

Abstract
Severe asthma accounts for almost half of the cost associated with asthma. Severe asthma is driven by heterogeneous molecular mechanisms. Conventional clinical trial design often lacks the power and efficiency to target subgroups with specific pathobiological mechanisms. Further, the validation and approval of new asthma therapies is a lengthy process. A large proportion of that time is taken by clinical trials to validate asthma interventions. The NIH Precision Medicine in Severe and/or Exacerbation Prone Asthma (PrecISE) program was established with the goal of designing and executing a trial that uses adaptive design techniques to rapidly evaluate novel interventions in biomarker-defined subgroups of severe asthma, while seeking to refine these biomarker subgroups, and to identify early markers of response to therapy. The novel trial design is an adaptive platform trial conducted under a single master protocol that incorporates precision medicine components. Further, it includes innovative applications of futility analysis, cross-over design with use of shared placebo groups and early futility analysis to permit more rapid identification of effective interventions. The development and rationale behind the study design are described. The interventions chosen for initial investigation and the criteria used to identify these interventions are enumerated. The biomarker-based adaptive design and analytic scheme are detailed as well as special considerations involved in the final trial design.
Keywords: Severe asthma, Therapy, Clinical Trial, Biomarkers, Precision medicine, Adaptive Design, Master Protocol, Platform Trial, Adaptive Enrichment
I. Goals of PrecISE
Severe asthma is driven by heterogeneous molecular underpinnings. The new class of asthma biologics aimed at inhibiting Type 2 inflammation are effective in only about half of severe asthma patients (1). Furthermore, the clinical response to these medications is highly variable. The development of new therapies takes many years, largely secondary to the time needed to complete clinical trials. Platform trials with adaptive study designs utilizing master protocols are promising alternatives to conventional study designs due to their capacity to efficiently study multiple interventions. Additionally, precision medicine approaches, utilizing phenotypic “biomarkers” to identify subgroups of patients more responsive to the study intervention, are now available.
The NIH Precision Medicine in Severe and/or Exacerbation Prone Asthma (PrecISE) program was established with the goal of creating a study that uses adaptive design techniques to rapidly evaluate novel interventions in biomarker-defined subgroups of severe asthma. Additionally, the program seeks to refine the biomarker subgroups and to possibly identify early markers of response to therapy.
As opposed to standard randomized trials in which the study procedures and outcomes are set at the beginning of the trial, and not modified after study initiation, adaptively designed studies allow prospectively planned modifications to one or more aspects of the design based on accumulating data from subjects in the trial and have been recognized as legitimate designs for drug approval by the FDA. (2). These adaptations are based on interim study data and have to be pre-specified. Adaptive components can include one, or several, of the following based on interim analyses: 1) Modifying allocation proportion to different arms; 2) Modifying the study population; 3) Modifying the endpoints; 4) Changing the sample size of the trial; 5) Dropping arms for futility; 6) Stopping the entire trial early for futility or efficacy;. Adaptive approaches allow for more efficient use of subjects since fewer subjects receive ineffective compounds and studies have the potential for producing results at smaller sample sizes than initially expected. In trials with multiple interventions this results in more rapid changing of treatments and reassignment of resources to more promising candidates.
To achieve these aims we designed a trial that incorporates an adaptive platform, a master protocol design, and precision medicine components. A platform design in a master protocol is a study designed in such a way that the basic structure can be used to perpetually evaluate multiple interventions for a disease process. Precision medicine approaches are adaptive approaches that allow for refinement of the patient subgroups targeted by interventions during a trial.
Although adaptive platform trials with precision medicine components have been utilized in cancer trials (3, 4), asthma is a temporally variable, complex, chronic inflammatory disease. Precision medicine trials in asthma present both opportunities and unique challenges for innovation in trial design. Herein we describe the development and structure of the PrecISE platform trial (Clinical Trials.gov NCT04129931). It includes innovative applications of futility analysis, cross-over design, and use of a shared placebo to permit more rapid identification of effective interventions and refinement of predictive biomarker subgroups. Our goal is to acquaint the reader with the trial and the innovative tools and analytic approaches it utilizes. We outline our objectives in adopting different components of adaptation and study structure and the framework and rationale we developed to achieve these objectives. We also describe the criteria used to identify our potential interventions and the biomarker-based adaptive design and analytic scheme. Additionally, we review special considerations relevant to the protocol and analysis, and implementation realities. A more detailed description of our statistical and analytic approach has been recently published (5).
II. Objectives and Framework and Rationale to Achieve Objectives
The Steering Committee (SC) and the Data, Modeling, and Coordination Center (DMCC) defined the objectives outlined in Table I. To achieve these objectives we agreed on the following conceptual approaches based on the provided rationales:
Using a master protocol to allow more rapid introduction of potential interventions rather than designing new protocols for each drug.
Utilizing a cross-over design with multiple periods in which subjects are treated with multiple interventions and the interventions share the same placebo for that subject, in order to maximally utilize information from recruited subjects. Such a design also provides subjects with an opportunity to “try” multiple interventions in the context of one overall trial.
