

Abstract
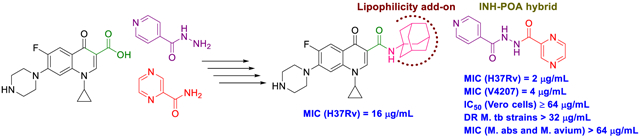
Several rationally designed isoniazid (INH), pyrazinamide (PZA) and ciprofloxacin (CPF) derivatives were conveniently synthesised and evaluated in vitro against H37Rv M. tb strain. CPF derivative 16 displayed a modest activity (MIC = 16 μg/mL) and was docked into the M. tb DNA gyrase. Isoniazid-pyrazinoic acid (INH-POA) hybrid 21a showed the highest potency in our study (MIC = 2 μg/mL). It also retained its high activity against the other tested M. tb drug sensitive strain (DS) V4207 (MIC = 4 μg/mL) and demonstrated negligible cytotoxicity against Vero cells (IC50 ≥ 64 μg/mL). Four tested drug-resistant (DR) M. tb strains were refractory to 21a, similar to INH, whilst being sensitive to CPF. Compound 21a was also inactive against two non-tuberculous mycobacterial (NTM) strains, suggesting its selective activity against M. tb. The noteworthy activity of 21a against DS strains and its low cytotoxicity highlight its potential to treat DS M. tb.
We report the synthesis of adamantane-containing and hybrid compounds of isoniazid, pyrazinamide and ciprofloxacin. The INH-POA hybrid was active against drug-sensitive M.tb strains and not cytotoxic to Vero cells. Its antimycobacterial profile suggests its selectivity towards drug sensitive and not the drug-resistant M.tb strains, or NTMs M.abs and M.avium.
Keywords: Tuberculosis, Isoniazid, Pyrazinamide, Ciprofloxacin, Indoleamides, Hybrid molecules
Graphical Abstract
1. Introduction
Tuberculosis (TB) is a highly contagious airborne disease that has existed for millennia and continues to pose a major threat to human health. It is one of the oldest life-threatening and leading deadliest diseases known to mankind, claiming more than 1 million lives annually throughout the world (Barberis et al., 2017; WHO, 2020). TB is caused by Mycobacterium tuberculosis (M. tb) organism that has the ability to stay dormant for years, persisting in the host body without any indication of disease, causing many people to become symptom-free carriers (inactive TB) (Boon & Dick, 2012). Once the immune system of these latently infected people become compromised due to, for instance, co-infection with HIV, this silent warfare of the bacteria will ultimately transform into the active form of TB (Pawlowski et al., 2012; Shankar et al., 2014). According to the World Health Organisation (WHO), one quarter of the human population harbor a latent M. tb infection with around 10 million people falling ill with TB every year. In 2018, TB caused an estimated 1.2 million deaths among HIV-negative people in addition to 0.25 million deaths among HIV-positive patients. This inexorable burden ranks TB as the number one cause of mortality/morbidity from a single infectious agent (surpassing HIV/AIDS) (WHO, 2020). The WHO directly observed treatment, short course (TB-DOTS) requires a six-month minimum treatment for drug-sensitive (DS) TB with the first-line anti-TB drugs divided into two phases. The first two months are the intensive phase treatment with a cocktail of four drugs (Figure 1), including isoniazid (INH, 1), rifampin (RIF), pyrazinamide (PZA, 3) and ethambutol (EMB). The subsequent four months represent the continuation phase treatment with INH and RIF to kill the dormant bacteria (Tiberi et al., 2017; WHO, 2020). Poor patient compliance to the lengthy duration of therapy, high pill count, and drug side-effects, contributed to the emergence of drug-resistant (DR) TB (Dheda et al., 2014; Lange et al., 2018; Seung et al., 2015; Sotgiu & Migliori, 2015; Velayati et al., 2013). Hydrophilic properties, charge, size and/or stability under physiological conditions account for inefficient cellular penetration of many anti-TB drugs and sub-optimal treatment. Accordingly, high drug doses are prescribed to compensate for the reduced penetration and bioavailability, taking its toll on the host’s vital organs, causing strong adverse effects. Therefore, highly efficacious anti-TB drugs are urgently needed. A common design tactic is to modify old drugs or existing compounds with an established bioactivity in order to attain, ideally, an enhanced anti-TB potency, efficacy against DR strains and short duration of treatment.
Figure 1.
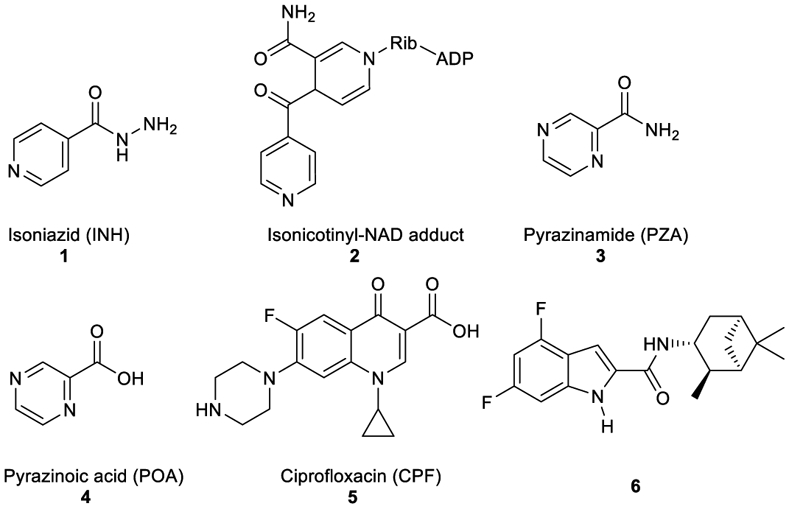
Structures of INH, isonicotinyl-NAD complex, POA, PZA, and ciprofloxacin.
Among the front-line TB antibiotics for treatment of DS strains, INH 1 is a prodrug that requires activation by the mycobacterial catalase-peroxidase enzyme (KatG) to the reactive free radical form which then gets implicated in a series of reactions, forming the isonicotinyl-NAD complex 2. This complex is a potent inhibitor to the enoyl-acyl carrier protein reductase InhA, a key enzyme in the biosynthesis of mycolic acids (MAs) (Vidossich et al., 2014). MAs are the integral building blocks of the mycobacterial cell wall and the primary mediators of the notorious impermeability and hydrophobic characters of the outer coating (Alsayed et al., 2019). Therefore, inhibiting InhA eventually leads to collapsing of the mycobacterial outer coating that serves as a protective permeability barrier from many antibiotics (Vilcheze & Jacobs, 2019). However, serum concentration of INH is greatly influenced by its acetylation via N-acetyltransferase enzyme (NAT) which constitutes the main metabolic pathway of INH in humans (Erwin et al., 2019). The generated N-acetylisoniazid metabolite is devoid of anti-TB activity, leading to a significant decrease in the INH bioavailability. Additionally, in M. tb, NAT is implicated in the resistance mechanism to INH (Unissa et al., 2016). For these reasons, chemically modifying the hydrazine unit in INH via incorporating a functional group is commonly used to avoid the N-acetylation process, and thereupon improve the drug bioavailability and the curative outcomes (Y. Q. Hu et al., 2017). Moreover, appending lipophilic moieties to the INH core can impart enhanced cell wall permeation to the drug. Hence, INH analogues with greater lipophilicity have emerged as potential anti-TB agents (Y. Q. Hu et al., 2017).
Like INH (Figure 1), PZA 3 is a prodrug which diffuses into the M. tb granuloma, where it gets activated by the pyrazinamidase enzyme to the active form of the drug pyrazinoic acid (POA, 4) (Miotto et al., 2014). However, the exact mechanism of PZA is still ambiguous. After penetrating the TB lesion, the active form of the drug POA accumulates inside the bacillus and kills the bacterial cell in the acidic environment of the TB granuloma (Njire et al., 2016). On the other hand, fluoroquinolones, such as ciprofloxacin (CPF, 5), have become a mainstay in treating multi-drug resistant (MDR) TB. The mechanism of action of this class of antibiotics is distinct from the first line drugs in which they inhibit DNA gyrases and in turn prevent bacterial DNA synthesis (Aldred et al., 2016; Schluger, 2013). When CPF 5 was rendered more hydrophobic by attaching alkyl substituents to the piperazine NH, the resulting N-alkylated ciprofloxacins were more active against M. tb than CPF 5 (Haemers et al., 1990).
The present study entails a simple and efficient synthesis of a number of INH, POA and CPF derivatives as well as their in vitro anti-TB activity. All final compounds were screened in vitro against M. tb H37Rv strain. The most potent compound 21a in our study was further evaluated for its antimycobacterial activity against another DS strain and four DR M. tb strains in addition to Mycobacterium abscessus (M. abs) and Mycobacterium avium (M. avium). In parallel, this compound was also tested in Vero cells for cytotoxicity evaluation.
