

Abstract
"Life is an instantaneous encounter of circulating matter and flowing energy" (Jean Giaja, Serbian physiologist), is one of the most elegant definitions not only of life but the relationship of redox biology and metabolism. Their evolutionary liaison has created inseparable yet dynamic homeostasis in health, which, when disrupted, leads to disease. This interconnection is even more pertinent today, in an era of increasing metabolic diseases of epidemic proportions such as obesity, metabolic syndrome, and diabetes. Despite great advances in understanding the molecular mechanisms of redox and metabolic regulation, we face significant challenges in preventing, diagnosing, and treating metabolic diseases. The etiological association and temporal overlap of these syndromes present significant challenges for the discrimination of appropriate clinical biomarkers for diagnosis, treatment, and outcome prediction. These multifactorial, multiorgan metabolic syndromes with complex etiopathogenic mechanisms are accompanied by disturbed redox equilibrium in target tissues and circulation. Free radicals and reactive species are considered both a causal factor and a consequence of disease status. Thus, determining the subtypes and levels of free radicals and reactive species, oxidatively damaged biomolecules (lipids, proteins, and nucleic acids) and antioxidant defense components as well as redox-sensitive transcription factors and fluxes of redox-dependent metabolic pathways will help define existing and establish novel redox biomarkers for stratifying metabolic diseases. This review aims to discuss diverse redox/metabolic aspects in obesity, metabolic syndrome, and diabetes, with the imperative to help establish a platform for emerging and future redox-metabolic biomarkers research in precision medicine. Future research warrants detailed investigations into the status of redox biomarkers in healthy subjects and patients, including the use of emerging 'omic' profiling technologies (e.g., redox proteomes, lipidomes, metabolomes, and transcriptomes), taking into account the influence of lifestyle (diet, physical activity, sleep, work patterns) as well as circadian ~24h fluctuations in circulatory factors and metabolites.
Keywords: Redox biomarkers, Obesity, Metabolic syndrome, Diabetes, Lipofuscin, NRF2, Circadian rhythms, Cancer
Graphical abstract
Highlights
-
•
Obesity, metabolic syndrome, and diabetes are redox diseases.
-
•
The redox-metabolic interface is a fertile ground for redox biomarkers research.
-
•
Validation of redox biomarkers could be instrumental in metabolic disease diagnostics, prognostics, and therapy.
-
•
Circadian rhythms affect the redox-metabolic integrity of metabolic diseases.
-
•
Redox biomarkers link cancer with obesity, metabolic syndrome, and diabetes.
Abbreviations
- 4-HNE
4-hydroxynonenal
- 8-epi-PGF2α
8-epi-prostaglandin F2α
- 8-oxo-dG
8-oxo-2'-deoxyguanosine
- 8-oxo-Guo
8-oxo-7,8-dihydroguanosine
- AGE
advanced glycation end products
- AREs
antioxidant response elements
- BMI
body mass index
- CA
carnosic acid
- CAT
catalase
- CEBPα
CCAAT enhancer binding protein α
- CS
carnosol
- CuZnSOD
copper-zinc superoxide dismutase
- DAG
diacylglycerol
- ETC
electron transport chain
- ERK
extracellular signal-regulated kinase
- F2-IsoPs
F2-Isoprostanes
- FABP4
fatty acid-binding protein 4
- FFA
free fatty acids
- FPG
fasting plasma glucose
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GSH
glutathione
- GSH-Px
glutathione peroxidase
- GSSG
oxidized GSH
- GST
glutathione S-transferase
- H2O2
hydrogen peroxide
- H2S
hydrogen sulfide
- HbA1c
glycosylated hemoglobin
- HDL
high-density lipoprotein
- HFD
high-fat diet
- HO•
hydroxyl radical
- HOMA-IR
Homeostatic Model Assessment for Insulin Resistance
- IFG
impaired fasting glucose
- IFN-γ
interferon-γ
- IGT
impaired glucose tolerance
- iNOS
inducible nitric oxide synthase
- IR
insulin resistance
- IRS
insulin receptor substrate
- JNK
NH2-terminal Jun kinases
- KEAP1
Kelch-like ECH-associated protein 1
- KD
knockdown
- KO
knockout
- LDL
low-density lipoprotein
- MDA
malondialdehyde
- MnSOD
manganese superoxide dismutase
- mtDNA
mitochondrial DNA
- NF-κB
nuclear factor-kappa B
- nNOS
neuronal nitric oxide synthase
- NO
nitric oxide
- NOD
non-obese diabetic
- NOS
nitric oxide synthase
- NRF2
nuclear factor (erythroid-derived 2)-like 2
- NQO1 NAD(P)H
quinone oxidoreductase 1
- O2•−
superoxide anion radical
- OGTT
oral glucose tolerance test
- p38 MAPK
p38 mitogen-activated protein kinases
- PCO
protein carbonyl
- PKC
protein kinase C
- PPARγ
peroxisome proliferator activated receptor γ
- PPP
pentose phosphate pathway
- Prdx
peroxiredoxin
- RAGE
receptor for advanced glycation end products
- RCS
reactive carbonyl species
- RNS
reactive nitrogen species
- RO(N)S
reactive oxygen and nitrogen species
- ROS
reactive oxygen species
- SAPK
stress-activated protein kinases
- SAT
subcutaneous adipose tissue
- SCN
suprachiasmatic nucleus
- SNF
sulforaphane
- SOD
superoxide dismutase
- T1DM
type 1 diabetes mellitus
- T2DM
type 2 diabetes mellitus
- TAG
triacylglycerol
- TBARS
thiobarbituric acid reactive substances
- TR
thioredoxin reductase
- Trx
thioredoxin
- VAT
visceral adipose tissue
- WAT
white adipose tissue
- WC
waist circumference
- WHR
waist-to-hip ratio
1. Introduction
Modern human medicine faces numerous challenges, despite the explosive progress of diagnostic and therapeutic approaches. Our ability to gain an in-depth understanding of the molecular basis of various pathological conditions has opened diverse research avenues and many questions that require urgent answers. If we were to allow ourselves some scientific skepticism, we would ascertain the fact that even after almost a hundred years since the discovery of insulin and despite much scientific progress, we still live in a pandemic of diabetes, especially type 2. The forecasts for the coming decades are not promising either. In parallel with type 2 diabetes mellitus (T2DM), another two conditions strongly associated with it – obesity and metabolic syndrome, paint the same picture of a pandemic with predictions of exponential growth in the future.
Each of these three conditions is complex in itself. The challenges we face are further compounded because these conditions can occur concomitantly while their underlying mechanisms often overlap. The complexity is multifactorial, whether we consider the etiology, clinical manifestations, or complex therapeutic approaches. For these reasons, we do not regard diabetes and obesity as classic diseases but rather as syndromes, including metabolic syndrome. When we take a comprehensive look at this triad, the scope of the problem reaches the level of multi-metabolic disorder/syndrome. Systemic metabolic dysregulation is initiated by the disturbances in several physiological systems, primarily those with endocrine functions. This includes various organs, predominantly white adipose tissue (WAT, specific morpho-functional depots), brown adipose tissue, pancreas, liver, and muscles.
The modern concept of redox biology and medicine also faces a number of challenges. The traditional concept where reactive species are viewed primarily in the light of oxidative damage they inflict on biomolecules is "distant" history. Indeed, reactive species are essential signaling molecules involved in almost every physiological process, from cell division and differentiation to metabolic regulation. Through direct and indirect action, they modulate the activity and function of biomolecules (proteins, lipids, nucleic acids) and redox-sensitive transcription factors, inducing an endogenous adaptive response in cells, including the antioxidant defense. The redox homeostasis of cells and their extracellular environments is determined by the degree of reactive species production and neutralization that are firmly coupled with (oxidative) metabolism. At the regulatory level, the relationship between reactive species production and metabolism is bidirectional and dynamic. Setting the redox state of cells is vital in health as much as in disease and opens redox-based therapeutic avenues for treating metabolic diseases such as obesity, metabolic syndrome, and diabetes.
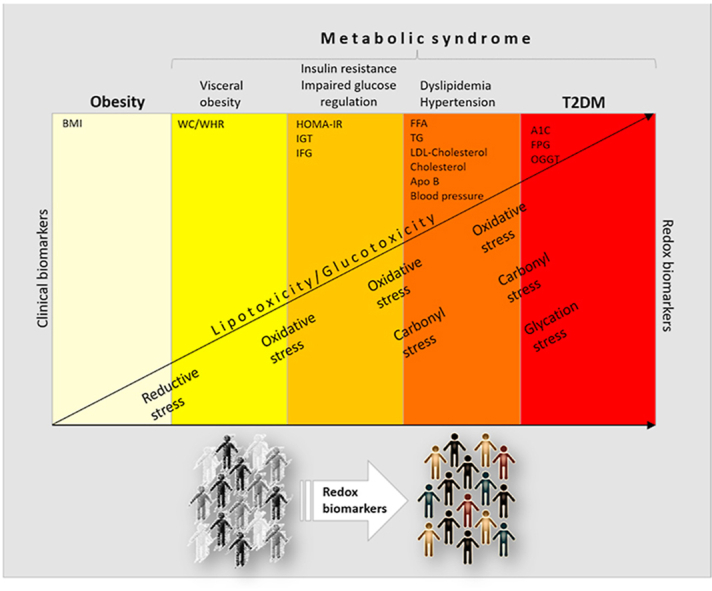
2. "Clinical" versus redox biomarkers
Ever since Claude Bernard postulated the "constancy of the internal environment" [1], we have been searching for (bio)markers of homeostasis whose change indicates disease. Many of them in obesity, metabolic syndrome, and diabetes (i.e., levels of insulin, glucose, free fatty acids, triacylglycerol, cholesterol, glycosylated hemoglobin-HbA1c) are clinical biomarkers that are routinely used in diagnostics in human medicine. Their changes are a reflection of complex metabolic disturbances, predominantly in the most affected tissues. Because their circulatory levels are dynamic in healthy individuals, it is not always straightforward to relate their circulatory changes in patients to pathological conditions.
The search for redox biomarkers that are clinically relevant is an imperative task of redox biology in regards to human medicine. However, when we discuss the utility of redox biomarkers, some considerations deserve attention. First of all, what is considered under the term redox biomarkers? Precisely what and where do we measure? What can we feasibly measure in humans? Redox biomarkers can include a diverse range of measurements from reactive species (that are often difficult to determine due to their nature and short life) and antioxidant defense components (enzymatic and non-enzymatic) to stable end-products of the oxidative modifications of proteins, lipids, and nucleic acids as well as concentrations of iron (free and bound) and those of other transition metals. They are primarily determined in the circulation (plasma/serum, erythrocytes, monocytes, platelets), urine, or saliva. Changes in their status are a reflection of corresponding changes in their concentration, expression, activity, or localization in affected tissues. Outstanding reviews of methodological aspects and theoretical considerations for potential redox-dependent biomarkers are provided by Halliwell [2], Poulsen et al. [3], and Wang et al. [4].
When it comes to metabolic diseases, we face many difficulties in defining and validating redox biomarkers. To precisely define existing and discover novel redox biomarkers, it is necessary to achieve a better understanding of the molecular mechanisms of redox-dependent metabolic processes in target tissues and organs as well as those involved in inter-organ communication.
3. Clinical criteria to define a complex landscape of obesity, metabolic syndrome, and diabetes
Body mass index (BMI) is a useful population-level measure of overweight state and obesity in medical practice. However, metabolic alterations at the core of insulin resistance (IR) and T2DM pathophysiology may develop over the broad continuum of BMI. Nevertheless, it is now well accepted that obesity becomes a metabolic disease once the adaptive lipid-storing function of adipose tissue fails [5]. To emphasize the importance of lipid-storage malfunction, as much as the overall excess of adipose tissue in metabolic disorders, overweight state and obesity are now defined as "excessive or abnormal fat accumulation that presents a health risk" by the World Health Organisation [6]. Heterogeneity of the obese state was attributed to the differences in adipose tissue accumulation back in 1947 when it was first noticed that apple-shaped (mostly men) subjects are more likely to develop diabetes and other health complications than their pear-shaped (mostly women) counterparts [7]. Today still, the anthropometric measurements of BMI, waist circumference (WC), and waist-to-hip ratio (WHR) are used by most healthcare institutions to define abdominal obesity with high metabolic risk [8]. Apart from the genetic background, the upsurge in abdominal obesity during the last several decades is a direct result of an excess in available energy that impairs the metabolic function of adipose tissue. Exceeded functional capacity of highly insulin-sensitive adipose tissue (subcutaneous) to store lipids causes visceral and ectopic lipid deposition in muscle, liver, and other organs, further aggravating insulin resistance and perpetuating prediabetic conditions (Fig. 1).
Fig. 1.

Adipose tissue - centric pathogenesis of type 2 diabetes. In health, postprandially, excess fat is stored in adipose tissue, limiting its deposition in other tissues. In obesity (1), due to energy excess, the number of hypertrophic adipocytes increases when counteractive metabolic and hyperplastic responses are surpassed, and the expanding and lipid-buffering capacity of adipose tissue fails (2). Unrestrained postprandial lipolysis and low oxidative capacity drive systemic free fatty acid (FFA) (3) and pro-inflammatory adipocytokine delivery to non-adipose tissues (4). Consequently, muscles, liver, and pancreas are faced with ectopic lipid deposition and lipotoxicity (5), which ultimately interferes with multiple regulatory mechanisms of glucose storage in muscles and liver. In parallel, the pancreas releases more insulin to sustain normoglycaemia (6). If such conditions persist, compensatory pancreatic hypersecretion of insulin fails, and signs of hyperglycemia occur. Initially only transiently, in postprandial state (defined as impaired glucose tolerance), and later persistently, regardless of the nutritional state, resulting in overt type 2 diabetes mellitus (T2DM) (7). Subjects with an excess of upper-body or visceral adiposity (right) are more prone to develop metabolic syndrome and type 2 diabetes for a given body mass index (BMI), as these "metabolically obese" individuals suffer from a low expanding capacity of lower-body or subcutaneous adiposity. In clinical practice, this state is crudely defined by a higher waist-to-hip ratio (WHR).
According to the American Diabetes Association, a prediabetic state is clinically defined by impaired fasting glucose (IFG) level and impaired glucose tolerance (IGT) test. IFG level of 100–125 mg/dL and IGT of 140–199 mg/dL plasma glucose, respectively, indicate prediabetes. However, the prediabetic state is characterized by a combination of different and overlapping clinical conditions that often congregate, thereby increasing the risk of diabetes onset. The gathering of interrelated metabolic risk factors such as atherogenic dyslipidemia (elevated serum triacylglycerol and apolipoprotein B), increased small low-density lipoprotein (LDL) particles, reduced level of high-density lipoprotein (HDL) and cholesterol, and elevated blood pressure and plasma glucose, is known as metabolic syndrome. This pleiad of metabolic risk factors often develops due to another set of underlying risk factors – obesity and insulin resistance. Although the risk of T2DM generally increases with increasing insulin resistance and obesity, it has been widely accepted that T2DM development depends on adipose tissue function rather than obesity per se. Accordingly, some people that appear normal-weight are insulin resistant and are thereby described as "metabolically obese" [9]. Similarly, obese people that are insulin sensitive are described as "metabolically healthy obese" [10]. These transient metabolic phenotypes - with or without IR, and with or without obesity (defined by BMI) complicate metabolic syndrome diagnostics and T2DM risk predictions (Fig. 2).
Fig. 2.

Types of obesity in respect to metabolic phenotype and respective risk of type 2 diabetes. According to body composition (defined by BMI and WHR) and metabolic profile (defined by HOMA-IR index and lipid profile), at least 4 types of metabolic states/phenotypes can be defined (I-IV). People who appear normal-weight can be insulin resistant and therefore designated as "metabolically obese" normal-weight (III). In addition, obese people who are insulin sensitive are designated as "metabolically healthy" obese (II). Transient metabolic phenotypes - with or without insulin resistance and with or without obesity should be considered in predictions of T2DM development. T2DM: type 2 diabetes mellitus; BMI: body mass index; HOMA-IR: Homeostatic Model Assessment of Insulin Resistance; WHR: waist-to-hip ratio.
Diabetes mellitus is a chronic metabolic disease characterized by hyperglycemia, defined either by HbA1c ≥ 6.5%, or fasting plasma glucose (FPG) FPG ≥126 mg/dL or the 2h plasma glucose after a 75 g oral glucose tolerance test (OGTT) ≥200 mg/dL. If left untreated, hyperglycemia leads to severe health complications increasing all-cause mortality. Diabetes can be broadly classified into two main categories: type 1 diabetes mellitus (T1DM), which occurs due to autoimmune or idiopathic β-cell destruction, usually leading to absolute insulin deficiency (accounts for ~24% cases), and T2DM, which develops due to a progressive insulin secretory defect in the background of insulin resistance, from a "stealthy" prediabetic state (accounts for ~76% cases) [11].
Obesity-associated T2DM, as well as the other obesity-associated diseases, can be prevented. The earlier the disease is recognized, the greater the successes of prevention and treatment. However, early symptoms are often missed. The global prevalence of diabetes is rising, currently affecting ~451 million adults worldwide, while according to the Center for Disease Control and Prevention, more than 84% of prediabetic patients are not even aware of their condition [12]. Since T2DM is strongly related to obesity, the risk of diabetes is easily overlooked in subjects who appear to be non-obese (according to BMI). Approximately 23% of the non-obese population is, however, metabolically obese [13]. These non-obese subjects are metabolically obese since they have visceral obesity, lower energy expenditure, dyslipidemia, insulin resistance, and higher diabetic risk than some obese counterparts, but may escape detection because they have normal BMI [14]. Therefore, new diagnostic approaches should be implemented in diabetes diagnostics, especially in complex prediabetic conditions.
