

Abstract
Diabetic retinopathy (DR) is a leading cause of blindness. It has long been regarded as vascular disease, but work in the past years has shown abnormalities also in the neural retina. Unfortunately, research on the vascular and neural abnormalities have remained largely separate, instead of being integrated into a comprehensive view of DR that includes both the neural and vascular components. Recent evidence suggests that the most predominant neural cell in the retina (photoreceptors) and the adjacent retinal pigment epithelium (RPE) play an important role in the development of vascular lesions characteristic of DR. This review summarizes evidence that the outer retina is altered in diabetes, and that photoreceptors and RPE contribute to retinal vascular alterations in the early stages of the retinopathy. The possible molecular mechanisms by which cells of the outer retina might contribute to retinal vascular damage in diabetes also are discussed. Diabetes-induced alterations in the outer retina represent a novel therapeutic target to inhibit DR.
Keywords: Diabetes, Diabetic retinopathy, Outer retina, Photoreceptors, RPE, Vasculature, Phototransduction, Visual cycle
1. Introduction
Diabetic retinopathy (DR) and diabetic macular edema (DME) are common microvascular complications in patients with diabetes. DR is a leading cause of visual impairment and blindness in people aged 24 to 64 years old, and it develops, to at least some degree, in the majority of patients who have had diabetes for 10 or more years (Engelgau et al., 2004). Clinically, there are two defined stages of DR. The first stage is non-proliferative DR (NPDR), which is characterized by microaneurysms, increased permeability, and vascular occlusion and degeneration. Subsequently, proliferative diabetic retinopathy (PDR) develops in some patients, and this is characterized by formation of new blood vessels of the retinal vasculature that can migrate out of the retina into the vitreous, where they impair vision secondary to leakage and fibrosis of the abnormal new vessels. Especially at later stages of DR, patients with diabetes can also develop DME, which involves retinal thickening in the macular area of the retina due to leaking blood vessels, and is the leading cause of visual impairment in diabetic patients (Cunha-Vaz et al., 2014; Daruich et al., 2018; Klaassen et al., 2013; Stitt et al., 2016).
The complications of diabetes within the eye develop gradually, and can go unnoticed by patients until there is impairment or loss of vision. Once diabetes has damaged the retina, vision can be lost permanently, so it is important to detect and treat the retinopathy as early as possible. Current therapies to treat existing DR (including laser photocoagulation, intravitreal injections of antibodies against Vascular Endothelial Growth Factor (VEGF) or corticosteroids, and surgery) are effective, but not all patients respond to them and there can be undesirable side effects. Good glycemic control has been shown to inhibit the development and progression of DR in diabetic dogs and patients (Diabetes Control and Complications Trial Research Group, 1993; Engerman et al., 1977; United Kingdom Prospective Diabetes Study, 1998), but it remains difficult to achieve and maintain such glycemic control for many patients. There remains a great need for therapies that prevent the retinopathy from ever developing (Stitt et al., 2016).
Most studies of DR to date have focused on vascular abnormalities, which is appropriate considering that diabetes-induced abnormalities of the retinal vasculature (i.e. capillary degeneration, permeability, and neovascularization) have been directly implicated in vision loss and impairment. Nevertheless, alterations in the neural retina also have been detected, and their contribution to vision loss is under study (Simo et al., 2018). Abundant evidence indicates that all retinal cell-types are affected by diabetes, including loss of inner retinal neurons and their projections (Barber et al., 1998; Gastinger et al., 2001; Gastinger et al., 2008; Gastinger et al., 2006), dysfunction of Müller cells and astrocytes (Barber et al., 2000; Mizutani et al., 1998; Puro, 2002; Rungger-Brandle et al., 2000), activation of microglia (Gaucher et al., 2007; Omri et al., 2011; Zeng et al., 2008; Zeng et al., 2000), and dysfunction or degeneration of the RPE (Aizu et al., 2002; Bensaoula and Ottlecz, 2001; Grimes and Laties, 1980; Omri et al., 2011; Samuels et al., 2015) (Fig 1). Although we acknowledge that diabetes affects all cell-types in the retina and it is difficult to discuss photoreceptors and RPE without considering also the cells of the inner retina and their relation to the outer retina, there exists already a substantial literature focusing on the inner retina in DR. Thus in this review, we focus on cells of the outer retina (photoreceptors and RPE) and their alterations in diabetes, and summarize evidence that the outer retina contributes to the development of the retinal vascular and neural lesions that are characteristic of DR. We focus mainly on in vivo studies, due to the growing recognition that development of DR is influenced by multiple cell-types, a complexity that is difficult to reproduce in vitro.
Fig 1.

Drawing of retinal architecture and the relation of the retinal vasculature to retinal photoreceptor cells. The retina is highly organized, with nuclei in the ganglion cell layer, inner nuclear layer and outer nuclear layer appearing in discrete layers. Between these nuclear layers are plexiform layers where processes from neural and glial cell-types intermix. Outer segments of the retinal photoreceptors interdigitate with the retinal pigment epithelium (RPE) to maintain the visual cycle and vision. The vasculature supplying the retina comes from two different sides, with the photoreceptors supplied by choroidal vessels below the retina, and the inner retina supplied by vascular networks within the inner retinas. Retinal microvessels do not directly interact with photoreceptor cells. Drawing of retina showing neural cells and the 3 layers of the retinal vasculature (left side of figure) is reproduced from Z. Fu et al, International Journal of Molecular Sciences 2020, 21, 1503 under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium.
2. Outer retina
2.1. Photoreceptor cells
Photoreceptor cells are specialized neurons in the retina that convert light into neural signals that are sent to the brain for image processing (Fain et al., 2010; Lagnado and Baylor, 1992). They are the most abundant cells in the retina (about 6 million cells in mice (Jeon et al., 1998) and 120 million cells in humans (Kolb, 1995)), and the most metabolically active cells in the body (Ames, 1992; Ames et al., 1992; Hoang et al., 2002). There are two types of photoreceptor cells, rods and cones. Rods have low spatial resolution but are very light sensitive, while cones have high spatial resolution but are less light sensitive. Rods represent about 90% of all photoreceptors in the human and 97% in the mouse retina (Jeon et al., 1998), whereas cones represent about 5–10% of all photoreceptors in humans and 3% of all photoreceptors in mice (Carter-Dawson and LaVail, 1979; Jeon et al., 1998). In humans, rods dominate the peripheral retina, whereas cone density increases towards the macula, reaching the highest density in the foveola. Mice lack a macula, and the density of cones increases dorso-ventrally (Ortin-Martinez et al., 2014).
Rods and cones have four primary structural/functional regions: outer segment, inner segment, cell body, and synaptic terminal. The outer segment is filled with a dense stack of membrane disks which contain the visual pigment (rhodopsin in rods and cone pigment in cones) and other proteins related to phototransduction or disk structure. Humans have a single rod pigment, rhodopsin, and usually have three kinds of cones that provide color vision by responding differently to light of different wavelengths (S-cones, M-cones and L-cones, responding to short-, medium- and long-wavelength light) based on the opsin present. Mice have a single rod pigment, rhodopsin, and two cone pigments (S- and M-cone pigments), with some individual cone cells expressing both S- and M-cone pigments (Applebury et al., 2000).
Photons cause conversion of 11-cis retinal to all-trans retinal, leading to rhodopsin activation and downstream signaling initiated by the G protein, transducin (Fain et al., 2010; Filipek et al., 2003; Lagnado and Baylor, 1992; Palczewski, 2006). Subsequent signaling leads to the hydrolysis of cGMP to GMP, resulting in the closing of cGMP-gated ion channels, ultimately causing hyperpolarization and less glutamate release from those cells (Lagnado and Baylor, 1992). The RPE works with photoreceptor cells to regenerate 11-cis retinal via the classical visual cycle to support function of both rods and cones (Felius et al., 2002; Saari, 2000; Samardzija et al., 2008). In the face of high rates of photon capture, cones are supplemented with additional 11-cis-retinal production also from Mueller cells (Palczewski and Kiser, 2020; Wang and Kefalov, 2011).
Phototransduction and replacement of shed photoreceptor outer segments during the day is very energy intensive (Ng et al., 2015), but photoreceptor cells have been reported to use more energy at night when photoreceptor ion channels are open (dark current) than in the day (Arden et al., 2005; Arden et al., 1998; Haugh et al., 1990; Linsenmeier, 1986; Okawa et al., 2008; Wang et al., 2010). In darkness, the outer segments of rod cells are depolarized, and energy and oxygen are consumed within the inner segments to support ion pumping (Ames et al., 1992), resulting in almost anoxic conditions at the proximal side of the inner segments (Wangsa-Wirawan and Linsenmeier, 2003). Likewise, a significant portion of rod cell ATP consumption in the dark occurs from Na/K-ATPase pumping out excess Na+ entering the photoreceptors via cGMP-gated channels in the outer segments, thereby maintaining the dark current and intracellular ion levels (Hagins et al., 1970; Lee et al., 2018; Okawa et al., 2008). An inhibitor of RPE65 (and thus the visual cycle), emixustat, reduced oxygen consumption and ion pumping in the dark (Kubota et al., 2019), a condition in which visual cycle activity already is low. Additional research is needed to determine the effect of such therapies on visual cycle activity under light-adapted conditions.
Light produces a large decrease in ATP consumption in photoreceptors resulting mostly from the decrease in ion influx through the cGMP-gated and Ca2+ channels, which is not compensated by the ATP needed for transduction (Okawa et al., 2008). Thus, the amount of ATP and oxygen consumed during phototransduction is small compared to that consumed as a result of the dark current (Lau and Linsenmeier, 2012; Linsenmeier, 1986; Okawa et al., 2008). In bright light, individual cone cells use much more ATP than individual rods (Medrano and Fox, 1995), but in darkness, ATP utilization by individual cones is regarded as similar to that in individual rod cells, because the amplitude and voltage-dependence of the dark current is similar in rods and cones (Nikonov et al., 2006; Yagi and Macleish, 1994).
Most of the glucose that reaches the retina is consumed by glycolysis and converted to lactate (Krebs, 1927; Warburg et al., 1924), and concentrations of lactic acid in the subretinal space can reach 19 mM (Adler and Southwick, 1992). Early evidence suggested that lactate released by Mueller glial cells was metabolized by photoreceptor cells (Poitry-Yamate et al., 1995), but other evidence suggests that photoreceptor cells themselves are the major site of aerobic glycolysis (Chinchore et al., 2017; Du et al., 2016; Lindsay et al., 2014; Medrano and Fox, 1995; Wang et al., 1997; Winkler, 1981). The purpose of aerobic glycolysis in photoreceptor cells has been proposed to be for anabolic metabolism and to release lactate to fuel MGC’s and suppress glycolysis in the RPE so that sufficient glucose can flow through the RPE to support metabolism by the photoreceptor cells (Chinchore et al., 2017; Kanow et al., 2017; Lindsay et al., 2014; Rajala et al., 2016). Both photoreceptors and RPE express monocarboxylic acid transporters that permit the transport of lactic acid between the two cell types (and other retinal cells).
In the mammalian retina, many cellular phenomena, including neuronal activity, are regulated by a circadian clock. Evidence generated in nondiabetic rabbits indicates that a circadian clock regulates the extracellular pH of the retina so that the pH is lower at night than in the day, especially the vicinity of the inner segments of photoreceptor cells, supporting the idea that photoreceptors serve as the primary source of protons (Dmitriev and Mangel, 2001). Assuming that the drop in pH reflects increased production of lactic acid via glycolysis, then the proportion of ATP generation by aerobic and anaerobic metabolism during the dark phase might not strongly dependent on the metabolic need of the photoreceptor in the dark. Perhaps the circadian clock increases photoreceptor cell glycolysis and lactic acid production independent of illumination.