2. Design
The first design strategy was to incorporate an adamantane ring as a hydrophobic moiety into the framework of INH, POA and CPF. Adamantane is considered an add-on “lipophilic bullet” that has a multifaceted value in medicinal chemistry (Wanka et al., 2013). The hydrophobic substituent constant of this carbocyclic group is estimated as Πadamantyl = 3.1, therefore integrating an adamantane motif into the structure of highly water-soluble molecules can shift their ClogP to a region that is more clinically useful (Liu et al., 2011). Hence, our first approach is based on endowing the aforementioned drugs with increased lipophilicity which in turn may enhance their penetration through the mammalian host tissues and the lipid-rich mycobacterial cell wall. Beyond enhancing the partition coefficient, the adamantyl group may also improve the stability of drugs and their pharmacokinetics and modulate their therapeutic index (Liu et al., 2011; Wanka et al., 2013). For instance, an inserted adamantane to the INH skeleton might serve as a protecting group, chemically modifying the hydrazine unit and blocking the N-acetylation process (Y. Q. Hu et al., 2017). Towards this, five derivatives 10a,b, 13a,b and 16 were synthesised to investigate the effect of these modifications on the in vitro biological activity compared to the parent drug.
Our group has previously reported several indole-2-carboxamides, such as 6, as potent anti-TB agents targeting the mycobacterial membrane protein large 3 (MmpL3) (Lun et al., 2013; Onajole et al., 2013; Stec et al., 2016). MmpL3 is a crucial transporter implicated in the flipping and release of the MAs precursors across the plasma membrane (Xu et al., 2017). Thereafter, MAs get accumulated in the M. tb cell envelope, forming a bilayer barrier, standing out as key players in the infection process. Accordingly, we synthesised and scrutinised the biological activity of two conjugates 18 and 20 in which we integrated INH and POA, respectively into the indoleamide architecture. Another hybrid conjugating both INH and POA 21a was also evaluated. The foregoing compounds design strategy is based on conjugating two different pharmacophore moieties of diverse bioactive substances known to inhibit different targets in M. tb to develop a new chemical entity. The new molecule might be capable of simultaneously hitting different targets, exerting multiple drug actions or one part can offset the adverse effects caused by the other part. The aforementioned approach of forming hybrids of bioactive molecules via conjugating efficacious drug fragments is well-known in drug design (Panda et al., 2016; Panda et al., 2019). Finally, we replaced the pyridine ring in 21a with a benzene ring, forming the analogous derivative 21b which is more lipophilic than INH-POA hybrid 21a to compare their activities.
3. Chemistry
The synthesis of target compounds 10a,b, 13a,b, 16, 18, 20, and 21a,b was accomplished as depicted in Schemes 1 - 4 using commercially available INH (1), POA (4), CPF (5) and 4,6-difluoroindole-2-carboxylic acid (17). Adamantane derivative 7 was esterified with methanol to give the diester derivative 8. Subsequent mono hydrolysis with sodium hydroxide of the diester 8 yielded the carboxylic acid 9. INH 1 was then coupled with either 1-adamantanecarboxylic acid or 9, following either amide coupling procedure A [1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC.HCl), hydroxybenzotriazole hydrate (HOBt) and N,N-diisopropylethylamine (DIPEA)] or coupling conditions B [EDC.HCl and 4-dimethylaminopyridine (DMAP)] to give the requisite final compounds 10a,b (Scheme 1). Although EDC.HCl/HOBt-mediated coupling is typically conducted at room temperature (rt) in the literature (Onajole et al., 2013), even after 48 h of stirring very low yields were obtained. Hence, we applied relatively harsh conditions via heating EDC.HCl/HOBt amide coupling reactions at 50 °C for 18 h which resulted in improved yield. Similar EDC-coupling protocol was previously adopted in which the reaction was heated at 45 °C in a microwave (Bahde et al., 2011). Alternatively, EDC.HCl/DMAP were also used as coupling reagents which generally provided better yield than EDC.HCl/HOBt in the derivatives in which both methods were employed. In Scheme 2, 1-adamantanecaboxylic acid 11 was treated with hydrazine hydrate in the presence of 1,1'-carbonyldiimidazole (CDI) to render the hydrazide intermediate 12. Then, similar to 10a,b, under standard amide coupling conditions A, compounds 13a,b were obtained via reacting POA 4 with 1-adamantylamine and 12, respectively. Both compounds 10a and 13a were previously reported, but their synthetic protocols were different from ours and they have never been evaluated against M. tb (Harikishore et al., 2013; Naredla et al., 2013). On the other hand, the synthesis of N-adamantyl CPF derivative 16 is delineated in Scheme 3, accomplished in three steps. The pyrazine NH of CPF 5 was initially protected using di-tert-butyl dicarbonate (Boc)2O to form the N-Boc derivative 14 which was then subjected to coupling procedure A with 1-adamantylamine to provide the amide 15. The Boc group in the crude amide 15 was then cleaved using trifluoroacetic acid (TFA) to afford the desired amine 16.
Scheme 1.
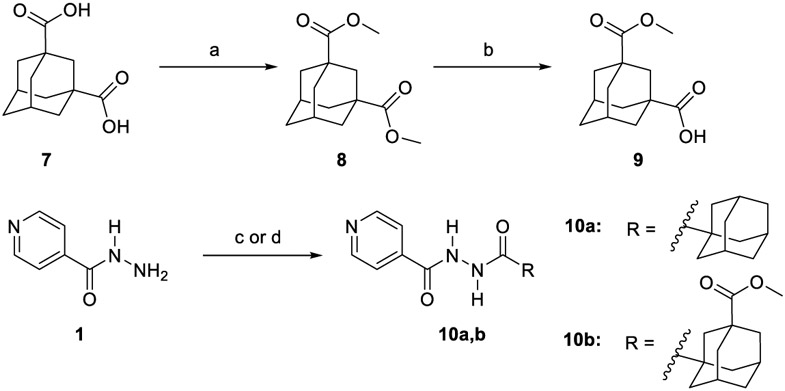
Synthetic pathway for compounds 10a,b.
Reagents and conditions:
(a) MeoH, conc. H2SO4, reflux, 16 h, 98%; (b) NaOH (1.1 equiv.), THF, MeoH, rt, 24h, 70%; (c) EDC.HCl, HOBt, 1-adamantanecarboxyclic acid or 9, DIPEA, DMF, 50 °C, 18h, 30-87%; (d) EDC.HCl, DMAP, 9, THF, DCM, rt, 72 h, 45%.
Scheme 4.
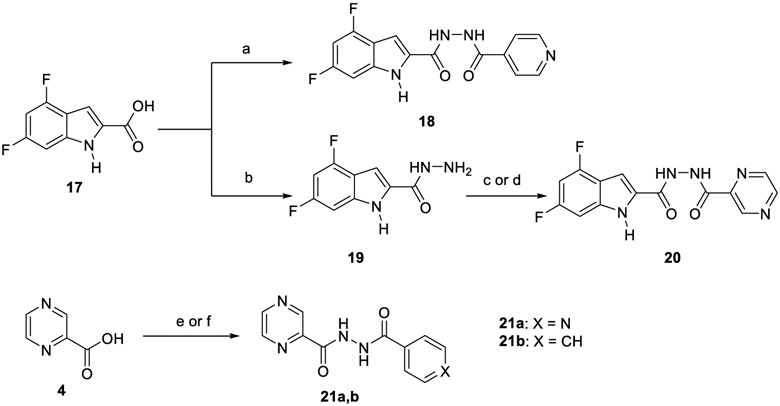
Synthetic pathway for compounds 18, 20 and 21a,b.
Reagents and conditions:
EDC.HCl, HOBt, INH, DIPEA, DMF, 50 °C, 18 h, 64%; (b) CDl, NH2NH2H2O, DMF, rt, 16 h, 74%; (c) EDC.HCl, HOBt, POA, DIPEA, DMF, 50 °C, 18 h, 32%; (d) EDC.HCl, DMAP, POA, THF, DCM, rt, 72 h, 70%; (e) EDC.HCl, DMAP, INH, THF, DCM, rt, 72 h, 66%; (f) EDC.HCl, HOBt, benzohydrazide, DIPEA, DMF, 50 °C, 18 h, 41%.
Scheme 2.
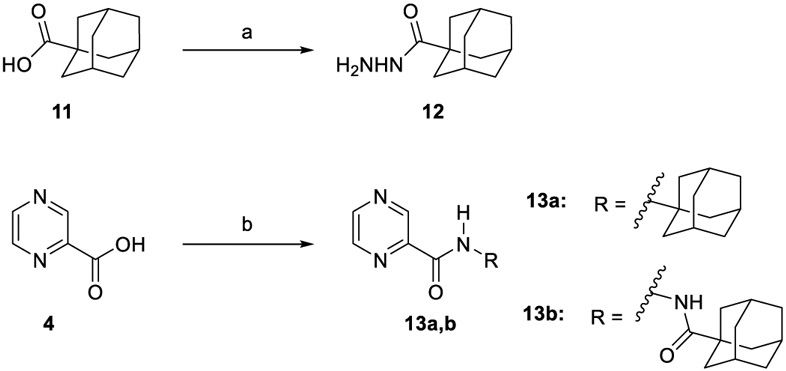
Synthetic pathway for compounds 13a,b.
