

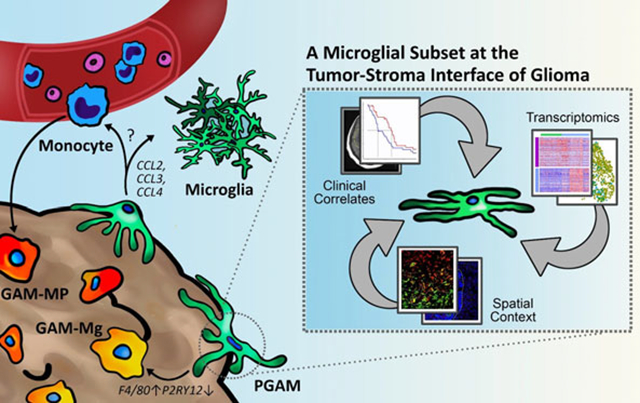
Abstract
The characterization of the tumor microenvironment (TME) in high grade gliomas (HGG) has generated significant interest in an effort to understand how neoplastic lesions in the central nervous system (CNS) are supported and to devise novel therapeutic targets. The TME of the CNS contains unique and specialized cells, including the resident myeloid cells, microglia. Myeloid involvement in HGG, such as glioblastoma, is associated with poor outcomes. Glioma-associated microglia and infiltrating monocytes/macrophages (GAM) accumulate within the neoplastic lesion where they facilitate tumor growth and drive immunosuppression. However, it has been difficult to differentiate whether microglia and macrophages have similar or distinct roles in pathology, and if the spatial organization of these cells informs outcomes. Here, we characterize the tumor–stroma border and identify peritumoral GAM (PGAM) as a unique subpopulation of GAM. Using data mining and analyses of samples derived from both murine and human sources we show that PGAM exhibit a pro-inflammatory and chemotactic phenotype that is associated with peripheral monocyte recruitment, and decreased overall survival. PGAM act as a unique subset of GAM at the tumor–stroma interface. We define a novel gene signature to identify these cells and suggest that PGAM constitute a cellular target of the TME.
Keywords: CCL2, GAM, glioma, leading edge, microglia, P2RY12
Graphical Abstract
1 |. INTRODUCTION
Infiltrating myeloid cells can comprise up to a third of high grade glioma (HGG) lesions (da Fonseca & Badie, 2013; Hambardzumyan, Gutmann, & Kettenmann, 2016). As gliomas arise from central nervous system (CNS) tissue, the tumor microenvironment (TME) is unique in that it contains neurons, astrocytes, and the resident myeloid cells, microglia. Microglia act as innate immune hubs of the CNS and influence many neuropathological settings. Inflammation and disruption of the blood–brain barrier result in the recruitment of monocytes/macrophages which leads to worsened disease outcomes. Despite their distinct cellular ontology (Ginhoux et al., 2010; Ginhoux, Lim, Hoeffel, Low, & Huber, 2013), microglia and macrophages have been described as virtually indistinguishable in pathological microenvironments in the CNS because they express similar biological markers and phenotypic properties. In HGG, it has become well recognized that glioma-associated microglia/macrophages (GAM) are associated with tumor support, therapeutic resistance, and poor survival (Hewedi, Radwan, Shash, & Elserry, 2013; Quail & Joyce, 2017; Sorensen, Dahlrot, Boldt, Hansen, & Kristensen, 2017). However, it is still not fully understood whether there are distinct roles for microglia and macrophages in HGG pathology and what these roles may be.
Recent application of powerful sequencing technologies such as single cell and bulk RNA sequencing have identified key transcripts that differentiate microglia from macrophages, for example, P2RY12, TMEM119, and OLFML3 (M. L. Bennett et al., 2016; Darmanis et al., 2015; Haage et al., 2019; Zhang et al., 2014), and additional comprehensive analyses have been able to parse differences between microglia and macrophages in HGG (Bowman et al., 2016; Z. Chen et al., 2017; Muller et al., 2017; Venteicher et al., 2017). Interestingly, these transcriptional identities are paralleled in human and mouse myeloid cells (M. L. Bennett et al., 2016; Masuda et al., 2019; Szulzewsky et al., 2015). As a result, several GAM therapeutic targets have been proposed, although their effectiveness has yet to be determined (Μ. L. Bennett & Bennett, 2019; Choi, Mai, Jackson, Belcaid, & Lim, 2018; Jackson, Lim, & Drake, 2014; Pyonteck et al., 2013).
Another factor complicating GAM transcriptional identities is the presence of unique local TME niches such as hypoxic cores, necrotic lesions, hyper-proliferative vasculature, and the expanding tumor margins. Recently, several microglia-specific genes including P2RY12, CX3CR1, and SIGLEC8 were found to associate with the leading edge of glioma12. Microglia in HGG are not uniform and rather exhibit a spectrum of activation, that is, a percentage of cells express relatively high levels of inflammatory markers (CCL3, CCL4, CCL3L1, CCL3l3, IL1A, IL1B, IL8, TNF) while the remainder resemble resting/homeostatic microglia (Levitin et al., 2019; Sankowski et al., 2019; Szulzewsky et al., 2015; Tirosh et al., 2016; Zhang et al., 2014). One study suggests that this heightened state of activation in HGG may be inherent to age-related microglia changes (Sankowski et al., 2019). Nevertheless, this pro-inflammatory status contradicts the global GAM assumption which describes GAM as universally anti-inflammatory and pro-tumorigenic. Taken together, an interesting question arises regarding the role of microglia at the leading edge of glioma: are microglia of this TME niche unique, and if so, what consequences does their presence have on malignant pathologies?
Until recently, molecular analysis of patient bulk samples has been confounded by the compilation of biological samples deriving from mixed sources, barring high-resolution analysis of the leading edge and tumor margins. In a recent publication Darmanis et al. sampled matched tumor core and peritumoral tissue from four subjects diagnosed with primary glioblastoma (GB/GBM) and performed single-cell RNA sequencing (scRNAseq) (Darmanis et al., 2017). Neoplastic cells of the invading glioma front were found to possess unique transcriptional and genetic profiles ultimately supporting invasive phenotypes. When the myeloid population was characterized, the peritumoral samples were enriched for microglia-specific transcripts and inflammatory markers CCL2, CCL3, CCL4, IL1A, IL1B, TNF, CXCL12, and IL6ST (Darmanis et al., 2017). This finding suggests that the pro-inflammatory and chemotactic signature consistently observed in tumor-associated microglia could be resultant from cells residing at the tumor leading edge. However, these peri-tumoral GAM (PGAM) have not been defined and their contributions to HGG pathology are still unknown.
Through data mining and integration of human databases, as well as the use of HGG murine models, we demonstrate that PGAM constitute a distinct microglial subset of GAM and are located at the leading-edge niche of HGG. We carefully define a transcriptional fingerprint of PGAM and use this information to correlate with disease outcomes.
2 |. RESULTS
2.1 |. Identification of peripheral glioma-associated microglia
We analyzed the myeloid cells of the Darmanis et al.’s dataset (Darmanis et al., 2017) (Figure 1a). In agreement with published results, our analysis identified a discrete cluster of myeloid cells in peri-tumoral tissue samples (Figure 1b) based on the expression of previously reported microglia-specific genes (M. L. Bennett et al., 2016; Darmanis et al., 2017) (TMEM119, P2RY12, GPR34, OLFML3, SLC2A5, SALL1, and ADORA3, Figure 1b). Then, 92.4% of myeloid cells from the peri-tumoral tissue were positive for microglia selection criteria. The remaining cells expressed markers for microglia and macrophages which represented classically described GAM populations. Expression of the seven microglia-specific transcripts was mapped across all myeloid cells (Figure 1c, Supplemental Figure 1); notably, P2RY12 localized almost exclusively to the PGAM group. Cells labeled as “Distant” represent nonlocal samples taken from the Darmanis et al.’s study. They were removed from our subsequent analyses. These results separate peri-tumoral microglia from microglia/macrophages of the tumor core. Moreover, peritumoral and adjacent CNS myeloid populations of cells are almost exclusively microglia.