4. Redox basis of glucotoxicity and lipotoxicity
Physical inactivity, aging, and obesity present major risk factors for T2DM development. Continuous high-calorie food intake creates chronic hyperglycemia and hyperlipidemia that gradually deteriorates insulin sensitivity and secretion, leading to overt T2DM. During T2DM pathogenesis, especially in advanced stages of T2DM, hyperglycemia-related oxidative stress is one of the leading causes of metabolic and endocrine disturbances as well as cellular damage in affected tissues. Glucose overload increases all pathways of glucose breakdown, leading to the accumulation of NADH and FADH2. This puts pressure on the electron transport chain (ETC) complexes and increases their reduction state. Subsequently, the electrochemical potential across the inner mitochondrial membrane causes electron leakage at complexes I, II, and III, leading to superoxide anion radical (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (HO•) production (Fig. 3). The elevated levels of reduced NADH as well as reactive oxygen and nitrogen species (ROS and RNS, respectively) inactivate glyceraldehyde 3-phosphate dehydrogenase (GAPDH), resulting in blockage of the glycolytic pathway and glyceraldehyde 3-phosphate accumulation. Glyceraldehyde 3-phosphate is a glycolytic intermediate and the main precursor for methylglyoxal synthesis. The accumulation of antecedent glycolytic metabolites, in turn, activates alternative pathways of glucose metabolism (polyol-, hexosamine- and advanced glycation pathways), additionally increasing reactive oxygen and nitrogen species (RO(N)S) production. The polyol pathway causes NADPH depletion associated with low glutathione reductase (GR) activity [15], while diminished levels of reduced glutathione (GSH) compromise cellular antioxidant capacity resulting in elevated levels of RO(N)S and oxidative damage. Therefore, reductive stress followed by oxidative stress is a leading hypothesis on the mechanism of hyperglycemia-induced metabolic syndrome [16]. Oxidative, carbonyl, and glycation stress induced by advanced glycation end products (AGE) and reactive dicarbonyls foster pathogenesis and complications of T2DM. AGEs bind to specific cell surface receptors of AGE (RAGE), causing alterations in respective signaling pathways and promoting intracellular RO(N)S production. Methylglyoxal is a strong glycating agent that reacts with arginine residues in proteins. Higher plasma concentrations of methylglyoxal underlie carbonyl stress-induced cardiovascular complications in patients with advanced diabetes [17] and "metabolically healthy obese" subjects [18]. High levels of methylglyoxal result from increased production and lower levels/activity of glyoxalases, GSH-dependent enzymes that metabolize methylglyoxal, and other reactive oxoaldehyde metabolites. This is usually accompanied by a decrease in aldehyde reductase, the enzyme responsible for the detoxification of carbonyl compounds [19]. In general, glucotoxicity implies activation of alternative glucose oxidation pathways that induce oxidative, carbonyl, and nitrosative stresses by enhancing free radical production or impairing antioxidant defense. Detailed mechanisms of impaired redox homeostasis in T2DM due to hyperglycemia are reviewed in recent papers [16,[19], [20], [21]].
Fig. 3.

From redox physiology to redox medicine in obesity, metabolic syndrome, and type 2 diabetes. In physiological states, the production of reactive species, metabolic processes, antioxidant defense responses, and repair/removal of oxidatively damaged biomolecules are highly synchronized processes. In metabolic diseases, this homeostasis is disturbed. Nutrient overload disrupts redox/metabolic equilibrium. Gluco- and lipo-toxicity are an integrative part of the overproduction of reactive species, forming a vicious circle in which oxidative stress is central. Induction of secondary pathways of glucose and lipid metabolism strongly enhances these processes, leading to oxidative stress and oxidative modification of all biomolecules, further inducing alterations in redox-metabolic regulation. T2DM: type 2 diabetes mellitus; ETC: electron transport chain; GSH: glutathione; GSH-Px: glutathione peroxidase; GR: glutathione reductase; NOS: nitric oxide synthase; NOX: NADPH oxidase; PPP: pentose phosphate pathway; Prdx: peroxiredoxin; SOD: superoxide dismutase; TR: thioredoxin reductase; Trx: thioredoxin.
As a consequence of adipose tissue lipid storage dysfunction in obesity-associated metabolic syndrome, increased circulatory FFA reposit in visceral fat, liver, muscle, and pancreatic β-cells, forming ectopic lipid deposits and causing lipotoxicity [22]. More than 50 years ago, it was postulated that plasma FFA delivery and fatty acid oxidation in muscles increase the mitochondrial acetyl CoA/CoA and NADH/NAD+ ratio, which in turn inactivates rate-controlling enzymes in glycolysis, ultimately resulting in the accumulation of glucose 6-phosphate [23]. It has been hypothesized that glucose 6-phosphate inhibits hexokinase II activity, increasing intracellular glucose concentrations and finally leading to decreased glucose uptake and glycogen synthesis. The subsequent studies gave complementary evidence that impaired glucose transport activity precedes fatty acid oxidation and represents the primary defect in insulin insensitivity and impaired glycogen synthesis in muscles [[24], [25], [26], [27]]. It appears that fatty acids taken from the circulation are first esterified and deposited as triacylglycerols and, after that, retrieved and oxidized [28]. When this ectopic pool of intracellular fat in muscles and other organs like the liver or pancreas exceeds critical levels, it causes lipotoxicity. In lipotoxicity, fatty acids and other lipid metabolites, such as diacylglycerol (DAG), ceramide, long-chain acyl-coenzyme A, lipid aldehydes, increase oxidative stress via numerous intracellular mechanisms. DAGs act as direct activators of protein kinase C (PKC) and other stress-sensitive serine/threonine kinases that impair tyrosine phosphorylation of insulin receptor substrate (IRS) and insulin signaling [[29], [30], [31]]. PKC and other stress-activated protein kinases (SAPK) increase the overproduction of nitric oxide (NO) via inducible nitric oxide synthase (iNOS), resulting in nitrosative/nitrative stress [32]. Notably, one of the main features of lipotoxicity implies the accumulation of reactive lipid aldehydes, including the 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA) and subsequent protein carbonylation [33]. In addition to the described mechanisms, there are numerous cellular sources of oxidative stress, including the endoplasmic reticulum stress, several stress-activated kinases, AGEs, iNOS, NADPH oxidases, xanthine oxidase, mitochondrial ETC, peroxisomes, lipid peroxidation, carbonylation related to lipotoxicity, and calcium signaling. Intensive research indicates a strong bidirectional link between calcium levels and RO(N)S production, pointing out that this interplay is disrupted in various pathophysiological conditions. Likewise, several oxidative stress sources converge and foster each other, creating a vicious cycle that results in insulin insensitivity and impaired insulin secretion (in the case of β cells). A comprehensive discussion of these potential mechanisms has been provided elsewhere [[34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44], [45]].
In addition to chronic elevations in circulating glucose and free fatty acid (FFA) levels, T2DM patients suffer from late diabetic complications across peripheral tissues [46,47]. Recent evidence suggests that common stress-activated signaling pathways such as nuclear factor-kappa B (NF-κB), p38 mitogen-activated protein kinases (p38 MAPK), and NH2-terminal Jun kinases (JNK)/SAPK underlie the development of most diabetic complications. Moreover, a chronic increase of FFA in obesity instigates similar redox-dependent stress pathways impairing insulin sensitivity and secretion [31].
5. Redox diseases need redox biomarkers
Advances in redox biology unequivocally indicate that all metabolic diseases, obesity, and related metabolic syndrome, as well as diabetes, can be considered redox diseases. Impaired redox regulation is coupled with metabolic dysregulation and vice versa. Both are at the core of the disease pathogenesis, initiators of dysfunction, and mediators of progression that contribute to further complications. It is necessary to develop functional redox biomarkers that would reflect adipose tissue function to screen a "crime scene" where many morbidities actually begin. Current research focuses on identifying redox biomarkers, specific for etiology and pathogenesis of distinct metabolic diseases, standardization of their measurement, and selection as clinically acceptable biomarkers. Underlying oxidative stress in metabolic diseases is often evident as concomitant changes in both levels of RO(N)S and lipid, protein, and nucleic acid oxidation products as well as changes in levels of antioxidant defense components [[48], [49], [50], [51], [52]]. Therefore, all redox parameters that determine the level of prooxidants - RO(N)S and their biological effects can potentially be relevant redox biomarkers of metabolic diseases. Measurement of these parameters in biofluids such as serum, plasma, urine, and saliva can help predict, diagnose, treat, and monitor the progression of metabolic diseases as well as differentiate and provide a functional link between obesity, metabolic syndrome, and diabetes [53].
Altered activity, expression, and specific gene variants of antioxidant enzymes, and oxidative modifications of mitochondrial DNA (mtDNA) have been investigated as potential biomarkers of metabolic diseases. Reduced activity of antioxidant enzymes, namely superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GSH-Px), and a significant decrease in the GSH/GSSG ratio, were found in peripheral blood mononuclear cells in obese individuals [54]. Additionally, these redox parameters are associated with metabolic functions of adipose tissue and the overall risk of metabolic syndrome. Higher basal GSH level and GSH-dependent enzyme activities were found in visceral adipose tissue (VAT) than in subcutaneous adipose tissue (SAT), while these intercepted differences in thiol-redox parameters reflect a higher triacylglycerol turnover in VAT depots [55]. In obese individuals, lower GSH levels in VAT were coupled with lower manganese superoxide dismutase (MnSOD) and higher NADPH oxidase protein expression. Interestingly, low CAT protein expression in VAT was specific to "metabolically obese" individuals (based on the BMI, Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) index, triacylglycerol, total-, LDL- and HDL-cholesterol levels), in both lean and obese subjects with high metabolic risk. In contrast, increased MnSOD activity associated with a higher degree of mtDNA oxidative damage was found in peripheral blood mononuclear cells of T2DM patients with late diabetic complications [56]. Increased glutathione S-transferase (GST) activity was found in the plasma of individuals with T2DM [57], while increased plasma GSH-Px activity was associated with improved renal function in T2DM patients with hypertension as a result of vitamin E and/or selenium supplementation [58]. Copper-zinc superoxide dismutase (CuZnSOD) polymorphisms were found to be associated with increased plasma copper concentrations in patients with newly diagnosed impaired glucose regulation and T2DM [59]. Furthermore, studies of the MnSOD, extracellular SOD, and CAT polymorphisms showed specific molecular association with diabetic neuropathy in T1DM patients [60].
Oxidation products of lipids, fatty acids, and nucleic acids are useful in characterizing and monitoring obesity as well as detecting prediabetic state and predicting associated health complications [61]. Higher levels of MDA in monocytes and F2-Isoprostanes (F2-IsoPs) and 8-oxo-2'-deoxyguanosine (8-oxo-dG) in urine were found in obese patients, where 8-oxo-dG showed potential for metabolic status monitoring since it correlated directly with glucose, insulin, HOMA index, and body weight and inversely with weight loss and antioxidant activities in morbidly obese patients undergoing bariatric surgery [54]. Moreover, serum levels of 8-oxo-dG and MDA were found to be increased in the serum of prediabetic subjects with high sensitivity for C-reactive protein and reduced GSH levels, implicating these redox markers as candidates for the diagnosis of prediabetes, as they correlated with glucose intolerance [62]. Similar disturbances in redox homeostasis are found in T2DM. High levels of oxidative DNA damage in lymphocytes of diabetic patients were found to be directly associated with serum lipid peroxidation and protein oxidation levels and inversely with SOD activity and protein thiol group content [63]. In another study, increased excretion of urinary markers of DNA (8-oxo-dG) and RNA (8-oxo-Guo) oxidation was found in children and adults, which particularly correlated with age and female sex. The authors reported a very interesting finding that obesity in children with insulin resistance correlates with higher levels of oxidation RNA markers, which could be predictive of T2DM [64].
Other redox biomarkers showed promise in monitoring the development of insulin resistance and diabetes progression. Long-term treatment of human primary preadipocytes from obese SAT with physiological concentrations of 4-HNE caused increased oxidative stress, inhibition of cell growth, loss of adipogenic capacity, and impairment of insulin sensitivity [65]. A higher level of 4-HNE was found in VAT of overweight/obese premenopausal women, in comparison to normal-weight women, irrespectively of metabolic syndrome risk factors [55]. Such changes were associated with lower levels of GSH and MnSOD and high NADPH oxidase protein expression levels. In contrast, a significant increase in 4-HNE was found in the SAT of morbidly obese premenopausal women, but again irrespective of metabolic syndrome risk factors [65]. Therefore, 4-HNE in VAT is related to VAT dysfunction and oxidative stress even at low BMI, leading to restricted lipid buffering function of adipose tissue and increased lipid peroxidation as a function of BMI increase. In T2DM patients, higher serum levels of 4-HNE were found to directly correlate with clinical prognostic parameters (glycated hemoglobin, fasting glucose) [66]. However, the role of 4-HNE in metabolic diseases has not yet been fully elucidated and is probably concentration-dependent. At low physiological concentrations, it plays a crucial role in redox homeostasis, acting as a signaling molecule and inducing an adaptive response in diabetes, while at high concentrations, it exhibits cytotoxicity due to its high reactivity and can cause adipocyte dysfunction and insulin resistance [67,68]. This is true not only in diabetes but in other pathophysiological conditions, such as Rett syndrome and aging. For example, high levels of plasma 4-HNE protein adducts were found to increase with disease progression in subjects affected by Rett syndrome [69]. Moreover, our group previously showed that high levels of 4-HNE correlate with degenerative cutaneous tissue changes and oxidative DNA damage with advanced age, which is not the case in young animals [70].
Many of the commonly analyzed lipid peroxidation products are often found in different metabolic diseases. However, additional research will be necessary to assess their utility in differentiating between obesity, metabolic syndrome, and diabetes or their potential to predict the development of diabetes in patients with obesity and metabolic syndrome. Importantly, several emerging studies highlight that specific parameters of lipid peroxidation could be used to achieve such stratifications. The development of metabolic syndrome in obese individuals can be predicted from a linear combination of visceral fat thickness and serum thiobarbituric acid reactive substances (TBARS):cholesterol ratio [50]. Low levels of plasma vitamin C and nitrosoglutathione were found to be inversely correlated with the grade of hypertension in patients with metabolic syndrome, possibly discriminating between patients with low and high risk for diabetes and cardiovascular complications [50]. Interestingly, the results from the Framingham study indicated that urinary 8-epi-prostaglandin F2α (8-epi-PGF2α) levels were highly associated with BMI and cardiovascular complications, but not with diabetes [71]. Moreover, these redox changes seem to be closely related to the type of obesity. Furukawa et al. were the first to demonstrate that obesity exacerbates systemic markers of oxidative stress in a feedback loop leading to metabolic syndrome [72]. Several studies since then found correlations of lipid peroxidation markers with abdominal [73], but not with general obesity [74]. Moreover, it was found that increased fat mass with visceral distribution is specifically associated with systemic chronic low-grade inflammation and oxidative stress markers [50,71,75], highlighting the importance of redox and inflammatory pathways cross-talk, a phenomenon recently termed "OxInflamation" [76]. For example, urinary 8-epi-PGF2 levels were particularly associated with the visceral fat area, even in non-obese subjects. Similarly, a significant correlation of urine 8-epi-PGF2α and inflammation associated with lipid peroxidation-mediated platelet activation was seen in obese women with the android type of obesity, indicating a higher risk of developing hypercholesterolemia, diabetes mellitus, and vascular complications [77].
In a hyperglycemic environment, reactive carbonyl species (RCS) are readily generated, and subsequently, advanced glycation end products and protein carbonyl (PCO) content represent important biomarkers in diabetes and associated syndromes [78]. High levels of PCO, advanced oxidation protein products, and lipid hydroperoxides accompanied by a lower total thiol content in T2DM patients with poor glycaemic control were identified as important prognostic redox biomarkers for the development of diabetic complications [79]. In an exciting study of nutrition-mediated redox control in T2DM, a high-protein diet was found to reduce plasma MDA and protein carbonyls levels but increase nitrotyrosine levels [80]. A direct association of carbonyl stress with increased risk of the acute coronary syndrome was found in T2DM patients, evident as an increase in methylglyoxal, methylglyoxal derived hydroimidazolones-1, and Nε-carboxymethyl-lysine serum levels [81]. Additionally, lower levels of GAPDH, glyoxalase 1, and aldehyde reductase suggest that metabolic disturbance precedes the development of acute coronary syndrome through RCS, while elevated levels of AGEs and protein carbonyls in vitreal fluid of T1DM and T2DM patients suggests their role in proliferative diabetic retinopathy development [82].
