

Abstract
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are fatal neurodegenerative disorders that are thought to exist on a clinical and pathological spectrum. FTD and ALS are linked by shared genetic causes (i.e. C9orf72 hexanucleotide repeat expansions) and neuropathology, such as inclusions of ubiquitinated, misfolded proteins (i.e. TAR DNA-binding protein 43; TDP-43) in the CNS. Furthermore, some genes that cause FTD or ALS when mutated encode proteins that localize to the lysosome or modulate endosome-lysosome function, including lysosomal fusion, cargo trafficking, lysosomal acidification, autophagy, or TFEB activity. In this review, we summarize evidence that lysosomal dysfunction, caused by genetic mutations (i.e. C9orf72, GRN, MAPT, TMEM106B) or toxic-gain of function (i.e. aggregation of TDP-43 or tau), is an important pathogenic disease mechanism in FTD and ALS. Further studies into the normal function of many of these proteins are required and will help uncover the mechanisms that cause lysosomal dysfunction in FTD and ALS. Mutations or polymorphisms in genes that encode proteins important for endosome-lysosome function also occur in other age-dependent neurodegenerative diseases, including Alzheimer’s (i.e. APOE, PSEN1, APP) and Parkinson’s (i.e. GBA, LRRK2, ATP13A2) disease. A more complete understanding of the common and unique features of lysosome dysfunction across the spectrum of neurodegeneration will help guide the development of therapies for these devastating diseases.
Keywords: Amyotrophic Lateral sclerosis (ALS), Frontotemporal dementia (FTD), Frontotemporal lobar degeneration (FTLD), Alzheimer’s disease and related dementias (ADRD), lysosome dysfunction, autophagy, neurodegeneration, ubiquitin, C9orf72, transactive response DNA binding protein 43 kDa (TDP-43), progranulin (PGRN), granulins (GRNs), Transmembrane protein 106B (TMEM106B), Microtubule-associated protein tau (MAPT), Transcription factor EB (TFEB)
1. Introduction
1.1. Amyotrophic lateral sclerosis (ALS) and Frontotemporal dementia (FTD)
ALS is a neurodegenerative disorder characterized by degeneration of the upper and lower motor neurons in the brain and spinal cord that causes respiratory paralysis and death (Brown and Al-Chalabi, 2017). With a typical age of onset between 51 and 66 years of age, patients experience worsening paralysis in voluntary muscles leading to death around 2 to 5 years after disease onset (Al-Chalabi et al., 2016; Longinetti and Fang, 2019). The incidence of ALS is estimated to be between 0.6 and 3.8 per 100,000 persons per year (Longinetti and Fang, 2019). However, symptom presentation is highly variable between ALS patients depending on which populations of neurons survive, leading to difficulty in diagnosing and treating the disorder (Al-Chalabi et al., 2016). Approximately 10% of patients have a family history of ALS and are labeled familial ALS (fALS) (Mejzini et al., 2019). The remaining ~90% of ALS cases occur without a family history and are considered idiopathic or sporadic ALS (sALS) (Chen et al., 2013). sALS and fALS have similar clinical presentations, and mutations in genes that cause fALS have also been discovered in sALS cases (Chen et al., 2013). Therefore, understanding the mechanisms of toxicity associated with fALS may uncover new disease treatments for all ALS patients.
Frontotemporal dementia (FTD) is the most common cause of dementia in people under the age of 60 and encompasses a heterogeneous group of disorders that affect the frontal and temporal regions of the brain (Bang et al., 2015). FTD is the clinical manifestation of frontotemporal lobar degeneration (FTLD) neuropathology. The broad clinical phenotype of FTD is divided into language or progressive deficits in behavior and executive function (Deleon and Miller, 2018). FTD has an estimated prevalence of ~0.02 to 0.22 cases per 1,000 (Bang et al., 2015; Hogan et al., 2016). Approximately 40% of individuals diagnosed with FTD have a family history of a neurodegenerative disease and are considered familial FTD (fFTD) (Deleon and Miller, 2018). The remaining cases are considered sporadic FTD (sFTD), although they may still harbor a mutation in a known gene that can cause FTD.
FTD has two distinct clinical syndromes: behavioral-variant FTD (bvFTD) and primary progressive aphasia (PPA) (Ljubenkov and Miller, 2016). Behavioral variant FTD is the most common (~60–80%) and is characterized by behavioral disinhibition, apathy, and socially inappropriate behavior. FTD with PPA is divided into non-fluent/agrammatic-variant primary progressive aphasia (nfvPPA) and semantic-variant primary progressive aphasia (svPPA) (Bang et al., 2015; Erkkinen et al., 2018; Hogan et al., 2016; Johnson et al., 2005). Interestingly, as the disorder progresses, symptoms from the three subtypes can converge, and sometimes patients develop global cognitive impairments and motor deficits (Kertesz et al., 2005). Although 40% of cases report a family history of dementia, only ~15% show clear autosomal dominant patterns (Rohrer et al., 2009). Because there are no disease-modifying therapies target yet, all current FTD treatments only target symptoms (Erkkinen et al., 2018).
FTD and ALS overlap at the neuropathological and genetic level (Abramzon et al., 2020). In 2006, the connection between FTD and ALS was solidified when it was discovered that TAR DNA-binding protein 43 kDa (TDP-43), an RNA/DNA binding protein encoded by the TARDBP gene, was a major component of the insoluble aggregates (inclusions) in ~97% of ALS and ~50% of FTD post-mortem brains (Arai et al., 2006; Neumann et al., 2006). Moreover, mutations in TARDBP cause ALS (Zou et al., 2017) as well as FTD, although this is less common (Caroppo et al., 2016). Subsequently, mutations in the same genes (i.e. C9orf72, OPTN, VCP) were discovered in patients with FTD, ALS, or a presentation of both FTD and ALS symptoms. It is estimated that around 15% of FTD patients will develop ALS symptoms and up to 48% of ALS patients show some degree of cognitive impairment consistent with FTD (Lomen-Hoerth et al., 2002; Portet et al., 2001; Ringholz et al., 2005).
In the last few years, new evidence has associated lysosomal dysfunction with the onset and development of FTD and ALS (Table 1). At a genetic level, mutations in multiple genes related to lysosome and autophagy function have been linked to both FTD and ALS, including C9orf72, SQSTM1/p62, UBQLN2, DCTN1, TBK1, OPTN, and VCP (Casterton et al., 2020). Mutations in other genes seem to exclusively cause FTD (i.e. MAPT and GRN) or ALS (i.e. SOD1) (Ling et al., 2013; Mackenzie and Neumann, 2016). Nevertheless, impaired lysosomes, autophagy, and vesicle trafficking occurs in both FTD and ALS post-mortem tissue (Farg et al., 2014; Lie and Nixon, 2019). Additionally, C9orf72 mutations that can cause FTD or ALS affect Rab protein activity, induce dysfunction of the endo-lysosomal pathway, and lead to the accumulation of protein aggregates (Farg et al., 2014). Moreover, mutations in genes encoding lysosomal proteins such as GRN, TMEM106B, and CHMP2 affect lysosome acidification and reduce lysosomal enzymatic activity (Brady et al., 2013; Tanaka et al., 2017). Thus, defects in lysosome function and associated pathways such as autophagy, which relies on proper lysosome function, may broadly contribute to FTD and ALS pathogenesis. In this review, we will focus on five important proteins (progranulin, C9orf72, TDP-43, TMEM106B, and tau), which are intimately involved in pathogenesis across the FTD and ALS disease spectrum, and summarize how they are implicated in lysosome function and dysfunction.
Table 1 -.
Summary of genes associated with FTD and ALS and their role in lysosome function
| Gene | Protein | Genome location | Role(s) in lysosome function | Type of mutation | Clinical Pheno-types(s) | Mutation Frequency | Effect of genetic mutation/variant | References |
|---|---|---|---|---|---|---|---|---|
| C9orf72 | C9orf72 | 9p21.2 | GTPase-activating protein (GAP) complex with SMCR9-WD41; lysosome trafficking; mTORC1 signaling | G4C2-repeat expansion | FTD, ALS | Common; Varies by population; ~42.1%* | Loss of C9orf72 function; gain of DPR and RNA toxicity | (DeJesus-Hernandez et al., 2011; Le Ber et al., 2009; Shao et al., 2020; Su et al., 2020; Tang et al., 2020) |
| GRN | Progranulin | 17q21.31 | Lysosome homeostasis; Hydrolase trafficking and function | Haplo-insufficiency; nonsense | FTD, NCL (CLN11) | Common; Varies by population; ~34.6%* | Loss of function; Lysosome impairment | (Arai et al., 2006; Baker et al., 2006; Holler et al., 2017; Neumann et al., 2006) |
| MAPT | Tau | 17 | Trafficking of autophagic vesicles and autolysosome fusion | Missense/deletion Intronic | FTD | Common; Varies by population; ~23.2%* | Tau hyperphosphorylation and protein aggregation | (Hutton et al., 1998; Lim et al., 2001; Pacheco et al., 2009) |
| TARDBP | TDP-43 | 1p36 | Lysosomal fusion and regulation of lysosomal protein expression | Mainly missense Truncating | FTD, ALS | ~3% | TDP-43 hyperubiquitination and aggregate formation | (Arai et al., 2006; Gao et al., 2018; Neumann et al., 2006)[14, 15, 216] |
| TMEM 106B | Transmembrane protein 106B | 7p21.3 | Lysosomal membrane protein, lysosome trafficking | Polymorphism; Missense (HLD) | FTD, HLD | Risk factor; Rare (HLD) | Increased TMEM106B levels?; impaired Lysosome: Loss of function (HLD) | (Brady et al., 2013; Nicholson et al., 2013; Simons et al., 2017; Van Deerlin et al., 2010) |
| CHMP2B | Charged multivesicular body protein 2B | 3p11.2 | Endocytic multivesicular body formation | C-terminal truncation | FTD | Rare | Loss of function | (Lindquist et al., 2008; Skibinski et al., 2005) |
| TBK1 | TANK Binding Kinase 1 | 12q14.2 | autophagy; lysophagy; phosphorylates OPTN | Missense; truncation | FTD, ALS | ~1.0% ALS; ~1.8% FTD | Loss of function | (Abramzon et al., 2020; Cirulli et al., 2015; Freischmidt et al., 2015) |
| OPTN | Optineurin | 10p13 | Autophagy receptor; Clearance protein aggregates; damaged organelles | Missense | FTD, ALS | Rare | Loss of function | (Bussi et al., 2018; Maruyama et al., 2010; Moore and Holzbaur, 2016; Pottier et al., 2015) |
| VCP | Valosin containing protein | 9p13.3 | Lysophagy; Sorting of proteins/aggregates to lysosomes | Missense | FTD, ALS MSP | Rare | Loss of function | (Johnson et al., 2010) |
| MFSD8 | Major facilitator superfamily domain containing 8 | 4q28.2 | Likely transports solutes across lysosome membrane; substrate unknown | Missense | FTD, NCL (CLN7) | Rare | Loss of function | (Geier et al., 2019) |
| CTSF | Cathepsin F | 11q13 | Lysosomal cysteine protease; multiple substrates | Missense | FTD, NCL (CLN13) | Rare | Loss of function | (van der Zee et al., 2016) |
| SQSTM1/p62 | Sequestosome-1/p62 | 5q35.3 | Facilitating protein aggregate degradation in lysosome; Autophagosome; mTORC1 regulation | Missense; nonsense | FTD, ALS | ~3% | Loss of function | (Hardy and Rogaeva, 2014; Le Ber et al., 2013) |
Population described in (Moore et al., 2020)
1.2. Lysosomal Biology
Lysosomes are membrane enclosed organelles that degrade a variety of macromolecules and were first described by Christian de Duve in 1955 (de Duve, 2005). Lysosomes have a single lipid bilayer, which contains over 100 membrane proteins, enclosing an acidic lumen that hosts 50 or more lysosomal hydrolases (Braulke and Bonifacino, 2009). Lysosomes are critically involved in the degradation and recycling of intracellular and extracellular material including lipids, proteins, nucleic acids, and carbohydrates. Historically, lysosomes were viewed as static organelles that represented the terminal endpoint of degradation following endocytosis or phagocytosis. In fact, lysosomes are dynamic, highly regulated, and can vary in their pH (Johnson et al., 2016), sub-cellular location (Li et al., 2016), and morphology (Saric et al., 2016). Moreover, lysosomes do more than simply degrade molecules and have other critical roles in cellular signaling, metabolism, membrane repair, homeostasis, and the immune response (Settembre et al., 2013; Xu and Ren, 2015).
The biogenesis of lysosomes is a complex, regulated process, and our understanding of the pathway is still evolving. Briefly, newly synthesized proteins can be delivered directly to a lysosome from the trans-Golgi network (TGN) or through early endosomes (EE), recycling endosomes (RE), late endosomes (LE), or multivesicular bodies (MVBs) (Luzio et al., 2014). Lysosomes also undergo frequent fusion-fission cycles, which serve to maintain steady-state levels of lysosome number, size, and function (Saffi and Botelho, 2019). The classical pathway of sorting proteins to the lysosome is based on the recognition of mannose-6 phosphate residues on oligosaccharide chains of hydrolases, which are trafficked by mannose-6 phosphate receptors (M6PRs) to the lysosome (Braulke and Bonifacino, 2009; Ghosh et al., 2003). Several M6PR-independent mechanisms play important roles in delivering cargo to the lysosome. For instance, the lysosomal integral membrane protein 2 (LIMP2) traffics β-glucocerebrosidase to the lysosome through binding cation-independent M6PR to form a heterotrimeric complex (Reczek et al., 2007; Zhao et al., 2014). Furthermore, Sortillin-1 (SORT1) traffics several proteins to the lysosome, including cathepsins D and H (CTSD and CTSH) as well as progranulin (Canuel et al., 2008; Hu et al., 2010). The LDL receptor related protein (LRP1) also contributes to the routing of CTSD and other proteins to the endo-lysosomal pathway (Derocq et al., 2012; Markmann et al., 2015).
Lysosomes are also critical for autophagy, another important degradation pathway in cells. Autophagy is a cellular process through which long-lived proteins, dysfunctional organelles, or invading pathogens are sequestered and degraded (Dikic and Elazar, 2018). In the autophagic pathway, cargo is enclosed in an autophagosome, which later merges with a late endosome to form an amphisome, or with a mature lysosome to form an autolysosome, which ultimately degrades the enclosed cargo (Berg et al., 1998; Gordon and Seglen, 1988). Autophagosome formation is initiated by unc-like kinase 51 (ULK1), and the elongation and closure of the vesicle is mediated by two autophagy related gene (ATG) pathways ATG5-ATG12 and ATG8, which is also known as microtubule-associated proteins 1A/1B light chain 3B (MAP1LC3 or LC3) (Mizushima et al., 2011; Reggiori and Ungermann, 2017). The cytoplasmic form of LC3 (termed LC3-I) is conjugated to phosphatidylethanolamine (PE) to form LC3-II, which is associated with the membrane surface of autophagosomes. The lipidation of LC3 changes its migration pattern in SDS/PAGE, enabling measurement of the conversion of LC3-I (~19 kDa) to LC3-II (~17 kDa) (Kabeya et al., 2000; Tanida et al., 2005). Measurement of the abundance and ratio of LC3-I to LC3-II is frequently used to monitor autophagy in cells and tissue, although complementary approaches are needed to fully understand how flux through the autophagy-lysosome pathway is altered in different contexts (for summary see (Bonam et al., 2020; Klionsky et al., 2016).
Many lysosomal functions rely on the acidification of the lysosome lumen, especially the activity of the hydrolytic enzymes present in the lysosome, which are optimally active at an acidic pH (pH 4.5–5.0) (Perera and Zoncu, 2016). The acidic pH is generated and maintained in the lysosome by the vacuolar ATPase (V-ATPase), a membrane transporter that uses ATP hydrolysis to pump protons against their electrochemical gradient into the lysosome, with counter ion contributions from the chloride channel CLC-7/Ostm1 (Mindell, 2012; Ohkuma et al., 1982). Vesicular acidification regulates the dissociation of cargoes from the mannose 6-phosphate receptors delivered to LEs and the maturation of immature hydrolases (Richo and Conner, 1994). The acidic environment is also critical for the modification of cargoes delivered to the lysosome, including lipids, ions, and proteins (Asano et al., 2011; Singh et al., 2009; Yambire et al., 2019). Furthermore, alkalinization of the lysosomal lumen through chemicals that inhibit the V-ATPase (i.e. bafilomycin A1 (Bowman et al., 1988)), or lysosomotropic agents, (i.e. chloroquine (CQ)), impair lysosome function, mTORC1 signaling, and autophagy, mimicking many features of lysosomal storage disorders (LSDs). Long-term treatment with chloroquine or bafilomycin A1 leads to toxicity and death of primary neurons, neural precursor cells, and multiple immortalized neural cell lines. (Fedele and Proud, 2020; Pasquier, 2016). Additionally, the lysosome also helps communicate the overall metabolic state of the cell through its interaction with the mechanistic target of rapamycin complex 1 (mTORC1), which localizes to Rab7 and LAMP2 positive vesicles (Harms et al., 1981; Sancak et al., 2010; Sancak et al., 2008; Settembre et al., 2013). MTORC1 is recruited to the lysosomal membrane under conditions of amino acid depletion by the coordination of Rag GTPases (Kim et al., 2008). In addition to catabolic regulation, the lysosome also participates in anabolic processing through its interaction with AMP-activated protein kinase (AMPK) (Zhang et al., 2014a). Lastly, the lysosome participates in signaling via the degradation of EGFR signals (Tomas et al., 2014) and processing of toll-like receptors involved in the innate immune response (Lee and Barton, 2014). These, and other, findings have expanded the view of the lysosome from a simple organelle designed for macromolecule degradation to a critical hub that integrates cellular signals (Ballabio and Bonifacino, 2019). Thus, the perturbation of lysosomal function at multiple levels from trafficking, acidification, cargo recruitment, cargo degradation, or signaling, could contribute to FTD and ALS, which we will discuss next.
1.3. Lysosome and Neurodegeneration.
Under physiological conditions, the lysosome participates in the degradation and recycling of numerous macromolecules and organelles throughout the cell (Martinez-Vicente et al., 2005). However, dysfunction of components of the lysosomal system is deleterious and causes a variety of fatal diseases. Lysosomal storage disorders (LSDs) are a group of more than 50 inherited diseases with genetic defects in various components of the lysosomal system, including membrane proteins, transporters, lysosomal hydrolases, and receptors, which cause the accumulation of specific substrates (Menzies et al., 2015; Wilcox, 2004). Interestingly, many LSDs have substantial central nervous system (CNS) manifestations (Prada and Grabowski, 2013). The CNS is particularly vulnerable to lysosome dysfunction, causing impairment of neuronal and glial function, which ultimately leads to neurodegeneration. For example, autophagosomes and lysosomes accumulate in Lewy Bodies, the intraneuronal inclusions that are characteristic of PD neuropathology (Mahul-Mellier et al., 2020; Shahmoradian et al., 2019). Additionally, damaging variants in multiple genes that cause LSDs are enriched in PD cases (Hopfner et al., 2020; Robak et al., 2017).
Lysosome dysfunction has been observed in the tissue of FTD and ALS patients. Neurons and microglia in the frontal cortex brain tissue of FTD patients have increased lipofuscin deposits, a diagnostic feature of many LSDs (Clayton et al., 2015; Ward et al., 2017). Lipofuscin is an auto-fluorescent aggregate composed of undegraded lipids, proteins, and metals, which is thought to develop from defective lysosome function over time (Moreno-Garcia et al., 2018). These protein deposits co-localize with LAMP1 and LAMP2 (Clayton et al., 2015), two well-characterized lysosome markers, suggesting that lipofuscin deposits may be derived from lysosomal compartments. Additionally, enlarged endosomes occur in primary fibroblasts and frontal cortical neurons of FTD (Urwin et al., 2010). Further, large autophagic vesicles containing CTSB activity accumulate in myoblasts from FTD patients (Tresse et al., 2010). A lipidomic analysis also found evidence that lysosomal degradation of triacylglycerides (TAGs) was impaired in human FTD brain tissue and a mouse model of FTD (Evers et al., 2017).
In ALS, motor neurons in the spinal cord have increased levels of glycosphingolipids (GSL), a characteristic feature of Gaucher disease (GD) (Dodge et al., 2015), and inclusion bodies composed of autophagosomes and autolysosomes (Sasaki, 2011). Furthermore, LAMP1 positive vesicles are decreased in induced pluripotent stem cell (iPSC)-derived motor neurons from ALS patients that carry the C9orf72 mutation, suggesting a reduced number of functional lysosomes (Shi et al., 2018). Finally, post-mortem ALS brain tissue has a ~62% reduction in the nuclear localization of transcription factor EB (TFEB), a master transcriptional regulator of lysosomal genes, suggesting that the regulation of the lysosome-autophagy pathway is impaired (Wang et al., 2016). Together, these data strongly support the idea that lysosomal dysfunction is a critical pathogenic cause of FTD and ALS. In this review, we will explore the importance of proper lysosome function to prevent the development of FTD and ALS, with a focus on the roles of progranulin, C9orf72, TDP-43, TMEM106B, and tau.