FIGURE 1.

Identification of peripheral glioma-associated microglia. Single-cell RNA sequencing (scRNAseq) analysis from Darmanis et al.’s dataset. (a) Following dimensional reduction, cells were annotated based on canonical marker expression (left). Myeloid cells segregate with respect to samples from tumor core, or adjacent central nervous system (CNS) tissue (right). (b) Myeloid cells from the peripheral tissue sample were selected for microglial core gene expression (TMEM119, P2RY12, GPR34, OLFML3, SLC2A5, SALL1, and ADORA3), and annotated as peritumoral glioma-associated microglia (PGAM) (n = 570). All other myeloid cells were labeled as bona fide glioma-associated microglia/macrophages (GAM) (n = 1,254). Samples were allowed to recluster via UMAP. (c) Microglial core gene expression plotted across the new myeloid clustering from Panel (b). The color scale indicates expression levels
We next sought to identify transcriptional differences between GAM of the tumor core and PGAM. Differential expression (DE) analysis identified several biologically relevant transcripts in PGAM. The DE transcripts were ranked and Gene Set Enrichment Analysis (GSEA) was performed (Subramanian et al., 2005). PGAM were enriched for chemotactic and pro-inflammatory programs carrying high relative expression of CCL2, CCL3, CCL4, CCL3L1, CXCL12, CSF1, TNF, and IL1B (Figure 2a). Based on relative expression, cytokine and chemokine receptor-ligand pairs were predicted which revealed elevated patterns of ligand expression in PGAM, extensive crosstalk between GAM and PGAM, as well as crosstalk between myeloid and neoplastic cells (Figure 2b). These results suggest the existence of a unique subpopulation of microglial cells in GBM, which reside in the peritumoral space and express high levels of chemotactic and pro-inflammatory genes.
FIGURE 2.

Peritumoral glioma-associated microglia (PGAM) are chemotactic and pro-inflammatory, and PGAM markers localize at the tumor–stroma interface in glioma. (a) Transcripts identified from differential gene expression are plotted against their associated GO terms from Gene Set Enrichment Analysis (GSEA) p val <.01 FDR <0.05. Axes are sorted such that highly overlapping transcripts and GO terms group together. Tiles are colored by fold change rank metric, relative to tumor core glioma-associated microglia/macrophages (GAM) expression. (b) Using the iTALK package, receptor-ligand pairs are predicted based on relative expression, between PGAM, GAM, and neoplastic cells. Arrows are colored by cellular population. Arrow lines are scaled by ligand expression, arrow heads are scaled by receptor expression. Inner circle: light blue denotes ligands; dark blue denotes receptor. (c) Left, representative H&E image of GL261 murine glioma, indicating where contralateral healthy, tumor core, and tumor peripheral tissues were sampled. Right, qRT-PCR log transformed ΔCT values of PGAM markers detected in (a), across sample region. n = 2–3. (d) CCL2+ cells populate peritumoral space in GL261 glioma samples. F4/80 is a pan macrophage/microglia marker. n = 3. (e) Bulk RNAseq samples from Bowman et al, and Haage et al. were combined, and PGAM markers were plotted against FACS sorted cell types, and murine glioma models GL261 and RCAS. PGAM markers associate with microglia populations, over BMDM and monocytes in both models. n = 3–6. (f) P2RY12 staining at the tumor–stroma border in GL261 murine glioma, Day 21. White dotted line represents tumor leading edge, white boxes highlight P2RY12+F4/80High microglia. Scale bar = 100 μm. (g) Semiquantitative analysis of spatial distribution of P2RY12 expression across the tumor–stroma interface in glioma, as in (f). Spatial signal from imaging technical replicates were scaled and combined, generating an average expression curve. The highly nucleated tumor, marked by high DAPI expression, correlates with the tumor border. n = 4. Scale bar = 50 μm. (h) Semiquantitative analysis of spatial distribution of TMEM119 expression inside the tumor core (left) and across the tumor–stroma interface (right) in glioma, as in (g,f). White boxes highlight patches of intratumoral TMEM119+F4/80High microglia. White arrows mark TMEM119+F4/80High microglia at tumor–stroma interface. n = 3. Scale bar = 250 and 100 μm
To examine if the chemotactic and pro-inflammatory phenotype observed in peri-tumoral microglia uncovered from the scRNAseq analysis of the human datasets is present in murine systems as well, we introduced orthotopic gliomas in C57Bl6 mice using 2 syngeneic glioma cell lines (GL261 and KR158B) (Chae et al., 2015; Reilly, Loisel, Bronson, McLaughlin, & Jacks, 2000; Seligman & J, 1939). Gliomas were microdissected and samples from the core, peri-tumoral space, and contralateral hemisphere (representing healthy tissue) were obtained (Figure 2c, left). Quantitative real-time PCR was performed on cDNA libraries generated from these tissue samples and gene expression of top PGAM markers was analyzed (Figure 2c). Relative expression of pro-inflammatory markers Ccl2, Ccl3, Ccl4, Il1b, and Tnf was all found to be elevated in the peritumoral sample corroborating the results from DE and GSEA.
Immunofluorescent staining of fixed tissue sections from the murine samples confirmed a high density of CCL2+/F4/80Low cells at the tumor–stroma border compared to cells of the tumor core in both glioma lines used (Figure 2d, Supplemental Figure 2a). To explore if these CCL2+/F4/80Low levels were associated with microglia, we utilized recently published FAC-sorted GAM bulk RNAseq datasets derived from the GL261 and RCAS/tv-a murine glioma models (Bowman et al., 2016; Haage et al., 2019). Bulk microglia are distinguished from macrophages based on CD11b and CD45 expression. Analysis of these datasets demonstrated that expression of Ccl2, Ccl3, and Ccl4 were markedly elevated in microglia (CD11b+/CD45int) over macrophages (CD11b+/CD45hi) and peripheral blood monocyte populations (Figure 2e), suggesting that microglia are the major source of these signaling molecules. It should be noted, however, that homeostatic microglia express these genes as well. Indeed, CCL2+ cells were present in contralateral healthy tissue sections of our samples (Supplemental Figure 2b). Similarly, across all cell types in the Darmanis et al.’s single cell dataset, CCL2, CCL3, and CCL4 were elevated in the myeloid cell populations of the peripheral tissue sample (Supplemental Figure 2c). Together, these data suggest that the primary source of these chemoattractants are the peritumoral microglia. As myeloid cells comprise the majority of noncancerous cell types found in individual biopsies (Supplemental File 1), this result also indicates both a relative and absolute increase of the expression of these molecules. The use of several orthogonal methods corroborates the existence of this unique chemotactic and pro-inflammatory PGAM subpopulation at the tumor–stroma interface in glioma.
Among cell signaling molecules differentially expressed between PGAM and tumor core GAM, P2RY12 was ranked as the sixth most DE gene consistent with its role as a microglia-specific marker (M. L. Bennett et al., 2016; Gu et al., 2016). To investigate the spatial distribution of P2RY12+ microglia in glioma, and interrogate its relevance as a potential marker of PGAM, immunostaining for P2RY12 was conducted in the murine glioma tissue (Figure 2f, Supplemental Figure 3a,b). A distinct double positive population of P2RY12+F4/80High cells was visualized exclusively at the tumor–stroma interface, in contrast to P2RY12+F4/80Low microglia in the adjacent CNS tissue, and P2RY12−F4/80High GAM of the tumor core (Figure 2f, Supplemental Figure 3b–d). Semiquantitative spatial analysis of P2RY12 signal across the tumor–stroma border identified a zone, approximately 400 μm in width, surrounding the glioma leading edge harboring double positive P2RY12+F4/80High cells (Figure 2g).
When TMEM119 was probed in the same samples as an additional microglia-specific transcript (M. L. Bennett et al., 2016), a different staining pattern was observed. TMEM119+ cells were visualized both within the tumor core as well as along the tumor border (Figure 2h). These results demonstrate that microglia retain TMEM119 expression after populating the neoplastic lesion, while P2RY12 marks microglia exclusively across the tumor–stroma interface and characterizes changes in microglia-specific genes HGG (Jordao et al., 2019).
