

Abstract
Lactate dehydrogenase (LDH) is a critical enzyme in the glycolytic metabolism pathway that is used by many tumor cells. Inhibitors of LDH may be expected to inhibit the metabolic processes in cancer cells and thus selectively delay or inhibit growth in transformed versus normal cells. We have previously disclosed a pyrazole-based series of potent LDH inhibitors with long residence times on the enzyme. Here, we report the elaboration of a new subseries of LDH inhibitors based on those leads. These new compounds potently inhibit both LDHA and LDHB enzymes, and inhibit lactate production in cancer cell lines.
Keywords: Medicinal chemistry, LDH inhibitor, Structure Activity Relationships, Cancer metabolism
Graphical Abstract
The well-known “Warburg effect” is based on the observation that the metabolic processes of cancer cells are quite distinct relative to that of untransformed tissues.1 Indeed, cancer cells tend to rely not upon mitochondrial oxidative phosphorylation, but instead on comparatively inefficient, non-oxidative glycolysis-based energy production.2 Glycolytic metabolism hinges upon the production of lactate from pyruvate, via the lactate dehydrogenase enzymes LDHA and LDHB, either of which can be overexpressed in cancers.3
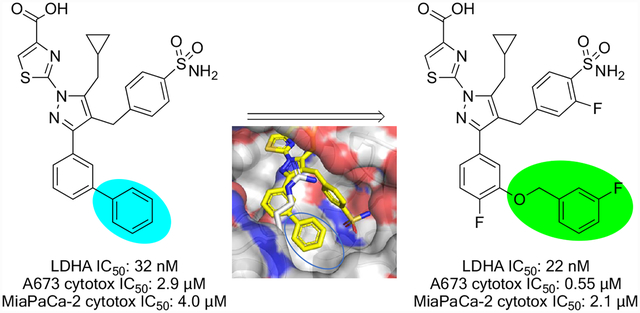
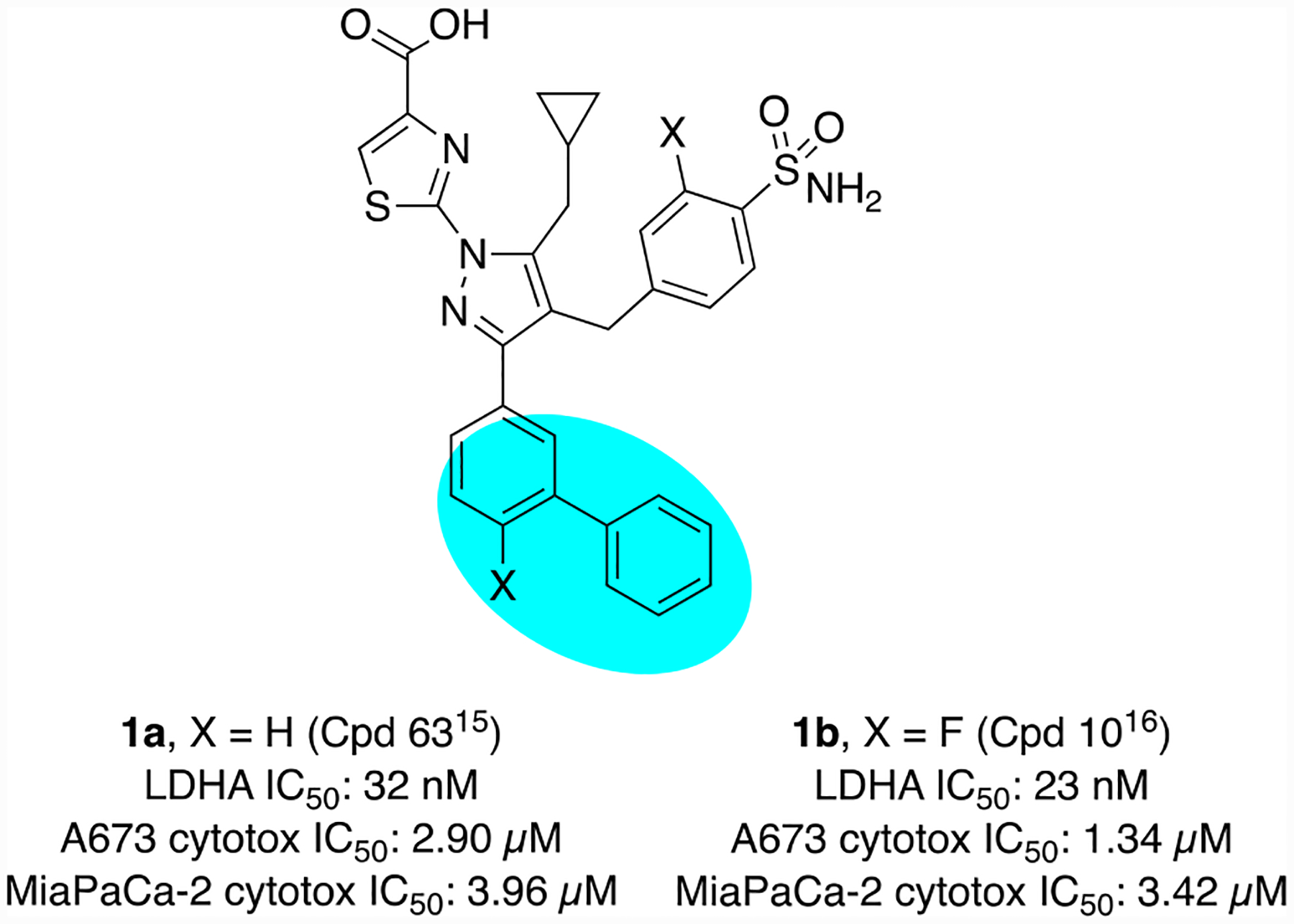
Supporting the hypothesis that inhibition of LDH function might selectively affect the growth of cancer cells due to this altered metabolism, several reports have demonstrated that ablation of LDHA activity can affect cancer cell growth in vitro and in vivo.5–9 These reports of biological validation have been accompanied by reports of a number of compounds aimed at the inhibition of LDH function.6,10–14 We have previously described a series of pyrazole-based inhibitors of LDH.15 While these compounds display potent inhibition of LDH at the enzymatic and cellular levels, as well as show promising ADME attributes, we have engaged in ongoing Structure Activity Relationship (SAR) studies to identify improved compounds, primarily via additional diversification around the meta substitution of the phenyl ring attached to the pyrazole (blue oval, Figure 1).