2. Evidence of lysosome dysfunction in FTD and ALS.
2.1. C9orf72
For years, multiple groups reported a strong genetic association between a locus on chromosome 9 and development of FTD or ALS, but the causative gene was unclear (Le Ber et al., 2009; Morita et al., 2006; Valdmanis et al., 2007; Vance et al., 2006). In 2011, two laboratories discovered that a hexanucleotide (GGGGCC; G4C2) repeat expansion (HRE) in the non-coding region of the chromosome 9 open reading frame 72 gene (C9orf72) was the underlying cause of disease (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The C9orf72 gene of most healthy individuals contains between two and twenty G4C2 repeats. In FTD or ALS patients with a C9orf72 HRE, an expansion of 100 or more G4C2 repeats is considered pathogenic (Rademakers et al., 2012). Intermediate expansions in C9orf72 between 24 and 30 repeats are also associated with an increased risk for ALS (Iacoangeli et al., 2019). Following the initial discovery in 2011, many labs rapidly replicated the identification of the C9orf72 mutant G4C2 repeat expansions in independent FTD and ALS patient populations throughout the world (Chio et al., 2012; Gijselinck et al., 2012; Ishiura et al., 2012). The G4C2 repeat expansion in C9orf72 is now widely recognized as the most common inherited genetic cause of both FTD and ALS (Rademakers et al., 2012).
When C9orf72 G4C2 expansion mutations were first discovered, the biological function of the C9orf72 protein was unknown. Moreover, it was unclear how the G4C2 repeat ultimately caused neurodegeneration. Researchers proposed three, non-mutually exclusive, pathogenic mechanisms caused by the C9orf72 G4C2 repeat. First, the production of mRNA containing large G4C2 repeats may be toxic by sequestering RNA-binding proteins and impairing their normal function. Second, dipeptide repeat proteins (DPRs) generated from non-conventional repeat associated non-ATG (RAN) translation may be toxic. Many papers have been published supporting the idea that RNA and DPR toxicity is a major source of pathology in various C9orf72 models (Balendra and Isaacs, 2018; Deng et al., 2013; Haeusler et al., 2016; Swinnen et al., 2020; Taylor et al., 2016). In this review, we will focus on the third mechanism: the possibility that the G4C2 repeat expansion leads to decreased transcription and levels of the C9orf72 protein, thus leading to partial loss-of-function.
We focus on the possibility of C9orf72 loss-of-function because a) C9orf72 protein levels are decreased in G4C2 repeat expansion carriers and b) recent data that the C9orf72 protein plays a critical role in lysosome function and autophagy, both of which are broadly implicated in FTD and ALS pathogenesis. First, C9orf72 mRNA and protein levels are consistently decreased in multiple brain regions, myeloid cells, and iPSC-derived neurons from G4C2 repeat expansion positive patients (DeJesus-Hernandez et al., 2011; Donnelly et al., 2013; Frick et al., 2018; van Blitterswijk et al., 2015; Viode et al., 2018). Importantly, insufficient levels of the C9orf72 protein can cause neurodegeneration in human iPSC-motor neurons, zebrafish, and C. elegans (Ciura et al., 2013; Shi et al., 2018; Therrien et al., 2013). Interestingly, knockout mice lacking the murine C9orf72 gene orthologue, 3110043O21Rik, do not have overt neurodegeneration, but instead have inflammation and a dysregulated immune system (Atanasio et al., 2016; Burberry et al., 2016; Burberry et al., 2020; McCauley et al., 2020; O’Rourke et al., 2016). However, decreased levels of C9orf72 protein can synergize with DPR toxicity and the combination may be necessary to induce neurodegeneration in C9orf72 G4C2 repeat in vitro cell or mouse models (Boivin et al., 2020; Shao et al., 2019). Furthermore, axonal trafficking was found to be disrupted in motor neurons with a C9orf72 HRE, and this phenotype was exacerbated by deletion of C9ORF72 (Abo-Rady et al., 2020). Additionally, extensive work in multiple mouse models found that reduction of C9ORF72 function increases toxicity of the C9orf72 HRE (Zhu et al., 2020).
The precise function(s) of the C9orf72 protein has been a mystery since the G4C2 repeat expansions in C9orf72 were found to cause FTD and ALS. Now, it is clear C9orf72 plays an important role in the function and homeostasis of the lysosome (Amick and Ferguson, 2017). For example, deletion of the C. elegans C9orf72 orthologue disrupts endo-lysosomal degradation and lysosome function (Corrionero and Horvitz, 2018). Further, human and mouse cell models lacking C9orf72 have swollen lysosomes, impaired mTORC1 activation, and defects in autophagy, lysosomal degradation, and exocytosis (Amick et al., 2016; O’Rourke et al., 2016; Shao et al., 2019; Shao et al., 2020; Shi et al., 2018). Additionally, C9orf72 knockout mice have defective autophagy and lysosome function throughout the body and particularly in brain-resident microglia and macrophages (Ho et al., 2019; O’Rourke et al., 2016; Shao et al., 2020). Because the C9orf72 HRE can cause disease through gain and loss of function, there is concern that it may be difficult to develop effective therapies targeting the expansion. Excitingly, apilimod, a small molecule inhibitor of PIKfyve kinase, has been shown recently to rescue both loss- and gain-of-function C9ORF72 disease mechanisms in vivo (Staats et al., 2019). Further studies are warranted to determine if apilimod, related small molecule kinase inhibitors, or other therapies like antisense oligonucleotide (ASO) can effectively treat C9orf72-mediated neurodegenerative diseases (Donnelly et al., 2013; Guo et al., 2020; Jiang et al., 2016; McCampbell et al., 2018).
A key question remains. How does loss of C9orf72 cause impaired lysosomal function and autophagy (Fig. 1)? One possibility is that C9orf72 directly binds to lysosomes and modulates their activity. Early studies examining the sub-cellular localization of C9orf72 were inconclusive due to low abundance of C9orf72 and non-specific antibodies. This issue was first solved by using a CRISPR-Cas9 approach to knock-in a HA epitope tag into the endogenous locus. Immuno-staining revealed that C9orf72 localizes to the lysosome, especially following amino-acid depletion (Amick et al., 2016). Further studies in human iPSC-derived neurons and U2OS cells with novel monoclonal antibodies confirmed that C9orf72-associated with lysosomes as well as undefined vesicles in human iPSC-derived neurons (Frick et al., 2018; Laflamme et al., 2019). This localization was present regardless of whether cells were amino acid deprived (Laflamme et al., 2019). C9orf72 also co-localizes to lysosomes and phagolysosomes in human macrophages, which express high levels of C9orf72 (Laflamme et al., 2019). Additionally, C9orf72 has also been reported to co-localize with the early endosomal proteins RAB5 and EEA1 (Shi et al., 2018). Together these studies reveal that C9orf72 is enriched within the endosome-lysosome network and likely plays an important role modulating lysosome function and related pathways.
Figure 1. Overview of the function of C9orf72 in the lysosome-autophagy pathway.
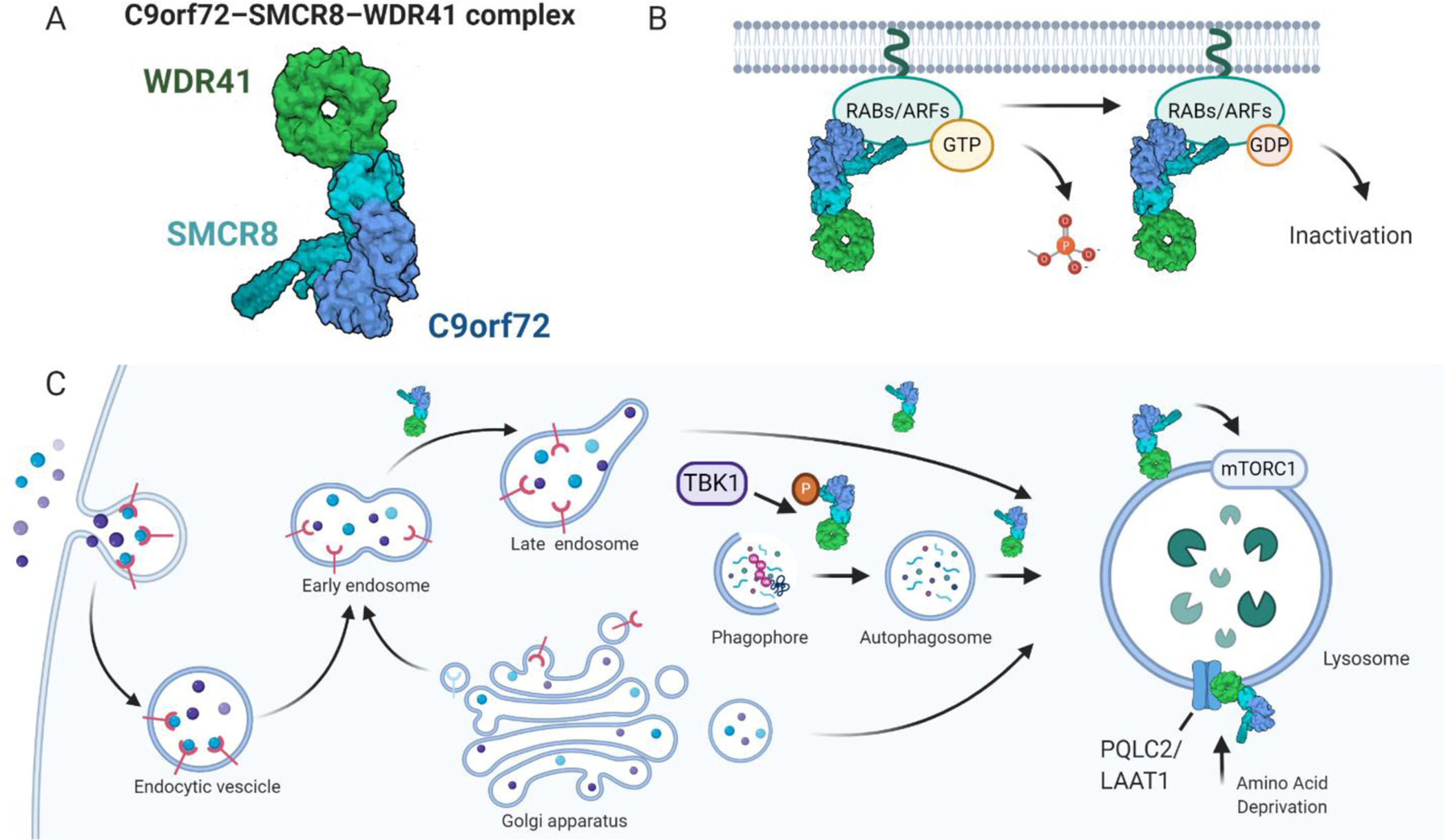
(A) C9orf72 forms a stable complex with SMCR8 and WDR41, shown here as a surface representation of a cryo-EM structure (PDB 6V4U), colored by chain (WDR41-green, SMCR8-cyan, C9orf72-blue). (B) The C9orf72-SMCR8-WDR41 complex functions as a GTPase activating protein (GAP) for RABs, ARFs, and potentially other GTPases, leading to conversion of GTP to GDP, and GTPase inactivation. (C) C9orf72 alone, or in complex with SMCR8-WDR41, has been functionally implicated in multiple parts of the lysosome-autophagy pathway, including vesicle fusion, phagophore formation, autophagosome maturation, and lysosome function, possibly by regulating mTORC1 activity. Finally, the C9orf72-SMCR8-WDR41 complex can be recruited to the surface of lysosomes by PQLC2/LAAT1, a lysosomal cationic amino acid transporter, during amino acid deprivation. Haploinsufficiency of C9orf72 induced by hexanucleotide repeat expansion (HRE) mutations in C9orf72 may impair multiple aspects of lysosome autophagy function, thus contributing to FTD and ALS pathogenesis. Created with BioRender.com.
The molecular function of C9orf72 at the lysosome is becoming clearer. An initial clue was the realization that C9orf72 has structural homology to the differentially expressed in normal and neoplasia (DENN) protein family (Levine et al., 2013; Zhang et al., 2012). Proteins with DENN domains typically function as Rab guanine nucleotide exchange factors (GEFs), which activate Rab-GTPases by facilitating the exchange of GDP for GTP, and serve to regulate membrane trafficking in cells (Marat et al., 2011). Indeed, one report found that C9orf72 could act on its own as a GEF for Rab5A, Rab7A, and Rab11A using in vitro assays (Iyer et al., 2018).
Recent work from multiple labs reveals a more complex picture of C9orf72 function in cells. Specifically, C9orf72 does not appear to act alone, but is part of a larger protein complex (Fig. 1) composed of Smith-Magenis syndrome chromosome region, candidate 8 (SMCR8) and WD repeat-containing protein 41 (WDR41) (Amick et al., 2016; Amick et al., 2018; Jung et al., 2017; Sellier et al., 2016; Sullivan et al., 2016; Xiao et al., 2016; Yang et al., 2016). The C9orf72-SMCR8-WDR41 complex binds tightly to one another and may work together as a GEF for RAB8A and RAB39B (Yang et al., 2016). However, a recent cryogenic electron microscopy (cryo-EM) structure of C9orf72-SMCR8-WDR41 suggests the complex functions as a GTPase activating protein (GAP) for Rab8a and Rab11a and not a GEF (Tang et al., 2020). Similarly, a different cryo-EM structure of C9orf72-SMCR8-WDR41 was unable to detect GEF activity and instead found GAP activity for the Arf family small GTPases (Su et al., 2020). It is unclear why there is a discrepancy between the initial report that C9orf72 is a GEF. Additional studies are needed to uncover the functions and molecular targets of the C9orf72-SMCR8-WDR41 complex.
Rabs and other binding partners, like ATG1/ULK1 kinase, may be important for mediating the diverse effects the C9orf72-SMCR8-WDR41 complex has on autophagy in experimental systems (Jung and Behrends, 2020). Moreover, all members of the C9orf72-SMCR8-WDR41 complex localize to the lysosome membrane and can influence mTORC1 signaling, which is critical for modulating lysosome function (Amick et al., 2016; Amick et al., 2018). Recently, PQLC2 was found to mediate the recruitment of the C9orf72 complex to lysosomes when cells are starved of cationic amino acids (Amick et al., 2020). PQLC2 is a lysosomal lysine/arginine transporter, also known as LAAT-1 (Liu et al., 2012), that is important for lysosomal amino acid recycling, proper endoplasmic reticulum (ER) quality control, and the unfolded protein response (UPR), which is an important pathway for preventing neurodegeneration (Hetz et al., 2020; Higuchi-Sanabria et al., 2020; Jezegou et al., 2012). C9orf72 can also bind to inactive Rag GTPases leading to decreased mTORC1 activity and alterations of the autophagy pathway and lysosomes (Wang et al., 2020). The connection between C9orf72 and the mTOR pathway is intriguing and may open up new areas of investigation in the FTD and ALS field, because dysregulated mTOR signaling is involved in multiple neurodegenerative diseases and an active target for drug development (Liu and Sabatini, 2020; Mazucanti et al., 2015; Schmeisser and Parker, 2019).
Together these studies strongly support a critical role for C9orf72 in maintenance of lysosome function and autophagy. Exciting experiments lie ahead to dissect how C9orf72 and its binding partners carry out such critical functions, which are important for neuronal health and survival. Future studies are also necessary to understand the degree that C9orf72 loss-of-function contributes to FTD or ALS pathogenesis and how it synergizes with other pathogenic mechanisms, such as RNA and DPR toxicity. These data will be critical for designing therapeutic strategies to target the C9orf72 gene and HRE to treat FTD and ALS.
2.2. Progranulin
Progranulin (Fig. 2; PGRN) is an ~88 kDa secreted, multi-functional glycoprotein (Bateman et al., 2018). In 2006, mutations in the granulin gene (GRN), which encodes PGRN, were found to cause FTD with ubiquitinated inclusions that contained an unknown protein (Baker et al., 2006; Cruts et al., 2006). Subsequently, TDP-43 was discovered to be the major ubiquitinated protein found in the neuronal inclusions of FTD patients with GRN mutations (Arai et al., 2006; Neumann et al., 2006). Pathogenic GRN mutations cause disease through haploinsufficiency and lead to ~50% reduction in PGRN mRNA and protein (Gass et al., 2006). Depending on the patient population, GRN mutations account for 5–20% of patients with a positive history of FTD and 1–5% of sporadic FTD patients (Baker et al., 2006; Rademakers et al., 2012). For the most part, pathogenic GRN mutations cause pure FTD and are not thought to cause ALS (Cannon et al., 2013; Del Bo et al., 2011; Schymick et al., 2007; Yu et al., 2010). Beyond FTD, a common genetic variant in GRN, the T allele of rs5848, has been associated with an increased risk of developing AD (Lee et al., 2011; Sheng et al., 2014; Xu et al., 2017) and PD (Chang et al., 2013; Chen et al., 2015).
Figure 2. Model of lysosome dysfunction caused by progranulin deficiency due to pathogenic GRN mutations.
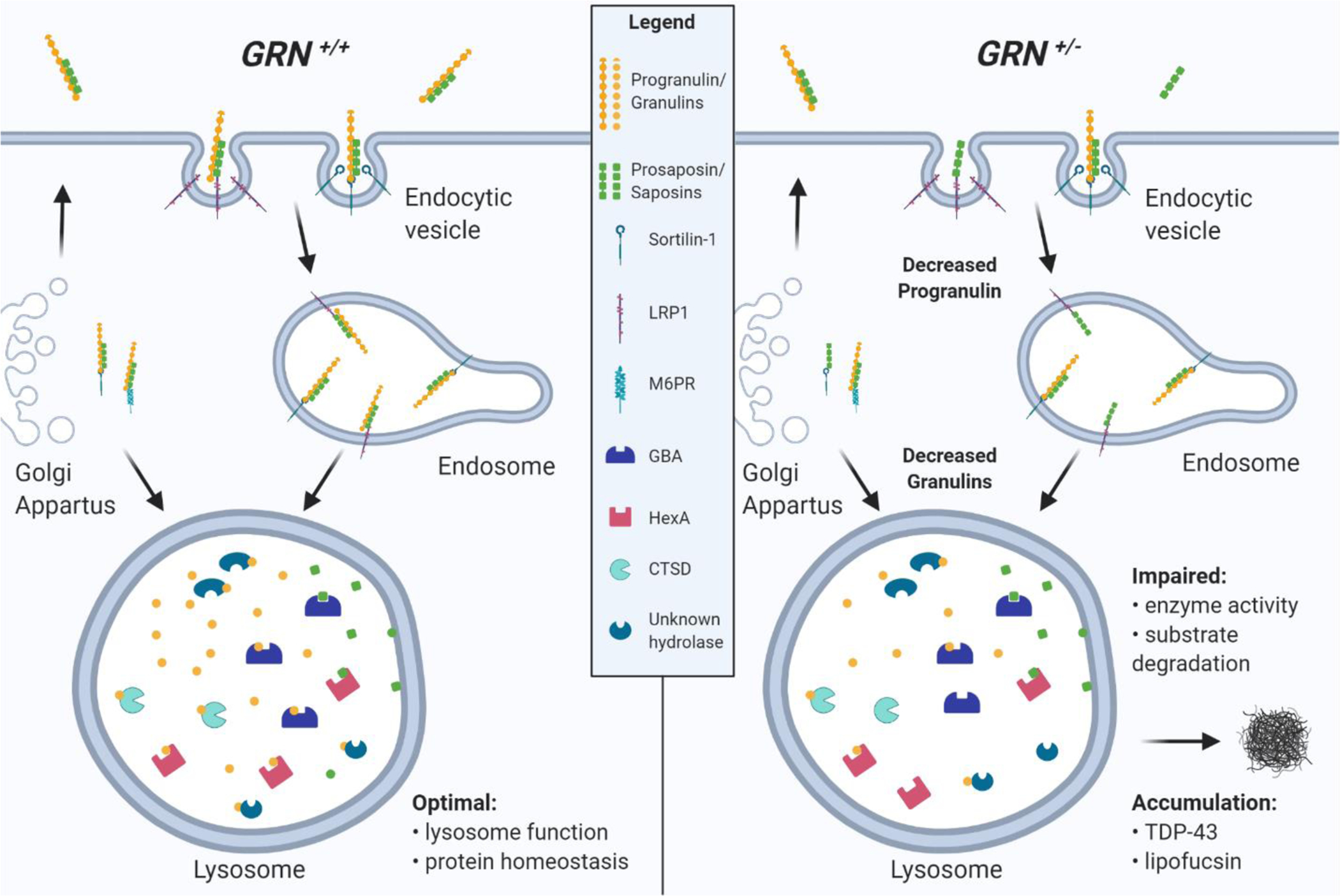
(Left panel) Under normal conditions (GRN+/+, no mutations) progranulin (PGRN) can be trafficked to the lysosome by sortilin-1 directly from the Golgi apparatus or following endocytosis. PSAP also binds PGRN and facilitates trafficking to the lysosome via LRP1 or M6PR pathways. In the lysosome, PGRN and PSAP are rapidly cleaved into individual granulins and saposins, which contribute to optimal lysosome function and protein homeostasis, potentially through regulation of lysosomal enzymes, such as GBA, HexA, CTSD, or other unknown lysosomal hydrolases. (Right panel) Pathogenic GRN mutations that cause FTD lead to haploinsufficiency (GRN+/−) resulting in an overall 50% reduction of PGRN in the cell and granulins in the lysosome, which may also decrease PSAP endocytosis and lysosomal saposins. Reduction of granulins may cause reduced lysosomal enzyme activity and substrate degradation, which ultimately leads to accumulation of undigested material and accumulation of phosphorylated-TDP-43 and lipofuscin.