Allowing interventions to demonstrate efficacy on any one of multiple efficacy outcomes (e.g. exacerbations, airway function, symptoms). Simultaneously testing for efficacy on multiple outcomes allows the use of the same trial design across interventions, even though we might be testing interventions that address different components of asthma pathobiology.
Reducing drug exposure time to the minimum duration thought to provide a likelihood of response to allow testing of multiple agents in a shorter time frame. For this reason, we adopted a substitute for exacerbations (CompEx) (see below) to evaluate efficacy in regard to exacerbations utilizing a reduced intervention duration.
Performing an early futility analysis to reduce the time committed to testing of interventions with a lower likelihood of efficacy, thus allowing more rapid accrual to the remaining interventions and/or replacement of those ineffective interventions with “back-up” interventions.
Preferentially assigning subjects to interventions based on pre-specified biomarker thresholds to allow rapid assessment of whether a drug is effective in the pre-specified biomarker subgroup.
Assigning some subjects to interventions even if they are outside the initial pre-specified biomarker group to permit adjustment of biomarker thresholds during the study.
Table I.
PrecISE Objectives
| Primary |
| 1. To identify novel therapies that are efficacious in pre-defined biomarker-based subgroups of severe asthma patients |
| 2. To optimize the subgroups targeted for treatment by refining the biomarkers and subgroup definitions |
| Secondary |
| 1. To gain information about potential monitoring biomarkers for selected therapies |
| 2. To explore the safety and effectiveness of selected therapies in adolescent patients with severe asthma. |
III. Criteria used to identify the top candidate interventions
Each clinical center application to join the network had proposed at least 3 specific interventions. At the initial SC meeting, all interventions were discussed and each was subsequently scored based upon the following criteria: 1) scientific basis for potential efficacy in severe asthma; 2) definition of a target biomarker subgroup likely to respond to the therapy; 3) feasibility of drug acquisition and administration (oral, injection, etc.); 4) cost of therapy (if not donated by industry); 5) known safety profiles and potential considerations in severe asthma; and 6) likelihood of obtaining approval for use in adolescents from the U.S. Food and Drug Administration. Those that were deemed to be safe and potentially accessible, with the greatest overall scores, were then pursued as the initial group of candidate interventions to be further investigated. Those finally chosen are listed in Table II. The hypothesized mechanisms and pharmacology of these interventions will be detailed in future publications.
Table II.
PrecISE Interventions and Biomarkers
| Investigative Approach (Agent) | Target Population | Predictive Biomarker & Threshold | Exploratory Monitoring Biomarkers | Secondary & Exploratory Predictive Biomarkers |
|---|---|---|---|---|
| Kit receptor inhibitor (imatinib) | T2 low | Blood Eos <300/ul | Serum tryptase Urinary PGD2 |
Serum tryptase Sputum PMNs Urinary PGD2 Sputum tryptase Airway wall thickness on HRCT Sputum mast cell gene expression |
| Anti-IL6 (clazakizumab) | Obesity/Metabolic dysfunction | Plasma IL-6 ≥ 3.1 pg/ul | hsCRP | hsCRP Fat density on HRCT |
| Jak inhibitor (itacitinib) | T2 high | Blood Eos ≥ 300/ul or FeNO > 20 ppb | FeNO Blood Eos |
hsCRP Sputum eosinophils Plasma IL-6 Mucus score on HRCT |
| GSNO Reductase Inhibitor Genotype (cavosonstat) | Increased GSNOR activity by genotype | GSNOR genotype | EBC- formate to formaldehyde ratio | Change in maximal post-BD FEV1 Air trapping on HRCT |
| Bacterial extract (Broncho-Vaxom) | Eosinophilic | Blood Eos ≥ 300/ul | Blood Eos Stool microbiome |
Sputum eosinophils Mucus score on HRCT |
| Nutritional ketosis (medium chain triglycerides) | High arginine metabolic asthma | FeNO ≥ 15 ppb | Blood ketones | Urinary bromotyrosine Fat distribution on HRCT |
BD = bronchodilator; EBC = exhaled breath condensate; Eos = eosinophils; FeNO = fractional exhaled nitric oxide; GSNOR = S-nitrosoglutathione reductase; HRCT = high resolution chest CT; hsCRP = high sensitivity C-reactive protein; T2 = type 2 inflammation
Each intervention required identification of proposed predictive biomarkers (up to two) which would define the clinical subgroup, hypothesized a priori, to most likely respond to each intervention. Those chosen are listed in Table II. Several secondary and exploratory predictive biomarkers were also identified for each intervention. In addition to using biomarkers to predict intervention response, each intervention required proposed biomarkers that would serve as monitoring biomarkers, intended to help identify early signs of efficacy (or lack thereof) (Table II). Several additional interventions are being similarly assessed to serve as alternatives should any of the initially chosen agents be eliminated due to futility or safety concerns.