2.2. Retinal pigment epithelium
The RPE is a highly specialized monolayer of pigmented cells, located between the choriocapillaris and the outer segment of photoreceptors (Marmorstein, 2001; Miller and Steinberg, 1977; Rizzolo, 1997; Strauss, 2005). The apical side of the RPE faces the outer segment of photoreceptors, while the basolateral side faces Bruch’s membrane, which separates the RPE from the choriocapillaris (Strauss, 2005). The interaction between the RPE and photoreceptor cells is important in maintaining the structural and functional integrity of the photoreceptors, including maintenance of the visual cycle in which all-trans retinal is recycled to 11-cis retinal. The RPE also helps maintain the photoreceptors by the diurnal phagocytosis of shed photoreceptor outer segments, by transport of water, ions, and metabolic products from the sub-retinal space to the blood, by secretion of neurotrophic factors, and by transport of nutrients (such as glucose and fatty acids) from the blood to photoreceptors (Strauss, 2005). The outer blood-retinal barrier exists at the RPE due to the tight junctions between the epithelial cells that control the movement of fluid and metabolites into and out of the neural retina.
2.3. Interphotoreceptor matrix
The interphotoreceptor matrix (IPM) is an extracellular matrix that lies in the subretinal space between the photoreceptor cells and the RPE, and it plays important roles in photoreceptor cell survival and dysfunction (Ishikawa et al., 2015). Functional roles of the IPM include retinal adhesion to the RPE, providing receptors for growth factor presentation, facilitating retinoid transport between photoreceptor cells and RPE, and transport of oxygen and nutrients to the photoreceptor cells. Some genes encoding proteins that are localized to the IPM have been associated with human inherited retinal diseases, including autosomal-recessive retinitis pigmentosa (Bandah-Rozenfeld et al., 2010), autosomal-dominant and -recessive forms of Best disease (Manes et al., 2013), Sorsby’s fundus dystrophy (Weber et al., 1994), and Doyne honeycomb retinal dystrophy (Marmorstein et al., 2002).
2.4. Müeller cells
Müeller cells, the major type of glial cells in the retina, are known to maintain metabolic support of retinal neurons via effects on ions, water, and bicarbonate regulation, regulation of glutamate-mediated retinal synaptic activity, and anti-oxidative support and regulation of the tightness of the blood-retinal barrier and neurovascular coupling (Reichenbach and Bringmann, 2013). Müeller cells touch every cell type in the retina, and are the partners with the photoreceptors in making the outer limiting membrane (OLM) with their tight junctions (Omri et al., 2010). Müeller cells also have been shown to participate in daylight vision by regeneration of 11-cis retinal in cone cells via a process that is independent of the classical visual cycle (Wang and Kefalov, 2011).
2.5. Photoreceptors and RPE as a unit
RPE and photoreceptors in the outer retina normally function as a unit for the maintenance of proper visual function, and disturbance of that close relationship (such as in retinal detachment) can ultimately lead to retinal degeneration and blindness. Likewise, mutations occurring in either the photoreceptor cells or the RPE can lead to retinal degeneration (Bramall et al., 2010).
3. Effect of diabetes on cells of the outer retina
3.1. Methods used to evaluate photoreceptor survival.
Studies generally have relied on a limited number of methodologies to evaluate the loss of cells of the outer retina. These techniques historically have been ophthalmoscopic and histological methods, and more recently, also optical coherence tomography (OCT), adaptive optics (AO), and 2-photon microscopy (Fig 2). Electrophysiology studies (see section 4.1) are less reliable to assess photoreceptor cell survival in diabetes, because pre-clinical studies clearly show diabetes-induced reductions in function despite the continued survival of essentially all photoreceptor cells.
Fig 2.
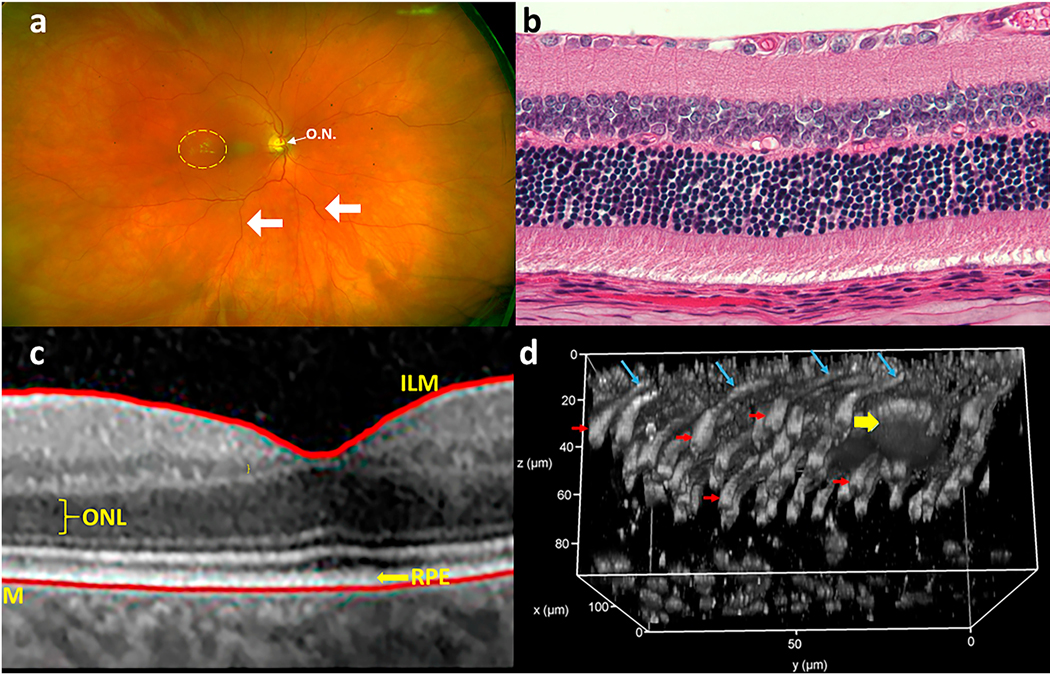
Methods currently used to assess retinal morphology, including the outer retina. (a) Retinal cross-section (hematoxylin and eosin stained retina); (b) optical coherence tomography (OCT) showing various retinal layers (ILM, Inner Limiting membrane; ONL, Outer Nuclear membrane; BM, Bruch’s membrane; RPE, retinal pigment epithelium); (c) color fundus photography (optic nervehead is indicated by O.N.; retinal vasculature is indicated by arrows; exudates are seen to the left of the fovea (yellow dashed circle)); (d) 2-photon microscopy with adaptive optics to generate a 3-dimensional picture of the retina/RPE interface in wildtype mice after intravitreal injection with peanut agglutinin tagged with rhodamine. RPE is at the top of the image at z=0 μm, cone synaptic terminals are at the bottom, around z=80 μm. A macrophage, just under the RPE and around 30 μm in diameter, is indicated with yellow arrow. Cone outer segments are indicated with blue arrows and cone inner segments are indicated with red arrows.
Because of the transparency of photoreceptor cells, conventional ophthalmoscopy has been of limited value in evaluation of photoreceptors in vivo, but loss of RPE cells can be monitored ophthalmoscopically. Histology of retinal tissue sections obviously is conducted on autopsy tissue, but it has the advantages of high resolution and the ability to identify specific cell-types and molecules by immunohistochemistry. OCT and AO, on the other hand, can be used non-invasively in vivo. State-of-the-art spectral domain-OCT devices provide quick, non-invasive and reproducible high-definition images of the anatomical changes in the laminar structure of the retinal layers and pathological modifications occurring in vivo. The images obtained allow discrimination and measurement of individual retinal layers (including the outer nuclear layer (ONL) and RPE), identification and location of cystoid bodies in the retina and vitreal-retinal attachments, and generation of 3D reconstructions of the tissue, but it provides essentially no functional information. AO is a technique allowing in vivo visualization and detection of changes in individual cone photoreceptors, including cone density, interphotoreceptor distance, and cone packing regularity (Lammer et al., 2016; Zaleska-Zmijewska et al., 2017). Adaptive optics scanning laser ophthalmoscopy (AO-SLO) offers high transverse resolution noninvasively, as well as the ability to detect fluorescent signals.
An indirect measure that has been reported to be relevant to outer retinal health is reflectivity. A strong relationship was recently established between outer retinal reflectivity and cone density measured with high-resolution adaptive optics (Flores et al., 2015; Saleh et al., 2017). Moreover, a relationship was observed between reflectivity and visual acuity in eyes having DME (Guyon et al., 2017). OCT image analysis in patients with DR showed that the photoreceptor inner segment ellipsoid layer had significantly lower reflectivity in eyes with mild NPDR compared with that of the control eyes, whereas the reflectivities of RPE and external limiting membrane did not differ between the groups. This was interpreted by the authors as possibly indicating early photoreceptor degeneration or outer segment disorganization (Guyon et al., 2017; Toprak et al., 2015). Unoki et al (Unoki et al., 2007) reported that within areas of nonperfusion of retinal capillaries, high-reflectivity deposits were found between the outer segments of photoreceptor and the RPE, and Bek (Bek, 1994) reported that a homogenous eosinophilic substance accumulated between the photoreceptor outer segments and the RPE corresponding to the areas of vascular nonperfusion and inner retinal changes in patients with DR.
Two-photon microscopy is offering a new noninvasive approach to characterize the structure, metabolic state and function of the retina (outer retina as well as inner retina). Infra-red (IR) light, such as used in two-photon microscopy, offers several advantages for characterization of the retina because IR is absorbed and scattered less than visible light by biological materials (Hammer et al., 1995), and thus an IR light probing beam can be focused at deeper levels in tissues. Second, the front of the human eye becomes progressively less transparent to visible light than to IR with aging (Boettner and Wolter, 1962). To date, two-photon microscopy has elucidated Vitamin A storage compartments within RPE and their role in the visual retinoid cycle (Golczak et al., 2005a; Imanishi et al., 2004a; Imanishi et al., 2004b), the efficiency of visual retinoid cycle in vivo as determined by changes in fluorescence of photoreceptor cells following light stimuli (Sharma et al., 2017), and the impact of candidate drugs on retinal diseases (Zhang et al., 2015). More recent advances have reduced the IR light power needed for safe application of two-photon excitation to the human retina (Palczewska et al., 2020; Palczewska et al., 2018).
3.2. Diabetes-induced changes in photoreceptor cell shape or cell death.
Conflicting results have been reported with respect to whether or not photoreceptor cells die in diabetes; some groups have presented evidence that some photoreceptor cells do degenerate in diabetes (Table 1), whereas others have presented evidence showing little to no loss of photoreceptor cells in diabetes (Table 2). Until recently, there has been little attempt to differentiate effects of diabetes on rods versus cones, but it seems likely that most reports have focused on rod cells due to their greater number in mammalian retinas,.
Table 1.
Studies that report structural changes in the outer retina in diabetes.
| Species | Strain | Duration of diabetes | Type of Diabetes | Photoreceptor abnormality | RPE abnormality | Reference |
|---|---|---|---|---|---|---|
| Rats | Wistar | 6 weeks | Streptozotocin (Type I diabetes) | Reduced photoreceptor cell density | - | (Bueno et al., 2020) |
| Rats | Wistar & Sprague-Dawley | 12 weeks | Streptozotocin (Type I diabetes) | OS shortening in rods, M-cones, and S-cones. | Reduced RPE thickness | (Enzsoly et al., 2014) |
| Rats | Sprague-Dawley | 1– 24 weeks | Streptozotocin (Type I diabetes) | Apoptosis | - | (Park et al., 2003) |
| Rats | Goto-Kakizaki rat | 6–12 months | Type II diabetes | Outer retinal edema and loss of S-cone photoreceptors | - | (Omri et al., 2013) |
| Rats | Otsuka Long‐ Evans Tokushima Fatty (OLETF) | ~14 months | Type II diabetes | Reduction in number of nuclei in ONL | Decreased height of RPE | (Lu et al., 2003) |
| Mice | BALB/c | Unspecified | Streptozotocin & high fat diet (Type II diabetes) | Reduction in ONL thickness | - | (Ren et al., 2016) |
| Mice | C57BL/6J | 8 weeks | Streptozotocin (Type I diabetes) | Loss of rods but not cones | - | (Piano et al., 2016) |
| Mice | C57BKS/J | 8 weeks | db/db (Type II diabetes) | Reduced ONL thickness | - | (Tang et al., 2011) |
| Mice | C57BKS/J | 8–24 weeks | db/db (Type II diabetes) | Reduced ONL thickness | - | (Bogdanov et al., 2014) |
| Mice | C57BL/6J | 3–9 months | Ins2Akita (Type I diabetes) | Modest cone photoreceptor | - | (Hombrebueno et al., 2014) |
| Mice | Not stated | 7–8 months | Ins2Akita (Type I diabetes) | Disorganization and thinning of inner and outer segments | - | (Fu et al., 2018) |
| Rodent | Meriones shawi | Up to 7 months | High fat diet (Type II diabetes) | Decrease in number of cones and photoreceptor OS | Damaged RPE | (Hammoum et al., 2017) |
| Human | - | 18 ± 5 years | Type I or II diabetes | Outer retina layer thinner in DME | - | (Eliwa et al., 2018) |
| Human | - | 22±13 years | Type I or II diabetes | Decreased cone density | - | (Lammer et al., 2016) |
| Human | - | 13 years | Type I or II diabetes | Abnormal cone packing arrangement | - | (Nesper et al., 2017) |
| Human | - | Unspecified | Type I or II diabetes | Selective S-cone loss | - | (Cho et al., 2000) |
| Human | - | Unspecified | Type I or II diabetes | Thinning of RPE in PDR | (Boynton et al., 2015) | |
| Human | - | 8–37 years | Type I diabetes | Reduced cone density | - | (Lombardo et al., 2014; Lombardo et al., 2016) |
| Human | - | Unspecified | Unspecified | Increase in ONL thickness | - | (Wanek et al., 2016) |
| Human | - | 16–18 years | Type II diabetes | Reduced cone density in the moderate/severe NPDR and PDR | - | (Soliman et al., 2016) |
Key:OS, Outer Segment; IS, Inner Segment; ONL, Outer Nuclear Layer; RPE, Retinal Pigment Epithelium; DME, diabetic macular edema; DR, diabetic retinopathy, PDR, proliferative diabetic retinopathy;
Table 2.
