

Abstract
Background.
Half of adults with cystic fibrosis (CF) develop CF-related diabetes (CFRD). CFRD contributes to worsened pulmonary function and malnutrition. We undertook this study to determine the effect of cystic fibrosis transmembrane regulator (CFTR) modulators on CRFD.
Methods.
We reviewed the medical records of adults with CF who followed in the CF clinic at Oklahoma University Medical Center. We collected data for age at diagnosis of CF and CFRD, CF mutations present, first date of ivacaftor therapy either alone or in combination, insulin use, pulmonary function, body mass index data, and home glucose monitoring results. Clinical resolution of CFRD was taken as discontinuation of routine insulin and resolution of high interstitial home glucose values.
Results.
We identified 69 adult CF patients, of whom 31 had CFRD. Among these 14 CFRD patients taking ivacaftor alone or in combination, four patients completely stopped using insulin. Another patient went from three times a day pre-prandial insulin to using insulin once a week. Home blood glucose and hemoglobin A1c values supported resolution of CFRD. Three patients continued to have hypoglycemia despite stopping insulin. No CFRD patient not taking CFTR modulators markedly changed the insulin regimen. Pulmonary function was preserved in those patients with resolved CFRD (FEV1 +6.75% ±7.6), whereas it worsened in CFRD patients who either were not taking CFTR modulators (FEV1 −2.09% ±3.9) or who had no response of CFRD status (FEV1 −4.9% ±7.6).
Conclusions.
About one-third of patients on CFTR modulator therapy had resolution or near resolution of CFRD.
Keywords: Cystic fibrosis, CF, cystic fibrosis-related diabetes, CFRD, ivacaftor
Introduction
Cystic fibrosis (CF) is a common autosomal recessive genetic disorder with life-limiting implications. Patients may have chronic progressive lung disease, recurrent pulmonary infections, pancreatic insufficiency, malnutrition, and ultimately death (1). The prognosis for patients with CF has improved remarkably since its recognition as a disease entity in 1938, at which time the predicted survival of an infant with CF was only six months of life (2). With increasing life expectancy and a shift in demographics of the CF population, comorbidities associated with the adult CF patient have become more prevalent. Half of all adults with CF develop cystic fibrosis related diabetes (CFRD), making it the most common comorbid condition (3).
CFRD is a unique subtype of diabetes mellitus, distinct from type 1 or type 2 (4). The hallmark of this disease is the loss of first-phase insulin secretion in the setting of CF, resulting in post-prandial hyperglycemia. Because CFRD contributes to worsened pulmonary function and malnutrition in the CF patient, current guidelines recommend annual screening with oral glucose tolerance testing beginning at age ten years (5, 6). Treatment of CFRD consists of insulin therapy almost exclusively. The mechanism of CFRD is poorly understood.
CF results from impairment of the cystic fibrosis transmembrane conductance regulator (CFTR) protein. More than 1500 variations of the CFTR gene (OMIN 602421) have been identified, and a minority of these are the most common disease-causing mutations (7). CFTR serves as a low-conductance, regulated chloride channel. In respiratory epithelial cells, dysfunction of CFTR results in abnormal gating of sodium and chloride, producing viscous secretions in dependent tissues (1, 2).
CFTR modulators are new drugs that target the defective CFTR protein such that lung function improves in patients with certain mutations (8). All current CFTR modulator therapies include ivacaftor. Ivacaftor is a potentiator of the defective CFTR chloride channel, increasing chloride conductance across dependent cell membranes. Few studies have been conducted to assess the effect of CFTR modulators on CFRD. After noting marked reduction of insulin requirements in long-standing CFRD patients following institution of CFTR modulator therapy, we undertook the present study to determine the effect of these drugs on CFRD in an adult CF population followed at a multidisciplinary CF Clinic.
Methods
CF Clinic.
The University of Oklahoma Health Science Center Comprehensive Cystic Fibrosis Clinic is housed at OU Medical Center, and is a joint effort of the Departments of Pediatrics and Internal Medicine. The clinic provides multidisciplinary care to both children and adults, including in-clinic adult endocrinology. The clinic is supported in part by a grant from the Cystic Fibrosis Foundation.
Records Review.
In this retrospective study, we reviewed the electronic medical records of all adults seen at the clinic between November 2016 and November 2018. We defined adults as persons over age 21 years. We ascertained the age, sex, age of onset of CF and CFRD, pulmonary function and body mass index data (BMI), as well as the CF genotype of each patient. Changes in diabetes management (insulin regimen, other glycemic management) as well as glycemic control along with adverse events (hypoglycemia) were ascertained. All procedures were approved by the Oklahoma University Health Science Center Institutional Review Board.
