

Abstract
Auranofin (AF) is an FDA-approved antirheumatic drug with anticancer properties that acts as a thioredoxin reductase 1 (TrxR) inhibitor. The exact mechanisms through which AF targets cancer cells remain elusive. To shed light on the mode of action, this study provides an in-depth analysis on the molecular mechanisms and immunogenicity of AF-mediated cytotoxicity in the non-small cell lung cancer (NSCLC) cell line NCI–H1299 (p53 Null) and its two isogenic derivates with mutant p53 R175H or R273H accumulation.
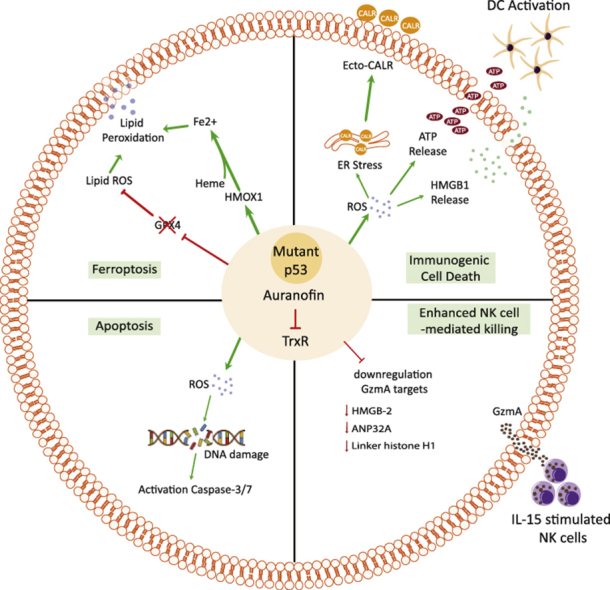
TrxR is highly expressed in a panel of 72 NSCLC patients, making it a valid druggable target in NSCLC for AF. The presence of mutant p53 overexpression was identified as an important sensitizer for AF in (isogenic) NSCLC cells as it was correlated with reduced thioredoxin (Trx) levels in vitro. Transcriptome analysis revealed dysregulation of genes involved in oxidative stress response, DNA damage, granzyme A (GZMA) signaling and ferroptosis. Although functionally AF appeared a potent inhibitor of GPX4 in all NCI–H1299 cell lines, the induction of lipid peroxidation and consequently ferroptosis was limited to the p53 R273H expressing cells. In the p53 R175H cells, AF mainly induced large-scale DNA damage and replication stress, leading to the induction of apoptotic cell death rather than ferroptosis. Importantly, all cell death types were immunogenic since the release of danger signals (ecto-calreticulin, ATP and HMGB1) and dendritic cell maturation occurred irrespective of (mutant) p53 expression. Finally, we show that AF sensitized cancer cells to caspase-independent natural killer cell-mediated killing by downregulation of several key targets of GZMA.
Our data provides novel insights on AF as a potent, clinically available, off-patent cancer drug by targeting mutant p53 cancer cells through distinct cell death mechanisms (apoptosis and ferroptosis). In addition, AF improves the innate immune response at both cytostatic (natural killer cell-mediated killing) and cytotoxic concentrations (dendritic cell maturation).
Keywords: Auranofin, Thioredoxin reductase, Non-small cell lung cancer, Mutant p53, Oxidative stress, Cancer cell death
Graphical abstract
Abbreviations
- AF
Auranofin
- CALR
Calreticulin
- CI
Confidence interval
- CTL
Cytotoxic T lymphocyte
- DAMPs
Damage-associated molecular patterns
- DC
Dendritic cells
- DFO
Deferoxamine
- E:T
Effector:Target
- EMT
Epithelial-mesenchymal transition
- FBS
Fetal bovine serum
- FDA
Food and Drug Administration
- FDR
False discovery rate
- Fer1
Ferrostatin-1
- FFPE
Formalin fixed paraffin embedded
- FPKM
Fragment per kilobase million
- GCU
Green calibrated unit
- GPX4
Glutathione peroxidase 4
- GSEA
Gene Set Enrichment analysis
- GSH
Glutathione
- GZMA
Granzyme A
- HDAC
Histone deacetylase
- HMGB1
High mobility group
- HMOX1
Heme oxygenase-1
- HR
Hazard ratio
- ICD
Immunogenic cell death
- IFNγ
Interferonγ
- IHC
Immunohistochemistry
- IL
Interleukin
- IPA
Ingenuity pathway analysis
- IRP
Iron regulatory protein
- IRS
Immunoreactivity scoring
- LINCS
The Library of Integrated Network-Based Cellular Signatures
- LUAD
Lung adenocarcinoma
- MFI
Mean Fluorescence intensity
- MHC-II
Major histocompatibility complexes class II
- NAC
n-acetyl-cysteine
- Nec1s
Necrostatin-s1
- NGS
Next generation sequencing
- NK
Natural killer cells
- NRF2
Nuclear factor erythroid 2-related factor 2
- NSCLC
Non-small cell lung cancer
- OS
Overall survival
- PBMC
Peripheral blood mononuclear cells
- PBS
Phosphate buffer saline
- PDAC
Pancreatic ductal adenocarcinoma
- PFA
Paraformaldehyde
- PFS
Progression-free survival
- PI
Propidium iodide
- PPARG
Peroxisome proliferator-activated receptor gamma
- PRDX
Peroxiredoxin
- RA
Rheumatoid arthritis
- ROIs
Regions of interests
- ROS
Reactive oxygen species
- RT
Room temperature
- SLC7A11
Solute carrier family 7 member 11
- SOD1
Superoxide dismutase 1
- SRB
Sulforhodamine B
- TCGA
The cancer genome atlas
- TGF-β
transforming growth factor-β
- TNF-α
tumor necrosis factor- α
- Trx
Thioredoxin
- TrxR
Thioredoxin reductase
- WT
Wild type
- ZEB1
Zinc finger E-box-binding homeobox 1
- αToco
Alpha-tocopherol
1. Introduction
Lung cancer is one of the most common malignancies and the leading cause of cancer-related death throughout the world. Approximately 85% of all lung cancers are categorized as non-small cell lung cancer (NSCLC), accounting for more than 1.8 million deaths per year [1]. Despite important advancements in treatment modalities, NSCLC patients have a 5-year overall survival rate below 15% [2].
The worldwide increase in NSCLC incidence and mortality rates is associated with resistance to standard chemotherapy and targeted therapies, which resulted in the search for alternative treatment methods [[3], [4], [5]]. Despite huge investments of biopharmaceutical companies, the number of new drugs introduced into clinical practice has not increased significantly [6], primarily due to a lack of efficacy and serious adverse effects in Phase II and III clinical trials [7]. Consequently, there is an increasing interest in repurposing well-known and well-characterized licensed non-cancer drugs to the oncology domain, as underscored by the Repurposing Drugs in Oncology project [8]. One compound that is receiving increasing interest as an object of repurposing strategies in cancer is auranofin (AF). AF is an orally available, lipophilic, organogold compound with a well-known safety profile that was approved by the U.S. Food and Drug Administration (FDA) for the treatment of rheumatoid arthritis (RA). AF has gained interest as an anticancer agent since it acts as a thioredoxin reductase 1 (TrxR) inhibitor – through formation of a stable coordinative bond between its gold(I) center and the active site selenocysteine residues [9,10] – and thereby perturbing cellular redox balance. TrxR was indeed identified as the main target of AF by different chemical proteomics tools [11].
The difference in intrinsic levels of reactive oxygen species (ROS) and redox status between normal and malignant cancer cells represents a potential therapeutic window for ROS-inducing therapies [12]. Cellular ROS levels are strictly controlled by balancing ROS-generating and scavenging systems, which includes the redox protein thioredoxin (Trx). The TrxR enzyme catalyzes the reduction of Trx with electrons from NADPH in the cell's Trx system [13]. Since many cancer cells depend on a functional Trx system, TrxR and Trx overexpression often participate in carcinogenesis, cancer progression, protection against apoptosis and poor prognosis [14,15]. NSCLC tumors have a significant elevation of TrxR activity both in vitro and in vivo [16]. Furthermore, high levels of Trx are associated with resistance to various standard chemotherapeutic agents, including the first-line treatment of advanced NSCLC cisplatin [17,18].
Preclinical evidence of the antineoplastic activity of AF in various tumor models have led to four ongoing clinical cancer trials for AF in chronic lymphocytic leukemia (NCT01419691, NCT01747798), ovarian cancer (NCT03456700) and lung cancer (NCT01737502) [19], which further strengthen the translational value of TrxR as a target of AF [[20], [21], [22]]. This potential is further supported by the recently released Project Score database, a genome-scale CRISPR-Cas9 drop-out screen to prioritize cancer therapeutic targets, showing antitumoral effects in 21% and 76% of 324 cell lines tested with knock-out of TXNRD1 and TXN genes, respectively [23].
Taken together, targeting the Trx/TrxR system via AF may have value for the treatment of NSCLC. However, the exact molecular mechanisms by which AF induces cancer cell death remain elusive. Therefore, we first studied the potential of targeting the Trx system using data extracted from the cancer genome atlas (TCGA) database and NSCLC patient samples. Next, we investigated potential biomarkers for AF treatment and identified mutant p53 overexpression as an important sensitizer. Mutations in the tumor suppressor gene TP53 are the most common genetic lesions found in NSCLC patients resulting in the aberrant accumulation of mutant p53 protein and poor patient survival [24]. Therapeutic strategies to efficiently target mutant p53 NSCLC are pressingly needed. Therefore, an isogenic NSCLC cell panel was used consisting of the parental p53 Null NCI–H1299 cell line and its two isogenic derivates with mutant p53 R175H or R273H overexpression, which are by far the best characterized p53 hotspot mutations in vitro and in vivo [25]. NCI-H1299 cell line with knock-in of selective p53 mutants is the gold-standard model in the field of p53 research to study the gain-of-function (GOF) effects of specific mutant p53 proteins [24,26,27], either for the basal phenotype of the cancer cells or their effect on therapy response. Amino acid changes in the R175 position are conformational mutations that cause a full or partial distortion of the correct folding and structure of p53, indirectly affecting the DNA-binding capacity of the mutant protein. On the other hand, amino acid changes in the R273 position are DNA-contact mutations, which directly affect the domains of p53 that interact with DNA [25,28,29]. A subsequent transcriptome-wide analysis revealed that in this isogenic setting, mutant p53 accumulation sensitizes NSCLC cells to AF-induced cell death, albeit via distinct pathways. NSCLC cells with medium levels of the p53 R175H mutant were prone to undergo caspase-dependent apoptosis, while p53 R273H mutant cells were more vulnerable to ferroptotic cell death. Irrespective of p53 expression levels, both AF-induced cell death mechanisms initiated the release of immunogenic cell death (ICD) related damage-associated molecular patterns (DAMPs) and dendritic cell (DC) maturation in vitro. In addition, AF primed mutant p53 NSCLC cells for caspase-independent natural killer (NK) cell-mediated killing by downregulation of several key targets of GZMA. Our study thus indicates that AF represents a potential novel therapeutic strategy to efficiently kill mutant p53 NSCLC tumor cells through distinct immunogenic cell death pathways.
2. Methods
Supplementary Table 1 contains all relevant information regarding the source and identifier of the antibodies, reagents, assays, etc. used throughout this study.
2.1. Cell lines and cell culture
A panel of eight NSCLC and pancreatic ductal adenocarcinoma (PDAC) cell lines with differing p53 status was used in this study to cover a large subset of patients (Suppl. Table 2). The human pancreatic cell lines included Mia-PaCa-2, Panc-1, Capan-2 and BxPC-3. The human NSCLC cell lines included NCI–H1975, NCI–H2228, NCI–H596 and A549. All these cell lines were purchased from the American Type Culture Collection, except for Mia-PaCa-2 (DSMZ-German collection of Microorganisms and Cell Cultures).