Studies that report no structural changes in the outer retina in diabetes.
| Species | Strain | Duration of diabetes | Type of Diabetes | Photoreceptor abnormality | RPE abnormality | Reference |
|---|---|---|---|---|---|---|
| Rats | Sprague-Dawley | 12 weeks | Streptozotocin (Type I diabetes) | No apoptosis in ONL | - | (Enzsoly et al., 2014) |
| Rats | Sprague-Dawley | 4–5 weeks | TetO rats (Type II diabetes) | No change in ONL nuclear density | - | (Reichhart et al., 2017) |
| Mice | C57BL/6J | 6 weeks | Streptozotocin (Type I diabetes) | No change in ONL cell number | - | (Moore-Dotson et al., 2016) |
| Mice | C57BL/6J | 2–14 weeks | Alloxan (Type I diabetes) | No apoptosis or reduction in thickness of the outer retina | - | (Martin et al., 2004) |
| Mice | C57BL/6J | 15–90 days | Alloxan (Type I diabetes) | No apoptosis or reduced thickness in the outer retina | - | (Gaucher et al., 2007) |
| Mice | C57BL/6J | 3–9 months | Ins2Akita (Type I diabetes) | No rod photoreceptor loss | - | (Hombrebueno et al., 2014) |
| Mice | C57BL/6J | 8 months | Streptozotocin (Type I diabetes) | No change in ONL thickness; no ONL cell loss | - | (Liu et al., 2016) |
| Mice | C57BL/6J | 8 months | Streptozotocin (Type I) | No change in ONL thickness; no ONL cell loss | - | (Tonade et al., 2017) |
| Canine (Dogs) | Beagle | 5 years | Alloxan (Type I diabetes) | No change in ONL, IS/OS thickness | - | (Tonade and Kern, 2017) |
| Human | Unspecified | Unspecified | ONL thickness increased in no DR diabetics; no change in NPDR | - | (Wanek et al., 2016) | |
| Human | - | ≥ 5 years | Type I diabetes | No change in cone density | - | (Tan et al., 2015) |
| Human | - | Unspecified | Unspecified | No reduction in ONL thickness | - | (Wanek et al., 2016) |
| Human | - | 5–10 years or more | Type II diabetes | No change in photoreceptor ONL | No reduction in RPE thickness | (Tavares Ferreira et al., 2016) |
| Human | - | 9–15 years | - | No change in ONL | - | (Gerhardinger et al., 1998) |
Key:OS, Outer Segment; IS, Inner Segment; ONL, Outer Nuclear Layer; RPE, Retinal Pigment Epithelium
Even though diabetes-associated changes in the inner retina are more apparent and better studied than those in the outer retina, visible retinopathy has been found to be negatively associated with thickness of the retina and photoreceptor layer (Tavares Ferreira et al., 2017). In another cross-sectional study by the same group (Tavares Ferreira et al., 2016), the thickness of the photoreceptor layer (measured by OCT using automatic segmentation software) was significantly thinner in patients with durations of less than 5 years or greater than 10 years compared to control patients. In contrast to this retinal thinning at <5 years and >10 years of diabetes, however, the thickness of the photoreceptor layer for patients who had been diabetic for an intermediate duration (between 5 and 10 years) was not significantly thinner. The authors concluded that the thinning of the photoreceptor layer was not linear with duration of diabetes. This finding has not been replicated, however, so it is not clear whether this might have been due to a sampling error. In a cross-sectional spectral domain-OCT study of diabetic patients, the photoreceptor outer segment layer at the foveal center was thinner in patients who had DR or DME than in either healthy volunteers and diabetic patients without retinopathy, thus indicating that the severity of DR is associated with changes in the photoreceptor layer (Ozkaya et al., 2017). This apparent loss of photoreceptors did not occur generally across the retina, because no significant differences between groups were found 750 μm temporal to and nasal to the center (Ozkaya et al., 2017). Other investigators likewise observed that abnormal packing of cone (increase in 4- and 8-sided cones compared to 6-sided cones (Lombardo et al., 2016)) within the photoreceptor layer occurred in tiny local foci that were geographically associated with defects in the deep capillary network of the retinal vasculature (Nesper et al., 2017; Scarinci et al., 2015; Scarinci et al., 2016). Some evidence even suggests that the photoreceptor layer undergoes shrinkage before the onset of hyperglycemia and overt diabetes; patients with metabolic syndrome were found to have a thinner than normal photoreceptor layer via OCT segmentation analysis, raising a possibility that insulin resistance or adipose tissue-derived inflammation might have an adverse effect on photoreceptor cell integrity independent of the hyperglycemia (Karaca and Karaca, 2018).
Photoreceptor alterations that are less severe than outright cell loss are represented by disruption of the external limiting membrane or the junction between the inner and outer segments (Table 3), and this alteration has been correlated with visual impairment. Hyper-reflective foci in the outer retinal layers on spectral domain-OCT images, another marker of visual disturbance, have been associated with foveal photoreceptor damage (Murakami and Yoshimura, 2013).
Table 3.
Reported changes in shape of outer retinal cells in diabetes.
| Species | Strain | Duration of diabetes | Type of Diabetes | Photoreceptor abnormality | RPE abnormality | Reference |
|---|---|---|---|---|---|---|
| Rats | Wistar | 3 weeks | Streptozotocin (Type I diabetes) | Reduced cone cell OS and number | - | (Szabadfi et al., 2012) |
| Human | - | 5–25 years | Unspecified | PDR patients having abnormal RPE fluorescence had thinner outer retina than patients with normal RPE fluorescence | PDR patients having abnormal RPE fluorescence had thinner outer retina than patients with normal RPE fluorescence | (Kang et al., 2016) |
| Human | - | Unspecified | Type I or II diabetes | Thinning of Outer Segments at foveal center in DME | - | (Ozkaya et al., 2017) |
| Human | - | IS/OS damage due to DME | - | (Muftuoglu et al., 2017a) | ||
| Human | - | Unspecified | Type II diabetes | Reduced thickness of ONL | - | (Tavares Ferreira et al., 2017) |
| Human | - | 5–10 years or more | Type II diabetes | Reduced thickness of IS/OS | - | (Tavares Ferreira et al., 2016) |
| Human | - | Unspecified | Type I or II diabetes | Selective S-cone loss; Reduced ONL, IS, and OS thickness | Thinning of RPE | (Cho et al., 2000) |
| Human | - | Unspecified | Type I or II diabetes | Selective S-cone loss; Reduced ONL, IS, and OS thickness | Thinning of RPE | (Boynton et al., 2015) |
| Human | - | ≥ 5 years | Type I and II diabetes | Reduced thickness in photoreceptor IS and OS | Thinning of RPE | (Bavinger et al., 2016) |
The majority of pre-clinical studies investigating photoreceptor cell health in diabetes have been conducted in rodents, and accordingly, for only a relatively short duration of diabetes. In some studies of diabetic rats, photoreceptor outer segments, which normally are in contact with the RPE, were found to be disorganized and reduced in number (Enzsoly et al., 2014; Tso et al., 1980), but these studies suffered from methodological or design weaknesses (brief fixation, a small number of animals, and/or very short duration of diabetes) that make their conclusions suspect. In Sprague–Dawley (SD) rats diabetic for one month, the thickness of the photoreceptor segment layers was significantly reduced by almost 19% compared to nondiabetics (Aizu et al., 2002). Wistar rats diabetic for 6 weeks showed an average of 31% lower density of photoreceptor cells across all retinal quadrants (Bueno et al., 2020). A model of type 2 diabetes, the BTBR ob/ob mouse, showed a modest, but statistically significant, reduction in thickness in the ONL at 20 weeks of age, but not at 6 weeks of age (Lee et al., 2018). In another model of type 2 diabetes (Otsuka Long‐Evans Tokushima Fatty (OLETF) rats), the number of photoreceptor cell nuclei decreased, RPE decreased in height and basal infoldings were poorly developed by 19 months of age (approximately 14 months duration of diabetes) (Lu et al., 2003). Another study, however, evaluated photoreceptor survival in dogs which had been maintained in intentionally good glycemic control or intentionally poor glycemic control for five years (Tonade and Kern, 2017). Retinas from these dogs previously had been evaluated with respect to retinal vascular pathology of DR, and the vascular pathology had been reported to be significantly worsened by poorly controlled glycemia, and significantly inhibited by good glycemic control (Engerman et al., 1977). In contrast to the demonstrable effects of glycemic control on the retinal vasculature, however, the five years of chronic hyperglycemia did not cause any reduction in thickness of the ONL (assessed in tissue sections of non-tapetal inferior, nasal retina), thus showing no evidence of loss of retinal photoreceptors due to diabetes in this large animal. In retinas of a single hypertensive monkey who spontaneously developed type 2 diabetes, a severe decline in number of photoreceptor inner and outer segments was observed (Johnson et al., 2005).
Cones also are affected in diabetes. Compared with age-matched control subjects, Lombardo et al (Lombardo et al., 2014) found a 10% decrease of parafoveal cone density in type 1 diabetic patients without maculopathy (mean duration of diabetes of 13 years) via high-resolution adaptive optics retinal imaging. Cone density, dispersion index, and packing index also were significantly different between patients lacking or those having mild NPDR, with these differences increasing with duration of diabetes (Lombardo et al., 2016; Soliman et al., 2016). Some diabetic patients showed localized areas of either missing or nonreflecting cones that persisted over time (Sawides et al., 2017). The data suggests more than merely an association between these cone cell defects and diabetes, because cone density was not significantly lower in prediabetic patients than that in a group of nondiabetic control patients (Zaleska-Zmijewska et al., 2017), but was significantly less than normal in patients with mild or moderate nonproliferative DR (Zaleska-Zmijewska et al., 2019). Decreased regularity of cone arrangement in the macula was correlated with presence of diabetes, increasing severity of DR, and presence of DME (Lammer et al., 2016), but investigators have not found a correlation between cone density and the level of severity of glycemic control (as estimated from HbA1c) or the duration of diabetes (Lammer et al., 2016; Soliman et al., 2016).