Reagents and conditions:
(a) CDI, NH2NH2H2O, DMF, rt, 12-16 h, 78%; (b) EDC.HCl, HOBt, 1-adamantylamine or 12, DIPEA, DMF, 50 °C, 18 h, 50-76%.
Scheme 3.
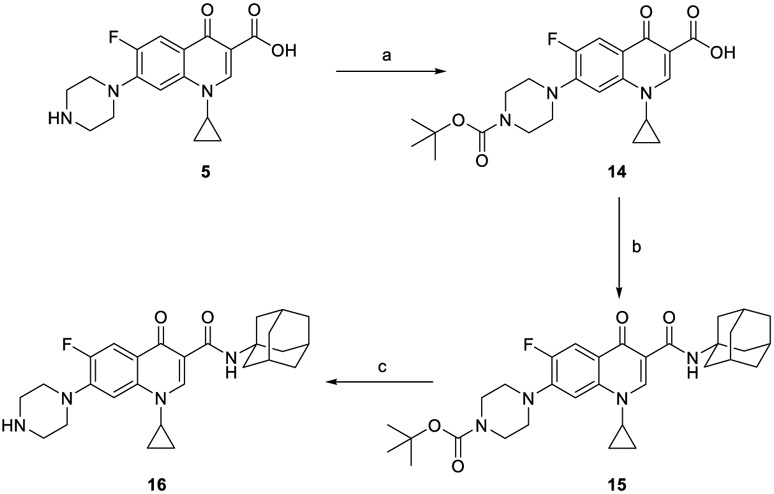
Synthetic pathway for compound 16.
Reagents and conditions:
(a) Di-tert-butyl dicarbonate, 2.0 M aqueous NaOH, dioxane, H2O, rt, 48 h, 99%; (b) EDC.HCl, HOBt, 1-adamantylamine, DIPEA, DMF, 50 °C, 18 h; (c) TFA, DCM, rt, 12 h 40% (over two steps).
In the second part of our study, various conjugates were synthesised as described in Scheme 4. For the preparation of hybrid 18, amide coupling of 4,6-difluoroindole-2-carboxylic acid 17 with INH 1 was conducted under coupling conditions A. Compound 17 was initially converted into the hydrazide derivative 19 using CDI and hydrazine hydrate, and was followed by amide coupling of 19 with POA 4 to furnish conjugate 20. Treatment of INH 1 with POA 4 using coupling method B delivered the desired hybrid 21a. This INH-POA conjugate was previously prepared using different synthetic strategies (Miniyar & Bhat, 1999; Panda et al., 2019). It was evaluated against H37Rv M. tb strain, albeit without specifying its exact minimum inhibitory concentration (MIC) (Miniyar & Bhat, 1999). Finally, analogue 21b was obtained via reacting INH 1 with benzohydrazide following coupling conditions A.
4. Results and Discussion
Final compounds 10a,b, 13a,b, 16, 18, 20, and 21a,b were evaluated for their antimycobacterial activity in vitro against M. tb H37Rv strain using the microplate alamarBlue assay (MABA) to obtain their corresponding MIC values as shown in Table 1. First, with the aim of increasing lipophilicity, an adamantyl group was appended to INH, PZA and CPF, whereupon five compounds 10a,b, 13a,b and 16 were evaluated. Both adamantane-based INH derivatives 10a,b were less active (MIC > 64 and = 32 μg/mL, respectively) than INH (MIC = 0.04 μg/mL) although they exhibited ClogP values of 2.22 and 1.84, respectively, higher than that of INH (ClogP = −0.67). It is noteworthy that the methyl ester adamantane derivative 10b was more potent than PZA (MIC = 200 μg/mL (Y. Hu et al., 2017; Werngren et al., 2012; Zhang et al., 2002)) and the unsubstituted adamantane derivative 10a. PZA derivatives 13a (MIC = 32 μg/mL) and 13b (MIC ≥ 32 μg/mL) were more potent compared to PZA. The higher lipophilicity of 13a,b (ClogP = 2.33 and 1.78, respectively), emanated from the adamantyl motif, compared to PZA (ClogP = −0.68) appeared to positively modulate the activity of this lead. It is also conceded that PZA displays poor activity against M. tb in vitro, notwithstanding its potent effect in vivo (Gopal et al., 2019). Additionally, our attempt to enhance the lipophilicity of CPF, resulted in compound 16 which exhibited moderate anti-TB activity (MIC = 16 μg/mL) in comparison to its precursor CPF (MIC = 0.25 μg/mL). Importantly, when docked into M. tb DNA gyrase (PDB ID: 5BTC) (Blower et al., 2016), the keto-amide moiety of 16 was anchored in the gyrase binding pocket via hydrated magnesium ion bridge (Figure 2). Several studies substantiated the importance of this bridge in connecting the fluoroquinolones to the DNA gyrase enzyme, foregrounding it as the primary conduit for the enzyme-drug interactions (Aldred et al., 2014; Aldred et al., 2013; Wohlkonig et al., 2010). Two crucial amino acids Ser90 and Asp94 were found to interact with this bridge, providing further support to the hydrogen-bonding network (Figure 3) (Aldred et al., 2016; Blower et al., 2016; Wohlkonig et al., 2010). When 16 was docked to the gyrase active site, the hydroxyl group of Ser90 was directly linked to the magnesium ion associated waters, similar to CPF. Because of the absence of Ser90 in the wild type (WT) M. tb gyrase, it was proved that the critical water-metal ion bridge that govern the interaction between the fluoroquinolones and the enzyme is primarily anchored by one amino acid Asp94 (Aldred et al., 2016). This is in accordance with previous results showing fluoroquinolone resistance in M .tb brought forth by various mutations at Asp94 in GyrA which could be attributed to the disrupted bridge function (Aldred et al., 2016; Maruri et al., 2012). Unlike CPF, the water-mediated magnesium ion network that bridges the quinolone core of 16 to the enzyme did not seem to interact with Asp94 (Figure 2). Compound 16 was also aligned in a slightly different fashion from CPF in the gyrase binding pocket as portrayed in Figure 3. All of these may contribute to the reduced activity of 16 compared to CPF.
Table 1.
In vitro anti-TB activity of target compounds 10a,b, 13a,b, 16, 18, 20, and 21a,b as well as reference compounds INH, PZA, CPF, and 6.
| Analogue | Structure | MICa (μg/mL) |
ClogPb |
|---|---|---|---|
| 10a | 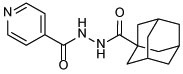 |
> 64 | 2.22 |
| 10b | 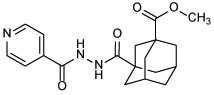 |
32 | 1.84 |
| 13a | 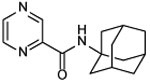 |
≥ 32 | 2.33 |
| 13b | 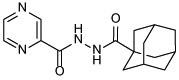 |
32 | 1.78 |
| 16 | 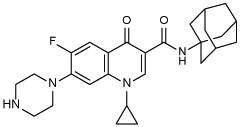 |
16 | 2.99 |
| 18 | 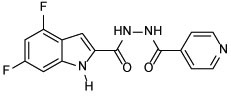 |
> 64 | 2.37 |
| 20 | 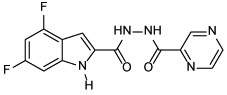 |
> 64 | 1.96 |
| 21a | 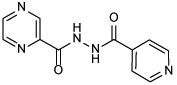 |
2 | −0.32 |
| 21b | 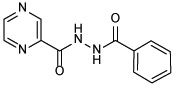 |
> 64 | 1.03 |
| INH 1 | 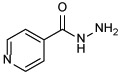 |
0.04 | −0.67 |
| PZA 3 | 200 | −0.68 | |
| CPF 5 | 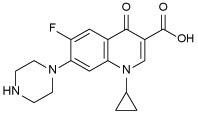 |
0.25 | −0.72 |
| 6 | 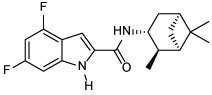 |
0.004 | 5.74 |
The lowest concentration of drug causing at least 90% reduction of bacterial growth by the Microplate alamarBlue assay (MABA). The reported MIC values are an average of three individual measurements.
Calculated using ChemDraw 16.0.
Figure 2.

2D representation of the putative binding interactions of ciprofloxacin (A) and compound 16 (B) with DNA
Figure 3.