Statistical Methods.
Based on data review, we divided patients into three groups. Group 1 includes patients with CFRD taking ivacaftor alone or in combination, who demonstrated clinical improvement in diabetes; Group 2 includes patients with CFRD, taking ivacaftor alone or in combination, without clinical improvement in diabetes; and Group 3 includes patients with CFRD, not taking CFTR modulators. The primary outcome of the study was resolution of CFRD as defined by discontinuation of anti-diabetic medications (insulin in all but one patient) along with non-elevated home glucose monitoring results.
Because CFRD adversely affects both pulmonary and metabolic status, we examined pulmonary function by recorded forced expiratory volume at 1 second as a percentage of predicted value (FEV1) and body mass index (BMI) data for all Groups. The first values for FEV1 and BMI were taken from the beginning of the study window (November 2016), and the comparison point was from the close of the study window (November 2018). We compared changes in FEV1 and BMI between the groups.
We removed the cases with unavailable FEV1 and BMI values and calculated the relative change in FEV1 and BMI by using (2018 value - 2016 value)/2016 value. We used relative difference instead of raw difference to account for baseline FEV1 or BMI. Given the small sample sizes for the groups, we used Exact Wilcoxon rank test to compare the differences of relative differences between Group 1 and Group 2, and between Group 1 and Group 3, separately. Changes in pulmonary function and BMI were secondary outcomes in the study.
Results
A total of 69 adults with CF were followed at the clinic during the study window. Thirty-one (45%) of these had CFRD confirmed by oral glucose tolerance testing. Of these 31 with CFRD, 14 were taking CTFR modulators (two patients were on ivacaftor alone, eight patients used the combination of ivacaftor and lumacaftor, and four patients used the combination of ivacaftor and tezacaftor). There were 17 CFRD patients not taking CFTR modulators. The reasons for not taking CFTR modulators included three patients with previous lung transplant, eight with ineligible mutations, three with adverse drug reaction to ivacaftor, one with concurrent lymphoma, and two newly-established patients not yet started.
Among the 14 patients taking CFTR modulators, we found five individuals with marked changes in the management of CFRD (Table 1). Patient 1 had an eight-year history of CFRD and developed hypoglycemia nine months after starting ivacaftor. He stopped using insulin completely and continued to have mild hypoglycemia as low as 40mg/dL with no further hyperglycemia by home finger stick blood sugar monitoring during the two years of follow up. Patient 2 had CFRD diagnosed two years before but was not treated with insulin prior to transferring care to our center. She had hypoglycemia while taking the anti-diabetic medication metformin, which was worsened by CFTR modulators. Upon her care being assumed by us, the metformin was discontinued. Subsequent continuous glucose monitor results implied resolution of CFRD. Patient 3 had a 14-year history of CFRD and had been on insulin since diagnosis. Six months after starting CFTR modulators he developed hypoglycemia. Insulin was tapered and stopped at 8 months post CFTR modulator initiation. More than two years later, he has normal fasting and post-prandial glucose values while still on CFTR modulators. Patient 4 started CFTR modulators two years after being diagnosed with CFRD. Twelve months later, she tapered and stopped insulin therapy. Finally, Patient 5 had CFRD for nine years before starting ivacaftor. After 12 months, he had tapered his insulin from using three pre-prandial injections daily to using pre-prandial insulin only one or two times weekly, and he reported occasional hypoglycemia.
Table 1.