Diabetes has been shown to damage cone photoreceptors also in some animal studies. At 12 weeks of diabetes, Wistar and Sprague Dawley rats showed morphologic abnormalities in the outer segments of rods, most M-cones, and some S-cones (Enzsoly et al., 2014), although retinal thickness and the density of cones expressing M- and S-sensitive opsins was unchanged, and no significant increase in the number of apoptotic cells was detected. Retinas of spontaneously diabetic Goto-Kakazaki rats (a model of Type 2 diabetes) were found to have a 20% decrease in cell cone density compared to controls by 12 months of age (Omri et al., 2013). Cone photoreceptor outer and inner segments were disorganized in Akita diabetic mice, and the thickness of rod photoreceptor segments was reduced, but administration of a long-acting FGF21 analog (PF-05231023) inhibited these defects (Fu et al., 2018). Hyperglycemia in zebrafish (induced by oscillating levels of glucose in the tank for 30 days) disrupted cone photoreceptor neurons, as evidenced by prominent morphological degeneration and dysfunctional cone-mediated ERG (Alvarez et al., 2010).
3.3. Which photoreceptors die in diabetes?
Although some studies have reported thinning of the ONL or the presence of TUNEL-positive cells in the ONL, positive identification of which cell-types undergo the cell death are incomplete. The only photoreceptor cell-type that has positively been identified as being lost in diabetes is the S-cone (Fig 3).
Fig 3.

Reduction in number of S-cones in retinas from diabetic patients. Enzyme histochemical reaction for carbonic anhydrase produces a black reaction product which labels L/M-cones, but not S-cones (arrows) or rods. Figs a and b are from the photoreceptor layer of the retina. Compared to nondiabetic patients (a), retinas from diabetic patients have a relative absence of carbonic anhydrase-negative S-cones (b). A summary of the density of (c) S-cones and (d) L/M cones at various retinal eccentricities shows significantly lower mean S-cone density at most eccentricities in diabetic patients compared to nondiabetic controls. Asterisks (*) indicate that differences in means are significant different between diabetic and nondiabetic patients. This figure is modified from figures 9, 12 and 13 in an article entitled, “Acquired color vision loss and a possible mechanism of ganglion cell death in glaucoma” in the Trans Am Ophthalmol Soc 2000;98: 331–63, and is republished with permission of the American Ophthalmological Society.
Loss of S-cones in diabetes has been detected in several studies of post-mortem retinae of human subjects with background or proliferative DR (Adams et al., 1987; Cho et al., 2000; Yamamoto et al., 1996). Analysis revealed incomplete and patchy loss of S-cones only in retinas from diabetics, and this cell loss did not occur in L/M cones. A statistically significant reduction in the density of S-cones was found at nearly all foveal eccentricities across the tissue, and the fraction of S-cones relative to the total number of cones was decreased by more than 20% with respect to the controls (Cho et al., 2000). A significant disruption of some S-cone outer segments was observed in rats by 6 months of diabetes, whereas discontinuities additionally were observed at the OLM at 12 months of diabetes, and the organized honeycomb pattern of tight junctions in the OLM around photoreceptor inner segments was markedly disrupted in diabetes (Omri et al., 2013).
It is not known why blue cones seem selectively affected in diabetes, but blue-sensitive cones differ appreciably from L- or M-cones. More than 95% of the amino acid sequences of L- or M-cone opsins are homologous, compared with only a 43% identity with S-cone opsin (Nathans et al., 1986).
Not all investigators have found evidence of photoreceptor cell loss in diabetes, or that it was consistent between types of diabetes. ONL thickness was found to be increased compared to controls in diabetic patients without retinopathy (Wanek et al., 2016), and others found that the outer retina seemed not to be affected at early stages of diabetes (Vujosevic and Midena, 2013). In another study, ONL thickness was decreased in parts of the pericentral and peripheral areas in the patients with Type 1 diabetes, but increased in patients having Type 2 diabetes (Chen et al., 2016). Studies conducted using Spontaneously Diabetic Torii (SDT) fatty rats reported that retinal thickness was significantly thicker than normal in this model, and this thickening occurred in both the ONL and all layers of the inner retina (Motohashi et al., 2018). Obviously, thickening of retinal layers might be due to accumulation of fluid within those layers (edema).
3.4. Photoreceptors and retinal edema.
DME, characterized by retinal thickening resulting from leaky blood vessels is more prevalent during the later phases of the retinopathy (Antonetti et al., 2012; Cunha-Vaz et al., 2014). It significantly impacts the photoreceptor layer in affected diabetic patients. Patients having DME had subnormal thickness of the outer retinal layer, and there was a stronger correlation between thickness of the outer retinal layer and vision than between central foveal point thickness and vision (Eliwa et al., 2018). In another cross-sectional clinical study, the mean thickness of the macular outer retinal layers were decreased significantly in both DME and non-DME groups compared with the control group (Wang et al., 2018).
Several studies have reported foveal microstructural defects of the photoreceptor layer occurring after DME that are closely associated with impaired visual acuity (Maheshwary et al., 2010; Oster et al., 2010; Otani et al., 2010; Sakamoto et al., 2009; Shen et al., 2016; Uji et al., 2012; Yanyali et al., 2011). These foveal microstructural abnormalities in photoreceptor cells have prognostic value. About half of eyes from patients who had DME with IS/OS damage in DME achieved complete restoration of IS/OS after resolution of DME, but those eyes in which IS/OS was not restored had more IS/OS damage at baseline, a longer duration of DME, and ended up with worse vision at follow-up (Muftuoglu et al., 2017a; Muftuoglu et al., 2017b).
Evidence of retinal edema in diabetic animal models has been infrequently reported, but noninvasive quantitative MRI measurements of whole central retinal thickness, intraretinal water content and apparent diffusion coefficients (water material) showed that diabetes caused an increase in whole central retinal thickness and water content metrics in male Sprague Dawley rats after as little as 2–4 months of diabetes (Berkowitz et al., 2012b). This is relevant to the present discussion because significant differences between water mobility (apparent diffusion coefficient) between diabetic and controls were restricted to the far outer half of the retina. Likewise at 2 months of diabetes, others (Danilova et al., 2018) reported partial destruction of photoreceptors as well as interstitial retinal edema, resulting in a reduction in the thickness of the retina in rats experimentally diabetic for 2 months. Retinal vascular hyperpermeability and thickening of the retina and layers between the retinal internal limiting membrane and the outer nuclear layer were reported in spontaneously diabetic Torii fatty rats at longer than 16 weeks of age (Motohashi et al., 2018; Toyoda et al., 2016). In spontaneously diabetic (type 2 diabetes) Goto-Kakizaki rats at 12 months of age, Omri et al (Omri et al., 2013) found the outer retina to be swollen and disorganized, notably having increased extracellular spaces between nuclei of photoreceptors.
3.5. Retinal pigment epithelium
Diabetes-induced alterations in the RPE have been reported (Omri et al., 2011; Vinores et al., 1988; Xu et al., 2011). Twelve weeks of diabetes in Wistar and Sprague Dawley rats was reported to cause RPE thinning and reduced RPE65 protein immunoreactivity (Enzsoly et al., 2014). Electron microscopy of diabetic rats showed disruptions in the RPE layer, shrunken nuclei, dilated and reduced endoplasmic reticulum, in-folding of cell membrane, altered melanosome distribution, as well as loss/degeneration of RPE cells (Blair et al., 1984; Tarchick et al., 2016; Tso et al., 1980; Wallow, 1983). A retrospective comparison of PDR patients having granular hypo- or hyper-autofluorescence (identified as diabetic retinal pigment epitheliopathy) with age- and sex-matched PDR patients lacking the autofluorescence defects found that eyes having the epitheliopathy had a thinner outer retina and thicker choroid, and had a worse logMAR (Logarithm of the Minimum Angle of Resolution) visual acuity than eyes in the PDR controls (Kang et al., 2016). In a study of diabetic patients with and without DME, RPE thickness was decreased in both groups compared with the control group, except in the macular and central ring (Wang et al., 2018).
3.6. Molecular alterations in photoreceptor cells and RPE in diabetes
3.6.1. Oxidative stress
Diabetes gives rise to increased oxidative stress in the retina (Al-Shabrawey et al., 2008a; Al-Shabrawey et al., 2008b; Berkowitz et al., 2015c; Caldwell et al., 2005; Du et al., 2013; Kanwar et al., 2007; Kowluru et al., 2015; Kowluru et al., 2001b). This stress plays a critical role in the development of DR, because vascular lesions such as capillary degeneration are inhibited by administration of anti-oxidants (Kowluru et al., 2001b) or over-expression of anti-oxidant enzymes such as superoxide dismutase (Berkowitz et al., 2009a; Kanwar et al., 2007). In vitro studies have shown that cultured endothelial cells, pericytes, and Müller cells can contribute to superoxide production, but the relative amount of oxidative stress coming from those cells is minor compared to that coming from photoreceptors at 8 weeks of diabetes (Du et al., 2013; Lee et al., 2012). Adherent or diapedesed leukocytes in retinas from diabetics might contribute to the diabetes-induced increase in retinal superoxide, but dicholorofluorescein- or dihydroethidium staining of retinal cryosections clearly shows that the majority of the diabetes-induced retinal oxidative stress comes from photoreceptors (Du et al., 2013; Liu et al., 2019), at least in early diabetes (Fig 4). Superoxide generation in diabetic db/db mice was greater at 8 weeks of age than that in nondiabetic controls (notably in the ONL), and it continued to progress and involve additional retinal cell-types by 20 weeks of age (Xiao et al., 2012). Consistent with photoreceptor cells being major contributors to the retinal oxidative stress of diabetes, the diabetes-induced increase in retinal superoxide was inhibited in retinas in which photoreceptor cells had degenerated due to opsin deficiency or chemically (via iodoacetic acid) (Du et al., 2013). Also, when photoreceptors are still present but “stressed” (such as in diabetes) or degenerating (such as in opsin−/−rodent models), they produce supranormal levels of superoxide in the retina (Liu et al., 2016). For reasons that remain unexplained, superoxide levels in retinas in which photoreceptor cells expressed a mutant opsin (P23H) became greater than normal and remained elevated even after the mutant photoreceptors had degenerated (Liu et al., 2016). Rod dysfunction and oxidative stress in diabetes are partially corrected by acute 11-cis-retinaldehyde treatment (Berkowitz et al., 2015c), which may be due to antioxidant properties. Nrf2 likewise is an important endogenous protective factor against oxidative stress in photoreceptor cells in light-induced damage and diabetes (Chen et al., 2017; Xu et al., 2014).
Fig 4.

Diabetes increases oxidative stress in the outer retina of diabetic rodents. (a) Oxidative stress was detected particularly in inner/outer segments of retinal photoreceptor cells of mice diabetic for 2 months compared to age-matched nondiabetic mice. Oxidative stress was detected in cryosections stained with dichlorofluorescein, with green indicates sites of oxidative stress, and blue staining of nuclei stained (DAPI). Retinal layers are indicated by abbreviations between the two figures. (b) Diabetes (D) of 2 months duration also decreased retinal activity of superoxide dismutase compared to that in nondiabetic (N) rats, and this decrease was inhibited by dietary supplementation with antioxidants. (c) Continuously produced paramagnetic free radicals from the outer retina measured in vivo using high-resolution 1/T1 magnetic resonance imaging (MRI) shows that only the outer retinal 1/T1 values from diabetic animals were significantly greater than normal, and were corrected to baseline with antioxidant therapy. Fig 4b reproduces a portion of Fig 2 in Free Radic Biol Med, Vol 26, Kowluru RA, Engerman RL, Kern TS, 1999. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. VI. Comparison of retinal and cerebral cortex metabolism, and effects of antioxidant therapy. Pages 371–378,1999, with permission from Elsevier. Fig 4c was reprinted with permission from Berkowitz BA, Bredell BX, Davis C, Samardzija M, Grimm C, Roberts R. Measuring In Vivo Free Radical Production by the Outer Retina. Invest Ophthalmol Vis Sci. 2015;56:7931–7938. © 2015 ARVO.
Leukocytes circulating in the blood also play a role in the diabetes-induced increase in retinal superoxide generation. Deletion of iNOS, PARP1, TLR2/4, IL-1β, or MyD88 only from marrow-derived cells inhibited the diabetes-induced increase in superoxide production in the retina (Li et al., 2012a; Tang et al., 2013). In addition, an example of cell-to-cell communication between leukocytes and retinal cells can be exemplified with 5-lipoxygenase (5-Lox). 5-Lox is found in leukocytes but not in retinal cells, and the deletion of 5-Lox solely from myeloid-derived cells significantly inhibited the increased generation of superoxide by the retina in diabetes (Gubitosi-Klug et al., 2008). Based on the evidence that the excess superoxide produced in the retina is generated mainly by photoreceptor cells, the release of leukotrienes and perhaps other lipid mediators or soluble factors from myeloid-derived cells are strong candidates to influence superoxide production by photoreceptor cells in diabetes.