IV. Protocol Design and Analytic Scheme
The study aims to randomize 800 subjects (650 adults and 150 adolescents (12–17 years of age)). The general structure of the study is summarized and illustrated in Figure 1. Briefly, over an approximately 8-week period, patients with reported asthma are screened to be sure they meet criteria for severe asthma (6). During that period their biomarkers for all possible interventions are ascertained. Where possible, subjects’ inhaled corticosteroid/long-acting beta agonist (ICS/LABA) controller inhaler is changed to a standard high dose ICS/LABA combination. Baseline symptoms and lung function are measured, and safety labs are assessed. Based on a subject’s biomarker profile, the intervention specific safety measurement thresholds, the available interventions, and the progress of the study, subjects are randomized to their first intervention, with an increased probability (but not a certainty) that they will be randomized to a treatment “best” for them based on their biomarker profile.
Figure 1. PrecISE Study Structure.

See Section IV – Protocol Design and Analytic Scheme.
As illustrated in Figure 1, each period of treatment consists of 4 months of treatment and a 2-month washout (except for clazakizumab treatment which requires a 4-month washout due to a long half-life). The first 2 periods of treatment correspond to a double-blind placebo controlled 2-period cross-over design for the assigned intervention, such that a subject will receive placebo during one of the initial 2 treatment periods. These two periods were incorporated so that patients who are not able to participate after these first two periods are guaranteed to have a placebo period. After the initial 2-period cross-over, each participant continues with repeated re-randomizations to sequential 4-month treatment periods and 2- (or 4-) month washout periods, for up to a total of 4 additional treatments. In each of these subsequent periods, subjects are assigned to interventions or a matching placebo (approximately 7:1 ratio during each period) based on biomarker profile, their history of prior treatments including placebo, the intervention-specific safety measurement thresholds, and the overall enrollment in each of the interventions at the point of each re-randomization. As discussed below (Section V), no subject will be randomized to more than two placebo periods during the trial.
Subjects enrolled at the beginning of this 42-month study can potentially receive a maximum of 5 different active treatments of the six listed in Table II (as long as none had a prolonged washout period). Subjects all will have at least one required placebo (during the 2-period cross-over) and a maximum of two placebo periods.
The analytic approach and biomarker adaptation are illustrated in Figure 2. Specifics of the analysis plan and the thresholds used are discussed in our statistical design paper (4). Briefly, as illustrated in Figure 2, after 60 subjects are enrolled and complete an intervention in the pre-specified biomarker-defined target subgroup, a futility analysis will be undertaken. If a drug is determined to be futile, that intervention is immediately discontinued, thereby freeing up patient resources to more rapidly complete investigation of other interventions or introduction of a new intervention with its attendant predictive biomarkers. If futility criteria are not met for the intervention, then a biomarker subgroup refinement is undertaken (see Figure 2) based on the outcomes in the 60 patients and 30 additional subjects that have been enrolled in that intervention outside the initial target subgroup. If necessary, the biomarker cut-off will be adjusted so that the next 45 subjects will be enrolled using the new threshold with ~20 additional subjects enrolled outside the new target threshold. Once completed, a second biomarker subgroup optimization will be performed (see Figure 2), and a final 45 subjects will be enrolled in the new, refined target subgroup. Final efficacy analyses will be performed on the 150 subjects who were in the target subgroups at any point in the study (Green band in Figure 2).