Summary of CFRD patients with significant changes in blood glucose and diabetes management after institution of ivacaftor.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
|---|---|---|---|---|---|
| Age at CF diagnosis | 2 years | 5 years | 5 weeks | 8 years | 2 years |
| Mutations | Δ508/G551D | Δ508 | Δ508 | Δ508 | Δ508/S549R |
| Age at CFRD diagnosis | 20 | 29 | 16 | 30 | 20 |
| Age at ivacaftor initiation | 28 | 30 | 29 | 32 | 29 |
| Specific drug | ivacaftor | combined** | combined | combined | ivacaftor |
| Time to CFRD resolution | 9 months | 1 month | 8 months | 12 months | 12 months* |
| Continued hypoglycemia | Yes | Yes | No | No | Yes |
| Follow up post CFRD resolution | 5 years | 1 year | 8 months | 2 years | 4 years* |
| Change in FEV1 (% predicted) | ND*** | +3% | +3% | +13% | +8% |
| Change in BMI | ND | −1.0 | +1.5 | +0.7 | 0 |
Patient had reduction in insulin use from short-acting insulin three times a day to once a week, not complete resolution
lumacaftor/ivacaftor combination drug
ND = no data
Thus, 36% of CFRD patients treated with CFTR modulators had markedly improved disease status (Group 1). Three patients developed persistent hypoglycemia. Notably, the reduction or cessation of insulin therapy did not occur for an average of 8·4 months following initiation of CFTR modulators. The average time of having CFRD prior to resolution was 7·2 years. In the 2·5-year mean follow-up time, all four patients with resolution of CFRD remained off insulin therapy, and the fifth patient continued to use a significantly-reduced dose of insulin on an intermittent basis (Table 1). Of the nine patients with CFRD taking CFTR modulators who showed no improvement in CFRD (Group 2), all had f508/f508 mutation, four took tezacaftor/ivacaftor, and five more took lumacaftor/ivacaftor (Table 2). Of the 17 patients with CFRD not taking CFTR modulators (Group 3), none demonstrated improvement in diabetes (zero of 17 versus 5 of 14 improved on CFTR modulators, p=0.012 by Fisher’s exact test) indicating statistically significant difference in CFRD improvement among those taking CFTR modulators. With elimination of the three patients not taking CFTR modulators after lung transplant, since these patients had multiple factors such as use of glucocorticoids that are expected to impact CFRD, the results remained statistically significant (p=0.04).
Table 2.
Summary of CFTR mutations and treatment in the three groups: resolved CFRD on modulator therapy, no change (sustained) CFRD on modulator, or sustained CFRD not on modulator.
| modulator/resolved CFRD | modulator/sustained CFRD | no modulator/sustained CFRD | |
|---|---|---|---|
| n=5 | n=9 | n=17 (16 genotyped) | |
| Δ508/Δ508 | 3 (60%) | 9 (100%) | 8 (53.3%) |
| Δ508/other | 2 (40%) | 0 (0%) | 7 (40%)* |
| G542X/G542X | 0 (0%) | 0 (%) | 1 (6.7%) |
| Ivacaftor alone | 2 (40%) | 0 (0%) | N/A |
| Combination | 3 (60%) | 9 (100%) | N/A |
Δ508/R553X, Δ508/R553X, Δ508/R553X, Δ508/1471delA, Δ508/N1303K, Δ508/A455E, Δ508/G542X
Another manner of considering the CF patients is the presence of a gating mutation versus Δ508. As can be seen in Table 2, 25% of the 14 with Δ508 ceased insulin. This falls to 17% if we omit patient 2 (Table 1) in whom the diagnosis of CFRD was made outside our center. Both the patients with gating mutations and on ivacaftor alone ceased insulin and had apparent resolution of CFRD (Table 2).
For FEV1, when we compared Group 1 versus Group 2, the one-sided p value was 0.0121, which indicates a significant difference between the two groups. Specifically, Group 1 had higher relative difference (0.2086) compared with Group 2 (−0.1003), corresponding to improved pulmonary function among those who also had improvement in CFRD, whereas those with worsened or unchanged CFRD on CFTR modulators had an average decline in pulmonary function. When we compared Group 1 versus Group 3, the one-sided p value was 0.0291, which again indicates significant difference between the two groups. Specifically, Group 1 has higher relative difference (0.2086) compared with Group 3 (0.00793). For BMI, Group 1 experienced a small average rise in BMI, though when we compared Group 1 versus Group 2, the one-sided p value was not statistically significant at 0.1576. Similarly, when we compared BMI data for Group 1 versus Group 3, the one-sided p value was 0.2231 (Figures 1 and 2).
Figure 1.