RPE contribution to retinal oxidative stress in diabetes has also been studied in vivo and in vitro. Li et al showed in vitro that RPE cells cultured in diabetes-like conditions produced increased levels of reactive oxygen species compared to those incubated under nondiabetic conditions (Li et al., 2012b). Although not directly studying oxidative stress in the RPE, administration of retinylamine (an inhibitor of RPE65 in RPE) to mice inhibited the diabetes-induced increase of superoxide in the retina (Liu et al., 2015), suggesting that the visual cycle in RPE cells contributes to the increase in retinal (and especially photoreceptor cell) oxidative stress in diabetes. In that study, retinylamine was administered at low doses only once per week, so direct anti-oxidant effects seem unlikely (Liu et al., 2015). The effect of superoxide on the RPE has also been demonstrated by Bailey and colleagues, who showed that chronic oxidative stress can disrupt RPE cell junctions and barrier integrity (Bailey et al., 2004). Oxidative stress is reported to cleave RPE65 in RPE cells in vitro, but whether or not this occurs also in vivo remains unclear (Lee et al., 2010).
Several relatively noninvasive methods to assess retinal oxidative stress or free radical generation recently have been described. A reactive oxygen species-activated, near-infrared hydrocyanine-800CW fluorescent probe was tested in light-induced retinal degeneration mouse models (detected by scanning laser ophthalmoscopy), and was found to appear in photoreceptor inner segments, similar to the established marker for reactive oxygen species, dichlorofluorescein (Prunty et al., 2015). Likewise, L-012, a chemiluminescent probe, was shown to safely demonstrate reactive oxygen species in the mouse retina following optic nerve crush or retinal ischemia/reperfusion injury models (Fan et al., 2017). Berkowitz and coworkers have used high-resolution 1/T1 magnetic resonance imaging (MRI) to noninvasively measure and localize paramagnetic free radicals generated in the retina. In both diabetes and the sodium iodate model, 1/T1 values in the outer retina were significantly greater than normal, and corrected to baseline with the combination of methylene blue and α-lipoic acid (Berkowitz et al., 2015a; Berkowitz et al., 2016b). Likewise, OCT studies have demonstrated that exposure to light causes an expansion of the outer retina, which can be inhibited by diltiazem (Berkowitz et al., 2019), which has been reported to cause retinal oxidative stress in retinal photoreceptor cells (Berkowitz et al., 2018). Inhibition of the diltiazem effect on retinal expansion by lipoic acid and methylene blue led the authors to conclude that the light-induced expansion of the outer retina was inhibited by oxidative stress, and that this phenomena might be used noninvasively to assess oxidative stress in the outer retina (Berkowitz et al., 2019). Nevertheless, the multitude of actions of diltiazem, lipoic acid and methylene blue suggest that additional research is warranted before attributing the inhibition of light-induced thickening of the outer retina solely to oxidative stress.
3.6.2. Inflammation
Induction of pro-inflammatory proteins, such as iNOS, ICAM1, and TNF-α, have been shown to be induced in the retina in diabetes, and to contribute to the development of microvascular lesions characteristic of DR, including vascular degeneration and permeability (Huang et al., 2011; Joussen et al., 2009; Joussen et al., 2004; Zheng et al., 2007). Recent evidence indicates that photoreceptor cells contribute to the diabetes-induced increases in inflammatory proteins in the retina (de Gooyer et al., 2006b; Du et al., 2013), and photoreceptor cells themselves are a source of increased inflammatory proteins (e.g. IL-1α, IL-1β, IL-6, IL-12, chemokine C-X-C motif ligand 1 (CXCL1), monocyte chemoattractant protein 1 (MCP-1), CXCL12a, I-309, chemokine ligand 25 (CCL25) and TNF-α) in diabetes (Tonade et al., 2017). Scuderi et al showed that photoreceptor cells are a source of IL-1β and its component receptors in the retina in diabetes (Scuderi et al., 2015). Evidence that photoreceptor cells can in fact be a source of pro-inflammatory proteins should not be surprising, since photoreceptor cells were shown to produce TNF-α and iNOS, respectively, in sympathetic ophthalmia and experimental uveitis (Parikh et al., 2008; Rajendram et al., 2007). ICAM-1 is elevated also in the retinal and choroidal vasculatures of diabetic patients (McLeod et al., 1995).
Inflammation within photoreceptor cells might be regulated by NF-κB signaling. When incubated under diabetes-like conditions, NF-κB signaling was activated in 661W cells (which show properties of both retinal ganglion and cone photoreceptor cells (Sayyad et al., 2017)), and transforming growth factor β-activated kinase 1 (TAK1) and NADPH oxidase were involved in that hyperglycemia-induced activation of NF-κB signaling and the release of soluble inflammatory products in those cells (Tonade et al., 2017). TAK1 is known to regulate IKK/NF-κB and MAPK pathways (Ajibade et al., 2013; Landstrom, 2010; Wang et al., 2001). Whether hyperglycemia causes activation of NF-κB signaling in photoreceptor or RPE cells in vivo is not yet known.
There is evidence that the RPE also contributes to retinal inflammation in diabetes. Administration of the RPE65 inhibitor, retinylamine, mitigated the diabetes-induced increase in expression of inflammatory proteins in the neural retina (Liu et al., 2015). How metabolic activity in RPE cells in diabetes influences expression of inflammatory proteins in retinal cells is not yet clear, but RPE cells have been shown to produce inflammatory proteins in other retinal diseases, such as proliferative vitreoretinopathy and uveitis (Jaffe et al., 1995; Ponnalagu et al., 2017). It was reported that activated monocytes release products that induce the expression of IL-1β, IL-6, IL-8, and other factors in human RPE cells (Jaffe et al., 1995).
3.6.3. Ion flux in photoreceptors
Ion flux into photoreceptor cells plays a critical role in processes that lead to vision, and these processes become disturbed in diabetes. Subnormal ion movement into photoreceptor cells of diabetic rodents has been demonstrated noninvasively using manganese-enhanced MRI (described below in section 4.3). Diabetes-induced decreases in activities of enzymes regulating ion movement in cells, such as calcium ATPase and Na/K-ATPase, in retina and RPE have been described (Bensaoula and Ottlecz, 1995, 2001; Crider et al., 1997; Kern et al., 1994; Kowluru, 2002; Kowluru et al., 1999; Kowluru et al., 1998; MacGregor and Matschinsky, 1986a; MacGregor and Matschinsky, 1986b; Ottlecz and Bensaoula, 1996; Ottlecz et al., 1993). Inasmuch as photoreceptor cells represent the majority of the cellular mass of the retina, the majority of the diabetes-induced decrease in Na/K-ATPase activity might occur in photoreceptor cells, but that assumption has not been directly verified to date. The diabetes-induced defect in ion movement into photoreceptor cells has been found to be inhibited by the antioxidants, lipoic acid or catalase, deletion of the inducible isoform of nitric oxide synthase (iNOS), or by photobiomodulation therapy with brief daily treatment with far-red light (Berkowitz et al., 2009a; Berkowitz et al., 2007b; Giordano et al., 2015; Saliba et al., 2015; Zheng et al., 2007).
3.6.4. Insulin signaling
The insulin receptor is known to be present in multiple layers of the human retina, including the outer nuclear layer, inner segments of rods and cones, the outer limiting membrane and in the pigment epithelium, and diabetes reduced expression of the receptor, particularly in the inner segments of the rods and cones (Albert-Fort et al., 2014; Naeser, 1997; Natalini et al., 2016; Rajala et al., 2007; Rajala et al., 2013; Rajala et al., 2008; Rajala et al., 2006; Samuels et al., 2015). Reiter and colleagues have reported that diabetes-induced losses of insulin receptor and Akt kinase activity in the retina were inhibited by systemic insulin treatment, and acute intravitreal insulin restored insulin receptor kinase activity (Imai et al., 2015; Reiter et al., 2003; Reiter et al., 2006). Various approaches have been reported to address the insulin resistance in the retina and retinal cells (Jiang et al., 2018; Jiang et al., 2015; Jiang et al., 2014a; Jiang et al., 2014b; Jiang et al., 2014c), but whether this resistance occurs in photoreceptors is not clear.
Exogenous insulin has been reported to disrupt tight junctions on cultured RPE cells, leading to an increase in permeability across the RPE (Sugimoto et al., 2013), but this has not yet been confirmed in vivo. A recent study showed evidence of endogenous insulin signaling in the RPE that is relevant to diabetes. Use of an RPE-specific insulin receptor knockout resulted in lower ERG a- and b-wave amplitudes in diabetic mice as compared to diabetic mice that expressed the insulin receptor on the RPE (Tarchick et al., 2019). Moreover, loss of insulin receptor-mediated signaling in the RPE reduced levels of reactive oxygen species and the expression of pro-inflammatory cytokines in the retinas of the diabetic mice. Apparently, insulin receptor-mediated signaling in the RPE regulates photoreceptor function and may play a role in the generation of oxidative stress and inflammation in the retina in diabetes.
3.6.5. Hypoxia
Photoreceptor cells have very high levels of mitochondria in their inner segment, and consume most of the oxygen in the retina (Ahmed et al., 1993; Linsenmeier and Braun, 1992), especially in the dark (Arden et al., 2005; Arden et al., 1998; Wang et al., 2010), when rod dark current becomes maximal (Haugh et al., 1990; Linsenmeier, 1986). Hypoxia-inducible factor 1 alpha (HIF-1α), a master regulator of cellular responses, becomes activated during hypoxic events, and has been shown to be increased in the photoreceptors and other layers in diabetes (D’Amico et al., 2015). HIF-1α has been implicated in hypoxia-driven abnormalities in DR, but the contribution of the outer retina to HIF-1α-driven events is unclear. Intraretinal oxygen (PO2) profiles in nondiabetic and diabetic Long-Evans rats indicated that diabetes did not affect oxygen consumption in the photoreceptors in either dark or light adaptation at 4 and 12 weeks of diabetes (Lau and Linsenmeier, 2014).
Retinal hypoxia or ischemia in diabetes (and other conditions) is known to cause the excessive release of VEGF, thus leading to diabetes-induced retinal vascular permeability and neovascularization (Barham et al., 2017; Campochiaro et al., 2016; Simo et al., 2014). Current therapies that use repeated intravitreal injections of VEGF inhibitors to lower intra-ocular levels of VEGF have become a mainstream therapy for the management of DME. Unfortunately, VEGF also mediates protective actions in cells, and Ins2(Akita) mice treated with anti-VEGF therapy exhibited reduced scotopic a- and b-wave and oscillatory potentials, a reduction in the length of the cone photoreceptor segments, and a reduction in cone photoreceptor cell density (Hombrebueno et al., 2015). Despite the widespread use of anti-VEGF therapies clinically, additional evidence on effects of this therapy on the outer retina is lacking.
3.6. Therapies reported to protect photoreceptors in diabetes
A number of therapies have been reported to have beneficial effects on preservation of photoreceptor cell survival or function in diabetes. These reports are not yet sufficient to draw conclusions about the mechanisms responsible for the photoreceptor abnormalities caused by diabetes, but they can serve as starting points to identify potential mechanisms and targets to further study the role of the outer retina in DR.
Selective inactivation of the insulin receptor gene in rod photoreceptors (Rajala et al., 2008) resulted in a reduction in phosphoinositide 3-kinase and Akt survival signal in rod photoreceptors, and resulted in loss of photoreceptor cells and decreased retinal function in mice exposed to bright light stress. These data suggest that the insulin receptor and its downstream signaling are important for photoreceptor metabolism and survival (Rajala et al., 2013; Rajala et al., 2008; Rajala and Anderson, 2010). Since abnormalities in insulin release or signaling are fundamental abnormalities that underlie the development of diabetes, these findings certainly seem relevant to photoreceptor function in diabetes.