Most pathogenic GRN mutations are small insertions, deletions, or duplications that affect the GRN reading-frame, splice-sites, or non-sense mutations, which lead to a premature stop codon and degradation of the GRN mRNA transcript (Nguyen et al., 2018a; Yu et al., 2010). An example of this phenomenon is the GRN R493X mutation, which is one of the most common GRN mutations that cause FTD (Chen-Plotkin et al., 2011). The GRN R493X mutation is predicted to produce a truncated protein, however, the mRNA is rapidly degraded through non-sense mediated decay, and no appreciable truncated PGRN R493X protein is made (Nguyen et al., 2018a). Other common mutations are c.813_816del, which is frequent in FTD patients from Italy (Benussi et al., 2013), as well as c.709–1G>A and c.26C> (p.A9D), which is more broadly distributed around the globe (Moore et al., 2020). The rs5848 T-allele in GRN, which is located in the 3’-untranslated region in a binding-site for miR-659, is thought to increase disease risk by decreasing PGRN levels, albeit to a lesser degree than pathogenic GRN mutations. In summary, heterozygous pathogenic GRN mutations cause disease through a shared loss-of-function pathogenic mechanism (Rademakers et al., 2012).
In the brain, PGRN is predominantly expressed in microglia and neurons, including purkinje cells, pyramidal cells of the hippocampus, and cerebral cortical neurons (Daniel et al., 2000; Zhang et al., 2014b; Zhou et al., 2017c). PGRN is composed of seven full-length granulin domains and one half-length paragranulin connected by peptide linker regions. These domains are proteolytically cleaved to release 7 individual ~6 kDa granulin peptides (Bateman and Bennett, 1998), named granulins 1 through 7, according to their position within PGRN starting at the amino (N)-terminus. Previously, it was thought PGRN was primarily cleaved into granulins in the extra-cellular space (Zhu et al., 2002). However, recent data from our lab and others suggest that in fact, the lysosome is a major site where PGRN is processed into stable granulins by cysteine proteases, such as cathepsins B and L (CTSB and CTSL) (Holler et al., 2017; Lee et al., 2017; Zhou et al., 2017b).
Over the decades since PGRN was first discovered, it has been implicated in many physiological processes ranging from cell-cycle progression, cell migration, neurotrophic signaling, wound repair, modulation of inflammation, and tumorigenesis (Bateman and Bennett, 2009; Van Damme et al., 2008). It is still mechanistically unclear how PGRN is involved in so many diverse pathways. One possibility from recent publications suggests PGRN, and individual granulins in particular, are critical to maintain healthy lysosomes (Holler et al., 2017; Lui et al., 2016; Paushter et al., 2018). The precise role of full-length PGRN and granulins within the lysosome is still a mystery, and further studies are needed to address this question. From a therapeutic perspective, overexpression of PGRN has been shown to decrease amyloid beta aggregation and toxicity in mouse models of AD, suggesting that increasing PGRN levels may provide therapeutic benefit for FTD, NCL, as well as AD (Minami et al., 2014).
Because PGRN is a secreted protein, reduced levels of PGRN can be detected in plasma or cerebrospinal fluid (CSF) samples from GRN mutation carriers and used as a diagnostic biomarker (Finch et al., 2009; Galimberti et al., 2018; Ghidoni et al., 2008; Sleegers et al., 2009). Interestingly, fibroblasts and post-mortem cortical brain tissue from FTD-GRN patients are haploinsufficient in both full-length PGRN and individual granulins (Holler et al., 2017). Although it is unclear if granulins are secreted, it would be important to know if they are, and if granulins levels are reduced in the plasma or CSF of GRN mutation carriers. This information could help provide insight into the role of granulins in normal physiology and in disease. Beyond PGRN and granulins, other proteins, such as glial fibrillary acidic protein (GFAP)(Heller et al., 2020), chitotriosidase (Woollacott et al., 2020), neuronal pentraxin (van der Ende et al., 2020), or transmembrane glycoprotein NMB (GPNMB) (Huang et al., 2020) may serve as additional biomarkers of disease progression in FTD.
One of the first pieces of evidence that PGRN may be involved in lysosome function arose in 2012 when homozygous GRN mutations were discovered to cause neuronal ceroid lipofuscinosis (NCL) type 11 (CLN11) (Smith et al., 2012). These NCL cases carried two copies of the GRN c.813_816del mutation (p. Thr272Serfs*10) and produced no detectable PGRN. NCLs are neurodegenerative disorders characterized by the accumulation of abnormal lipopigment in lysosomes in parallel with the clinical features of cerebellar ataxia, seizures, and progressive decline in cognitive and motor functions (Mole et al., 2019). Since the original report, other cases of CLN11 caused by homozygous GRN mutations have been discovered, further validating the importance of PGRN to lysosome function (Almeida et al., 2016; Canafoglia et al., 2014; Faber et al., 2017; Kamate et al., 2019). Recently, six new patients with homozygous GRN mutations were discovered that had variable clinical phenotypes (Huin et al., 2020). The majority had symptoms of juvenile onset NCL, however three individuals developed symptoms of bvFTD, including behavioral disinhibition and apathy. Taken together, this suggests that FTD and NCL caused by GRN mutations are clinical phenotypes that exist along a spectrum of severity that correlates with lysosome dysfunction (Huin et al., 2020; Smith et al., 2012).
Studies in PGRN-deficient mice (Grn−/−) further supports the important role of PGRN in lysosomal function. An NCL-like phenotype has been consistently observed in the brains of multiple Grn−/− mouse lines (Ahmed et al., 2010; Filiano et al., 2013; Petkau et al., 2012; Wils et al., 2012; Zhou et al., 2017d) including p62-positive protein aggregates (Chang et al., 2017; Tanaka et al., 2014; Wils et al., 2012), as well as enhanced levels of lysosomal proteins such as cathepsin D (CTSD) and LAMP1/2 (Gotzl et al., 2018; Gotzl et al., 2014; Tanaka et al., 2014). Importantly, the accumulation of lipofuscinosis and intracellular NCL-like storage material was discovered in postmortem cortical brain tissue and cells from FTD patients with pathogenic GRN mutations (Ward et al., 2017). Taken together, these data raise the possibility that PGRN and/or granulins have an important function within the lysosome and that loss of this activity may contribute to the development of FTD, ALS, and related neurodegenerative disorders. Further studies are necessary to uncover the precise lysosomal function of PGRN and granulins.
Following the initial finding by Ward et al., increasing evidence supports the potential role of PGRN and individual granulins within the lysosome (Ward et al., 2017). First, after transcription and biosynthesis, a portion of PGRN is secreted to the extracellular space, but the majority of intracellular PGRN localizes within lysosomes (Holler et al., 2017; Hu et al., 2010; Naphade et al., 2010). The SORT1 membrane receptor facilitates PGRN trafficking to lysosomes (Fig. 2) through a) binding and endocytosis of extracellular PGRN or b) binding PGRN as it migrates through the TGN (Hu et al., 2010). Another reported binding partner of PGRN is prosaposin (PSAP) (Zhou et al., 2015), a secreted glycoprotein predominantly localized to lysosomes, which is essential for the lysosomal degradation of sphingolipids (Schulze and Sandhoff, 2014). PGRN is thought to “piggyback” on PSAP’s route to the lysosome using M6PR and LRP1 as trafficking receptors in a SORT1-independent pathway (Zhou et al., 2015). Surprisingly, it appears that PGRN can also facilitate the lysosomal trafficking of PSAP through its receptor SORT1 (Zhou et al., 2017c). Higher levels of PSAP are found in the serum of Grn−/− mice and FTD-GRN patients have reduced levels of PSAP in neurons from postmortem cortex (Zhou et al., 2017c). Although the binding of PGRN to SORT1 appears to be important for trafficking to lysosomes, there are multiple reports that PGRN is bioactive in a SORT1-independent manner (De Muynck et al., 2013), suggesting that SORT1 is not required for all of PGRN’s functions (Gass et al., 2012). Abnormal accumulation of phosphorylated TDP-43 is a neuropathological hallmark of FTD patients with GRN mutations (Arai et al., 2006; Neumann et al., 2006) and phosphorylated TDP-43 accumulates in Grn−/− mice at an advanced age (Wils et al., 2012). Interestingly, loss of PSAP in mice produced an FTD-like phenotype with accumulation of hyperphosphorylated TDP-43 in the insoluble fraction of brain lysates (Zhou et al., 2017c). These findings suggest a potential role of PGRN in the trafficking of other lysosomal proteins, which may further exacerbate lysosome dysfunction in FTD patients with PGRN haploinsufficiency.
Mounting evidence suggests that PGRN can play a direct or indirect role on the activity of lysosomal enzymes. Once PGRN is within the lysosome, it is rapidly processed into individual granulins (Holler et al., 2017; Lee et al., 2017; Zhou et al., 2017b). Several groups find that PGRN and/or granulins can regulate the enzymatic activity of CTSD, a lysosomal protease that is important for activation of lysosomal hydrolases and general protein degradation (Beel et al., 2017; Valdez et al., 2017; Ward et al., 2017; Zhou et al., 2017a). Both PGRN and GRN-7 can bind directly to CTSD, modulating its enzymatic activity (Beel et al., 2017; Valdez et al., 2017; Zhou et al., 2017a). Decreased enzymatic activity of CTSD also was reported in Grn−/− mice and FTD-GRN patients (Beel et al., 2017; Valdez et al., 2017; Zhou et al., 2017a) despite an increase in the levels of CTSD protein (Beel et al., 2017; Gotzl et al., 2014), suggesting that PGRN could be necessary for the proper function of CTSD. Interestingly, PGRN can also regulate other lysosomal enzymes including glucocerebrosidase (GBA) (Arrant et al., 2019; Jian et al., 2016a; Jian et al., 2016b; Krainc et al., 2020; Zhou et al., 2019), and β-hexosaminidase A (HexA) (Chen et al., 2018), suggesting that PGRN may modulate lysosome function through direct regulation of multiple lysosomal enzymes. PGRN is reported to bind directly to GBA (Arrant et al., 2019) and HexA (Chen et al., 2018) to regulate their enzymatic activity. Both enzymes help degrade sphingolipids in the lysosome: GBA degrades glucocerebroside (also called glucosylceramide) and HexA degrades GM2 gangliosides (Schulze and Sandhoff, 2014). PGRN has also been proposed to directly influence lysosome acidification and enzymatic activity (Tanaka et al., 2017). Together, this evidence suggests that the haploinsufficiency of PGRN may decrease lysosomal enzymatic activity, leading to lysosomal dysfunction, substrate accumulation, and eventually FTD. Further studies are required to determine the individual role each granulin may play within the lysosome.
Interestingly, PGRN shares structural features and neurotrophic properties with PSAP. Like PGRN, PSAP is also cleaved within the lysosome, to generate 4 individual subunits, called saposins (Sap A, B, C, and D). Mutations in specific saposin domains of PSAP affect their unique lysosomal function in different pathological conditions such as Gaucher disease (Sap C), metachromatic leukodystrophy (Sap B), or Krabbe disease (Sap A) (Ferrini et al., 1989; Hulkova et al., 2001; Kuchar et al., 2009), suggesting a crucial role of individual saposins in the lysosome. Although the function of individual granulins is unclear, there is evidence that they are active within the lysosome. First, granulin-7 can bind the lysosomal protease CTSD (Beel et al., 2017), suggesting individual granulins may have different roles independent of full-length PGRN. Moreover, haploinsufficiency of individual granulins within the lysosome was observed in samples from FTD patients (Holler et al., 2017), suggesting a potential role of granulin proteins in the regulation of lysosomal function. Recent data comparing individual human granulins reveal that each granulin shares no more than 60% sequence identity and that each granulin exhibits different electrostatic charges at neutral pH (Butler et al., 2019). Differences in electrostatic charges and protein sequences between granulins support the hypothesis that individual granulins could perform unique functions in the lysosome, which remains to be determined.
Finally, an important piece of evidence indicating a key role for PGRN in the lysosome is that GRN expression is modulated by TFEB (Belcastro et al., 2011), which regulates other lysosomal genes such as CTSD. TFEB activates a response known as the Coordinated Lysosomal Expression and Regulation (CLEAR), which induces the expression of genes involved in lysosomal function and autophagy, including PGRN (Settembre et al., 2011). This observation suggests that PGRN is an essential protein for lysosomal homeostasis, and PGRN haploinsufficiency due to heterozygous GRN mutations may cause lysosomal dysfunction and ultimately cause neurodegeneration.
Considering the summarized evidence, PGRN and granulins may play an essential role in lysosomal function by regulating enzymatic activity, maintaining the acidification of the lysosome, and participating in the lysosomal trafficking of key lysosomal regulators, such as PSAP. Therefore, the decrease of PGRN and granulins caused by mutations in GRN could induce lysosomal dysfunction and lead to the development of neurodegenerative diseases. Future investigation of the mechanism(s) by which PGRN or granulins regulate lysosomal functions are necessary to elucidate the role of PGRN and the lysosome in the development of FTD. Furthermore, understanding the role of individual granulins in lysosomal function may enable the development of novel therapeutic approaches for FTD, ALS, AD, and other LSDs.
2.3. TDP-43
Transactivation-response DNA-binding protein of 43 kDa (TDP-43) is a ubiquitously expressed protein encoded by the TARDBP gene on chromosome 1 (Ayala et al., 2005; Neumann et al., 2006; Ou et al.). TDP-43 was first described as a transcriptional repressor that binds TAR DNA to repress human immunodeficiency virus 1 (HIV-1) gene transcription. TDP-43 is now appreciated as an RNA/DNA binding protein with diverse functions including DNA and RNA homeostasis, transcriptional activation, mRNA splicing, transport, translation, and degradation (Gao et al., 2018; Ou et al., 1995). The discovery in 2006 that TDP-43 was a novel and frequent component of ubiquitin-positive inclusions in both ALS and FTD revolutionized the field (Arai et al., 2006; Neumann et al., 2006). Subsequent studies have validated that TDP-43 pathology is a common neuropathological feature of multiple neurodegenerative diseases, leading to the creation of a subset of neurological diseases termed TDP-43 proteinopathies (Tan et al., 2015).
Causative genetic mutations in TARDBP were first linked to ALS in 2008 (Sreedharan et al., 2008). To date, over 50 autosomal dominant mutations have been identified, a majority of which cluster toward the C-terminal low complexity domain. Even so, only ~5% of ALS cases have a TARDBP mutation (Kapeli et al., 2017; Mann and Snowden, 2017; Tan et al., 2017a; Tan et al., 2017b). Furthermore, at least two mutations in TARDBP have been linked to FTD, accounting for <1% of FTD cases (Kapeli et al., 2017). TDP-43 pathology is widespread among both ALS and FTD cases, where it is estimated to occur in ~97% of ALS cases and ~45% of FTD cases (Gao et al., 2018; Prasad et al., 2019). Mutations in many genes that are linked to ALS and/or FTD such as GRN, C9orf72, and VCP also cause TDP-43 proteinopathy, suggesting TDP-43 accumulation and dysfunction is a common link between both ALS cases and FTD cases (Bang et al., 2015).
Understanding the normal function of TDP-43 may provide insight into why TDP-43 inclusions are common in multiple neurodegenerative diseases and how they contribute to pathogenesis. TDP-43 is a 414 amino acid protein comprised of a N-terminal domain, a nuclear localization sequence (NLS), two RNA recognition motifs (RRM), a predicted nuclear export signal (NES), and a low complexity glycine-rich domain thought to facilitate various protein-protein interactions (Prasad et al., 2019). Although TDP43 is primarily a nuclear protein, ~27% of TDP-43 protein is found in the cytoplasm (Barmada et al., 2010). The ability of TDP-43 to shuttle between the cytoplasm and the nucleus is important for TDP-43’s pleiotropic functions (Barmada et al., 2010). Nucleocytoplasmic shuttling of TDP-43 is dependent on various factors, including transcription, cellular activity, and stress events (Ayala et al., 2008; Diaper et al., 2013; Khalfallah et al., 2018). Thus, many have proposed that TDP-43 pathology may be a result of deficits in nucleocytoplasmic shuttling or degradation (Ayala et al., 2008; Chou et al., 2018; Scotter et al., 2015; Sugai et al., 2018; Winton et al., 2008).
The precise neuropathological signature of TDP-43 proteinopathies can vary between ALS and FTD cases (Cairns et al., 2007; Laferriere et al., 2019; Tan et al., 2017b). Common features between pathogenic subtypes include the accumulation of insoluble TDP-43, C-terminal TDP-43 cleavage products, hyperphosphorylation, ubiquitination, and formation of insoluble inclusions (Neumann et al., 2006). These inclusions are primarily in the cytoplasm, but are also found in the nucleus and/or neurites of neurons and glial, and are directly correlated with disease severity and neuronal death (Barmada et al., 2010; Lee et al., 2019; Neumann et al., 2007a; Neumann et al., 2006; Scotter et al.; Valdez et al., 2017). TDP-43 inclusion formation is thought to cause both loss- and gain-of-function toxicity (Barmada et al., 2010; Diaper et al., 2013; Vanden Broeck et al., 2015; Zhang et al., 2009). These include changes in splicing profile, impairment of mitochondria, dysregulation of metal ion homeostasis, impaired nucleosomes, dysregulation of autophagy, and disruption of lysosomal dynamics (Dang et al., 2014; Highley et al., 2014; Prasad et al., 2019; Sugai et al., 2018; Wang et al., 2019). Thus, TDP-43 protein homeostasis is important to maintain overall health of cells and may be particularly important in post-mitotic and long-lived neuronal cells.
TDP-43 protein levels are tightly regulated, likely because excess TDP-43 is toxic in many organisms. In particular, TDP-43 regulates production of its own mRNA through a negative feedback loop mechanism in which increased TDP-43 protein downregulates the generation of its own mRNA (Avendano-Vazquez et al., 2012; Ayala et al., 2011). Both genetic and non-genetic models of ALS and FTD show that formation of TDP-43 inclusions alters TDP-43 autoregulation (Sugai et al., 2018; Sugai et al., 2019; White et al., 2018). For example, expression of the ALS-FTD-linked mutation Q331K dysregulates TDP-43 autoregulation leading to a ~14% increase in TARDBP transcript expression (White et al., 2018). This finding is important because increased TDP-43 protein levels cause deficits in neuronal dendritic complexity, increased phosphorylation of TDP-43, and neuronal death (Herzog et al., 2017; Lu et al., 2016; Vogt et al., 2018; Xu et al., 2010). Thus, maintenance of proper levels of TDP-43 is necessary for cellular health and occurs through three main degradation pathways: 1) the ubiquitin-proteasome system (UPS), 2) chaperone-mediated autophagy (CMA) that occurs in lysosomes, and 3) the autophagosome-lysosome pathway (Fig. 3). In this review, we will focus on discussing lysosomal pathways (CMA and autophagy) but will briefly discuss the key contributions of the UPS system.
Figure 3. Aggregation of TDP-43 impairs lysosome-autophagy function.
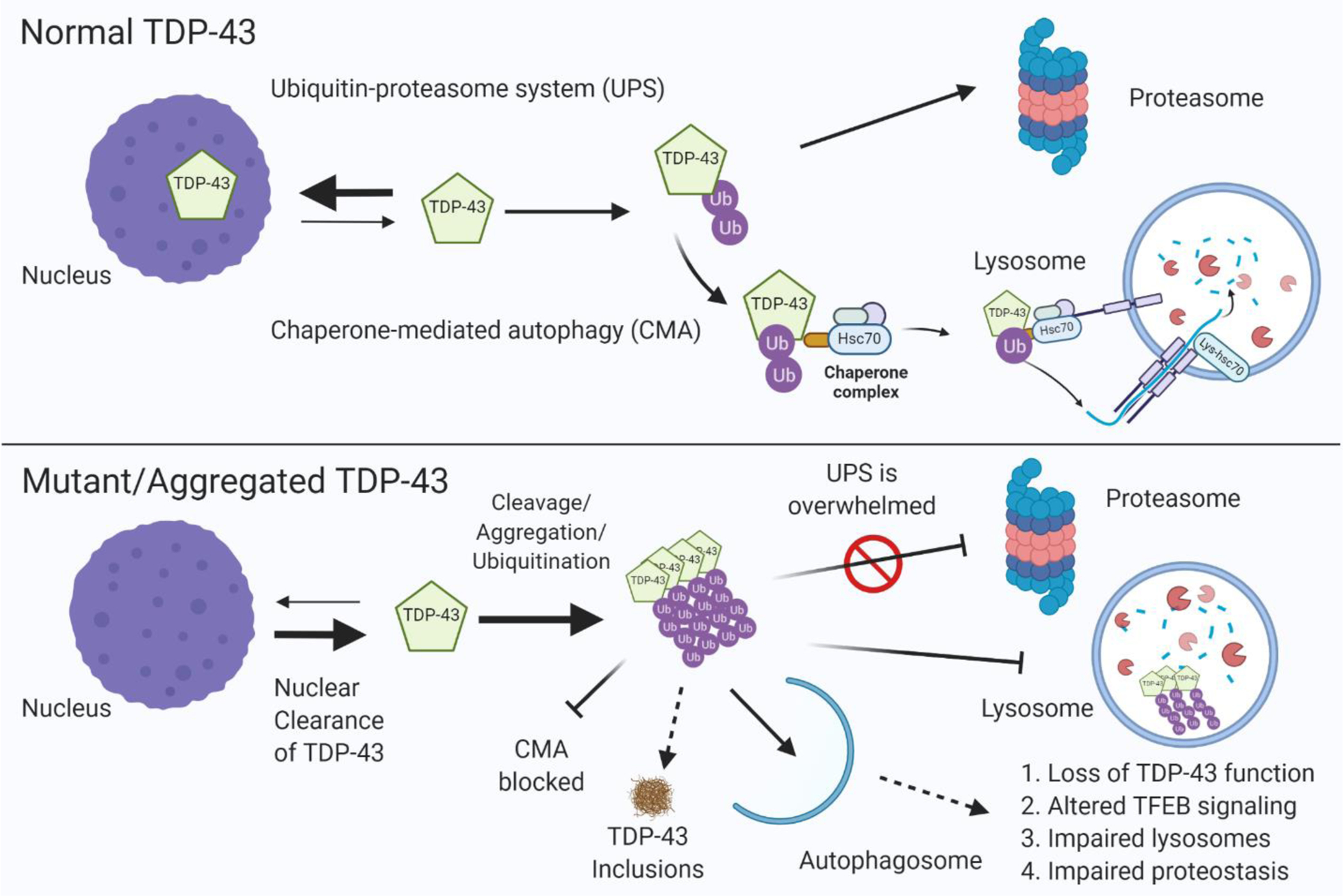
(Upper panel) In healthy cells, optimum levels of TDP-43 are maintained by 1) degradation through the ubiquitin–proteasome system (UPS) or 2) degradation through chaperone mediated autophagy (CMA) by binding to Hsc70 and import into the lysosome through LAMP2A. (Lower panel) TARDBP gene mutations or other causes (i.e. lysosome dysfunction cause by GRN or C9orf72 mutations) lead to accumulation and aggregation of TDP-43, which ultimately form insoluble inclusions. TDP-43 aggregates are not efficiently cleared by the proteasome which leads to their clearance through autophagosomes that required fusion with lysosomes. TDP-43 aggregates can block CMA and impair lysosome function, leading to a feed-forward toxic mechanism causing loss of TDP-43 function, TDP-43 inclusion formation, lysosome dysfunction, impaired protein homeostasis in the cell, and ultimately neurodegeneration.