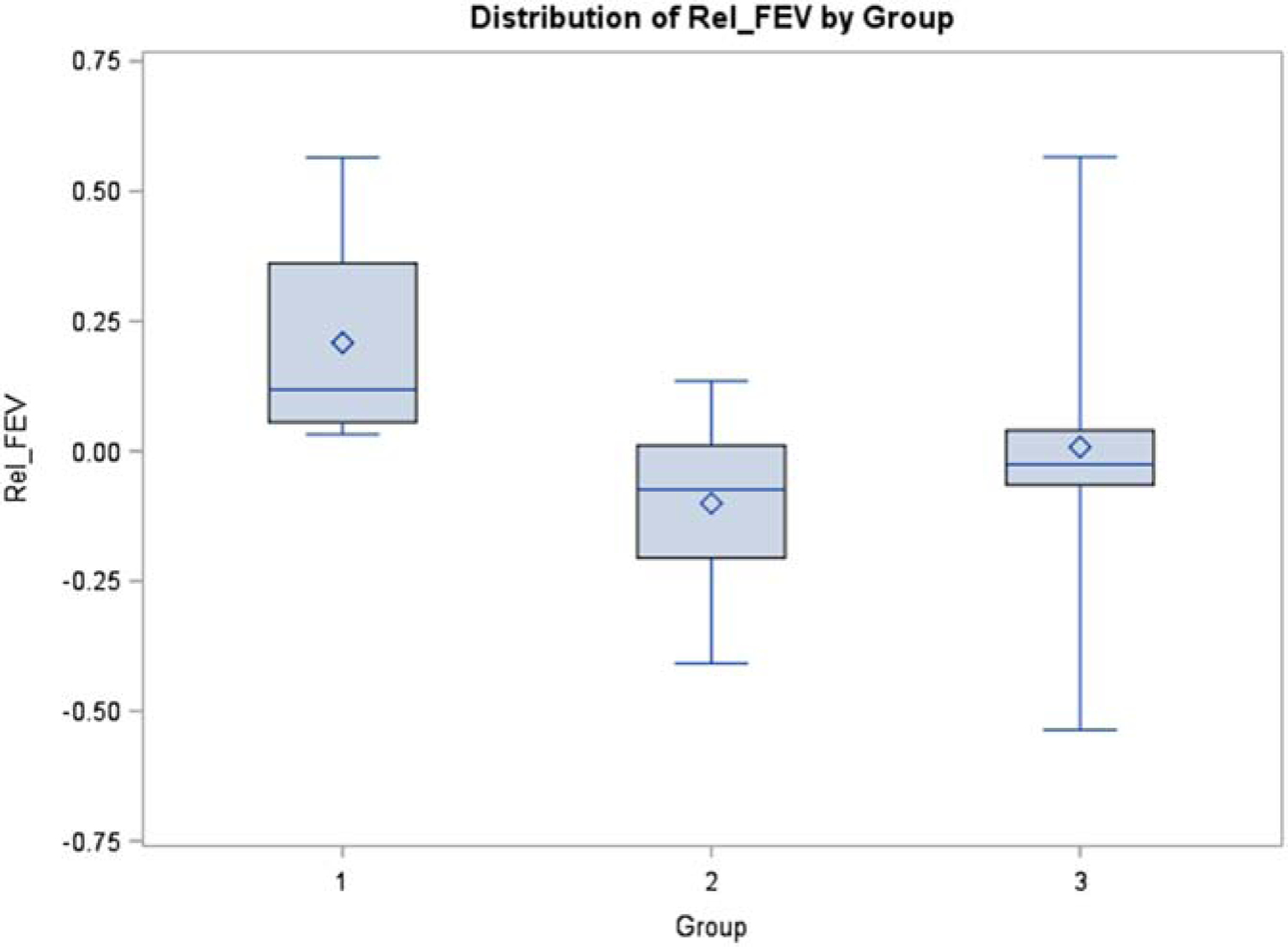
Distribution of relative difference in FEV1 by Group number. Values are derived using formula: [(2018 FEV1 value) – (2016 FEV1 value)]/(2016 FEV1 value). Exact Wilcoxon rank test shows significant difference (one-sided p value = 0.012) for Group 1 (+0.2086) vs Group 2 (−0.1003), and significant difference (one-sided p value = 0.0291) for Group 1 vs Group 3 (0.00793).
Figure 2.