Figure 2. Futility Analysis, Biomarker Adaptation, and Study Analysis for an Intervention in PrecISE.
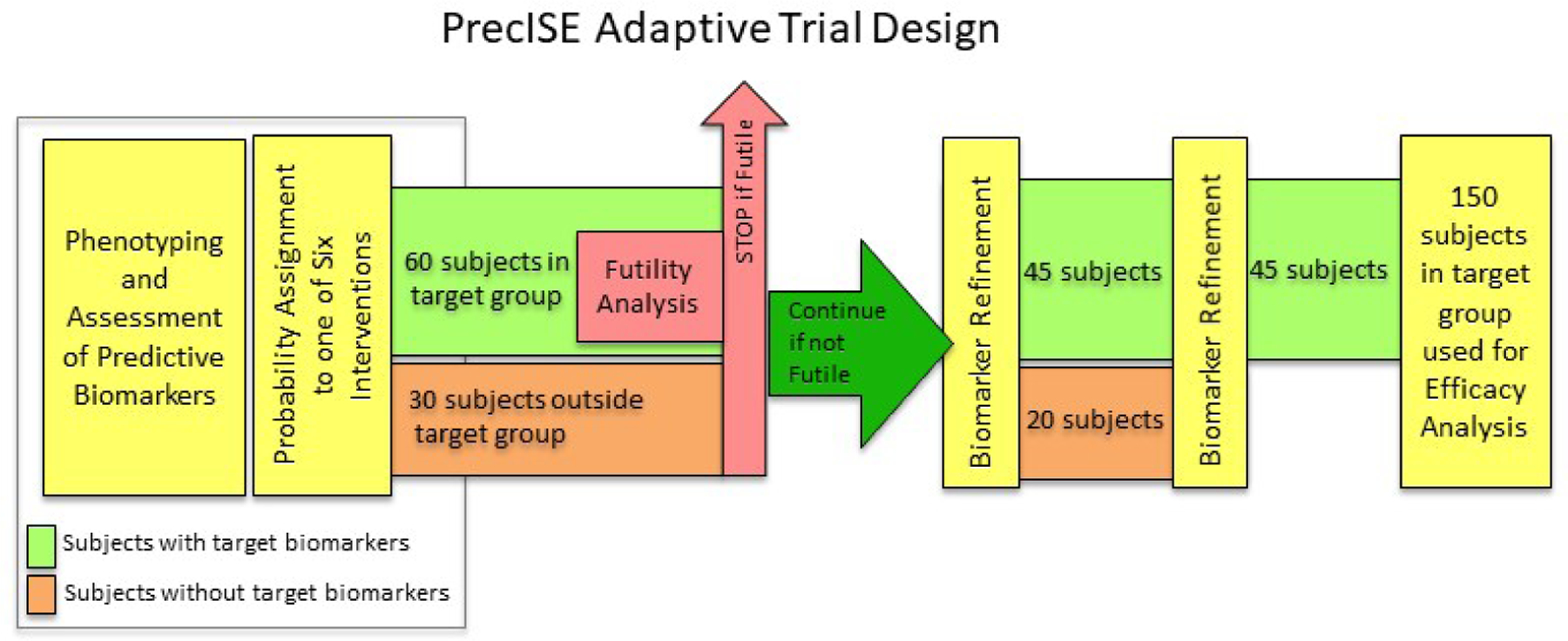
Patients are characterized, and their biomarker characteristics are determined and assigned during the run-in (see text). Based on these biomarker characteristics (Table II) they are preferentially (but not exclusively) assigned to interventions and placebos throughout their participation in the trial (Figure 1). As seen in Figure 2, patients within the target biomarker subgroup (green band) and those outside the target subgroup (orange band) are assigned to each intervention. After an intervention has accumulated 60 patients within the target biomarker subgroup (green band) and 30 subjects outside the biomarker subgroup (orange band), a futility analysis is performed restricted to the within-target 60 patients (see text). If the intervention is dropped for futility, all patients still receiving that intervention are assigned to alternative interventions. If the intervention is not dropped for futility, subgroups are refined and subjects within and outside the newly refined biomarker thresholds are enrolled and evaluated as outlined with an additional scheduled refinement as shown. When 150 patients are enrolled within the varying refined subgroups a final efficacy analysis is performed restricted to the 150 patients within the refined subgroups.
V. Specific Considerations Regarding Study Design and Analysis
The study design and analysis plan outlined in Figure 1 and 2 require several areas of explication.
Outcomes.
Since the study structure is designed to facilitate screening of multiple interventions with different mechanisms, the selected interventions may impact differing aspects of asthma control. We therefore chose three primary efficacy outcomes to address this possibility. Specifically, we chose to assess: 1) Airway function (FEV1); 2) Symptoms (ACQ-6); 3) Exacerbations and loss-of-control events as a substitute for exacerbations, using the CompEx instrument (7). These outcomes will enable us to determine the effect of each intervention on the three key components of asthma morbidity. Importantly, we considered the possibility of including asthma exacerbations (defined as a treatment with systemic corticosteroids) as a primary outcome measurement. However, the sample size required to appropriately test a reasonable effect size prohibited using asthma exacerbations as a primary endpoint. Due to this limitation the investigators elected to use the CompEx instrument as a substitute for asthma exacerbations. Steroid-requiring exacerbations will be examined as a secondary outcome and safety measures will be in place to handle exacerbations. The effect sizes for a difference in treatment responses relative to placebo that we believe we will be able to detect with 80% power in a final sample of approximately 150 subjects within the target biomarker subgroups are listed in Table III. In all these calculations, data from adolescents will be combined with data from the adults.
Table III.
PrecISE Primary Outcomes
| Primary Outcome | Minimally Detectable Treatment Difference from Placebo (80% Power) |
|---|---|
| Airway function (FEV1)a | 4.3% predicted |
| Symptoms (ACQ-6)b | 0.3 score |
| Loss of Asthma Control (CompEx)C | 0.66 event rate |
FEV1 measured prior to bronchodilator administration (Assumed SD = 14.5%)
Asthma Control Questionnaire (Assumed SD = 1)
CompEx (Assumed SD = 2)
Upfront Assignment to Interventions Based on Phenotypic Biomarkers.