Close-up view of the DNA gyrase active site (retrieved from PDB ID: 5BTC) in complex with CPF (magenta) and compound 16 (light brown, docked in silico in the active site), showing their overlay and different alignment. Green circled is the chelation/hydrogen-bonding network, designating the water/magnesium ion bridge (red/yellow spheres) coordinating the keto-acid in CPF. The putative binding profile of CPF also shows an increased support of the hydrogen-bond interactions by Ser90 and Asp94.
Next, we conjugated pharmacophoric units of different anti-TB substances with distinct mechanism of actions. The first two hybrids, comprising the 4,6-difluoroindole nucleus linked to INH or POA to give 18 and 20, respectively, showed a dramatic loss of activity (MIC > 64 μg/mL) compared to the original agents. The inactivity of 18 and 20 relative to the 4,6-difluoroindoleamide 6 (MIC = 0.004 μg/mL) is likely ascribed to the diminished lipophilicity of these two analogues (ClogP = 2.37 and 1.96, respectively) compared to 6 (ClogP = 5.74). In addition, extending the amide linker of the indoleamide analogues in our previous study was unfavourable (Alsayed et al., 2020), resonating with the reduced activities of 18 and 20. On the other hand, tethering INH with POA, which generated conjugate 21a, led to the most active compound in our current study (MIC = 2 μg/mL). In fact, this hybrid was previously reported to exhibit > 70% growth inhibition at 3 μg/mL against the highly virulent M. tb Erdman strain (Panda et al., 2019). Evidently, this hybrid exhibited higher anti-TB activity than PZA but less than INH. Meanwhile, the homologous derivative 21b, in which we replaced the pyridine moiety with benzoic acid, was bereft of anti-TB activity (MIC > 64 μg/mL) despite its increased lipophilicity compared to 21a (ClogP = 1.03 and −0.32). The discrepancy between the activities of 21a and 21b could be attributed to the presence/lack of the INH prodrug moiety, respectively. In other words, removal of the INH pharmacophore in 21b likely accounted for its attenuated activity compared to 21a, as both compounds retained PZA which displayed poor in vitro activity (MIC = 200 μg/mL (Y. Hu et al., 2017; Werngren et al., 2012; Zhang et al., 2002)). Indeed, this was further corroborated by the previous findings of Judge et al in which when they coupled INH with benzoic acid, the resulting N'-benzoylisonicotinohydrazide compound showed some potency against the H37Rv strain (Judge et al., 2013). Overall, it seems that lipophilicity is not the sole driving force for anti-TB activity in our study. The higher anti-TB activity demonstrated by the more hydrophilic anti-TB drugs, currently in the market, compared to our relatively lipophilic analogues, gives validity to the notion that the positive correlation between lipophilicity and anti-TB activity is not a clear-cut phenomenon (Machado et al., 2018; Piccaro et al., 2015; Tong et al., 2017).
The most potent INH-POA hybrid 21a, together with CPF as a positive control, were further evaluated for their cytotoxicity against mammalian cells. In addition, these two compounds were tested for their antimycobacterial activity against another DS and four DR M. tb strains, as well as M. abs and M. avium (Table 2). Both 21a and CPF exhibited the same high IC50 against Vero cells (IC50 ≥ 64 μg/mL), indicating their limited cytotoxicity. We further assessed 21a and CPF against a panel of clinical isolates of M. tb, originally procured from TB patients. Compound 21a retained its activity against DS M. tb strain (V4207), albeit less active than CPF (MIC = 4 and 0.25 μg/mL, respectively). This hybrid was stripped of its potency when tested against two multi-drug resistant (MDR) M. tb strains (V2475, KZN494) and two extensively-drug resistant (XDR) M. tb strains (R506, TF274). Notably, the anti-TB activity of CPF against the tested MDR strains was 8-fold higher than the XDR ones (MIC = 0.25 and 2 μg/mL, respectively). Panda et al evaluated hybrid 21a against tuberculous and non-tuberculous mycobacterial (NTM) strains, wherein it exhibited activity at 20 μg/mL concentration against Mycobacterium bovis (M. bovis) and at 10 μg/mL against Mycobacterium marinum (M. marinum) and Mycobacterium fortuitum (M. fortuitum) (Panda et al., 2019). Hence, in line with our pursuit to scrutinise the antimycobacterial activity of 21a, we further evaluated both 21a and CPF against another two NTM strains, namely M. abs and M. avium. Surprisingly, compound 21a was devoid of activity against both strains (MIC > 64 μg/mL). CPF, however, displayed a 32-fold surge in potency against M. avium compared to M. abs (MIC = 0.25 and 8, respectively). This in turn suggests the selective activity of 21a against DS M. tb.
Table 2.
Cytotoxicity [IC50 (μg/mL)] against Vero cells of 21a and CPF (positive control) and their activity [MIC (μg/mL)] on selected clinical isolates of M. tb and two NTM strains.
| IC50 | M. tb | M. abs | M. avium | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Vero cells | SIa | V4207/DS | V2475/MDRb | KZN494/MDRb | R506/XDRc | TF274/XDRc | |||
| 21a | ≥ 64 | ≥ 32 | 4 | > 32 | > 32 | > 32 | > 32 | > 64 | > 64 |
| CPF | ≥ 64 | ≥ 256 | 0.25 | 0.25 | 0.25 | 2 | 2 | 8 | 0.25 |
Selectivity index (SI) = IC50(Vero)/MIC(H37Rv)
Resistant to INH and rifampin (RIF)
Resistant to INH, RIF, levofloxacin, ofloxacin, and kanamycin
5. Conclusion
In summary, we rationally designed, concisely synthesised and evaluated the anti-TB activity of several INH, PZA and CPF derivatives against H37Rv DS M. tb strain. The adamantane-dependent modulation of the preceding drugs resulted in enhanced lipophilicity (higher ClogP) in compounds 10a,b, 13a,b, and 16. The noteworthy activity of the adamantane-derived INH compound 10b set this derivative forth for future modifications in which the integrated methyl ester functional group can serve as a building block amenable to further structure changes. The higher lipophilicity of 13a,b, with respect to the reference standard PZA, was likely responsible for their improved potency. CPF derivative 16 manifested modest activity and was docked into M. tb DNA gyrase enzyme. In derivatives 18, 20 and 21a, pharmacophores from various anti-TB ligands working through different mechanisms were conjugated. INH-POA hybrid 21a displayed the most potent activity in our study which was replicated against V4207 DS M. tb strain. Compound 21a and CPF showed low cytotoxicity in mammalian cells (Vero Cells). Unlike CPF, when tested against four MDR- and XDR-TB strains and two NTM strains, 21a was devoid of activity. This in turn suggests the selective activity of 21a against DS M. tb without any significant effects on the tested DR M. tb strains and the NTM strains M. abs and M. avium. These findings provide valuable information for future elaboration on the adamantane-based compounds, especially the methyl ester derivative 10b, to attain higher potency and drug-like molecules. Additionally, the low cytotoxicity of hybrid 21a and its high in vitro potency merit further studies on its in vivo anti-TB activity against DS strains.
6. Experimental Details
6.1. Chemistry
6.1.1. General Information
1H NMR and 13C NMR spectra were recorded on a Bruker Avance III spectrometer at 400 and 100 MHz, respectively, with TMS as an internal standard. Standard abbreviations indicating multiplicity were as follows: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, td = triplet of doublets, q = quadruplet, m = multiplet and br = broad. HRMS experiments were performed on a Thermo Scientific Q-Exactive Orbitrap mass spectrometer. TLC was carried out on Analtech silica gel TLC plates (200 μm, 20 X 20 cm). Flash chromatography was performed using a Teledyne Isco CombiFlash Rf system with RediSep columns or manually using SiliCycle SiliaFlash® P60 Silica Gels [40-63 μm (230-400 mesh)]. Final compounds were purified by preparative HPLC unless otherwise stated. The preparative HPLC employed a Phenomenex Luna® Omega 5 μm Polar C18 100A (21.2 mm × 150 mm) column, with detection at 254 and 280 nm on a Shimadzu SPD-20A detector, flow rate = 25.0 mL/min. Method 1: 5–50% acetonitrile/H2O in 15 min; 50–50% acetonitrile/H2O in 10 min; 50–5% acetonitrile/H2O in 10 min. Method 2: 20–50% acetonitrile/H2O in 15 min; 50–70% acetonitrile/H2O in 10 min; 70–20% acetonitrile/H2O in 10 min. Both solvents contained 0.05 vol % of trifluoroacetic acid (TFA). Purities of final compounds were established by analytical HPLC, which was carried out using Waters 1525 binary pump, 717 plus autosampler, and 2487 dual wavelength absorbance detector, with a Phenomenex Luna® 5μ C18(2) 100 Å (150 X 4.6 mm) column. Analytical HPLC method: flow rate = 1 mL/min; gradient elution over 30 min. Gradient: 100% H2O to 100% acetonitrile in 10 min; 100% acetonitrile in 10 min; 100% acetonitrile to 100% H2O in 10 min. Both solvents again incorporated 0.05% TFA. The purity of all tested compounds was > 95% as determined by the HPLC method described above.