It should be noted that the immunofluorescent staining marking these double positive peritumoral microglia was not homogeneously detected around the entire glioma leading edge but rather was evident in patches along the invading margin. This finding is consistent with recent reports which identify community-level behaviors along the leading edge of glioma (Alieva et al., 2019; Hide et al., 2018). Similarly, cell bodies of mature neurons among neoplastic cells were detected at different areas along the tumor–stroma interface (Supplemental Figure 3e,f).
Given the expression change of P2YR12 in the murine glioma models (Figure 2), we sought to examine whether P2RY12 is expressed similarly in human glioma. Immunostaining was conducted on xenografts from primary and recurrent GBM samples. Xenograft tissues provided tumor–stroma leading edge context. Only very low expression of P2YR12 was visualized in the bulk tumor (Supplementary Figure 4). Patient information can be found in Supplemental File 1 and (deCarvalho et al., 2018). We also used the microglia fluorescent lineage reporter model (Parkhurst et al., 2013) to examine the microglial P2RY12 expression over time. Double positive CX3CR1YFPtdTomato+ recombinant microglia were found within the tumor core at early (Supplemental Figure 5b) and peak disease time points (Supplemental Figure 5a), although the number of double positive cells in the tumor core at day 21 was low. P2RY12 expression was detected at both time points (Figure 2f, Supplemental Figure 5), but was associated to a higher degree with the tumor periphery rather than the tumor core. The changes in P2RY12 signal across the tumor–stroma interface point toward the dynamic relationship between the two cell types.
These results validate the presence of chemotactic/pro-inflammatory PGAM at the tumor invasive margin, offer P2YR12 as a microglia-specific cell surface protein as a marker to identify PGAM, and characterize changes in microglia-specific gene expression with spatial context in HGG. Our data from qPCR and immunofluorescence experiments corroborate information from scRNAseq, and bulk RNAseq analyses, which report a transcriptional landscape similar to what is exhibited at the protein level in vivo. Additionally, we show that the signature of this peritumoral microglial population is evident in three immunocompetent mouse models of HGG (GL261, KR158B, and RCAS/tv-a from (Bowman et al., 2016; Haage et al., 2019)), supporting the translational accuracy of these markers and phenotypes.
2.2 |. A novel transcriptional signature marks PGAM
While the above analyses define several specific molecular markers of PGAM, a more robust and comprehensive signature is needed to define PGAM across datasets and sampling modalities. Then, 189 candidate genes for PGAM were identified between the scRNAseq DE analysis and our literature review. To develop a faithful and spatially conscious transcriptional signature of PGAM, the selection was manually curated to render a list of 47 genes (Supplementary File 1) and their levels of expression were paneled across three anatomically annotated HGG datasets for validation. Using the myeloid cells from the Darmanis scRNAseq data in Figure 1, relative expression of the 47 proposed PGAM-specific genes and the top 100 DE genes of tumor core GAM were plotted and cells were grouped horizontally by tissue sample (tumor core vs. periphery) (Figure 3a). In line with the previous results, cells of the peripheral tissue samples express relatively high levels of PGAM genes. Signature scores were generated by calculating the average expression of PGAM and GAM gene lists for each cell. Plotting the signature scores indicated that cells of the peripheral tumor sample were enriched for PGAM gene signature where cells of the tumor core were enriched for the GAM signature (Figure 3a).
FIGURE 3.

A novel peritumoral glioma-associated microglia (PGAM) gene signature. (a–c) Left, heatmap of relative expression levels of PGAM and tumor core glioma-associated microglia/macrophages (GAM) genes, grouped horizontally by anatomical sample location. Rows are scaled by z score. Right, violin plots of signature scores generated by averaging PGAM or GAM gene expression across samples. Dataset derived from (a) Darmanis et al.; (b) Puchalski et al. (IVY GAP); and (c) Gill et al. Y-axes represent scores specific to each dataset, and are not on the same scale between gene signatures, or across datasets
The same analysis was carried out on two additional datasets both containing spatially annotated samples derived from human HGG. The Ivy Glioblastoma Atlas Project (Puchalski et al., 2018) contains matched histologically annotated and bulk RNAseq samples derived from microdissection of fixed human specimens. The heatmap and gene scoring process revealed that the PGAM gene signature was exclusively enriched in leading edge and infiltrating tumor samples (Figure 3b). Conversely, tumor core GAM genes were expressed at relatively low levels in these same border regions. Importantly, both signatures were detected at moderate levels in cellular/core tumor samples depicting that the PGAM gene list is capturing these cells across the tumor–stroma interface. Using a third spatially annotated bulk RNAseq dataset derived from MRI-guided biopsies of GBM (Gill et al., 2014), the PGAM gene signature was enriched in noncontrast enhancing/peritumoral samples whereas contrast enhancing samples representing the tumor core correlated with GAM signature scores (Figure 3c). This result supports the spatial accuracy of the PGAM gene list and suggests transcriptional correlations. The matched healthy reference tissue from patients in this dataset also shared positivity for PGAM-specific gene expression which is in-line with the spatial overlap between CNS adjacent and peritumoral microglia. However, the presence of the PGAM signature score in tumor core samples and the wide range of scores in noncontrast enhancing/peritumoral samples suggest that this gene list accounts for microglia across the tumor–stroma interface. This idea is supported by the range of microglia activation states seen in HGG (Muller et al., 2017; Sankowski et al., 2019; Tirosh et al., 2016).
Thus, the PGAM-specific signature gene list influenced by the expression of chemotactic/pro-inflammatory molecules, microglia-specific gene expression patterns, and border-associated transcripts reported in literature faithfully identify PGAM in scRNAseq and bulk RNAseq samples with spatial correspondence. The refined PGAM gene list proposed here expands on the 14 genes previously proposed (Muller et al., 2017) and includes additional 33 markers which comprehensively identify this novel subpopulation of GAM at the tumor–stroma interface (Supplemental File 1).
2.3 |. Comparison of PGAM across glioma
There is a growing body of transcriptomic data made publicly available by research groups worldwide. We took advantage of this large pool of data to test the robustness of the PGAM signature across healthy and pathological samples. To capture the heterogeneity surrounding HGG, we collected data from primary and recurrent Grade IV GBM, Grade III HGG, as well as IDH mutant astrocytoma and oligodendroglioma (Darmanis et al., 2017; Neftel et al., 2019; Tirosh et al., 2016; Venteicher et al., 2017; Yuan et al., 2018). It has been proposed that the observed activated microglial profiles reflect aging-related changes of microglia (Sankowski et al., 2019). Although these conclusions were made using reference samples, the reference biopsies were taken from patients experiencing pathologies other than HGG, namely, epilepsy or undefined glioma/carcinoma (Sankowski et al., 2019). After applying a quality control workflow to these reference data, over 90% of microglia cells were removed (the presence of mitochondrial genes in the majority of detected transcripts indicated compromised cell viability). To get a clearer transcriptional profile of only healthy microglia at single cell resolution, and given the presence of the PGAM gene signature in the murine HGG models, we sourced homeostatic, healthy microglia transcriptomes from a murine dataset (Huang et al., 2018). In this dataset, scRNAseq was performed on FAC-sorted murine microglia under physiological control conditions, as well as on microglia repopulating the CNS following a chemo-depletion regiment using the CSF1R inhibitor PLX3397 (Huang et al., 2018). These conditions not only reflect resting CNS microglia but also capture putative transcriptional programs of microglia during proliferation in the healthy CNS, thus spanning the normal physiology of healthy microglia.