We have pre-defined the initial “target” biomarker subgroup that we will test for each intervention as outlined in Table II. In this study, subjects who fall within the parameters of a biomarker-defined subgroup will be preferentially assigned to the intervention(s) expected to be effective for that biomarker profile (Figure 2). We point this principle out to contrast it with the typical post-hoc biomarker discovery approach. In the latter approach, subjects may be randomly assigned to therapies and post-hoc analysis identifies biomarker profiles that associate with improved responses to specific therapies. In our case, we have initially pre-specified the target biomarker subgroup for each intervention (Table II) and if an intervention is not found to be futile (after enrollment of the first 60 subjects in the target biomarker subgroup), we will refine the target biomarker group and assign additional subjects to the intervention (Figure 2). At this point, we continue to include some subjects outside the (newly) defined target subgroup, so that we can perform a second biomarker refinement step for the subgroup after additional patients have been enrolled (Figure 2). We will perform post-hoc analyses to see if we can identify other biomarkers that define responder subject subgroups. In Ivanova et al (5), we consider the statistical implications of upfront adaptive assignment vs. unbiased assignment in more detail.
As a corollary, we were faced with the issue of when to assign a subject’s predictive biomarker characteristics. We have chosen to assign predictive biomarker status based on biomarker determinations during the run-in period, prior to any treatment intervention. We have done so to decrease issues related to possible carryover effects from a prior treatment during the short interval of time available for the washout (see below 4-month intervention periods and 2-month washout, except for clazakizumab’s 4-month washout). While we will not be reassigning subject predictive biomarker status during the process of preferential randomization assignments, we will be reassessing these predictive biomarkers prior to each randomization period. In a secondary analysis, we will examine whether predictive biomarkers obtained just prior to each intervention period are better in defining target response subgroups.
Crossing Subjects Over to Multiple Treatments to Maximize the Information Collected in the Study.
We adopted a cross-over design for multiple reasons: 1) Since the subject population is limited, crossing subjects reduces the need to recruit a new population for each intervention and increases power for the sample size since each subject is used as their own control; 2) Since subjects may qualify for multiple interventions based on overlapping target subgroup definitions, crossing subjects over from one treatment to another affords subjects the opportunity to be assigned to multiple drugs with a greater opportunity to identify interventions effective in a particular subject (in effect “precision” medicine at the subject level within the context of the trial); 3) Multiple cross-overs allow us to “share” placebo periods providing increased efficiency to compare an intervention with placebo (See below Placebo Considerations).
Early Futility Analysis and Futility Boundaries.
All of our currently proposed interventions are novel in severe asthma. Many have not been tested in humans as regards their efficacy in severe asthma. Thus, there is a likelihood that several may be ineffective. We recognized that enrollment of a full cohort along with biomarker profiling specific to an intervention that was not effective (as judged by our outcomes in our trial) would divert subjects and resources from potentially informative interventions. To make the trial adaptive in this regard, we strove to introduce early futility analysis. Further, we set moderately wide boundaries in these analyses (please see Statistical Analysis Publication (5)). We recognized that setting wider futility boundaries may result in a higher likelihood of discontinuing “effective” therapies, and therefore tried to balance this consideration against our goals of testing multiple interventions.
4-Month Intervention Periods and 2-Month Washout.
The study timelines allow us a maximum of 42 to 44 months of study participation in order to have a sufficient number of cross-over treatments per subject to meet our study objectives, we designed a 4-month treatment period and 2-month washout period for each intervention. Our rationale for choosing 4 months and 2 months is as follows: 1) Nearly all effective asthma treatments to date have shown the majority of their effect within 3–4 months; 2) While effects on yearly exacerbations cannot be assumed to be reflected accurately within 4 months, we are using the recently reported instrument known as the CompEx which, with 3 months of measurement, generally yields similar power to that obtained measuring annual asthma exacerbation rates over a year;(7) 3) As regards the washout, 2 months exceeds 5 times the known pharmacokinetic half-life of the drugs under consideration (except for clazakizumab, for which we introduced a 4-month washout). In regard to this latter point, we recognized that it is possible that the pharmacodynamic half-life of an intervention could exceed the 2-month washout. To account for this possibility, we are not assessing efficacy (at the end of each period) relative to baseline at the start of each period, but rather comparing interventions with placebo based on the outcomes measured at the end of each treatment period. To assess if our washouts are long enough, we will model baseline efficacy outcomes as a function of previous treatment to shed light on the presence of carry-over effects in the trial.
Placebo Considerations.
To assess whether an intervention is effective within its biomarker subgroup, interventions will be compared to a placebo administered during the double-blind placebo cross-over phase (Figure 1) and additional placebos administered during subsequent treatment assignments. The latter additional placebos have been inserted to preserve blinding in the subsequent periods, and to assess for period, seasonal, and secular temporal effects over this ~3.5-year study. As described in Section IV, after the initial double-blind 2-period cross-over period, subjects will be randomized to active intervention or matching placebo at an approximately 7 to 1 ratio, with the caveat that no subject can receive placebo in more than two periods during the trial. Throughout the trial subjects assigned to placebo will receive a single dummy appropriate to their assigned therapy for that period. We did not introduce simultaneous dummies for all the interventions (potentially up to 6 interventions and dummies at the same time) due to subject burden.
VI. Implementation Reality and Challenges of Developing a Platform Protocol
Implementation of our design has presented several challenges.