Furthermore, reduced NO bioavailability is recognized as one of the key factors that contribute to disturbed redox homeostasis in metabolic diseases, especially diabetes. In an excellent review of human studies investigating NO metabolism in metabolic diseases, Kobayashi summarizes the effects of endothelial nitric oxide synthase (eNOS) polymorphisms in a wide range of conditions (T2DM, IR, obesity) with modifications at the molecular level [83]. There is a clear association between eNOS polymorphisms with altered NOS activity, increased IR, and the incidence of metabolic syndrome and T2DM. In contrast, the level of asymmetric dimethylarginine, an endogenous NOS inhibitor found in all cells and plasma, is increased, while NO production is decreased in insulin resistance. In addition, specific alternations in NO metabolism were found in T2DM (decreased neuronal nitric oxide synthase-nNOS protein expression in skeletal muscle) and obesity (reduced NO availability and reduced eNOS expression in skeletal muscle). Although rare, existing human studies confirm the role of the l-arginine/NO pathway in diabetes, showing that long-term oral l-arginine supplementation improves endothelial function, oxidative stress, and adipokine release in obese T2DM patients with insulin resistance [84]. Besides, an increased level of NO (measured indirectly) correlates with T1DM (saliva and plasma) [85,86] and T2DM (saliva) [86], while salivary NO level is lower in T2DM subjects with xerostomia [87]. Demonstrated reduced NO bioavailability in diabetes [88] could be influenced by another important player in diabetes pathogenesis that has biomarker potential, calcium. Indeed, the overall calcium metabolism was found to be disturbed in T1DM and T2DM, from decreased intestinal absorption to increased urinary excretion [89]. However, there is currently no consensus on the functional significance of plasma calcium alterations in diabetic patients. Interestingly, all three NOS isoforms are regulated by calcium, of which eNOS and nNOS directly via calcium-calmodulin [90], where an increase in calcium concentration is linked directly to NO production by eNOS [91]. However, this link is still controversial since other calcium-independent mechanisms are involved in regulating NOS activity: phosphorylation, availability of l-arginine, other reactive species, and oxidatively damaged biomolecules [90], many of which are disturbed in diabetes. Therefore, the synergistic effects of calcium-dependent and independent mechanisms likely impact altered NO homeostasis in diabetes. In addition to the NO and NO metabolites, hydrogen sulfide (H2S) should be further investigated as a potential redox biomarker in diabetes since H2S concentrations, similarly to NO, regulate a wide range of functions and are reduced in the plasma of patients with T2DM [92].
6. Lipofuscin as a novel redox biomarker of insulin-related metabolic diseases
Lipofuscin is a non-degradable cellular material composed of oxidized proteins, lipids, and metals stored in lysosomes, whose accumulation has been widely regarded as a hallmark of cellular aging [93]. The question has been posed whether lipofuscin can be repurposed as a biomarker of metabolic diseases that could predict the progression of obesity, metabolic syndrome, and diabetes. Nowadays, the contribution of pathological accumulation of lipofuscin is highlighted for many age-related diseases and complex conditions, which can also be regarded as metabolic diseases, including Alzheimer's and Parkinson's disease [94], denervation atrophy [95], inherited juvenile form of macular degeneration [96], lipid myopathy [97], chronic obstructive pulmonary disease [98], and Melanosis coli [99].
In the search for novel redox biomarkers reflecting insulin-related metabolic diseases, lipofuscin could be a promising and reliable candidate. Namely, lipofuscinogenesis, the formation of lipofuscin, occurs mainly in metabolically sensitive organs such as the brain, heart, liver, and adipose tissue (Fig. 4). The presence, subcellular distribution, and lipofuscin content vary greatly in tissue- and cell-specific manner. Besides, the amount of lipofuscin in specimens from individuals of the same age varies and could be linked to disease or diet variation, which are all associated with the rate of oxidative protein damage, mitochondrial functionality, proteasomal system, and lysosomal functionality [100].
Fig. 4.

Lipofuscinogenesis in obesity and type 2 diabetes. Lipofuscinogenesis in hepatocytes (A - arrow) and cardiomyocytes (D- LF) of obese mice and brown adipocytes of hyperinsulinemic rats (B -rectangle, E − LF) results in gradual lipofuscin accumulation (C). In hyperinsulinemia, a state preceding obesity and diabetes, excessive production of H2O2 and higher import of iron drive peroxidative damage of mitochondria (mth) and lipid bodies (LB) and lead to the formation of lipofuscin (LF) containing their undigested remnants and iron. White star – damaged, structurally altered lipid body (B) and mitochondria (D). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
The concept that the accumulation of lipofuscin in the cell is not only age-, but also metabolism-dependent is not new [100,101], given close relations between metabolic disorders and aging as well as the role of oxidative stress in metabolic syndrome [102,103]. Hyperinsulinemia is implicated in metabolic syndrome as a condition that precedes obesity and diabetes and is associated with pancreatic islet hyperplasia, and is thus recognized as an early indicator of metabolic dysfunction [104]. The association of hyperinsulinemia and insulin resistance to mitochondrial dysfunction [105] and iron levels through rapid and pronounced stimulation of iron uptake and loading by adipocytes [[106], [107], [108]] is well established. Furthermore, transferrin receptors colocalize with glucose transporters and insulin-like growth factor II receptors in the microsomal membranes of cultured adipocytes, suggesting that regulation of iron uptake by insulin occurs in parallel with its effects on glucose transport [109]. Indeed, obese people have increased iron levels [110,111] and become prone to IR and potentially harmful lipofuscinogenesis.
Hyperinsulinemia represents a risk factor for cardiovascular disease, neurodegeneration, diabetes, and cancer [112], while this type of dysfunction is a commonly observed feature in several animal models for obesity and diabetes [113,114]. Diabetes results in lipofuscin accumulation in various tissues and organs [115]. Since hyperinsulinemia is also part of those metabolic diseases [116,117], the molecular events linking insulin and lipofuscin accumulation warrant further investigation. Hyperinsulinemia drives an overproduction of hydrogen peroxide and increased iron uptake, creating a harmful milieu, which leads to oxidative damage. As a result, damaged organelles and macromolecules accumulate inside cells in the form of lipofuscin [100]. Since lipofuscin accumulates within lysosomes as an end product, it is assumed that it could not be removed [118,119] and could thus represent a hallmark redox biomarker of cellular aging [[120], [121], [122], [123]].
Interestingly, some data shows that lipofuscin could be found in a soluble state in bodily fluids and quantified. Hegedus et al. [124] documented an increase of plasma lipofuscin levels in stored blood and quantified human plasma lipofuscin [125]. Recently, lipofuscin was measured in human plasma and saliva [126]. This data raises the question of the existence of extracellular lipofuscin and its origins. Extraneuronal lipofuscin was found in the brain samples, suggesting a possible physiological removal from the nerve cells to the capillary endothelium and its later elimination via phagocytosis by glia cells [127]. Indeed, recent findings have shown that lipofuscin could be exocytosed from the cardiomyocytes into circulation through capillary endothelium [128]. Hence, lipofuscin could be regarded as a promising redox biomarker of obesity and diabetes. The revival of non-invasive lipofuscin measurements in bodily fluids with accurate and reliable methods shows promise for monitoring insulin-related metabolic diseases [129].
7. The NRF2 redox pathway at the crossroads between the antioxidant defense and energy metabolism
The NRF2 pathway, a master regulator of cellular defense against oxidative and xenobiotic stresses, has emerged as a critical target of energy metabolism [130]. Nuclear factor (erythroid-derived 2)-like 2 (NRF2), a cap-n-collar basic leucine zipper transcription factor, serves to mount a cellular defense response against environmental and endogenous stressors. Under conditions of lower stress, NRF2 is sequestered in the cytoplasm by associating with Kelch-like ECH-associated protein 1 (KEAP1) and Cullin 3 ubiquitin E3 ligase. Upon exposure to electrophilic and oxidative stresses, NRF2 becomes released from its cytosolic repressor KEAP1 and translocates to the nucleus, where it forms a complex with MAF and JUN proteins. This leads to NRF2 binding to antioxidant response elements (AREs) in numerous gene promoters and subsequent activation of diverse transcriptional programs, including activation of genes encoding phase 2 detoxification and antioxidant enzymes [131]. In addition, NRF2 regulates a large number of genes involved in gluconeogenesis and lipid metabolism as well as β-oxidation, mitochondrial biogenesis, and inflammation, processes which show robust diurnal variation (Fig. 5) [130,[132], [133], [134]]. Whilst there is strong evidence for the role of NRF2/KEAP1 pathway in modulating adipogenesis, responses to high-fat diet (HFD), insulin sensitivity, and metabolic health, the direction of this control is conflicting in the literature. Previous research has shown that NRF2 inhibits adipogenic differentiation via activation of the aryl hydrocarbon receptor (AHR) pathway. Adipogenic stimulation of Nrf2 knockout (KO) mouse embryonic fibroblasts (MEFs) led to increased intracellular lipid accumulation and elevated expression of peroxisome proliferator-activated receptor gamma and CCAAT/enhancer binding protein α (Pparγ and Cebpα, respectively), whilst delayed adipogenesis was seen in Keap1 knockdown (KD) MEFs [135]. In line with these data, Xu et al. demonstrated that in MEFs isolated from mouse models of NRF2 activation, Keap1-KD, and leptin (ob/ob)-Keap1-KD, adipogenesis was suppressed, and adipogenic genes, such as Pparγ, Cebpα, and fatty acid-binding protein 4 (Fabp4) were decreased [136]. The same study demonstrated that pharmacological NRF2 activation by sulforaphane (SFN) prevented adipogenesis and lipid accumulation [136]. Additional NRF2 inducers such as carnosic acid and carnosol originating from dried rosemary leaves were also shown to inhibit adipogenic differentiation by stimulating glutathione metabolism [137].
Fig. 5.

Schematic diagram depicting the interconnected roles of NRF2 and circadian clock machinery in diverse transcriptional programs implicated in obesity, metabolic syndrome, and diabetes. Several different transcriptional programs are regulated by both NRF2 and the circadian molecular clock through positive and negative feedback control in various cell types and tissues. NRF2 and the molecular clock are also involved in the bidirectional regulation of each other. The target transcriptional programs display ~24h oscillations in various components and range from antioxidant/detoxification and inflammatory responses to pathways involved in mitochondrial biogenesis/β-oxidation, adipogenesis/lipogenesis, as well as insulin and glucose-regulated pathways. NRF2: nuclear factor (erythroid-derived 2)-like 2.
In contrast, other research has shown that Nrf2 KO mice have lower amounts of adipose tissue and that mouse and human preadipocytes show suppressed adipocyte differentiation resulting from the downregulation of Pparγ and Cebpα expression following Nrf2 shRNAs whilst enhanced adipogenesis was seen with Keap1 shRNAs [138]. In line with this work, another study demonstrated NRF2 as a positive regulator of adipogenesis using preadipocytes derived from white adipose tissue [139].
Further to its role in adipogenesis and adipose tissue maintenance, NRF2 also plays an important role in modulating obesity. CDDO-Im, a potent NRF2 activator, was shown to prevent body weight gain and WAT increase in response to HFD in wild type mice [140]. Similar findings were found using another NRF2 inducer, oltipraz, which prevented insulin resistance and weight gain in response to HFD [141]. Consistently, a number of other NRF2 inducers such as quercetin, curcumin, resveratrol, and chromium histidinate have all shown anti-obesity effects at least partially through NRF2 induction (reviewed in Ref. [142]).
Surprisingly, some in vivo animal studies show a complex role of NRF2 in influencing responses to HFD. For example, Nrf2 KO mice were found to show blunted fat pad increases following HFD compared to wild-type mice, and this was independent of differences in food consumption, intestinal fat absorption, and physical activity [138]. Similarly, another study found that Nrf2 deficiency protects against HFD-induced obesity through fibroblast growth factor 21 and improves glucose tolerance and insulin sensitivity [143]. On the other hand, Keap1-KD mice, which have increased NRF2 levels, also had lower body weights and fat masses than wild-type mice on HFD [136]. In line with this work, mice over-expressing NRF2 target, NAD(P)H quinone dehydrogenase 1 (Nqo1), conferred protection from diet-induced metabolic defects through the preservation of glucose homeostasis, insulin sensitivity, and lipid handling with improved physiological outcomes [144]. NQO1-overexpressing mice on HFD had lower adipose tissue macrophages and enhanced expression of lipogenic enzymes together with the reduction in circulating and hepatic lipids.
In contrast, some reports have shown that NRF2/KEAP1 pathway has no effects on body weight in response to HFD feeding; however, Nrf2 KO mice were protected from HFD-induced hyperglycemia [145]. Whereas Nrf2 KO mice were resistant to HFD-induced glucose intolerance, Keap1 KD mice exhibited prolonged elevation of circulating glucose during a glucose tolerance test, even on a control diet. Indeed, some reports agree that whole-body Nrf2 KO mice are more glucose-tolerant and insulin-sensitive than wild-type mice [143], whilst Keap1 KD mice on a background of leptin deficiency (a genetic model of obesity) showed higher glucose concentration than control mice [136]. On the other hand, mice with myeloid-selective Nrf2 deficiency were less sensitive to insulin, suggesting that NRF2 in myeloid cells acts negatively against the development of insulin resistance [146]. Interestingly, Nrf2 deletion impairs glucose tolerance and exacerbates hyperglycemia in type 1 diabetic mice [147]. Indeed, NRF2 activation plays an important role in protection against autoreactive T-cell-induced T1DM using non-obese diabetic (NOD) mice, a polygenic model of human type 1 diabetes [148]. Keap1 KD mice inhibited T-cell infiltration, ameliorated impairment of insulin secretion, and prevented the development of diabetes mellitus in NOD mice. This occurred through decreasing both plasma interferon-γ (IFN-γ) levels and IFN-γ-positive cell numbers in the pancreatic islets.
To resolve some of the conflicting data on the role of NRF2/KEAP1 pathway in obesity and metabolic health, studies have investigated the effect of Nrf2 deletion in a tissue-specific manner. Nrf2 deletion from adipocytes, but not hepatocytes, potentiated systemic metabolic dysfunction after long-term HFD-induced obesity in mice [149]. Mice with Nrf2 loss in adipocytes showed a partially deteriorated glucose tolerance, higher fasting glucose levels, and higher levels of cholesterol and non-esterified fatty acids. In contrast, mice with Nrf2 loss in hepatocytes had lower insulin levels and improved insulin sensitivity without changes to liver triglyceride accumulation. Furthermore, another study investigated the role of NRF2 in influencing the risk of metabolic syndrome by using a whole-body or adipocyte-specific Nrf2 KO mice on a leptin-deficient ob/ob background, a model of extreme positive energy balance [150]. On ob/ob background, loss of Nrf2, globally or in adipocytes, showed reduced WAT mass but led to a more severe metabolic syndrome with aggravated insulin resistance, hyperglycemia, and hypertriglyceridemia. These new studies have revealed that NRF2/KEAP1 pathway has differential effects on weight control and metabolic health in a cell-type-specific manner.
Pharmacological NRF2 activation has shown more consistent outcomes regarding their protective effects on metabolic health. An NRF2 inducer SFN reversed disease signature in diabetic livers and attenuated exaggerated glucose production and glucose intolerance. Importantly, SFN reduced fasting blood glucose and HbA1c in obese patients with dysregulated T2DM [132]. Another NRF2 inducer, oltipraz, improved glucose metabolism and prevented insulin signaling impairments in mice on HFD [141]. Similarly, curcumin improved insulin sensitivity through the regulation of muscular mitochondrial redox balance; ellagic acid protected against HFD-induced metabolic syndrome and NRF2 agonist Dh404 prevented insulin resistance (reviewed in Ref. [142]). Although pharmacological NRF2 activation has shown promising results in several animal and human studies, the synthetic NRF2 inducers have caused adverse cardiac events and gastrointestinal toxicities in clinical trials [151]. Alternative supplementation with glucoraphanin, a stable precursor of SFN, attenuated weight gain, increased energy expenditure, and core body temperature led to improved glucose tolerance and insulin sensitivity in HFD-fed wild-type mice. This occurred via activating insulin-stimulated Akt phosphorylation in the liver, muscle, and WAT and preventing uncoupling protein 1 protein reduction in WAT. Glucoraphanin was also able to reduce circulatory levels of lipopolysaccharide, attenuate inflammatory signaling pathways such as JNK and extracellular signal-regulated kinase (ERK), and inflammation-activated M1-like macrophage accumulation in liver and adipose tissue. Finally, glucoraphanin was able to reduce hepatic elevated lipogenic gene expression, lipid peroxidation levels of MDA, and expression of NADPH oxidase subunits [151].
Overall, whilst some of the discrepancies in the above studies may result from the use of different cell types (e.g., primary vs. immortalized, MEFs vs. adipocytes), mouse genetic backgrounds as well as the length or types of HFD (milk fat vs. lard), it is important to note that most studies do not specify the gender or age of animal models used. Moreover, responses to HFD as well as various outputs of energy metabolism show profound time-of-day circadian differences, which is another important confounding factor to take into consideration. Therefore, future studies are urgently needed to decipher the precise temporal and cell-type-specific role of NRF2 and its target molecular mechanisms in regulating adipose tissue homeostasis and responses to HFD and determine how gender and age modulate this regulation.
8. Energy metabolism and redox regulation as a function of ~24h circadian rhythms
Circadian rhythms govern ~24h rhythms in daily human and mammalian physiology, metabolism, and behavior both in health and disease, including the daily rhythms in feeding activity, hormone production, and glucose metabolism [152]. The circadian timing coordinates temporal variation in biochemical processes with changes in nutrient intake in order to optimize energy balance and maintain metabolic homeostasis [153]. The circadian timing system consists of a central brain clock in the anterior hypothalamic suprachiasmatic nucleus (SCN) and various peripheral tissue clocks, which drive positive/negative feedback loops in transcription/translation and metabolite abundance [154]. The central clock regulates food intake, energy expenditure, whole-body insulin sensitivity, and glucose homeostasis, whilst the local peripheral clocks also contribute to this regulation and fine-tune these processes. Some examples include the peripheral clock in the gut, which regulates glucose absorption, peripheral clocks in muscle, adipose tissue, and liver, which regulate local insulin sensitivity, and the peripheral clock in the pancreas which regulates insulin secretion [[155], [156], [157]].
Misalignment between circadian timing system and daily rhythms of sleep-wake behavior or food intake as a result of genetic, environmental, or behavioral circadian disruptions is associated with metabolic disorders such as obesity, insulin resistance, and diabetes [158]. Indeed, disrupted circadian rhythms of body temperature, heart rate, and fasting blood glucose are seen in both prediabetes and T2DM [159]. Moreover, emerging work has shown that chrono-based nutritional interventions such as time-restricted feeding can rescue obesity and metabolic syndrome in mice lacking a genetic circadian clock [156,157].