We integrated these data to remove batch-related artifacts using the recently published tool LIGER (Welch et al., 2019). LIGER applies an integrative non-negative matrix factorization (iNMF) approach to reduce batch and sample-related artifacts and captures relevant biological signal across datasets. The algorithm then builds on the strengths of NMF for clustering, dimensional reduction, and downstream analysis. LIGER asserts strengths in analyzing CNS tissue as well as integrating data of different types and across species (Welch et al., 2019). It is also computationally efficient and scalable compared to other scRNAseq integration methods (Tran et al., 2020). LIGER was supplied with 48 samples and over 11,000 myeloid/microglia cells for integration and dimensional reduction. PGAM-labeled cells (annotated from the Darmanis dataset) separated from homeostatic microglia, repopulating microglia, as well as the bulk of tumor core GAM (Figure 4a). We identified key transcripts that define clusters using the feature loadings produced by the NMF reduction (Figure 4b). PGAM loaded on top PGAM signature genes including IPCEF1, CCL2, CCL3, and CCL4, and also loaded on transcripts DUSP6 and A2M. Two GAM subclusters were also identified. GAM subcluster 1 was enriched in several transcripts involved in active DNA remodeling and repair, including ZBTB8A, ZNF850, POLH, and PDE4C, whereas GAM subcluster 2 expressed various interferon-related genes, including IFIT3, IFIT1, IFIT2, and ISG15. Interestingly, GAM subcluster 1 was composed solely of cells from IDH mutant samples while nearly all of GAM subcluster 2 was composed of cells from IDH wild-type patients. In combination with the IDH status of these tumor biopsies, the distinct transcripts may be characteristic of unique metabolic phenotypes in these cells.
FIGURE 4.

Glioma-associated microglia/macrophages (GAM) across health and glioma. (a) Myeloid cells from 48 samples were integrated using LIGER. UMAP was performed to plot the cells, which are colored by annotated populations. (b) Feature loadings derived from LIGER were used to identify top transcripts contributing to cluster formation. The top five transcripts for each cluster are plotted, colored by the average expression across clusters, and sized by the percentage of cells expressing the transcripts. (c) Cells form the integrated data were scored for peritumoral glioma-associated microglia (PGAM) gene signature, and the signature scores were plotted as an independent feature. (d) PGAM scores for each cell were plotted against patient age. Raincloud plots show the distribution of cell scores across each age bin. (e) Violin plots of relative expression of the putative markers of repopulating microglia, across the annotated populations. GAM-Other contains all unidentified GAM and subclusters. (f) Murine glioma sections were triple stained for Ki67, P2RY12, and F4/80. Ki67 positivity is primarily within the tumor core GAM, and neoplastic cells. White arrows mark triple positive cells. White dotted line marks tumor border. (g) Outline of the predicted pattern of transcriptional evolution in peritumoral microglia. (h) Pseudotime analysis of myeloid cells, following integration. “r” marks selected homeostatic microglia acting as a root starting point for graph learning (blue line). Cells are colored by predicted pseudotime. (i) Expression of chemotactic/pro-inflammatory genes are plotted in pseudotime. The expression is fitted with a smooth generalized linear model
These data indicate that the PGAM transcriptional signature distinguishes tumor-associated microglia from microglia of the healthy brains at the single cell level and is preserved across heterogenous disease states. When the scoring approach was used as in Figure 3, the PGAM signature faithfully identified these cells and predicted nearby PGAM-like cells (Figure 4c). Notably, microglia of non-pathological sources were negative for the PGAM gene signature. We tested the hypothesis that the chemotactic/pro-inflammatory phenotype observed was related to aged microglia and found no association between PGAM gene expression and patient age (Figure 4d). This finding suggests that the pro-inflammatory phenotype of microglia results from phenotypic changes at the leading-edge niche.
As indicated by the dimensional reduction, repopulating microglia and PGAM form distinct clusters across the projected dimensions. Additionally, PGAM were negative for transcripts putatively marking the repopulating microglia cohort MKI67, CDK1, CD36, and NES43 (Figure 4e). To validate this finding, tissue sections from the murine HGG models were probed for the proliferation marker Ki67. Only a few double positive P2RY12+Ki67+ cells were present at the tumor–stroma interface (<5%) and most of the Ki67 signal was associated with tumor core GAM and neoplastic cells (Figure 4f). This result supports the ability of LIGER analysis to accurately capture inherent differences between microglial populations across species. Collectively, these data indicate that microglia are accurately compared across healthy CNS parenchyma and glioma, and that PGAM transcriptional signatures coincide with a novel subpopulation of chemotactic and pro-inflammatory microglial that is maintained across heterogeneous disease. The data are available for exploration at https://mcap91.shinyapps.io/GAMHealthandDisease/.
2.4 |. PGAM constitute an intermediate between healthy microglia and GAM
To further elucidate the cellular origins and dynamics of PGAM, bioinformatic analysis was employed using the pseudotime learning algorithm provided by Monocle3 (Qiu et al., 2017; Trapnell et al., 2014). To account for phenotypic variability, we removed cells of the GAM subcluster 1 and repopulating microglia groups. This allowed for a more relevant organization of clusters. Remaining cells were reclustered with UMAP. Cells of the homeostatic microglial cluster were selected as a physiological “root” representing a starting point from which all microglia could transcriptionally deviate. The pseudotime trajectory progressed from homeostatic microglia to the human derived healthy reference microglia and then to the PGAM cluster (Figure 4g,h)). This organization proposes that both sources of reference microglia serve as a precursor to PGAM transcriptional states. The trajectory continues into the bulk GAM cluster, where bifurcations occur between GAM subcluster 2, and other potentially distinct GAM substates. The expression of CCL2, CCL3, CCL4, IL1B, and TNF across pseudotime was mapped resulting in peak expression levels corresponding to PGAM cells (Figure 4i).
The results from pseudotime analysis give insights on the observed activation spectrum of microglia and potentially implicate PGAM as a microglia subpopulation at the tumor–stroma interface. The current analysis, however, does not delineate the fate or migration of PGAM; that is, whether or not these cells are drawn into the tumor or are “casualties” of the expanding margin. Future work incorporating a temporal/spatial dimension of PGAM behavior would more equivocally assign such behaviors.
2.5 |. PGAM enrichment correlates with MRI, immune signatures, and poor prognosis in GBM
We next investigated the relationship between the PGAM gene signature and clinical covariates of disease using the REMBRANDT database, a collection of clinical, transcriptional, and radiomic data (Madhavan et al., 2009). Twenty-eight patients with radiomic and transcriptomic data were scored for PGAM enrichment and split into either high or low groups based on median gene signature score. Using the 30 reported radiomic features, a Chi-squared test was performed between the two PGAM groups. Although no correlation was found between PGAM signature and tumor volume (as the biopsies used for the correlations were not spatially annotated in the REMBRANDT database), several statistically significant associations were revealed between PGAM enrichment and poor enhancement quality of HGG lesions (Supplementary File 1). Notably, high levels of PGAM significantly correlated with absence of contrast-enhancing tumor regions and margin in patients as well as a strong, but not significant, increase in noncontrast enhancing proportions of tumor (Figure 5a, Supplemental File 1). This finding corroborates the earlier results in which PGAM scoring was greater in MRI-guided biopsies of noncontrast enhancing samples which represented the peritumoral space (Gill et al., 2014) (Figure 3c). Together these joint bulk RNAseq and radiomic analyses propose that PGAM can be associated with MRI contrast enhancing features in HGG.
FIGURE 5.