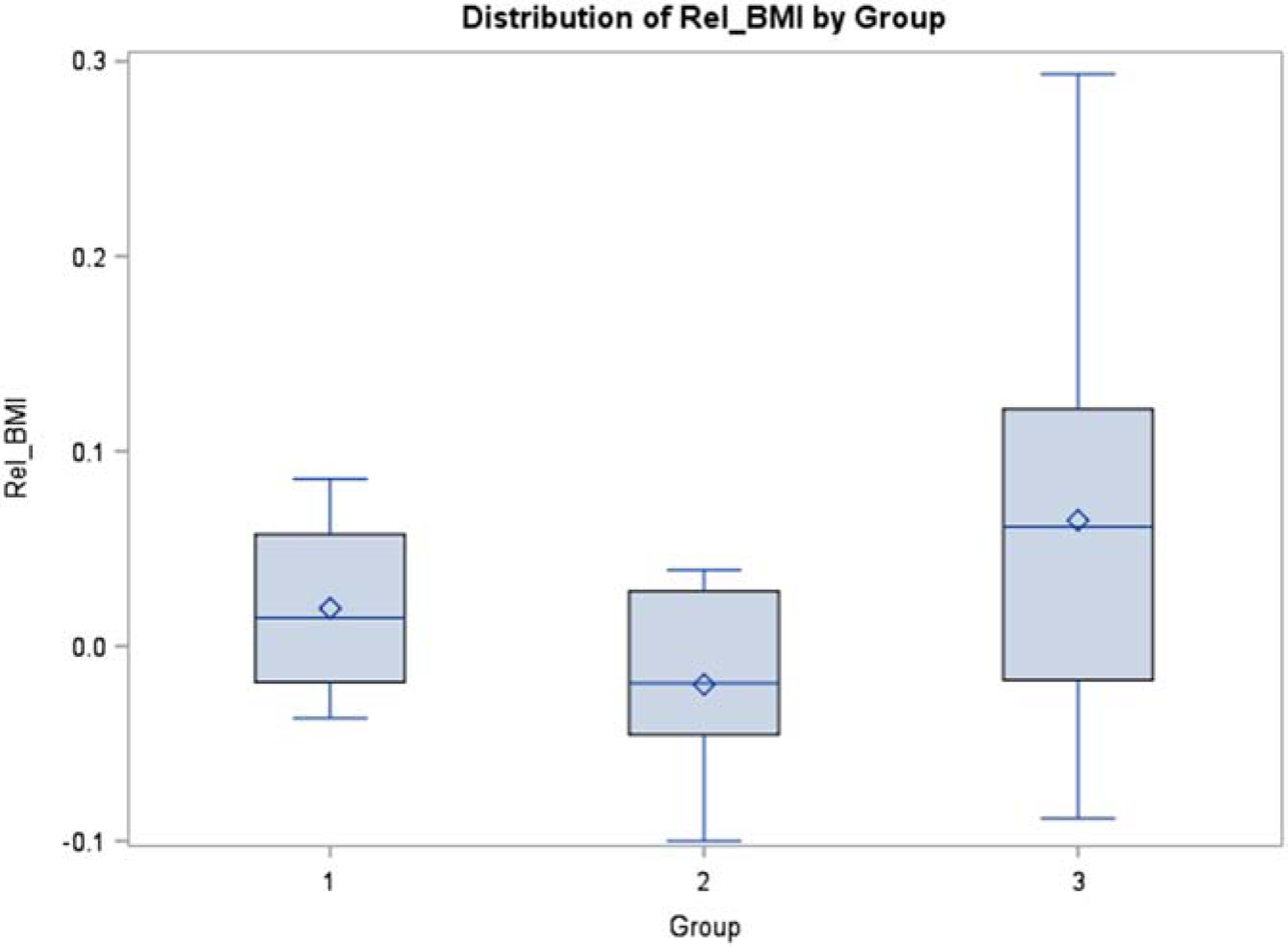
Distribution of relative difference in BMI by Group number. Values are derived using formula: [(2018 BMI value) – (2016 BMI value)]/(2016 BMI value). Exact Wilcoxon rank test shows no significant difference for Group 1 vs Group 2 (one-sided p value is 0.1576), and no significant difference for Group 1 vs group 3 (one-sided p value is 0.2231).
Discussion
Cystic fibrosis has shifted to a disease of both children and adults, and CFRD represents the most common comorbidity in adult CF patients (1, 9). CFRD contributes to worsened pulmonary function and nutritional status and increases morbidity in CF patients. We herein present the clinical effect of CFTR modulator drugs on CFRD (Table 1). No patients evaluated were treated with elexacaftor/tezacaftor/ivacaftor as this therapy was not available during the course of our study.
CF results from a genetic defect in CFTR. The CFTR is expressed in multiple tissues such that dysfunction of the CFTR results in a spectrum of multi-organ disease. Mutations of the CFTR are divided into five classes: defects in I) protein production, II) protein processing, III) regulation, IV) conduction, V) reduced amounts of functional protein, and VI) accelerated turnover (10). Several new drugs have been introduced to treat CF by targeting specific classes of CFTR mutations.