An additional mutant p53 cell line panel was used for the majority of experiments performed in this study and consisted of the parental NSCLC NCI–H1299 cells (homozygous partial p53 deletion, Null) and its isogenic derivates stably transfected to express either mutant p53 R175H or R273H protein, as per established protocols [30] (provided by prof. Dr. Haupt at the Peter MacCallum Cancer Centre, Australia). Mutant p53 R175H and R273H are overexpressing mutant p53 compared to p53 Null parental line for which no protein was detected by Western blotting. Mia-PaCa-2 and Panc-1 were cultured in the required medium being DMEM (Life Technologies) supplemented with 10% fetal bovine serum (FBS, Life Technologies), 1% penicillin/streptomycin (Life technologies) and 2 mM l-glutamine (Life technologies). Capan-2, BxPC-3, NCI–H1975, NCI–H2228, NCI–H596 and the isogenic NCI–H1299 cell lines were cultured in RPMI (Life Technologies) supplemented as described above. To rule out differences in sensitivity to AF due to variations in selenium concentration [31], all experiments were performed using the same supplier of FBS (Gibco, 10270-106) and triplicates of the RNA sequencing, TrxR activity, GSH content and Western blot protein level validation experiments (Fig. 3A–E) were performed with the same batch of treated cells and growth medium. Cells were grown as monolayers and maintained in exponential growth phase in 5% CO2/95% air in a humidified incubator at 37 °C. A separate batch of the isogenic mutant p53 NCI–H1299 cell lines was stably transduced with the Incucyte® NucLight Red Lentivirus reagent with a puromycin gene to allow cell tracking on the IncuCyte ZOOM system (Sartorius). All cell lines were confirmed as mycoplasma-free using the MycoAlert Mycoplasma Detection Kit.
Fig. 3.

Effect of AF on mRNA and protein targets related to antioxidant mechanisms, ferroptosis and DNA damage. (A) TrxR activity in μMol/Minute/mgProtein after treatment of the isogenic NCI–H1299 cell lines with a cytostatic (1 μM) and cytotoxic (5 μM) AF concentration for six and 24 hours. (B) Relative GSH protein content after treatment of the isogenic NCI–H1299 cells with a cytostatic (1 μM) and cytotoxic (5 μM) AF concentration for six and 24 hours, relative to untreated. Error bars represent the standard deviation. *p ≤ 0.05 significant differences compared with untreated control. (C) Ingenuity pathway analysis of affected pathways after treatment with 1 μM AF for 24 hours in the isogenic NCI–H1299 cell lines. Color key legend represents -log (p-value). (D) Differentially expressed genes after AF treatment, relative to untreated, in the isogenic NCI–H1299 cell lines. Color key legend represents expression log2 ratio (red = upregulation; green = downregulation). (E) Western blot was used to determine the protein levels of NRF2, HMOX1, GPX4, SOD1 and Trx in the isogenic NCI–H1299 cells after treatment with 1 μM AF for 24 hours of two independent replicates (REP1 and REP2). -actin was used as internal control. Molecular weights of the proteins are represented in kilodalton (kDa). (F–G) Box plot representing H2AX spot occupancy (F) and pRPA32 nuclear intensity (G) calculated using an algorithm on Image J software, in the isogenic NCI–H1299 cell lines treated with PBS, 1 μM and 2 μM AF for 24 hours. Representative images are presented in Supplementary Fig. 8. Box boundaries represent the upper and lower quartiles and the black line within represents the median. The whiskers display the 95% confidence interval, while the dots outside the whiskers represent outliers. Experiments were performed at least in triplicate. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
2.2. NSCLC patient cohort and tissue specimens
The study cohort consisted of 72 patients diagnosed with stage I-IV NSCLC adenocarcinoma. Tissue samples from tumor resections were obtained from the Antwerp University Hospital Tumorbank as formalin fixed paraffin embedded (FFPE) specimens. Tissue samples were prepared as described by Deben et al. [32,33]. The patients’ main clinical and pathological parameters are summarized in Suppl. Table 3. The study was approved by the Ethics committee of the Antwerp University Hospital (reference number 17/30/339).
2.3. Immunohistochemistry
Five m-thick sections were prepared from FFPE specimens. Sections were subjected to heat-induced antigen retrieval by incubation in a low pH buffer (anti-TrxR) and a high pH buffer (anti-p53) for 20 minutes at 97 °C (PT-Link) (DAKO). Subsequently, endogenous peroxidase activity was quenched by incubating the slides in peroxidase blocking buffer (DAKO) for 5 minutes. Incubation with primary monoclonal antibodies anti-p53 (ready to use for 30 minutes) and anti-TrxR (1:50 for 40 minutes) was performed on a DAKO autostainer Link 48 instrument. For anti-TrxR and anti-p53, the Envision FLEX + detection kit was used according to the instructions of the manufacturer. Sections were counterstained with hematoxylin, dehydrated and mounted. Positive controls were included in each staining run and consisted of tonsil tissue (anti-p53 and anti-TrxR) and A549 cells (anti-TrxR). Scoring was performed by two independent observers, including an experienced pathologist. To account for heterogeneity of TrxR, an immunoreactivity scoring (IRS) system was applied. Staining intensity was designated as either no staining (0), weak (+), moderate (++) and strong staining (+++) and the percentage positive tumor cells were scored 1 to 4 (<10%, 10–50%, 51–80% and >80% of tumor cells stained). Afterwards, the overall IRS was calculated by multiplication of these two parameters. Cases were grouped in four categories as TrxR negative (0, IRS 0), mild (1, IRS 1–3), moderate (2, IRS 4–7) and strong (3, IRS 8–12) positive (Fig. 1C). p53 protein accumulation was scored as null, wild type or aberrant overexpression (Suppl. Fig. 5).
Fig. 1.

Expression of Trx system in NSCLC patients. (A) Expression of the thioredoxin pathway genes, thioredoxin (TXN), thioredoxin reductase 1–2 (TXNRD1-2) and peroxiredoxin 1-6 (PRDX1-6) represented as fragments per kilobase million (FPKM). FPKM values were extracted from the LUAD cohort of the cancer genome atlas (TCGA) RNA-seq dataset using the Human Protein Atlas. (B) Kaplan-Meier survival curves showing the correlation of TXN, TXNRD1-2 and PRDX4 and 6 gene expression with the survival of LUAD patients. Curves were plotted based on the best expression cut-off, i.e. the FPKM value that yields maximal difference with regard to survival using the Human Protein Atlas. Number of LUAD patients with low or high gene expression were included between brackets. (C) Representative sections of NSCLC tumor samples classified as negative (IRS0), weak (IRS1), moderate (IRS2) and strong (IRS3) TrxR protein expression using immunohistochemistry staining. (D) Pie chart representing the percentage of NSCLC patients with IRS0 to IRS3 for TrxR protein expression. (E) Kaplan-Meier survival curves for overall survival (OS) and progression-free survival (PFS) based on IRS of TrxR protein. Crosses signify censored events where a patient's life ended. Significance was reached when p < 0.05 determined by log rank. IRS: immunoreactivity scoring.
2.4. TCGA database
Fragment per kilobase million (FPKM) values were extracted from 500 lung adenocarcinoma (LUAD-cohort) patients of TCGA RNA-seq dataset using the Human Protein Atlas [34,35]. Kaplan-Meier survival curves were plotted based on the best expression cut-off, i.e. the FPKM value that yields maximal difference with regard to survival using the Human Protein Atlas. A cut-off of a log-rank p-value < 0.01 was used.
2.5. Sulforhodamine B cytotoxicity assay
A colometric sulforhodamine B (SRB) assay was used to measure the treatment-induced cytotoxicity of AF. Optimal seeding density of each cell line for each plate type was determined in order to ensure exponential growth during the whole duration of the assay. The different cell lines were seeded in 96-well plates, incubated overnight and exposed to 0–10 μM AF for 72 hours. Cell monolayers were then fixed with 10% trichloroacetic acid for 1 hour at 4 °C and stained with 100 L 0.1% SRB, as previously described [36]. The IC50 value (i.e. drug concentrations causing 50% growth inhibition) for every cell line was calculated using WinNolin® Software (Pharsight).
2.6. Western blotting
Cells were seeded at 2.3 x 106 cells in T175 flasks. The next day, cells were treated with 1 μM AF, 5 μM AF or 0.06% DMSO for 24 hours. This batch of cells was used in different set-ups throughout this study. For the protein based experiments, cells were lysed in lysis buffer (10 mM TrisHCl, 400 mM NaCl, 1 mM EDTA, 0.1% NP40 and protease inhibitor). After centrifugation (10 min, 13 000 rpm, 4 °C), cleared lysates containing the isolated proteins were harvested and kept at −80 °C. Protein concentrations were determined using the Pierce BCA protein kit, according to the manufacturer's instructions. To determine baseline protein levels, cells were collected after subculturing and lysed as described above. Western blot was performed as described previously [37]. Blocking, primary and secondary antibody incubation were performed using the SNAP id 2.0 protein detection system, according to the manufacturer's instructions. Membranes were incubated with following antibodies: anti-Nuclear factor erythroid 2-related factor 2 (NRF2, 1:2000), anti-p53 (1:1000), anti-glutathione peroxidase 4 (GPX4, 1:2000), anti-solute carrier family 7 member 11 (SLC7A11, 1:1000), anti-TrxR1 (1:2000), anti-heme oxygenase-1 (HMOX1, 1:1000), anti-Trx (1:2000), anti-oxidative stress cocktail (catalase, superoxide dismutase 1 (SOD1), Trx and smooth muscle actin, 1:350) and anti-β-actin (1:2500, internal control). Goat anti-rabbit (1:10 000) or goat anti-mouse (1:10 000) fluorescently labeled secondary antibodies were used. Fluorescent detection was performed using the Odyssey imaging system (Li-Cor). Image Studio Lite software (Li-Cor) was used to perform pixel quantification of the images. Intra-run normalization against the internal actin control was performed for each sample. Inter-replicate normalization for each protein of interest was performed based on the average actin normalized signal for each blot.
2.7. ROS
Cells were seeded in 96-well plates, incubated overnight and exposed to 2 μM AF or vehicle (phosphate buffer saline, PBS). Immediately after treatment, 2.5 μM CellROX Green reagent was added to the cells. Afterwards, the plate was transferred to the temperature- and CO2-controlled IncuCyte ZOOM (10X magnification, Essen BioScience). ROS was monitored over time by pictures that were taken every four hours to limit phototoxicity. For analysis, average green calibrated unit (GCU) was plotted for every cell line after 16 hours for at least three repeats.
2.8. Thioredoxin reductase activity assay
The batch of vehicle- and AF-treated protein lysates was used to measure TrxR activity using the Thioredoxin Reductase Colorimetric Assay Kit, according to the manufacturer's protocols. Absorbance was recorded at 405 nm with an iMARK® microplate absorbance reader (Bio-Rad) during the initial 15 minutes of the reaction. TrxR activity was calculated using the formula provided by the protocol, whereby background measurements were subtracted from all values. An equal amount of protein was loaded for each condition as determined by the Pierce BCA protein kit.
2.9. Glutathione level quantification
The batch of vehicle- and AF-treated cells was used to determine cellular concentrations of glutathione (GSH), quantified using the GSH/GSSG-Glo™ Assay kit, according to the manufacturer's protocols. Therefore, an equal amount of cells (7500 cells/well) was added to a white 96-well plate in the presence of GSH lysis reagent. Luminescent intensity was measured using a VICTOR™ plate reader (PerkinElmer), which was proportional to the amount of GSH.
2.10. RNA isolation
The same batch of vehicle- and AF-treated cells was collected and pelleted to isolate total RNA using the RNA Plus Mini Kit, according to the manufacturer's instructions. RNA concentration was checked using the Qubit RNA BR Assay Kit on Qubit 4 Fluorometer (ThermoFisher). The quality of the RNA samples was determined using the Fragment Analyzer Automated CE System (Agilent, DNF-471 Standard Sensitivity RNA Analysis Kit) with the High Sensitivity NGS Fragment Analysis Kit. No samples had to be excluded based on low quantity or quality.