Figure 1.
Analysis of crystal structures of our reported compounds bound to human LDHA (Figure 2) reveals that a considerable amount of hydrophobic space is available in the binding pocket occupied by the meta phenyl of 1 (blue oval, Figure 2). This hypothesis is confirmed by the reported SAR that shows generally superior cellular potency for compounds with substitutions that fill in this space. We sought to further occupy this pocket with alternative substitutions, in a quest to further improve compound potency and/or pharmacokinetic profiles while reducing lipophilicity. One such SAR campaign is described elsewhere.16 Here, we describe another exploration that focuses on the incorporation of heteroatoms such as oxygen and nitrogen at the meta position; these have the significant additional advantage of providing a convenient synthetic handle for elaboration.
Figure 2.
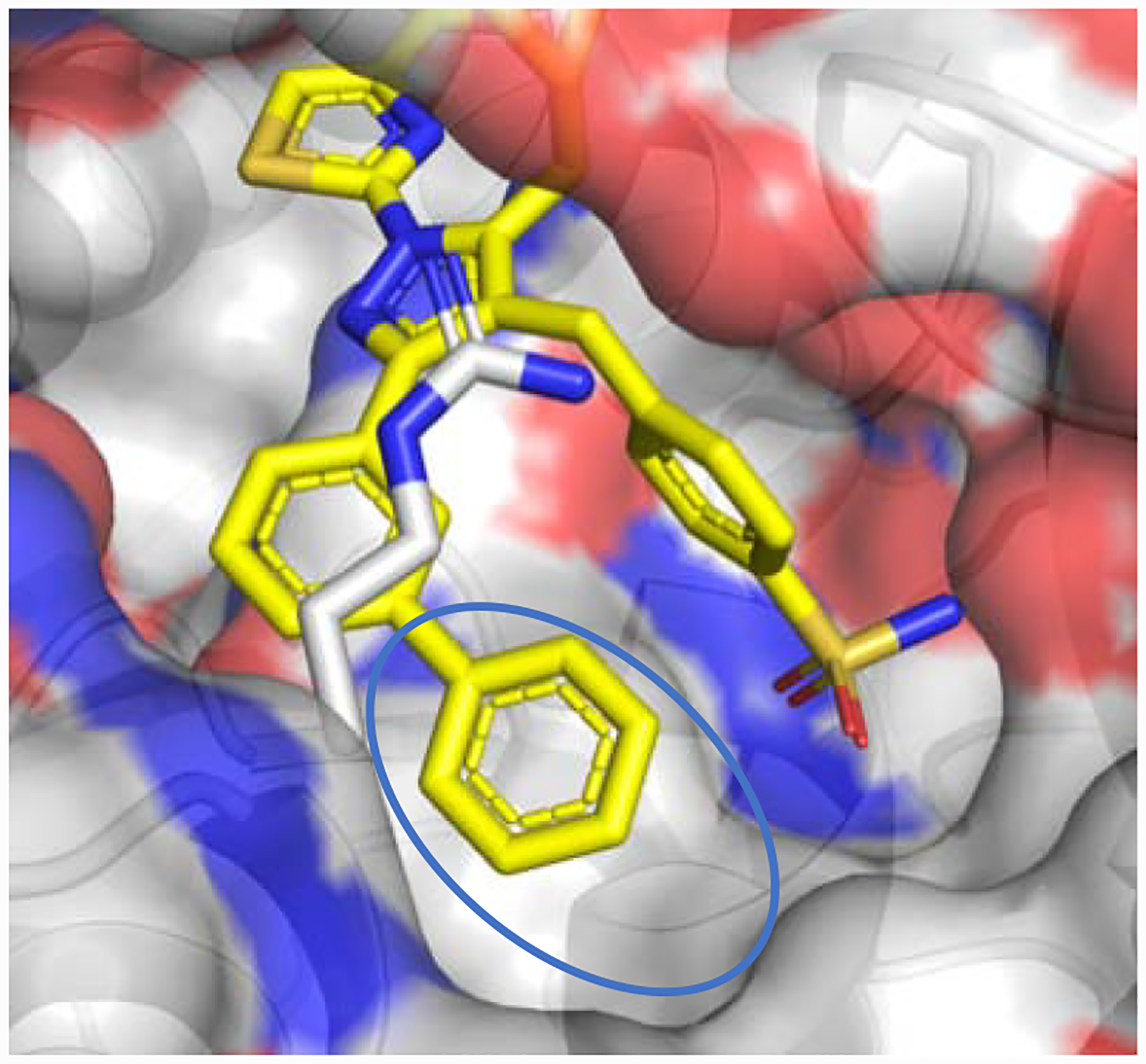
X-ray co-crystal structure of 1a bound to LDHA (PDB code 5W8L).15 Oxygen atoms are shown in red, nitrogens in blue. Lipophilic space is highlighted by a blue oval. Surface representation of Lys105 omitted for clarity.