Ivacaftor works in patients with class III mutations by potentiating the defective CFTR protein, thus increasing chloride conductance across cell membranes (8). The known clinical effect is significant improvement in pulmonary function (11, 12). However, the role of ivacaftor and other CFTR modulators in CFRD remains unclear. All our patients with clinical resolution of CFRD were taking ivacaftor either alone or in combination. Those on CFTR modulator therapy were more likely to stop insulin and no longer have elevated home glucose values than those who were not on such therapy. This was the primary outcome of our valuation.
Recently, Thomassen, et al. assessed insulin secretion and glucose metabolism in five patients with CF following 6–8 weeks of therapy with lumacaftor/ivacaftor. In response to IV glucose tolerance testing, two patients had improved, but three had worsened acute insulin secretion. The study did not assess efficacy of lumacaftor/ivacaftor in CFRD patients (13). In a prior study, Bellin et al. conducted glucose tolerance testing at baseline and after one month of ivacaftor therapy in five CF patients. Four of five had an increase in first phase insulin secretion following intravenous glucose loads, including two patients who previously had no detectable insulin secretory response to glucose loading (14).
Both of these studies of the effect of ivacaftor on insulin and glucose in glucose tolerance testing were conducted after only weeks of taking the drugs. We found that patients stopped insulin many months after starting CFTR modulators. Thus, we conclude these studies of glucose tolerance were likely performed too early to demonstrate the full effect. Future studies should allow for several months of therapy prior to investigation of glucose tolerance and insulin secretion, and should focus on larger sample size.
In terms of length of time to effect of ivacaftor on CFRD, other publications document similar findings to ours, with improvement of insulin secretion or discontinuation of insulin therapy in CFRD patients following months of ivacaftor therapy. A recent study of 12 CF patients, 7 with normal glucose tolerance and 5 with abnormal glucose tolerance, showed increased early phase c-peptide secretion four months after starting ivacaftor (15). The authors concluded that, based on the disposition (of glucose) index, the results were consistent with an effect on beta cell function. In a case report, a 25-year-old man (Δ508/G551D) with longstanding CFRD stopped insulin after 13 months of ivacaftor therapy (16). In two brothers, one with CFRD and one with impaired glucose tolerance, oral glucose tolerance testing improved after 16 weeks of ivacaftor for both patients. Both had Δ508/S549R mutations (17). Two patients in the ivacaftor arm of a randomized, placebo-controlled trial had hypoglycemia (12). One of these had CRFD and one did not. Resolution of CFRD was not mentioned in this paper.
There is an ongoing debate regarding the pathophysiology of CFRD. Many authors attribute CFRD to progressive pancreatic beta cell destruction (10, 18), while others entertain the theory that CFRD results from defective or deficient CFTR in the pancreatic beta cell (19–21). The results of our study do not support the beta cell destruction theory. For example, patients 1, 3, and 5 (Table 1) experienced marked improvement of CFRD after having had the disease for 8, 13, and 10 years, respectively. We would not expect this to be possible if the patients’ long-standing CFRD was attributable to loss of beta cell mass. Other plausible explanations include reduction in overall systemic and islet inflammation (18, 22), which is consistent with the long interval between initiation of CFTR modulators and CFRD resolution, as well as confounding development of insulin resistance in the aging CFRD patient (23). Alternatively, there may be an effect of CFTR modulators on pancreatic beta cells that is independent of CFTR.
In porcine models of CF, knockout of CFTR results in impaired glucose homeostasis (24). In a ferret model with CFTR gene exon 10 disrupted, newborns exhibit impaired regulation of insulin secretion in the absence of overt pancreatic structural pathology (25). Both these models demonstrate expression of CFTR in pancreatic beta cells. There is debate regarding methods used to ascertain the expression of CFTR in human pancreatic beta cells (22), though some authors propose that CFTR plays a role in human insulin secretion, exocytosis, and regulation (21). Chloride channel activity in both wild-type and mutated CFTR protein was directly modified by VX-770 (ivacaftor) in Sf9 cells with induced CFTR overexpression (8). In the present study, all patients on ivacaftor (alone or in combination) therapy with sustained CFRD were homozygous for Δ508; thus, there may be a differential response of CFRD based on mutations carried, but sample size was too small to make any conclusion. Of course, the mutational profile of the CF patients eligible for specific CFTR modulator therapy depends upon the specific CFTR genotypes. CF patients with more severe CFTR genotypes are more likely to develop CFRD and have a higher mortality than those patients with mild genotypes. Furthermore, this effect was more pronounced among men (26). Thus, our data may be confounded by the observed difference in CFTR genotypes between the groups.