Current high-throughput technologies have allowed for the dynamic monitoring of diverse cellular processes over ~24h daily cycle, ranging from gene and protein expression to metabolite abundance, thus discovering potential circadian biomarkers. Indeed, ~40% of the genome is under circadian control across different mammalian tissues [160]. For example, circadian metabolomes have been reported for human serum, saliva, breath, and urine, as well as tissues from several species in different disease conditions and following genetic disruption [161]. Circadian proteomics of plasma proteins from human participants undergoing simulated night shift work showed 24h time-of-day patterns in 573 of 1129 proteins analyzed and revealed that circadian misalignment altered multiple proteins known to regulate glucose homeostasis and/or energy metabolism, with implications for altered metabolic physiology [162]. Social jetlag, a measure of disruption of the circadian system, has been associated with increased fasting glucose levels and a higher risk of overweight and metabolically obese status in a study with ~792 Brazilian participants [163]. Elevation of circulating FFA levels is associated with insulin resistance and diabetes risk [164]. FFA levels in healthy individuals show strong circadian variation, and sleep restriction relative to normal sleep led to its increased levels in nocturnal and early morning hours. This was associated with prolonged secretion of nocturnal growth hormone levels and early morning noradrenaline levels [164]. A recent genome-wide association study of reported day-time sleepiness analyzing 452,071 individuals from the UK Biobank has identified 42 genetic loci that clustered into specific biological subtypes and shared genetic links with several chronic diseases, including diabetes [165].
Previous work has characterized ~24h time-of-day variation and the effects of sleep deprivation on urinary metabolite profiles in healthy male participants under highly controlled environmental conditions by 1H NMR spectroscopy [166]. Significant changes were observed with respect to both time-of-day and sleep deprivation, indicating that sampling time and sleep deprivation are highly relevant when identifying biomarkers in metabolic profiling studies. Further work by the same group using targeted liquid chromatography/mass spectrometry metabolomics identified 130 plasma metabolites every 2h over 24h, and showed that average metabolite concentrations were significantly altered by increased body mass (90 of 130) and T2DM (56 of 130). Moreover, ~40% of plasma metabolites showed daily rhythms, highlighting the importance of controlling the time-of-day for diagnosis and biomarker discovery [167]. In addition, a complementary study identified using untargeted and targeted liquid chromatography (LC)/MS metabolomics that acute sleep deprivation reduced amplitude of rhythms of >50% of rhythmic plasma metabolites identified [168]. The altered metabolite rhythms in overweight and T2DM individuals is consistent with previous work showing that individuals with T2DM have lower night-time serum melatonin levels and an increased risk of comorbid sleep disturbances compared with healthy individuals [169].
Moreover, the effects of sleep restriction and loss were also examined using RNAseq profiling of WAT transcriptome between day and night. This has shown that sleep restriction blunts the normal daily variation in WAT transcriptome and that sleep deprivation led to further dampening [170]. This resulted in uncoupling of the clock-controlled regulated pathways of carbohydrate and lipid metabolism from the adipose tissue clock, leading to increased carbohydrate turnover and impaired glucose homeostasis [170]. In line with this work, human mature adipocytes show altered circadian rhythms in obese patients with age [171]. Similarly, a further study investigated whether the expression of clock genes in peripheral blood cells is impaired in T2DM and whether inflammatory markers are associated with circadian clock gene expression in T2DM patients. This study revealed that a number of core clock genes was decreased in leukocytes of T2DM patients, which was correlated with elevated levels of plasma inflammatory markers such as interleukin 6 and tumor necrosis factor α [172].
Circadian rhythms are highly interconnected to redox homeostasis, and this is demonstrated to be a bidirectional process as many metabolites and products of oxidative metabolism, as well as components of redox pathways, can feedback and influence circadian timing [[173], [174], [175], [176], [177]]. Recently we and others demonstrated that circadian molecular machinery regulates endogenous rhythms in NRF2 protein and transcript levels in several metabolic tissues, including the liver, lung, pancreas, and heart [158,[178], [179], [180]]. Moreover, we have shown that target antioxidant genes oscillate in tissues and cells in an NRF2-dependent manner [179], which is coupled to the daily oscillatory changes in cellular and tissue ROS levels [175]. In addition, we and others have observed redox oscillations in protein carbonylation marker and glutathione redox states in mouse tissues as well as human cells and bodily fluids [179,[181], [182], [183], [184]].
A number of additional potential redox biomarkers have also been shown to be under circadian control. Circadian variation in lipid peroxidation marker MDA is observed in mouse erythrocytes [185] as well as in healthy human volunteers. Circadian control of plasma lipid peroxides (MDA) and serum ascorbic acid and uric acid levels were reported [186]. Night shift workers have lower levels of antioxidant defense components and higher levels of TBARS when compared to day workers [187]. Plasma TBARS concentrations are increased in the fasting state in obese men [188] whilst bariatric surgery reduced both TBARS and protein carbonyl concentrations in obese individuals [189]. Furthermore, the measurement of ~24h urinary excretion of 8-oxo-G oxidative damage marker suggests that obesity in men is also associated with increased oxidative damage to RNA [190]. The removal of oxidative damage to DNA/RNA such as 8-oxoguanine via the DNA base excision repair pathway is also under the circadian influence [191]. Altogether, these findings urge further work to examine the circadian rhythmicity of diverse redox biomarkers and antioxidant defense components in human research and animal models of metabolic diseases as well as test the potential of chrono-nutritional interventions in human clinical trials.
9. Cancer as a metabolic disease: deciphering the intriguing relationship with obesity, metabolic syndrome, and diabetes through redox biomarkers
The cumulative burden of neoplastic and metabolic diseases is hard to grasp, but current predictions do not seem optimistic [192,193]. Cancer is predicted to pose a higher threat to human health than cardiovascular diseases by 2030 [194]. Whilst obesity and diabetes take on epidemic proportions, evidence supporting the association between metabolic disorders and cancer has also accumulated. Prospective observational studies and meta-analyses show that obesity increases incidence [195,196] and mortality [197] for many cancers. Likewise, there is substantial evidence linking diabetes and cancer [198], although the association is more complicated to dissect, primarily due to the confounding influence of obesity on both diabetes and cancer development. In general, the association of site-specific cancers with obesity further increases in diabetes, including colorectal, pancreatic, liver, gall bladder, kidney, breast, and endometrial [199,200]. The relationship becomes even more complicated when different antidiabetic and antineoplastic treatments are considered [201,202].
In light of the abundant epidemiological data and critical research highlighting metabolic reprogramming and redox regulation as two inseparable phenomena in cancer, the view has gradually shifted towards contemplation of cancer as a redox-metabolic disease [[203], [204], [205], [206]]. Thus, obesity, metabolic syndrome, and diabetes could be regarded as potential driving forces for cancer development, acting through systemic and local influences on the tumor microenvironment [207,208]. Within this framework, it is not surprising that cancer-associated adipocytes emerged as critical players in cancer progression, especially for site-specific cancers strongly associated with obesity, such as colorectal [209], breast [210], and prostate [211].
Obesity-associated circulating levels of glucose, free fatty acids, insulin, insulin-like growth factor 1 as well as adipose tissue inflammation and dysregulated cytokine and adipokine signaling are some of the mechanisms that link obesity and cancer and are extensively reviewed elsewhere [212,213]. More recently, research focus has shifted towards metabolic cross-talk as one of the integral aspects of cancer-adipose tissue two-way communication [214,215]. Several exciting studies, including our own, showed not only that metabolic support provided by adipocytes is instrumental for cancer progression and metastasis but that it is redox-sensitive [[216], [217], [218]] and obesity-mediated [[219], [220], [221]] (Fig. 6). Thus, disturbed redox regulation in cancer-adipose tissue cross-talk could provide one of the missing links between metabolic diseases and cancer.
Fig. 6.

Schematic representation of obesity-driven cancer-adipose tissue cross-talk. Cancer cells exist within an intricate tumor milieu comprised of stromal constituents, namely extracellular matrix components and numerous stromal cell types, including fibroblasts, adipocytes, mast cells, dendritic cells, immune cells, and vascular endothelial cells, among others. Obesity can shape the tumor microenvironment through systemic and local influences acting on both cancer cells and adipocytes, thus (re)framing their cross-talk and (re)directing cancer proliferation, metastasis, and therapeutic resistance. Recently, a concept of metabolic cooperation was introduced to describe the nutritional and metabolic support provided by adipocytes through the supply of substrates for carbohydrate and lipid metabolism. Moreover, this cooperation seems to be supported by redox regulatory mechanisms and may include redox synchronization between cancer cells and adipocytes. Redox-metabolic profiling of cancer and adipose tissue could be instrumental in advancing personalized therapeutic approaches, especially for individuals with concomitant metabolic diseases such as obesity, metabolic syndrome, and diabetes. Glu: glucose; FFA: free fatty acids; IGF-1: insulin-like growth factor 1; 4-HNE: 4-hydroxynonenal; ROS: reactive oxygen species; RNS: reactive nitrogen species.
Great efforts were invested in identifying redox biomarkers that could predict cancer risk, stage of progression, and therapeutic outcome. Most commonly analyzed redox biomarkers are oxidation and glycation products of nucleic acids, proteins, and lipids in plasma, serum, urine, and saliva [222]. Results from prospective, case-control, and nested case-control studies provide valuable information about the potential of these biomarkers to serve as clinical parameters but often yield inconclusive or contradictory results. A crucial consideration is their usefulness in individuals with concomitant metabolic and neoplastic diseases since similar parameters are often used to differentiate obesity, metabolic syndrome, diabetes, and several types of cancer.
One of the most studied lipid peroxidation products, MDA, was found to be consistently elevated in plasma/serum of patients with breast [223], prostate [224,225], and colon cancer [226]. Among markers of nucleic acid oxidation products, serum level of 8-oxo-dG was found to be inversely associated with lymph node metastasis in breast cancer patients [227], while urinary levels were inversely associated with longer survival in colon cancer patients [228]. Measurement of advanced glycation products also showed predictive potential in several types of cancer. A prospective nested case-control study showed an association between plasma Nε-carboxymethyl-lysine and the risk of prostate cancer [229], while Nε-carboxymethyl-lysine content in breast cancer tissue was associated with better prognosis with tamoxifen treatments and worse prognosis with chemotherapy [230]. Urinary levels of isoprostanes were generally associated with the cancer risk and stage of progression for breast [231], prostate [232,233], and colon cancer [234], although a recent large follow-up study found no association with these malignancies [235].
One of the challenges in the biomarker research field is that previously found associations are often lost when larger sample sizes are considered. For example, a case-control study found a direct association of urinary levels of 15-F2t-isoprostane, and an inverse association of 8-oxo-dG with breast cancer risk [231]. Two years later, the same group found no difference in urinary 15-F2t-isoprostane and 8-oxo-dG between breast cancer patients and controls in a large population-based case-control study [236]. Another issue concerns the uniformity of study cohorts, especially regarding confounding factors such as obesity. This is nicely illustrated in an example where a large prospective nested case-control study found no differences in urinary levels of 15-F2t-isoprostane and 2,3-dinor-5,6-dihydro-15-F2t-IsoP between breast cancer patients and controls, but further analysis according to BMI revealed that high levels of isoprostanes increase the cancer risk for obese and decrease the cancer risk for normal-weight women [237]. Moreover, the same redox biomarkers may be indicative of some but not other types of cancer; such is the case with plasma protein carbonyls. Several studies found an increase in breast cancer risk in women with higher levels of plasma protein carbonyls [238,239], while a large randomized, placebo-controlled trial found no association with a prostate cancer risk [240].
We are beginning to appreciate the importance of complex metabolic diseases in shaping the tumor microenvironment, while the accompanying research of underlying redox regulatory mechanisms is truly in its infancy. Nevertheless, results from recent comprehensive transcriptomic, proteomic, and metabolomic studies emphasize the enormous potential of individual profiling for cancer diagnostic and prognostic advancements, highlighting that study of redox-metabolic networks is a fertile ground for the search for biomarkers that will guide our approach in personalized therapy designs. More extensive population-based prospective and nested control-case studies employing multivariate analysis are needed to draw definite conclusions.
10. Conclusions and future perspectives
"All right, said the Cat; and this time it vanished quite slowly, beginning with the end of the tail, and ending with the grin, which remained some time after the rest of it had gone".
Lewis Carroll (Alice in Wonderland)
This quote illustrates the duality of present perspectives when we talk about redox biomarkers in complex disease conditions such as those related to obesity, metabolic syndrome, and diabetes. Currently, we are beginning to understand the redox bases of metabolic diseases but are encountering numerous challenges in validating redox biomarkers. These include overlapping of metabolic conditions, differential clinical presentation and progression, the involvement of a wide range of organs and organ systems, limitations in the availability of human biological samples, and the absence of reference values of redox biomarkers in healthy subjects, including diurnal variations. On the other hand, after decades of testing redox biomarkers in plasma/serum, blood cells, urine, saliva, and other bodily fluids and available biological material, some redox biomarkers are approaching the fulfillment of strict criteria [2]. The development of various redox methodologies and less invasive approaches in recent years is encouraging. In addition to measuring markers of oxidative damage to biomolecules and alterations in antioxidant defense components, direct electrochemical measurement of reactive species (NO, H2O2, and even H2S) has become a real possibility today. The selection of representative sets of biomarkers for specific metabolic diseases and their standardization is one of the first steps warranting further research. Rigorous evaluation of redox proteomes, lipidomes, metabolomes, and transcriptomes will be of great help in deciphering the complex redox-metabolic landscapes. This should be preceded by the validation of redox biomarkers in healthy subjects, taking into account the influence of lifestyle (diet, physical activity, sleep, work patterns) and diurnal fluctuations in metabolites influenced by circadian rhythms. The development of new methodologies for determining redox biomarkers in tissue biopsies, such as adipose tissue, could eliminate some of the challenges related to the standardization of redox biomarkers in obesity, metabolic syndrome, and diabetes. Emerging advances in redox biology, both research-wise and methodologically, allow us to be optimistic about the future of redox disease biomarkers in clinical medicine.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgments
The authors are grateful for the support from the Ministry of Education, Science and Technological Development of the Republic of Serbia [grant numbers 451-03-68/2020-14/200007 and 451-03-68/2020-14/200178] and the Science Fund of the Republic of Serbia, PROMIS [grant number 6066747] as well as from the MRC New Investigator Research Grant, the Medical Research Council (MRC)-Versus Arthritis Centre for Integrated Research on Musculoskeletal Ageing, The Rosetrees Trust and Wellcome Trust Institutional Strategic Fund Fellowship (VPV).
Contributor Information
Bato Korac, Email: koracb@ibiss.bg.ac.rs.
Aleksandra Jankovic, Email: aleksandra.jankovic@ibiss.bg.ac.rs.