Peritumoral glioma-associated microglia (PGAM) correlate with clinical covariates of Glioma-associated microglia/macrophages (GBM). (a) Patients were split into high/low PGAM signature scoring groups. Left, pie charts of feature scores, by PGAM group. n represents the number of patients with each feature score, and are colored by increasing feature score severity. Proportion of enhancing, and noncontrast enhancing tumor regions are shown. Right, representative MRI images from each PGAM group, and their corresponding feature score. CE, contrast enhancing; nCE, noncontrast enhancing. Yellow dotted line demarks primary lesion. n = 14 per group. (b–d) Primary diagnosed GBM IDH wild-type patients from TCGA-GBM cohort were scored for PGAM gene signature, and split into high/low scoring groups. n = 35 per group. (b) Boxplot of ESTIMATE immune and stromal scores, stratified by PGAM signature scoring. (c) Patient transcriptomes from PGAM high/low groups were analyzed by CIBERSORT. Plotted are the predicted relative fractions of 22 immune cell subpopulations ±SEM. Two-way analysis of variance (ANOVA) with Tukey’s post hoc comparison. p < .001***. (d) Top, Kaplan–Meier survival curve, stratified by PGAM group. Dotted line marks median survival. MS PGAM low = 5.7, MS PGAM high = 13.3. Log rank p val = .028. Bottom, Cox proportional hazards regression model was conducted, using covariates of survival. Forrest plot of the resulting corrected hazard ratios, following regression
We then examined whether these pro-inflammatory peritumoral microglia are associated with immune signatures of the TME. Using the TCGA-GBM cohort (Cancer Genome Atlas Research, 2008), 138 primary diagnosed and IDH wild-type patients were scored for PGAM enrichment. Patients with highest PGAM gene expression exhibited markedly higher ESTIMATE scores for immune and stromal signatures (Yoshihara et al., 2013) (Figure 5b), likely reflecting high numbers of peritumoral microglia and other glial cell involvement at the tumor leading edge and an association with systemic immune cell infiltrates. Using the CIBERSORT in silico deconvolution algorithm immune cell populations present in these samples were characterized (B. Chen, Khodadoust, Liu, Newman, & Alizadeh, 2018). Of the 22 paneled cell types in CIBERSORT, infiltrating monocytes were enriched in highly scoring PGAM patients while resting macrophage populations were enriched in PGAM-low samples (Figure 5c). As PGAM are defined by elevated levels of CCL2, CCL3, and CCL4 expression, these results suggest that PGAM may represent a potent source of monocyte/macrophage recruitment to the TME (Chang et al., 2016; Cho et al., 2019; Flores-Toro et al., 2020).
GAM recruitment negatively impacts patient survival in HGG, particularly by presenting tumor promoting properties and sculpting an immunosuppressive TME (Quail & Joyce, 2017; Sorensen et al., 2017). The currently prevalent hypothesis is that polarization of GAM to a pro-inflammatory phenotype may facilitate tumor abrogation (Biswas & Mantovani, 2010; Poh & Ernst, 2018; Pyonteck et al., 2013; Yan et al., 2017). To examine if the chemotactic and pro-inflammatory phenotype of PGAM that correlates with monocyte recruitment also correlates with GBM outcomes, Kaplan–Meier survival analysis was performed using primary diagnosed, IDH-wild-type GBM patient data from TCGA and stratified by PGAM gene signature scoring (Figure 5d). A survival disadvantage was revealed when patients scored high for PGAM genes (median survival ~5.7 months, vs. ~13.3 median survival of PGAM low group). Multivariate cox-proportional hazards analysis was performed to regress out potentially confounding factors of survival in this patient cohort. Against several diagnostic and investigational clinical covariates of GBM, PGAM signature scoring was significantly associated with poorer overall survival (Figure 5d, bottom).
3 |. DISCUSSION
Over the past decade, there has been great focus on defining and modulating the TME of solid tumors to deplete pro-tumorigenic support and foster therapeutic sensitivity. Success in the immunotherapeutic space has supported this effort; however, there have been limitations to this strategy as the TME is not fully characterized. Specifically for GBM, although many immunotherapy-based clinical trials are underway little success in extending survival has been achieved. The progression and recurrence of GBM have been attributed to the highly invasive phenotype of neoplastic cells, their chemo- and radiotherapy resistance, and the support of the CNS TME. As such, myeloid cells of the TME have become a focus of investigation. Description of local TME niches will help decipher the roles of these cells in disease progression and may uncover novel therapeutic targets.
Using several approaches, we characterize here a previously undescribed microglia population in GBM which comprise a subpopulation of GAM. PGAM are peritumoral microglia marked by chemotactic and pro-inflammatory gene enrichment, as well as robust P2YR12 expression. The cells spatially correlate with the leading edge of glioma. The spatial context provided and the changes in P2RY12 expression may reflect a transition of local, resting/homeostatic microglia, which initially populate the tumor, toward traditional GAM phenotypes. Alternatively, it is possible that blood-derived macrophages, not expressing P2RY12, are the main cellular constituent of the core of the growing neoplasm. Nonetheless, the changes in expression do point to a dynamic nature of the relationship between the cell types.
This novel gene signature serves to define PGAM in both scRNAseq and bulk RNAseq data and unmasks these cells in both human and mouse derived samples. The PGAM signature associated with clinical and radiomic features of HGG. As MRI is used for patient follow-up to detect HGG progression/recurrence, integrating this transcriptional signature with radiomics may prove informative in the clinical setting.
Potent inflammatory and chemotactic molecules make up the PGAM gene signature. A shift toward pro-inflammatory GAM polarization is traditionally thought to be beneficial in cancer (Biswas & Mantovani, 2010; Cassetta & Pollard, 2018; Poh & Ernst, 2018). However, the results presented here caution that high levels of pro-inflammatory chemokines and cytokines may need to be carefully examined. If a primary function of the innate immune system is to further recruit innate and adaptive immune cells, we predict that unchecked recruitment of inflammatory monocytes to the neoplastic site may result in the eventual prevalence of pro-tumorigenic and immunosuppressive programs (Hambardzumyan et al., 2016; Quail & Joyce, 2017). In line with this idea, a recent report has demonstrated that TLR2 activation of myeloid cells at the glioma border confers a pro-inflammatory and pro-tumorigenic shift which ultimately prevents antigen presentation and supports tumor growth (Qian et al., 2018). Thus, It may be critical to incorporate the idea of “myeloid checkpoint inhibition” (Awad, de Vlaeminck, Maebe, Goyvaerts, & Breckpot, 2018; Biswas & Mantovani, 2010; van den Berg & Valerius, 2019) which describes strategies aiming to modulate pro-tumorigenic functions of GAM. Such strategies would limit the effector and pro-tumorigenic functions of macrophages and thus attenuate the immunosuppressive and tumor-supportive functions of the TME. In the presented analysis, several targets have been identified which align with this idea.
There is accumulating evidence to suggest that P2RY12 dynamically regulates glioma/microglial interactions and modulates the proliferative capacity of glioma cells. A series of elegant studies by Chia et al. demonstrated that glioma cells adapt neuron-microglial signaling mechanisms to fuel their own proliferation (Chia, Keatinge, Mazzolini, & Sieger, 2019). Akt1+ preneoplastic cells exhibit higher cytosolic calcium levels via activation of NMDA receptors. Increased cytosolic calcium levels trigger ATP release, which activates P2RY12 receptors on microglia, leads to microglial process extension and an increase in Akt1+/microglial cell interactions (Chia et al., 2019). NMDA antagonism and CRISPR/Cas9-mediated deletion of P2RY12 reduced interactions and significantly perturbed Akt1+ cell proliferation. Antagonizing P2RY12 receptors on microglia may therefore abolish such pro-proliferative interactions.
Microglia P2RY12 signaling has been linked to aberrant seizure-induced neurogenesis (Eyo et al., 2014; Mo et al., 2019). As recent studies have outlined substantial neuronal-neoplastic communication in GBM (Venkataramani et al., 2019; Venkatesh et al., 2019), the presence of non-uniform populations of P2RY12+F4/80High peritumoral microglia, and mature neurons at the glioma leading edge suggests that a potential tripartite system exists including neurons, glioma, and myeloid cells. Future work should further detail P2RY12 molecular involvement in HGG.
Microglial transcriptomes change with aging. Among these transcriptional changes P2RY12 has been shown to be significantly down-regulated; however, direct phenotypic effects of these changes have not been adequately studied (Galatro et al., 2017; Hickman et al., 2013; Ximerakis et al., 2019). Since age is a primary clinical covariate in HGG, the patterns and consequences of microglial P2RY12 expression and resulting GAM phenotypes need further investigation (Ladomersky et al., 2019; Zhu et al., 2017). Although we did not observe any correlation between the PGAM gene signature (which includes P2RY12) and aging, it is possible that rapid changes in expression result from underlying shifts in response to the TME rather than age per se.