Given the improvement of CFRD in some of our patients, we hypothesize that CFTR modulators may prevent development of CFRD. Our data do not address this issue. A recent study of nine pediatric patients found no significant difference in continuous glucose monitoring variables pre-and post-lumacaftor/ivacaftor during a median 26 weeks prior to and 29 weeks following initiation of therapy (27). Larger studies of CFRD status, glucose tolerance, and insulin secretion in children starting CFTR modulators prior to the onset of CFRD will be needed to further assess this possibility. Additionally, given the possible role of CFTR in normal pancreatic beta cells and the known effect of CFTR modulators on wild-type CFTR, the effect of CFTR modulators could be studied in patients with type 2 diabetes mellitus.
Our study is inherently limited by its retrospective design and size, as well as by the unavoidable differences in VFTR genotypes as discussed above. In addition, we were unable to obtain follow up OGTT data in patients with apparent resolution of CFRD status, as it was not performed in the clinic for various individual reasons. Of course, it is not possible to conduct a randomized controlled study in which patients who otherwise qualify for CFTR modulator therapy would be denied the drug for the purpose of study control. After lung transplant CF patient do not take CFTR modulators and we had three such patients. However, the results remained statistically significant when the data were analyzed without these three.
Conclusion
Our results suggest that the use of ivacaftor alone or in combination is associated with improvement in CFRD such that insulin is no longer required in one-quarter to one-third patients perhaps depending upon the CFRT mutation. In addition, symptomatic hypoglycemia persisted in a 40% of those who stopped or markedly reduced insulin. While these patients may have even had the resolution of CFRD, vigilance will be required to monitor for worsening blood sugar and return of CFRD requiring insulin therapy. Since these drugs also increase conductance of chloride across normal CFTR channels, this study may carry further-reaching implications for the treatment of patients with type 2 diabetes mellitus. Patients commencing CFTR modulators with CFRD should be alerted to the potential for hypoglycemia as well as the possibility of improvement or possible resolution of CFRD even months after starting treatment.
Highlights.
Diabetes is a common complication in adults with cystic fibrosis
Cystic fibrosis related diabetes (CFRD) is distinct from both type 1 and 2 diabetes
Ivacaftor is a cystic fibrosis transmembrane modulator
Initiation of ivacaftor resulted in about one-third of CFRD patients stopping insulin
Funding
This study was supported in part by National Institute of General Medical Sciences grant GM104938. The funding source had no role in the execution of the study, writing of the manuscript or decision to submit.
RHS reports NIH funding (GM104938) supported this work in part.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare no conflicts of interest.
Author state
The authors have no conflict of interests to report.
References
- 1.Elborn JS. Cystic fibrosis. Lancet (London, England). 2016;388(10059):2519–31. [DOI] [PubMed] [Google Scholar]
- 2.Davis PB. Cystic fibrosis since 1938. American journal of respiratory and critical care medicine. 2006;173(5):475–82. [DOI] [PubMed] [Google Scholar]
- 3.Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes care. 2009;32(9):1626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Konrad K, Scheuing N, Badenhoop K, Borkenstein MH, Gohlke B, Schofl C, et al. Cystic fibrosis-related diabetes compared with type 1 and type 2 diabetes in adults. Diabetes/metabolism research and reviews. 2013;29(7):568–75. [DOI] [PubMed] [Google Scholar]
- 5.Bismuth E, Laborde K, Taupin P, Velho G, Ribault V, Jennane F, et al. Glucose tolerance and insulin secretion, morbidity, and death in patients with cystic fibrosis. The Journal of pediatrics. 2008;152(4):540–5, 5.e1. [DOI] [PubMed] [Google Scholar]
- 6.Moran A, Brunzell C, Cohen RC, Katz M, Marshall BC, Onady G, et al. Clinical Care Guidelines for Cystic Fibrosis–Related Diabetes. A position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. 2010;33(12):2697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castellani C, Cuppens H, Macek M Jr., Cassiman JJ, Kerem E, Durie P, et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2008;7(3):179–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eckford PDW, Canhui Li, Ramjeesingh M, Bear CE. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Potentiator VX-770 (Ivacaftor) Opens the Defective Channel Gate of Mutant CFTR in a Phosphorylation-dependent but ATP-independent Manner. Journal of Biological Chemistry. 2012;287(44):36639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgel PR, Bellis G, Olesen HV, Viviani L, Zolin A, Blasi F, et al. Future trends in cystic fibrosis demography in 34 European countries. The European respiratory journal. 2015;46(1):133–41. [DOI] [PubMed] [Google Scholar]