References
- 1.Claude B. JB Baillière; Paris: 1865. Introduction á l’étude de la medicine. [Google Scholar]
- 2.Halliwell B. The wanderings of a free radical. Free Radic. Biol. Med. 2009;46:531–542. doi: 10.1016/j.freeradbiomed.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Poulsen H.E., Weimann A., Henriksen T., Kjær L.K., Larsen E.L., Carlsson E.R., Christensen C.K., Brandslund I., Fenger M. Oxidatively generated modifications to nucleic acids in vivo: measurement in urine and plasma. Free Radic. Biol. Med. 2019;145:336–341. doi: 10.1016/j.freeradbiomed.2019.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Wang J., Schipper H.M., Velly A.M., Mohit S., Gornitsky M. Salivary biomarkers of oxidative stress: a critical review. Free Radic. Biol. Med. 2015;85:95–104. doi: 10.1016/j.freeradbiomed.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Wilkin T.J., Voss L.D. Metabolic syndrome: maladaptation to a modern world. [Review] [83 refs] J. R. Soc. Med. 2004;97:511–520. doi: 10.1258/jrsm.97.11.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waxman A. WHO global strategy on diet, physical activity and health. Food Nutr. Bull. 2004;25:292–302. doi: 10.1177/156482650402500310. [DOI] [PubMed] [Google Scholar]
- 7.Vague J. La différenciation sexuelle; facteur déterminant des. Presse Med. 1947;55:339. [PubMed] [Google Scholar]
- 8.Nishida C., Ko G.T., Kumanyika S. Body fat distribution and noncommunicable diseases in populations: overview of the 2008 WHO expert consultation on waist circumference and waist-hip ratio. Eur. J. Clin. Nutr. 2010;64:2–5. doi: 10.1038/ejcn.2009.139. [DOI] [PubMed] [Google Scholar]
- 9.Ruderman N., Chisholm D., Pi-Sunyer X., Schneider S. The metabolically obese, normal-weight individual revisited. Diabetes. 1998;47:699–713. doi: 10.2337/diabetes.47.5.699. [DOI] [PubMed] [Google Scholar]
- 10.Karelis A.D., Messier V., Brochu M., Rabasa-Lhoret R. Metabolically healthy but obese women: effect of an energy-restricted diet. Diabetologia. 2008;51:1752–1754. doi: 10.1007/s00125-008-1038-4. [DOI] [PubMed] [Google Scholar]
- 11.American Diabetes Association 2. Classification and diagnosis of diabetes. Diabetes Care. 2015;38:S8–S16. doi: 10.2337/dc15-S005. [DOI] [PubMed] [Google Scholar]
- 12.Lin X., Xu Y., Pan X., Xu J., Ding Y., Sun X., Song X., Ren Y., Shan P.F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: an analysis from 1990 to 2025. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-71908-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruderman N.B., Schneider S.H., Berchtold P. The “metabolically-obese,” normal-weight individual. Am. J. Clin. Nutr. 1981;34:1617–1621. doi: 10.1093/ajcn/34.8.1617. [DOI] [PubMed] [Google Scholar]
- 14.Conus F., Rabasa-Lhoret R., Péronnet F. Characteristics of metabolically obese normal-weight (MONW) subjects. Appl. Physiol. Nutr. Metabol. 2007;32:4–12. doi: 10.1139/H06-092. [DOI] [PubMed] [Google Scholar]
- 15.Cumbie B.C., Hermayer K.L. Current concepts in targeted therapies for the pathophysiology of diabetic microvascular complications. Vasc. Health Risk Manag. 2007;3:823–832. [PMC free article] [PubMed] [Google Scholar]
- 16.Yan L. Redox imbalance stress in diabetes mellitus: role of the polyol pathway. Anim. Model Exp. Med. 2018;1:7–13. doi: 10.1002/ame2.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanssen N.M.J., Scheijen J.L.J.M., Jorsal A., Parving H.H., Tarnow L., Rossing P., Stehouwer C.D.A., Schalkwijk C.G. Higher plasma methylglyoxal levels are associated with incident cardiovascular disease in individuals with type 1 diabetes: a 12-year follow-up study. Diabetes. 2017;66:2278–2283. doi: 10.2337/db16-1578. [DOI] [PubMed] [Google Scholar]
- 18.Masania J., Malczewska-Malec M., Razny U., Goralska J., Zdzienicka A., Kiec-Wilk B., Gruca A., Stancel-Mozwillo J., Dembinska-Kiec A., Rabbani N., Thornalley P.J. Dicarbonyl stress in clinical obesity. Glycoconj. J. 2016;33:581–589. doi: 10.1007/s10719-016-9692-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rabbani N., Thornalley P.J. Glyoxalase 1 modulation in obesity and diabetes. Antioxidants Redox Signal. 2019;30:354–374. doi: 10.1089/ars.2017.7424. [DOI] [PubMed] [Google Scholar]
- 20.Manna P., Jain S.K. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: causes and therapeutic strategies. Metab. Syndr. Relat. Disord. 2015;13:423–444. doi: 10.1089/met.2015.0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyer F., Vidot J.B., Dubourg A.G., Rondeau P., Essop M.F., Bourdon E. Oxidative stress and adipocyte biology: focus on the role of AGEs. Oxid. Med. Cell. Longev. 2015;2015 doi: 10.1155/2015/534873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravussin E., Smith S.R. Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann. N. Y. Acad. Sci. 2002;967:363–378. doi: 10.1111/j.1749-6632.2002.tb04292.x. [DOI] [PubMed] [Google Scholar]
- 23.Randle P.J., Garland P.B., Hales C.N., Newsholme E.A. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;281:785–789. doi: 10.1016/S0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 24.Boden G., Chen X., Ruiz J., White J.V., Rossetti L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J. Clin. Invest. 1994;93:2438–2446. doi: 10.1172/JCI117252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roden M., Price T.B., Perseghin G., Petersen K.F., Rothman D.L., Cline G.W., Shulman G.I. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dresner A., Laurent D., Marcucci M., Griffin M.E., Dufour S., Cline G.W., Slezak L.A., Andersen D.K., Hundal R.S., Rothman D.L., Petersen K.F., Shulman G.I. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Invest. 1999;103:253–259. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciaraldi T.P., Abrams L., Nikoulina S., Mudaliar S., Henry R.R. Glucose transport in cultured human skeletal muscle cells: regulation by insulin and glucose in nondiabetic and non-insulin-dependent diabetes mellitus subjects. J. Clin. Invest. 1995;96:2820–2827. doi: 10.1172/JCI118352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Unger R.H., Orci L. Lipotoxic diseases of nonadipose tissues in obesity. Int. J. Obes. 2000;24:S28–S32. doi: 10.1038/sj.ijo.0801498. [DOI] [PubMed] [Google Scholar]
- 29.Jornayvaz F.R., Shulman G.I. Diacylglycerol activation of protein kinase Cε and hepatic insulin resistance. Cell Metabol. 2012;15:574–584. doi: 10.1016/j.cmet.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soumura M., Kume S., Isshiki K., Takeda N., Araki S.I., Tanaka Y., Sugimoto T., Chin-Kanasaki M., Nishio Y., Haneda M., Koya D., Kashiwagi A., Maegawa H., Uzu T. Oleate and eicosapentaenoic acid attenuate palmitate-induced inflammation and apoptosis in renal proximal tubular cell. Biochem. Biophys. Res. Commun. 2010;402:265–271. doi: 10.1016/j.bbrc.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 31.Evans J.L., Goldfine I.D., Maddux B.A., Grodsky G.M. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 32.Shimabukuro M., Zhou Y.T., Levi M., Unger R.H. Fatty acid-induced β cell apoptosis: a link between obesity and diabetes. Proc. Natl. Acad. Sci. U. S. A. 1998;95:2498–2502. doi: 10.1073/pnas.95.5.2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hauck A.K., Bernlohr D.A. Thematic review series: lipotoxicity: Many roads to cell dysfunction and cell death: oxidative stress and lipotoxicity. J. Lipid Res. 2016;57:1976–1986. doi: 10.1194/jlr.R066597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solinas G., Karin M. JNK1 and IKKβ: molecular links between obesity and metabolic dysfunction. Faseb. J. 2010;24:2596–2611. doi: 10.1096/fj.09-151340. [DOI] [PubMed] [Google Scholar]
- 35.Henriksen E.J., Diamond-Stanic M.K., Marchionne E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011;51:993–999. doi: 10.1016/j.freeradbiomed.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Görlach A., Bertram K., Hudecova S., Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol. 2015;6:260–271. doi: 10.1016/j.redox.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madreiter-Sokolowski C.T., Thomas C., Ristow M. Interrelation between ROS and Ca2+ in aging and age-related diseases. Redox Biol. 2020;36 doi: 10.1016/j.redox.2020.101678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han J., Kaufman R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016;57:1329–1338. doi: 10.1194/jlr.R067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaneki M., Shimizu N., Yamada D., Chang K. Nitrosative stress and pathogenesis of insulin resistance. Antioxidants Redox Signal. 2007;9:319–329. doi: 10.1089/ars.2006.1464. [DOI] [PubMed] [Google Scholar]
- 40.Di Meo S., Iossa S., Venditti P. Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J. Endocrinol. 2017;233 doi: 10.1530/JOE-16-0598. R15–42. [DOI] [PubMed] [Google Scholar]
- 41.Jankovic A., Korac A., Buzadzic B., Stancic A., Otasevic V., Ferdinandy P., Daiber A., Korac B. Targeting the NO/superoxide ratio in adipose tissue: relevance to obesity and diabetes management. Br. J. Pharmacol. 2017;174:1570–1590. doi: 10.1111/bph.13498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu J., Lu W., Shi B., Klein S., Su X. Peroxisomal regulation of redox homeostasis and adipocyte metabolism. Redox Biol. 2019;24 doi: 10.1016/j.redox.2019.101167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ly L.D., Xu S., Choi S.-K., Ha C.-M., Thoudam T., Cha S.-K., Wiederkehr A., Wollheim C.B., Lee I.-K., Park K.-S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017;49 doi: 10.1038/emm.2016.157. e291–e291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shao B., Bayraktutan U. Hyperglycaemia promotes human brain microvascular endothelial cell apoptosis via induction of protein kinase C-βI and prooxidant enzyme NADPH oxidase. Redox Biol. 2014;2:694–701. doi: 10.1016/j.redox.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Desco M.C., Asensi M., Márquez R., Martínez-Valls J., Vento M., Pallardó F.V., Sastre J., Viña J. Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes. 2002;51:1118–1124. doi: 10.2337/diabetes.51.4.1118. [DOI] [PubMed] [Google Scholar]
- 46.Fraze E., Donner C.C., Swislocki A.L.M., Chiou Y.A.M., Chen Y.D.I., Reaven G.M. Ambient plasma free fatty acid concentrations in noninsulin-dependent diabetes mellitus: evidence for insulin resistance. J. Clin. Endocrinol. Metab. 1985;61:807–811. doi: 10.1210/jcem-61-5-807. [DOI] [PubMed] [Google Scholar]
- 47.Miles J.M., Wooldridge D., Grellner W.J., Windsor S., Isley W.L., Klein S., Harris W.S. Nocturnal and postprandial free fatty acid kinetics in normal and type 2 diabetic subjects: effects of insulin sensitization therapy. Diabetes. 2003;52:675–681. doi: 10.2337/diabetes.52.3.675. [DOI] [PubMed] [Google Scholar]
- 48.Yesilbursa D., Serdar Z., Serdar A., Sarac M., Coskun S., Jale C. Lipid peroxides in obese patients and effects of weight loss with orlistat on lipid peroxides levels. Int. J. Obes. 2005;29:142–145. doi: 10.1038/sj.ijo.0802794. [DOI] [PubMed] [Google Scholar]
- 49.Weber D., Milkovic L., Bennett S.J., Griffiths H.R., Zarkovic N., Grune T. Measurement of HNE-protein adducts in human plasma and serum by ELISA-Comparison of two primary antibodies. Redox Biol. 2013;1:226–233. doi: 10.1016/j.redox.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palmieri V.O., Grattagliano I., Portincasa P., Palasciano G. Systemic oxidative alterations are associated with visceral adiposity and liver steatosis in patients with metabolic syndrome. J. Nutr. 2006;136:3022–3026. doi: 10.1093/jn/136.12.3022. [DOI] [PubMed] [Google Scholar]
- 51.Karbownik-Lewinska M., Szosland J., Kokoszko-Bilska A., Stepniak J., Zasada K., Gesing A., Lewinski A. Direct contribution of obesity to oxidative damage to macromolecules. Neuroendocrinol. Lett. 2012;33:453–461. [PubMed] [Google Scholar]
- 52.Adnan M.T., Amin M.N., Uddin M.G., Hussain M.S., Sarwar M.S., Hossain M.K., Uddin S.M.N., Islam M.S. Increased concentration of serum MDA, decreased antioxidants and altered trace elements and macro-minerals are linked to obesity among Bangladeshi population. Diabetes Metab. Syndr. Clin. Res. Rev. 2019;13:933–938. doi: 10.1016/j.dsx.2018.12.022. [DOI] [PubMed] [Google Scholar]
- 53.Cazzola R., Rondanelli M., Russo-Volpe S., Ferrari E., Cestaro B. Decreased membrane fluidity and altered susceptibility to peroxidation and lipid composition in overweight and obese female erythrocytes. J. Lipid Res. 2004;45:1846–1851. doi: 10.1194/jlr.M300509-JLR200. [DOI] [PubMed] [Google Scholar]
- 54.Monzo-Beltran L., Vazquez-Tarragón A., Cerdà C., Garcia-Perez P., Iradi A., Sánchez C., Climent B., Tormos C., Vázquez-Prado A., Girbés J., Estáñ N., Blesa S., Cortés R., Chaves F.J., Sáez G.T. One-year follow-up of clinical, metabolic and oxidative stress profile of morbid obese patients after laparoscopic sleeve gastrectomy. 8-oxo-dG as a clinical marker. Redox Biol. 2017;12:389–402. doi: 10.1016/j.redox.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jankovic A., Korac A., Srdic-Galic B., Buzadzic B., Otasevic V., Stancic A., Vucetic M., Markelic M., Velickovic K., Golic I., Korac B. Differences in the redox status of human visceral and subcutaneous adipose tissues - relationships to obesity and metabolic risk. Metabolism. 2014;63:661–671. doi: 10.1016/j.metabol.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 56.García-Ramírez M., Francisco G., García-Arumí E., Hernández C., Martínez R., Andreu A.L., Simó R. Mitochondrial DNA oxidation and manganese superoxide dismutase activity in peripheral blood mononuclear cells from type 2 diabetic patients. Diabetes Metab. 2008;34:117–124. doi: 10.1016/j.diabet.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 57.Sharma M., Gupta S., Singh K., Mehndiratta M., Gautam A., Kalra O.P., Shukla R., Gambhir J.K. Association of glutathione-S-transferase with patients of type 2 diabetes mellitus with and without nephropathy. Diabetes Metab. Syndr. Clin. Res. Rev. 2016;10:194–197. doi: 10.1016/j.dsx.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 58.Zitouni K., Steyn M.R.C.P., Lyka E., Kelly F.J., Cook P., Ster I.C., Earle K.A. Derepression of glomerular filtration, renal blood flow and antioxidant defence in patients with type 2 diabetes at high-risk of cardiorenal disease. Free Radic. Biol. Med. 2020;161:283–289. doi: 10.1016/j.freeradbiomed.2020.10.005. [DOI] [PubMed] [Google Scholar]
- 59.Yin J., Wang X., Li S., Zhu Y., Chen S., Li P., Luo C., Huang Y., Li X., Hu X., Yang W., Bao W., Shan Z., Liu L. Interactions between plasma copper concentrations and SOD1 gene polymorphism for impaired glucose regulation and type 2 diabetes. Redox Biol. 2019;24 doi: 10.1016/j.redox.2019.101172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chistiakov D.A., Zotova E.V., Savost’anov K.V., Bursa T.R., Galeev I.V., Strokov I.A., Nosikov V.V. The 262T>C promoter polymorphism of the catalase gene is associated with diabetic neuropathy in type 1 diabetic Russian patients. Diabetes Metab. 2006;32:63–68. doi: 10.1016/S1262-3636(07)70248-3. [DOI] [PubMed] [Google Scholar]
- 61.Agarwal A., Hegde A., Yadav C., Ahmad A., Manjrekar P.A., Srikantiah R.M. Assessment of oxidative stress and inflammation in prediabetes—a hospital based cross-sectional study. Diabetes Metab. Syndr. Clin. Res. Rev. 2016;10:S123–S126. doi: 10.1016/j.dsx.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 62.Mahat R.K., Singh N., Rathore V., Arora M., Yadav T. Cross-sectional correlates of oxidative stress and inflammation with glucose intolerance in prediabetes. Diabetes Metab. Syndr. Clin. Res. Rev. 2019;13:616–621. doi: 10.1016/j.dsx.2018.11.045. [DOI] [PubMed] [Google Scholar]
- 63.Arif M., Islam M.R., Waise T.M.Z., Hassan F., Mondal S.I., Kabir Y. DNA damage and plasma antioxidant indices in Bangladeshi type 2 diabetic patients. Diabetes Metab. 2010;36:51–57. doi: 10.1016/j.diabet.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 64.Jørs A., Lund M.A.V., Jespersen T., Hansen T., Poulsen H.E., Holm J.C. Urinary markers of nucleic acid oxidation increase with age, obesity and insulin resistance in Danish children and adolescents Nucleotide oxidation in childhood obesity. Free Radic. Biol. Med. 2020;155:81–86. doi: 10.1016/j.freeradbiomed.2020.05.009. [DOI] [PubMed] [Google Scholar]
- 65.Elrayess M.A., Almuraikhy S., Kafienah W., Al-Menhali A., Al-Khelaifi F., Bashah M., Zarkovic K., Zarkovic N., Waeg G., Alsayrafi M., Jaganjac M. 4-Hydroxynonenal causes impairment of human subcutaneous adipogenesis and induction of adipocyte insulin resistance. Free Radic. Biol. Med. 2017;104:129–137. doi: 10.1016/j.freeradbiomed.2017.01.015. [DOI] [PubMed] [Google Scholar]