Drug Acquisition.
Since almost all our therapies are novel interventions in asthma, most of these drugs are not available for purchase over the counter. As a result, we needed to negotiate with each manufacturer to obtain drug. All the manufacturers were interested in participating but contract negotiations related to intellectual property and drug manufacturing timelines frequently caused delays. In some cases, production of placebo has also contributed to delays. At the time of this writing we have reached agreement with manufacturers of all 6 initial interventions but some of these negotiations have taken more than a year.
Regulatory Approval.
Almost all of our interventions are investigational new drugs (not currently on the market) or not approved for use in asthma and therefore require FDA approval for study in severe asthma. The FDA agreed that we could submit a single IND for the PrecISE master protocol that included all interventions available at study start, with IND amendments submitted as other interventions are added to the study. The FDA also gave us guidance that in order to approve interventions for adolescents the intervention would have to have minimal toxicity or, if there were potential for toxicity, we would need to show adequate evidence to suggest effectiveness in severe asthma. As a result, of the six interventions listed in Table II, it appears at this time that only two will be able to be used in adolescents.
Based on availability, we have initiated the trial with medium-chain triglycerides and clazakizumab. At the time of this writing, we have received FDA approval to add imatinib. Approval to add cavosonstat, BronchoVaxom, and itacitinib will hopefully follow in short order.
Overlapping Target Biomarker Groups and Prevalence of the Target Biomarker Group in the Population.
Some of the interventions chosen have overlapping biomarker-defined target subgroups as can be seen in Table II. We realized that if we attempted to enroll subjects in these interventions simultaneously accrual would be slow and thus a futility analysis would be delayed. As a result, if a subject is eligible for two or more interventions, the randomization algorithm favors the intervention closest to achieving the enrollment target for the 1st interim analysis (the futility interim analysis).
VII. Conclusion
The PrecISE trial represents an innovative approach to trial design in severe asthma. By bringing together experts in clinical trial design, adaptive trial design, and development of master protocols, we have developed a platform that we believe will allow us to rapidly evaluate novel interventions in severe asthma. The design also simultaneously allows us to define and refine potential predictive biomarkers and to explore biomarkers of response. The trial structure also permits subjects to receive multiple interventions with reduced placebo periods, making the approach more attractive to potential participants.
Declaration of Funding Sources
The PrecISE study is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, grants U24 HL138998, 1UG1HL139054, 1UG1HL139098, 1UG1HL139106, 1UG1HL139117, 1UG1HL139118, 1UG1HL139119, 1UG1HL139123, 1UG1HL139124, 1UG1HL139125, 1UG1HL139126. Support for site institutional infrastructure came from National Institute of Health Clinical & Translational Science Award grants UL1TR002451 (Harvard), UL1TR000427 (University of Wisconsin), UL1TR002366 (University of Kansas), UL1TR002389 (University of Chicago), UL1TR002489 (University of North Carolina), UL1TR001857 (University of Pittsburgh), UL1TR001442 (University of California, San Diego), and UL1TR001872 (University of California, San Francisco). The single IRB is supported by 5U24TR001608, the Duke/Vanderbilt Trial Innovation Center. The following endowed chair positions contributed additional support: Gloria M. and Anthony C. Simboli Distinguished Chair in Asthma Research (EI), William W. and Judith H. Busse Professor of Allergy & Asthma Research (LCD). The study also gratefully acknowledges receiving contributed product from Vitaeris, owned and operated by CSL group (clazakizumab), Vitaflo (MCT), Sun Pharma (imatinib), OM Pharma, a Vifor Pharma Group Company (OM-85, BronchoVaxom), Incyte (itacitinib), Laurel Venture (cavosonstat) and GlaxoSmithKline (Advair Diskus and Ventolin).