6.1.2. General procedure for amide coupling (Method A)
To a solution of the appropriate carboxylic acid (1 equiv.) in anhydrous dimethylformamide (DMF, 10 mL/mmol), hydroxybenzotriazole hydrate (HOBt, 2 equiv.) and 1-ethyl-3-(3-(dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl, 2 equiv.) were added at room temperature (rt). After stirring for 10 min, the corresponding amine (1.5 equiv.) and N,N-diisopropylethylamine (DIPEA, 3 equiv.) were added, and the reaction mixture was stirred at 50 °C for 18 h. After this time NaHCO3 solution (25 mL) was added, and the mixture was extracted with EtOAc (3 × 25 mL). The combined organic layers were washed with NaHCO3 solution (5 × 25 mL), brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using dichloromethane/methanol (DCM/MeOH) gradient prior to further preparative HPLC purification unless otherwise stated.
6.1.3. General procedure for amide coupling (Method B)
To a stirred solution of carboxylic acid (1 mmol) in a 1:1 mixture of tetrahydrofuran (THF) and DCM, EDC.HCl (1.2 mmol), the corresponding amine (1.2 mmol), and 4-dimethylaminopyridine (DMAP, 0.3 mmol) were added and the reaction mixture was stirred at room temperature for 72 h. The solvent was then removed under vacuum and the residue was purified by flash chromatography using DCM/MeOH gradient prior to further HPLC purification unless otherwise stated.
6.1.4. General procedure for amide coupling (Method C)
A mixture of carboxylic acid (1 mmol) and 1,1'-carbonyldiimidazole (CDI) (1.5 mmol) in anhydrous DMF (10 mL) was stirred at rt for two hours, followed by the addition of hydrazine hydrate solution (5 mmol) and stirring was continued for 16 h at rt. Water (25 mL) was then added to the reaction mixture and the formed precipitate was filtered off, washed with water (4 × 25 mL) and dried. The crude product was used directly in the next step without additional purification as they were already pure according to the crude 1H/13C NMR spectra.
6.1.5. Preparation of intermediates and final compounds 8, 9, 10a,b, 12, 13a,b, 14, 16, 18 – 20, and 21a,b
Dimethyl adamantane-1,3-dicarboxylate (8)
To a solution of 1,3-adamantanedicarboxylic acid in methanol, 0.5 mL of conc. H2SO4 was added and the reaction mixture was refluxed for 16 h and then concentrated in vacuo. The residue was slowly quenched with saturated NaHCO3 solution, followed by extraction with DCM (3 × 25 mL). The combined organic layers were washed with brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and evaporated under vacuum. 1H NMR data of 8 matched that reported in the literature (Averina et al., 2014). White solid, yield: 98%. 1H NMR (DMSO-d6) δ 3.59 (s, 6H), 2.12 – 2.03 (m, 2H), 1.90 (s, 2H), 1.84 – 1.70 (m, 8H), 1.63 (s, 2H).
3-(Methoxycarbonyl)adamantane-1-carboxylic acid (9)
To a solution of compound 8 (1 mmol) in dry THF, a solution of NaOH (1.1 mmol) in dry methanol was added dropwise under an argon atmosphere. After 24 h of stirring at rt, the reaction mixture was concentrated in vacuo, diluted with water (1 × 25 mL), and washed with DCM (1 × 25 mL). The aqueous layer was subsequently acidified with HCl until the pH value dropped to 1-2 and extracted with DCM (4 × 25 mL). The combined organic layers were dried over anhydrous Na2SO4 filtered and evaporated under reduced pressure. The crude product was used for the next reaction without further purification. White solid, yield: 70%. 1H NMR (CDCl3) δ 3.67 (s, 3H), 2.17 (s, 2H), 2.06 (s, 2H), 1.88 (s, 8H), 1.69 (s, 2H); 13C NMR (CDCl3) δ 183.3, 177.2, 51.8, 40.9, 40.8, 39.5, 37.9, 37.7, 35.3, 27.7.
N'-(adamantane-1-carbonyl)isonicotinohydrazide (10a)
The title compound was synthesised from INH and 1-adamantanecarboxylic acid according to general procedure A. White solid, yield: 87%. 1H NMR (DMSO-d6) δ 10.57 (s, 1H), 9.59 (s, 1H), 8.82 (s, 2H), 7.86 (d, J = 5.8 Hz, 2H), 2.01 (s, 3H), 1.88 (d, J = 2.6 Hz, 6H), 1.70 (d, J = 2.2 Hz, 6H); 13C NMR (DMSO-d6) δ 176.7, 164.0, 149.6, 141.5, 122.5, 40.1, 39.0, 36.5, 28.0; HRMS (ESI) m/z calcd for C17H21N3O2 ([M + H]+) m/z 300.1707; found 300.1700.
Methyl-3-(2-isonicotinoylhydrazine-1-carbonyl)adamantane-1-carboxylate (10b)
The title compound was obtained from reacting INH and compound 9 following method A or B. After flash chromatography, the product was further purified via crystallization from diethyl ether. White solid, yield: 30% (method A) and 45% (method B). 1H NMR (DMSO-d6) δ 10.52 (s, 1H), 9.67 (s, 1H), 8.76 (dd, J = 4.6, 1.3 Hz, 2H), 7.77 (dd, J = 4.5, 1.5 Hz, 2H), 3.61 (s, 3H), 2.11 (s, 2H), 1.96 (s, 2H), 1.91 – 1.73 (m, 8H), 1.65 (s, 2H); 13C NMR (DMSO-d6) δ 177.0, 176.0, 164.4, 150.9, 140.1, 121.7, 52.0, 41.0, 40.3, 38.04, 37.99, 35.3, 27.9; HRMS (ESI) m/z calcd for C19H23N3O4 ([M + H]+) m/z 358.1761; found 358.1754.
Adamantane-1-carbohydrazide (12)
This compound was obtained from 1-adamantanecarboxylic acid employing method C and its 1H NMR data matched that reported in the literature (Seliverstova et al., 2018). White solid, yield: 78%. 1H NMR (DMSO-d6) δ 8.67 (s, 1H), 4.10 (s, 2H), 1.94 (s, 3H), 1.75 (d, J = 2.7 Hz, 6H), 1.71 – 1.58 (m, 6H).
N-(1-adamantyl)pyrazine-2-carboxamide (13a)
The title compound was synthesised from POA and 1-adamantylamine according to general procedure A and its 1H NMR data matched that reported in the literature (Naredla et al., 2013). The purity of the compound was > 95% after flash chromatography. White solid, yield: 76%. 1H NMR (DMSO-d6) δ 9.16 (s, 1H), 8.81 (s, 1H), 8.63 (s, 1H), 7.74 (s, 1H), 2.08 (s, 9H), 1.67 (s, 6H).
N'-(adamantane-1-carbonyl)pyrazine-2-carbohydrazide (13b)
The title compound was obtained from POA and compound 12 employing method A and further purified via crystallization from ethanol after flash chromatography. Buff solid, yield: 50%. 1H NMR (DMSO-d6) δ 10.48 (s, 1H), 9.54 (s, 1H), 9.16 (d, J = 1.3 Hz, 1H), 8.91 (d, J = 2.4 Hz, 1H), 8.77 (dd, J = 2.3, 1.5 Hz, 1H), 2.00 (s, 3H), 1.88 (d, J = 2.1 Hz, 6H), 1.70 (s, 6H); 13C NMR (DMSO-d6) δ 176.5, 162.5, 148.3, 144.9, 144.1, 144.0, 40.1, 39.0, 36.5, 28.0; HRMS (ESI) m/z calcd for C16H20N4O2 ([M + H]+) m/z 301.1659; found 301.1651.
7-(4-(tert-butoxycarbonyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (14)
Commercially available CPF (2 mmol) was dissolved in 20 mL of water:dioxane (1:1) containing 4 mL of 2.0 M aqueous NaOH solution. Di-tert-butyl dicarbonate (Boc2O, 3 mmol) was then added and the reaction mixture was stirred at rt until completion (48 h). Three quarters of the solvent was evaporated in vacuo, followed by acidification with aqueous 2.0 M HCl solution and the formed precipitate was filtered off, extensively washed with water (2 × 50 mL), and dried. 1H NMR data of the product matched that reported in the literature (Tehler et al., 2013). White solid, yield: 99%. 1H NMR (CDCl3) δ 8.78 (s, 1H), 8.05 (d, J = 12.9 Hz, 1H), 7.37 (d, J = 7.1 Hz, 1H), 3.67 (t, J = 5.0 Hz, 4H), 3.60 – 3.43 (m, 1H), 3.29 (t, J = 5.1 Hz, 4H), 1.50 (s, 9H), 1.43 – 1.36 (m, 2H), 1.24 – 1.18 (m, 2H).