2.11. 3’ mRNA sequencing and data processing
RNA samples were amplified using the QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina, following the manufacturer's protocol for long fragments. The PCR Add-On kit for Illumina (Lexogen GmbH, 020.96) was incorporated in order to determine the optimal number of PCR cycles. Following quantitative (Qubit dsDNA HS Assay Kit) and qualitative analysis, the resulting cDNA libraries were equimolarly pooled. Sequencing was performed on a NextSeq 500 (Illumina) using a NextSeq 500/550 High Output Kit v2.5 (150 cycles). RNA sequencing analysis was initiated using the BlueBee platform (www.bluebee.com). First, the four FASTQ files per samples were merged using the pipeline “Bluebee FASTQ merging (input: files) 1.2.0”. Next, the merged FASTQ files were trimmed, the reads were mapped to the human reference genome build 38 and a quality control was performed using the pipeline “FWD Human (GRCh38.77) Lexogen QuantSeq 2.2.3”. Next, differential expression analysis was carried out in R with the DESeq2 v1.28.1 package [38]. Transcriptome profiles were functionally analyzed using ingenuity pathway analysis (IPA) Core Analysis [39]. A fold change cut-off of 0.5 and p-value cut-off of 0.05 was used for the three cell lines. Next, Core Analysis of each cell line were subjected to Comparison Analysis based on -log(p-value) and activation z-score. Expression log ratio values were extracted from affected genes in the top-ranked pathways and presented in a heatmap. Gene Set Enrichment Analysis (GSEA) was performed on full normalized RNA sequencing data using the GSEA software package [40], developed by the Broad Institute (Maltham). GSEA was performed with the computationally generated gene set database representing genes involved in epithelial-mesenchymal transition (EMT, standard name: Hallmark_epithelial_mesenchymal_transition, systematic name: M5930, Molecular Signature Database v7.1 database) [41]. Gene sets with a false discovery rate (FDR) 0.05 and p-value 0.05 were considered.
2.12. Immunocytochemistry
Cells were seeded in a black μClear 96-well plate, and afterwards treated with 1 μM and 2 μM AF for 24 hours. Cultures were fixed in 2% paraformaldehyde (PFA) for 20 minutes at room temperature (RT). Fixed cultures were permeabilized with 0.3% Triton X-100 in PBS (0.1 M, pH 7.4) for 8 minutes, followed by one hour incubation with the primary antibodies against replication stress phospho-RPA32 (pRPA32, 1:1000), DNA damage gamma-H2AX (γH2AX, 1:1000) and p53 (7F5, 1:1600) at RT in 50% FBS in PBS. After washing with PBS, secondary antibodies GAR-Cy3 (1:400), GAM-488 (1:1000), Alexa Fluor 488 (1:400) were added for one hour at RT to the respective primary antibodies. Finally, DAPI was applied to the cultures for 20 minutes at a concentration of 5 μg/mL. Images were acquired with a Nikon Ti fluorescence microscope using a 20x dry lens (numerical aperture 0.75). Per well, 16 frames were acquired in three channels (395, 470 and 555 mm excitation). Image processing was performed in Fiji, a packaged version of ImageJ freeware [42]. The number of pRPA32 and γH2AX spots was quantified using an image analysis pipeline that has been described before (Cellblocks.ijm) [43]. In brief, the analysis detects regions of interest (ROIs) corresponding to the complete nuclei in the field of view and subsequently detects nuclear spots per marker of interest and measures their intensity and morphological features. Quantification of p53 was based on the measurement of signal intensities within the nuclear ROIs in the maker channel. Downstream data analysis was performed in R studio.
2.13. Analysis of cell death pathways
NucLight Red transduced isogenic NCI–H1299 cell panel was seeded in a 96-well plate. The next day, cells were preincubated with the desired cell death pathway inhibitors for 1 hour (5 mM n-acetyl-cysteine (NAC), 1 μM ferrostatin-1 (Fer1), 100 μM alpha-tocopherol (Toco), 10 μM Z-VAD-FMK and 10 μM necrostatin-s1 (Nec1s)) or 4 hours (100 μM Deferoxamine, DFO) and then treated with 2 μM AF in the presence of the IncuCyte Cytotox Green Reagent (50 nM) or Caspase-3/7 Green Apoptosis Reagent (2.5 μM). Afterwards, the plate was transferred to the temperature- and CO2-controlled IncuCyte ZOOM for 72 hours. Cell death was monitored by pictures (10X magnification) that were taken every 24 hours to limit phototoxicity. For analysis, green object count (1/mm2), red object count (1/mm2), green-red overlapping object count (1/mm2) and Caspase-3/7 Green GCU x μm2/image were determined with the IncuCyte basic analyzer. The percentage of cell death and Caspase-3/7 positive cells was calculated with the formula: .
2.14. Lipid peroxidation
Cellular lipid ROS was measured using the Image-iT™ Lipid Peroxidation Kit, according to the manufacturer's instructions. Therefore, isogenic NCI–H1299 cell panel were treated with 2 μM AF for 48 hours or the positive control (cumene hydroperoxide) for 2 hours. Afterwards, 10 μM of the C11-BODIPY dye was added to the culture and incubated for 30 minutes at 37 °C. Acquisition was performed on a CytoFLEX (BD) and FlowJo v10.1 software (TreeStar) was used to calculate the ratios C11-BODIPY red over green mean fluorescence intensity (MFI) signals.
2.15. Analysis of ICD-related DAMPs
Isogenic NCI–H1299 cells were seeded and treated with 2 μM AF, but analysis of ICD-related markers occurred at different time points. (i) After 24 hours of treatment in cell culture media supplemented with heat inactivated FBS, supernatant was collected. On the freshly collected supernatant, extracellular ATP levels (nmol) were assessed with the ENLITEN® ATP assay system, according to the manufacturer's protocol. The bioluminescent signal was measured using a VICTOR™ plate reader (PerkinElmer). (ii) After 48 hours of treatment, cells were harvested and incubated with 5% normal goat serum. After washing, cells were stained for ecto-calreticulin (CALR) expression using a AF488-conugated anti-human CALR antibody (1:100). Prior to analysis, cells were stained with Annexin V APC and propidium iodide (PI) to distinguish between early apoptotic and necrotic cells. Cell debris and necrotic cells (PI+) were excluded from analysis. Isotype control (rabbit IgG, 1:100) was included to correct for aspecific staining. Flow cytometric data was acquired on a BD Accuri™ C6 instrument (BD Biosciences) and analyzed using FlowJo v10.1 software (TreeStar). (iii) To determine the release of high mobility group box 1 (HMGB1, ng/mL), supernatant was collected after 48 hours of AF treatment, centrifuged and stored at −80 °C. An enzyme-linked immunosorbent (ELISA) assay was performed, according to the manufacturer's protocol, and absorption was measured using an iMARK™ plate reader (Bio-Rad).
2.16. In vitro generation of human monocyte-derived DCs
Human peripheral blood mononuclear cells (PBMC) were isolated by LymphoPrep gradient separation (Sanbio) out of buffy coats from healthy donors (Red Cross Flanders Blood Service, Belgium). Then, monocytes were isolated from these PBMCs using CD14 microbeads, according to manufacturer's protocol with purity >90% after isolation. CD14+ cells were plated at a density of 1.25–1.35 x 106 cells/mL in RPMI supplemented with 2.5% human albumin, 800 U/mL granulocyte-macrophage colony stimulating factor and 50 ng/mL interleukin (IL)-4 at day 0, as previously described [44]. Immature DCs were harvested on day five.
2.17. Maturation status of DCs
After in vitro generation of DCs, isogenic NCI–H1299 cells were seeded in 6-well plates on day four and incubated overnight. On day five, tumor cells were treated with 2 μM of AF for six hours. Afterwards, immature DCs were labeled with 2 μM of cytoplasmic violet-fluorescent CellTracker Violet BMQC dye at a concentration of 106 cells per mL and placed in co-culture at an effector (E) to target (T) ratio of 1:1 (E:T). After 48 hours in co-culture, supernatant was stored at −20 °C for future analysis and DCs were immediately used for flowcytometric detection of DC maturation markers on day seven. Binding of anti-CD86 PECy7 (1:25) and anti-major histocompatibility complexes class II (MHC-II) FITC (1:50) was measured on the violet + viable (Live/Dead Near IR+, 1:50) DC population. For every marker, an isotype control was used to subtract aspecific signals. Results are represented as and as the percentage of DCs double positive for MHC-II and CD86. Acquisition was performed on a FACSAria II (BD Biosciences) and data analysis was performed using FlowJo v10.1 software (TreeStar).
2.18. Cytokine and chemokine secretion profile
A multiplex electrochemiluminescent assay on a SECTOR3000 (MesoScale Discovery) was used for the detection of IL-6, tumor necrosis factor (TNF)-α and transforming growth factor (TGF)-β, as previously described [45]. Standards and samples (supernatant collected from NCI–H1299:DC co-cultures) were measured in duplicate and the assay was performed according to the manufacturer's protocol. Data analysis was performed using the MSD Discovery workbench 4.0 software.
2.19. NK cell isolation and stimulation
Cryopreserved PBMCs were thawed and incubated overnight in complete RPMI medium with 5 μL DNase. Subsequently, primary NK cells were negatively isolated from PBMCs using a NK isolation kit for negative selection, according to the manufacturer's instructions. Purity of the NK cells was measured using anti-CD3 FITC (1:20) and anti-CD56 PE (1:20) and was above 90%. NK cells were split in two equal portions: one to stimulate with 10 ng/mL recombinant human IL-15, while the other portion was left unstimulated. Both conditions were incubated overnight at 37 °C and 5% CO2.
2.20. NK cell-mediated cytotoxicity assay
NucLight Red transduced isogenic NCI–H1299 cells were seeded in a 96-well plate. After overnight incubation, appropriate conditions were preincubated with 50 μM Z-VAD-FMK for 1 hour and then treated with a non-toxic dose of 0.55 μM AF in the presence of the IncuCyte Cytotox Green reagent (50 nM) or Caspase-3/7 Green Apoptosis Reagent (2.5 μM). After 24 hours, the AF primed target cells were co-cultured with (un)stimulated effector NK cells at a E:T ratio of 5:1. Tumor cells incubated without NK cells served as controls. The plate was then transferred to the temperature- and CO2- controlled IncuCyte ZOOM for 48 hours. Cell death was monitored by pictures that were taken every 24 hours to limit phototoxicity. For analysis, green object count (1/mm2), red object count (1/mm2), green-red overlapping object count (1/mm2) and Caspase-3/7 Green GCU x μm2/image were determined with the IncuCyte ZOOM analyzer. NK cells were filtered out based on size. The percentage of tumor cell death was calculated with the formula: .
2.21. GZMA dependency and NK cell activation
NCI–H1299 cells were seeded in 24-well plates, incubated overnight and treated with 0.55 μM AF. The NK cells were isolated and stimulated as described before. After 24 hours of treatment, AF primed tumor cells were co-cultured with the (un)stimulated NK cells at a 5:1 E:T ratio. Anti-CD107a-FITC antibodies (1:50) were added after 30 minutes of co-culture. Monensin (1:1000) was added after an additional 30 minutes and co-culture continued for an additional 4 hours to enhance intracellular cytokine staining. After washing, cells were stained with the following antibodies: anti-CD45 APC-Cy7 (1:50), anti-CD3 PE-Cy7 (1:100), anti-CD56 PE-CF594 (1:25), anti-CD69 PerCP-Cy5.5 (1:100) and Live/Dead Fixable Aqua (1:50) for 30 minutes at 4 °C. Afterwards, co-cultures were fixed, permeabilized and stained for intracellular cytokines using anti-GZMA PE (1:25) and anti-interferon γ (IFNγ) APC (1:12.5) using the FOXP3 Fix/Perm Buffer Set, according to the manufacturer's instructions. Acquisition was performed on a FACSAria II (BD BioSciences). The percentage of NK cells positive for a given marker was calculated using the overton histogram subtraction tool in FlowJo v10.1 software (TreeStar), subtracting the control histogram (e.g. NK cells) from the sample histogram (e.g. AF + NK cells in co-culture).