The UPS system is a major pathway of TDP-43 degradation. Ubiquilin-1 binds ubiquitinated and non-ubiquitinated TDP-43 to facilitate clearance of TDP-43 aggregates through the autophagosome and/or proteasome (Kim et al., 2009). Further,TDP-43 insoluble aggregates are ubiquitinated and contain proteins associated with degradation, including ubiquilin-1, ubiquilin-2, and sequestosome-1 (Arai et al., 2006; Deng et al., 2011; Neumann et al., 2006). TDP-43 is ubiquitinated at residues K48 and K63 by parkin, an E3 ubiquitin ligase that forms a multiprotein complex with HDAC6 (Hebron et al., 2013; Urushitani et al., 2010). Interestingly, TDP-43 is primarily ubiquitinated in the nucleus while aggregation-prone TDP-43 is ubiquitinated in the cytosol (Scotter et al., 2014; Urushitani et al., 2010). Given this, cytoplasmic mislocalization of TDP-43 may lead to increased levels of ubiquitinated TDP-43 that is inefficiently cleared from the cell. Further evidence for this idea arises from the observation that pharmacological inhibition of the proteasome also increases full-length and cleaved, aggregation prone TDP-43 (Wang et al., 2010a). Lastly, pathogenic TDP-43 mutations increase the ubiquitin-proteasome mediated turnover of TDP-43 (Araki et al., 2014).
Evidence suggests that once the proteasome system is overwhelmed, excess levels of TDP-43 impairs upstream and downstream stages of lysosome-mediated autophagy (Fig. 3). In fact, boosting autophagy in cellular models can alleviate TDP-43 induced neurotoxicity by reducing the cytoplasmic mislocalization and aggregation of TDP-43 (Barmada et al., 2014; Caccamo et al., 2009; Wang et al., 2012). Conversely, pharmacological inhibition of autophagy increases toxic levels of TDP-43 by impairing the ability of cells to clear large aggregates of TDP-43 (Scotter et al., 2014; Wang et al., 2010a). Further, loss of TDP-43 function in the nucleus decreases autophagosome–lysosome fusion leading to an accumulation of autophagosomes and lysosomes that may represent a feed-forward loop leading to neurodegeneration (Skoko et al., 2016).
Beyond the UPS, CMA also contributes to the constitutive degradation of TDP-43. The CMA pathway utilizes a chaperone protein to bind soluble proteins and direct them to the lysosome for degradation. Typically, the chaperone heat shock cognate 71 kDa protein (Hsc70) binds protein targets with a KFERQ-like recognition motif (Chiang et al., 1989) and directly traffics cargo to the lysosomal-associated membrane protein 2A (LAMP2A), which acts as a receptor to translocate proteins to the lysosomal lumen for degradation (Tekirdag and Cuervo, 2018). The QVKKD sequence in TDP-43 appears to mediate binding to Hsc70 and directs lysosomal degradation through CMA (Huang et al., 2014). Further, 25 and 35 kDa fragments of TDP are degraded through the UPS and CMA pathways (Huang et al., 2014). Recent work provides additional evidence that endogenous TDP-43 is a bona fide CMA substrate in vitro and in vivo (Ormeno et al., 2020). In particular, genetic downregulation of CMA by decreasing LAMP2a levels increases TDP-43 levels (Huang et al., 2014; Ormeno et al., 2020).
Additionally, the health and integrity of lysosomes themselves is impaired when TDP-43 expression is decreased. Genetic knockout (KO) of TDP-43 using CRISPR-Cas9 induces widespread changes in lysosomal function, including abnormal accumulation of lysosomes at the perinuclear region, decreased localization of CTSL and PGRN to the lysosome, abnormal CTSB processing, and higher secretion of unprocessed proteins from the lysosome (Roczniak-Ferguson and Ferguson, 2019). Furthermore, expression of aggregation prone TDP-43 causes Galectin-3 to translocate into lysosomes, which is an indicator of lysosomal damage (Ormeno et al., 2020). Moreover, ALS-linked TDP-43 mutations decrease the size and trafficking of lysosomes and increase neurodegeneration in aged iPSC-derived motor neurons (Kreiter et al., 2018). This finding is supported by a subsequent stud, which found that excess TDP-43 disrupted function of lysosomes and triggered cytotoxic autophagy (Leibiger et al., 2018).
Given multiple reports that pathogenic TDP-43 aggregates are preferentially cleared through the autophagy-lysosome pathway, have led to the suggestion that stimulating autophagy could target TDP-43 aggregates and enhance survival of affected neurons (Wang et al., 2010a). In support of this idea, treatment with rapamycin, which activates autophagy by inhibiting mTOR, enhanced degradation of toxic C-terminal TDP-43 fragments, and promoted the proper nuclear localization of TDP-43 (Caccamo et al., 2009). Full length TDP-43 can also be degraded through the autophagy-lysosome pathway (Barmada et al., 2014).
TDP-43 toxicity is also modulated by lysosome fusion. Specifically, loss of Vps-16, 18, 39, and 41, which are known to facilitate late endosome and lysosome fusion through the multi-subunit homotypic fusion and protein sorting (HOPS) core, exacerbates TDP-43 induced toxicity (Balderhaar and Ungermann, 2013; Liu et al., 2017). Lastly, TDP-43 can directly influence the activity of TFEB, a master transcription factor of lysosomal genes (Skoko et al., 2016). Depletion of TDP-43 causes a global increase in both the mRNA and protein expression of TFEB-regulated genes which impairs the fusion of autophagosomes with lysosomes, ultimately leading to accumulation of immature autophagic vesicles (Skoko et al., 2016). The regulation of TFEB activity may occur through direct binding of TDP-43 to TFEB mRNA (Xiao et al., 2011).
Taken together, TDP-43 has an important function in the normal, homeostatic function of lysosomes (Fig. 3). TDP-43 maintains lysosome function through multiple mechanisms including modulation of lysosomal function and the expression of genes critical for the proper function of autophagy and lysosome pathways. Conversely, misfolding and aggregation of TDP-43 directly impairs autophagy and lysosome homeostasis, potentially contributing to a toxic feed-forward mechanism that contributes to neurodegeneration. Therefore, approaches to restore proper function of the lysosome may provide novel therapies to treat FTD, ALS, and other neurodegenerative disease linked to TDP-43 mislocalization and aggregation.
2.4. TMEM106B
TMEM106B is a lysosomal transmembrane protein of unknown function that has been implicated in FTD and related disorders (Fig. 4). The TMEM106B protein first came to the attention of the neurodegenerative field in 2010 following the publication of a large, international collaborative genome wide association study (GWAS) (Van Deerlin et al., 2010). This study found that the occurrence of FTLD with TDP-43 inclusions (FTLD-TDP) was associated with multiple single-nucleotide polymorphisms (SNPs), which mapped to a single linkage disequilibrium block on chromosome 7p21 that contained the TMEM106B gene. Three SNPs (rs6966915, rs1020004, and rs1990622) retained genome-wide significance following Bonferroni correction. For the most significant SNP, rs1990622, the major T allele was associated with an increased risk (odds ratio 1.64) for developing FTLD-TDP, while the minor C allele was associated with a reduced risk of developing FTLD-TDP (odds ratio 0.61). Intriguingly, genetic variants in TMEM106B do not appear to increase the risk of developing ALS (Vass et al., 2011).
Figure 4. Dysfunction of TMEM106B impairs formation, transport, and function of lysosomes.
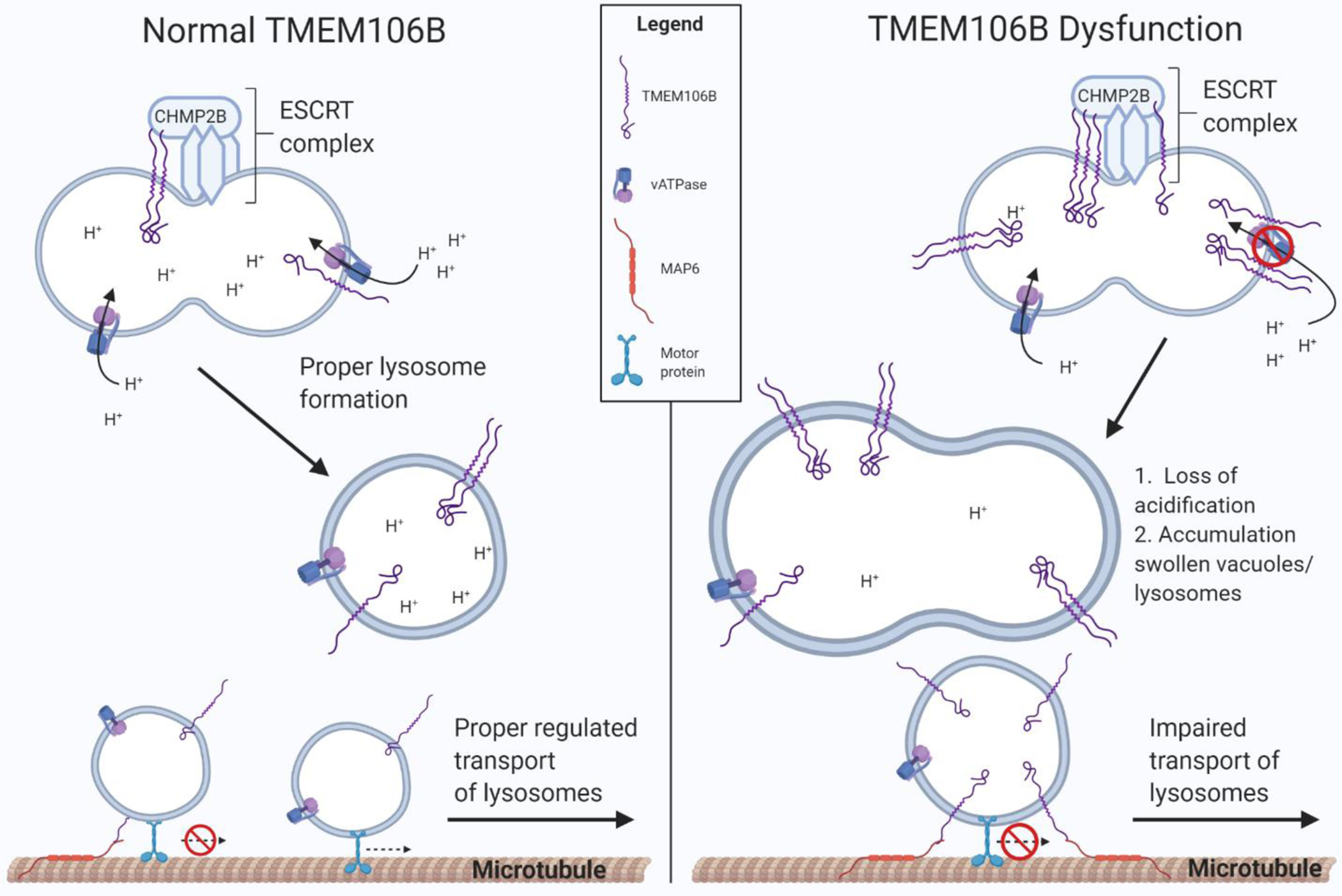
(Left panel) TMEM106B plays an important regulatory role in lysosome function, possibly through interaction with CHMP2B, a member of the ESCRT-III complex, and with V-ATPase, which is critical for proper lysosome formation. TMEM106B is also thought to bind microtubule-associated protein 6 (MAP6), a protein that stabilizes microtubules and regulates lysosome trafficking in neurons. (Right panel) Elevated TMEM106B levels, thought to be increased by the TMEM106B risk allele, or loss of TMEM106B function, causes impaired lysosome acidification and transport, leading to dysfunction and accumulation of swollen vacuoles and lysosomes.
Following the initial GWAS study, the association between variants in TMEM106B and the risk of developing FTD has been reproduced by multiple labs (Finch et al., 2011; van der Zee et al., 2011). Furthermore, studies have also associated variants in TMEM106B with the development of hippocampal sclerosis of aging (Murray et al., 2014; Nelson et al., 2015), the accumulation of TDP-43 inclusions in Alzheimer’s disease (AD) (Rutherford et al., 2012) or older people without FTLD (Yu et al., 2015), and a recently described disease named limbic-predominant age-related TDP-43 encephalopathy (LATE) (Hokkanen et al., 2020; Nelson et al., 2019). Intriguingly, a mutation in TMEM106B (p. Asp252Asn) was found to cause a novel form of hypomyelinating leukodystrophy (HLD), a heterogeneous group of disorders caused by defects in myelination (Simons et al., 2017). The genetics of TMEM106B and the role of TMEM106B in neurodegeneration has been recently reviewed (Jain and Chen-Plotkin, 2018; Nicholson and Rademakers, 2016; Zhou et al., 2021).
Two major questions regarding the role of TMEM106B in FTD remain. First, what is the specific SNP or SNPs in the linkage disequilibrium block spanning the TMEM106B locus that is responsible for altering disease risk? Second, how do TMEM106B variants alter the risk of developing neurodegeneration? Multiple studies support the idea that TMEM106B variants associated with an increased risk of disease may cause increased levels of the TMEM106B protein (Gallagher et al., 2017; Van Deerlin et al., 2010). Alternatively, the rs3173615 SNP in TMEM106B that is associated with FTD risk causes a change in amino acid sequence that may be mechanistically important (Cruchaga et al., 2011; Deming and Cruchaga, 2014; Nicholson et al., 2013; van Blitterswijk et al., 2014). Specifically, the variant leads to a coding change from the more common, highly conserved, threonine (Thr185; rs3173615 (C); risk allele) to a serine (Ser185; rs3173615 (G); protective allele) at position 185 in the TMEM106B protein. In theory, this amino acid change could alter the levels, or function, of the TMEM106B protein and might explain its impact on disease risk. In support of this idea, in vitro cell-based studies found that the protective TMEM106B Ser185 isoform (protective variant) was degraded more rapidly compared to TMEM106B Thr185 (risk variant) (Nicholson et al., 2013).
An alternative explanation is that rs1990622 or other variants in the TMEM106B region might affect splicing or gene expression. In support of this possibility, the non-coding risk-associated allele of rs1990622 was found to recruit CCCTC-binding factor (CTCF) to the TMEM106B promoter causing increased expression of TMEM106B, which was correlated with increased cytotoxicity and risk of neurodegeneration (Gallagher et al., 2017). In contrast, a recent study found that the rs1990622 allele had a dominant effect on the aging brain’s transcriptome that was not dependent on TMEM106B mRNA levels (Yang et al., 2020). Instead, the authors propose that the TMEM106B p.S185T haplotype increases disease risk by causing TMEM106B hyperfunction, similar to overexpression of TMEM106B, which causes dysregulation of lysosomes and myelination that ultimately causes pathologic aggregation of TDP-43 (Yang et al., 2020). Further studies are needed to understand the genetics and mechanisms more completely behind the association of TMEM106B with brain health and neurodegeneration.
TMEM106B is a type-II single pass transmembrane protein that localizes to endosomal and lysosomal membranes (Brady et al., 2013; Chen-Plotkin et al., 2012; Schwenk et al., 2014; Stagi et al., 2014). The carboxy (C)-terminal domain of TMEM106B projects into the lysosomal lumen while the smaller amino (N)-terminal domain extends into the cytosol (Brady et al., 2013; Lang et al., 2012). TMEM106B is part of a larger protein family (pfam07092; (Lu et al., 2020)) with unknown function, which includes TMEM106A, TMEM106B, and TMEM106C (Satoh et al., 2014). TMEM106B is expressed in neurons, glia, and in endothelial cells and pericytes lining the central nervous system(Busch et al., 2013). Within neurons, it maintains a somatodendritic distribution (Brady et al., 2013; Chen-Plotkin et al., 2012; Lang et al., 2012). A comprehensive report of the glycosylation sites in TMEM106B found five N-glycosylation sites at residues N145, N151, N164, N183, and N256, each with an N-X-T/S consensus sequence (Lang et al., 2012). Residues N183 and N256 are necessary for lysosomal/endosomal localization (Lang et al., 2012), and N183 glycosylation contributes to the stabilization of TMEM106B (Nicholson et al., 2013). TMEM106B is cleaved by the enzyme signal peptide peptidase-like 2A and other unknown lysosomal proteases in a process called regulated intramembrane proteolysis (Brady et al., 2014).
Emerging data describing the protein binding partners of TMEM106B are beginning to provide insight into the function of TMEM106B. First, the N-terminus of TMEM106B can interact with itself as well as TMEM106C to form homo- and hetero-multimers at the surface of the lysosome (Stagi et al., 2014). The same study found that the N-terminus of TMEM106B interacts with the endocytic adaptor proteins, clathrin heavy chain and, the μ1 subunit of adipocyte protein 2, suggesting that TMEM106B may play an important role in the endolysosomal sorting pathway and lysosome trafficking. This idea is supported by a study that found TMEM106B binds to the C-terminus of microtubule-associated protein 6 (MAP6) (Schwenk et al., 2014). Additionally, TMEM106B directly binds to CHMP2B, a member of the endosomal sorting complexes required for transport III (ESCRT-III) (Henne et al., 2011; Jun et al., 2015). Interestingly, the TMEM106B T185 (risk allele) binds to CHMP2B more tightly compared to S185 (protective allele), which correlates with impaired autophagic flux and increased neurotoxicity (Jun et al., 2015). Whether the binding of TMEM106B and CHMP2B directly contributes to cellular dysfunction remains unclear. TMEM106B is also reported to directly bind to the AP1 subunit of V-ATPase (Klein et al., 2017). The precise contributions of these interactions to proper lysosome function or dysfunction in neurodegeneration are unknown but are an important focus of future studies.
TMEM106B mRNA and protein expression are increased in FTD-TDP brain tissue, especially in cases with GRN mutations (Chen-Plotkin et al., 2012; Finch et al., 2011; Gotzl et al., 2014; Van Deerlin et al., 2010). Additionally, disease associated TMEM106B variants are thought to increase TMEM106B expression. Thus, several groups have explored models using TMEM106B overexpression. Overexpression of TMEM106B causes translocation of TFEB to the nucleus (Stagi et al., 2014), accumulation of large LAMP1+/TMEM106B+ vacuoles (Busch et al., 2016; Chen-Plotkin et al., 2012; Gallagher et al., 2017), loss of lysosome acidification (Chen-Plotkin et al., 2012; Nicholson and Rademakers, 2016), disruption of lysosomal trafficking and function (Busch et al., 2016; Chen-Plotkin et al., 2012), and cytotoxicity in a variety of cellular models (Busch et al., 2016; Gallagher et al., 2017; Suzuki and Matsuoka, 2016). TMEM106B overexpression also decreases PGRN secretion and increases intracellular PGRN levels (Brady et al., 2013; Chen-Plotkin et al., 2012; Nicholson et al., 2013), potentially through TFEB-induced upregulation of the CLEAR network genes, which includes GRN (Tanaka et al., 2017). Further, TMEM106B overexpression decreases the production of granulins, suggesting that TMEM106B could increase FTD risk by inhibiting the processing of PGRN into granulins through lysosome dysfunction (Holler et al., 2017). Another lab found that overexpression of TMEM106B exacerbated lysosome dysfunction in Grn-deficient mice (Zhou et al., 2017d). Site-directed mutagenesis to prevent TMEM106B lysosomal localization ablated TMEM106B-mediated impairment of lysosome function, indicating lysosomal localization of TMEM106B is necessary to induce dysfunction (Busch et al., 2016; Suzuki and Matsuoka, 2016). Moreover, the cytotoxic response of TMEM106B overexpression was completely ablated by the addition of Bcl-xL (Suzuki and Matsuoka, 2016), an anti-apoptotic protein that blocks caspase-dependent mitochondrial-mediated apoptosis (Kim, 2005). Together these findings suggest that overabundance of TMEM106B can cause lysosome dysfunction that can lead to mitochondrial-mediated apoptosis. Indeed, the role of lysosome dysfunction upstream of the apoptotic cascade is well-supported (Guicciardi et al., 2004).