The basic synthetic routes used to construct these analogs are depicted in Schemes 1 and 2. For pyrazoles bearing no substitution at the 5-position, we utilized sequential functionalization of a bromopyrazole. For most analogs, this compound was synthesized by condensation of hydrazine onto an aldol product of 2, followed by bromination and installation of the thiazole by SNAr reaction. Deprotection of the methyl ether to a phenol allowed installation of diversity using straightforward alkylation procedures. The phenyl sulfonamide was incorporated from the corresponding pinacol boronate, and saponification of the ester yielded analogs 6 b-g. In some cases (6a, 6h), the corresponding commercially available acetophenones were carried through the sequence.
Scheme 1.
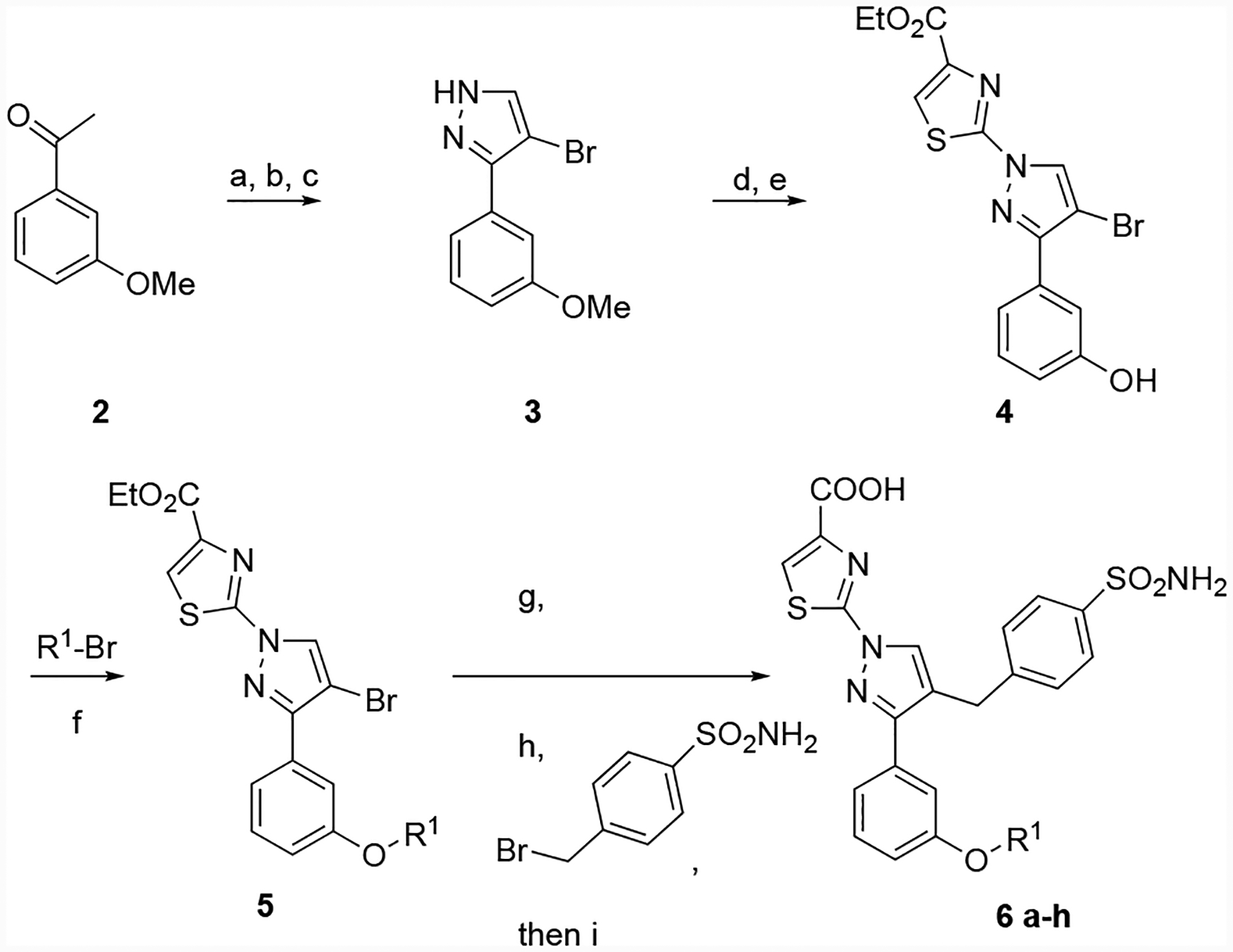
Preparation of 5-H pyrazole analogs. Reagents and conditions: (a) C2H5OCHO, NaH; (b) hydrazine, ethanol, 92 % (two steps); (c) NBS, DMF, 70 %; (d) ethyl 2-bromothiazole-4-carboxylate, K2CO3, DMSO, 73%; (e) BBr3, DCM, 60 %; (f) K2CO3, DMF; (g) 4,4,4’,4’,5,5,5’,5’-octamethyl-2,2’-bi(1,3,2-dioxaborolane), PdCl2(dppf), 1,4-dioxane; (h) Pd(PPh3)4, K2CO3, dioxane, water; (i) 1 M LiOH, THF, methanol.