2.22. Statistics
All experiments were performed at least in triplicate. All statistical computations were conducted using SPSS version 26, unless otherwise stated, and significance for all statistics was reached if p ≤ 0.05. Prism 8.2.1 software (GraphPad) was used for data comparison and graphical data representations. The associations between clinicopathological parameters and TrxR or Trx protein expression levels were investigated by 2 analysis for categorical variables. Differences in overall survival (OS) and progression-free survival (PFS) between patient groups were determined by Kaplan-Meier analysis. A Multivariate Cox Proportional Hazard model was fitted to identify independent prognostic markers, presented as a hazard ratio (HR) and its 95% confidence interval (CI). The non-parametric Mann-Whitney U test was used to compare means between two groups (untreated vs. AF-treated). To compare means between different treatment groups (e.g. NK cell-mediated killing), statistical significance was determined by a one-way ANOVA, followed by Tukey's post hoc test. Spearman correlation coefficients were calculated to investigate the correlation between the different antioxidant related proteins, AF IC50 values and p53 protein levels in different NSCLC and PDAC cell lines. Analysis of H2AX and pRPA32 spots was performed using linear mixed models, after which significance was determined using multiple comparisons of means with Tukey contrasts.
3. Results
3.1. TrxR as a potential druggable target for NSCLC
Since the Trx system is involved in many cancers, we determined the gene expression of the Trx antioxidant pathway members in the lung adenocarcinoma (LUAD)-cohort using the publicly available TCGA database. Data analysis showed that nearly all members of the Trx system were present in LUAD patients at the mRNA level (Fig. 1A). In addition, high expression of TXN, TXNRD1, peroxiredoxin 4 (PRDX4) and PRDX6 genes was correlated with the outcome of the LUAD patients (Fig. 1B).
At the protein level, immunohistochemical (IHC) analysis was performed to evaluate TrxR expression in 72 NSCLC adenocarcinoma patients (Fig. 1C–E, Suppl. Table 4-5). TrxR protein was detected in 95.8% of the NSCLC tissues, with varying expression in all stages of our patient population (Fig. 1D, Suppl. Table 4). In the non-neoplastic lung tissue, TrxR was detected in cylindrical epithelium, lymphocytes and stromal cells (Suppl. Table 4). No significant association was found between TrxR and any clinicopathological feature (Suppl. Table 5), and univariate survival analysis using Kaplan-Meier showed that TrxR was no significant predictor of worse OS or PFS (Fig. 1E). Overall, TrxR was expressed in NSCLC patients both at the mRNA and protein level suggesting that the Trx/TrxR system represents a valid druggable target in NSCLC for AF.
3.2. Mutant p53 accumulation sensitizes NSCLC cell lines to AF treatment
The cytotoxic effect of AF was investigated in a panel of eight NSCLC and PDAC cells with a different p53 background. Our main focus was on NSCLC adenocarcinoma, but we included PDAC cell lines to determine if AF can be used as a tumor-agnostic therapy with a broader application radius. Dose-response survival curves and their corresponding IC50 values showed variable sensitivity to AF with the wild type (WT) p53 cell line A549 being the least sensitive one (Fig. 2A and B). The strong variability in response between the other cell lines, indicated that TP53 mutation status alone cannot explain sensitivity to AF. When analyzing p53 protein levels, we found an inverse correlation with AF IC50 values in NSCLC and PDAC cells (r = −0.925; p = 0.001) (Fig. 2C–E, Suppl. Fig 1 and 2). This suggests that high levels of mutant p53 protein sensitize cancer cells to AF.
Fig. 2.

Cytotoxic response to AF in NSCLC and PDAC cell lines and their p53 expression levels. (A) Dose-response survival curves after 72 hours of AF treatment (0–8 μM) in eight NSCLC and PDAC cell lines. (B) IC50 values (μM) after 72 hours of AF treatment in eight NSCLC and PDAC cell lines with different p53 backgrounds. (C) Spearman's r correlation between baseline p53 protein expression (determined by Western blotting) and AF IC50 values (μM). (D) Western blots representing the baseline protein expression of NRF2, catalase, TrxR1, p53, SLC7A11, GPX4, SOD1 and Trx in eight NSCLC and PDAC cell lines. -actin was used as internal control. Molecular weights of the proteins are represented in kilodalton (kDa). Quantification of triplicates is presented in Supplementary Fig. 2. (E) Spearman's r correlation coefficient between baseline mutant p53 protein and AF IC50 values, and baseline expression of TrxR1, Trx, GPX4, SLC7A11, NRF2, SOD1, catalase (determined by Western blotting), GSH (determined by luminescence) and ROS (determined by IncuCyte ZOOM analysis) in eight NSCLC and PDAC cell lines. (F) p53 protein expression in the isogenic mutant p53 R175H and R273H cells (determined by Western blotting). (G) IC50 values (μM) after 72 hours of AF treatment in the isogenic NCI–H1299 cell panel (determined by SRB assay). (H) Fold change of Trx protein expression, relative to NCI–H1299 Null control cells (determined by Western blotting using two different anti-Trx antibodies, red dots), in the isogenic NCI–H1299 cell line panel. Experiments were performed at least in triplicate. Error bars represent the standard deviation. *p ≤ 0.05 significant differences. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Since inhibition of TrxR modulates the intracellular redox state due to increased ROS levels [46,47], we sought to investigate potential antioxidant related biomarkers for AF treatment. The identification of a predictive biomarker for therapy response can greatly improve patient stratification and clinical response. Therefore, data extracted from the TCGA database showed the expression of different antioxidant related genes in 500 LUAD patients (Suppl. Fig. 3). In addition, the baseline levels of these antioxidant related proteins were determined and correlated to the AF IC50 values of eight NSCLC and PDAC cell lines (Fig. 2D and E, Suppl. Fig. 1 and 2). High levels of NRF2 positively correlated with higher AF IC50 values (r = −0.738; p = 0.046), while no significant correlation was observed for Trx (r = 0.643; p = 0.096) and SLC7A11 (r = 0.643; p = 0.096) (Fig. 2E, Suppl. Fig. 1). Additionally, no significant correlation was found between mutant p53 expression levels and expression of the other antioxidant related proteins (Fig. 2D and E, Suppl. Fig. 1).
In order to validate these findings, we used the isogenic mutant p53 NSCLC NCI–H1299 cell panel (Fig. 2F–H, Suppl. Fig. 4). The p53 R273H mutant showed significantly higher levels of mutant p53 protein compared to R175H cells (Fig. 2F, Suppl. Fig. 4A and B) and was the most sensitive to AF (Fig. 2G, Suppl. Fig. 4C). Compared to R273H mutant, the R175H mutant contained medium levels of mutant p53 protein and had subsequently a higher AF IC50 value (Fig. 2F and G, Suppl. Fig. 4A and B). In relation to the antioxidant related proteins, we found that Trx protein levels were significantly lower in mutant p53 expressing cells (Fig. 2H). These results contradict the findings of Lisek et al., and therefore we validated these findings with an additional anti-Trx antibody (Fig. 2H, marked in red) [48]. Similarly, the two isogenic mutant p53 cell lines had lower SLC7A11 protein levels (Suppl. Fig. 4D) consistent with the findings of Liu et al. [24]. GPX4 levels were significantly higher in the R273H cell line compared to p53 Null and p53 R175H cells (Suppl. Fig. 4D).
Interestingly, in our NSCLC patient population, not all TP53 mutant patients showed aberrant overexpression of the mutant p53 protein, indicating that expression level is not a valid marker to distinguish WT from mutant p53 tumors (Suppl. Fig. 5–6; Suppl. Table 6).
Overall, we identified that the expression levels of p53 protein in NSCLC were determinant for AF treatment sensitivity in vitro.
3.3. AF treatment rewires the transcriptome of NSCLC cells towards antioxidant defense, DNA damage repair and ferroptosis induction
To validate AF's action as TrxR inhibitor and oxidative stress mediator, we examined its dose-dependent effects on the TrxR activity and GSH content using cytostatic (1 μM) and cytotoxic (5 μM) concentrations. TrxR activity was fully depleted with both concentrations of AF already after six hours of incubation (Fig. 3A). A six hour treatment with 1 μM AF did not alter GSH content significantly, but a prolonged treatment time of 24 hours resulted in a significant increase in GSH. This most likely represents a counter mechanism to balance the perturbed redox status (Fig. 3B). At 5 μM, AF significantly lowered intracellular GSH protein after six hours and no viable cells were present after 24 hours (Fig. 3B). No differences were observed based on p53 protein levels.
To further elucidate the mode of action of AF in mutant p53 NSCLC, we performed a shotgun transcriptome analysis in the isogenic NCI–H1299 cell line panel following 24 hours treatment with 1 μM (cytostatic) AF. Pathway analysis (IPA) revealed the NRF2-mediated Oxidative Stress Response as the top signaling pathway affected by AF in all three cell lines, independent of p53 protein levels (Threshold of -log(p-value) > 1.3, Fig. 3C), with HMOX1 being the strongest differentially expressed gene (Fig. 3D). AF-mediated upregulation of HMOX1, NRF2 and two other antioxidant related genes TXN and SOD1 were confirmed at the protein level using Western blotting (Fig. 3E). Consistent with the cytostatic concentration of 1 μM, G2/M DNA Damage Checkpoint Regulation was the second most affected pathway (Fig. 3C and D).
In line with the increased GSH content (Fig. 3B), several GSH biosynthesis related genes (SLC7A11, GCLM and GCLC) were upregulated (Fig. 3D). Interestingly, the GPX4 gene was the only gene of the GSH pathway significantly downregulated in all three cell lines after AF treatment (Fig. 3D), which was confirmed at the protein level (Fig. 3E). Decreased activity of GPX4 is one of the key features of ferroptosis and closely related to the upregulation of the HMOX1, FTH1 and FTL genes, since they are all linked to the iron homeostasis signaling and ferroptotic pathways.
Finally, IPA analysis revealed that the Granzyme A (GZMA) signaling pathway was highly affected by AF treatment with the strongest effect in the p53 R175H mutant cells (Fig. 3C and D). GZMA is the most abundant serine protease in the cytotoxic granules of cytotoxic T lymphocytes and NK cells [49]. Several key targets of GZMA were significantly downregulated in the presence of mutant p53 R175H cells, including histones H1 (H1), High-mobility group protein B2 (HMGB2) and Acidic Nuclear Phosphoprotein 32 Family Member A (ANP32A) genes (Fig. 3D, Suppl. Fig. 7). We hypothesize that AF treatment could enhance the susceptibility of mutant p53 NSCLC cells towards NK cell-mediated killing, operated via the release of cytotoxic granules containing GZMA.
To discover similarities in the mode of action of AF and other drugs, the transcriptional signature of (cytostatic) AF treatment was compared to The Library of Integrated Network-Based Cellular Signatures (LINCS) chemical perturbagen signature database. We found a drug activity that is closely related to NF-kB, proteasome, AKT, JAK/STAT and histone deacetylase (HDAC) inhibitors and the ferroptosis inducer withaferin A (Suppl. Table 7).
Since inhibition of the Trx and GSH system (Fig. 3A and B) results in a perturbation of the redox balance and increased ROS levels (Fig. 4D), we next investigated whether the isogenic NCI–H1299 cell line panel was more prone to experience DNA damage upon treatment with AF. A dose-dependent induction of replication stress (pRPA32) and overall DNA damage (H2AX) was observed in all three cell lines (Fig. 3F and G, Suppl. Fig. 8). We found a clear p53-dependent difference since accumulation of H2AX was significantly higher in the AF treated mutant p53 R175H cells compared to Null and R273H cells (p < 0.001) (Fig. 3F, Suppl. Fig. 8).
Fig. 4.