Approaches to reduce TMEM106B expression have been proposed as potential therapies for FTD because increasing TMEM106B levels induce toxicity, and protective TMEM106B variants are associated with decreased TMEM106B expression. Some initial reports supported this idea; however, subsequent work did not. Three recent publications from independent labs, described below, appears to resolve these discordant results. In 2017, genetic deletion of TMEM106B was reported to ameliorate many lysosomal and FTD-related phenotypes in Grn−/− mice (Klein et al., 2017). Elevated levels of Dipeptidyl peptidase 2 (DPP2), CTSB, and LAMP1 were normalized along with reduced retinal degeneration and amelioration of aberrant behavior in the open field and elevated plus maze test in Grn−/−; Tmem106B−/− mice compared to Grn−/− mice (Klein et al., 2017). Remarkably, lipofuscin accumulation, CD68+ microglia abundance, or complement C1q levels were not rescued in the double KO mice.
In contrast, partial knockout (KO) of TMEM106B did not rescue increased LAMP1 levels, increased β-Hexosaminidase, or β-Glucocerebrosidase activities, or reduced social dominance in Grn+/− mice (Arrant et al., 2018). Further, neither knockdown nor KO of TMEM106B rescued behavioral deficits, RNA foci, DPR formation, or phosphorylated TDP-43 accumulation in an AAV-based mouse model of C9orf72-repeat induced toxicity (Nicholson et al., 2018). Intriguingly, knockdown of C9orf72 did increase TMEM106B expression and other lysosomal proteins, suggesting that both genes interact in some fashion (Nicholson et al., 2018).
In early 2020, a new Tmem106b KO mouse generated using CRISPR-Cas9 was reported that developed swollen axons, impaired lysosomal function, and accumulation of lipofuscin, ubiquitinated substrates, and autophagosomes, particularly in motor neurons (Luningschror et al., 2020). These data strongly suggest TMEM106B is important for transporting lysosomes from the axons to the soma. However, it was unclear why the phenotype of these Tmem106b KO was so starkly different than the original report by Klein et al. A trio of subsequent papers has helped resolve this issue. They find that double knockout of Tmem106b and Grn consistently produced motor deficits, neurodegeneration, lysosomal dysfunction, accumulation of autophagosomes, and robust phosphorylated TDP-43 pathology that was absent in mice missing either gene (Feng et al., 2020b; Werner et al., 2020; Zhou et al., 2020). It appears the milder phenotypes in Tmem106B deficient mice that were first reported were a result of incomplete deletion of TMEM106b expression caused by a gene trap approach, which can allow leaky expression of the full-length gene (Luningschror et al., 2020; Zhou et al., 2020). Taken together, these new studies highlight a functional link between TMEM106B, PGRN, and the lysosome, but raise questions about the viability of altering levels of TMEM106B as a therapeutic approach.
Overall, a large body of evidence demonstrates that TMEM106B directly affects lysosome function in the brain. In addition, TMEM106B functionally intersects with GRN and C9orf72 at the lysosome. The nature of these interactions is unclear but an active area of investigation. Although the precise molecular function of TMEM106B is not known, TMEM106B clearly plays a critical role in the proper sorting and transport of endosomes and lysosomes in neurons (Fig. 4). Further work is necessary to understand how variants in TMEM106B alter the risk of developing FTD and neurodegeneration in general. The importance of TMEM106B to neurodegeneration clearly warrants deeper studies to uncover the molecular function of TMEM106B.
2.5. MAPT
Microtubule-associated protein tau (MAPT), which encodes the protein tau, was the first gene identified with mutations that cause FTD (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998). Since then, over 60 dominantly inherited mutations MAPT have been identified accounting for 5%−20% of fFTD and 0%−3% of sFTD (Greaves and Rohrer, 2019; Pottier et al., 2016; Rohrer and Warren, 2011). Tau is a protein highly expressed in neurons with the ability to bind and stabilize microtubules (Murphy et al., 1977) (Fig. 5). Mutations can affect either the protein level or mRNA splicing sites, resulting in a decreased ability of tau to bind microtubules (LeBoeuf et al., 2008), or increased tendency to aggregate and form hyperphosphorylated filaments (Goedert and Spillantini, 2011; Hong et al., 1998). Furthermore, intra-neuronal accumulation of hyperphosphorylated tau in neurofibrillary tangles (NFTs) is present in 45% of all FTD cases (Halliday et al., 2012; Shi et al., 2005).
Figure 5. Tau aggregation and accumulation impairs lysosome function through multiple mechanisms.
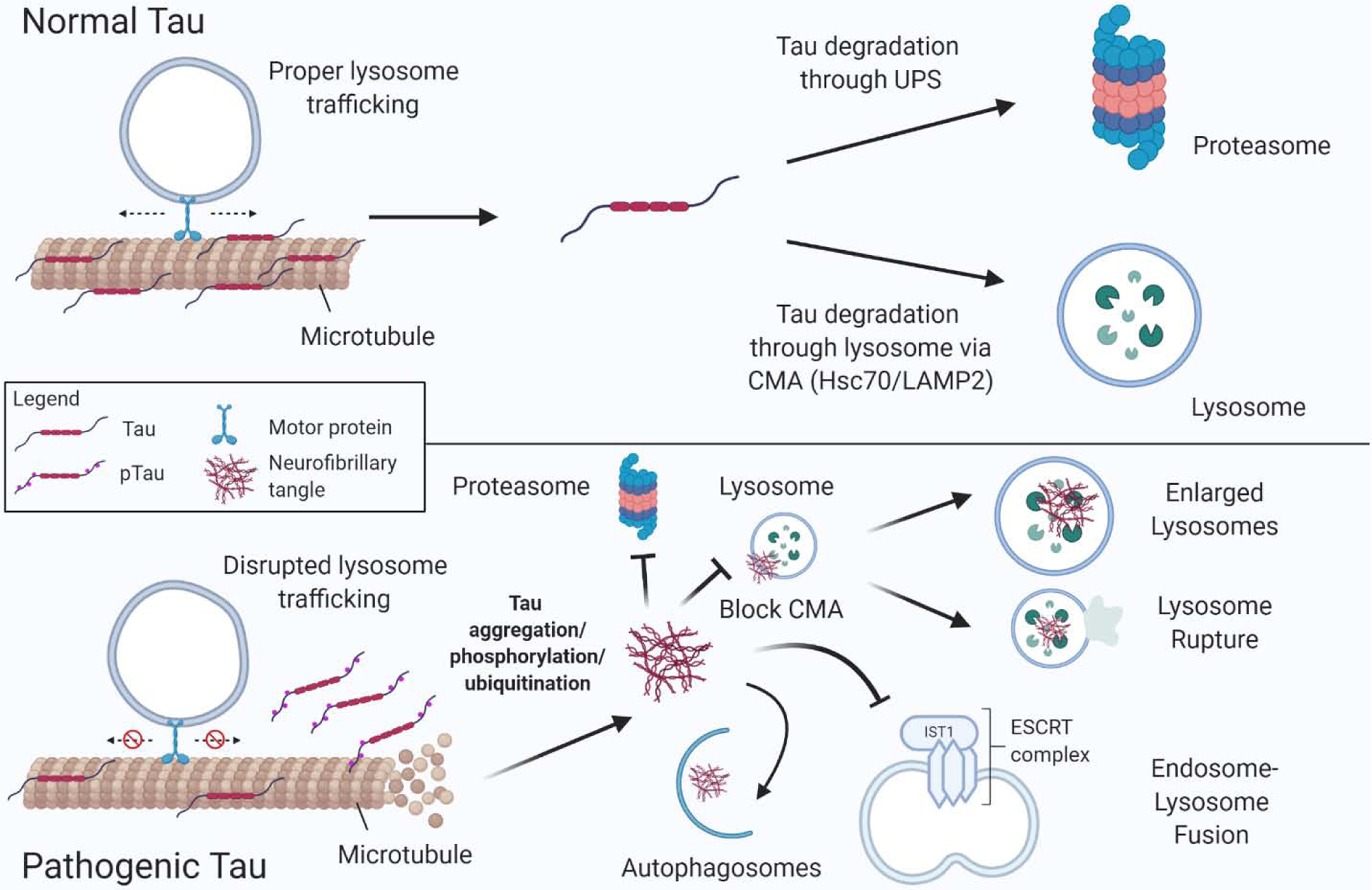
(Upper panel) In normal, healthy conditions, one function of tau is to stabilize microtubules, which facilitates proper trafficking and maintenance of lysosomes. Homeostatic levels of tau are maintained by degradation through two pathways: 1) the proteasome via the ubiquitin–proteasome system (UPS) and 2) in the lysosome through chaperone mediated autophagy (CMA) via Hsc70/LAMP2A. (Lower panel) In pathogenic conditions, such as FTD caused by MAPT mutations, tau is prone to aggregation leading to hyper-phosphorylation, ubiquitination, and microtubule destabilization. Smaller tau aggregates and larger neurofibrillary tangles (NFTs) inhibit the proteasome and are shunted to autophagosomes for degradation. Tau aggregates also inhibit IST1 of the ESCRT complex, block CMA, impair lysosome function leading to enlarged, dysfunctional lysosomes, and lysosome rupture.
Phenotypic and pathological heterogeneity within FTD-tau is common. Pathological subtypes of FTD-tau include Pick’s Disease (PiD), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and argyrophilic grain disease (AGD) (Bahia et al., 2013). Pathologically, FTD-tau brains are characterized by the intracellular accumulation of NFTs, composed of hyperphosphorylated tau, which occurs predominantly in neurons, and glial cells more rarely (Goedert and Spillantini, 2011). In addition to the presence of NTFs, there is also evidence of dysfunctional autophagy-lysosome pathways in the tissue of FTD-tau cases. The brains of patients with PSP and CBD have a marked increase in p62-positive aggregates or inclusions, and well as an increase in the autophagosome marker LC3 (Piras et al., 2016). Additionally, LAMP1 immunostaining is abnormal across FTD-tau subtypes (Bain et al., 2019; Piras et al., 2016), suggesting that protein degradation through the lysosome-autophagy pathway is compromised in FTD-tau brains. Interestingly, tau NTFs are also present in Niemann Pick type C brains, a lysosomal storage disease (Pacheco et al., 2009), further suggesting that lysosomal dysregulation contributes to tauopathy.
Tau is expressed throughout the CNS, including astrocytes and oligodendrocytes (LoPresti et al., 1995; Muller et al., 1997). Tau is highly expressed in neurons (Binder et al., 2020), and the maintenance of proper tau levels by regulating the translation and degradation of tau is critical for the function and survival of neurons. For example, aggregation of tau is toxic, and activation of autophagy facilitates the clearance of tau aggregates in tauopathy models (Mohamed et al., 2014). Treatment of JNPL3 mice, which overexpress human P301L tau (Lewis et al., 2000), with methylene blue (Congdon et al., 2012), and treatment of mice expressing P301S with trehalose (Schaeffer et al., 2012), or rapamycin (Ozcelik et al., 2013; Siman et al., 2015) decreases insoluble tau, suggesting tau is degraded through lysosome-dependent autophagy (Schaeffer and Goedert, 2012). Furthermore, tau clearance is regulated by TFEB. Overexpression of TFEB decreases NFT pathology in the Tg4510 tau mouse model and reduces phosphorylated tau in AD patient derived neurons (Binder et al., 2020; Polito et al., 2014). Recently it has been shown that treatment with lonafarnib, a farnesyltransferase inhibitor, decreases tau and phospho-tau accumulation in rTg4510 mouse brain as well as hiPSC neurons expressing MAPT with a R406W mutation. The beneficial effect of lonafarnib on tau pathology was mediated by inhibition of Rhes, which activated lysosomes and autophagy (Hernandez et al., 2019). These studies highlight the important role of the autophagy and lysosomal system in clearance and degradation of tau in cellular and animal models of tauopathy.
Several autophagy receptors are known to play a role in influencing the intraneuronal levels of tau. SQSTM1/p62, also known as sequestosome-1, is an autophagy receptor involved in lysosomal degradation of protein targets (Komatsu et al., 2007). Hyperphosphorylated tau is increased in Sqstm1 knockout mice compared to wild type mice (Ramesh Babu et al., 2008). Furthermore, the overexpression of p62 in a MAPT mouse model ameliorated insoluble neurofibrillary tangles and reduced the pathologic spreading of tau (Xu et al., 2019). Taken together, these data suggest that p62 may play a role in the autophagic degradation of pathogenic tau. Furthermore, tau can also be degraded in the lysosomes through CMA via its interaction with Hsc70 (Petrucelli et al., 2004). Tau harbors two imperfect motifs in its fourth repeat domain which are recognized by hsc70 (Wang et al., 2010b; Wang et al., 2010c). In vitro studies demonstrate that wild type, but not mutant, tau is degraded in the lysosome through CMA (Caballero et al., 2018). Moreover, tau is also a cargo of the chaperone protein Bcl-2-associated athanogene 3 (BAG3) (Lei et al., 2015), which functions as a co-chaperone in both hsc70 (Behl, 2016) and p62 (Ji et al., 2019) related lysosome-autophagic degradation pathways.
Proteins are degraded within the lysosome by a variety of lysosomal hydrolases and proteases. Lysosomal cathepsins can proteolytically cleave and help degrade tau. There is evidence that CTSD cleaves recombinant tau in vitro (Kenessey et al., 1997). Additionally, CTSL can cleave mutant tau at the lysosomal membrane, after initial processing by an unknown protease (Wang et al., 2009). Inhibition of serine and cysteine proteases, including cathepsins, with leupeptin increases tau secretion (Mohamed et al., 2014), while chloroquine treatment, which inhibits CTSB, CTSD, and CTSL, increases tau accumulation (Hamano et al., 2008), providing additional evidence that cathepsins play an important role in the degradation and clearance of tau. While there is evidence that cathepsins interact and co-localize with NTFs (Akwa et al., 2018), it remains unclear what role cathepsin cleaved tau fragments play in human tauopathies.
A major role of tau is to stabilize microtubules (MTs) and prevent depolymerization by decreasing the dissociation of tubulin subunits (Murphy et al., 1977; Trinczek et al., 1995) (Fig. 5). Several studies have found that mutant tau decreases the ability of tau to bind MT, thereby modulating the ability of the protein to stabilize and maintain homeostatic MT dynamics (Elbaum-Garfinkle et al., 2014; LeBoeuf et al., 2008). In relation to the lysosome, stable microtubules are important for the transport of autophagic vesicles and autolysosome fusion (Kochl et al., 2006), and phosphorylation of tau alters axonal transport (Rodriguez-Martin et al., 2013). Mature autophagosomes traffic to the soma where they merge with lysosomes to form autolysosomes (Maday et al., 2012). Inhibition of autophagy has been shown to change the primary localization of tau containing vesicles from the soma to neurites (Akwa et al., 2018). Furthermore, studies of transgenic mice expressing FTD-tau related mutations found an enlargement of lysosomal compartments and an increase in autophagic vesicles, suggesting a dysregulation in autophagic flux (Lim et al., 2001; Lin et al., 2003).
Several components of the endosomal sorting complex required for transport (ESCRT) are implicated in tau pathology. Different species of tau traffic through the endosomal system (Michel et al., 2014; Wu et al., 2013). Further, impaired endosome function, via the simultaneous knockdown of CHMP6 or CHMP2A and CHMP2B, leads to the escape of tau seeds, causing increased tau aggregation, and formation of galectin-3 positive puncta, which is indicative of lysosomal damage (Chen et al., 2019). Interestingly, mutations in CHMP2B itself can cause FTD (Skibinski et al., 2005). Modulation of the protein RAB7A, another protein involved in lysosomal biogenesis and endosome-lysosome formation, regulates tau secretion (Rodriguez et al., 2017). Furthermore, it was recently shown that the accumulation of tau inhibits IST1, a component of the ESCRT pathway important for autophagosome and lysosome fusion (Feng et al., 2020a). Taken together these data suggest that vesicle fusion and trafficking could influence tau pathology and lead to escape of tau seeds and tau aggregate formation. Tau aggregates and NFTs may also directly impair the endo-lysosomal pathway, causing additional neurotoxicity.
Although MAPT mutations have only been rarely identified in patients with classic ALS (Origone et al., 2018), MAPT mutations have been implicated in a family with lower motor neuron disease (Di Fonzo et al., 2014). Furthermore, several recent studies have proposed that mutations in MAPT increase the risk of developing ALS and increase disease severity. In a cohort of Han Chinese patients with sporadic ALS, an analysis of MAPT UTRs and introns 9–12 identified two variants rs105788 A > G and rs123972 T > A that were enriched in ALS cases, but not controls (Fang et al., 2013). These variants were associated with an earlier age of disease onset and a higher frequency of bulbar palsy and breathing difficulties, however further studies are required to investigate a possible link to lysosomal function.
One study of 3540 European ALS patients found no association between disease and the MAPT H1/H2 haplotype (Taes et al., 2010). However, a larger study identified an association between the H1 haplotype and disease using eQTL analysis of several GWAS studies carried out on European cohorts of several ALS-FTD spectrum disorders (Karch et al., 2018). They found that the MAPT haplotype H1 increased ALS risk, particularly variant rs199533, which is in high linkage disequilibrium (D=0.97) with the identifying variant for the H1 haplotype rs7224296. Both variants and the MAPT H1 haplotype are associated with alternative splicing of MAPT exons 2 and 3 leading to decreased MAPT mRNA with exons 2 and 3 (Karch et al., 2018).
The H1 haplotype is also associated with several other neurodegenerative disorders with evidence of lysosomal dysfunction, including FTD, Parkinson’s Disease, ALS/Parkinson’s complex from Guam ALS/Parkinson’s complex from Guam, and Alzheimer’s Disease (Caparros-Lefebvre et al., 2002; Martin et al., 2001; Sanchez-Juan et al., 2019; Yokoyama et al., 2017). Further study is needed to investigate the relationship between the H1 haplotype, neurodegeneration, and autophagy. While tau is not thought to have a normal function within the lysosome, tau aggregates are degraded through the lysosome-autophagy pathway. Additionally, the accumulation of tau aggregates and NFTs are associated with dysregulated autophagy and abnormal lysosomal morphology in human tissue. Furthermore, tau may indirectly influence the activity, biogenesis, and trafficking of lysosomes through its action on microtubules and the cytoskeleton (Fig. 5).
2.6. Additional genes implicating lysosome dysfunction in FTD and ALS.
In addition to the genes discussed previously, there are several genes (Table 1) that harbor rare mutations, accounting for less than 5% of fFTD and less than 1% of fALS cases, that play important roles in the formation, trafficking, or function of lysosomes (Cirulli et al., 2015; Greaves and Rohrer, 2019). Those genes are CHMP2B (Lindquist et al., 2008; Skibinski et al., 2005), valosin containing protein (VCP/p97) (Johnson et al., 2010b), SQSTM1/p62 (Hardy and Rogaeva, 2014; Le Ber et al., 2013), cathepsin F (CTSF) (van der Zee et al., 2016), and major facilitator superfamily domain 8 (MFSD8) (Geier et al., 2019). We will briefly discuss their role in the lysosome below.
CHMP2B is a component of the endosomal sorting complex required for transport-III (ESCRT-III), which is critical for the formation of endocytic multi-vesicular bodies (Tsang et al., 2006). Like FTD-GRN cases, human brain tissue from FTD patients with CHMP2B mutations have pathologic lipofuscin accumulation in the frontal cortex. Furthermore, disruption of CHMP2B can prevent the recruitment of the late endosomal protein Rab7, which inhibits the maturation of endosomes to lysosomes (Urwin et al., 2010). In addition, a mouse model of FTD-CHMP2B displays LAMP1 and LAMP2 enriched intraneuronal inclusions and lipofuscin, which is indicative of lysosome dysfunction (Clayton et al., 2015).
VCP is a ubiquitously expressed protein that is a member of the AAA+ (ATPases Associated with diverse cellular Activities) family involved in DNA replication, cell cycle regulation, organelle biogenesis, and protein degradation (Kakizuka, 2008; van den Boom and Meyer, 2018). VCP is involved in the extraction of ubiquitinated substrates from membranes and participates in the sorting of proteins to the lysosome as well as clearance of damaged lysosomes (Arhzaouy et al., 2019; Papadopoulos et al., 2017; Ramanathan and Ye, 2012; Ritz et al., 2011). Mutations in VCP can cause various autosomal-dominant neurodegenerative disorders (Tang and Xia, 2016) including FTD (Saracino et al., 2018; Wong et al., 2018) and ALS (Abramzon et al., 2012; Johnson et al., 2010a; Koppers et al., 2012). VCP mutations were first discovered to cause inclusion body myopathy (IBM) with Paget’s disease of the bone (PDB) and frontotemporal dementia, abbreviated IBMPFD, but now more commonly called multisystem proteinopathy (MSP) (Korb et al.; Watts et al., 2004) In vitro studies suggest that VCP mutations underlying MSP lead to a toxic gain-of-function by accelerating protein unfoldase activity (Blythe et al., 2019). Ubiquitinated TDP-43 inclusions are the predominant neuropathology found in FTD or ALS associated with VCP mutations (Matsubara et al.; Neumann et al., 2007b). However, a novel mutation in VCP (p.Asp395Gly) was described recently that causes fFTD with a unique pathology defined by accumulation of neuronal vacuoles and neurofibrillary tau tangles, not TDP-43 (Darwich et al., 2020). Intriguingly, a VCP-binding co-factor named SVIP (Small VCP-Interacting Protein) was recently discovered that recruits VCP to lysosomes and is important for lysosome homeostasis (Johnson et al., 2021). Moreover, the authors found a SVIP mutation in an FTD patient that was pathogenic in a Drosophila model system. Although this mutation was from a single FTD patient, and needs to be replicated in larger genetic studies, the mechanistic link between VCP, SVIP, and lysosome function is compelling.