Disclosure of potential conflict of interest:
Dr. Israel reports grants from Gossamer Bio; grants and non-financial support from Circassia; grants and personal fees from Amgen, AstraZeneca, Avillion, Merck, Novartis, Sanofi Genzyme; grants, personal fees and non-financial support from Genentech, GlaxoSmithKline, and TEVA; personal fees from AB Science, Biometry, Equillium, 4D Pharma, Pneuma Respiratory, PPS Health, Regeneron, and Sienna Biopharmaceutical; and other considerations from Vorso Corp, outside the submitted work. Dr. Denlinger reports grants from NIH-NHLBI, during the conduct of the study; grants and personal fees from AstraZeneca, personal fees from Sanofi-Regeneron, outside the submitted work. Dr. Bacharier reports grants from NIH/NHLBI, during the conduct of the study; personal fees from GlaxoSmithKline, Genentech/Novartis, DBV Technologies, AstraZeneca, WebMD/Medscape, Sanofi/Regeneron, Vectura, and Circassia, and personal fees and non-financial support from Merck, Teva, Boehringer Ingelheim, outside the submitted work. Dr. LaVange reports grants from NHLBI, during the conduct of the study. Dr. Moore reports grants from NHLBI during the conduct of the study; grants and personal fees from Astrazeneca, GlaxoSmithKline, and Sanofi Regeneron, and grants from Gossamer Bio Inc, Cumberland Pharmaceuticals, and Genentech, outside the submitted work. Dr. Peters reports grants from Astrazeneca, Boehringer-Ingelheim, Genentech, GlaxoSmithKline, Sanofi-Genzyme-Regeneron, and Teva, personal fees from OrbiMed, outside the submitted work. Dr. Wright reports grants from National Institutes of Health, during the conduct of the study. Dr. Mauger reports grants from NHLBI, during the conduct of the study; grants from GSK, TEVA, AstraZeneca, Genentech, NHLBI, Boehringer-Ingelheim, and Sanofi-Genzyme-Regeneron, outside the submitted work. Dr. Akuthota reports grants from NIH, during the conduct of the study; personal fees from WebMD/Medscape, AHK, Prime CME, UpToDate, Rockpointe, Projects in Knowledge, MJH LifeSciences, and Vindico CME, and grants and personal fees from GlaxoSmithKline and AstraZeneca, outside the submitted work. Ms. Bach reports grants from NHLBI, during the conduct of the study. Dr. Bleecker reports personal fees and other considerations from AstraZeneca, MedImmune, Boehringer Ingelheim, Regeneron, and Sanofi Genzyme; other considerations from Genentech, Johnson and Johnson (Janssen), and Novartis; and personal fees from GlaxoSmithKline, outside the submitted work. Dr. Cardet reports personal fees from AstraZeneca, personal fees from Genentech, outside the submitted work. Dr. Carr reports grants from NIH/NHLBI, during the conduct of the study; grants and personal fees from AstraZeneca and Novartis, and personal fees from GSK, Regeneron, and UpToDate, outside the submitted work. Dr. Castro reports grants from NIH, ALA, PCORI, Pulmatrix, and Shionogi; grants and personal fees from AstraZeneca, Novartis, GlaxoSmithKline, and Sanofi-Aventis; and personal fees from Teva, Genentech, and Regeneron, outside the submitted work. Dr. Comhair reports grants NHLBI, during the conduct of the study, and a patent, “Capric Acid and Myristic Acid Compositions for Treatment Conditions (Publication number 20200230094),” pending. Dr. Covar reports grants from NHLBI, during the conduct of the study; grants from GSK, Sanofi Regeneron, Teva, and ALA ACRC, outside the submitted work. Dr. Erzurum reports grants from National Institutes of Health (NIH), during the conduct of the study. Dr. Fahy reports personal fees from Boehringer Ingelheim, Pieris, Arrowhead Pharmaceuticals, Gossamer, Ionis Pharmaceuticals, and Suzhou Connect Biopharmaceuticals, Ltd., outside the submitted work; in addition, Dr. Fahy has a patent WO2014153009A2, “Thiosaccharide mucolytic agents,” issued, and a patent WO2017197360, “CT Mucus Score - A new scoring system that quantifies airway mucus impaction using CT lung scans,” pending. Dr. Fajt reports personal fees from American Academy of Allergy, Asthma and Immunology, and grants from Breathe PA Organization, outside the submitted work. Dr. Gaston reports grants from NIH during the conduct of the study, and personal fees from Laurel and Respiratory Research, Inc., outside the submitted work. Dr. Hoffman is a founder and shareholder of VIDA Diagnostics, a company commercializing lung image analysis software developed, in part, at the University of Iowa. Dr. Jackson reports grants from NIH - ECHO/CREW, NHLBI, and NIAID, during the conduct of the study; personal fees from Sanofi-Regeneron, Pfizer, AstraZeneca, Novartis, and Vifor Pharma, and grants and personal fees from GlaxoSmithKline, outside the submitted work. Dr. Jain reports grants from NIH, during the conduct of the study. Dr. Jarjour reports grants from NIH-NHLBI during the conduct of the study; grants and personal fees from AstraZeneca, and personal fees from GSK and Boehringer Ingelheim for consultations outside the submitted work. Dr. Ji reports personal fees and other considerations from Bayesoft, other considerations from Laiya Consulting, and personal fees from Astella, outside the submitted work. Dr. Kraft reports grants from NIH, Sanofi, ALA, Chiesi, and Astra-Zeneca, and other considerations from Elsevier, Sanofi, and Astra-Zeneca, outside the submitted work. Dr. Krishnan reports grants from NIH-NHLBI during