Induction of apoptotic and ferroptotic cell death after AF treatment of mutant p53 NSCLC cells. (A–C) Percentage of cell death after treatment with 2 μM AF for 72 hours in the absence or presence of apoptosis, necroptosis and ferroptosis inhibitors and ROS scavenger NAC in the isogenic p53 Null (A), p53 R175H (B) and p53 R273H mutated NCI–H1299 cells (C). (D) Fold change of the CellROX Green Calibration Units (GCU), relative to untreated, of the three isogenic NCI–H1299 cell lines after treatment with 2 μM AF for 16 h. (E–F) Percentage of Caspase-3/7 green positive cells (E) and Caspase-3/7 total green objected integrated intensity (GCU x μm2/image) (F) in the isogenic NCI–H1299 cell lines after treatment with 2 μM AF for 72 hours. Error bars represent the standard error of the mean. (G) Overlay histograms of C11 BODIPY Green 581/591 signal after treatment of the isogenic NCI-H1299 cell lines with PBS, 2 μM AF (48 hours) or cumene hydroperoxide (positive control, 2 hours). (H) Relative ratio of red over green mean fluorescent intensity (MFI) signal of the C11-BODIPY 581/591 reagent after treatment with 2 μM AF (48 hours) or cumene hydroperoxide (positive control, 2 hours) of the isogenic NCI–H1299 cell lines. Error bars represent the standard deviation. Experiments were performed at least in triplicate. *p ≤ 0.05 significant differences compared to untreated control. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.4. AF kills mutant p53 NSCLC cells via distinct cell death pathways
DNA damage is a well-known trigger of cell death and transcriptome analysis suggested the involvement of ferroptotic pathways. To identify the molecular determinants of AF-induced cell death, we made use of a set of specific cell death pathway inhibitors. In the absence of inhibitors, the sensitizing effect of mutant p53 overexpression to AF-induced cell death was recapitulated as evidenced by the more than 2-fold increase in cell death for the p53 R175H and p53 R273H cell lines compared to the p53 Null cells (Fig. 4A–C, Suppl. Fig. 9). Co-treatment with the ROS scavenger NAC completely protected all cell lines for AF-induced cell death, supporting the driving influence of ROS in cell death induction (Fig. 4A–C). Indeed, quantitative analysis of the intracellular ROS levels by means of the fluorescent sensor dye CellROX green, revealed that all cells showed increased levels of ROS after AF treatment (Fig. 4D). Interestingly, cell death was significantly reduced by the pan-caspase inhibitor Z-VAD-FMK only in p53 R175H cells, while inhibitors of ferroptosis (Fer1, DFO, toco) partially prevented cell death in p53 R273H cells (Fig. 4B and C). The significantly higher induction of Caspase 3/7 activity in the R175H cell line (Fig. 4E and F) and the characteristic induction of lipid peroxidation in the R273H cell line (Fig. 4G and H) validates that both cell lines die via distinct pathways, namely apoptosis for the R175H cell line and ferroptosis for the R273H cell line.
Based on Principal Component Analysis of our RNA-seq data, the mutant p53 R175H cells and p53 Null cells clustered separately from the mutant p53 R273H cells with high degree of variance, reflecting the differences in phenotypic baseline characteristics (Suppl. Fig 10). As previously discussed, R273H cells also expressed significantly higher levels of mutant p53 compared to the R175H cell line (Fig. 2F, Suppl. Fig. 4A and B) which could sensitize the cell for AF treatment according to our hypothesis. However, we do not believe this explains the difference in cell death mechanisms. We hypothesize that the mesenchymal-like state of the R273H cells sensitizes them for ferroptotic cell death, as ferroptosis inducers that can target GPX4 or regulate GSH levels are able to eliminate drug resistance in mesenchymal cancer cells [50]. However, Gene Set Enrichment Analysis (GSEA) of our RNAseq data showed that epithelial-mesenchymal transition (EMT) signature genes were not enriched in the p53 R273H cells over the p53 Null or p53 R175H cells (Suppl. Fig. 11A). In addition, the R273H cells did not display a higher migratory and invasive capacity compared to p53 R175H cells (Suppl. Fig. 11B–D). Yet, Viswanathan et al. recently reported that zinc finger E-box-binding homeobox 1 (ZEB1), a regulator of EMT and cellular lipid metabolism via peroxisome proliferator-activated receptor gamma (PPARG), is causally linked to lipid peroxide vulnerability and GPX4 dependency in high-mesenchymal state cells [51]. Consistent with these findings, GPX4 protein levels were significantly higher in R273H cells (Suppl. Fig. 4D); ZEB1, PPARG, CDH2 (N-cadherin) mRNA expression levels were upregulated compared to R175H and Null cells and CDH1 (E-cadherin) expression was downregulated (Suppl. Fig. 11E). Moreover, the NCI–H1975 cell line harbors the same mutant p53 R273H protein, but is classified as epithelial related to its EGFR mutations [52]. Consistent with our hypothesis, ferroptosis inhibitors failed to rescue NCI–H1975 cells from AF-induced cell death, which acted through caspase-dependent apoptosis (Suppl. Fig. 12A–B).
3.5. AF induces immunogenic cell death in mutant p53 NSCLC cell lines
Depending on the initiating stimulus, cancer cell death can be immunogenic. ICD is characterized by the expression and release of DAMPs by dying tumor cells, which function either as adjuvant or danger signals to activate DCs and initiate an effective antitumor immune response [53]. Hence, we next investigated the immunogenic potential of AF-induced cell death in the isogenic NCI–H1299 cell line panel. A cytotoxic concentration of AF (2 μM) significantly induced canonical DAMPs including surface exposure of ecto-CALR (Fig. 5A, Suppl. Fig. 13), secretion of ATP (Fig. 5B) and release of HMGB1 into the supernatant (Fig. 5C) in all three cell lines. We checked if these DAMPs in turn triggered DC maturation to establish an effective adaptive immune response. Indeed, we observed a significant increase in the mature CD86+/MHC-II+ DC population (Fig. 5D and E, Suppl. Fig. 14) whereby the expression levels of both markers strongly increased (Suppl. Fig. 15) without inducing DC-mediated cytotoxicity towards the NCI–H1299 tumor cells (Suppl. Fig. 16). Since the maturation of DCs is associated with changes in the production of proinflammatory and immunosuppressive cytokines [54], we analyzed the secretion of IL-6, TNF-α as well as TGF-β in co-cultures, respectively. The interaction between DCs and AF-treated isogenic cells significantly decreased the release of TNF-α (Fig. 5F), IL-6 (Fig. 5G) and TGF-β (Fig. 5H), independent of (mutant) p53 expression levels. Collectively, these in vitro results showed that AF treatment induced the release of the most important hallmarks of ICD (ecto-CALR, ATP and HMGB1) and DC maturation (CD86 and MHC-II), independent of (mutant) p53 expression or cell death pathway, and dampened the release of cytokines.
Fig. 5.

AF treatment of mutant p53 NSCLC cells induces the release of ICD markers and DC maturation. (A) Percentage of surface-exposed ecto-calreticulin (ecto-CALR) positive NCI–H1299 Null, p53 R175H and p53 R273H cells after treatment with 2 μM AF for 48 hours. Gating strategy of CALR+ cells represented in Supplementary Fig. 13. (B) Secretion of ATP in the supernatant of the isogenic NCI–H1299 cell lines 24 hours after treatment with 2 μM AF (nM range). (C) HMGB1 secretion induced 48 hours after treatment with 2 μM AF in the isogenic NCI–H1299 cells (ng/mL range). (D) Percentage of MHC-II and CD86 double positive DCs after 48 hours of co-culture with AF-treated H1299 null, p53 R175H and p53 R273H cells (E:T ratio 1:1) using flow cytometry, relative to untreated. Gating strategy represented in Supplementary Fig. 14. Error bars represent the standard deviation. (E) Contour plots of the DC population double positive for MHC-II and CD86 in co-culture with either PBS- or AF-treated mutant p53 R175H cells. (F–H) Fold change of the cytokines TNF-α (F), IL-6 (G) and TGF-β (H) released by DCs in supernatant of co-cultures with PBS- or AF-treated NCI–H1299 cells. Error bars represent the standard error of the mean with every dot representing a different healthy donor. Experiments were performed in triplicate. *p ≤ 0.05 significant differences compared with untreated control. DCs: dendritic cells.
3.6. AF primes mutant p53 NSCLC cells for IL-15-stimulated NK cell-mediated killing
Transcriptome analysis showed that AF downregulated key targets of GZMA (HMGB2, ANP32A and linker histone H1; Fig. 3C and D and Suppl Fig. 7) in p53 R175H and R273H cell lines. These targets play a critical role in GZMA-induced nuclear damage via NK cells [49]. Therefore, we hypothesized that AF treatment could enhance the susceptibility of mutant p53 NSCLC cells towards NK cell-mediated killing, operated via the release of cytotoxic granules containing GZMA. To test this, we co-cultured the isogenic NCI–H1299 cells with IL-15-primed NK cells (10 ng/mL). In this setting, exposure of the NSCLC cells to a non-toxic dose (0.55 μM) of AF significantly increased cell death, especially in p53 R175H cells (Fig. 6A and B). Contrary to Granzyme B, GZMA activates caspase-independent programmed cell death via its unique substrates and mediators [49]. When significantly blocking of caspase-dependent cell death via Z-VAD-FMK (Fig. 6C), NK cell-mediated killing of AF-treated R175H cells was not significantly decreased compared to unblocked conditions (Fig. 6D). These data suggest a potential role for caspase-independent GZMA in the augmented killing of AF-treated NCI–H1299 cells.
Fig. 6.

Effect of AF treatment on co-cultures of mutant p53 NSCLC cells and natural killer cells. (A) Percentage of death NSCLC cells in co-cultures of (IL-15) NK cells and NCI–H1299 Null, p53 R175H and p53 R273H cells exposed to a non-toxic concentration of 0.55 μM AF for 72 hours. Every dot represents NK cells isolated from a different healthy donor added to the co-culture in 5:1 E:T ratio. Error bars represent the standard error of the mean with every dot representing a different healthy donor. *p ≤ 0.01, **p ≤ 0.001. (B) Representative IncuCyte ZOOM images show the number of NucLight Red positive viable p53 R175H cells and Cytotox Green positive dead p53 R175H cells. (C) Caspase-3/7 total green objected integrated intensity (GCU x μm2/image) in co-cultures of (IL-15) NK cells and p53 R175H cells exposed to Pan-caspase inhibitor Z-VAD-FMK (50 μM) and a non-toxic concentration of 0.55 μM AF for 48 hours. *p ≤ 0.05 (D) Percentage of death NSCLC cells in co-cultures of (IL-15) NK cells and p53 R175H cells exposed to Pan-caspase inhibitor Z-VAD-FMK (50 μM) and a non-toxic concentration of 0.55 μM AF for 72 hours. Error bars represent the standard error of the mean with every dot representing a different healthy donor. (E) Overton percentage of CD107a, IFNγ, CD69 and GZMA positive unstimulated NK cells in co-culture with AF-treated NCI–H1299 Null, p53 R175H and p53 R273H cells, relative to unstimulated NK cells in co-culture with PBS-treated cells. (F) Overton percentage of CD107a, IFNγ, CD69 and GZMA positive IL-15 stimulated NK cells in co-culture with AF-treated NCI–H1299 Null, p53 R175H and p53 R273H cells, relative to IL-15 stimulated NK cells in co-culture PBS-treated cells. IL-15 NK cells were stimulated with 10 ng/mL IL-15 overnight. Gating strategy represented in Supplemental Fig. 17. Error bars represent the standard deviation with every dot representing a different healthy donor. NK cells: natural killer cell. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To further unravel the underlying mechanism via which the combination of AF + IL-15 NK cells increased the killing of mutant p53 NSCLC cells, we asked whether AF treatment either stimulated the activation of NK cells or rather made tumor cells more vulnerable to NK cell-mediated killing, corresponding with our GZMA dependent hypothesis (Fig. 6E and F, Suppl. Figs. 17 and 18). IL-15 is a cytokine that is known to increase the proliferation and activation of NK cells [55], as shown by the upregulation of CD69, CD107a and IFNγ expression on NK cells (Suppl. Fig 18A–C). Since the expression of intracellular GZMA was present in the entire NK cell population (Suppl. Fig. 18C), the effect of IL-15 stimulation and AF treatment on this expression was limited (Suppl. Fig. 18A–C). Importantly, AF treatment did not significantly change the expression of any marker on unstimulated (Fig. 6E) or IL-15 stimulated (Fig. 6F) NK cells, indicating that AF has limited effect on the function and activation of NK cells.