Interestingly, patients with SQSTM1/p62 mutations may also develop PDB (Hocking et al., 2002; Laurin et al., 2002). p62 is a scaffold protein and was the first autophagy receptor identified in mammals (Bjorkoy et al., 2005). p62 interacts with misfolded or ubiquitinated proteins and targets them for degradation through autophagic and proteasomal pathways (Long et al., 2008; Tan et al., 2008). p62 also plays a role in autophagosome formation (Su and Wang, 2011) and mTORC1 regulation (Jain et al., 2010). Notably, rare mutations in SQSTM1/p62 can cause FTD and present with FTD-TDP pathology (Kovacs et al., 2016).
Mutations in CTSF (van der Zee et al., 2016) and MFSD8 (Geier et al., 2019) are the most recent genes encoding lysosomal proteins to be linked to FTD. CTSF encodes cathepsin F, a lysosomal cysteine protease (Tang et al., 2006; Wang et al., 1998). Homozygous mutations in CTSF were found to cause autosomal recessive adult-onset neuronal ceroid lipofuscinosis (ANCL or Kufs disease [KD]) in a Belgian family. Whereas a heterozygous CTSF p.Arg245His mutation was found in two FTD patients (van der Zee et al., 2016). The neuropathology of one CTSF mutation case revealed accumulation of autofluorescent lipofuscin, abnormal neuronal axons, and hyperphosphorylated tau. Subsequently, a missense variant (p.L465W) in trans with a macro-deletion in CTSF were discovered in an individual diagnosed with FTD (Blauwendraat et al., 2018). Additional studies are needed to validate the pathogenicity of these specific CTSF mutations. Nevertheless, when considered together, these studies support the concept that mutations in CTSF may contribute to the development of FTD.
MFSD8 encodes the Major Facilitator Superfamily Domain Containing 8 (MFSD8) protein, which is a ubiquitous integral membrane protein of unknown function that localizes to the lysosome (Siintola et al., 2007). MFSD8 mutations linked to FTD cause an increase in lysosomal proteins, such as LAMP2 and CTSD, accumulation of autophagosomes, and a decrease in the rate of protein degradation (Geier et al., 2019). Notably, homozygous loss-of-function mutations in both CTSF and MFSD8 cause NCL (Aiello et al., 2009; Smith et al., 2013) providing further evidence that lysosomal dysfunction can cause FTD.
In addition to the genes detailed in the preceding sections, a few additional genes of note involved in lysosome/autophagy function have been implicated in FTD or ALS pathogenesis including optineurin (OPTN), tank-binding kinase 1 (TBK1), and ubiquilin 2 (UBQLN2). The function of the proteins encoded by these three genes will be briefly discussed here, but have reviewed in more detail recently (Mathis et al., 2019; Nguyen et al., 2018b; Sirkis et al., 2019). Optineurin (OPTN) is a multi-functional adaptor protein that regulates vesicular trafficking, intracellular signaling, and binds ubiquitinated cargo proteins to facilitate autophagy (Ryan and Tumbarello, 2018). OPTN has also been shown to play an important role in clearance of damaged lysosomes that functions to restore lysosomal quality control (Bussi et al., 2018). Tank-binding kinase 1 (TBK1) is a ubiquitously expressed multi-functional serine-threonine kinase, belonging to the family of non-canonical IκB kinase (IKK) (Fitzgerald et al., 2003), that plays many important roles, including the initiation and completion of autophagy (Pilli et al., 2012). In fact, TBK1 phosphorylates OPTN to regulate autophagy as well as recycling of damaged lysosomes (Bussi et al., 2018). Ubiquilin 2 (UBQLN2) is a member of the ubiquitin-like protein family (ubiquilins). UBQLN2 binds ubiquitinated proteins through a C-terminal ubiquitin-associated (UBA) domain and an N-terminal ubiquitin-like (UBL) domain and facilitates protein degradation through the UPS (Seok Ko et al., 2004). Recently, UBQLN2 has been found to regulate mTOR activity, autophagy, and lysosomal acidification, which is impaired by UBQLN2 mutations that cause ALS (Şentürk et al., 2019). Our understanding of the role of the lysosome and autophagy in FTD and ALS continues to develop and novel genes encoding proteins involved in these processes are still being identified (Nakamura et al., 2020). Taken together, these findings present additional evidence that genetic mutations that cause FTD or ALS initiate disease through defects in the lysosome-autophagy pathway.
3. Conclusion
After decades of research, it is increasingly clear that lysosomal dysfunction is an important pathogenic mechanism leading to neurodegeneration in FTD and ALS. Multiple genes that cause, or increase the risk of developing, FTD, ALS, or a combined FTD-ALS phenotype, functionally converge at the lysosome. For example, PGRN, granulins, TMEM106B, MFSD8, CTSF, and C9orf72, all localize within the lumen or membrane of the lysosome. Thus, mutations or altered function of these proteins could disrupt lysosomal homeostasis and contribute to neurodegeneration. When considered together, many of the proteins we have summarized contribute to lysosome homeostasis through modulation of lysosomal pH, lysosomal trafficking, lysosomal fusion, or activity of hydrolases within the lysosome.
The impairment of lysosome function and protein clearance over time could cause the accumulation of protein aggregates (i.e. TDP-43), which are defining neuropathological features of FTD and ALS, and decrease neuronal and glial health and survival (Boland et al., 2018). The precise mechanisms leading from lysosomal impairments in FTD or ALS to overt neurodegeneration are beginning to be elucidated but are still an active area of study. In particular, multiple possibilities have been proposed that suggest lysosome dysfunction can directly or indirectly lead to synaptic defects and impaired synaptic transmission (Para et al., 2020), impaired proteostasis and increased proteotoxicity (Hipp et al., 2019), autophagic blockade (Darios and Stevanin, 2020; Menzies et al., 2017), mitochondrial impairment (Moore and Holzbaur, 2016; Plotegher and Duchen, 2017; Richter et al., 2016), impaired nucleocytoplasmic transport (Chou et al., 2018), and neuroinflammation (Fiorenza et al., 2018; Huang et al., 2020; Zhang et al., 2020), which may ultimately cause neurodegeneration. Despite the complicated downstream pathways, numerous studies support the idea that activation of lysosome and autophagy can help clear protein aggregates found in FTD and ALS, and improve neuronal survival (Barmada et al., 2014; Castillo et al., 2013; Chen et al., 2020; Congdon et al., 2012; Holler et al., 2016; Ozcelik et al., 2013; Schaeffer et al., 2012; Wang et al., 2012; West et al., 2020). Future studies should focus on developing therapeutic interventions that restore lysosomal health and function as a method to promote neuron survival and prevent neurodegeneration in FTD and ALS (Bonam et al., 2019; Chen et al., 2020; Djajadikerta et al., 2020; Peng et al., 2019).
Acknowledgements
We thank the generous support that made this work possible including the National Institutes of Health (NIH)/NINDS grants (R01 NS093362, R01 NS105971), a New Vision Award (Donors Cure Foundation), an Emory Alzheimer’s Disease Center Pilot Grant P50AG025688, the Alzheimer’s Drug Discovery Foundation, the Association for Frontotemporal Degeneration (ADDF/AFTD), the Bluefield Project to Cure Frontotemporal Dementia, and the BrightFocus Foundation. We apologize to any colleagues that were not cited due to space and time constraints.
Abbreviations
- UPS
ubiquitin-proteasome system
- CMA
chaperone mediated autophagy
- NCL
neuronal ceroid lipofuscinosis
- CLEAR
Coordinated Lysosomal Expression and Regulation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
4. References
- Abo-Rady M, et al. , 2020. Knocking out C9ORF72 Exacerbates Axonal Trafficking Defects Associated with Hexanucleotide Repeat Expansion and Reduces Levels of Heat Shock Proteins. Stem Cell Reports. 14, 390–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramzon Y, et al. , 2012. Valosin-containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosis. Neurobiology of Aging. 33, 2231.e1–2231.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramzon YA, et al. , 2020. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Frontiers in Neuroscience. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z, et al. , 2010. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol. 177, 311–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello C, et al. , 2009. Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum Mutat. 30, E530–40. [DOI] [PubMed] [Google Scholar]
- Akwa Y, et al. , 2018. Synaptic activity protects against AD and FTD-like pathology via autophagic-lysosomal degradation. Mol Psychiatry. 23, 1530–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Chalabi A, et al. , 2016. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 15, 1182–94. [DOI] [PubMed] [Google Scholar]
- Almeida MR, et al. , 2016. Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging. 41, 200 e1–200 e5. [DOI] [PubMed] [Google Scholar]
- Amick J, Ferguson SM, 2017. C9orf72: At the intersection of lysosome cell biology and neurodegenerative disease. Traffic. 18, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amick J, et al. , 2016. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell. 27, 3040–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amick J, et al. , 2018. WDR41 supports lysosomal response to changes in amino acid availability. Mol Biol Cell. 29, 2213–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amick J, et al. , 2020. PQLC2 recruits the C9orf72 complex to lysosomes in response to cationic amino acid starvation. J Cell Biol. 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, et al. , 2006. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 351, 602–11. [DOI] [PubMed] [Google Scholar]
- Araki W, et al. , 2014. Disease-associated mutations of TDP-43 promote turnover of the protein through the proteasomal pathway. Mol Neurobiol. 50, 1049–58. [DOI] [PubMed] [Google Scholar]
- Arhzaouy K, et al. , 2019. VCP maintains lysosomal homeostasis and TFEB activity in differentiated skeletal muscle. Autophagy. 15, 1082–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrant AE, et al. , 2018. Partial Tmem106b reduction does not correct abnormalities due to progranulin haploinsufficiency. Mol Neurodegener. 13, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrant AE, et al. , 2019. Impaired beta-glucocerebrosidase activity and processing in frontotemporal dementia due to progranulin mutations. Acta Neuropathol Commun. 7, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano T, et al. , 2011. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol. 31, 2040–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasio A, et al. , 2016. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 6, 23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avendano-Vazquez SE, et al. , 2012. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 26, 1679–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala YM, et al. , 2011. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30, 277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala YM, et al. , 2005. Human, Drosophila, and C.elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol. 348, 575–88. [DOI] [PubMed] [Google Scholar]
- Ayala YM, et al. , 2008. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 121, 3778–85. [DOI] [PubMed] [Google Scholar]
- Bahia VS, et al. , 2013. Neuropathology of frontotemporal lobar degeneration: a review. Dement Neuropsychol. 7, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain HDC, et al. , 2019. The role of lysosomes and autophagosomes in frontotemporal lobar degeneration. Neuropathol Appl Neurobiol. 45, 244–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, et al. , 2006. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 442, 916–9. [DOI] [PubMed] [Google Scholar]
- Balderhaar HJ, Ungermann C, 2013. CORVET and HOPS tethering complexes - coordinators of endosome and lysosome fusion. J Cell Sci. 126, 1307–16. [DOI] [PubMed] [Google Scholar]
- Balendra R, Isaacs AM, 2018. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. 14, 544–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabio A, Bonifacino JS, 2019. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nature Reviews Molecular Cell Biology. 21, 101–118. [DOI] [PubMed] [Google Scholar]
- Bang J, et al. , 2015. Frontotemporal dementia. Lancet. 386, 1672–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, et al. , 2014. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 10, 677–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, et al. , 2010. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 30, 639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Bennett HP, 1998. Granulins: the structure and function of an emerging family of growth factors. J Endocrinol. 158, 145–51. [DOI] [PubMed] [Google Scholar]
- Bateman A, Bennett HP, 2009. The granulin gene family: from cancer to dementia. Bioessays. 31, 1245–54. [DOI] [PubMed] [Google Scholar]
- Bateman A, et al. , 2018. A Brief Overview of Progranulin in Health and Disease. Methods Mol Biol. 1806, 3–15. [DOI] [PubMed] [Google Scholar]
- Beel S, et al. , 2017. Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum Mol Genet. 26, 2850–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl C, 2016. Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharmacol Sci. 37, 672–688. [DOI] [PubMed] [Google Scholar]
- Belcastro V, et al. , 2011. Transcriptional gene network inference from a massive dataset elucidates transcriptome organization and gene function. Nucleic Acids Res. 39, 8677–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benussi L, et al. , 2013. Estimating the age of the most common Italian GRN mutation: walking back to Canossa times. J Alzheimers Dis. 33, 69–76. [DOI] [PubMed] [Google Scholar]
- Berg TO, et al. , 1998. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem. 273, 21883–92. [DOI] [PubMed] [Google Scholar]
- Binder JL, et al. , 2020. Optical induction of autophagy via Transcription factor EB (TFEB) reduces pathological tau in neurons. PLoS One. 15, e0230026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, et al. , 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 171, 603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwendraat C, et al. , 2018. The wide genetic landscape of clinical frontotemporal dementia: systematic combined sequencing of 121 consecutive subjects. Genet Med. 20, 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe EE, et al. , 2019. Multisystem Proteinopathy Mutations in VCP/p97 Increase NPLOC4·UFD1L Binding and Substrate Processing. Structure. 27, 1820–1829.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin M, et al. , 2020. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 39, e100574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, et al. , 2018. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov. 17, 660–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonam SR, et al. , 2020. Progress and Challenges in the Use of MAP1LC3 as a Legitimate Marker for Measuring Dynamic Autophagy In Vivo. Cells. 9, 1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonam SR, et al. , 2019. Lysosomes as a therapeutic target. Nat Rev Drug Discov. 18, 923–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman EJ, et al. , 1988. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 85, 7972–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady OA, et al. , 2013. The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet. 22, 685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady OA, et al. , 2014. Regulated intramembrane proteolysis of the frontotemporal lobar degeneration risk factor, TMEM106B, by signal peptide peptidase-like 2a (SPPL2a). J Biol Chem. 289, 19670–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braulke T, Bonifacino JS, 2009. Sorting of lysosomal proteins. Biochim Biophys Acta. 1793, 605–14. [DOI] [PubMed] [Google Scholar]
- Brown RH Jr., Al-Chalabi A, 2017. Amyotrophic Lateral Sclerosis. N Engl J Med. 377, 1602. [DOI] [PubMed] [Google Scholar]
- Burberry A, et al. , 2016. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 8, 347ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burberry A, et al. , 2020. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature. 582, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch JI, et al. , 2013. Expression of TMEM106B, the frontotemporal lobar degeneration-associated protein, in normal and diseased human brain. Acta Neuropathol Commun. 1, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch JI, et al. , 2016. Increased expression of the frontotemporal dementia risk factor TMEM106B causes C9orf72-dependent alterations in lysosomes. Hum Mol Genet. 25, 2681–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi C, et al. , 2018. Alpha-synuclein fibrils recruit TBK1 and OPTN to lysosomal damage sites and induce autophagy in microglial cells. J Cell Sci. 131, jcs226241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler VJ, et al. , 2019. Age- and stress-associated C. elegans granulins impair lysosomal function and induce a compensatory HLH-30/TFEB transcriptional response. PLoS Genet. 15, e1008295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero B, et al. , 2018. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell. 17, e12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, et al. , 2009. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J Biol Chem. 284, 27416–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, et al. , 2007. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 171, 227–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canafoglia L, et al. , 2014. Recurrent generalized seizures, visual loss, and palinopsia as phenotypic features of neuronal ceroid lipofuscinosis due to progranulin gene mutation. Epilepsia. 55, e56–9. [DOI] [PubMed] [Google Scholar]
- Cannon A, et al. , 2013. Clinicopathologic variability of the GRN A9D mutation, including amyotrophic lateral sclerosis. Neurology. 80, 1771–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canuel M, et al. , 2008. Sortilin mediates the lysosomal targeting of cathepsins D and H. Biochem Biophys Res Commun. 373, 292–7. [DOI] [PubMed] [Google Scholar]
- Caparros-Lefebvre D, et al. , 2002. Guadeloupean parkinsonism: a cluster of progressive supranuclear palsy-like tauopathy. Brain. 125, 801–11. [DOI] [PubMed] [Google Scholar]
- Caroppo P, et al. , 2016. Defining the spectrum of frontotemporal dementias associated with TARDBP mutations. Neurol Genet. 2, e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casterton RL, et al. , 2020. Pathomechanism Heterogeneity in the Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Disease Spectrum: Providing Focus Through the Lens of Autophagy. J Mol Biol. 432, 2692–2713. [DOI] [PubMed] [Google Scholar]
- Castillo K, et al. , 2013. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 9, 1308–20. [DOI] [PubMed] [Google Scholar]
- Chang KH, et al. , 2013. Association between GRN rs5848 polymorphism and Parkinson’s disease in Taiwanese population. PLoS One. 8, e54448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MC, et al. , 2017. Progranulin deficiency causes impairment of autophagy and TDP-43 accumulation. J Exp Med. 214, 2611–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, et al. , 2011. Genetic and clinical features of progranulin-associated frontotemporal lobar degeneration. Arch Neurol. 68, 488–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, et al. , 2012. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci. 32, 11213–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, et al. , 2019. Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J Biol Chem. 294, 18952–18966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, et al. , 2013. Genetics of amyotrophic lateral sclerosis: an update. Mol Neurodegener. 8, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, et al. , 2020. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol Neurodegener. 15, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, et al. , 2018. Progranulin associates with hexosaminidase A and ameliorates GM2 ganglioside accumulation and lysosomal storage in Tay-Sachs disease. J Mol Med (Berl). 96, 1359–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, et al. , 2015. Association of progranulin polymorphism rs5848 with neurodegenerative diseases: a meta-analysis. J Neurol. 262, 814–22. [DOI] [PubMed] [Google Scholar]
- Chiang HL, et al. , 1989. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 246, 382–5. [DOI] [PubMed] [Google Scholar]
- Chio A, et al. , 2012. Extensive genetics of ALS: a population-based study in Italy. Neurology. 79, 1983–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CC, et al. , 2018. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 21, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, et al. , 2015. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 347, 1436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciura S, et al. , 2013. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. 74, 180–7. [DOI] [PubMed] [Google Scholar]
- Clayton EL, et al. , 2015. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol. 130, 511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon EE, et al. , 2012. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 8, 609–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrionero A, Horvitz HR, 2018. A C9orf72 ALS/FTD Ortholog Acts in Endolysosomal Degradation and Lysosomal Homeostasis. Curr Biol. 28, 1522–1535 e5. [DOI] [PubMed] [Google Scholar]
- Cruchaga C, et al. , 2011. Association of TMEM106B Gene Polymorphism With Age at Onset in Granulin Mutation Carriers and Plasma Granulin Protein Levels. Archives of Neurology. 68, 581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, et al. , 2006. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 442, 920–4. [DOI] [PubMed] [Google Scholar]
- Dang TN, et al. , 2014. Increased metal content in the TDP-43(A315T) transgenic mouse model of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Front Aging Neurosci. 6, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel R, et al. , 2000. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 48, 999–1009. [DOI] [PubMed] [Google Scholar]
- Darios F, Stevanin G, 2020. Impairment of Lysosome Function and Autophagy in Rare Neurodegenerative Diseases. J Mol Biol. 432, 2714–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwich NF, et al. , 2020. Autosomal dominant VCP hypomorph mutation impairs disaggregation of PHF-tau. Science. eaay8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Duve C, 2005. The lysosome turns fifty. Nat Cell Biol. 7, 847–9. [DOI] [PubMed] [Google Scholar]
- De Muynck L, et al. , 2013. The neurotrophic properties of progranulin depend on the granulin E domain but do not require sortilin binding. Neurobiol Aging. 34, 2541–7. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, et al. , 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 72, 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bo R, et al. , 2011. No major progranulin genetic variability contribution to disease etiopathogenesis in an ALS Italian cohort. Neurobiol Aging. 32, 1157–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleon J, Miller BL, 2018. Frontotemporal dementia. Handb Clin Neurol. 148, 409–430. [DOI] [PubMed] [Google Scholar]
- Deming Y, Cruchaga C, 2014. TMEM106B: a strong FTLD disease modifier. Acta Neuropathol. 127, 419–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, et al. , 2011. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 477, 211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, et al. , 2013. Defects in RNA Metabolism links FTD and ALS Pathogenesis: TDP-43, FUS, and C9orf72. Current Enzyme Inhibition. 9, 28–40. [Google Scholar]
- Derocq D, et al. , 2012. Cathepsin D is partly endocytosed by the LRP1 receptor and inhibits LRP1-regulated intramembrane proteolysis. Oncogene. 31, 3202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo A, et al. , 2014. Lower motor neuron disease with respiratory failure caused by a novel MAPT mutation. Neurology. 82, 1990–8. [DOI] [PubMed] [Google Scholar]
- Diaper DC, et al. , 2013. Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum Mol Genet. 22, 1539–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I, Elazar Z, 2018. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 19, 349–364. [DOI] [PubMed] [Google Scholar]
- Djajadikerta A, et al. , 2020. Autophagy Induction as a Therapeutic Strategy for Neurodegenerative Diseases. J Mol Biol. 432, 2799–2821. [DOI] [PubMed] [Google Scholar]
- Dodge JC, et al. , 2015. Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 112, 8100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, et al. , 2013. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 80, 415–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S, et al. , 2014. Tau mutants bind tubulin heterodimers with enhanced affinity. Proc Natl Acad Sci U S A. 111, 6311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkkinen MG, et al. , 2018. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb Perspect Biol. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers BM, et al. , 2017. Lipidomic and Transcriptomic Basis of Lysosomal Dysfunction in Progranulin Deficiency. Cell Rep. 20, 2565–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber I, et al. , 2017. A new phenotype associated with homozygous GRN mutations: complicated spastic paraplegia. Eur J Neurol. 24, e3–e4. [DOI] [PubMed] [Google Scholar]
- Fang P, et al. , 2013. MAPT as a predisposing gene for sporadic amyotrophic lateral sclerosis in the Chinese Han population. Neural Regen Res. 8, 3116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farg MA, et al. , 2014. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 23, 3579–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele AO, Proud CG, 2020. Chloroquine and bafilomycin A mimic lysosomal storage disorders and impair mTORC1 signalling. Biosci Rep. 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, et al. , 2020a. MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: a vicious cycle in Alzheimer neurodegeneration. Autophagy. 16, 641–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T, et al. , 2020b. Loss of TMEM106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep. 21, e50219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrini S, et al. , 1989. Morphologic and functional characterization of human peripheral blood T cells expressing the T cell receptor gamma/delta. Eur J Immunol. 19, 1183–8. [DOI] [PubMed] [Google Scholar]