the conduct of the study, and grants from PCORI, Regeneron, and Sergey Brin Family Foundation outside the submitted work. Dr. Kumar reports grants from NHLBI, during the conduct of the study; and personal fees from Regeneron outside the submitted work. Dr. Andrew Liu reports grants from National Institutes of Health, during the conduct of the study; and other considerations from Propeller Health, outside the submitted work. Dr. Mark Liu reports grants from NIH, during the conduct of the study; personal fees from Astra Zeneca, grants and personal fees from Gossamer Bio, and grants from GlaxoSmithKline, Boehringer Ingelheim, Mereo BioPharma, and MedImmune, outside the submitted work. Dr. Ly reports grants from Vertex and Gilead, outside the submitted work. Ms. Marquis reports grants from NHLBI, during the conduct of the study. Dr. Martinez reports grants from NIH/NHLBI, NIH/NIEHS, NIH/NIAID, and NIIH/Office of Director, and personal fees from Copeval, outside the submitted work. Dr. O’Neal reports grants from NHLBI, during the conduct of the study. Dr. Ortega reports personal fees from Regeneron and Sanofi, outside the submitted work. Dr. Phipatanakul reports grants from NIH, and other considerations from CSL Behring, Vitaflo, Astra Zeneca, Merck, GSK, Sun Pharma, Vifor, Circassia, during the conduct of the study; grants, personal fees and other considerations from Genentech/Novartis and Regeneron/Sanofi, and other considerations from Thermo Fisher, Lincoln Diagnostics, and Kaleo, outside the submitted work. Dr. Ross reports grants from NIH, during the conduct of the study; grants and non-financial support from TEVA, grants from Astra Zeneca, Boehringer Ingelheim, and Novartis, and non-financial support from GlaxoSmithKline and Merck, outside the submitted work. Dr. Smith reports grants from NHLBI, during the conduct of the study; and personal fees from GSK, outside the submitted work. Dr. Szefler reports grants from Propeller Health, and other considerations from Boehringer-Ingelheim, GlaxoSmithKline, Astra Zeneca, Sanofi, and Regeneron, outside the submitted work. Dr. Teague reports grants from NIH/NHLBI, during the conduct of the study; grants from NIH/NHLBI R21, outside the submitted work. Dr. Wechsler reports grants, personal fees and non-financial support from Teva; grants and personal fees from Novartis, Sanofi, and GlaxoSmithKline; personal fees from Regeneron, Mylan, Genentech, Sentien, Restorbio, Equillium, GALA Therapeutics, Pulmatrix, and Cohero Health; personal fees and non-financial support from Boehringer Ingelheim and AstraZeneca, and non-financial support from Merck, outside the submitted work. Dr. Wenzel reports grants from Boehringer-Ingelheim and TEVA; and grants and personal fees from AstraZeneca, GSK, Sanofi Genzyme, and Novartis, outside the submitted work. Dr. White reports grants from NHLBI, during the conduct of the study; personal fees from Regeneron, AstraZeneca, CHEST Foundation, and Sanofi, outside the submitted work. Dr. Zeki reports other considerations from InStatin, Inc., outside the submitted work. In addition, Dr. Zeki has a patent PCT/US2020/025543 pending (no sponsor). Dr. Ivanova reports other considerations from CSL Behring, outside the submitted work. All other authors have nothing to disclose.
Abbreviations
- ACQ-6
Asthma Control Questionnaire – 6 question version
- DMCC
Data, Modeling, and Coordination Center
- FDA
Food and Drug Administration
- ICS/LABA
Inhaled corticosteroid and long-acting beta-agonist
- IND
Investigational New Drug Application
- NHLBI
National Heart, Lung, & Blood Institute
- NIH
National Institutes of Health
- PrecISE
Precision Medicine in Severe and/or Exacerbation Prone Asthma
- SC
Steering Committee
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Israel E, Reddel HK. Severe and Difficult-to-Treat Asthma in Adults. N Engl J Med. 2017;377(10):965–76. [DOI] [PubMed] [Google Scholar]
- 2.US Food and Drug Administration (FDA). Adaptive Designs of Clinical Trials of Drugs and Biologics: Guidance for Industry. Docket Number FDA-2018-D-3124. Silver Spring, MD: U.S. Food and Drug Administration; 2019. Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/adaptive-design-clinical-trials-drugs-and-biologics-guidance-industry. Accessed Jan 14, 2021. [Google Scholar]
- 3.Park JW, Liu MC, Yee D, Yau C, van ‘t Veer LJ, Symmans WF, et al. Adaptive Randomization of Neratinib in Early Breast Cancer. N Engl J Med. 2016;375(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rugo HS, Olopade OI, DeMichele A, Yau C, van ‘t Veer LJ, Buxton MB, et al. Adaptive Randomization of Veliparib-Carboplatin Treatment in Breast Cancer. N Engl J Med. 2016;375(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivanova A, Israel E, LaVange LM, Peters MC, Denlinger LC, Moore WC, et al. The precision interventions for severe and/or exacerbation-prone asthma (PrecISE) adaptive platform trial: statistical considerations. J Biopharm Stat. 2020:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J. 2014;43(2):343–73. [DOI] [PubMed] [Google Scholar]
- 7.Fuhlbrigge AL, Bengtsson T, Peterson S, Jauhiainen A, Eriksson G, Da Silva CA, et al. A novel endpoint for exacerbations in asthma to accelerate clinical development: a post-hoc analysis of randomised controlled trials. The Lancet Respiratory medicine. 2017;5(7):577–90. [DOI] [PubMed] [Google Scholar]