To broaden the applicability of AF's induced susceptibility towards NK cell-mediated killing, an additional WT p53 NSCLC cell line (A549) and two mutant p53 PDAC cell lines (MiaPaca and BxPC3) were included (Suppl. Fig. 19). Comparable to p53 Null NSCLC cells, AF treatment did not affect NK cell-mediated killing of the WT p53 A549 cells. On the other hand, exposure of both mutant p53 expressing PDAC cells MiaPaca and BxPC3 to AF increased NK cell-mediated tumor cell killing (Suppl. Fig. 19).
In conclusion, AF primed mutant p53 NSCLC for killing by IL-15 stimulated NK cells, without affecting NK cell activation, degranulation or intracellular GZMA expression.
4. Discussion
The therapeutic efficacy of AF against cancer together with its relative safety profile in patients highlights its potential as an attractive drug for clinical treatment of cancer [9]. This study emphasizes the versatility of this compound to be repurposed as an anticancer therapy in mutant p53 expressing NSCLC, highlighted by five key aspects.
First, our study and others have shown that AF's main mechanism of action, at both cytostatic and cytotoxic concentrations, is the inhibition of the redox enzyme TrxR [56,57]. To confirm the targetability of the Trx system in NSCLC, strong TrxR expression was detected in the majority of our NSCLC patient group at the protein level and in the LUAD TCGA-cohort at the mRNA level. Similarly, Soini et al. reported a significantly increased expression of both Trx and TrxR in 89 NSCLC cases with only 3 and 8 negative cases, respectively [58]. In general, NSCLC has been identified as one of the tumor types with the highest TrxR expression both in vitro and in vivo [16,58,59]. Since TrxR protein was also strongly expressed in all stages of our NSCLC patient population, targeting TrxR/Trx system via AF will have a broad application in the treatment of NSCLC, which was further exploited in this study.
Second, out of ten potential antioxidant predictive biomarker proteins, we are the first to identify mutant p53 abundance rather than status as an important sensitizer for AF treatment in NSCLC and PDAC cell lines due to reduced Trx expression and overall lower expression of several antioxidant proteins in vitro. Our main focus was on NSCLC adenocarcinoma, but we included mutant p53 PDAC cell lines to determine if AF can be used as a tumor-agnostic therapy with a broader application radius.
Trx and TrxR are major proteins taking part in the regulation of many transcription factors, including p53, and thus the redox state of cells [60,61]. The interaction between TrxR and p53 was already exploited by studies using the p53-reactivating compounds RITA or APR-246. These therapies also inhibited TrxR activity in a tumor-dependent and p53-dependent manner, whereby the dual targeting of p53 and TrxR resulted in ROS-induced cytotoxicity [[62], [63], [64]]. In addition, previous results of our group have demonstrated that inactivating TP53 mutations were associated to a shortened time to progression and decreased patient survival time in the NSCLC patient population [32,33]. More than 75% of these mutations result in aberrant accumulation of a full length p53 protein, typically with only a single amino acid substitution. In most cases, these mutant p53 proteins lost their WT tumor suppressor function and acquired an oncogenic GOF, as in the mutant p53 R175H and R273H cells [65,66], which could be therapeutically exploited by AF as shown in this study.
However, our in vitro results contradict with the report of Lisek et al. showing that induction of mutant p53 variants in breast and lung cancer (R175H) cells leads to higher expression of the TXN gene and corresponding Trx protein [48]. To detect Trx protein, researchers of this report used a now discontinued anti-Trx antibody (sc-20146, Santa Cruz Biotechnology). Therefore, we used two Trx antibodies of different companies to observe that isogenic mutant p53 overexpression was negatively correlated with AF IC50 values and Trx levels in vitro.
Using the same isogenic mutant p53 NCI–H1299 setting in vitro, Liu and colleagues demonstrated that mutant p53 accumulation diminished GSH synthesis by blocking NRF2-dependent transcription of SLC7A11, a unit of the cystine/glutamate antiporter system xC- [24]. Contrary, we observed that mutant p53 was no limiting factor in the activation of NRF2 and GSH levels, despite reduced expression of SLC7A11 in the mutant p53 NCI–H1299 cells. It has already been reported that loss of the Trx system upregulates the GSH system as a backup role to prevent cancer cell death [56,67]. Therefore, in an attempt to overcome the toxicity of AF-mediated TrxR inhibition, our data showed that all mutant p53 NSCLC cells first boosted their antioxidant defense capacities by upregulation of pro-survival molecules, such as NRF2 and GSH, to maintain their redox balance. However, in spite of these feedback mechanisms, simultaneous depletion of GSH levels and inhibition of TrxR at a cytotoxic concentration of 5 μM AF further pushed cancer cells to cross the threshold of cell death, independent of their (mutant) p53 expression levels. This is in line with the report of Hatem et al. showing that AF targeted cooperatively the GSH and Trx systems in NSCLC and breast cancer cell lines [68].
Altogether, we showed that AF attenuated mutant p53 GOF effects by killing mutant p53 cells with higher p53 protein levels more efficiently.
Third, we discovered that AF sensitized mutant p53 R175H NSCLC cells for DNA damage, replication stress and Caspase-3/7-dependent apoptosis while p53 R273H cells were more vulnerable to AF-induced ferroptotic cell death due to downregulation of GPX4 and lipid peroxidation.
Findings of multiple reports already linked ferroptosis to p53 signaling, which is reported to depend on p53 mutation status, cell-type and cellular context [[69], [70], [71]]. In addition, Liu and colleagues proposed that accumulation of p53 R175H and R273H protein sensitized lung cancer cells to ferroptosis by transcriptional suppression of SLC7A11 [24]. Ferroptosis is triggered by inactivation of GPX4, which leads to the accumulation of toxic lipid hydroperoxides, iron-mediated lipid peroxidation and cell death [70]. To our knowledge, we are the first to demonstrate that AF decreases the expression of both GPX4 mRNA and GPX4 protein levels in an isogenic mutant p53 NSCLC setting. However, only AF-mediated cell death of the R273H cells was inhibited by specific ferroptosis inhibitors and induced lipid peroxidation. As proposed by a report of Viswanathan et al. [50], we showed that increased expression of ZEB1 and CDH2 genes, downregulation of CDH1 and higher basal GPX4 levels, potentially made mutant p53 R273H cells more vulnerable to ferroptotic triggers, compared to its isogenic counterparts. Contrary to several reports [[72], [73], [74]], p53 R273H did not exhibit EMT characteristics that promoted cellular migration and invasion. In addition, Thompson et al. reported that both mutant p53 expressing NCI–H1299 cells and mutant p53 cells from other malignancies displayed an increased sensitivity for the ferroptosis inducer erastin, compared to WT p53 cells. Contrary to our results, lipid peroxidation in the mutant p53 R273H NCI–H1299 cells did not increase in response to erastin, due to their higher levels of basal lipid peroxidation. Altogether, the mechanisms contributing to increased ferroptotic sensitivity in mutant p53 expressing cells are complex and may not be consistent between distinct TP53 mutation types [71,75]. However, with AF, we were able to kill p53 R273H overexpressing cells in a ferroptotic fashion via GPX4 inactivation and lipid peroxidation.
According to the LINCS chemical perturbagen signature database, AF's drug activity is closely related to the ferroptosis inducer withaferin A. The natural phytochemical withaferin A has been proposed as ferroptosis-inducing agent in neuroblastoma by targeting GPX4 and/or enhancing of the labile Fe(II) pool through excessive HMOX1 activation [76]. The latter is part of the noncanonical ferroptotic induction characterized by activated NRF2 pathway. Similarly, we demonstrated that a cytostatic AF concentration upregulated NRF2-mediated oxidative stress response with increased expression of NRF2 target genes HMOX1 and FTH1. HMOX1 induction was already shown by others to be dependent on TrxR inhibition [[77], [78], [79]].
Fourth, we are the first to demonstrate the immunogenic potential of AF-induced apoptotic and ferroptotic cell death in isogenic mutant p53 NSCLC cell lines in vitro.
Although the anti-inflammatory effect of AF has been widely exploited for the treatment of RA, little is known on the immunomodulatory effects of AF-treated tumor cells. The ability of cancer therapies to induce ICD depends on their ability to induce ER stress and ROS production [80]. Increased ROS following AF treatment in lung cancer has already been well established by our study and others [81], and the induction of ER stress has been described in chronic lymphocytic leukemia [79] and gastric cancer [47]. Our data is the first to show a significant increase in ICD-related DAMPs and DC maturation following AF treatment in mutant p53 NSCLC in vitro. Interestingly, DC-mediated secretion of the immunosuppressive TGF- cytokine decreased in co-cultures with AF-treated NSCLC cells, compared to their untreated counterparts. TGF- plays a major role in immunosuppression within the tumor microenvironment through the prevention of immune infiltration into tumor tissue and promotion of tumor cell proliferation [82]. Both DAMPs and DC maturation occurred independent of the mutant p53 expression and the type of AF-induced cell death, with which we are one of the first studies to describe the immunogenicity of ferroptosis in vitro [[83], [84], [85]]. We are aware that the golden standard assay for validating ICD is the in vivo vaccination assay. However, we believe that this assay is of little value, since it does not take into account the effect of AF on immune cells in vivo. Therefore, this will be studied in depth in a future study to accurately map the influence of AF on the antitumor immunity cycle in vivo. The cooperation between AF and anti-PD-L1 therapy in triple-negative breast cancer mouse model supports the use of AF as an immunomodulating agent [86].
Fifth, we observed that AF primed mutant p53 NSCLC cells for caspase-independent NK cell-mediated killing by downregulation of GZMA targets in vitro, without affecting NK cell activation, degranulation or intracellular GZMA expression.
GZMA is the most abundant serine protease in the cytotoxic granules of cytotoxic T lymphocytes (CTL) and NK cells activating a programmed cell death pathway that morphologically resembles apoptosis but has unique substrates and mediators. NK cells are functionally similar to CTL, but are able to kill target cells without any antigen priming or prior activation. Current cancer immunotherapies mainly focus on CTL, but have also many toxicity and efficacy problems [87]. Therefore, NK cells were chosen as proof of concept to study the effect of AF treatment on immune cell-mediated cancer cell killing in vitro. Since AF was used as an antirheumatic drug in the 1980's, various older studies already reported an augmentation of NK cell activity by lower doses of AF in vitro and in RA patients [[88], [89], [90]]. In our study, we are the first to demonstrate that AF increases caspase-independent NK cell-mediated killing of mutant p53 NSCLC cells.
Compared to the daily dosage of 6 mg AF in RA patients, it has been shown that much higher doses of AF must be given in vivo to inhibit TrxR activity in the tumor in mice [91,92] and a relatively high concentration of AF is required in vitro to induce cancer cell death as shown by our study and others [22]. Consequently, it might prove challenging to obtain sufficiently high concentrations of AF in the patient's tumor without increasing unwanted side-effects [9,93,94]. However, new technological innovations such as AF-loaded nanoparticles could offer a possible solution since they are able to achieve a desired effect at the right place in the body and minimize systemic cytotoxicity [95,96]. Alternatively, well-designed combination strategies might limit the need for high AF doses.