- Filiano AJ, et al. , 2013. Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J Neurosci. 33, 5352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch N, et al. , 2009. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 132, 583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch N, et al. , 2011. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 76, 467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorenza MT, et al. , 2018. The pathogenesis of lysosomal storage disorders: beyond the engorgement of lysosomes to abnormal development and neuroinflammation. Hum Mol Genet. 27, R119–R129. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, et al. , 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 4, 491–6. [DOI] [PubMed] [Google Scholar]
- Frick P, et al. , 2018. Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol Commun. 6, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti D, et al. , 2018. Progranulin plasma levels predict the presence of GRN mutations in asymptomatic subjects and do not correlate with brain atrophy: results from the GENFI study. Neurobiol Aging. 62, 245 e9–245 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MD, et al. , 2017. A Dementia-Associated Risk Variant near TMEM106B Alters Chromatin Architecture and Gene Expression. Am J Hum Genet. 101, 643–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, et al. , 2018. Pathomechanisms of TDP-43 in neurodegeneration. J Neurochem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass J, et al. , 2006. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Human Molecular Genetics. 15, 2988–3001. [DOI] [PubMed] [Google Scholar]
- Gass J, et al. , 2012. Progranulin regulates neuronal outgrowth independent of sortilin. Mol Neurodegener. 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geier EG, et al. , 2019. Rare variants in the neuronal ceroid lipofuscinosis gene MFSD8 are candidate risk factors for frontotemporal dementia. Acta Neuropathol. 137, 71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghidoni R, et al. , 2008. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. 71, 1235–9. [DOI] [PubMed] [Google Scholar]
- Ghosh P, et al. , 2003. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol. 4, 202–12. [DOI] [PubMed] [Google Scholar]
- Gijselinck I, et al. , 2012. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 11, 54–65. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, 2011. Pathogenesis of the tauopathies. J Mol Neurosci. 45, 425–31. [DOI] [PubMed] [Google Scholar]
- Gordon PB, Seglen PO, 1988. Prelysosomal convergence of autophagic and endocytic pathways. Biochemical and Biophysical Research Communications. 151, 40–47. [DOI] [PubMed] [Google Scholar]
- Gotzl JK, et al. , 2018. Early lysosomal maturation deficits in microglia triggers enhanced lysosomal activity in other brain cells of progranulin knockout mice. Mol Neurodegener. 13, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotzl JK, et al. , 2014. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 127, 845–60. [DOI] [PubMed] [Google Scholar]
- Greaves CV, Rohrer JD, 2019. An update on genetic frontotemporal dementia. J Neurol. 266, 2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guicciardi ME, et al. , 2004. Lysosomes in cell death. Oncogene. 23, 2881–90. [DOI] [PubMed] [Google Scholar]
- Guo W, et al. , 2020. The multifaceted role of kinases in amyotrophic lateral sclerosis: genetic, pathological and therapeutic implications. Brain. 143, 1651–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, et al. , 2016. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 17, 383–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday G, et al. , 2012. Mechanisms of disease in frontotemporal lobar degeneration: gain of function versus loss of function effects. Acta Neuropathol. 124, 373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamano T, et al. , 2008. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci. 27, 1119–30. [DOI] [PubMed] [Google Scholar]
- Hardy J, Rogaeva E, 2014. Motor neuron disease and frontotemporal dementia: sometimes related, sometimes not. Exp Neurol. 262 Pt B, 75–83. [DOI] [PubMed] [Google Scholar]
- Harms E, et al. , 1981. Lysosomal pool of free-amino acids. Biochem Biophys Res Commun. 99, 830–6. [DOI] [PubMed] [Google Scholar]
- Hebron ML, et al. , 2013. Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J Biol Chem. 288, 4103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller C, et al. , 2020. Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia. J Neurol Neurosurg Psychiatry. 91, 263–270. [DOI] [PubMed] [Google Scholar]
- Henne WM, et al. , 2011. The ESCRT pathway. Dev Cell. 21, 77–91. [DOI] [PubMed] [Google Scholar]
- Hernandez I, et al. , 2019. A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci Transl Med. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog JJ, et al. , 2017. TDP-43 misexpression causes defects in dendritic growth. Sci Rep. 7, 15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, et al. , 2020. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 21, 421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highley JR, et al. , 2014. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol Appl Neurobiol. 40, 670–85. [DOI] [PubMed] [Google Scholar]
- Higuchi-Sanabria R, et al. , 2020. Lysosomal recycling of amino acids affects ER quality control. Sci Adv. 6, eaaz9805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp MS, et al. , 2019. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 20, 421–435. [DOI] [PubMed] [Google Scholar]
- Ho WY, et al. , 2019. The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy. 15, 827–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking LJ, et al. , 2002. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet. 11, 2735–9. [DOI] [PubMed] [Google Scholar]
- Hogan DB, et al. , 2016. The Prevalence and Incidence of Frontotemporal Dementia: a Systematic Review. Can J Neurol Sci. 43 Suppl 1, S96–S109. [DOI] [PubMed] [Google Scholar]
- Hokkanen SRK, et al. , 2020. Putative risk alleles for LATE-NC with hippocampal sclerosis in population-representative autopsy cohorts. Brain Pathol. 30, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler CJ, et al. , 2017. Intracellular Proteolysis of Progranulin Generates Stable, Lysosomal Granulins that Are Haploinsufficient in Patients with Frontotemporal Dementia Caused by GRN Mutations. eNeuro. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler CJ, et al. , 2016. Trehalose upregulates progranulin expression in human and mouse models of GRN haploinsufficiency: a novel therapeutic lead to treat frontotemporal dementia. Mol Neurodegener. 11, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, et al. , 1998. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 282, 1914–7. [DOI] [PubMed] [Google Scholar]
- Hopfner F, et al. , 2020. Rare Variants in Specific Lysosomal Genes Are Associated With Parkinson’s Disease. Movement Disorders. 35, 1245–1248. [DOI] [PubMed] [Google Scholar]
- Hu F, et al. , 2010. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 68, 654–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, et al. , 2014. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J Cell Sci. 127, 3024–38. [DOI] [PubMed] [Google Scholar]
- Huang M, et al. , 2020. Network analysis of the progranulin-deficient mouse brain proteome reveals pathogenic mechanisms shared in human frontotemporal dementia caused by GRN mutations. Acta Neuropathologica Communications. 8, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huin V, et al. , 2020. Homozygous GRN mutations: new phenotypes and new insights into pathological and molecular mechanisms. Brain. 143, 303–319. [DOI] [PubMed] [Google Scholar]
- Hulkova H, et al. , 2001. A novel mutation in the coding region of the prosaposin gene leads to a complete deficiency of prosaposin and saposins, and is associated with a complex sphingolipidosis dominated by lactosylceramide accumulation. Hum Mol Genet. 10, 927–40. [DOI] [PubMed] [Google Scholar]
- Hutton M, et al. , 1998. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 393, 702–5. [DOI] [PubMed] [Google Scholar]
- Iacoangeli A, et al. , 2019. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol Commun. 7, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiura H, et al. , 2012. C9ORF72 repeat expansion in amyotrophic lateral sclerosis in the Kii peninsula of Japan. Arch Neurol. 69, 1154–8. [DOI] [PubMed] [Google Scholar]
- Iyer S, et al. , 2018. C9orf72, a protein associated with amyotrophic lateral sclerosis (ALS) is a guanine nucleotide exchange factor. PeerJ. 6, e5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, et al. , 2010. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 285, 22576–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N, Chen-Plotkin AS, 2018. Genetic Modifiers in Neurodegeneration. Current genetic medicine reports. 6, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezegou A, et al. , 2012. Heptahelical protein PQLC2 is a lysosomal cationic amino acid exporter underlying the action of cysteamine in cystinosis therapy. Proc Natl Acad Sci U S A. 109, E3434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, et al. , 2019. BAG3 and SYNPO (synaptopodin) facilitate phospho-MAPT/Tau degradation via autophagy in neuronal processes. Autophagy. 15, 1199–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian J, et al. , 2016a. Progranulin Recruits HSP70 to beta-Glucocerebrosidase and Is Therapeutic Against Gaucher Disease. EBioMedicine. 13, 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian J, et al. , 2016b. Association Between Progranulin and Gaucher Disease. EBioMedicine. 11, 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, et al. , 2016. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron. 90, 535–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AE, et al. , 2021. SVIP is a molecular determinant of lysosomal dynamic stability, neurodegeneration and lifespan. Nature Communications. 12, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DE, et al. , 2016. The position of lysosomes within the cell determines their luminal pH. J Cell Biol. 212, 677–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JK, et al. , 2005. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol. 62, 925–30. [DOI] [PubMed] [Google Scholar]
- Johnson JO, et al. , 2010a. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron. 68, 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JO, et al. , 2010b. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 68, 857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun MH, et al. , 2015. TMEM106B, a frontotemporal lobar dementia (FTLD) modifier, associates with FTD-3-linked CHMP2B, a complex of ESCRT-III. Mol Brain. 8, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Behrends C, 2020. Multifaceted role of SMCR8 as autophagy regulator. Small GTPases. 11, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, et al. , 2017. Multiplex image-based autophagy RNAi screening identifies SMCR8 as ULK1 kinase activity and gene expression regulator. Elife. 6, e23063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, et al. , 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizuka A, 2008. Roles of VCP in human neurodegenerative disorders. Biochem Soc Trans. 36, 105–8. [DOI] [PubMed] [Google Scholar]
- Kamate M, et al. , 2019. Neuronal ceroid lipofuscinosis type-11 in an adolescent. Brain Dev. 41, 542–545. [DOI] [PubMed] [Google Scholar]
- Kapeli K, et al. , 2017. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum Genet. 136, 1193–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, et al. , 2018. Selective Genetic Overlap Between Amyotrophic Lateral Sclerosis and Diseases of the Frontotemporal Dementia Spectrum. JAMA Neurol. 75, 860–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenessey A, et al. , 1997. Degradation of tau by lysosomal enzyme cathepsin D: implication for Alzheimer neurofibrillary degeneration. J Neurochem. 69, 2026–38. [DOI] [PubMed] [Google Scholar]
- Kertesz A, et al. , 2005. The evolution and pathology of frontotemporal dementia. Brain. 128, 1996–2005. [DOI] [PubMed] [Google Scholar]
- Khalfallah Y, et al. , 2018. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci Rep. 8, 7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, et al. , 2008. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 10, 935–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R, 2005. Unknotting the roles of Bcl-2 and Bcl-xL in cell death. Biochem Biophys Res Commun. 333, 336–43. [DOI] [PubMed] [Google Scholar]
- Kim SH, et al. , 2009. Potentiation of amyotrophic lateral sclerosis (ALS)-associated TDP-43 aggregation by the proteasome-targeting factor, ubiquilin 1. J Biol Chem. 284, 8083–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein ZA, et al. , 2017. Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron. 95, 281–296 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, et al. , 2016. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochl R, et al. , 2006. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 7, 129–45. [DOI] [PubMed] [Google Scholar]
- Komatsu M, et al. , 2007. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 131, 1149–63. [DOI] [PubMed] [Google Scholar]
- Koppers M, et al. , 2012. VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 33, 837.e7–13. [DOI] [PubMed] [Google Scholar]
- Korb MK, et al. , 2020. Multisystem proteinopathy: Where myopathy and motor neuron disease converge. Muscle Nerve. n/a. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, et al. , 2016. Clinicopathological description of two cases with SQSTM1 gene mutation associated with frontotemporal dementia. Neuropathology. 36, 27–38. [DOI] [PubMed] [Google Scholar]
- Krainc D, et al. , 2020. Progranulin mutations result in impaired processing of prosaposin and reduced glucocerebrosidase activity. Human Molecular Genetics. 29, 716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiter N, et al. , 2018. Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol Dis. 115, 167–181. [DOI] [PubMed] [Google Scholar]
- Kuchar L, et al. , 2009. Prosaposin deficiency and saposin B deficiency (activator-deficient metachromatic leukodystrophy): report on two patients detected by analysis of urinary sphingolipids and carrying novel PSAP gene mutations. Am J Med Genet A. 149a, 613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laferriere F, et al. , 2019. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat Neurosci. 22, 65–77. [DOI] [PubMed] [Google Scholar]
- Laflamme C, et al. , 2019. Implementation of an antibody characterization procedure and application to the major ALS/FTD disease gene C9ORF72. Elife. 8, e48363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CM, et al. , 2012. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem. 287, 19355–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurin N, et al. , 2002. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 70, 1582–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Ber I, et al. , 2009. Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology. 72, 1669–76. [DOI] [PubMed] [Google Scholar]
- Le Ber I, et al. , 2013. SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 70, 1403–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBoeuf AC, et al. , 2008. FTDP-17 mutations in Tau alter the regulation of microtubule dynamics: an “alternative core” model for normal and pathological Tau action. J Biol Chem. 283, 36406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BL, Barton GM, 2014. Trafficking of endosomal Toll-like receptors. Trends Cell Biol. 24, 360–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, et al. , 2017. The lysosomal protein cathepsin L is a progranulin protease. Mol Neurodegener. 12, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, et al. , 2011. rs5848 variant of progranulin gene is a risk of Alzheimer’s disease in the Taiwanese population. Neurodegener Dis. 8, 216–20. [DOI] [PubMed] [Google Scholar]
- Lee SM, et al. , 2019. TDP-43 cytoplasmic inclusion formation is disrupted in C9orf72-associated amyotrophic lateral sclerosis/frontotemporal lobar degeneration. Brain Commun. 1, fcz014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z, et al. , 2015. BAG3 facilitates the clearance of endogenous tau in primary neurons. Neurobiol Aging. 36, 241–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibiger C, et al. , 2018. TDP-43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum Mol Genet. 27, 1593–1607. [DOI] [PubMed] [Google Scholar]
- Levine TP, et al. , 2013. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 29, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, et al. , 2000. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 25, 402–5. [DOI] [PubMed] [Google Scholar]
- Li X, et al. , 2016. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol. 18, 404–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie PPY, Nixon RA, 2019. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol Dis. 122, 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim F, et al. , 2001. FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol Cell Neurosci. 18, 702–14. [DOI] [PubMed] [Google Scholar]
- Lin WL, et al. , 2003. Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol. 32, 1091–105. [DOI] [PubMed] [Google Scholar]
- Lindquist SG, et al. , 2008. Frontotemporal dementia linked to chromosome 3 (FTD-3)--current concepts and the detection of a previously unknown branch of the Danish FTD-3 family. Eur J Neurol. 15, 667–70. [DOI] [PubMed] [Google Scholar]
- Ling SC, et al. , 2013. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 79, 416–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, et al. , 2012. LAAT-1 is the lysosomal lysine/arginine transporter that maintains amino acid homeostasis. Science. 337, 351–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, et al. , 2017. Endocytosis regulates TDP-43 toxicity and turnover. Nat Commun. 8, 2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GY, Sabatini DM, 2020. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 21, 183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubenkov PA, Miller BL, 2016. A Clinical Guide to Frontotemporal Dementias. Focus (Am Psychiatr Publ). 14, 448–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomen-Hoerth C, et al. , 2002. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 59, 1077–9. [DOI] [PubMed] [Google Scholar]
- Long J, et al. , 2008. Ubiquitin recognition by the ubiquitin-associated domain of p62 involves a novel conformational switch. J Biol Chem. 283, 5427–40. [DOI] [PubMed] [Google Scholar]
- Longinetti E, Fang F, 2019. Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr Opin Neurol. 32, 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPresti P, et al. , 1995. Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci U S A. 92, 10369–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, et al. , 2020. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, et al. , 2016. The Overexpression of TDP-43 Protein in the Neuron and Oligodendrocyte Cells Causes the Progressive Motor Neuron Degeneration in the SOD1 G93A Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. Int J Biol Sci. 12, 1140–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui H, et al. , 2016. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell. 165, 921–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luningschror P, et al. , 2020. The FTLD Risk Factor TMEM106B Regulates the Transport of Lysosomes at the Axon Initial Segment of Motoneurons. Cell Rep. 30, 3506–3519 e6. [DOI] [PubMed] [Google Scholar]
- Luzio JP, et al. , 2014. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb Perspect Biol. 6, a016840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, 2016. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem. 138 Suppl 1, 54–70. [DOI] [PubMed] [Google Scholar]
- Maday S, et al. , 2012. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 196, 407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahul-Mellier AL, et al. , 2020. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A. 117, 4971–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DMA, Snowden JS, 2017. Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathol. 27, 723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marat AL, et al. , 2011. DENN domain proteins: regulators of Rab GTPases. J Biol Chem. 286, 13791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmann S, et al. , 2015. Lrp1/LDL Receptor Play Critical Roles in Mannose 6-Phosphate-Independent Lysosomal Enzyme Targeting. Traffic. 16, 743–59. [DOI] [PubMed] [Google Scholar]
- Martin ER, et al. , 2001. Association of single-nucleotide polymorphisms of the tau gene with late-onset Parkinson disease. JAMA. 286, 2245–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, et al. , 2005. Protein degradation and aging. Exp Gerontol. 40, 622–33. [DOI] [PubMed] [Google Scholar]
- Mathis S, et al. , 2019. Genetics of amyotrophic lateral sclerosis: A review. J Neurol Sci. 399, 217–226. [DOI] [PubMed] [Google Scholar]
- Matsubara T, et al. , An autopsy report of a familial amyotrophic lateral sclerosis case carrying VCP Arg487His mutation with a unique TDP43 proteinopathy. Neuropathology. n/a. [DOI] [PubMed] [Google Scholar]
- Mazucanti CH, et al. , 2015. Longevity Pathways (mTOR, SIRT, Insulin/IGF-1) as Key Modulatory Targets on Aging and Neurodegeneration. Curr Top Med Chem. 15, 2116–38. [DOI] [PubMed] [Google Scholar]
- McCampbell A, et al. , 2018. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest. 128, 3558–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley ME, et al. , 2020. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. 585, 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejzini R, et al. , 2019. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front Neurosci. 13, 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, et al. , 2017. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron. 93, 1015–1034. [DOI] [PubMed] [Google Scholar]
- Menzies FM, et al. , 2015. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 16, 345–57. [DOI] [PubMed] [Google Scholar]
- Michel CH, et al. , 2014. Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J Biol Chem. 289, 956–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami SS, et al. , 2014. Progranulin protects against amyloid beta deposition and toxicity in Alzheimer’s disease mouse models. Nat Med. 20, 1157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, 2012. Lysosomal acidification mechanisms. Annu Rev Physiol. 74, 69–86. [DOI] [PubMed] [Google Scholar]
- Mizushima N, et al. , 2011. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 27, 107–32. [DOI] [PubMed] [Google Scholar]
- Mohamed NV, et al. , 2014. Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci Rep. 4, 5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mole SE, et al. , 2019. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. 18, 107–116. [DOI] [PubMed] [Google Scholar]
- Moore AS, Holzbaur EL, 2016. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A. 113, E3349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KM, et al. , 2020. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 19, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Garcia A, et al. , 2018. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front Neurosci. 12, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, et al. , 2006. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 66, 839–44. [DOI] [PubMed] [Google Scholar]
- Muller R, et al. , 1997. Expression of microtubule-associated proteins MAP2 and tau in cultured rat brain oligodendrocytes. Cell Tissue Res. 288, 239–49. [DOI] [PubMed] [Google Scholar]
- Murphy DB, et al. , 1977. Role of tubulin-associated proteins in microtubule nucleation and elongation. J Mol Biol. 117, 33–52. [DOI] [PubMed] [Google Scholar]
- Murray ME, et al. , 2014. Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol. 128, 411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura R, et al. , 2020. A multi-ethnic meta-analysis identifies novel genes, including ACSL5, associated with amyotrophic lateral sclerosis. Commun Biol. 3, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naphade SB, et al. , 2010. Progranulin expression is upregulated after spinal contusion in mice. Acta Neuropathol. 119, 123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 2019. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 142, 1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 2015. Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J Neuropathol Exp Neurol. 74, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, et al. , 2007a. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol. 66, 177–83. [DOI] [PubMed] [Google Scholar]
- Neumann M, et al. , 2007b. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 66, 152–7. [DOI] [PubMed] [Google Scholar]
- Neumann M, et al. , 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 314, 130–3. [DOI] [PubMed] [Google Scholar]
- Nguyen AD, et al. , 2018a. Murine knockin model for progranulin-deficient frontotemporal dementia with nonsense-mediated mRNA decay. Proc Natl Acad Sci U S A. 115, E2849–e2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HP, et al. , 2018b. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 34, 404–423. [DOI] [PubMed] [Google Scholar]
- Nicholson AM, et al. , 2013. TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem. 126, 781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson AM, Rademakers R, 2016. What we know about TMEM106B in neurodegeneration. Acta Neuropathol. 132, 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson AM, et al. , 2018. Loss of Tmem106b is unable to ameliorate frontotemporal dementia-like phenotypes in an AAV mouse model of C9ORF72-repeat induced toxicity. Acta Neuropathol Commun. 6, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke JG, et al. , 2016. C9orf72 is required for proper macrophage and microglial function in mice. Science. 351, 1324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuma S, et al. , 1982. Identification and characterization of a proton pump on lysosomes by fluorescein-isothiocyanate-dextran fluorescence. Proc Natl Acad Sci U S A. 79, 2758–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origone P, et al. , 2018. Role of MAPT in Pure Motor Neuron Disease: Report of a Recurrent Mutation in Italian Patients. Neurodegener Dis. 18, 310–314. [DOI] [PubMed] [Google Scholar]