N-(1-adamantyl)-1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxamide (16)
Compound 14 and 1-adamantylamine were reacted following general procedure A. The residue obtained after evaporating the EtOAc extract in vacuo was dissolved in 20 mL DCM, followed by the addition of 4 mL TFA. The reaction mixture was stirred for 12 h and concentrated in vacuo. NaHCO3 solution was then added to the residue, followed by extraction with DCM (3 × 50 mL). The combined organic phases were washed with brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using DCM/MeOH gradient prior to further HPLC purification. White solid, yield: 40%. 1H NMR (DMSO-d6) δ 9.89 (s, 1H), 8.59 (s, 1H), 7.82 (d, J = 13.6 Hz, 1H), 7.45 (d, J = 7.5 Hz, 1H), 3.80 – 3.61 (m, 1H), 3.18 (t, J = 4.4 Hz, 4H), 2.90 (s, 4H), 2.04 (s, 9H), 1.66 (s, 6H), 1.32 – 1.24 (m, 2H), 1.14 – 1.03 (m, 2H); 13C NMR (DMSO-d6) δ 174.8, 163.0, 152.9 (d, J = 247.0 Hz), 147.1, 143.5 (d, J = 10.6 Hz), 138.8, 121.9 (d, J = 6.9 Hz), 111.9 (d, J = 22.6 Hz), 111.4, 107.0, 51.0, 47.0 (d, J = 4.2 Hz), 43.2, 41.9, 36.5, 35.5, 29.3, 8.0; HRMS (ESI) m/z calcd for C27H33FN4O2 ([M + H]+) m/z 465.2660; found 465.2659.
4,6-difluoro-N'-isonicotinoyl-1H-indole-2-carbohydrazide (18)
The title compound was obtained from commercially available 4,6-difluoroindole-2-carboxylic acid (17) and INH employing method A. White solid, yield: 64%. 1H NMR (DMSO-d6) δ 12.22 (s, 1H), 10.95 (s, 1H), 10.77 (s, 1H), 8.87 (s, 2H), 7.92 (d, J = 4.9 Hz, 2H), 7.39 (d, J = 2.0 Hz, 1H), 7.07 (dd, J = 9.3, 1.7 Hz, 1H), 6.95 (td, J = 10.4, 1.9 Hz, 1H); 13C NMR (DMSO-d6) δ 164.5, 160.4, 160.0 (dd, J = 239.3, 12.1 Hz),156.3 (dd, J = 249.2, 15.6 Hz), 149.9, 140.9, 138.4 (dd, J = 15.2, 12.9 Hz), 130.8 (d, J = 3.2 Hz), 122.5, 113.6 (d, J = 21.7 Hz), 99.9, 96.0 (dd, J = 29.7, 23.2 Hz), 95.2 (dd, J = 25.9, 4.4 Hz); HRMS (ESI) m/z calcd for C15H10F2N4O2 ([M + H]+) m/z 317.0845; found 317.0839.
4,6-difluoro-1H-indole-2-carbohydrazide (19)
This compound was synthesised from 4,6-difluoroindole-2-carboxylic acid (17) according to method C. Buff solid, yield: 74%. 1H NMR (DMSO-d6) δ 12.04 (s, 1H), 9.86 (s, 1H), 7.16 (s, 1H), 7.01 (dd, J = 9.4, 1.4 Hz, 1H), 6.87 (td, J = 10.4, 1.9 Hz, 1H), 4.52 (s, 2H); 13C NMR (DMSO-d6) δ 160.8, 159.5 (dd, J = 238.4, 12.3 Hz), 156.1 (dd, J = 248.5, 15.5 Hz), 138.0 (dd, J = 15.2, 13.2 Hz), 132.0 (d, J = 3.3 Hz), 113.6 (dd, J = 21.8, 0.7 Hz), 98.0, 95.6 (dd, J = 29.7, 23.3 Hz), 95.0 (dd, J = 25.9, 4.5 Hz).
4,6-difluoro-N'-(pyrazine-2-carbonyl)-1H-indole-2-carbohydrazide (20)
The title compound was obtained from POA and intermediate 19 employing method A or B. The product was further crystallised from DCM to attain > 95% purity. Light buff solid, yield: 32% (method A) and 70% (method B). 1H NMR (DMSO-d6) δ 12.22 (br s, 1H), 10.72 (br s, 2H), 9.23 (s, 1H), 8.94 (d, J = 1.8 Hz, 1H), 8.81 (s, 1H), 7.41 (s, 1H), 7.07 (d, J = 9.0 Hz, 1H), 6.92 (t, J = 10.1 Hz, 1H); 13C NMR (DMSO-d6) δ 162.9, 160.1, 159.9 (dd, J = 239.1, 12.1 Hz), 156.3 (dd, J = 249.2, 15.6 Hz), 148.6, 144.6, 144.2, 144.1, 138.3 (dd, J = 15.2, 13.0 Hz), 131.0 (d, J = 3.2 Hz), 113.6 (d, J = 21.8 Hz), 99.9, 96.0 (dd, J = 29.7, 23.3 Hz), 95.2 (dd, J = 25.9, 4.3 Hz); HRMS (ESI) m/z calcd for C14H9F2N5O2 ([M + H]+) m/z 318.0797; found 318.0793.
N'-isonicotinoylpyrazine-2-carbohydrazide (21a)
The title compound was prepared from POA and INH employing method B and further purified via recrystallization from diethyl ether after flash chromatography. White solid, yield: 66%. 1H NMR (DMSO-d6) δ 10.96 (s, 2H), 9.22 (s, 1H), 8.94 (d, J = 2.3 Hz, 1H), 8.79 (d, J = 6.0 Hz, 3H), 7.82 (d, J = 5.8 Hz, 2H); 13C NMR (DMSO-d6) δ 164.7, 162.8, 150.9, 148.6, 144.5, 144.2, 144.1, 140.0, 121.9; HRMS (ESI) m/z calcd for C11H9N5O2 ([M + H]+) m/z 244.0829; found 244.0824.
N'-benzoylpyrazine-2-carbohydrazide (21b)
The title compound was obtained from benzohydrazide and POA employing method A. White solid, yield: 41%. 1H NMR (DMSO-d6) δ 10.85 (s, 1H), 10.60 (s, 1H), 9.22 (d, J = 1.2 Hz, 1H), 8.94 (d, J = 2.4 Hz, 1H), 8.80 (dd, J = 2.4, 1.5 Hz, 1H), 7.93 (d, J = 7.1 Hz, 2H), 7.61 (t, J = 7.3 Hz, 1H), 7.53 (t, J = 7.4 Hz, 2H); 13C NMR (DMSO-d6) δ 167.2, 163.3, 148.3, 144.4, 144.3, 143.7, 132.8, 132.2, 129.2, 127.9; HRMS (ESI) m/z calcd for C12H10N4O2 ([M + H]+) m/z 243.0877; found 243.0870.
6.2. Biology
MIC was determined using Microplate alamarBlue assay (MABA) as previously reported (Collins & Franzblau, 1997; Pieroni et al., 2011). Modified MABA was used to determine the MIC of PZA. Briefly, 7H9 (Difco™ Middlebrook 7H9 Broth, BD) without Tween 80 but containing 10% OADC (BBL™ Middlebrook OADC Enrichment, BD) was prepared fresh with the pH value was adjusted to 5.45. Assay was set up as previously reported (Collins & Franzblau, 1997; Pieroni et al., 2011). After 15 days of incubation, 15 μl of 7H9 (pH = 10.75) was added to each well and immediately followed by addition of 32.5 μL of alamar blue. Plate was incubated overnight before reading. MABA format was also used in the cytotoxicity evaluation on Vero Cells (Lun et al., 2013).
6.3. Molecular docking protocol
In silico molecular modelling analysis was undertaken using the Molecular Operating Environment MOE software version 2008.10 (Chemical Computing Group, Montreal, Canada). The X-ray crystal structure of M. tb DNA gyrase in complex with CPF (5BTC) (Blower et al., 2016) was retrieved from the protein data bank (PDB). The binding pocket was ready for docking after 3D protonating the enzyme, whereby hydrogens and partial charges were added to the system for optimisation. In order to validate the docking protocol, the co-crystallised CPF was docked into the active site. Next, the structure of 16 was drawn in ChemDraw Ultra 16.0, saved as .mol file, opened inside the MOE program, 3D protonated and geometrically optimised using MMFF94x forcefield with gradient 0.05. Compound 16 was then docked into the same binding site of CPF using MOE-DOCK function, employing Triangle Matcher placement method and scored using London dG scoring methodology. Then, molecular mechanics forcefield was applied to relax the generated poses which were further ranked using London dG scoring function and the top 30 poses were retained. The best-ranked pose with the lowest binding free energy (i.e the smallest docking score) was selected.