Scheme 2.
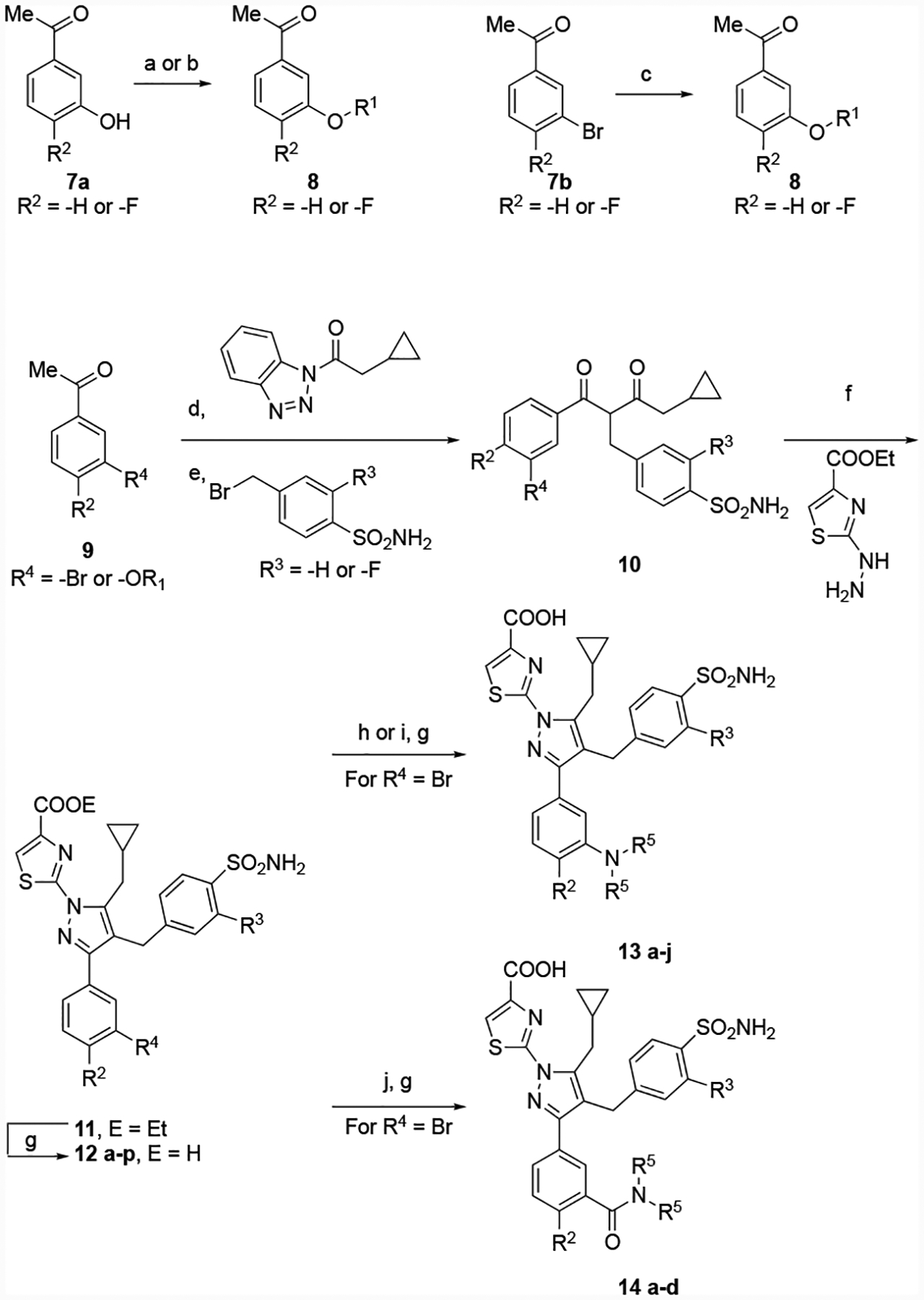
Preparation of 5-methylene cyclopropyl pyrazole analogs. Reagents and conditions: (a) R1Br, K2CO3, DMF, 100–160 °C; b) R1OH, PPh3, di-t-butyl diazocarboxylate THF, 0 °C. 95%; c) R1B(OH)2, Cu(OAc)2, pyridine, DCM; (d) MgBr2-OEt2, DCM, DIPEA; e) Cs2CO3, KI, DMSO; (f) pyrrolidine, EtOH, pTSA 90°C; (g) LiOH or NaOH, dioxane, MeOH, water; (h) R5NC, PdCl2(Ph3P)2, CsF, DMSO, water; (i) NH2R5, K3PO4, Pd(P(tBu)3)2, DMA, 53 %; (j) HN(R5)2, Mo(CO)6, Pd(OAc)2, T-BINAP, Cs2CO3, MeCN, toluene.