- Ormeno F, et al. , 2020. Chaperone Mediated Autophagy Degrades TDP-43 Protein and Is Affected by TDP-43 Aggregation. Front Mol Neurosci. 13, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou SH, et al. , 1995. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. Am Soc Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcelik S, et al. , 2013. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One. 8, e62459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco CD, et al. , 2009. Tau normal function influences Niemann-Pick type C disease pathogenesis in mice and modulates autophagy in NPC1-deficient cells. Autophagy. 5, 548–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, et al. , 2017. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 36, 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Para C, et al. , 2020. Neuropathophysiology of Lysosomal Storage Diseases: Synaptic Dysfunction as a Starting Point for Disease Progression. J Clin Med. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier B, 2016. Autophagy inhibitors. Cell Mol Life Sci. 73, 985–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushter DH, et al. , 2018. The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 136, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, et al. , 2019. Preserving Lysosomal Function in the Aging Brain: Insights from Neurodegeneration. Neurotherapeutics. 16, 611–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Zoncu R, 2016. The Lysosome as a Regulatory Hub. Annu Rev Cell Dev Biol. 32, 223–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkau TL, et al. , 2012. Synaptic dysfunction in progranulin-deficient mice. Neurobiol Dis. 45, 711–22. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, et al. , 2004. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 13, 703–14. [DOI] [PubMed] [Google Scholar]
- Pilli M, et al. , 2012. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 37, 223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piras A, et al. , 2016. Autophagic and lysosomal defects in human tauopathies: analysis of postmortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun. 4, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotegher N, Duchen MR, 2017. Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders. Trends Mol Med. 23, 116–134. [DOI] [PubMed] [Google Scholar]
- Polito VA, et al. , 2014. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med. 6, 1142–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorkaj P, et al. , 1998. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 43, 815–25. [DOI] [PubMed] [Google Scholar]
- Portet F, et al. , 2001. Cognitive impairment in motor neuron disease with bulbar onset. Amyotroph Lateral Scler Other Motor Neuron Disord. 2, 23–9. [DOI] [PubMed] [Google Scholar]
- Pottier C, et al. , 2016. Genetics of FTLD: overview and what else we can expect from genetic studies. J Neurochem. 138 Suppl 1, 32–53. [DOI] [PubMed] [Google Scholar]
- Prada CE, Grabowski GA, 2013. Neuronopathic lysosomal storage diseases: clinical and pathologic findings. Dev Disabil Res Rev. 17, 226–46. [DOI] [PubMed] [Google Scholar]
- Prasad A, et al. , 2019. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front Mol Neurosci. 12, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, et al. , 2012. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 8, 423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan HN, Ye Y, 2012. The p97 ATPase associates with EEA1 to regulate the size of early endosomes. Cell Res. 22, 346–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh Babu J, et al. , 2008. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 106, 107–20. [DOI] [PubMed] [Google Scholar]
- Reczek D, et al. , 2007. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell. 131, 770–83. [DOI] [PubMed] [Google Scholar]
- Reggiori F, Ungermann C, 2017. Autophagosome Maturation and Fusion. J Mol Biol. 429, 486–496. [DOI] [PubMed] [Google Scholar]
- Renton AE, et al. , 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 72, 257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richo GR, Conner GE, 1994. Structural requirements of procathepsin D activation and maturation. J Biol Chem. 269, 14806–12. [PubMed] [Google Scholar]
- Richter B, et al. , 2016. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 113, 4039–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringholz GM, et al. , 2005. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 65, 586–90. [DOI] [PubMed] [Google Scholar]
- Ritz D, et al. , 2011. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat Cell Biol. 13, 1116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robak LA, et al. , 2017. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain. 140, 3191–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, Ferguson SM, 2019. Pleiotropic requirements for human TDP-43 in the regulation of cell and organelle homeostasis. Life Sci Alliance. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Martin T, et al. , 2013. Tau phosphorylation affects its axonal transport and degradation. Neurobiol Aging. 34, 2146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez L, et al. , 2017. Rab7A regulates tau secretion. J Neurochem. 141, 592–605. [DOI] [PubMed] [Google Scholar]
- Rohrer JD, et al. , 2009. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 73, 1451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JD, Warren JD, 2011. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol. 24, 542–9. [DOI] [PubMed] [Google Scholar]
- Rutherford NJ, et al. , 2012. TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology. 79, 717–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TA, Tumbarello DA, 2018. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Frontiers in Immunology. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffi GT, Botelho RJ, 2019. Lysosome Fission: Planning for an Exit. Trends Cell Biol. 29, 635–646. [DOI] [PubMed] [Google Scholar]
- Sancak Y, et al. , 2010. Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell. 141, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, et al. , 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 320, 1496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Juan P, et al. , 2019. The MAPT H1 Haplotype Is a Risk Factor for Alzheimer’s Disease in APOE epsilon4 Non-carriers. Front Aging Neurosci. 11, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saracino D, et al. , 2018. Novel VCP mutations expand the mutational spectrum of frontotemporal dementia. Neurobiology of Aging. 72, 187.e11–187.e14. [DOI] [PubMed] [Google Scholar]
- Saric A, et al. , 2016. mTOR controls lysosome tubulation and antigen presentation in macrophages and dendritic cells. Mol Biol Cell. 27, 321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, 2011. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 70, 349–59. [DOI] [PubMed] [Google Scholar]
- Satoh J, et al. , 2014. TMEM106B expression is reduced in Alzheimer’s disease brains. Alzheimers Res Ther. 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer V, Goedert M, 2012. Stimulation of autophagy is neuroprotective in a mouse model of human tauopathy. Autophagy. 8, 1686–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer V, et al. , 2012. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain. 135, 2169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser K, Parker JA, 2019. Pleiotropic Effects of mTOR and Autophagy During Development and Aging. Front Cell Dev Biol. 7, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze H, Sandhoff K, 2014. Sphingolipids and lysosomal pathologies. Biochim Biophys Acta. 1841, 799–810. [DOI] [PubMed] [Google Scholar]
- Schwenk BM, et al. , 2014. The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J. 33, 450–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymick JC, et al. , 2007. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry. 78, 754–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotter EL, et al. , 2015. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics. 12, 352–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotter EL, et al. , 2014. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J Cell Sci. 127, 1263–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, et al. , 2016. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Şentürk M, et al. , 2019. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nature Cell Biology. 21, 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok Ko H, et al. , 2004. Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Letters. 566, 110–114. [DOI] [PubMed] [Google Scholar]
- Settembre C, et al. , 2011. TFEB links autophagy to lysosomal biogenesis. Science. 332, 1429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, et al. , 2013. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 14, 283–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahmoradian SH, et al. , 2019. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci. 22, 1099–1109. [DOI] [PubMed] [Google Scholar]
- Shao Q, et al. , 2019. C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta Neuropathol Commun. 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Q, et al. , 2020. C9orf72 and smcr8 mutant mice reveal MTORC1 activation due to impaired lysosomal degradation and exocytosis. Autophagy. 16, 1635–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng J, et al. , 2014. Progranulin polymorphism rs5848 is associated with increased risk of Alzheimer’s disease. Gene. 542, 141–5. [DOI] [PubMed] [Google Scholar]
- Shi J, et al. , 2005. Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol. 110, 501–12. [DOI] [PubMed] [Google Scholar]
- Shi Y, et al. , 2018. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 24, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siintola E, et al. , 2007. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am J Hum Genet. 81, 136–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, et al. , 2015. The mTOR Inhibitor Rapamycin Mitigates Perforant Pathway Neurodegeneration and Synapse Loss in a Mouse Model of Early-Stage Alzheimer-Type Tauopathy. PLoS One. 10, e0142340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons C, et al. , 2017. A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain. 140, 3105–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, et al. , 2009. Autophagy regulates lipid metabolism. Nature. 458, 1131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirkis DW, et al. , 2019. Recent advances in the genetics of frontotemporal dementia. Curr Genet Med Rep. 7, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G, et al. , 2005. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 37, 806–8. [DOI] [PubMed] [Google Scholar]
- Skoko N, et al. , 2016. Absence of TDP-43 is difficult to digest. EMBO J. 35, 115–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleegers K, et al. , 2009. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol. 65, 603–9. [DOI] [PubMed] [Google Scholar]
- Smith KR, et al. , 2013. Cathepsin F mutations cause Type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum Mol Genet. 22, 1417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, et al. , 2012. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 90, 1102–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, et al. , 1998. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 95, 7737–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, et al. , 2008. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 319, 1668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staats KA, et al. , 2019. Small molecule inhibition of PIKFYVE kinase rescues gain- and loss-of-function C9ORF72 ALS/FTD disease processes in vivo. bioRxiv. 685800. [Google Scholar]
- Stagi M, et al. , 2014. Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B. Mol Cell Neurosci. 61, 226–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Wang X, 2011. Autophagy and p62 in cardiac protein quality control. Autophagy. 7, 1382–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M-Y, et al. , 2020. Structure of the lysosomal SCARF (L-SCARF) complex, an Arf GAP haploinsufficient in ALS and FTD. bioRxiv. 2020.04.15.042515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugai A, et al. , 2018. Robustness and Vulnerability of the Autoregulatory System That Maintains Nuclear TDP-43 Levels: A Trade-off Hypothesis for ALS Pathology Based on in Silico Data. Front Neurosci. 12, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugai A, et al. , 2019. Non-genetically modified models exhibit TARDBP mRNA increase due to perturbed TDP-43 autoregulation. Neurobiol Dis. 130, 104534. [DOI] [PubMed] [Google Scholar]
- Sullivan PM, et al. , 2016. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 4, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Matsuoka M, 2016. The Lysosomal Trafficking Transmembrane Protein 106B Is Linked to Cell Death. J Biol Chem. 291, 21448–21460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen B, et al. , 2020. RNA toxicity in non-coding repeat expansion disorders. EMBO J. 39, e101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taes I, et al. , 2010. Tau levels do not influence human ALS or motor neuron degeneration in the SOD1G93A mouse. Neurology. 74, 1687–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JM, et al. , 2008. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 17, 431–9. [DOI] [PubMed] [Google Scholar]
- Tan RH, et al. , 2017a. ALS/FTLD: experimental models and reality. Acta Neuropathol. 133, 177–196. [DOI] [PubMed] [Google Scholar]
- Tan RH, et al. , 2015. TDP-43 proteinopathies: pathological identification of brain regions differentiating clinical phenotypes. Brain. 138, 3110–22. [DOI] [PubMed] [Google Scholar]
- Tan RH, et al. , 2017b. Distinct TDP-43 inclusion morphologies in frontotemporal lobar degeneration with and without amyotrophic lateral sclerosis. Acta Neuropathol Commun. 5, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, et al. , 2014. Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol Commun. 2, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, et al. , 2017. Progranulin regulates lysosomal function and biogenesis through acidification of lysosomes. Hum Mol Genet. 26, 969–988. [DOI] [PubMed] [Google Scholar]
- Tang CH, et al. , 2006. Murine cathepsin F deficiency causes neuronal lipofuscinosis and late-onset neurological disease. Mol Cell Biol. 26, 2309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, et al. , 2020. Cryo-EM structure of C9ORF72-SMCR8-WDR41 reveals the role as a GAP for Rab8a and Rab11a. Proc Natl Acad Sci U S A. 117, 9876–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WK, Xia D, 2016. Mutations in the Human AAA(+) Chaperone p97 and Related Diseases. Frontiers in molecular biosciences. 3, 79–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, et al. , 2005. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 1, 84–91. [DOI] [PubMed] [Google Scholar]
- Taylor JP, et al. , 2016. Decoding ALS: from genes to mechanism. Nature. 539, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekirdag K, Cuervo AM, 2018. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J Biol Chem. 293, 5414–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therrien M, et al. , 2013. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One. 8, e83450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas A, et al. , 2014. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 24, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresse E, et al. , 2010. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 6, 217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinczek B, et al. , 1995. Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol Biol Cell. 6, 1887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang HT, et al. , 2006. A systematic analysis of human CHMP protein interactions: additional MIT domain-containing proteins bind to multiple components of the human ESCRT III complex. Genomics. 88, 333–46. [DOI] [PubMed] [Google Scholar]
- Urushitani M, et al. , 2010. Synergistic effect between proteasome and autophagosome in the clearance of polyubiquitinated TDP-43. J Neurosci Res. 88, 784–97. [DOI] [PubMed] [Google Scholar]
- Urwin H, et al. , 2010. Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum Mol Genet. 19, 2228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez C, et al. , 2017. Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum Mol Genet. 26, 4861–4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdmanis PN, et al. , 2007. Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol. 64, 240–5. [DOI] [PubMed] [Google Scholar]
- van Blitterswijk M, et al. , 2015. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 130, 863–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, et al. , 2014. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 127, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, et al. , 2008. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 181, 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin VM, et al. , 2010. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 42, 234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J, Meyer H, 2018. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol Cell. 69, 182–194. [DOI] [PubMed] [Google Scholar]
- van der Ende EL, et al. , 2020. Neuronal pentraxin 2: a synapse-derived CSF biomarker in genetic frontotemporal dementia. J Neurol Neurosurg Psychiatry. 91, 612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zee J, et al. , 2016. Mutated CTSF in adult-onset neuronal ceroid lipofuscinosis and FTD. Neurol Genet. 2, e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zee J, et al. , 2011. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain. 134, 808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, et al. , 2006. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain. 129, 868–76. [DOI] [PubMed] [Google Scholar]
- Vanden Broeck L, et al. , 2015. Functional complementation in Drosophila to predict the pathogenicity of TARDBP variants: evidence for a loss-of-function mechanism. Neurobiol Aging. 36, 1121–9. [DOI] [PubMed] [Google Scholar]
- Vass R, et al. , 2011. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 121, 373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viode A, et al. , 2018. New Antibody-Free Mass Spectrometry-Based Quantification Reveals That C9ORF72 Long Protein Isoform Is Reduced in the Frontal Cortex of Hexanucleotide-Repeat Expansion Carriers. Front Neurosci. 12, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt MA, et al. , 2018. TDP-43 induces p53-mediated cell death of cortical progenitors and immature neurons. Sci Rep. 8, 8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, et al. , 1998. Human cathepsin F. Molecular cloning, functional expression, tissue localization, and enzymatic characterization. J Biol Chem. 273, 32000–8. [DOI] [PubMed] [Google Scholar]
- Wang H, et al. , 2016. Transcription Factor EB Is Selectively Reduced in the Nuclear Fractions of Alzheimer’s and Amyotrophic Lateral Sclerosis Brains. Neurosci J. 2016, 4732837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, et al. , 2012. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A. 109, 15024–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, et al. , 2020. C9orf72 associates with inactive Rag GTPases and regulates mTORC1-mediated autophagosomal and lysosomal biogenesis. Aging Cell. 19, e13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, et al. , 2019. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 15, e1007947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. , 2010a. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett. 469, 112–6. [DOI] [PubMed] [Google Scholar]
- Wang Y, et al. , 2010b. Proteolytic processing of tau. Biochem Soc Trans. 38, 955–61. [DOI] [PubMed] [Google Scholar]
- Wang Y, et al. , 2009. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 18, 4153–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. , 2010c. Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy. 6, 182–3. [DOI] [PubMed] [Google Scholar]
- Ward ME, et al. , 2017. Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GD, et al. , 2004. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 36, 377–81. [DOI] [PubMed] [Google Scholar]
- Werner G, et al. , 2020. Loss of TMEM106B potentiates lysosomal and FTLD-like pathology in progranulin-deficient mice. EMBO Rep. 21, e50241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West RJH, et al. , 2020. Neuroprotective activity of ursodeoxycholic acid in CHMP2B(Intron5) models of frontotemporal dementia. Neurobiol Dis. 144, 105047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, et al. , 2018. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat Neurosci. 21, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox WR, 2004. Lysosomal storage disorders: the need for better pediatric recognition and comprehensive care. J Pediatr. 144, S3–14. [DOI] [PubMed] [Google Scholar]
- Wils H, et al. , 2012. Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J Pathol. 228, 67–76. [DOI] [PubMed] [Google Scholar]
- Winton MJ, et al. , 2008. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 283, 13302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TH, et al. , 2018. Three VCP Mutations in Patients with Frontotemporal Dementia. J Alzheimers Dis. 65, 1139–1146. [DOI] [PubMed] [Google Scholar]
- Woollacott IOC, et al. , 2020. Cerebrospinal Fluid YKL-40 and Chitotriosidase Levels in Frontotemporal Dementia Vary by Clinical, Genetic and Pathological Subtype. Dement Geriatr Cogn Disord. 49, 56–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, et al. , 2013. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem. 288, 1856–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, et al. , 2016. C9orf72 isoforms in Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. Brain Res. 1647, 43–49. [DOI] [PubMed] [Google Scholar]
- Xiao S, et al. , 2011. RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol Cell Neurosci. 47, 167–80. [DOI] [PubMed] [Google Scholar]
- Xu H, Ren D, 2015. Lysosomal Physiology. Annual Review of Physiology. 77, 57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HM, et al. , 2017. PGRN Is Associated with Late-Onset Alzheimer’s Disease: a Case-Control Replication Study and Meta-analysis. Mol Neurobiol. 54, 1187–1195. [DOI] [PubMed] [Google Scholar]
- Xu Y, et al. , 2019. The cargo receptor SQSTM1 ameliorates neurofibrillary tangle pathology and spreading through selective targeting of pathological MAPT (microtubule associated protein tau). Autophagy. 15, 583–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, et al. , 2010. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 30, 10851–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yambire KF, et al. , 2019. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. Elife. 8, e51031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HS, et al. , 2020. Genetics of Gene Expression in the Aging Human Brain Reveal TDP-43 Proteinopathy Pathophysiology. Neuron. 107, 496–508 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, et al. , 2016. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv. 2, e1601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama JS, et al. , 2017. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathologica. 133, 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CE, et al. , 2010. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol. 67, 161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, et al. , 2015. The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology. 84, 927–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CS, et al. , 2014a. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 20, 526–40. [DOI] [PubMed] [Google Scholar]
- Zhang D, et al. , 2012. Discovery of Novel DENN Proteins: Implications for the Evolution of Eukaryotic Intracellular Membrane Structures and Human Disease. Front Genet. 3, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, et al. , 2020. Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. , 2014b. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 34, 11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, et al. , 2009. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 106, 7607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, et al. , 2014. Lysosome sorting of β-glucocerebrosidase by LIMP-2 is targeted by the mannose 6-phosphate receptor. Nat Commun. 5, 4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, et al. , 2020. Loss of Tmem106b exacerbates FTLD pathologies and causes motor deficits in progranulin-deficient mice. EMBO Rep. 21, e50197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, et al. , Lysosomal Dysfunction and Other Pathomechanisms in FTLD: Evidence from Progranulin Genetics and Biology. In: Ghetti B, et al. , (Eds.), Frontotemporal Dementias : Emerging Milestones of the 21st Century. Springer International Publishing, Cham, 2021, pp. 219–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, et al. , 2017a. Regulation of cathepsin D activity by the FTLD protein progranulin. Acta Neuropathol. 134, 151–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, et al. , 2017b. Lysosomal processing of progranulin. Mol Neurodegener. 12, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, et al. , 2019. Progranulin deficiency leads to reduced glucocerebrosidase activity. PLoS One. 14, e0212382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, et al. , 2015. Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J Cell Biol. 210, 991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, et al. , 2017c. Impaired prosaposin lysosomal trafficking in frontotemporal lobar degeneration due to progranulin mutations. Nat Commun. 8, 15277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, et al. , 2017d. Elevated TMEM106B levels exaggerate lipofuscin accumulation and lysosomal dysfunction in aged mice with progranulin deficiency. Acta Neuropathol Commun. 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, et al. , 2002. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 111, 867–78. [DOI] [PubMed] [Google Scholar]
- Zhu Q, et al. , 2020. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 23, 615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou ZY, et al. , 2017. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 88, 540–549. [DOI] [PubMed] [Google Scholar]