Acknowledgments
SSRA is grateful for the support of Curtin International Postgraduate Research Scholarship (CIPRS). WRB is thankful for the support of NIH grants AI 37856 and HL 133190. HG acknowledges the ARC Discovery Early Career Researcher Award DE160100482.
References:
- Aldred KJ, Blower TR, Kerns RJ, Berger JM, & Osheroff N (2016). Fluoroquinolone interactions with Mycobacterium tuberculosis gyrase: Enhancing drug activity against wild-type and resistant gyrase. Proc. Natl. Acad. Sci. U S A, 113(7), E839–846. doi: 10.1073/pnas.1525055113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred KJ, Breland EJ, Vlckova V, Strub MP, Neuman KC, Kerns RJ, & Osheroff N (2014). Role of the water-metal ion bridge in mediating interactions between quinolones and Escherichia coli topoisomerase IV. Biochemistry, 53(34), 5558–5567. doi: 10.1021/bi500682e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred KJ, McPherson SA, Turnbough CL Jr., Kerns RJ, & Osheroff N (2013). Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: mechanistic basis of quinolone resistance. Nucleic Acids Res., 41(8), 4628–4639. doi: 10.1093/nar/gkt124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsayed SSR, Beh CC, Foster NR, Payne AD, Yu Y, & Gunosewoyo H (2019). Kinase Targets for Mycolic Acid Biosynthesis in Mycobacterium tuberculosis. Curr. Mol. Pharmacol, 12(1), 27–49. doi: 10.2174/1874467211666181025141114 [DOI] [PubMed] [Google Scholar]
- Alsayed SSR, Lun S, Luna G, Beh CC, Payne AD, Foster N, Bishai WR, & Gunosewoyo H (2020). Design, synthesis, and biological evaluation of novel arylcarboxamide derivatives as anti-tubercular agents. RSC Advances, 10(13), 7523–7540. doi: 10.1039/C9RA10663D [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averina EB, Sedenkova KN, Bakhtin SG, Grishin YK, Kutateladze AG, Roznyatovsky VA, Rybakov VB, Butov GM, Kuznetsova TS, & Zefirov NS (2014). symm-Tetramethylenecyclooctane: en route to polyspirocycles. J. Org. Chem, 79(17), 8163–8170. doi: 10.1021/jo501380y [DOI] [PubMed] [Google Scholar]
- Bahde RJ, Appella DH, & Trenkle WC (2011). A one-pot preparation of N-2-mercaptobenzoyl-amino amides. Tetrahedron Lett., 52(32), 4103–4105. doi: 10.1016/j.tetlet.2011.05.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberis I, Bragazzi NL, Galluzzo L, & Martini M (2017). The history of tuberculosis: from the first historical records to the isolation of Koch's bacillus. J. Prev. Med. Hyg, 58(1), E9–E12. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5432783/pdf/2421-4248-58-E9.pdf [PMC free article] [PubMed] [Google Scholar]
- Blower TR, Williamson BH, Kerns RJ, & Berger JM (2016). Crystal structure and stability of gyrase-fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U S A, 113(7), 1706–1713. doi: 10.1073/pnas.1525047113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon C, & Dick T (2012). How Mycobacterium tuberculosis goes to sleep: the dormancy survival regulator DosR a decade later. Future Microbiol., 7(4), 513–518. doi: 10.2217/fmb.12.14 [DOI] [PubMed] [Google Scholar]
- Collins L, & Franzblau SG (1997). Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother, 41(5), 1004–1009. doi: 10.1128/aac.41.5.1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dheda K, Gumbo T, Gandhi NR, Murray M, Theron G, Udwadia Z, Migliori GB, & Warren R (2014). Global control of tuberculosis: from extensively drug-resistant to untreatable tuberculosis. Lancet Respir. Med, 2(4), 321–338. doi: 10.1016/S2213-2600(14)70031-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin ER, Addison AP, John SF, Olaleye OA, & Rosell RC (2019). Pharmacokinetics of isoniazid: The good, the bad, and the alternatives. Tuberculosis (Edinb), 116S, S66–S70. doi: 10.1016/j.tube.2019.04.012 [DOI] [PubMed] [Google Scholar]
- Gopal P, Gruber G, Dartois V, & Dick T (2019). Pharmacological and Molecular Mechanisms Behind the Sterilizing Activity of Pyrazinamide. Trends Pharmacol. Sci, 40(12), 930–940. doi: 10.1016/j.tips.2019.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemers A, Leysen DC, Bollaert W, Zhang MQ, & Pattyn SR (1990). Influence of N substitution on antimycobacterial activity of ciprofloxacin. Antimicrob. Agents Chemother, 34(3), 496–497. doi: 10.1128/aac.34.3.496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikishore A, Leow ML, Niang M, Rajan S, Pasunooti KK, Preiser PR, Liu X, & Yoon HS (2013). Adamantyl derivative as a potent inhibitor of Plasmodium FK506 binding protein 35. ACS Med. Chem. Lett, 4(11), 1097–1101. doi: 10.1021/ml400306r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Wu X, Luo J, Fu Y, Zhao L, Ma Y, Li Y, Liang Q, Shang Y, & Huang H (2017). Detection of pyrazinamide resistance of Mycobacterium tuberculosis using nicotinamide as a surrogate. Clin. Microbiol. Infect, 23(11), 835–838. doi: 10.1016/j.cmi.2017.03.028 [DOI] [PubMed] [Google Scholar]
- Hu YQ, Zhang S, Zhao F, Gao C, Feng LS, Lv ZS, Xu Z, & Wu X (2017). Isoniazid derivatives and their anti-tubercular activity. Eur. J. Med. Chem, 133, 255–267. doi: 10.1016/j.ejmech.2017.04.002 [DOI] [PubMed] [Google Scholar]
- Judge V, Narasimhan B, Ahuja M, Sriram D, Yogeeswari P, De Clercq E, Pannecouque C, & Balzarini J (2013). Synthesis, antimycobacterial, antiviral, antimicrobial activity and QSAR studies of N(2)-acyl isonicotinic acid hydrazide derivatives. Med. Chem, 9(1), 53–76. doi: 10.2174/157340613804488404 [DOI] [PubMed] [Google Scholar]
- Lange C, Chesov D, Heyckendorf J, Leung CC, Udwadia Z, & Dheda K (2018). Drug-resistant tuberculosis: An update on disease burden, diagnosis and treatment. Respirology, 23(7), 656–673. doi: 10.1111/resp.13304 [DOI] [PubMed] [Google Scholar]
- Liu J, Obando D, Liao V, Lifa T, & Codd R (2011). The many faces of the adamantyl group in drug design. Eur. J. Med. Chem, 46(6), 1949–1963. doi: 10.1016/j.ejmech.2011.01.047 [DOI] [PubMed] [Google Scholar]
- Lun S, Guo H, Onajole OK, Pieroni M, Gunosewoyo H, Chen G, Tipparaju SK, Ammerman NC, Kozikowski AP, & Bishai WR (2013). Indoleamides are active against drug-resistant Mycobacterium tuberculosis. Nat. Commun, 4, 2907. doi: 10.1038/ncomms3907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado D, Girardini M, Viveiros M, & Pieroni M (2018). Challenging the Drug-Likeness Dogma for New Drug Discovery in Tuberculosis. Front. Microbiol, 9, 1367. doi: 10.3389/fmicb.2018.01367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruri F, Sterling TR, Kaiga AW, Blackman A, van der Heijden YF, Mayer C, Cambau E, & Aubry A (2012). A systematic review of gyrase mutations associated with fluoroquinolone-resistant Mycobacterium tuberculosis and a proposed gyrase numbering system. J. Antimicrob. Chemother, 67(4), 819–831. doi: 10.1093/jac/dkr566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miniyar P, & Bhat A (1999). Pyrazinoic acid hydrazide derivatives: synthesis and antimycobacterial activities. Indian J. Heterocy. Ch, 9(2), 155–156. [Google Scholar]
- Miotto P, Cabibbe AM, Feuerriegel S, Casali N, Drobniewski F, Rodionova Y, Bakonyte D, Stakenas P, Pimkina E, Augustynowicz-Kopec E, Degano M, Ambrosi A, Hoffner S, Mansjo M, Werngren J, Rusch-Gerdes S, Niemann S, & Cirillo DM (2014). Mycobacterium tuberculosis pyrazinamide resistance determinants: a multicenter study. mBio, 5(5), e01819–01814. doi: 10.1128/mBio.01819-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naredla RR, Dash BP, & Klumpp DA (2013). Preparation of pyrazine carboxamides: a reaction involving N-heterocyclic carbene (NHC) intermediates. Org. Lett, 15(18), 4806–4809. doi: 10.1021/ol402200x [DOI] [PubMed] [Google Scholar]