In a series of recent reports, the CCL2/CCR2-signaling axis in HGG and other solid cancers has been correlated with monocyte/macrophage recruitment to the TME, poor patient prognosis, and increased blood flow to the neoplasm (Chang et al., 2016; Cho et al., 2019; Flores-Toro et al., 2020; Yang et al., 2020). Targeting this pathway in preclinical mouse models of HGG has resulted in improved outcomes (da Fonseca & Badie, 2013; Ginhoux et al., 2010; Sorensen et al., 2017). One of the main pro-inflammatory cytokines associated with the PGAM signature is CCL2. As glioma cells secrete CCL20 which induces neighboring myeloid cells (PGAM) to secrete CCL2, the function of PGAM may be to attract peripheral monocytes and T regulatory cells through CCR2 and CCR4, respectively (Chang et al., 2016). Peripheral monocytes can differentiate into myeloid-derived suppressor cells (MDSCs) and, along with immunosuppressive Tregs, inhibit cytotoxic T-cell responses (Chang et al., 2016). The observation that CCL2 mediates recruitment of multiple immunosuppressive cell types is supported by the finding that dual CCR2 antagonism and PDL1 checkpoint blockade synergistically prolong the survival of glioma-bearing mice (Flores-Toro et al., 2020). While such results are encouraging, antagonizing CCR2 to slow the accumulation of immunosuppressive MDSCs does little to prevent the accumulation of CCR4+ Tregs, which suggests that sequestering CCL2 may be an alternate treatment modality. Our CIBERSORT data indicate that Tregs are not enriched in the PGAM high group, potentially because glioma cells may release Treg-specific chemoattractants, such as CCL2 and CCL22 (Jordan et al., 2008), which serve to recruit a sufficient amount of Tregs to the expanding lesion regardless of PGAM status. In a clinical study of diabetic nephropathy marked by infiltration of pro-inflammatory monocytes, CCL2 antagonism was accomplished by treating patients with the RNA aptamer emapticap pegol (NOX-E36) (Menne et al., 2017). Administration of the CCL2-sequestering ligand was well-tolerated and reduced albuminuria, a hallmark of diabetic nephropathy. Sequestering CCL2 in the bloodstream could also circumvent the common obstacle of designing drugs that can cross the blood–brain barrier.
4 |. CONCLUSIONS
In sum, PGAM constitute a subpopulation of microglia of the peritumoral space that have a unique gene signature and may influence disease outcomes by recruiting systemic monocytes to the TME. The possibility to detect these outcomes using a combination of radiomic and transcriptomic approaches is an attractive option. Profiling myeloid cells in further detail may uncover targets and signaling pathways which could be exploited in solid tumor malignancies, such as GBM. Our analysis identifies a subset of microglia present in glioma invading margins and suggests a role for them in tumor promotion. Potential targets could be leveraged to modify pro-tumorigenic functionality of GAM.
5 |. METHODS
5.1 |. Immunocompetent glioma cell lines
GL261 cells were obtained from Dr Michael Lim’s lab, and are derived from a chemically induced C57BL6 murine astroglioma (Seligman & J, 1939). KR158 cells were obtained from Drs Tyler Jacks’ and Behnam Badie’s labs, and are genetically engineered Nf1/Tp53 mutants (Chae et al., 2015). Cells were cultured in Dulbecco’s modified Eagle’s medium, 10% serum, 1% antibiotic, and 1% sodium pyruvate, at 37°C 5% CO2.
5.2 |. Microglia lineage reporter mouse line
To generate the lineage microglia fluorescent reporter mice Cx3CR1: CreER-IRES-YFP mice were crossed with the Rosa26:lsl-TdTomato line on the C57/Bl6 background (Parkhurst et al., 2013). Adult animals are treated with 1 mg Tamoxifen dissolved in corn oil via oral gavage once a day on Days 1 and 3 to induce the expression of Cre (thus inducing TdTomato expression). The animals were then housed normally for 30 days to allow for blood-derived monocyte turnover.
5.3 |. Murine glioma model
Orthotopic gliomas were established in adult mice. Briefly, mice were anesthetized, a midline incision was made on the scalp, and a small burr hole was drilled in the skull at stereotactic coordinates of bregma, −1 mm anteroposterior and +2 mm mediolateral. Total of 1 μl of 3 × 104 cells (GL261 or KR158) were injected over 2 min at a depth of 3 mm. After completion of the procedure, the incision was sutured and the mice were placed on a heated surface until fully recovered from anesthesia.
5.4 |. Immunofluorescent staining
Whole brains were collected from animals on Days 15 and 21 posttumor implantation. Animals were transcardially perfused with 4% paraformaldehyde, brains were dissected, and postfixed overnight, cryopreserved in 30% sucrose for 24 hr, and then embedded for cryosectioning. Then, 20 μm tissue sections were collected on Superfrost plus microscope slides, and stored at −80°C until use. For immunostaining, sections were brought to room temperature, and washed in PBS 3x. Slides were then blocked with 5% serum, 0.3% Triton X-100 in PBS for 1 hr at room temperature. Primary antibody was incubated overnight at 4°C. Slides were washed with PBS-T, and secondary antibody was incubated 1 hr at room temperature. Sliders were again washed with PBS-T, and mounted with Fluoromount-G (Thermo, #00–4958-02). See Supplemental Methods Table for dilutions. Confocal imaging was performed using the Leica Sp8-x system, with white light and argon lasers.
5.5 |. RNA isolation, cDNA synthesis, and quantitative real-time PCR
Whole brains were collected from animals on day 21 posttumor implantation. Microdissection was performed to sample ~100 mg of tissue from the tumor core, peritumoral tissue, and contralateral tissue. Total RNA was collected with TRIzol (Thermo, #15596026) following the manufacturer’s protocol. cDNA libraries were created using the High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems, #4368814). Real-time quantitative PCR was performed using a StepOnePlus thermocycler. A cutoff of 35 Ct was applied, and ΔCt values were calculated by normalizing to housekeeping gene GAPDH. Given that some transcripts were not detected in healthy tissue, ΔΔCt was not calculated. ΔCt values were log transformed and used for analysis.
5.6 |. Bioinformatic analysis of publicly available datasets
5.6.1 |. Bulk RNAseq
For all bulk RNAseq samples, data were downloaded from the GEO, and normalized using log2(FPKM+1). For murine analysis of pro-inflammatory chemokines, normalized datasets were merged by common gene ids, and processed for batch removal using Combat (Johnson, Li, & Rabinovic, 2007). For other data and analysis, heatmaps were created using a modified function of heatmap3.
5.6.2 |. scRNAseq
R Raw counts matrices were downloaded from GEO, and expression objects were created using Seurat (Butler, Hoffman, Smibert, Papalexi, & Satija, 2018; Stuart et al., 2019). Quality control was performed, removing cells with abnormally high or low feature counts, percent mitochondrial gene expression > = 10%, and percent ribosomal gene expression > = 25%. In accordance with [(Sankowski et al., 2019)], cells were also removed based on percent KCNQ1OT1 expression > = 2%. Following merging of samples, features expressed in a minimum of 40 cells were retained. After QC was applied, samples containing less than 20 cells were omitted. Myeloid cells were selected from datasets based on PTPRC, FCGR3A, CD14, as well as other canonical monocyte marker expression. To integrate the murine dataset, mouse transcripts were converted to human transcripts by selecting for homologs, derived from biomaRt (Durinck et al., 2005; Durinck, Spellman, Birney, & Huber, 2009) genes symbol lists. Only direct orthologs, or mouse transcripts mapping to unique human transcripts were retained.
Integration of single cell data was performed across patient/sample, using the following parameters for LIGER, and SeuratWrappers provided functions: variable features = 3,000, k = 13, lambda = 8. UMAP was performed with the following hyperparameters: n.neighbors = 40, min.dist = 0.6. iNMF projections were passed to Monocle3, and pseudotime graph was learned using the default parameters.