A survey of small functionalities at the 3-ether position (Table 1) revealed the potential for good LDHA inhibition in this series. Based on the methoxy-bearing 6a, a wide range of lipophilic substitutions effected a small improvement in potency of LDHA inhibition. Indeed, good tolerance is shown for a simple cyclic alkane (6b), several simply substituted benzyl systems (6c-e), and a phenyl ether (6h). Interestingly, a 3-pyridinylmethyl substitution (6g) was ~3 fold less potent compared with a 2-pyridinyl group (6f). Despite generally good enzyme inhibition, these compounds produced only modest inhibition of LDHA activity in cells, with none of the compounds demonstrating an IC50 for cellular lactate production of less than 5 μM.
Table 1.
Initial survey of ethers at the 3-position.
| Cpd | R1 | LDHA IC50 (nM)a | MaPaCa2 Lactate IC50 (μM)b |
|---|---|---|---|
| 6a | methyl | 90 | >57 |
| 6b | cyclopentylmethyl | 29 | 7.7 |
| 6c | 3-fluorobenzyl | 26 | 6.1 |
| 6d | 3-methoxybenzyl | 21 | 5.4† |
| 6e | 4-methylbenzyl | 39 | 7.5† |
| 6f | 2-pyridinylmethyl | 23 | >57 |
| 6g | 3-pyridinylmethyl | 77 | >57 |
| 6h | phenyl | 17 | 9.4† |
We hypothesized that, much like our previously reported compounds, application of the contemporaneously discovered SAR regarding pyrazole substutitions15 and phenyl sulfonamide fluorination16 would afford more potent analogs. Thus, we prepared compounds bearing an alkyl substituent at the 5-position of the pyrazole, using synthetic routes similar to those reported previously (Scheme 2)15,16 Elaboration of the acetophenones 7a and 7b to the desired ethers 8 proved easier than late-stage functionalization for most analogs. The derivatized acetophenones were alkylated twice using the previously described conditions15,16 to give the 1,3-diketones 10, which were cyclized to the corresponding pyrazoles 11. As reported, the cyclization produces a separable mixture of regioisomers; the desired isomer was cleanly saponified to give the ether analogs 12 a-p.
The activity data with these compounds (Table 2) was generally improved relative to the compounds in Table 1, with some analogs generating single digit nanomolar inhibition of the enzyme and submicromolar inhibition of lactate production in MiaPaCa-2 cells. Compounds without the sulfonamide ring fluorine tended toward lower activity (e.g. 12i). The matched pair 12m vs. 12l demonstrates an extreme example of this relationship. Interestingly, we found an imperfect correlation between enzyme and cellular inhibition data. For example, while the 5–20 nM inhibition data for analogs 12b-g demonstrates again a wide tolerance for alkyl ether groups in LDH inhibition, the accompanying lactate data varies in a different pattern. Analogs 12f and 12g are the most potent of this set in the lactate assay, despite higher enzyme IC50 values. We selected the benzyl substitution of 12g for further elaboration (selected compounds shown). Fluoro substitution on this benzyl (12h) was well tolerated, but replacement with a heterocycle (12j) diminished activity. We also elected to explore removal of the methylene with phenyl ethers 12 k-p. While enzyme inhibition for this phenyl ether subseries was generally weaker than with comparators bearing alkyl substituents, several analogs still showed lactate inhibition IC50 values near 1 μM.
Table 2.