Administration of the pyridoxamine, a form of vitamin B6 that can inhibit the formation of advanced glycation endproducts as well as form weak interactions with transition metal ions (Booth et al., 1996; Murakoshi et al., 2009; Stitt et al., 2002), inhibited photoreceptor loss in diabetic BALB/c mice exposed to bright light, potentially via upregulation of Trx, pErk1/2 and Nrf2 expression, and downregulation of ASK1 (Ren et al., 2016).
PKCzeta is required for NF-ĸB-mediated cell death, and a marked accumulation of the phosphorylated p65 subunit of NF-ĸB staining was detected in the photoreceptor layer of diabetic rodents (Omri et al., 2013). An inhibitor of PKCzeta was claimed to have inhibited photoreceptor cell death in diabetes, but no data was provided to support this claim.
Administration of pituitary adenylate cyclase activating polypeptide (PACAP) ameliorated the shortening of rod outer segments and degeneration of cone photoreceptor cells in diabetic rats (Szabadfi et al., 2012), resulting in terminals of the cone photoreceptors being better preserved, with a significant increase in outer segment length and number. The very short duration of diabetes (only 3 weeks) in this study, however, makes interpretation of the relevance of these results to long-standing diabetes difficult to assess.
FGF21 therapy, which decreases body weight and improves the lipid profile in type 2 diabetes (Talukdar et al., 2016), inhibited disorganization and thinning of photoreceptor inner and outer segments in streptozotocin diabetic mice and Ins2Akita diabetic mice (Fu et al., 2018).
Conclusions.
There is now abundant clinical evidence that there can be some photoreceptor cell loss in diabetes, but the extent of that loss is modest compared to that occurring in many genetic retinal degenerative diseases. Whether the degree of photoreceptor cell loss occurring in diabetic patients has a significant impact on visual function in diabetic patients currently is not clear, but the continued presence of most photoreceptor cells makes this seem unlikely. Overall, the differences between studies that do and do not detect photoreceptor death seem not to be attributable simply to differences in duration of diabetes or species studied. Most clinical studies describing photoreceptor survival in diabetes have not differentiated between type 1 and type 2 diabetes, so there currently is not enough information to determine at present whether the type of diabetes has a particular effect on photoreceptor cell loss in diabetes. Animal studies where diabetes type is clearer do not show clear differences between diabetes type. Since the amount of thinning of the ONL seen in most studies related to diabetes is relatively modest, OCT image quality might contribute to differences between clinical studies, but this seems less likely in many animal studies that used histologic methods. Since retinal thickness differs in central and peripheral retina, differences in areas sampled or how photoreceptor death was assessed could affect both clinical and pre-clinical studies. The possibility that photoreceptor cell loss is patchy and does not occur uniformly across the retina also could be an important variable in the differences between reports.
4. Functional and molecular changes of the outer retina in diabetes
Numerous changes in retinal function have been reported in diabetes (Harrison et al., 2011; Holfort et al., 2010), but the present discussion will focus on those that are relevant to photoreceptors and RPE.
4.1. Electrophysiology and color perception
The electroretinogram (ERG) is a noninvasive method to evaluate the function of specific layers or neurons of the retina, and the multifocal ERG has been reported to predict the development of diabetic retinopathy (Bearse et al., 2006; Harrison et al., 2011). ERG defects in diabetes are especially notable in inner retina, as evidenced by the almost universal detection of diabetes-induced changes in the b-wave, but a-wave abnormalities also have been reported. Photoreceptors are the source of the negative a-wave (Penn and Hagins, 1969), and reductions in a-wave amplitude have been used as indications of photoreceptor cell dysfunction. Bresnick and Palta reported that a-wave was significantly delayed in the patients with diabetes compared to non-diabetic control patients (Bresnick and Palta, 1987; Liu and Deng, 2001) (Fig 5), and patients with diabetes and varying levels of retinopathy were detected to have rod and cone deficits in the a-wave compared to age matched control subjects (Holopigian et al., 1997). It is interesting that such inner-retina abnormalities have been reported to be fully accounted for by photoreceptor dysfunction in some, but not all, patients having DR (Holopigian et al., 1992). Amplitude of the a wave has not been found to be abnormal in some studies of diabetic patients (Holopigian et al., 1992; Lovasik and Spafford, 1988).
Fig 5.

Diabetes impairs rod cell function as assessed by ERG. Figure (a) illustrates how latency and amplitude (La and a, respectively) of the a-wave, as well as latency and amplitude (Lb and b respectively) of the b-wave of the ERG are determined. Figure (b) illustrates that diabetes significantly slows a-wave implicit time in diabetic patients (n=58) compared to nondiabetic patients (n=21). (a) is reproduced from Perlman, Ido. “The Electroretinogram: ERG ”. Webvision. Moran Eye Center, January 25, 2012. Web. June 10, 2020. http://webvision.med.utah.edu/book/Part XI: Electrophysiology /The Electroretinogram: ERG/ under a Attribution, Noncommercial 4.0 International (CC BY-NC) Creative Commons license. Fig 5b is drawn from data reported in (Bresnick and Palta, 1987).
Greenstein et al reported losses of S-cone pathway sensitivity (using an increment threshold technique) in diabetic patients with either early or no retinopathy (Greenstein et al., 1990). Bavinger et al showed that there were reductions in cone sensitivity and rod recovery rates (upon bleaching with stimulus light) in patients having moderate NPDR or PDR compared to control subjects (Bavinger et al., 2016). Subjects with untreated PDR compared with subjects treated with PRP exhibited similar rod recovery rates and cone sensitivities. Thinner RPE as assessed by OCT was associated with slower rod recovery and lower cone sensitivity, and thinner photoreceptor inner and outer segments were associated with lower cone sensitivity (Bavinger et al., 2016). The results suggested that RPE and photoreceptor cell dysfunction, as assessed by cone sensitivity level and rod- and RPE-mediated dark adaptation, progresses with worsening DR, and that rod recovery dysfunction occurs earlier than cone dysfunction. In contrast to reports of changes in photoreceptor function in diabetes, Longhin et al reported that diabetic patients without DR and with mild NPDR did not show alterations in rod-based function (as examined by microperimetry and ERG) compared to healthy subjects (Longhin et al., 2016).
Reduced cone sensitivity and attenuated high-frequency flicker ERGs have provided evidence for impaired cone function even in diabetics who have mild or no retinopathy (McAnany et al., 2020; McAnany and Park, 2019). Other tests now are being shown to provide new insight into the functional defects of the outer retina in diabetes (Bavinger et al., 2016; Boynton et al., 2015).
Multi-focal ERG (mfERG) simultaneously collects many local cone-driven ERG signals from the retina under light-adapted conditions (Hood et al., 2012). Waveforms in the mfERG are similar in shape to those of the light-adapted full-field ERG, with an initial negative deflection (termed N1) that has generators similar to those of the a-wave of the light-adapted full-field ERG (Kumar et al., 2013). The N1 component was found to have decreased amplitude in all regions across the retina in diabetic patients with little to no retinopathy (including normal BRB permeability without any clinical signs of DR), as well as in diabetics showing BRB breakdown with no clinical signs of DR or mild nonproliferative DR (Reis et al., 2014; Yu et al., 2002; Ziccardi et al., 2018), but there were no differences in amplitude or implicit time between these groups. DME causes functional impairment in the outer retina, as demonstrated by significantly reduced amplitudes and delayed latencies of mfERG components (Bearse and Ozawa, 2014; Nagesh et al., 2016; Tehrani et al., 2015; Xia et al., 2020).
Unlike the clinical studies using patients with long-standing diabetes, there has been disagreement regarding effects of diabetes on the function of the outer retina in animals. As assessed by amplitude and latency of the ERG a-wave, losses in the function of rod photoreceptors were seen in diabetic Sprague-Dawley rats as early as 2 days after the induction of diabetes, with recovery in some components by 4 weeks and a secondary loss of function at 12 weeks (Bui et al., 2003; Kusari et al., 2007; Phipps et al., 2004). Likewise, Type I and II diabetic mice models were shown to have a progressive decline in a-wave and b-wave amplitudes after the onset of hyperglycemia (Hancock and Kraft, 2004; Samuels et al., 2015). Reichhart et al showed evidence of photoreceptor malfunction (based on the ratio between scotopic a- and b-waves) in that there was a reduction in scotopic a-wave in TetO rats (a type II diabetes model in which the insulin receptor is knocked down) (Reichhart et al., 2017). Similarly, spontaneously diabetic db/db mice and Ins2Akita mice were shown to have defects in a-wave amplitude and implicit time compared to non-diabetic controls (Bogdanov et al., 2014; Hombrebueno et al., 2014).
In contrast, Kohzaki et al reported that there was no significant reduction in a-wave ERG in 11 weeks diabetic rats compared to non-diabetic rats (Kohzaki et al., 2008). Rats diabetic for 12 weeks were shown to have no change in a-wave at the brightest luminous energy (Bui et al., 2009). Ramsey et al observed no significant changes in the sensitivity or amplitude of a- and b-waves in diabetic rats compared to non-diabetic rats (Ramsey et al., 2006). Also, at 22 weeks of diabetes, C57Bl/6 mice were detected to have no significant differences in a-wave amplitudes compared to non-diabetic animals (Samuels et al., 2012), and Ins2Akita mice showed no defect in a-wave at 7–8 months of age (likely 5–6 months diabetes) (Fu et al., 2018).
In an effort to evaluate the cause of compromised neural retinal response to light in early diabetes, ERG studies of the dim light retinal rod pathway were conducted in C57BL/6J mice diabetic for 6 weeks. Those studies showed that rod bipolar cells had no change in excitatory input from rod photoreceptors, although they did show reduced light-evoked inhibitory input from amacrine cells. Although early diabetes causes deficits in the rod pathway, these results provided no evidence that the deficit arises in rods themselves (Moore-Dotson et al., 2016). Slower rod recovery or lower cone sensitivity was associated with thinner RPE or photoreceptor inner/outer segment layer (as assessed by OCT) in diabetes (Bavinger et al., 2016), suggesting that RPE and photoreceptor cell dysfunction progress in parallel with structural changes in the retina in diabetes.
Consistent with other published reports, Becker et al. found that transduction and transmission of light signals by photoreceptor cells (as reflected in amplitude and implicit time of the ERG a-wave) were compromised in db/db mice diabetic (type 2) for 6 months. Using an ex vivo ERG system that eliminates effects of external (systemic) factors, however, rod signaling in retinas from diabetic animals exposed to a normoglycemic environment was found to be similar to that in age-matched nondiabetic controls. Conversely, acutely elevated glucose ex vivo increased light-evoked photoreceptor responses in nondiabetic control mice, but did not affect light responses in diabetic mice. This data suggests that long-term diabetes does not irreversibly change the ability of rod photoreceptors to transduce and mediate light signals, but that rod cells from diabetic mice adapt to the abnormal environment of diabetes to make them less sensitive to increased availability of glucose. Thus, the dysfunction of the light signal transduction or transmission in diabetic rods might not be intrinsic, but instead secondary to systemic abnormalities of the diabetic mileau (Becker et al., 2020). Similarly, the scotopic ERG a- and b-waves were reported to be increased in Zucker diabetic fatty (ZDF) rats, but acute insulin treatment led to blood sugar reduction and a reduction in a-wave amplitudes (Johnson et al., 2013). Apparently, the photoreceptor cells could not quickly adapt to the lower availability of glucose.
Reports of a deterioration in color vision prior to the development of gross vascular pathology of DR are numerous (Green et al., 1985; Hardy et al., 1995; Hardy et al., 1992; Ismail and Whitaker, 1998; Kurtenbach et al., 1994; Moloney and Drury, 1982; Tregear et al., 1997; Trick et al., 1988), and loss of color discrimination for shades of blue and yellow have been reported at this early stage of the disease (Banford et al., 1994; Cho et al., 2000; Dean et al., 1997; Kurtenbach et al., 2002; North et al., 1997a; North et al., 1997b; Roy et al., 1986). This defect has been explained at least in part by selective cone loss (Cho et al., 2000).