5.6.3 |. Survival analysis
Transcriptomic and clinical data were downloaded from TCGA GBM cohort using TCGABiolinks. Healthy, recurrent, and IDH mutant samples were omitted. Patients were scored for gene signatures, and Kaplan–Meier survival analysis was performed. Multivariate cox regression analysis was conducted, using age, sex, MGMT status, and molecular subtype as covariates.
Supplementary Material
Microglia at the border of glioma exhibit a pro-inflammatory phenotype.
Microglia-specific gene expression dynamically changes at the tumor–stroma interface.
Microglia at the tumor–stroma interface correlate with clinical outcomes of glioblastoma.
ACKNOWLEDGMENTS
The authors would like to thank members of the Tsirka and Moffitt labs for helpful suggestions along the way. Artwork credit Lucie Chrastecka. This work was partially supported by T32GM127253 Scholars in BioMedical Sciences Program funds (M. D. C.), SBU URECA (N. S.), NIH T32GM007518 (M. M.), NIH T32GM008444 (K. O., D. R.), and SBU funding (S. E. T.).
Funding information
National Institute of General Medical Sciences, Grant/Award Numbers: T32GM007518, T32GM008444, T32GM127253; National Institutes of Health, Grant/Award Number: 3R01MH123093-01S1; Stony Brook University URECA
Footnotes
CONFLICT OF INTEREST
The authors have no competing interests, or other interests that might be perceived to influence the results and/or discussion reported in this article.
ETHICS STATEMENT
All work described in this manuscript was approved by the Stony Brook University IACUC committee (Protocol 246,938).
CONSENT FOR PUBLICATION
All authors are in agreement with the content of this manuscript.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article and can be found in the supplemental information. Data and code can be accessed via https://github.com/mcap91/PGAM and can be explored via https://mcap91.shinyapps.io/GAMHealthandDisease/.
REFERENCES
- Alieva M, Leidgens V, Riemenschneider MJ, Klein CA, Hau P, & van Rheenen J (2019). Intravital imaging of glioma border morphology reveals distinctive cellular dynamics and contribution to tumor cell invasion. Scientific Reports, 9(1), 2054. 10.1038/s41598-019-38625-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad RM, de Vlaeminck Y, Maebe J, Goyvaerts C, & Breckpot K (2018). Turn back the TIMe: Targeting tumor infiltrating myeloid cells to revert cancer progression. Frontiers in Immunology, 9, 1977. 10.3389/fimmu.2018.01977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, … Barres BA (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America, 113(12), E1738–E1746. 10.1073/pnas.1525528113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ΜL, & Bennett FC (2019). The influence of environment and origin on brain resident macrophages and implications for therapy. Nature Neuroscience, 23, 157–166. 10.1038/s41593-019-0545-6 [DOI] [PubMed] [Google Scholar]
- Biswas SK, & Mantovani A (2010). Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nature Immunology, 11(10), 889–896. 10.1038/ni.1937 [DOI] [PubMed] [Google Scholar]
- Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, … Joyce JA (2016). Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Reports, 17(9), 2445–2459. 10.1016/j.celrep.2016.10.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, & Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology, 36(5), 411–420. 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 455(7216), 1061–1068. 10.1038/nature07385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L, & Pollard JW (2018). Targeting macrophages: Therapeutic approaches in cancer. Nature Reviews. Drug Discovery, 17(12), 887–904. 10.1038/nrd.2018.169 [DOI] [PubMed] [Google Scholar]
- Chae M, Peterson TE, Balgeman A, Chen S, Zhang L, Renner DN, … Parney IF (2015). Increasing glioma-associated monocytes leads to increased intratumoral and systemic myeloid-derived suppressor cells in a murine model. Neuro-Oncology, 17(7), 978–991. 10.1093/neuonc/nou343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, … Lesniak MS (2016). CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Research, 76(19), 5671–5682. 10.1158/0008-5472.CAN-16-0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Khodadoust MS, Liu CL, Newman AM, & Alizadeh AA (2018). Profiling tumor infiltrating immune cells with CIBERSORT. Methods in Molecular Biology, 1711, 243–259. 10.1007/978-1-4939-7493-1_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, … Hambardzumyan D (2017). Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Research, 77(9), 2266–2278. 10.1158/0008-5472.CAN-16-2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia K, Keatinge M, Mazzolini J, & Sieger D (2019). Brain tumours repurpose endogenous neuron to microglia signalling mechanisms to promote their own proliferation. eLife, 8. 10.7554/eLife.46912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HR, Kumari N, Thi Vu H, Kim H, Park CK, & Choi SH (2019). Increased antiangiogenic effect by blocking CCL2-dependent macrophages in a rodent glioblastoma model: Correlation study with dynamic susceptibility contrast perfusion MRI. Scientific Reports, 9(1), 11085. 10.1038/s41598-019-47438-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Mai N, Jackson C, Belcaid Z, & Lim M (2018). It takes two: Potential therapies and insights involving microglia and macrophages in glioblastoma. Neuroimmunology and Neuroinflammation, 5(10), 42. 10.20517/2347-8659.2018.47 [DOI] [Google Scholar]
- da Fonseca AC, & Badie B (2013). Microglia and macrophages in malignant gliomas: Recent discoveries and implications for promising therapies. Clinical & Developmental Immunology, 2013, 264124. 10.1155/2013/264124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, … Quake SR (2017). Single-cell RNA-Seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Reports, 21(5), 1399–1410. 10.1016/j.celrep.2017.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, … Quake SR (2015). A survey of human brain transcriptome diversity at the single cell level. Proceedings of the National Academy of Sciences of the United States of America, 112(23), 7285–7290. 10.1073/pnas.1507125112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, Cherba D, … Verhaak RGW (2018). Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nature Genetics, 50(5), 708–717. 10.1038/s41588-018-0105-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, de Moor B, Brazma A, & Huber W (2005). BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics, 21(16), 3439–3440. 10.1093/bioinformatics/bti525 [DOI] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, & Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nature Protocols, 4(8), 1184–1191. 10.1038/nprot.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, & Wu LJ (2014). Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. The Journal of Neuroscience, 34(32), 10528–10540. 10.1523/JNEUROSCI.0416-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, … Harrison JK (2020). CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proceedings of the National Academy of Sciences of the United States of America, 117(2), 1129–1138. 10.1073/pnas.1910856117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, … Eggen BJL (2017). Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nature Neuroscience, 20(8), 1162–1171. 10.1038/nn.4597 [DOI] [PubMed] [Google Scholar]
- Gill BJ, Pisapia DJ, Malone HR, Goldstein H, Lei L, Sonabend A, … Canoll P (2014). MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proceedings of the National Academy of Sciences of the United States of America, 111(34), 12550–12555. 10.1073/pnas.1405839111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, … Merad M (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science, 330(6005), 841–845. 10.1126/science.1194637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Lim S, Hoeffel G, Low D, & Huber T (2013). Origin and differentiation of microglia. Frontiers in Cellular Neuroscience, 7, 45. 10.3389/fncel.2013.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Eyo UB, Murugan M, Peng J, Matta S, Dong H, & Wu LJ (2016). Microglial P2Y12 receptors regulate microglial activation and surveillance during neuropathic pain. Brain, Behavior, and Immunity, 55, 82–92. 10.1016/j.bbi.2015.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haage V, Semtner M, Vidal RO, Hernandez DP, Pong WW, Chen Z, … Gutmann DH (2019). Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathologica Communications, 7(1), 20. 10.1186/s40478-019-0665-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Gutmann DH, & Kettenmann H (2016). The role of microglia and macrophages in glioma maintenance and progression. Nature Neuroscience, 19(1), 20–27. 10.1038/nn.4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewedi IH, Radwan NA, Shash LS, & Elserry TH (2013). Perspectives on the immunologic microenvironment of astrocytomas. Cancer Management and Research, 5, 293–299. 10.2147/CMAR.S48942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, & El Khoury J (2013). The microglial sensome revealed by direct RNA sequencing. Nature Neuroscience, 16(12), 1896–1905. 10.1038/nn.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hide T, Komohara Y, Miyasato Y, Nakamura H, Makino K, Takeya M, … Yano S (2018). Oligodendrocyte progenitor cells and macrophages/microglia produce glioma stem cell niches at the tumor border. eBioMedicine, 30, 94–104. 10.1016/j.ebiom.2018.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, … Peng B (2018). Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nature Neuroscience, 21(4), 530–540. 10.1038/s41593-018-0090-8 [DOI] [PubMed] [Google Scholar]