- 66.Lou B., Boger M., Bennewitz K., Sticht C., Kopf S., Morgenstern J., Fleming T., Hell R., Yuan Z., Nawroth P.P., Kroll J. Elevated 4-hydroxynonenal induces hyperglycaemia via Aldh3a1 loss in zebrafish and associates with diabetes progression in humans. Redox Biol. 2020;37 doi: 10.1016/j.redox.2020.101723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dasuri K., Ebenezer P., Fernandez-Kim S.O., Zhang L., Gao Z., Bruce-Keller A.J., Freeman L.R., Keller J.N. Role of physiological levels of 4-hydroxynonenal on adipocyte biology: implications for obesity and metabolic syndrome. Free Radic. Res. 2013;47:8–19. doi: 10.3109/10715762.2012.733003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maulucci G., Daniel B., Cohen O., Avrahami Y., Sasson S. Hormetic and regulatory effects of lipid peroxidation mediators in pancreatic beta cells. Mol. Aspect. Med. 2016;49:49–77. doi: 10.1016/j.mam.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 69.Valacchi G., Pecorelli A., Cervellati C., Hayek J. 4-hydroxynonenal protein adducts: key mediator in Rett syndrome oxinflammation. Free Radic. Biol. Med. 2017;111:270–280. doi: 10.1016/j.freeradbiomed.2016.12.045. [DOI] [PubMed] [Google Scholar]
- 70.Jankovic A., Saso L., Korac A., Korac B. Relation of redox and structural alterations of rat skin in the function of chronological aging. Oxid. Med. Cell. Longev. 2019;2019 doi: 10.1155/2019/2471312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keaney J.F., Larson M.G., Vasan R.S., Wilson P.W.F., Lipinska I., Corey D., Massaro J.M., Sutherland P., Vita J.A., Benjamin E.J. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham study. Arterioscler. Thromb. Vasc. Biol. 2003;23:434–439. doi: 10.1161/01.ATV.0000058402.34138.11. [DOI] [PubMed] [Google Scholar]
- 72.Furukawa S., Fujita T., Shimabukuro M., Iwaki M., Yamada Y., Nakajima Y., Nakayama O., Makishima M., Matsuda M., Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shin M.J., Hyun Y.J., Kim O.Y., Kim J.Y., Jang Y., Lee J.H. Weight loss effect on inflammation and LDL oxidation in metabolically healthy but obese (MHO) individuals: low inflammation and LDL oxidation in MHO women. Int. J. Obes. 2006;30:1529–1534. doi: 10.1038/sj.ijo.0803304. [DOI] [PubMed] [Google Scholar]
- 74.Cervellati C., Bonaccorsi G., Cremonini E., Romani A., Castaldini C., Ferrazzini S., Giganti M., Fila E., Massari L., Bergamini C.M. Waist circumference and dual-energy X-ray absorptiometry measures of overall and central obesity are similarly associated with systemic oxidative stress in women. Scand. J. Clin. Lab. Invest. 2014;74:102–107. doi: 10.3109/00365513.2013.860618. [DOI] [PubMed] [Google Scholar]
- 75.Festa A., D'Agostino R., Williams K., Karter A.J., Mayer-Davis E.J., Tracy R.P., Haffner S.M. The relation of body fat mass and distribution to markers of chronic inflammation. Int. J. Obes. 2001;25:1407–1415. doi: 10.1038/sj.ijo.0801792. [DOI] [PubMed] [Google Scholar]
- 76.Valacchi G., Virgili F., Cervellati C., Pecorelli A. OxInflammation: from subclinical condition to pathological biomarker. Front. Physiol. 2018;9 doi: 10.3389/fphys.2018.00858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davì G., Guagnano M.T., Ciabattoni G., Basili S., Falco A., Marinopiccoli M., Nutini M., Sensi S., Patrono C. Platelet activation in obese women: role of inflammation and oxidant stress. J. Am. Med. Assoc. 2002;288 doi: 10.1001/jama.288.16.2008. 2008–14. [DOI] [PubMed] [Google Scholar]
- 78.Thornalley P.J. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems - role in ageing and disease. Drug Metabol. Drug Interact. 2008;23:125–150. doi: 10.1515/DMDI.2008.23.1-2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Çakatay U. Protein oxidation parameters in type 2 diabetic patients with good and poor glycaemic control. Diabetes Metab. 2005;31:551–557. doi: 10.1016/S1262-3636(07)70230-6. [DOI] [PubMed] [Google Scholar]
- 80.Pivovarova-Ramich O., Markova M., Weber D., Sucher S., Hornemann S., Rudovich N., Raila J., Sunaga-Franze D., Sauer S., Rohn S., Pfeiffer A.F.H., Grune T. Effects of diets high in animal or plant protein on oxidative stress in individuals with type 2 diabetes: a randomized clinical trial: high protein diet and oxidative stress. Redox Biol. 2020;29 doi: 10.1016/j.redox.2019.101397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bora S., Adole P.S., Motupalli N., Pandit V.R., Vinod K.V. Association between carbonyl stress markers and the risk of acute coronary syndrome in patients with type 2 diabetes mellitus–A pilot study. Diabetes Metab. Syndr. Clin. Res. Rev. 2020;14:1751–1755. doi: 10.1016/j.dsx.2020.08.037. [DOI] [PubMed] [Google Scholar]
- 82.Géhl Z., Bakondi E., Resch M.D., Hegedus C., Kovács K., Lakatos P., Szabó A., Nagy Z., Virág L. Diabetes-induced oxidative stress in the vitreous humor. Redox Biol. 2016;9:100–103. doi: 10.1016/j.redox.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kobayashi J. Nitric oxide and insulin resistance. Immunoendocrinology. 2015;2:e657. doi: 10.14800/Immunoendocrinology.657. [DOI] [Google Scholar]
- 84.Lucotti P., Setola E., Monti L.D., Galluccio E., Costa S., Sandoli E.P., Fermo I., Rabaiotti G., Gatti R., Piatti P.M. Beneficial effects of a long-term oral L-arginine treatment added to a hypocaloric diet and exercise training program in obese, insulin-resistant type 2 diabetic patients. Am. J. Physiol. Endocrinol. Metab. 2006;291:906–912. doi: 10.1152/ajpendo.00002.2006. [DOI] [PubMed] [Google Scholar]
- 85.Abdolsamadi H.R., Rezaei F., Goodarzi M.T., Moghimbeigi A., Jazaeri M., Asadi S., Ahmadi-Motamayel F. Comparison of salivary nitric oxide and epidermal growth factor level between diabetic patients and healthy individuals. Int. J. Diabetes Dev. Ctries. 2015;35:477–482. doi: 10.1007/s13410-014-0207-x. [DOI] [Google Scholar]
- 86.Astaneie F., Afshari M., Mojtahedi A., Mostafalou S., Zamani M.J., Larijani B., Abdollahi M. Total antioxidant capacity and levels of epidermal growth factor and nitric oxide in blood and saliva of insulin-dependent diabetic patients. Arch. Med. Res. 2005;36:376–381. doi: 10.1016/j.arcmed.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 87.Afsaneh Abadi P., Koopaie M., Montazeri R. Comparison of salivary nitric oxide and oral health in diabetic patients with and without xerostomia. Diabetes Metab. Syndr. Clin. Res. Rev. 2020;14:11–15. doi: 10.1016/j.dsx.2019.11.014. [DOI] [PubMed] [Google Scholar]
- 88.Tessari P., Cecchet D., Cosma A., Vettore M., Coracina A., Millioni R., Iori E., Puricelli L., Avogaro A., Vedovato M. Nitric oxide synthesis is reduced in subjects with type 2 diabetes and nephropathy. Diabetes. 2010;59:2152–2159. doi: 10.2337/db09-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wongdee K., Krishnamra N., Charoenphandhu N. Derangement of calcium metabolism in diabetes mellitus: negative outcome from the synergy between impaired bone turnover and intestinal calcium absorption. J. Physiol. Sci. 2017;67:71–81. doi: 10.1007/s12576-016-0487-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Förstermann U., Sessa W.C. Nitric oxide synthases: regulation and function. Eur. Heart J. 2012;33 doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Charoensin S., Eroglu E., Opelt M., Bischof H., Madreiter-Sokolowski C.T., Kirsch A., Depaoli M.R., Frank S., Schrammel A., Mayer B., Waldeck-Weiermair M., Graier W.F., Malli R. Intact mitochondrial Ca2+ uniport is essential for agonist-induced activation of endothelial nitric oxide synthase (eNOS) Free Radic. Biol. Med. 2017;102:248–259. doi: 10.1016/j.freeradbiomed.2016.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jain S.K., Bull R., Rains J.L., Bass P.F., Levine S.N., Reddy S., McVie R., Bocchini J.A. Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxidants Redox Signal. 2010;12:1333–1338. doi: 10.1089/ars.2009.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Terman A., Brunk U.T. Lipofuscin. Int. J. Biochem. Cell. Biol. 2004;36:1400–1404. doi: 10.1016/j.biocel.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 94.Moreno-García A., Kun A., Calero O., Medina M., Calero M. An overview of the role of lipofuscin in age-related neurodegeneration. Front. Neurosci. 2018;12 doi: 10.3389/fnins.2018.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Neel B.A., Zong H., Backer J.M., Pessin J.E. Identification of atypical peri-nuclear multivesicular bodies in oxidative and glycolytic skeletal muscle of aged and Pompe's disease mouse models. Front. Physiol. 2015;6 doi: 10.3389/fphys.2015.00393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Adler L., Boyer N.P., Chen C., Ablonczy Z., Crouch R.K., Koutalos Y. The 11-cis retinal origins of lipofuscin in the retina. Prog. Mol. Biol. Transl. Sci. 2015;134:e1–12. doi: 10.1016/bs.pmbts.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 97.Brunk U.T., Terman A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002;33:611–619. doi: 10.1016/S0891-5849(02)00959-0. [DOI] [PubMed] [Google Scholar]
- 98.Allaire J., Maltais F., Leblanc P., Simard P.M., Whittom F., Doyon J.F., Simard C., Jobin J. Lipofuscin accumulation in the vastus lateralis muscle in patients with chronic obstructive pulmonary disease. Muscle Nerve. 2002;25:383–389. doi: 10.1002/mus.10039. [DOI] [PubMed] [Google Scholar]
- 99.Morgan J.A., McFarlane M., Disney B.R. An unusually pigmented colon. Lancet Gastroenterol. Hepatol. 2018;3:884. doi: 10.1016/S2468-1253(18)30347-9. [DOI] [PubMed] [Google Scholar]
- 100.Jung T., Bader N., Grune T. Lipofuscin: formation, distribution, and metabolic consequences. Ann. N. Y. Acad. Sci. 2007;1119:97–111. doi: 10.1196/annals.1404.008. [DOI] [PubMed] [Google Scholar]
- 101.Sohal R.S. Relationship between metabolic rate, lipofuscin accumulation and lysosomal enzyme activity during aging in the adult housefly, Musca domestica. Exp. Gerontol. 1981;16:347–355. doi: 10.1016/0531-5565(81)90055-3. [DOI] [PubMed] [Google Scholar]
- 102.Salmon A.B. Beyond diabetes: does obesity-induced oxidative stress drive the aging process? Antioxidants. 2016;5 doi: 10.3390/antiox5030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bonomini F., Rodella L.F., Rezzani R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015;6:109–120. doi: 10.14336/AD.2014.0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Thomas D.D., Corkey B.E., Istfan N.W., Apovian C.M. Hyperinsulinemia: an early indicator of metabolic dysfunction. J. Endocr. Soc. 2019;3:1727–1747. doi: 10.1210/js.2019-00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rains J.L., Jain S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011;50:567–575. doi: 10.1016/j.freeradbiomed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Le Guenno G., Chanséaume E., Ruivard M., Morio B., Mazur A. Study of iron metabolism disturbances in an animal model of insulin resistance. Diabetes Res. Clin. Pract. 2007;77:363–370. doi: 10.1016/j.diabres.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 107.Markelic M., Velickovic K., Golic I., Klepal W., Otasevic V., Stancic A., Jankovic A., Vucetic M., Buzadzic B., Korac B., Korac A. The origin of lipofuscin in brown adipocytes of hyperinsulinaemic rats: the role of lipid peroxidation and iron. Histol. Histopathol. 2013;28:493–503. doi: 10.14670/HH-28.493. [DOI] [PubMed] [Google Scholar]
- 108.Korać A., Vereć M., Davidović V. Insulin-induced iron loading in the rat brown adipose tissue: histochemical and electron-microscopic study. Eur. J. Histochem. 2003;47:241–244. doi: 10.4081/833. [DOI] [PubMed] [Google Scholar]
- 109.Tanner L.I., Lienhard G.E. Localization of transferrin receptors and insulin-like growth factor II receptors in vesicles from 3T3-L1 adipocytes that contain intracellular glucose transporters. J. Cell Biol. 1989;108:1537–1545. doi: 10.1083/jcb.108.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dandona P., Flynn D.M., Hoffbrand A.V. Hyperinsulinism in iron overload. Diabetologia. 1984;27:610. doi: 10.1007/BF00276981. [DOI] [PubMed] [Google Scholar]
- 111.Adamska A., Łebkowska A., Krentowska A., Adamski M., Kowalska I. The association between serum ferritin concentration and visceral adiposity estimated by whole-body DXA scan in women with polycystic ovary syndrome. Front. Endocrinol. 2020;10 doi: 10.3389/fendo.2019.00873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Després J.P., Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881–887. doi: 10.1038/nature05488. [DOI] [PubMed] [Google Scholar]
- 113.Lowell B.B., S-Susulic V., Hamann A., Lawitts J.A., Himms-Hagen J., Boyer B.B., Kozak L.P., Flier J.S. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature. 1993;366:740–742. doi: 10.1038/366740a0. [DOI] [PubMed] [Google Scholar]
- 114.Hamann A., Flier J.S., Lowell B.B. Decreased brown fat markedly enhances susceptibility to diet-induced obesity, diabetes, and hyperlipidemia. Endocrinology. 1996;137:21–29. doi: 10.1210/endo.137.1.8536614. [DOI] [PubMed] [Google Scholar]
- 115.Jain S.K., Levine S.N., Duett J., Hollier B. Reduced vitamin E and increased lipofuscin products in erythrocytes of diabetic rats. Diabetes. 1991;40:1241–1244. doi: 10.2337/diab.40.10.1241. [DOI] [PubMed] [Google Scholar]
- 116.Kolb H., Stumvoll M., Kramer W., Kempf K., Martin S. Insulin translates unfavourable lifestyle into obesity. BMC Med. 2018;16 doi: 10.1186/s12916-018-1225-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kopp W. Development of obesity: the driver and the passenger. Diabetes, Metab. Syndrome Obes. Targets Ther. 2020;13:4631–4642. doi: 10.2147/DMSO.S280146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Terman A., Dalen H., Brunk U.T. Ceroid/lipofuscin-loaded human fibroblasts show decreased survival time and diminished autophagocytosis during amino acid starvation. Exp. Gerontol. 1999;34:943–957. doi: 10.1016/S0531-5565(99)00070-4. [DOI] [PubMed] [Google Scholar]
- 119.Terman A., Abrahamsson N., Brunk U.T. Ceroid/Lipofuscin-loaded human fibroblasts show increased susceptibility to oxidative stress. Exp. Gerontol. 1999;34:755–770. doi: 10.1016/S0531-5565(99)00045-5. [DOI] [PubMed] [Google Scholar]
- 120.Evangelou K., Lougiakis N., Rizou S.V., Kotsinas A., Kletsas D., Muñoz-Espín D., Kastrinakis N.G., Pouli N., Marakos P., Townsend P., Serrano M., Bartek J., Gorgoulis V.G. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell. 2017;16:192–197. doi: 10.1111/acel.12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pajares M., Cuadrado A., Engedal N., Jirsova Z., Cahova M. The role of free radicals in autophagy regulation: implications for ageing. Oxid. Med. Cell. Longev. 2018;2018:2450748. doi: 10.1155/2018/2450748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Woodall B.P., Gustafsson B. Autophagy—a key pathway for cardiac health and longevity. Acta Physiol. 2018;223 doi: 10.1111/apha.13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brunk U.T., Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 2002;269:1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 124.Hegedus Z.L., Altschule M.D., Frank H.A., Nayak U. Increase in plasma lipofuscin levels of stored blood. Crit. Care Med. 1985;13:155–159. doi: 10.1097/00003246-198503000-00003. [DOI] [PubMed] [Google Scholar]
- 125.Hegedus Z.L., Kuttab S.H., Altschule M.D., Nayak U. Isolation of melanin from human plasma lipofuscin. Arch. Physiol. Biochem. 1981;89:393–398. doi: 10.3109/13813458109069489. [DOI] [PubMed] [Google Scholar]
- 126.Feng F.K.E.L.L., Kong X.P., Wang D.S., Liu H.C. Lipofuscin in saliva and plasma and its association with age in healthy adults. Aging Clin. Exp. Res. 2015;27:573–580. doi: 10.1007/s40520-015-0326-3. [DOI] [PubMed] [Google Scholar]
- 127.El-Ghazzawi E.F., Malaty H.A. Electron microscopic observations on extraneuronal lipofuscin in the monkey brain. Cell Tissue Res. 1975;161:555–565. doi: 10.1007/BF00224144. [DOI] [PubMed] [Google Scholar]
- 128.Wang Lei, Chang-Yi Xiao, Li Jia-Hua T.G.-C.S.-S. Observation of the transport and removal of lipofuscin from the mouse myocardium 2 using transmission electron microscope. 2020. [DOI]
- 129.Rizou S.V., Evangelou K., Myrianthopoulos V., Mourouzis I., Havaki S., Athanasiou A., Vasileiou P.V.S., Margetis A., Kotsinas A., Kastrinakis N.G., Sfikakis P., Townsend P., Mikros E., Pantos C., Gorgoulis V.G. A novel quantitative method for the detection of lipofuscin, the main by-product of cellular senescence, in fluids. Methods Mol. Biol. 2019;1896:119–138. doi: 10.1007/978-1-4939-8931-7_12. [DOI] [PubMed] [Google Scholar]
- 130.Vasileva L.V., Savova M.S., Amirova K.M., Dinkova-Kostova A.T., Georgiev M.I. Obesity and NRF2-mediated cytoprotection: where is the missing link? Pharmacol. Res. 2020:156. doi: 10.1016/j.phrs.2020.104760. [DOI] [PubMed] [Google Scholar]