There is considerable evidence to suggest that there is a selective vulnerability of the short wavelength photoreceptors (S-cones) and their associated pathway in ocular and systemic disease (Greenstein et al., 1990). This has been demonstrated subjectively in diabetic subjects through psychophysical testing (Banford et al., 1994; North et al., 1997b; Roy et al., 1986) and objectively using the S-cone electroretinogram (Yamamoto et al., 1996; Yamamoto et al., 1997). A clinical test of blue cone pathway sensitivity showed that diabetics were 40 times less sensitive than a group of normal patients, whereas the diabetic group was only 2 times less sensitive than the nondiabetic controls with respect to sensitivity of the red and green cone pathways. The blue cone sensitivity loss was detectable in diabetics who had no macular edema, but was worsened in the presence of edema (Adams et al., 1987). Yamamoto et al (Yamamoto et al., 1996) recorded the S-cone ERG in a group of subjects with Type 1 or 2 diabetes mellitus having either no retinopathy or background DR. The amplitude of the S-cone ERG b-wave was found to be significantly reduced in both diabetic groups (while the amplitude of the L- and M-cone ERG was unaffected), but whether or not this was due to a photoreceptor cell defect per se is not clear. Changes in the S-cone ERG are not universal findings. Some studies report S-cone pathway deficits only apparent when background DR develops (Greenstein et al., 1990; Ismail and Whitaker, 1998; Mortlock et al., 2005).
Alterations in the RPE in diabetes affect both electrophysiology and integrity of the outer blood-retinal barrier (BRB). The c-wave of the ERG is a parameter of RPE function, resulting from the change in trans-epithelial potential due to the hyperpolarization at the apical membrane of the RPE cells (Fishman et al., 2001). Several investigators reported that there is a reduction of c-wave ERG in diabetic rodent models (Fishman et al., 2001; Reichhart et al., 2017; Samuels et al., 2015). Samuels et al demonstrated that different models of Type I diabetes (streptozotocin-induced) and Type II diabetes (mice having the diabetogenic mutation of the leptin receptor (Leprdb/db) on two different backgrounds) developed a reduction in c-wave ERG compared to non-diabetic controls (Samuels et al., 2015). Reichhart et al also showed a reduction in scotopic c-wave in TetO diabetic rats (Reichhart et al., 2017).
4.2. Permeability across the outer BRB.
The blood-retina barrier (BRB) occurs in the endothelial cells of the retinal microvasculature and at the RPE cell layer, and the RPE maintains fluid homeostasis in the retina by removing fluid that is extravasated by the retinal vasculature. Disruption of normal outer BRB function occurs during diabetes (Antonetti et al., 2008; Beasley et al., 2014; Xu et al., 2011), potentially allowing leakage of fluid into the outer retina from the choriocapillaris. Histology of post-mortem eyes from diabetic and control patients showed leakage of albumin from both the inner BRB and outer BRB (Vinores et al., 1989). Likewise, clinical study of patients having NPDR reported that all of the patients enrolled in the study appeared to have fluorescein leakage on fluorescein angiograms which appeared to diffuse from the RPE, suggesting that the outer blood-retinal barrier contributed to increased permeability and leakage in diabetes (Weinberger et al., 1995). In rodent models, diabetic animals showed increased tracer leakage through the RPE compared to controls (Grimes and Laties, 1980; Kirber et al., 1980; Wallow, 1983; Xu and Le, 2011). Diabetes leads to loss of tight junctions around photoreceptors at the OLM, including decreased expression of occludin and dissociation of junctions between photoreceptor cells (Omri et al., 2010; Scarinci et al., 2015).
The RPE also exhibits impaired fluid clearance from the neural retina in diabetes, which in combination with loss of outer BRB integrity, might contribute to retinal edema (Simo et al., 2010). The ability of RPE to pump fluid from the apical to the basal side of the RPE was measured by injecting a small amount of fluid into the subretinal space, and then measuring the rate at which that retinal bleb was resorbed using OCT (Dahrouj et al., 2013; Dahrouj et al., 2014; Desjardins et al., 2016a; Desjardins et al., 2016b). The time required for fluid resorption was longer in diabetic rats compared to controls, and possibly resulted from a reduction of active fluid transport by the RPE (or via a simple leakage of the fluid out of the bleb). Na/K-ATPase is one of the pumps that regulate cellular fluid levels, and the activity of this enzyme in the total retina and RPE is significantly decreased in diabetes (Bensaoula and Ottlecz, 1995; Crider et al., 1997; Kern et al., 1994; Kowluru, 2002; Kowluru et al., 1998; MacGregor and Matschinsky, 1986b; Ottlecz and Bensaoula, 1996). Inadequate activity of Na/K-ATPase in the photoreceptor cells or RPE could contribute to retinal edema, but several other molecular abnormalities, including altered paracellular diffusion, facilitated diffusion, and active transport, likely contribute also to the disruption of the outer BRB in diabetes (Xia and Rizzolo, 2017).
4.3. Ion transport in outer retina.
Ion movement mediates a variety of critical functions for cells, including signaling, pH balance, and volume regulation. Ion transport into photoreceptor cells and RPE has been evaluated in vivo using manganese-enhanced MRI (MEMRI) in vivo. MEMRI is a noninvasive imaging modality that uses manganese (Mn2+), a strong MRI contrast agent, as a surrogate to monitor movement of ionic calcium into excitable cells via L-type calcium channels (Berkowitz et al., 2012a; Berkowitz et al., 2016a; Berkowitz et al., 2009a; Berkowitz et al., 2014; Berkowitz et al., 2006; Berkowitz et al., 2007a; Berkowitz et al., 2009b; Berkowitz et al., 2007b). Using this method, rod cell ion channels, which are normally open in the dark, were found to become paradoxically closed in dark-adapted diabetic animals (Berkowitz et al., 2012a; Berkowitz et al., 2009a; Berkowitz et al., 2007b), and a single injection of 11-cis-retinaldehyde, which is normally produced by the visual cycle, caused the closed calcium channels in rod cells to partially open (Berkowitz et al., 2015c). Inasmuch as Na/K-ATPase is known to play an important role in ion movement across photoreceptors and the majority of retinal Na/K-ATPase activity resides in photoreceptor cells and RPE (Berkowitz et al., 2007a; Wong-Riley, 2010), the diabetes-induced reductions in activities of retinal Na/K-ATPase (Bensaoula and Ottlecz, 1995; Crider et al., 1997; Kern et al., 1994; Kowluru, 2002; Kowluru et al., 1998; MacGregor and Matschinsky, 1986b; Ottlecz and Bensaoula, 1996) and calcium ATPase (Kern et al., 1994; Kowluru et al., 1999) likely contribute to a reduction in ion transport in retinal photoreceptors in diabetes (Berkowitz et al., 2015d). This has not been directly demonstrated however.
4.4. Molecular abnormalities that contribute to dysfunction of the outer retina.
In the retinas of Sprague-Dawley rats diabetic for 6 weeks, the expression of rhodopsin kinase increased compared to that in nondiabetic animals, while that of transducin (alpha subunit) and recoverin decreased. The diabetes-induced increase in rhodopsin kinase immunostaining occurred in the outer limiting membrane, some rod cells in the outer segment layer, and at the tip of the OPL, whereas the expression of transducin immunoreactivity was significantly decreased in the OPL (Kim et al., 2005). Other molecular changes occurring in photoreceptor cells of diabetic rodents have been summarized elsewhere (Kern and Berkowitz, 2015).
Defects in insulin signaling can contribute to diabetes-induced defects in retinal function. Compared to results in normal nondiabetic controls, ERG studies of animals in which the insulin receptor was knocked down (whole body) showed evidence of photoreceptor malfunction (based on the ratio between photopic and scotopic a- and b-waves) and impaired RPE function (based on reduced c-wave amplitudes) which was largely inhibited by pharmacologic blockage of the angiotensin-II receptor 1 (Reichhart et al., 2017). The authors pointed out that the a/c-wave ratio remained unchanged compared to normal controls, which they interpreted as suggesting that the c-wave reduction was secondary to the a-wave reduction and, thus, of the functional loss of photoreceptors. Other evidence, however, suggests that insulin does affect RPE cells. In that study, experimental diabetes (streptozotocin) of 4 weeks duration did not result in a significant reduction in a- (or b-) wave amplitudes, but did cause significant reductions in those amplitudes if the insulin receptor also was deleted selectively from RPE cells. Knocking out the insulin receptor from RPE cells did not result in significant abnormalities in the amplitude of the RPE-dependent c-wave. This data suggests that photoreceptors were more severely affected by diabetes when RPE cells lacked the insulin receptor, but seems in contrast to evidence from the same animals that diabetes-induced increases in retinal oxidative stress and expression of pro-inflammatory cytokines in retina were inhibited if RPE cells lacked the insulin receptor (Tarchick et al., 2019). Both oxidative stress and expression of pro-inflammatory proteins have been implicated in the pathogenesis of DR.
RPE isolated from cadaver donors who had been diabetic showed that insulin-mediated stimulation of glucose uptake and lactate production by the RPE was decreased significantly compared with that in nondiabetic controls. Thus, diabetes-mediated effects on the RPE (previously thought to be insulin-independent) might occur at least in part via reduced insulin signaling (Miceli and Newsome, 1991). In contrast, oxygen consumption and the percentage of endogenous oxygen consumption from fatty acid oxidation were similar in both the diabetic and nondiabetic groups.
Access to glucose also can affect the function of photoreceptor cells and other retinal neurons. Transport of glucose across the BRB is regulated largely via facilitated glucose transporters (Gluts) that permit ATP- and sodium-independent movement of glucose across plasma membranes. Once glucose crosses the RPE or the retinal endothelial cells, it can reach other cell-types via additional glucose transporters (predominantly Glut1 in the outer segments and cell bodies of photoreceptors and most other cells of the retina, as well as via Glut3 found in retinal neurons (Wong-Riley, 2010)). In animal studies, diabetes reduced the expression of Glut1 in the retinal vasculature, but had no effect on the transporter in the RPE or on Glut 3 in retinal neurons (Badr et al., 2000). Glut4, a glucose transporter that differs from other glucose-transporters in that its transport of glucose is stimulated by insulin also has been detected in retinal photoreceptor cells (Sanchez-Chavez et al., 2012). Although expression of Glut4 was not modified in retinas of type 1 diabetic rats, activity of this transporter (and thus transport of glucose into retinal photoreceptors cells) would be expected to be subnormal in insulin-resistant or -deficient diabetes.
Immunohistochemical staining of eyes from patients with and without diabetes using an antibody against the Glut1 glucose transporter showed that the transporter was present in photoreceptor cells and RPE of the retina, as well as in other ocular cells. The pattern of Glut1 immunoreactivity in the diabetic eyes was virtually identical to that in the nondiabetic specimens (Kumagai et al., 1994). Results from an in vitro study of RPE under hyperglycemic conditions differed from the in vivo study described above, in that they showed Glut1 expression to be downregulated in high glucose (Kim et al., 2007). Another study found that elevated glucose increased the Vmax for glucose uptake without affecting glucose transporter-1 expression in primary cultures of human adult RPE (Busik et al., 2002).
Even acutely, oxygen and glucose levels have effects on photoreceptor cell function. Kurtenbach et al (Kurtenbach et al., 2006) reported that acute increases in oxygen and glucose cause a significant enhancement of cone metabolism compared with rod metabolism. These investigators also found that dark-adapted cone sensitivity was improved by an increase in blood oxygen saturation or by acute hyperglycemia in nondiabetic patients, whereas rod sensitivity was unchanged by these conditions. The dark-adapted cone threshold was comparable to that found in normal subjects, whereas the final rod threshold was impaired, suggesting that dark-adapted rods of diabetic patients are hypoxic before the onset of retinopathy. Likewise, retinal oxygen tension in dark-adapted diabetic cats reached a minimum PO2 of nearly 0 mmHg in the outer retina (Linsenmeier, 1986; Wangsa-Wirawan and Linsenmeier, 2003). Although photoreceptors have been known to utilize glucose for many years, recent evidence shows that they metabolize also fatty acids (Joyal et al., 2018). The importance of glucose as a critical regulator of photoreceptor cell metabolism and function needs to be re-interpreted in light of contributions also of fatty acid metabolism, especially in diseases like diabetes (Fu et al., 2017; Joyal et al., 2017; Joyal et al., 2018).