- Jackson CM, Lim M, & Drake CG (2014). Immunotherapy for brain cancer: Recent progress and future promise. Clinical Cancer Research, 20(14), 3651–3659. 10.1158/1078-0432.CCR-13-2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, & Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, 8(1), 118–127. 10.1093/biostatistics/kxj037 [DOI] [PubMed] [Google Scholar]
- Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, & Heimberger AB (2008). Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunology, Immunotherapy, 57(1), 123–131. 10.1007/s00262-007-0336-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordao MJC, Sankowski R, Brendecke SM, Sagar, Locatelli G, Tai YH, … Prinz M (2019). Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science, 363 (6425). 10.1126/science.aat7554 [DOI] [PubMed] [Google Scholar]
- Ladomersky E, Scholtens DM, Kocherginsky M, Hibler EA, Bartom ET, Otto-Meyer S, … Wainwright DA (2019). The coincidence between increasing age, immunosuppression, and the incidence of patients with glioblastoma. Frontiers in Pharmacology, 10, 200. 10.3389/fphar.2019.00200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitin HM, Yuan J, Cheng YL, Ruiz FJ, Bush EC, Bruce JN, … Sims PA (2019). De novo gene signature identification from single-cell RNA-seq with hierarchical Poisson factorization. Molecular Systems Biology, 15(2), e8557. 10.15252/msb.20188557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan S, Zenklusen JC, Kotliarov Y, Sahni H, Fine HA, & Buetow K (2009). Rembrandt: Helping personalized medicine become a reality through integrative translational research. Molecular Cancer Research, 7(2), 157–167. 10.1158/1541-7786.MCR-08-0435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L, Sagar, … Prinz M (2019). Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature, 566(7744), 388–392. 10.1038/s41586-019-0924-x [DOI] [PubMed] [Google Scholar]
- Menne J, Eulberg D, Beyer D, Baumann M, Saudek F, Valkusz Z, … Emapticap Study G (2017). C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrology, Dialysis, Transplantation, 32(2), 307–315. 10.1093/ndt/gfv459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo M, Eyo UB, Xie M, Peng J, Bosco DB, Umpierre AD, … Wu LJ (2019). Microglial P2Y12 receptor regulates seizure-induced neurogenesis and immature neuronal projections. The Journal of Neuroscience, 39(47), 9453–9464. 10.1523/JNEUROSCI.0487-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, … Diaz A (2017). Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biology, 18(1), 234. 10.1186/s13059-017-1362-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, … Suva ML (2019). An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell, 178(4), 835–849 e821. 10.1016/j.cell.2019.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR III, Lafaille JJ, … Gan WB (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell, 155(7), 1596–1609. 10.1016/j.cell.2013.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh AR, & Ernst M (2018). Targeting macrophages in cancer: From bench to bedside. Frontiers in Oncology, 8, 49. 10.3389/fonc.2018.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchalski RB, Shah N, Miller J, Dalley R, Nomura SR, Yoon JG, … Foltz GD (2018). An anatomic transcriptional atlas of human glioblastoma. Science, 360(6389), 660–663. 10.1126/science.aaf2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, … Joyce JA (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nature Medicine, 19(10), 1264–1272. 10.1038/nm.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Luo F, Yang J, Liu J, Liu R, Wang L, … Chu Y (2018). TLR2 promotes Glioma immune evasion by downregulating MHC class II molecules in microglia. Cancer Immunology Research, 6(10), 1220–1233. 10.1158/2326-6066.CIR-18-0020 [DOI] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma YA, & Trapnell C (2017). Single-cell mRNA quantification and differential analysis with census. Nature Methods, 14(3), 309–315. 10.1038/nmeth.4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, & Joyce JA (2017). The microenvironmental landscape of brain tumors. Cancer Cell, 31(3), 326–341. 10.1016/j.ccell.2017.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, & Jacks T (2000). Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nature Genetics, 26(1), 109–113. 10.1038/79075 [DOI] [PubMed] [Google Scholar]
- Sankowski R, Bottcher C, Masuda T, Geirsdottir L, Sagar, Sindram E, … Prinz M (2019). Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nature Neuroscience. 10.1038/s41593-019-0532-y [DOI] [PubMed] [Google Scholar]
- Seligman AMS, & J, M. (1939). Studies in carcinogenesis: VIII: Experimental production of brain tumors in mice with methylcholantherene. Cancer Research, 37. [Google Scholar]
- Sorensen MD, Dahlrot RH, Boldt HB, Hansen S, & Kristensen BW (2017). Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathology and Applied Neurobiology, 44, 185–206. 10.1111/nan.12428 [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM III, … Satija R (2019). Comprehensive integration of single-cell data. Cell, 177(7), 1888, e1821–1902. 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, … Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America, 102(43), 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, … Kettenmann H (2015). Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One, 10(2), e0116644. 10.1371/journal.pone.0116644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, … Suva ML (2016). Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature, 539(7628), 309–313. 10.1038/nature20123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HTN, Ang KS, Chevrier M, Zhang X, Lee NYS, Goh M, & Chen J (2020). A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biology, 21(1), 12. 10.1186/s13059-019-1850-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, … Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology, 32(4), 381–386. 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg TK, & Valerius T (2019). Myeloid immune-checkpoint inhibition enters the clinical stage. Nature Reviews. Clinical Oncology, 16(5), 275–276. 10.1038/s41571-018-0155-3 [DOI] [PubMed] [Google Scholar]
- Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T, … Kuner T (2019). Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature, 573 (7775), 532–538. 10.1038/s41586-019-1564-x [DOI] [PubMed] [Google Scholar]
- Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M, … Monje M (2019). Electrical and synaptic integration of glioma into neural circuits. Nature, 573(7775), 539–545. 10.1038/s41586-019-1563-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, … Suva ML (2017). Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science, 355 (6332), eaai8478. 10.1126/science.aai8478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, & Macosko EZ (2019). Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell, 177(7), 1873, e1817–1887. 10.1016/j.cell.2019.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ximerakis M, Lipnick SL, Innes BT, Simmons SK, Adiconis X, Dionne D, … Rubin LL (2019). Single-cell transcriptomic profiling of the aging mouse brain. Nature Neuroscience, 22(10), 1696–1708. 10.1038/s41593-019-0491-3 [DOI] [PubMed] [Google Scholar]
- Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL, & Joyce JA (2017). Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene, 36, 6049–6058. 10.1038/onc.2017.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, … Jia X (2020). CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Molecular Cancer, 19(1), 41. 10.1186/s12943-020-01165-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, … Verhaak RG (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nature Communications, 4, 2612. 10.1038/ncomms3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Levitin HM, Frattini V, Bush EC, Boyett DM, Samanamud J, … Sims PA (2018). Single-cell transcriptome analysis of lineage diversity in high-grade glioma. Genome Medicine, 10(1), 57. 10.1186/s13073-018-0567-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, … Wu JQ (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of Neuroscience, 34(36), 11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Kros JM, van der Weiden M, Zheng P, Cheng C, & Mustafa DA (2017). Expression site of P2RY12 in residential microglial cells in astrocytomas correlates with M1 and M2 marker expression and tumor grade. Acta Neuropathologica Communications, 5(1), 4. 10.1186/s40478-016-0405-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and can be found in the supplemental information. Data and code can be accessed via https://github.com/mcap91/PGAM and can be explored via https://mcap91.shinyapps.io/GAMHealthandDisease/.