration due to impaired telomere maintenance, not only in the lung parenchyma but also in the bronchioles, causes the multiple pathological findings of IP in DC patients.
Second, IP in a patient with DC would show rapid disease progression and be less responsive to therapy. In our case, during the 18 months from our initial visit to the start of treatment, the FVC decreased by 500 mL and the DLco (% of predicted) decreased by 20%. The rate of FVC decline in our case was more rapid than previously reported in IPF, which is the most progressive among the IIPs (16). A prior study reported that a decline of FVC and DLco in IPF with telomerase mutation (FVC -300 mL/year,%DLco -5.8%/year) was more rapid than that in IPF without mutation (17). This is similar to the results found in our patient. In our case, treatment with antifibrotic agents was selected because anti-inflammatory therapy with corticosteroids and immunosuppressants increases the risk of immune suppression by bone marrow failure of DC. However, our patient's respiratory function has continued to decrease following the initiation of pirfenidone. Although there are few reports on the treatment of interstitial pneumonia in DC patients, it is reported that interstitial pneumonia with a telomerase complex mutation may be less responsive to treatment with pirfenidone, and therefore, the role of antifibrotic therapy in these patients is still unclear (18). On the other hand, immunosuppressive therapy has also reportedly increased the harmful effects and worsened the prognosis in the patients with a short telomere length (19). In addition, a limited number of small cohort studies have reported that patients with a mutation in the telomerase gene had more complications and worse outcomes after lung transplantation (20,21). For that reason, it is important to carefully consider the indications for lung transplantation in our case due to his bone marrow failure and liver cirrhosis. In a recent prospective report, danazol was shown to increase the telomere length and appeared to stabilize the fibrosis score on HRCT and the DLco on PFT in seven patients with telomerase mutations (22). Although danazol has not been used in our case, the further accumulation of cases is required to establish an optimal treatment strategy for IP in DC patients.
In conclusion, we presented a case of interstitial pneumonia in a patient with dyskeratosis congenita. Although most case reports have described UIP as a common pattern of IP in DC patients, their pathological findings would be diverse, including f-NSIP, DIP, bronchiolitis. Additionally, IP in a patient with DC may show rapid disease progression and be less responsive to therapy. The accumulation of further cases is necessary to confirm our results.
The authors state that they have no Conflict of Interest (COI).
Acknowledgement
We thank the Research Center for Immunological Analysis, Inc. for performing the measurement of telomere length.
References
- 1. Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood 107: 2680-2685, 20061. [DOI] [PubMed] [Google Scholar]
- 2. Walne AJ, Marrone A, Dokal I. Dyskeratosis congenita: a disorder of defective telomere maintenance? Int J Hematol 82: 184-189, 2005. [DOI] [PubMed] [Google Scholar]
- 3. Utz JP, Ryu JH, Myers JL, Michels VV. Usual interstitial pneumonia complicating dyskeratosis congenita. Mayo Clin Proc 80: 817-821, 2005. [DOI] [PubMed] [Google Scholar]
- 4. Imokawa S, Sato A, Toyoshima M, et al. Dyskeratosis congenita showing usual interstitial pneumonia. Intern Med 33: 226-230, 1994. [DOI] [PubMed] [Google Scholar]
- 5. Safa WF, Lestringant GG, Frossard PM. X-linked dyskeratosis congenita: restrictive pulmonary disease and a novel mutation. Thorax 56: 891-894, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 198: e44-e68, 2018. [DOI] [PubMed] [Google Scholar]
- 7. Ichida F, Tsuji T, Omata M, et al. New Inuyama classification; new criteria for histological assessment of chronic hepatitis. International Hepatology Communications 6: 112-119, 1996. [Google Scholar]
- 8. Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol 110: 768-779, 2000. [DOI] [PubMed] [Google Scholar]
- 9. Kirwan M, Dokal I. Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet 73: 103-112, 2008. [DOI] [PubMed] [Google Scholar]
- 10. Fernández García MS, Teruya-Feldstein J. The diagnosis and treatment of dyskeratosis congenita: a review. J Blood Med 21: 157-167, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nelson ND, Bertuch AA. Dyskeratosis congenita as a disorder of telomere maintenance. Mutat Res 730: 43-51, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Knight S, Vulliamy T, Copplestone A, Gluckman E, Mason P, Dokal I. Dyskeratosis congenita (DC) registry: identification of new features of DC. Br J Haematol 103: 990-996, 1998. [DOI] [PubMed] [Google Scholar]
- 13. Yabe M, Yabe H, Hattori K, et al. Fatal interstitial pulmonary disease in a patient with dyskeratosis congenita after allogeneic bone marrow transplantation. Bone Marrow Transplant 19: 389-392, 1997. [DOI] [PubMed] [Google Scholar]
- 14. Paul SR, Perez-Atayde A, Williams DA. Interstitial pulmonary disease associated with dyskeratosis congenita. Am J Pediatr Hematol Oncol 14: 89-92, 1992. [PubMed] [Google Scholar]
- 15. Borie R, Tabèze L, Thabut G, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J 48: 1721-1731, 2016. [DOI] [PubMed] [Google Scholar]
- 16. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370: 2071-2082, 2014. [DOI] [PubMed] [Google Scholar]
- 17. Newton CA, Batra K, Torrealba J, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J 48: 1710-1720, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Justet A, Thabut G, Manali E, et al. Safety and efficacy of pirfenidone in patients carrying telomerase complex mutation. Eur Respir J. Forthcoming. [DOI] [PubMed] [Google Scholar]
- 19. Newton CA, Zhang D, Oldham JM, et al. Telomere length and use of immunosuppressive medications in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 200: 336-347, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tokman S, Singer JP, Devine MS, et al. Clinical outcomes of lung transplant recipients with telomerase mutations. J Heart Lung Transplant 34: 1318-1324, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Silhan LL, Shah PD, Chambers DC, et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J 44: 178-187, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Townsley DM, Dumitriu B, Young NS. Danazol treatment for telomere diseases. N Engl J Med 375: 1095-1096, 2016. [DOI] [PubMed] [Google Scholar]