- Njire M, Tan Y, Mugweru J, Wang C, Guo J, Yew W, Tan S, & Zhang T (2016). Pyrazinamide resistance in Mycobacterium tuberculosis: Review and update. Adv. Med. Sci, 61(1), 63–71. doi: 10.1016/j.advms.2015.09.007 [DOI] [PubMed] [Google Scholar]
- Onajole OK, Pieroni M, Tipparaju SK, Lun S, Stec J, Chen G, Gunosewoyo H, Guo H, Ammerman NC, Bishai WR, & Kozikowski AP (2013). Preliminary structure-activity relationships and biological evaluation of novel antitubercular indolecarboxamide derivatives against drug-susceptible and drug-resistant Mycobacterium tuberculosis strains. J. Med. Chem, 56(10), 4093–4103. doi: 10.1021/jm4003878 [DOI] [PubMed] [Google Scholar]
- Panda SS, Detistov OS, Girgis AS, Mohapatra PP, Samir A, & Katritzky AR (2016). Synthesis and molecular modeling of antimicrobial active fluoroquinolone-pyrazine conjugates with amino acid linkers. Bioorg. Med. Chem. Lett, 26(9), 2198–2205. doi: 10.1016/j.bmcl.2016.03.062 [DOI] [PubMed] [Google Scholar]
- Panda SS, Girgis AS, Mishra BB, Elagawany M, Devarapalli V, Littlefield WF, Samir A, Fayad W, Fawzy NG, Srour AM, & Bokhtia RM (2019). Synthesis, computational studies, antimycobacterial and antibacterial properties of pyrazinoic acid–isoniazid hybrid conjugates. RSC Advances, 9(35), 20450–20462. doi: 10.1039/C9RA03380G [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlowski A, Jansson M, Skold M, Rottenberg ME, & Kallenius G (2012). Tuberculosis and HIV co-infection. PLoS Pathog., 8(2), e1002464. doi: 10.1371/journal.ppat.1002464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccaro G, Poce G, Biava M, Giannoni F, & Fattorini L (2015). Activity of lipophilic and hydrophilic drugs against dormant and replicating Mycobacterium tuberculosis. J. Antibiot. (Tokyo), 68(11), 711–714. doi: 10.1038/ja.2015.52 [DOI] [PubMed] [Google Scholar]
- Pieroni M, Tipparaju SK, Lun S, Song Y, Sturm AW, Bishai WR, & Kozikowski AP (2011). Pyrido[1,2-a]benzimidazole-based agents active against tuberculosis (TB), multidrug-resistant (MDR) TB and extensively drug-resistant (XDR) TB. ChemMedChem, 6(2), 334–342. doi: 10.1002/cmdc.201000490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluger NW (2013). Fluoroquinolones in the treatment of tuberculosis: which is best? Am. J. Respir. Crit. Care Med, 188(7), 768–769. doi: 10.1164/rccm.201308-1446ED [DOI] [PubMed] [Google Scholar]
- Seliverstova DV, Suslonov VV, Zarubaev VV, & Trifonov RE (2018). Synthesis, Structure, and Anti-influenza Activity of 2-(Adamantan-1-yl)-5-aryl-1,3,4-oxadiazoles and 2-(Adamantan-1-yl)-5-aryltetrazoles. Russian Journal of Organic Chemistry, 54(4), 633–638. doi: 10.1134/s107042801804019x [DOI] [Google Scholar]
- Seung KJ, Keshavjee S, & Rich ML (2015). Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harb. Perspect. Med, 5(9), a017863. doi: 10.1101/cshperspect.a017863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar EM, Vignesh R, Ellegard R, Barathan M, Chong YK, Bador MK, Rukumani DV, Sabet NS, Kamarulzaman A, Velu V, & Larsson M (2014). HIV-Mycobacterium tuberculosis co-infection: a 'danger-couple model' of disease pathogenesis. Pathog. Dis, 70(2), 110–118. doi: 10.1111/2049-632X.12108 [DOI] [PubMed] [Google Scholar]
- Sotgiu G, & Migliori GB (2015). Facing multi-drug resistant tuberculosis. Pulm. Pharmacol. Ther, 32, 144–148. doi: 10.1016/j.pupt.2014.04.006 [DOI] [PubMed] [Google Scholar]
- Stec J, Onajole OK, Lun S, Guo H, Merenbloom B, Vistoli G, Bishai WR, & Kozikowski AP (2016). Indole-2-carboxamide-based MmpL3 Inhibitors Show Exceptional Antitubercular Activity in an Animal Model of Tuberculosis Infection. J. Med. Chem, 59(13), 6232–6247. doi: 10.1021/acs.jmedchem.6b00415 [DOI] [PubMed] [Google Scholar]
- Tehler U, Fagerberg JH, Svensson R, Larhed M, Artursson P, & Bergstrom CA (2013). Optimizing solubility and permeability of a biopharmaceutics classification system (BCS) class 4 antibiotic drug using lipophilic fragments disturbing the crystal lattice. J. Med. Chem, 56(6), 2690–2694. doi: 10.1021/jm301721e [DOI] [PubMed] [Google Scholar]
- Tiberi S, Scardigli A, Centis R, D'Ambrosio L, Munoz-Torrico M, Salazar-Lezama MA, Spanevello A, Visca D, Zumla A, Migliori GB, & Caminero Luna JA (2017). Classifying new anti-tuberculosis drugs: rationale and future perspectives. Int. J. Infect. Dis, 56, 181–184. doi: 10.1016/j.ijid.2016.10.026 [DOI] [PubMed] [Google Scholar]
- Tong AST, Choi PJ, Blaser A, Sutherland HS, Tsang SKY, Guillemont J, Motte M, Cooper CB, Andries K, Van den Broeck W, Franzblau SG, Upton AM, Denny WA, Palmer BD, & Conole D (2017). 6-Cyano Analogues of Bedaquiline as Less Lipophilic and Potentially Safer Diarylquinolines for Tuberculosis. ACS Med. Chem. Lett, 8(10), 1019–1024. doi: 10.1021/acsmedchemlett.7b00196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unissa AN, Subbian S, Hanna LE, & Selvakumar N (2016). Overview on mechanisms of isoniazid action and resistance in Mycobacterium tuberculosis. Infect. Genet. Evol, 45, 474–492. doi: 10.1016/j.meegid.2016.09.004 [DOI] [PubMed] [Google Scholar]
- Velayati AA, Farnia P, & Masjedi MR (2013). The totally drug resistant tuberculosis (TDR-TB). Int. J. Clin. Exp. Med, 6(4), 307–309. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3631557/pdf/ijcem0006-0307.pdf [PMC free article] [PubMed] [Google Scholar]
- Vidossich P, Loewen PC, Carpena X, Fiorin G, Fita I, & Rovira C (2014). Binding of the antitubercular pro-drug isoniazid in the heme access channel of catalase-peroxidase (KatG). A combined structural and metadynamics investigation. J. Phys. Chem. B, 118(11), 2924–2931. doi: 10.1021/jp4123425 [DOI] [PubMed] [Google Scholar]
- Vilcheze C, & Jacobs WR Jr. (2019). The Isoniazid Paradigm of Killing, Resistance, and Persistence in Mycobacterium tuberculosis. J. Mol. Biol, 431(18), 3450–3461. doi: 10.1016/j.jmb.2019.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanka L, Iqbal K, & Schreiner PR (2013). The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives. Chem. Rev, 113(5), 3516–3604. doi: 10.1021/cr100264t [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werngren J, Sturegard E, Jureen P, Angeby K, Hoffner S, & Schon T (2012). Reevaluation of the critical concentration for drug susceptibility testing of Mycobacterium tuberculosis against pyrazinamide using wild-type MIC distributions and pncA gene sequencing. Antimicrob. Agents Chemother, 56(3), 1253–1257. doi: 10.1128/AAC.05894-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. (2020). World Health Organisation Global Tuberculosis Report, Geneva. Retrieved from https://apps.who.int/iris/bitstream/handle/10665/336069/9789240013131-eng.pdf
- Wohlkonig A, Chan PF, Fosberry AP, Homes P, Huang J, Kranz M, Leydon VR, Miles TJ, Pearson ND, Perera RL, Shillings AJ, Gwynn MN, & Bax BD (2010). Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol, 17(9), 1152–1153. doi: 10.1038/nsmb.1892 [DOI] [PubMed] [Google Scholar]
- Xu Z, Meshcheryakov VA, Poce G, & Chng SS (2017). MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. U S A, 114(30), 7993–7998. doi: 10.1073/pnas.1700062114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Permar S, & Sun Z (2002). Conditions that may affect the results of susceptibility testing of Mycobacterium tuberculosis to pyrazinamide. J. Med. Microbiol, 51(1), 42–49. doi: 10.1099/0022-1317-51-1-42 [DOI] [PubMed] [Google Scholar]