Ether analogs (12) bearing the cyclopropylmethyl pyrazole substitution.
| Cpd | R2/3 | R4 | LDHA IC50 (nM)a | MaPaCa2 Lactate IC50 (μM)b |
|---|---|---|---|---|
| 12a | F/H | -O-methyl | 8 | 5.7 |
| 12b | F/F | -O-2,2-difluorocyclo-propyl)methyl | 5 | 1.4 |
| 12c | F/F | -O-3-tetrahydrofuranylmethyl | 5 | 3.0 |
| 12d | F/F | -O-2-tetrahydrofuranylmethyl | 5 | 2.9 |
| 12e | F/F | -O-cyclopropyl | 8 | 2.1 |
| 12f | H/F | -O-cyclopentyl | 13† | 0.71† |
| 12g | H/F | -O-benzyl | 19 | 0.69 |
| 12h | F/F | -O-3-fluorobenzyl | 22 | 0.67† |
| 12i | F/H | -O-4-fluorobenzyl | 84 | 3.3† |
| 12j | F/F | -O-(5-(trifluoromethyl)-2-furanyl)methyl | 23 | 1.0 |
| 12k | F/H | -O-phenyl | 34 | 1.7 |
| 12l | H/H | -O-3-fluorophenyl | 1060 | 19 |
| 12m | F/F | -O-3-fluorophenyl | 27 | 0.97 |
| 12n | F/F | -O-4-fluorophenyl | 41 | 1.7 |
| 12o | F/F | -O-4-trifluoromethylphenyl | 71 | 1.1 |
| 12p | F/F | -O-4-methylphenyl | 26 | 0.87† |
We also evaluated aniline derivatives (13 a-j, Table 3). These analogs were prepared either from acetophenones similar to 9 (Scheme 2), but bearing the required amines, or from a later stage palladium-mediated Buchwald-Hartwig style coupling reaction17 from the corresponding bromides (11, R4 = Br) that were carried through the synthetic route (Scheme 2) from the corresponding acetophenone. A selection of small saturated cyclic analogs 13 a-f were evaluated, showing a rather narrow SAR for LDHA inhibition and cellular effects. Reinforcing a trend observed in our earlier work, the cellular data did not track well with biochemical inhibition of LDHA. For example, the morpholine analog 13a showed reasonable LDHA inhibition, but poor cellular activity, perhaps as a result of the polar oxygen atom. Removing this polarity (13 b-f) generated mixed results in enzyme inhibition, but generally improved cellular IC50 values by ~3–10 fold. The more elaborated spirocyclic azetidine 13g and the aniline 13h displayed inhibition data within the same range as the initial amines.
Table 3.
Amine (13) and amide (14) analogs.
| Cpd | R2/3 | R5 | LDHA IC50 (nM)a | MaPaCa2 Lactate IC50 (μM)b |
|---|---|---|---|---|
| 13a | H/F | -CH2CH2OCH2CH2- | 27 | 23.4 |
| 13b | H/F | -CH2CH2CF2CH2CH2- | 17 | 8.7 |
| 13c | H/F | -CH2CH2CH(CF3)CH2CH2- | 47 | 2.4 |
| 13d | H/F | -CH2CF2CH2CH2CH2- | 18 | 8.6 |
| 13e | H/F | -CH2CH(CF3)CH2CH2CH2- | 59 | 6.9 |
| 13f | H/F | -CH2CF2CH2CH2- | 15 | 4.2 |
| 13g | F/F | 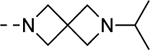 |
37 | 24.1† |
| 13h | H/H | H, phenyl | 314 | 6.7 |
| 13i | F/F | H, CO-phenyl | 7 | 5.0 |
| 13j | F/F | -COCH2CH2CH2- | 111 | 6.0† |
| 14a | F/F | H, -tBu | 6 | 11.7 |
| 14b | F/F | -CH2CH2CH2CH2- | 154† | 19.9† |
| 14c | F/F | -CH2CH2OCH2CH2- | 22 | 3.2† |
| 14d | F/F | H, benzyl | 8† | 9.1† |
We postulated that an amide bond would create a planar derivative that might match more closely with the biphenyl and alkynes previously reported, and thus have superior residence time and cellular activity.15,16 These analogs were synthesized from a bromide-bearing intermediate 11 (R4 = Br). The amides were installed with either a coupling of an isocyanate and subsequent hydrolysis and saponification (for nitrogen-attached amides 13 a-j) or by a molybdenum-mediated carbonylative amidation and saponification (to give carbonyl-attached amides 14 a-d). Ultimately, these analogs also failed to deliver a noticeable boost to biochemical or cellular activity. Despite certain analogs displaying potent LDHA inhibition, with IC50 values of less than 10 nM (14a, 14d), the amine and amide subseries as a whole was somewhat inferior in the cellular lactate inhibition assay compared to the ether subseries.
As we have previously reported15,16, compounds in the pyrazole chemotype tend to be potent inhibitors of both LDHA and LDHB. A similar relationship was found in this work, with LDHB IC50 values typically 2–4 fold better than those for LDHA (Table 4).
Table 4.