- 131.Hayes J.D., Dinkova-Kostova A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 132.Axelsson A.S., Tubbs E., Mecham B., Chacko S., Nenonen H.A., Tang Y., Fahey J.W., Derry J.M.J., Wollheim C.B., Wierup N., Haymond M.W., Friend S.H., Mulder H., Rosengren A.H. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aah4477. [DOI] [PubMed] [Google Scholar]
- 133.Vomhof-DeKrey E.E., Picklo M.J. The Nrf2-antioxidant response element pathway: a target for regulating energy metabolism. J. Nutr. Biochem. 2012;23:1201–1206. doi: 10.1016/j.jnutbio.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 134.Jo Early, Menon D., Wyse C.A., Cervantes-Silva M.P., Zaslona Z., Carroll R.G., Palsson-McDermott E.M., Angiari S., Ryan D.G., Corcoran S.E., Timmons G., Geiger S.S., Fitzpatrick D.J., O'Connell D., Xavier R.J., Hokamp K., O'Neill L.A.J., Curtis A.M. Circadian clock protein BMAL1 regulates IL-1β in macrophages via NRF2. Proc. Natl. Acad. Sci. U. S. A. 2018;115:E8460–E8468. doi: 10.1073/pnas.1800431115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shin S., Wakabayashi N., Misra V., Biswal S., Lee G.H., Agoston E.S., Yamamoto M., Kensler T.W. NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol. Cell Biol. 2007;27:7188–7197. doi: 10.1128/mcb.00915-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Xu J., Kulkarni S.R., Donepudi A.C., More V.R., Slitt A.L. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes. 2012;61:3208–3218. doi: 10.2337/db11-1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Takahashi T., Tabuchi T., Tamaki Y., Kosaka K., Takikawa Y., Satoh T. Carnosic acid and carnosol inhibit adipocyte differentiation in mouse 3T3-L1 cells through induction of phase2 enzymes and activation of glutathione metabolism. Biochem. Biophys. Res. Commun. 2009;382:549–554. doi: 10.1016/j.bbrc.2009.03.059. [DOI] [PubMed] [Google Scholar]
- 138.Pi J., Leung L., Xue P., Wang W., Hou Y., Liu D., Yehuda-Shnaidman E., Lee C., Lau J., Kurtz T.W., Chan J.Y. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J. Biol. Chem. 2010;285:9292–9300. doi: 10.1074/jbc.M109.093955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hou Y., Xue P., Bai Y., Liu D., Woods C.G., Yarborough K., Fu J., Zhang Q., Sun G., Collins S., Chan J.Y., Yamamoto M., Andersen M.E., Pi J. Nuclear factor erythroid-derived factor 2-related factor 2 regulates transcription of CCAAT/enhancer-binding protein β during adipogenesis. Free Radic. Biol. Med. 2012;52:462–472. doi: 10.1016/j.freeradbiomed.2011.10.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shin S., Wakabayashi J., Yates M.S., Wakabayashi N., Dolan P.M., Aja S., Liby K.T., Sporn M.B., Yamamoto M., Kensler T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-Imidazolide. Eur. J. Pharmacol. 2009;620:138–144. doi: 10.1016/j.ejphar.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yu Z., Shao W., Chiang Y., Foltz W., Zhang Z., Ling W., Fantus I.G., Jin T. Oltipraz upregulates the nuclear respiratory factor 2 alpha subunit (NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high-fat diet in C57BL/6J mice. Diabetologia. 2011;54:922–934. doi: 10.1007/s00125-010-2001-8. [DOI] [PubMed] [Google Scholar]
- 142.Seo H.A., Lee I.K. The role of NRF2: adipocyte differentiation, obesity, and insulin resistance. Oxid. Med. Cell. Longev. 2013;2013:184598. doi: 10.1155/2013/184598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chartoumpekis D.V., Ziros P.G., Psyrogiannis A.I., Papavassiliou A.G., Kyriazopoulou V.E., Sykiotis G.P., Habeos I.G. Nrf2 represses FGF21 during long-term high-fat diet - induced obesity in mice. Diabetes. 2011;60:2465–2473. doi: 10.2337/db11-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Di Francesco A., Choi Y., Bernier M., Zhang Y., Diaz-Ruiz A., Aon M.A., Kalafut K., Ehrlich M.R., Murt K., Ali A., Pearson K.J., Levan S., Preston J.D., Martin-Montalvo A., Martindale J.L., Abdelmohsen K., Michel C.R., Willmes D.M., Henke C. NQO1 protects obese mice through improvements in glucose and lipid metabolism. Npj Aging Mech. Dis. 2020;6 doi: 10.1038/s41514-020-00051-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang Y.K.J., Wu K.C., Liu J., Klaassen C.D. Nrf2 deficiency improves glucose tolerance in mice fed a high-fat diet. Toxicol. Appl. Pharmacol. 2012;264:305–314. doi: 10.1016/j.taap.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Meher A.K., Sharma P.R., Lira V.A., Yamamoto M., Kensler T.W., Yan Z., Leitinger N. Nrf2 deficiency in myeloid cells is not sufficient to protect mice from high-fat diet-induced adipose tissue inflammation and insulin resistance. Free Radic. Biol. Med. 2012;52:1708–1715. doi: 10.1016/j.freeradbiomed.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Aleksunes L.M., Reisman S.A., Yeager R.L., Goedken M.J., Klaassen C.D. Nuclear factor erythroid 2-related factor 2 deletion impairs glucose tolerance and exacerbates hyperglycemia in type 1 diabetic mice. J. Pharmacol. Exp. Therapeut. 2010;333:140–151. doi: 10.1124/jpet.109.162271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yagishita Y., Uruno A., Chartoumpekis D.V., Kensler T.W., Yamamoto M. Nrf2 represses the onset of type 1 diabetes in non-obese diabetic mice. J. Endocrinol. 2019;240:403–416. doi: 10.1530/JOE-18-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chartoumpekis D.V., Palliyaguru D.L., Wakabayashi N., Fazzari M., Khoo N.K.H., Schopfer F.J., Sipula I., Yagishita Y., Michalopoulos G.K., O'Doherty R.M., Kensler T.W. Nrf2 deletion from adipocytes, but not hepatocytes, potentiates systemic metabolic dysfunction after long-term high-fat diet-induced obesity in mice. Am. J. Physiol. Endocrinol. Metab. 2018;315:E180–E195. doi: 10.1152/ajpendo.00311.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Xue P., Hou Y., Chen Y., Yang B., Fu J., Zheng H., Yarborough K., Woods C.G., Liu D., Yamamoto M., Zhang Q., Andersen M.E., Pi J. Adipose deficiency of Nrf2 in ob/ob mice results in severe metabolic syndrome. Diabetes. 2013;62:845–854. doi: 10.2337/db12-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nagata N., Xu L., Kohno S., Ushida Y., Aoki Y., Umeda R., Fuke N., Zhuge F., Ni Y., Nagashimada M., Takahashi C., Suganuma H., Kaneko S., Ota T. Glucoraphanin ameliorates obesity and insulin resistance through adipose tissue browning and reduction of metabolic endotoxemia in mice. Diabetes. 2017;66:1222–1236. doi: 10.2337/db16-0662. [DOI] [PubMed] [Google Scholar]
- 152.Flanagan A., Bechtold D., Pot G.K., Johnston J.D. Chrono‐nutrition: from molecular and neuronal mechanisms to human epidemiology and timed feeding patterns. J. Neurochem. 2020;00:1–20. doi: 10.1111/jnc.15246. [DOI] [PubMed] [Google Scholar]
- 153.Reinke H., Asher G. Crosstalk between metabolism and circadian clocks. Nat. Rev. Mol. Cell Biol. 2019;20:227–241. doi: 10.1038/s41580-018-0096-9. [DOI] [PubMed] [Google Scholar]
- 154.Krishnaiah S.Y., Wu G., Altman B.J., Growe J., Rhoades S.D., Coldren F., Venkataraman A., Olarerin-George A.O., Francey L.J., Mukherjee S., Girish S., Selby C.P., Cal S., Er U., Sianati B., Sengupta A., Anafi R.C., Kavakli I.H., Sancar A. Clock regulation of metabolites reveals coupling between transcription and metabolism. Cell Metabol. 2017;25:961–974. doi: 10.1016/j.cmet.2017.03.019. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Stenvers D.J., Scheer F.A.J.L., Schrauwen P., la Fleur S.E., Kalsbeek A. Circadian clocks and insulin resistance. Nat. Rev. Endocrinol. 2019;15:75–89. doi: 10.1038/s41574-018-0122-1. [DOI] [PubMed] [Google Scholar]
- 156.Kolbe I., Leinweber B., Brandenburger M., Oster H. Circadian clock network desynchrony promotes weight gain and alters glucose homeostasis in mice. Mol. Metab. 2019;30:140–151. doi: 10.1016/j.molmet.2019.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chaix A., Lin T., Le H.D., Chang M.W., Panda S. Time-restricted feeding prevents obesity and metabolic syndrome in mice lacking a circadian clock. Cell Metabol. 2019;29:303–319. doi: 10.1016/j.cmet.2018.08.004. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.West A.C., Smith L., Ray D.W., Loudon A.S.I., Brown T.M., Bechtold D.A. Misalignment with the external light environment drives metabolic and cardiac dysfunction. Nat. Commun. 2017;8 doi: 10.1038/s41467-017-00462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gubin D.G., Nelaeva A.A., Uzhakova A.E., Hasanova Y.V., Cornelissen G., Weinert D. Disrupted circadian rhythms of body temperature, heart rate and fasting blood glucose in prediabetes and type 2 diabetes mellitus. Chronobiol. Int. 2017;34:1136–1148. doi: 10.1080/07420528.2017.1347670. [DOI] [PubMed] [Google Scholar]
- 160.Cederroth C.R., Albrecht U., Bass J., Brown S.A., Dyhrfjeld-Johnsen J., Gachon F., Green C.B., Hastings M.H., Helfrich-Förster C., Hogenesch J.B., Lévi F., Loudon A., Lundkvist G.B., Meijer J.H., Rosbash M., Takahashi J.S., Young M., Canlon B. Medicine in the fourth dimension. Cell Metabol. 2019;30:238–250. doi: 10.1016/j.cmet.2019.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dyar K.A., Eckel-Mahan K.L. Circadian metabolomics in time and space. Front. Neurosci. 2017;11 doi: 10.3389/fnins.2017.00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Depner C.M., Melanson E.L., McHill A.W., Wright K.P. Mistimed food intake and sleep alters 24-hour time-of-day patterns of the human plasma proteome. Proc. Natl. Acad. Sci. U. S. A. 2018;115 doi: 10.1073/pnas.1714813115. E5390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mota M.C., Silva C.M., Balieiro L.C.T., Fahmy W.M., Crispim C.A. Social jetlag and metabolic control in non-communicable chronic diseases: a study addressing different obesity statuses. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-06723-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Broussard J.L., Chapotot F., Abraham V., Day A., Delebecque F., Whitmore H.R., Tasali E. Sleep restriction increases free fatty acids in healthy men. Diabetologia. 2015;58:791–798. doi: 10.1007/s00125-015-3500-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang H., Lane J.M., Jones S.E., Dashti H.S., Ollila H.M., Wood A.R., van Hees V.T., Brumpton B., Winsvold B.S., Kantojärvi K., Palviainen T., Cade B.E., Sofer T., Song Y., Patel K., Anderson S.G., Bechtold D.A., Bowden J., Emsley R. Genome-wide association analysis of self-reported daytime sleepiness identifies 42 loci that suggest biological subtypes. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-11456-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Giskeødegård G.F., Davies S.K., Revell V.L., Keun H., Skene D.J. Diurnal rhythms in the human urine metabolome during sleep and total sleep deprivation. Sci. Rep. 2015;5 doi: 10.1038/srep14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Isherwood C.M., Van der Veen D.R., Johnston J.D., Skene D.J. Twenty-four-hour rhythmicity of circulating metabolites: effect of body mass and type 2 diabetes. Faseb. J. 2017;31:5557–5567. doi: 10.1096/fj.201700323R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Davies S.K., Ang J.E., Revell V.L., Holmes B., Mann A., Robertson F.P., Cui N., Middleton B., Ackermann K., Kayser M., Thumser A.E., Raynaud F.I., Skene D.J. Effect of sleep deprivation on the human metabolome. Proc. Natl. Acad. Sci. U. S. A. 2014;111:10761–10766. doi: 10.1073/pnas.1402663111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Forrestel A.C., Miedlich S.U., Yurcheshen M., Wittlin S.D., Sellix M.T. Chronomedicine and type 2 diabetes: shining some light on melatonin. Diabetologia. 2017;60:808–822. doi: 10.1007/s00125-016-4175-1. [DOI] [PubMed] [Google Scholar]
- 170.Wilms B., Leineweber E.M., Mölle M., Chamorro R., Pommerenke C., Salinas-Riester G., Sina C., Lehnert H., Oster H., Schmid S.M. Sleep loss disrupts morning-to-evening differences in human white adipose tissue transcriptome. J. Clin. Endocrinol. Metab. 2019;104:1687–1696. doi: 10.1210/jc.2018-01663. [DOI] [PubMed] [Google Scholar]
- 171.Kolbe I., Carrasco-Benso M.P., López-Mínguez J., Lujan J., Scheer F.A.J.L., Oster H., Garaulet M. Circadian period of luciferase expression shortens with age in human mature adipocytes from obese patients. Faseb. J. 2019;33:175–180. doi: 10.1096/fj.201800441R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yu R., Tian L., Ding Y., Gao Y., Li D., Tang Y. Correlation between inflammatory markers and impaired circadian clock gene expression in type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2019;156 doi: 10.1016/j.diabres.2019.107831. [DOI] [PubMed] [Google Scholar]
- 173.Edgar R.S., Green E.W., Zhao Y., Van Ooijen G., Olmedo M., Qin X., Xu Y., Pan M., Valekunja U.K., Feeney K.A., Maywood E.S., Hastings M.H., Baliga N.S., Merrow M., Millar A.J., Johnson C.H., Kyriacou C.P., O'Neill J.S., Reddy A.B. Peroxiredoxins are conserved markers of circadian rhythms. Nature. 2012;485:459–464. doi: 10.1038/nature11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Imamura K., Yoshitane H., Hattori K., Yamaguchi M., Yoshida K., Okubo T., Naguro I., Ichijo H., Fukada Y. ASK family kinases mediate cellular stress and redox signaling to circadian clock. Proc. Natl. Acad. Sci. U. S. A. 2018;115:3646–3651. doi: 10.1073/pnas.1719298115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wible R.S., Ramanathan C., Sutter C.H., Olesen K.M., Kensler T.W., Liu A.C., Sutter T.R. NRF2 regulates core and stabilizing circadian clock loops, coupling redox and timekeeping in mus musculus. eLife. 2018;7 doi: 10.7554/eLife.31656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pei J.F., Li X.K., Li W.Q., Gao Q., Zhang Y., Wang X.M., Fu J.Q., Cui S.S., Qu J.H., Zhao X., Hao D.L., Ju D., Liu N., Carroll K.S., Yang J., Zhang E.E., Cao J.M., Chen H.Z., Liu D.P. Diurnal oscillations of endogenous H2O2 sustained by p66Shc regulate circadian clocks. Nat. Cell Biol. 2019;21:1553–1564. doi: 10.1038/s41556-019-0420-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rey G., Valekunja U.K., Feeney K.A., Wulund L., Milev N.B., Stangherlin A., Ansel-Bollepalli L., Velagapudi V., O'Neill J.S., Reddy A.B. The pentose phosphate pathway regulates the circadian clock. Cell Metabol. 2016;24:462–473. doi: 10.1016/j.cmet.2016.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Xu Y.Q., Zhang D., Jin T., Cai D.J., Wu Q., Lu Y., Liu J., Klaassen C.D. Diurnal variation of hepatic antioxidant gene expression in mice. PLoS One. 2012;7 doi: 10.1371/journal.pone.0044237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pekovic-Vaughan V., Gibbs J., Yoshitane H., Yang N., Pathiranage D., Guo B., Sagami A., Taguchi K., Bechtold D., Loudon A., Yamamoto M., Chan J., van der Horst G.T.J., Fukada Y., Meng Q.J. The circadian clock regulates rhythmic activation of the NRF2/glutathionemediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. 2014;28:548–560. doi: 10.1101/gad.237081.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lee J., Moulik M., Fang Z., Saha P., Zou F., Xu Y., Nelson D.L., Ma K., Moore D.D., Yechoor V.K. Bmal1 and -cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced -cell failure in mice. Mol. Cell Biol. 2013;33:2327–2338. doi: 10.1128/mcb.01421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Desvergne A., Ugarte N., Petropoulos I., Friguet B. Circadian modulation of proteasome activities and removal of carbonylated proteins. Free Radic. Biol. Med. 2014;75:S18. doi: 10.1016/j.freeradbiomed.2014.10.631. [DOI] [PubMed] [Google Scholar]
- 182.Su H., Gornitsky M., Geng G., Velly A.M., Chertkow H., Schipper H.M. Diurnal variations in salivary protein carbonyl levels in normal and cognitively impaired human subjects. Age. 2008;30:1–9. doi: 10.1007/s11357-007-9042-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Blanco R.A., Ziegler T.R., Carlson B.A., Cheng P.Y., Park Y., Cotsonis G.A., Accardi C.J., Jones D.P. Diurnal variation in glutathione and cysteine redox states in human plasma1-3. Am. J. Clin. Nutr. 2007;86:1016–1023. doi: 10.1093/ajcn/86.4.1016. [DOI] [PubMed] [Google Scholar]