- 10.Moskowitz SM, Chmiel JF, Sternen DL, Cheng E, Gibson RL, Marshall SG, et al. Clinical practice and genetic counseling for cystic fibrosis and CFTR-related disorders. Genetics in medicine : official journal of the American College of Medical Genetics. 2008;10(12):851–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel S, Sinha IP, Dwan K, Echevarria C, Schechter M, Southern KW. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database of Systematic Reviews. 2015(3). [DOI] [PubMed] [Google Scholar]
- 12.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. The New England journal of medicine. 2011;365(18):1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomassen JC, Mueller MI, Alejandre Alcazar MA, Rietschel E, van Koningsbruggen-Rietschel S. Effect of Lumacaftor/Ivacaftor on glucose metabolism and insulin secretion in Phe508del homozygous cystic fibrosis patients. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2018;17(2):271–5. [DOI] [PubMed] [Google Scholar]
- 14.Bellin MD, Laguna T, Leschyshyn J, Regelmann W, Dunitz J, Billings J, et al. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediatric diabetes. 2013;14(6):417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly A, De Leon DD, Sheikh S, Camburn D, Kubrak C, Peleckis AJ, et al. Islet Hormone and Incretin Secretion in Cystic Fibrosis after Four Months of Ivacaftor Therapy. American journal of respiratory and critical care medicine. 2019;199(3):342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayes D Jr., McCoy KS, Sheikh SI. Resolution of cystic fibrosis-related diabetes with ivacaftor therapy. American journal of respiratory and critical care medicine. 2014;190(5):590–1. [DOI] [PubMed] [Google Scholar]
- 17.Tsabari R, Elyashar HI, Cymberknowh MC, Breuer O, Armoni S, Livnat G, et al. CFTR potentiator therapy ameliorates impaired insulin secretion in CF patients with a gating mutation. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2016;15(3):e25–7. [DOI] [PubMed] [Google Scholar]
- 18.Hart NJ, Aramandla R, Poffenberger G, Fayolle C, Thames AH, Bautista A, et al. Cystic fibrosis-related diabetes is caused by islet loss and inflammation. JCI insight. 2018;3(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo JH, Chen H, Ruan YC, Zhang XL, Zhang XH, Fok KL, et al. Glucose-induced electrical activities and insulin secretion in pancreatic islet beta-cells are modulated by CFTR. Nature communications. 2014;5:4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ntimbane T, Mailhot G, Spahis S, Rabasa-Lhoret R, Kleme ML, Melloul D, et al. CFTR silencing in pancreatic beta-cells reveals a functional impact on glucose-stimulated insulin secretion and oxidative stress response. American journal of physiology Endocrinology and metabolism. 2016;310(3):E200–12. [DOI] [PubMed] [Google Scholar]
- 21.Edlund A, Esguerra JL, Wendt A, Flodstrom-Tullberg M, Eliasson L. CFTR and Anoctamin 1 (ANO1) contribute to cAMP amplified exocytosis and insulin secretion in human and murine pancreatic beta-cells. BMC Med. 2014;12:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norris AW, Ode KL, Merjaneh L, Sanda S, Yi Y, Sun X, et al. Survival in a bad neighborhood: pancreatic islets in cystic fibrosis. The Journal of endocrinology. 2019. [DOI] [PMC free article] [PubMed]
- 23.Colomba J, Boudreau V, Lehoux-Dubois C, Desjardins K, Coriati A, Tremblay F, et al. The main mechanism associated with progression of glucose intolerance in older patients with cystic fibrosis is insulin resistance and not reduced insulin secretion capacity. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2019. [DOI] [PubMed]
- 24.Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. The New England journal of medicine. 2015;372(4):351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olivier AK, Yi Y, Sun X, Sui H, Liang B, Hu S, et al. Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. The Journal of clinical investigation. 2012;122(10):3755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis C, Blackman SM, Nelson A, Oberdorfer E, Wells D, Dunitz J, et al. Diabetes-related mortality in adults with cystic fibrosis. Role of genotype and sex. Am J Respir Crit Care Med.191(2):194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li A, Vigers T, Pyle L, Zemanick E, Nadeau K, Sagel SD, et al. Continuous glucose monitoring in youth with cystic fibrosis treated with lumacaftor-ivacaftor. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2019;18(1):144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]