Characterization of selected analogs.
| Cpd | LDHB IC50 (μM)a | MiaPaCa2 Lactate (μM)b | MiaPaCa2 cytotox (μM)c | A673 Lactate (μM)b | (A673) cytotox (μM)c | SPR KD (nM) / τ (s) | Rat Microsomal Stability t1/2 (min)d | PAMPA Perm (x10−6 cm/sec) | Kinetic solubility (μg / mL) |
|---|---|---|---|---|---|---|---|---|---|
| 12b | 3† | 1.4 | 2.4 | 1.1 | 0.90 | 0.008 / 21,052 | > 30 | 4.9 | 76.1 |
| 12e | 3 | 2.1 | 5.7 | 1.9 | 1.5 | 0.004 / 19,230 | > 30 | 5.8 | > 87 |
| 12g | 5 | 0.69 | 1.4 | 0.59 | 0.42 | 0.016 / 88,496 | 2.8 | 1.1 | 72.4 |
| 12h | 5 | 0.67† | 2.1 | 0.73 | 0.55 | 0.91 / 13,568 | < 1 | 2.9 | 10.7 |
| 12j | 6† | 1.0 | 2.1 | 1.1 | 0.44 | 0.001 / 100,000 | > 30 | 12.0 | 53.2 |
| 12p | 19 | 0.87† | 2.1† | 0.55 | 0.55 | 1.1 / 9,708 | 6.2 | 13.0 | 24.0 |
| 13a | 8† | 23.4 | 18.7 | 9.4† | 20.4 | 0.44 / 286 | > 30 | 1.6 | > 88 |
| 13h | 1 | 6.7 | 21.2† | 3.1 | 34.5† | 1.5 / 59 | 6.0 | 4.4 | 49.3 |
| 13i | 2 | 5.0 | 17.4 | 2.3 | 5.3 | - | > 30 | 6.4 | > 98 |
| 14b | 4 | 19.9† | 6.6† | 16.6 | > 50 | - | > 30 | 5.7 | > 93 |
| 14d | 2 | 9.1† | 15.7 | 4.7 | 5.1 | - | 14.9 | 8.7 | 81.0 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the previously reported assay,15,16 reported as a mean of 3 determinations. CV ≤ 0.2 unless otherwise noted by†.
IC50 values represent the half maximal (50%) inhibition of lactate,15,16 reported as a mean of 3 determinations. CV ≤ 0.33 unless otherwise noted by†.
IC50 values represent the half maximal (50%) inhibition of proliferation,15,16 reported as a mean of 3 determinations. CV≤ 0.33 unless otherwise noted by†.
Assessed using a high-throughput multi-time point stability assay.18
As a class, the ether compounds trend toward superior cellular potency compared to amine-based compounds, not only in the MiaPaCa2 lactate production assay, but also in an A673 Ewing’s Sarcoma line (Table 4). In general, the effects of the compounds on cellular proliferation showed a similar trend as the lactate production assay, with the ether compounds demonstrating low micromolar cytotoxicity in the MiaPaCa2 line. Notably, activity in A673 cells was typically superior to 1a or 1b for some of the ethers, with 12g, 12h, 12j, and 12p showing submicromolar proliferation IC50 values.
We assessed selected compounds for their binding to LDHA by SPR. Similar to previously reported compounds, some of the ether-based inhibitors (e.g. 12b, 12e, 12h, 12j, and 12p) with submicromolar inhibition of lactate production in the MiaPaCa2 cells showed very tight binding to LDHA and accompanying long residence times, whereas alkyl amines, such as 13a, and the aniline 13h, characteristically displayed faster dissociation kinetics, with inferior cellular data to match.
The compounds reported in this manuscript benefited from generally good solubility and low, but non-limiting, passive permeability characteristics (Table 4). However, the initial assessments of metabolic stability produced mixed results. Simple alkyl ethers (e.g. 12b, 12e) were typically reasonable, whereas a benzyl ether (12h) was quite poor, as was a biphenyl ether (12p). Interestingly, heteroaromatic moieties (12j) can rescue the stability of the benzyl subseries, indicating promise for the identification of more stable pseudo-benzylethers. Likewise, selected amine-based compounds showed some variability in the microsomal data. While amides in both configurations (13i and 14b) showed good stability, the introduction of a benzyl position (14d) presented a likely metabolic soft spot.
This paper presents alternative LDHA inhibitors with heteroatomic structural elaborations, based on the previously reported pyrazole series. Selected compounds achieved low nanomolar inhibition of LDHA and LDHB. However, cellular inhibition of LDH function was less consistent. From this study, ether analogs bearing a cyclopropylmethyl pyrazole substitution (scaffold 12) stood out, with some examples showing single digit picomolar binding affinity for LDHA by SPR and inhibition of proliferation of an Ewing’s Sarcoma line superior to earlier leads. Variable SAR with respect to metabolic stability provides a direction for additional optimization of this sub-class of inhibitors.
Supplementary Material
Acknowledgments
We thank David Myszka at Biosenor Tools for conducting the SPR experiments. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Compounds reported in this manuscript are also exemplified in patent applications that have been licensed. Under certain circumstances, co-authors on this manuscript may receive royalties or other payments through their respective employers.