5. Conclusion
We demonstrated the high effectiveness of AF in eradicating NSCLC cells via distinct molecular mechanisms, including apoptotic and ferroptotic cell death dictated by the overexpression of mutant p53 protein. Irrespective of (mutant) p53 expression, both cell death mechanisms resulted in a significant increase of DAMPs and maturation of DCs in vitro. In addition, AF primed mutant p53 expressing NSCLC cells for caspase-independent NK cell-mediated killing by downregulation of several key targets of GZMA, without affecting NK cell activation, degranulation or intracellular GZMA expression. Altogether, our study provides novel in vitro data on the underlying mechanisms of AF-mediated cell death and its positive effect on the innate immune response in p53 mutant NSCLC, supporting its use as a versatile anticancer agent.
Funding statement
L.F.B is a research fellow of the Research Foundation Flanders (FWO). This research is funded by FWO grant numbers 11E7719 N and 1520819 N, as well as by a BOF-KP grant of the University of Antwerp (promotor C.D.).
Author contributions
Conceptualization, L.F.B. and C.D.; Methodology, L.F.B., C.D., J.V.L., W.D.V. and T.F.; Formal analysis, L.F.B., C.D., B.C., K.L. and W.D.V.; Investigation L.F.B., C.D., J.V.L., T.F., C.H., H.L. and J.D.W.; Writing – original draft preparation, L.F.B.; Writing – review and editing, J.V.L., T.F., H.L., C.H., K.L., B.C., W.D.V., J.D.W., E.B., V.S., M.P., E.S. and C.D.; Visualization, L.F.B. and C.D.; Supervision, C.D. and E.S.; Project administration, C.D. and E.S. All authors have read and agreed to the published version of this article.
Declaration of competing interest
The authors declare no conflict of interest.
Ethics approval
The study was approved by the Ethics committee of the Antwerp University Hospital (reference number 17/30/339).
Acknowledgements
The authors express their gratitude to Céline Merlin and Hans de Reu for technical assistance. The authors also thank Willy Floren and the University Foundation of Belgium for their financial support.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.101949.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Torre L.A., Bray F., Siegel R.L., Ferlay J., Lortet-Tieulent J., Jemal A. Global cancer statistics. CA A Cancer J. Clin. 2012;65(2):87–108. doi: 10.3322/caac.21262. 2015. [DOI] [PubMed] [Google Scholar]
- 3.Groenendijk F.H., Bernards R. Drug resistance to targeted therapies: déjà vu all over again. Mol Oncol. 2014;8(6):1067–1083. doi: 10.1016/j.molonc.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sosa Iglesias V., Giuranno L., Dubois L.J., Theys J., Vooijs M. Drug resistance in non-small cell lung cancer: a potential for NOTCH targeting? Front Oncol. 2018;8:267. doi: 10.3389/fonc.2018.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsvetkova E., Goss G.D. Drug resistance and its significance for treatment decisions in non-small-cell lung cancer. Curr. Oncol. 2012;19(Suppl 1):S45–S51. doi: 10.3747/co.19.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shim J.S., Liu J.O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014;10(7):654–663. doi: 10.7150/ijbs.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fogel D.B. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: a review. Contemp Clin Trials Commun. 2018;11:156–164. doi: 10.1016/j.conctc.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pantziarka P., Verbaanderd C., Sukhatme V., Rica Capistrano I., Crispino S., Gyawali B. ReDO_DB: the repurposing drugs in oncology database. Ecancermedicalscience. 2018;12:886. doi: 10.3332/ecancer.2018.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roder C., Thomson M.J. Auranofin: repurposing an old drug for a golden new age. Drugs R. 2015;15(1):13–20. doi: 10.1007/s40268-015-0083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Onodera T., Momose I., Kawada M. Potential anticancer activity of auranofin. Chem. Pharm. Bull. 2019;67(3):186–191. doi: 10.1248/cpb.c18-00767. [DOI] [PubMed] [Google Scholar]
- 11.Saei A.A., Gullberg H., Sabatier P., Beusch C.M., Johansson K., Lundgren B. Comprehensive chemical proteomics for target deconvolution of the redox active drug auranofin. Redox Biol. 2020;32:101491. doi: 10.1016/j.redox.2020.101491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S.J., Kim H.S., Seo Y.R. Understanding of ROS-inducing strategy in anticancer therapy. Oxid Med Cell Longev. 2019;2019:5381692. doi: 10.1155/2019/5381692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ren X., Zou L., Zhang X., Branco V., Wang J., Carvalho C. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxidants Redox Signal. 2017;27(13):989–1010. doi: 10.1089/ars.2016.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmgren A., Lu J. Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010;396(1):120–124. doi: 10.1016/j.bbrc.2010.03.083. [DOI] [PubMed] [Google Scholar]
- 15.Yoo M.-H., Xu X.-M., Carlson B.A., Gladyshev V.N., Hatfield D.L. Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J. Biol. Chem. 2006;281(19):13005–13008. doi: 10.1074/jbc.C600012200. [DOI] [PubMed] [Google Scholar]
- 16.Ye S., Chen X., Yao Y., Li Y., Sun R., Zeng H. Thioredoxin reductase as a novel and efficient plasma biomarker for the detection of non-small cell lung cancer: a large-scale, multicenter study. Sci. Rep. 2019;9(1):2652. doi: 10.1038/s41598-018-38153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirsh V. Systemic therapies in metastatic non-small-cell lung cancer with emphasis on targeted therapies: the rational approach. Curr. Oncol. 2010;17(2):13–23. doi: 10.3747/co.v17i2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlenius T.C., Tonissen K.F. Thioredoxin and cancer: a role for thioredoxin in all states of tumor oxygenation. Cancers. 2010;2(2):209–232. doi: 10.3390/cancers2020209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.U.S. National Library of Medicine. [Accessed on 8 June 2020]. Available from: https://clinicaltrials.gov.
- 20.Nobili S., Mini E., Landini I., Gabbiani C., Casini A., Messori L. Gold compounds as anticancer agents: chemistry, cellular pharmacology, and preclinical studies. Med. Res. Rev. 2010;30(3):550–580. doi: 10.1002/med.20168. [DOI] [PubMed] [Google Scholar]
- 21.Madeira J.M., Gibson D.L., Kean W.F., Klegeris A. The biological activity of auranofin: implications for novel treatment of diseases. Inflammopharmacology. 2012;20(6):297–306. doi: 10.1007/s10787-012-0149-1. [DOI] [PubMed] [Google Scholar]
- 22.Li H., Hu J., Wu S., Wang L., Cao X., Zhang X. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget. 2015;7(3):3548–3558. doi: 10.18632/oncotarget.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Behan F.M., Iorio F., Picco G., Goncalves E., Beaver C.M., Migliardi G. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019;568(7753):511–516. doi: 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- 24.Liu D.S., Duong C.P., Haupt S., Montgomery K.G., House C.M., Azar W.J. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017;8(1):14844. doi: 10.1038/ncomms14844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roszkowska K.A., Gizinski S., Sady M., Gajewski Z., Olszewski M.B. Gain-of-Function mutations in p53 in cancer invasiveness and metastasis. Int. J. Mol. Sci. 2020;21(4):1334. doi: 10.3390/ijms21041334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalo E., Kogan-Sakin I., Solomon H., Bar-Nathan E., Shay M., Shetzer Y. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J. Cell Sci. 2012;125(Pt 22):5578–5586. doi: 10.1242/jcs.106815. [DOI] [PubMed] [Google Scholar]
- 27.Shakya R., Tarulli G.A., Sheng L., Lokman N.A., Ricciardelli C., Pishas K.I. Mutant p53 upregulates alpha-1 antitrypsin expression and promotes invasion in lung cancer. Oncogene. 2017;36(31):4469–4480. doi: 10.1038/onc.2017.66. [DOI] [PubMed] [Google Scholar]
- 28.Sabapathy K., Lane D.P. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018;15(1):13–30. doi: 10.1038/nrclinonc.2017.151. [DOI] [PubMed] [Google Scholar]
- 29.Stein Y., Rotter V., Aloni-Grinstein R. Gain-of-Function mutant p53: all the roads lead to tumorigenesis. Int. J. Mol. Sci. 2019;20(24):6197. doi: 10.3390/ijms20246197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blandino G., Levine A.J., Oren M. Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene. 1999;18(2):477–485. doi: 10.1038/sj.onc.1202314. [DOI] [PubMed] [Google Scholar]
- 31.Karlenius T.C., Shah F., Yu W.C., Hawkes H.J., Tinggi U., Clarke F.M. The selenium content of cell culture serum influences redox-regulated gene expression. Biotechniques. 2011;50(5):295–301. doi: 10.2144/000113666. [DOI] [PubMed] [Google Scholar]
- 32.Deben C., Van den Bossche J., Van Der Steen N., Lardon F., Wouters A., de Beeck K.O. Deep sequencing of the TP53 gene reveals a potential risk allele for non-small cell lung cancer and supports the negative prognostic value of TP53 variants. Tumour Biol. 2017;39(2) doi: 10.1177/1010428317694327. 1010428317694327. [DOI] [PubMed] [Google Scholar]
- 33.Deben C., Deschoolmeester V., Lardon F., Rolfo C., Pauwels P. TP53 and MDM2 genetic alterations in non-small cell lung cancer: evaluating their prognostic and predictive value. Crit. Rev. Oncol. Hematol. 2016;99:63–73. doi: 10.1016/j.critrevonc.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 34.Uhlen M., Zhang C., Lee S., Sjostedt E., Fagerberg L., Bidkhori G. A pathology atlas of the human cancer transcriptome. Science. 2017;357(6352) doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- 35.The Human Protein Atlas. [Accessed on 14 August 2019]. Available from: www.proteinatlas.org.
- 36.Pauwels B., Korst A.E., de Pooter C.M., Pattyn G.G., Lambrechts H.A., Baay M.F. Comparison of the sulforhodamine B assay and the clonogenic assay for in vitro chemoradiation studies. Canc. Chemother. Pharmacol. 2003;51(3):221–226. doi: 10.1007/s00280-002-0557-9. [DOI] [PubMed] [Google Scholar]
- 37.Pauwels B., Korst A.E., Andriessen V., Baay M.F., Pattyn G.G., Lambrechts H.A. Unraveling the mechanism of radiosensitization by gemcitabine: the role of TP53. Radiat. Res. 2005;164(5):642–650. doi: 10.1667/rr3445.1. [DOI] [PubMed] [Google Scholar]
- 38.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.QIAGEN Inc. [Accessed on 26 March 2020]. Available from: https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis.
- 40.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. Unit. States Am. 2005;102(43):15545. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gene Set Enrichment Analysis. Molecular Signature Database [Accessed on 3 April 2020]. Available from: https://www.gsea-msigdb.org/gsea/msigdb.
- 42.Fiji. [Accessed on 10 August 2020]. Available from: https://fiji.sc.