- 184.Wang T.A., Yu Y.V., Govindaiah G., Ye X., Artinian L., Coleman T.P., Sweedler J.V., Cox C.L., Gillette M.U. Circadian rhythm of redox state regulates excitability in suprachiasmatic nucleus neurons. Science (80- ) 2012;337:839–842. doi: 10.1126/science.1222826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sani M., Sebai H., Ghanem-Boughanmi N., Boughattas N.A., Ben-Attia M. Circadian (about 24-hour) variation in malondialdehyde content and catalase activity of mouse erythrocytes. Redox Rep. 2015;20:26–32. doi: 10.1179/1351000214Y.0000000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kushwaha R.S., Gupta R.C., Sharma J.P., Sharma S., Singh R.K., Cornelissen G. Circadian periodicity of circulating plasma lipid peroxides, uric acid and ascorbic acid in renal stone formers. Indian J. Clin. Biochem. 2017;32:220–224. doi: 10.1007/s12291-016-0594-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Teixeira K.R.C., dos Santos C.P., de Medeiros L.A., Mendes J.A., Cunha T.M., De Angelis K., Penha-Silva N., de Oliveira E.P., Crispim C.A. Night workers have lower levels of antioxidant defenses and higher levels of oxidative stress damage when compared to day workers. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-40989-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Montes-Nieto R., Insenser M., Murri M., Fernández-Durán E., Ojeda-Ojeda M., Martínez-García M.Á., Luque-Ramírez M., Escobar-Morreale H.F. Plasma thiobarbituric acid reactive substances (TBARS) in young adults: obesity increases fasting levels only in men whereas glucose ingestion, and not protein or lipid intake, increases postprandial concentrations regardless of sex and obesity. Mol. Nutr. Food Res. 2017;61 doi: 10.1002/mnfr.201700425. [DOI] [PubMed] [Google Scholar]
- 189.Bitencourt M.R., Patias L.D., Beck M., da C., Alvarez G., Diehl L.N., Duarte M.F., Schetinger M.R., Morsch V.M. Evaluation of the biochemical, inflammatory and oxidative profile of obese patients given clinical treatment and bariatric surgery. Clin. Chim. Acta. 2017;465:72–79. doi: 10.1016/j.cca.2016.12.012. [DOI] [PubMed] [Google Scholar]
- 190.Cejvanovic V., Asferg C., Kjær L.K., Andersen U.B., Linneberg A., Frystyk J., Henriksen T., Flyvbjerg A., Christiansen M., Weimann A., Jeppesen J., Poulsen H.E. Markers of oxidative stress in obese men with and without hypertension. Scand. J. Clin. Lab. Invest. 2016;76:620–625. doi: 10.1080/00365513.2016.1230776. [DOI] [PubMed] [Google Scholar]
- 191.Manzella N., Bracci M., Strafella E., Staffolani S., Ciarapica V., Copertaro A., Rapisarda V., Ledda C., Amati M., Valentino M., Tomasetti M., Stevens R.G., Santarelli L. Circadian modulation of 8-oxoguanine DNA damage repair. Sci. Rep. 2015;5 doi: 10.1038/srep13752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ligibel J.A., Alfano C.M., Courneya K.S., Demark-Wahnefried W., Burger R.A., Chlebowski R.T., Fabian C.J., Gucalp A., Hershman D.L., Hudson M.M., Jones L.W., Kakarala M., Ness K.K., Merrill J.K., Wollins D.S., Hudis C.A. American Society of Clinical Oncology position statement on obesity and cancer. J. Clin. Oncol. 2014;32:3568–3574. doi: 10.1200/JCO.2014.58.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Arnold M., Pandeya N., Byrnes G., Renehan A.G., Stevens G.A., Ezzati M., Ferlay J., Miranda J.J., Romieu I., Dikshit R., Forman D., Soerjomataram I. Global burden of cancer attributable to high body-mass index in 2012: a population-based study. Lancet Oncol. 2015;16:36–46. doi: 10.1016/S1470-2045(14)71123-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Trent L., The state of cancer care in America A report by the American society of clinical oncology. J. Oncol. Pract. 2014;10:119–142. doi: 10.1200/JOP.2014.001386. 2014. [DOI] [PubMed] [Google Scholar]
- 195.Renehan A.G., Tyson M., Egger M., Heller R.F., Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 196.Bhaskaran K., Douglas I., Forbes H., Dos-Santos-Silva I., Leon D.A., Smeeth L. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5·24 million UK adults. Lancet. 2014;384:755–765. doi: 10.1016/S0140-6736(14)60892-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Calle E.E., Rodriguez C., Walker-Thurmond K., Thun M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. Adults. N. Engl. J. Med. 2003;348:1625–1638. doi: 10.1056/nejmoa021423. [DOI] [PubMed] [Google Scholar]
- 198.Tsilidis K.K., Kasimis J.C., Lopez D.S., Ntzani E.E., Ioannidis J.P.A. Type 2 diabetes and cancer: umbrella review of meta-analyses of observationlal studies. BMJ. 2015;350:1–11. doi: 10.1136/bmj.g7607. [DOI] [PubMed] [Google Scholar]
- 199.Shikata K., Ninomiya T., Kiyohara Y. Diabetes mellitus and cancer risk: review of the epidemiological evidence. Canc. Sci. 2013;104:9–14. doi: 10.1111/cas.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.García-Jiménez C., Gutiérrez-Salmerón M., Chocarro-Calvo A., García-Martinez J.M., Castaño A., De La Vieja A. From obesity to diabetes and cancer: epidemiological links and role of therapies. Br. J. Canc. 2016;114:716–722. doi: 10.1038/bjc.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sullivan S.A., Tran A.M., Clark L.H., Bateman N.W., Hood B.L., Conrads T.P., Lee D., Zhou C., Makowski L., Bae-Jump V.L. Reversal of obesity-driven aggressiveness of endometrial cancer by metformin. Gynecol. Oncol. 2017;145:21. doi: 10.1016/j.ygyno.2017.03.064. [DOI] [Google Scholar]
- 202.Thakkar B., Aronis K.N., Vamvini M.T., Shields K., Mantzoros C.S. Metformin and sulfonylureas in relation to cancer risk in type II diabetes patients: a meta-analysis using primary data of published studies. Metabolism. 2013;62:922–934. doi: 10.1016/j.metabol.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 203.Panieri E., Santoro M.M. Ros homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016;7 doi: 10.1038/cddis.2016.105. e2253-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kalyanaraman B., Cheng G., Hardy M., Ouari O., Bennett B., Zielonka J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. 2018;15:347–362. doi: 10.1016/j.redox.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kalyanaraman B. Teaching the basics of cancer metabolism: developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. 2017;12:833–842. doi: 10.1016/j.redox.2017.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Li P., Zhang D., Shen L., Dong K., Wu M., Ou Z., Shi D. Redox homeostasis protects mitochondria through accelerating ROS conversion to enhance hypoxia resistance in cancer cells. Sci. Rep. 2016;6:1–13. doi: 10.1038/srep22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.O'Flanagan C.H., Rossi E.L., McDonell S.B., Chen X., Tsai Y.-H., Parker J.S., Usary J., Perou C.M., Hursting S.D. Metabolic reprogramming underlies metastatic potential in an obesity-responsive murine model of metastatic triple negative breast cancer. Npj Breast Canc. 2017;3:1–10. doi: 10.1038/s41523-017-0027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Madak-Erdogan Z., Band S., Zhao Y.C., Smith B.P., Kulkoyluoglu-Cotul E., Zuo Q., Casiano A.S., Wrobel K., Rossi G., Smith R.L., Kim S.H., Katzenellenbogen J.A., Johnson M.L., Patel M., Marino N., Storniolo A.M.V., Flaws11 J.A. Free fatty acids rewire cancer metabolism in obesity-associated breast cancer via estrogen receptor and mTOR signaling. Canc. Res. 2019;79:2494–2510. doi: 10.1158/0008-5472.CAN-18-2849. [DOI] [PubMed] [Google Scholar]
- 209.Kawashima K., Maeda K., Saigo C., Kito Y., Yoshida K., Takeuchi T. Adiponectin and intelectin-1: important adipokine players in obesity-related colorectal carcinogenesis. Int. J. Mol. Sci. 2017;18:1–12. doi: 10.3390/ijms18040866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wu Q., Li B., Li Z., Li J., Sun S., Sun S. Cancer-associated adipocytes: key players in breast cancer progression. J. Hematol. Oncol. 2019;12:1–15. doi: 10.1186/s13045-019-0778-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Uehara H., Kobayashi T., Matsumoto M., Watanabe S., Yoneda A., Bando Y. Adipose tissue critical contributor to the development of prostate cance - uehara - 2018 - J. Med. Invest. 2018;65 doi: 10.2152/jmi.65.9. [DOI] [PubMed] [Google Scholar]
- 212.Avgerinos K.I., Spyrou N., Mantzoros C.S., Dalamaga M. Obesity and cancer risk: emerging biological mechanisms and perspectives. Metabolism. 2019;92:121–135. doi: 10.1016/j.metabol.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 213.Quail D.F., Dannenberg A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2019;15:139–154. doi: 10.1038/s41574-018-0126-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wang Y.Y., Attané C., Milhas D., Dirat B., Dauvillier S., Guerard A., Gilhodes J., Lazar I., Alet N., Laurent V., Le Gonidec S., Biard D., Hervé C., Bost F., Ren G.S., Bono F., Escourrou G., Prentki M., Nieto L. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight. 2017;2 doi: 10.1172/jci.insight.87489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zhang X., Wang Z., Li J., Gu D., Li S., Shen C., Song Z. Increased 4-hydroxynonenal formation contributes to obesity-related lipolytic activation in adipocytes. PLoS One. 2013;8 doi: 10.1371/journal.pone.0070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Li Y.P., Tian F.G., Shi P.C., Guo L.Y., Wu H.M., Chen R.Q., Xue J.M. 4-Hydroxynonenal promotes growth and angiogenesis of breast cancer cells through HIF-1α stabilization. Asian Pac. J. Cancer Prev. APJCP. 2014;15:10151–10156. doi: 10.7314/APJCP.2014.15.23.10151. [DOI] [PubMed] [Google Scholar]
- 217.Huang Y., Li W., Kong A.N.T. Anti-oxidative stress regulator NF-E2-related factor 2 mediates the adaptive induction of antioxidant and detoxifying enzymes by lipid peroxidation metabolite 4-hydroxynonenal. Cell Biosci. 2012;2 doi: 10.1186/2045-3701-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Burlaka A.P., Ganusevich, Vovk A.V., Burlaka A.A., Gafurov M.R., Lukin S.N. Colorectal cancer and mitochondrial dysfunctions of the adjunct adipose tissues: a case study. BioMed Res. Int. 2018;2018 doi: 10.1155/2018/2169036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kalezic A., Udicki M., Galic B.S., Aleksic M., Korac A., Jankovic A., Korac B. Lactate metabolism in breast cancer microenvironment: contribution focused on associated adipose tissue and obesity. Int. J. Mol. Sci. 2020;21:1–13. doi: 10.3390/ijms21249676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Crujeiras A.B., Cabia B., Carreira M.C., Amil M., Cueva J., Andrade S., Seoane L.M., Pardo M., Sueiro A., Baltar J., Morais T., Monteiro M.P., Lopez-Lopez R., Casanueva F.F. Secreted factors derived from obese visceral adipose tissue regulate the expression of breast malignant transformation genes. Int. J. Obes. 2016;40:514–523. doi: 10.1038/ijo.2015.208. [DOI] [PubMed] [Google Scholar]
- 221.Laurent V., Toulet A., Attané C., Milhas D., Dauvillier S., Zaidi F., Clement E., Cinato M., Le Gonidec S., Guérard A., Lehuédé C., Garandeau D., Nieto L., Renaud-Gabardos E., Prats A.C., Valet P., Malavaud B., Muller C. Periprostatic adipose tissue favors prostate cancer cell invasion in an obesity-dependent manner: role of oxidative stress. Mol. Canc. Res. 2019;17:821–835. doi: 10.1158/1541-7786.MCR-18-0748. [DOI] [PubMed] [Google Scholar]
- 222.Frijhoff J., Winyard P.G., Zarkovic N., Davies S.S., Stocker R., Cheng D., Knight A.R., Taylor E.L., Oettrich J., Ruskovska T., Gasparovic A.C., Cuadrado A., Weber D., Poulsen H.E., Grune T., Schmidt H.H.H.W., Ghezzi P. Clinical relevance of biomarkers of oxidative stress. Antioxidants Redox Signal. 2015;23:1144–1170. doi: 10.1089/ars.2015.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Suzana S., Normah H., Fatimah A., Nor Fadilah R., Ahmad Rohi G., Amin I., Cham B.G., Mohd Rizal R., Fairulnizal M.N. Antioxidants intake and status, and oxidative stress in relation to breast cancer risks: a case-control study. Asian Pac. J. Cancer Prev. APJCP. 2008;9:343–350. [PubMed] [Google Scholar]
- 224.Hacer I.A., Zeynep A.A., Ö Can, Riza K.A., Dildar K., Tülay A. The effect of prostate cancer and antianrogenic therapy on lipid peroxidation and antioxidant systems. Int. Urol. Nephrol. 2003;36:57–62. doi: 10.1023/B:UROL.0000032676.31470.b2. [DOI] [PubMed] [Google Scholar]
- 225.Pande D., Negi R., Karki K., Dwivedi U.S., Khanna R.S., Khanna H.D. Simultaneous progression of oxidative stress, angiogenesis, and cell proliferation in prostate carcinoma. Urol. Oncol. Semin. Orig. Invest. 2013;31:1561–1566. doi: 10.1016/j.urolonc.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 226.Murlikiewicz L., Grzegorczyk K., Lewicka M., Buczyński A., Rutkowski M. Oxidative stress in colonic adenocarcinoma: an impact on the body's antioxidative status and oxidative protein damage. Adv. Clin. Exp. Med. 2018;27:77–82. doi: 10.17219/acem/67819. [DOI] [PubMed] [Google Scholar]
- 227.Sova H., Jukkola-Vuorinen A., Puistola U., Kauppila S., Karihtala P. 8-Hydroxydeoxyguanosine: a new potential independent prognostic factor in breast cancer. Br. J. Canc. 2010;102:1018–1023. doi: 10.1038/sj.bjc.6605565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dziaman T., Banaszkiewicz Z., Roszkowski K., Gackowski D., Wisniewska E., Rozalski R., Foksinski M., Siomek A., Speina E., Winczura A., Marszalek A., Tudek B., Olinski R. 8-Oxo-7,8-dihydroguanine and uric acid as efficient predictors of survival in colon cancer patients. Int. J. Canc. 2014;134:376–383. doi: 10.1002/ijc.28374. [DOI] [PubMed] [Google Scholar]
- 229.Yang S., Pinney S.M., Mallick P., Ho S.M., Bracken B., Wu T. Impact of oxidative stress biomarkers and carboxymethyllysine (an advanced glycation end product) on prostate cancer: a prospective study. Clin. Genitourin. Canc. 2015;13:e347–e351. doi: 10.1016/j.clgc.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Nass N., Ignatov A., Andreas L., Weißenborn C., Kalinski T., Sel S. Accumulation of the advanced glycation end product carboxymethyl lysine in breast cancer is positively associated with estrogen receptor expression and unfavorable prognosis in estrogen receptor-negative cases. Histochem. Cell Biol. 2017;147:625–634. doi: 10.1007/s00418-016-1534-4. [DOI] [PubMed] [Google Scholar]
- 231.Rossner P., Gammon M.D., Terry M.B., Agrawal M., Fang F.Z., Teitelbaum S.L., Eng S.M., Gaudet M.M., Neugut A.I., Santella R.M. Relationship between urinary 15-F2t-isoprostane and 8-oxodeoxyguanosine levels and breast cancer risk. Cancer Epidemiol. Biomark. Prev. 2006;15:639–644. doi: 10.1158/1055-9965.EPI-05-0554. [DOI] [PubMed] [Google Scholar]
- 232.Barocas D.A., Motley S., Cookson M.S., Chang S.S., Penson D.F., Dai Q., Milne G., Roberts L.J., Morrow J., Concepcion R.S., Smith J.A., Fowke J.H. Oxidative stress measured by urine F2-isoprostane level is associated with prostate cancer. J. Urol. 2011;185:2102–2107. doi: 10.1016/j.juro.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Brys M., Morel A., Forma E., Krzeslak A., Wilkosz J., Rozanski W., Olas B. Relationship of urinary isoprostanes to prostate cancer occurence. Mol. Cell. Biochem. 2013;372:149–153. doi: 10.1007/s11010-012-1455-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Rasool M., Malik A., Ghuman A.A., Basit Ashraf M.A., Arooj M., Waquar S., Zahid S., Shaheen S., Qazi A., Naseer M.I., Zamzami M.A., Al-Ghafari A., Baothman O.A., Zeyadi M., Helmi N., Choudhry H., Jamal M.S., Al-Qahtani M.H. Implications of isoprostanes and matrix metalloproteinase-7 having potential role in the development of colorectal cancer in males. Front. Oncol. 2018;8 doi: 10.3389/fonc.2018.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Gào X., Brenner H., Holleczek B., Cuk K., Zhang Y., Anusruti A., Xuan Y., Xu Y., Schöttker B. Urinary 8-isoprostane levels and occurrence of lung, colorectal, prostate, breast and overall cancer: results from a large, population-based cohort study with 14 years of follow-up. Free Radic. Biol. Med. 2018;123:20–26. doi: 10.1016/j.freeradbiomed.2018.05.065. [DOI] [PubMed] [Google Scholar]
- 236.Shen J., Gammon M.D., Terry M.B., Wang Q., Bradshaw P., Teitelbaum S.L., Neugut A.I., Santella R.G.M. Telomere length, oxidative damage, antioxidants and breast cancer risk. Int. J. Canc. 2009;124:1637–1643. doi: 10.1002/ijc.24105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Dai Q., Gao Y.T., Shu X.O., Yang G., Milne G., Cai Q., Wen W., Rothman N., Cai H., Li H., Xiang Y., Chow W.H., Zheng W. Oxidative stress, obesity, and breast cancer risk: results from the Shanghai women's health study. J. Clin. Oncol. 2009;27:2482–2488. doi: 10.1200/JCO.2008.19.7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Rossner P., Terry M.B., Gammon M.D., Agrawal M., Zhang F.F., Ferris J.S., Teitelbaum S.L., Eng S.M., Gaudet M.M., Neugut A.I., Santella R.M. Plasma protein carbonyl levels and breast cancer risk. J. Cell Mol. Med. 2007;11:1138–1148. doi: 10.1111/j.1582-4934.2007.00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zipprich J., Beth Terry M., Liao Y., Agrawal M., Gurvich I., Senie R., Santella R.M. Plasma protein carbonyls and breast cancer risk in sisters discordant for breast cancer from the New York Site of the breast cancer family registry. Canc. Res. 2009;69:2966–2972. doi: 10.1158/0008-5472.CAN-08-3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Hoque A., Ambrosone C.B., Till C., Goodman P.J., Tangen C., Kristal A., Lucia S., Wang Q., Kappil M., Thompson I., Hsing A.W., Parnes H., Santella R.M. Serum oxidized protein and prostate cancer risk within the prostate cancer prevention trial. Canc. Prev. Res. 2010;3:478–483. doi: 10.1158/1940-6207.CAPR-09-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]