References and notes
- 1.Warburg O; Posener K; Negelein E Biochem. Z 1924, 152, 319–344. [Google Scholar]
- 2.Vander Heiden MG; Cantley LC; Thompson CB Science 2009, 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu Y; Liao M; Liu R; Chen J; Feng H; Fu Z World J. Surg. Oncol 2014, 12, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rong Y; Wu W; Ni X; Kuang T; Jin D; Wang D; Lou W Tumour Biol. 2013, 34, 1523–1530. [DOI] [PubMed] [Google Scholar]
- 5.Allison SJ; Knight JRP; Granchi C; Minutolo F; Phillips RM Oncogensis 2014, 3, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le A; Cooper CR; Gouw AM; Dinavachi R; Maitra A; Deck LM; Royer RE; Vander Hagt DL; Semenza GL; Dang CV Proc. Natl. Acad. Sci 2010, 107, 2037–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheng SL; Liu JJ; Dai YH; Sun ZG; Xiong XP; Huang G FEBS Journal 2012, 279, 3898–3910. [DOI] [PubMed] [Google Scholar]
- 8.Fantin VR; St-Piere J; Leder P Canc. Cell, 2006, 9, 425–434. [DOI] [PubMed] [Google Scholar]
- 9.Xie H; Hanai J. -i.; Ren J-G; Kats L; Burgess K; Bhargava P; Signoretti S; Billiard J; Duffy KJ; Grant A; Wang X; Lorkiewicz PK; Schatzman S; Bousamra M; Lane AN; Higashi RM; Fan TWM; Pandolfi PP; Sukhatme VP; Seth P Cell Metab. 2014, 19, 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Granchi C; Roy S; Giacomelli C; Macchia M; Tuccinardi T; Martinelli A; Lanza A; Betti L; Giannaccini G; Lucachhini A; Funel N; León LG; Giovannetti E; Peters GJ; Palchaudhuri R; Calvaresi EC; Hergenrother PJ; Minutolo F J. Med. Chem 2011, 24, 1599–1612. [DOI] [PubMed] [Google Scholar]
- 11.Kohlmann A; Zech SG; Li F; Zhou T; Squillance RM; Commodore L; Greenfield MT; Lu X; Miller DP; Huang W-S; Qi G; Thomas RM; Wang Y; Zhang S; Dodd R; Liu S; Xu R; Xu Y; Miret JJ; Rivera V; Clackson T; Shakespeare WC; Zhu X; Dalgarno DC J. Med. Chem 2013, 56, 1023–1040. [DOI] [PubMed] [Google Scholar]
- 12.Ward RA; Brassington C; Breeze AL; Caputo A; Critchlow S; Davies G; Goodwin L; Hassall G; Greenwood R; Holdgate GA; Mrosek M; Norman RA; Pearson S; Tart J; Tucker JA; Vogtherr M; Whittaker D; Wingfield J; Winter J; Hudson K J. Med. Chem 2012, 55, 3285–3306. [DOI] [PubMed] [Google Scholar]
- 13.Billiard J; Dennison JB; Briand J; Annan RS; Chai D; Colón M; Dodson CS; Gilbert SA; Greshock J; Jing J; Lu H; McSurdy-Freed JE; Orband-Miller LA; Mills GB; Quinn CJ; Schneck JL; Scott GF; Shaw AN; Waitt GM; Wooster RF; Duffy KJ Cancer Metab. 2013, 1, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boudreau A; Purkey HE; Hitz A; Robarge K; Peterson D; Labadie S; Kwong M; Hong R; Gao M; Del Nagro C; Pusapati R; Ma S; Salphati L; Pang P; Zhou A; Lai T; Li Y; Chen Z; Wei B; Yen I; Sideris S; McCleland M; Firestein R; Corson L; Vanderbilt A; Williams S; Daemen A; Belvin M; Eigenbrot C; Jackson PK; Malek S; Hatzivassiliou G; Sampath D; Evangelista M; O’Brien T Nat. Chem. Biol 2016, 12, 779–786. [DOI] [PubMed] [Google Scholar]
- 15.Rai G; Brimacombe KR; Mott BR; Urban DJ; Hu X; Yang S-M; Lee TD; Cheff DM; Pohida K; Benavides GA; Darley-Usmar VM; Moore WJ;Stott G; Flint A; Hall MD; Neckers L; Van Dang C; Waterson AG; Simeonov A;Jadhav A; Maloney DJ J. Med. Chem 2017; 60: 9184–9204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rai G; Urban D; Mott B; Hu X; Yang S-M; Benavides G; Johnson M; Squadrito G; Brimacombe K; Lee T; Cheff D; Zhu H; Henderson MJ; Pohida Ka.; Sulikowski G; Dranow D; Kabir M; Shah P; Padilha E; Tao D; Fang Y; Christov P; Kim K; Jana S; Muttil P; Anderson T; Kunda N; Hathaway H; Kusewitt D; Oshima N; Cherukuri M; Davies D; Norenberg J; Sklar L; Moore Wi.; Dang C; Stott G; Neckers L; Flint A; Usmar VD; Simeonov A; Waterson A; Jadhav A; Hall M; Maloney D “Pyrazole-Based Lactate Dehydrogenase (LDH) Inhibitors with Optimized Cell Activity and Pharmacokinetic Properties” J. Med. Chem 2020, 63(19), 10984–11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruiz-Castillo P; Buchwald SL Chem. Rev 2016, 116(9), 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah P; Kerns E; Nguyen D-T; Obach RS; Wang AQ; Zakharov A; McKew J; Simeonov A; Hop CECA; Xu X Drug Metab. Dispos 2016, 44, 1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.