- 43.De Vos W.H., Van Neste L., Dieriks B., Joss G.H., Van Oostveldt P. High content image cytometry in the context of subnuclear organization. Cytometry A. 2010;77(1):64–75. doi: 10.1002/cyto.a.20807. [DOI] [PubMed] [Google Scholar]
- 44.Van Loenhout J., Flieswasser T., Freire Boullosa L., De Waele J., Van Audenaerde J., Marcq E. Cold atmospheric plasma-treated PBS eliminates immunosuppressive pancreatic stellate cells and induces immunogenic cell death of pancreatic cancer cells. Cancers. 2019;11(10) doi: 10.3390/cancers11101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Waele J., Marcq E., Van Audenaerde J.R., Van Loenhout J., Deben C., Zwaenepoel K. Poly(I:C) primes primary human glioblastoma cells for an immune response invigorated by PD-L1 blockade. OncoImmunology. 2017;7(3) doi: 10.1080/2162402X.2017.1407899. e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wen C., Wang H., Wu X., He L., Zhou Q., Wang F. ROS-mediated inactivation of the PI3K/AKT pathway is involved in the antigastric cancer effects of thioredoxin reductase-1 inhibitor chaetocin. Cell Death Dis. 2019;10(11):809. doi: 10.1038/s41419-019-2035-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zou P., Chen M., Ji J., Chen W., Chen X., Ying S. Auranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget. 2015;6(34):36505–36521. doi: 10.18632/oncotarget.5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lisek K., Campaner E., Ciani Y., Walerych D., Del Sal G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget. 2018;9(29):20508–20523. doi: 10.18632/oncotarget.24974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lieberman J. Granzyme A activates another way to die. Immunol. Rev. 2010;235(1):93–104. doi: 10.1111/j.0105-2896.2010.00902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Viswanathan V.S., Ryan M.J., Dhruv H.D., Gill S., Eichhoff O.M., Seashore-Ludlow B. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Viswanathan V.S., Ryan M.J., Dhruv H.D., Gill S., Eichhoff O.M., Seashore-Ludlow B. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453–457. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Byers L.A., Diao L., Wang J., Saintigny P., Girard L., Peyton M. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Canc. Res. 2013;19(1):279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kepp O., Senovilla L., Vitale I., Vacchelli E., Adjemian S., Agostinis P. Consensus guidelines for the detection of immunogenic cell death. OncoImmunology. 2014;3(9) doi: 10.4161/21624011.2014.955691. e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Showalter A., Limaye A., Oyer J.L., Igarashi R., Kittipatarin C., Copik A.J. Cytokines in immunogenic cell death: applications for cancer immunotherapy. Cytokine. 2017;97:123–132. doi: 10.1016/j.cyto.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Audenaerde J.R.M., De Waele J., Marcq E., Van Loenhout J., Lion E., Van den Bergh J.M.J. Interleukin-15 stimulates natural killer cell-mediated killing of both human pancreatic cancer and stellate cells. Oncotarget. 2017;8(34):56968–56979. doi: 10.18632/oncotarget.18185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Du Y., Zhang H., Lu J., Holmgren A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J. Biol. Chem. 2012;287(45):38210–38219. doi: 10.1074/jbc.M112.392225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fan C., Zheng W., Fu X., Li X., Wong Y.S., Chen T. Enhancement of auranofin-induced lung cancer cell apoptosis by selenocystine, a natural inhibitor of TrxR1 in vitro and in vivo. Cell Death Dis. 2014;5:e1191. doi: 10.1038/cddis.2014.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soini Y., Kahlos K., Näpänkangas U., Kaarteenaho-Wiik R., Säily M., Koistinen P. Widespread expression of thioredoxin and thioredoxin reductase in non-small cell lung carcinoma. Clin. Canc. Res. 2001;7(6):1750. [PubMed] [Google Scholar]
- 59.Lincoln D.T., Ali Emadi E.M., Tonissen K.F., Clarke F.M. The thioredoxin-thioredoxin reductase system: over-expression in human cancer. Anticancer Res. 2003;23(3b):2425–2433. [PubMed] [Google Scholar]
- 60.Eriksson S.E., Ceder S., Bykov V.J.N., Wiman K.G. p53 as a hub in cellular redox regulation and therapeutic target in cancer. J. Mol. Cell Biol. 2019;11(4):330–341. doi: 10.1093/jmcb/mjz005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cassidy P.B., Edes K., Nelson C.C., Parsawar K., Fitzpatrick F.A., Moos P.J. Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles. Carcinogenesis. 2006;27(12):2538–2549. doi: 10.1093/carcin/bgl111. [DOI] [PubMed] [Google Scholar]
- 62.Hedström E., Eriksson S., Zawacka-Pankau J., Arnér E.S., Selivanova G. p53-dependent inhibition of TrxR1 contributes to the tumor-specific induction of apoptosis by RITA. Cell Cycle. 2009;8(21):3584–3591. doi: 10.4161/cc.8.21.9977. [DOI] [PubMed] [Google Scholar]
- 63.Shi Y., Nikulenkov F., Zawacka-Pankau J., Li H., Gabdoulline R., Xu J. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. 2014;21(4):612–623. doi: 10.1038/cdd.2013.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peng X., Zhang M.Q.Z., Conserva F., Hosny G., Selivanova G., Bykov V.J.N. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013;4(10):e881–e. doi: 10.1038/cddis.2013.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muller P.A.J., Vousden K.H. p53 mutations in cancer. Nat. Cell Biol. 2013;15(1):2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 66.Oren M., Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010;2(2):a001107–a. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harris Isaac S., Treloar Aislinn E., Inoue S., Sasaki M., Gorrini C., Lee Kim C. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Canc. Cell. 2015;27(2):211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 68.Hatem E., Azzi S., El Banna N., He T., Heneman-Masurel A., Vernis L. Auranofin/vitamin C: a novel drug combination targeting triple-negative breast cancer. J. Natl. Cancer Inst. 2018 doi: 10.1093/ije/djy149. [DOI] [PubMed] [Google Scholar]
- 69.Gnanapradeepan K., Basu S., Barnoud T., Budina-Kolomets A., Kung C.-P., Murphy M.E. The p53 tumor suppressor in the control of metabolism and ferroptosis. Front. Endocrinol. 2018;9:124. doi: 10.3389/fendo.2018.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hassannia B., Vandenabeele P., Vanden Berghe T. Targeting ferroptosis to iron out cancer. Canc. Cell. 2019;35(6):830–849. doi: 10.1016/j.ccell.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 71.Liu J., Zhang C., Wang J., Hu W., Feng Z. The regulation of ferroptosis by tumor suppressor p53 and its pathway. Int. J. Mol. Sci. 2020;21(21) doi: 10.3390/ijms21218387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Coffill C.R., Muller P.A.J., Oh H.K., Neo S.P., Hogue K.A., Cheok C.F. Mutant p53 interactome identifies nardilysin as a p53R273H-specific binding partner that promotes invasion. EMBO Rep. 2012;13(7):638–644. doi: 10.1038/embor.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hosain S.B., Khiste S.K., Uddin M.B., Vorubindi V., Ingram C., Zhang S. Inhibition of glucosylceramide synthase eliminates the oncogenic function of p53 R273H mutant in the epithelial-mesenchymal transition and induced pluripotency of colon cancer cells. Oncotarget. 2016;7(37):60575–60592. doi: 10.18632/oncotarget.11169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao Y., Li Y., Sheng J., Wu F., Li K., Huang R. P53-R273H mutation enhances colorectal cancer stemness through regulating specific lncRNAs. J. Exp. Clin. Canc. Res. 2019;38(1):379. doi: 10.1186/s13046-019-1375-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thompson L.R., Oliveira T.G., Hermann E.R., Chowanadisai W., Clarke S.L., Montgomery M.R. Distinct TP53 mutation types exhibit increased sensitivity to ferroptosis independently of changes in iron regulatory protein activity. Int. J. Mol. Sci. 2020;21(18) doi: 10.3390/ijms21186751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hassannia B., Wiernicki B., Ingold I., Qu F., Van Herck S., Tyurina Y.Y. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Invest. 2018;128(8):3341–3355. doi: 10.1172/JCI99032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mostert V., Hill K.E., Burk R.F. Loss of activity of the selenoenzyme thioredoxin reductase causes induction of hepatic heme oxygenase-1. FEBS (Fed. Eur. Biochem. Soc.) Lett. 2003;541(1):85–88. doi: 10.1016/s0014-5793(03)00309-0. [DOI] [PubMed] [Google Scholar]
- 78.Dunigan K., Li Q., Li R., Locy M.L., Wall S., Tipple T.E. The thioredoxin reductase inhibitor auranofin induces heme oxygenase-1 in lung epithelial cells via Nrf2-dependent mechanisms. Am. J. Physiol. Lung Cell Mol. Physiol. 2018;315(4):L545–L552. doi: 10.1152/ajplung.00214.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fiskus W., Saba N., Shen M., Ghias M., Liu J., Gupta S.D. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Canc. Res. 2014;74(9):2520–2532. doi: 10.1158/0008-5472.CAN-13-2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krysko D.V., Garg A.D., Kaczmarek A., Krysko O., Agostinis P., Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Canc. 2012;12(12):860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 81.Hou G.-X., Liu P.-P., Zhang S., Yang M., Liao J., Yang J. Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis. Cell Death Dis. 2018;9(2):89. doi: 10.1038/s41419-017-0159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bai X., Yi M., Jiao Y., Chu Q., Wu K. Blocking TGF-β signaling to enhance the efficacy of immune Checkpoint inhibitor. OncoTargets Ther. 2019;12:9527–9538. doi: 10.2147/OTT.S224013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Friedmann Angeli J.P., Krysko D.V., Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Canc. 2019 doi: 10.1038/s41568-019-0149-1. [DOI] [PubMed] [Google Scholar]
- 84.Efimova I., Catanzaro E., Van der Meeren L., Turubanova V.D., Hammad H., Mishchenko T.A. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J. ImmunoTher. Canc. 2020;8(2) doi: 10.1136/jitc-2020-001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang D., Kepp O., Kroemer G. Ferroptosis becomes immunogenic: implications for anticancer treatments. OncoImmunology. 2020;10(1):1862949. doi: 10.1080/2162402X.2020.1862949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Raninga P.V., Lee A.C., Sinha D., Shih Y.-Y., Mittal D., Makhale A. Therapeutic cooperation between auranofin, a thioredoxin reductase inhibitor and anti-PD-L1 antibody for treatment of triple-negative breast cancer. Int. J. Canc. 2020;146(1):123–136. doi: 10.1002/ijc.32410. [DOI] [PubMed] [Google Scholar]
- 87.Rosenberg J., Huang J. CD8(+) T cells and NK cells: parallel and complementary soldiers of immunotherapy. Curr. Opin. Chem. Eng. 2018;19:9–20. doi: 10.1016/j.coche.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pedersen B.K., Abom B. Characterization of the in vitro effect of triethylphosphine gold (auranofin) on human NK cell activity. Clin. Exp. Rheumatol. 1986;4(3):249–253. [PubMed] [Google Scholar]
- 89.Russell A.S., Miller C. The augmentation of human NK-cell activity by auranofin compared to interferon. Int. J. Immunopharm. 1984;6(5):451–454. doi: 10.1016/0192-0561(84)90083-3. [DOI] [PubMed] [Google Scholar]
- 90.Abom B., Pedersen B.K. The in vivo effect of triethylphosphine gold (auranofin), sodium aurothiomalate and azathioprine on natural killer cell activity of patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 1987;5(1):47–52. [PubMed] [Google Scholar]
- 91.Fath M.A., Ahmad I.M., Smith C.J., Spence J., Spitz D.R. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin. Canc. Res. 2011;17(19):6206. doi: 10.1158/1078-0432.CCR-11-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luo M., Shang L., Brooks M.D., Jiagge E., Zhu Y., Buschhaus J.M. Targeting breast cancer stem cell state equilibrium through modulation of redox signaling. Cell Metabol. 2018;28(1):69–86. doi: 10.1016/j.cmet.2018.06.006. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Laxer R.M. Chapter 5 - pharmacology and drug therapy. In: Cassidy J.T., Petty R.E., Laxer R.M., Lindsley C.B., editors. Textbook of Pediatric Rheumatology. fifth ed. W.B. Saunders; Philadelphia: 2005. pp. 76–141. [Google Scholar]
- 94.Aronson J.K., editor. Meyler's Side Effects of Drugs. sixteenth ed. Elsevier; Oxford: 2016. Gold and gold salts; pp. 572–583. [Google Scholar]
- 95.Díez-Martínez R., García-Fernández E., Manzano M., Á Martínez, Domenech M., Vallet-Regí M. Auranofin-loaded nanoparticles as a new therapeutic tool to fight streptococcal infections. Sci. Rep. 2016;6(1):19525. doi: 10.1038/srep19525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Awasthi R., Roseblade A., Hansbro P.M., Rathbone M.J., Dua K., Bebawy M. Nanoparticles in cancer treatment: opportunities and obstacles. Curr. Drug Targets. 2018;19(14):1696–1709. doi: 10.2174/1389450119666180326122831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.