The diabetes-induced deterioration of the c-wave was arrested by treatment of the diabetic animals with either myo-inositol supplementation or an inhibitor of aldose reductase in diabetic rodents, suggesting that hyperglycemia-associated defects in myo-inositol or sorbitol metabolism may be involved in the pathogenesis of the electrophysiological deficits of the diabetic retina (Crider et al., 1997; Hotta et al., 1995; Hotta et al., 1997; MacGregor and Matschinsky, 1985, 1986a). Administration of FGF21 to diabetic mice improved a-wave in ERG to levels above normal (Fu et al., 2018). Studies of the Goto-Kakizaki rat model (type 2 diabetes) showed that PKCζ is localized predominantly in tight junction protein complexes of the RPE and in photoreceptor inner segments and outer segments of cone photoreceptors (Omri et al., 2013), and short-term hyperglycemia led to delocalization of the enzyme. In long-term diabetes, outer blood retinal barrier was disrupted, but reduction of the PKCζ over-activation using a PKCζ-specific inhibitory peptide inhibited the mis-localization of the kinase, improved the outer blood retinal barrier integrity, and inhibited photoreceptor cell death (Omri et al., 2013). Injection of Placental Growth Factor-1 (PLGF-1) into the vitreous of the rat eye induced an opening of the RPE tight junctions with subsequent sub-retinal fluid accumulation and retinal edema (Miyamoto et al., 2007).
A recent study found that intravitreal levels of retinol binding protein 3 (RBP3; also known as Interphotoreceptor retinol-binding protein (IRBP)) were elevated in patients having type 1 diabetes for 50+ years and no or only mild DR (Yokomizo et al., 2019). Moreover, the intravitreal RBP3 concentration in this population was inversely associated with DR severity, raising a possibility that RBP3 might be an endogenous protective factor in those patients. RBP3 is secreted mainly by retinal photoreceptor cells, and plays an important role in the visual cycle by shuttling retinols between the photoreceptor cells and the RPE (Gonzalez-Fernandez and Ghosh, 2008; Jin et al., 2009). Further experimental studies demonstrated that intravitreal injection or photoreceptor-specific overexpression of RBP3 in rodents inhibited the detrimental effects of vascular endothelial growth factor (VEGF) and hyperglycemia, in part by inhibiting VEGF receptor activation and decreasing glucose uptake into retinal cells (Yokomizo et al., 2019). Other investigators have reported that diabetes-induced oxidative stress impairs dark–light regulation of subretinal space hydration, which regulates the distribution of IRBP (Berkowitz, 2020).
Conclusions.
The function of cells in the outer retina is affected by diabetes. There is abundant evidence that visual function, as assessed by electrophysiology, becomes impaired in many diabetic patients. This functional abnormality involves cells of the outer retina, and the level of glycemia likely is one factor in the dysfunction. The failure to detect ERG functional defects seems to be most common at short durations of diabetes, but there is variability in the ability to detect a-wave defects in diabetic rodents.. Defects in visual function not uncommonly have been detected before the clinical manifestation of overt vascular pathology. Diabetes-induced increases in permeability across the outer BRB has been demonstrated in animals, but its significance in patients and in animals remains to be determined.
5. Can the outer retina affect the retinal vasculature?
The neurons, glia and vascular cells of the retina act together as a neurovascular unit, and each influences the metabolism and function of the others (Fu et al., 2020; Simo et al., 2018). The retina is served by multiple vascular beds, with three interconnected capillary plexuses (superficial plexus in the retinal nerve fiber layer, middle plexus at the inner border of the inner nuclear layer, and deep plexus at the outer border of the inner nuclear layer) serving predominantly the inner retina, and the choriocapillaris below the retina providing nutrients to the photoreceptors and RPE (Fig 1). Choroidal blood flow greatly exceeds that provided by the retinal vasculature (Nair et al., 2011), accounting for almost 80% of the glucose and oxygen metabolized by the retina (Alm and Bill, 1972; Tornquist and Alm, 1979). Diabetes frequently causes a progressive increase in the fraction of microvessels that are leaky, remodeled, nonperfused and degenerated in the retina (Kohner and Henkind, 1970; Shimizu et al., 1981; Sleightholm et al., 1988) as well as in the choriocapillaris (Cao et al., 1998; Lutty, 2017). In the early non-proliferative phases of the retinopathy, some retinal endothelial cells and pericytes undergo an apoptotic death in diabetes (Mizutani et al., 1996), which occurs at least in part secondary to oxidative stress (generated by mitochondria (Du et al., 2003; Kowluru, 2013; Kowluru and Mishra, 2015; Santos et al., 2013) and NADPH oxidase (Du et al., 2015; Sahajpal et al., 2019)), inflammation (Tang and Kern, 2011) and glucose modification of extracellular macromolecules (Hammes et al., 2011).
5.1. Methods used to evaluate the retinal vasculature.
Several methods have been used to evaluate the retinal vasculature in diabetes. In living patients and animals, techniques available to assess vascular structure and function include ophthalmoscopy, fluorescein angiography, and more recently, OCT-angiography, and adaptive optics-scanning laser ophthalmoscopy (Fig 6). From autopsy tissue, histologic techniques including immunohistochemical staining to identify microvessels in retinal wholemounts, and preparations of the isolated retinal microvasculature have provided great insight into the effects of diabetes on the microvessels. An important difference between clinical and histologic techniques to visualize the retinal vasculature is that clinically, microaneurysms are visible in the retina only for a period of time, but then disappear clinically as they become nonperfused, whereas all of the lesions that have developed in those microvessels over the lifetime of the subject remain visible in the histologic preparations because that methodology does not rely on intact perfusion to view the vasculature. Retinal blood flow can be determined in vivo in diabetes, but that topic is not directly related to the topic of this manuscript, and will not be discussed here. In the near future, advances in 2-photon microscopy also are expected to be suitable for analysis of the vasculature.
Fig 6.

Methods used to assess the retinal vasculature. (a) fluorescein angiogram, (b) optical coherence tomography-angiography (OCT-A), (c) trypsin digest preparation. In (a), optic nervehead is indicated by O.N., retinal arterioles (thinner arrows) and venules (thicker arrows) are indicated by yellow arrows, fovea is indicated by yellow dashed circle, and numerous microaneurysms are indicated by small fluorescent dots (thin white arrows). In (b), the yellow dashed circle outlines the fovea. Erythrocyte flow is indicated as white lines in this static image. There are no microvessels within the fovea. Red circles demonstrate microaneurysms, and yellow arrows point out areas of capillary nonperfusion in a diabetic patient. In isolated microvessels (c), endothelial cell nuclei (e; red arrows) are oval shape, whereas pericytes (p; black arrows) are darkly stained and protrude out from the vascular profile. Degenerated retinal capillaries (thick yellow arrows) occur in both nondiabetic and diabetic animals and humans, but are more numerous in diabetics.
Retinal wholemounts.
Immunostaining of the retinal vasculature within the intact retina in situ allows preservation of the 3-D structure of the vasculature, and allows study of the interaction of the microvessels with the surrounding neural environment. Because the retina remains intact, however, the immunohistochemical technique is time-consuming. Comparable methods have been developed for analysis of the morphology of the choriocapillaris (Cao et al., 1998; McLeod and Lutty, 1994).
“Trypsin digest” preparation.
Incubation of the formalin-fixed retina in a proteolytic solution (crude trypsin or elastase (Cogan et al., 1961; Kuwabara and Cogan, 1960; Laver et al., 1993; Veenstra et al., 2015)) results in the removal of retinal neurons and glia, leaving only the delicate “spider-web”-like network of the entire retinal vasculature that can be stained using conventional colorimetric stains or studied by immunohistochemistry. The result yields a high-resolution evaluation of the vascular morphology, however, the 3-D organization of the vascular bed in the retina is lost.
Fluorescein angiography (FA).
FA provides 2-D image sets that allow dynamic visualization of retinal vascular circulation with a wide field of view, as well as patterns of dye leakage and pooling, and the speed at which fluorescein transits from the arterial to venous portions of the retina (Kohner and Dollery, 1970; Sleightholm et al., 1988). FA resolution does not allow visualization of capillary beds, and thus it cannot separately visualize the different capillary networks within the retina. Other drawbacks that limit the use of FA include its invasive nature, its cost, and the time it takes to perform. Although considered safe, the dye poses risks ranging from nausea to allergic reactions, including, anaphylaxis. AO-SLO is able to image fine vessel structures like the capillary and the arteriole wall, and to detect fluorescence making it highly suitable for FA. Indocyanin green likewise has been used for clinical assessment of flow in the choriocapillaris (Weinberger et al., 1998).
OCT-angiography (OCT-A).
OCT-A is a noninvasive technique that acquires volumetric angiographic information without the use of a dye. It detects movement of red blood cells within the vessels, and uses that information to allow high-resolution visualization of the individual layers of the retinal vasculature as well as the choroidal vasculature. OCT-A can show both structural and blood flow information, and it provides a detailed view of the retinal vasculature, making it possible to accurately delineate microvascular abnormalities (Chalam and Sambhav, 2016). Unlike FA, however, OCT-A is a static image, and has a smaller field of view than FA.
Adaptive optics scanning laser ophthalmoscopy (AO-SLO).
AO-SLO has been used to visualize the retinal vasculature in vivo, demonstrating extensive capillary remodeling in mild or moderate NPDR and varying blood flow patterns (Burns et al., 2014). The authors concluded that existing clinical classifications based on lower magnification clinical assessment may not adequately measure key vascular differences among individuals with NPDR.
5.2. Photoreceptor cells contribute to the degeneration of retinal capillaries in the absence of diabetes.
A number of reports have shown that abnormalities in the outer retina correlate with or contribute to damage of the retinal vasculature (Bhutto and Amemiya, 2001; May and Narfstrom, 2012; Vessey et al., 2014; Wang et al., 2019), and that pharmacologic treatments, such as safranal (Fernandez-Sanchez et al., 2012; Fernandez-Sanchez et al., 2015b)) or RTL220 (Adamus et al., 2012) which inhibited outer retina degeneration in rodent models, also partially inhibited capillary degeneration. Intravitreal injection of tunicamycin into the eye induced photoreceptor ER stress and apoptosis, followed by a significant degeneration of retinal capillaries. The authors concluded that the retinal vascular degeneration was secondary to ER stress-mediated dysfunction and/or loss of photoreceptor cells (Wang et al., 2019), but effects of the drug on other cells (including capillary cells themselves) has not been ruled out. Since none of those pharmacologic treatments were specific for photoreceptor cells, however, the link between photoreceptor cells and vascular abnormalities was not clearly established in these studies.
Some studies provide a more direct link between photoreceptors and degeneration of the retinal vasculature. Overexpression of a mutant cilia gene (polycystin-2), a gene that is found in photoreceptor cells, resulted in a spatial-temporal correlation between the loss of photoreceptor cells and degeneration of capillaries in the retina (Feng et al., 2009). In addition, an established model for hereditary retinal degeneration (based on the mutation in CEP290, a gene in photoreceptor cells) showed outer retina degeneration which correlated with the degeneration of retinal capillaries (May and Narfstrom, 2012). Some studies indicate that photoreceptor cell loss in patients (Heegaard et al., 2003) or animal models (de Gooyer et al., 2006a; Fernandez-Sanchez et al., 2012; Penn et al., 2000) with retinitis pigmentosa is associated with reduced blood flow or atrophy of retinal capillaries (Beutelspacher et al., 2011).
Most convincingly, genetic alterations that affect only photoreceptor cells have been found to lead to appreciable degeneration of the retinal vasculature in nondiabetic animals (Caldwell et al., 1989; de Gooyer et al., 2006b; Fernandez-Sanchez et al., 2015a; Hanna et al., 2018; Ivanova et al., 2019; Liu et al., 2016). The temporal relationship between vascular and photoreceptor cell degeneration in the mouse was examined by Blanks & Johnson (Blanks and Johnson, 1986), who concluded that photoreceptor death in rd (retinal degeneration) mutant mice appeared to precede vascular regression. The diabetes-induced degeneration of the retinal vasculature that is characteristic of wildtype animals was not observed in diabetic mice lacking rhodopsin (and thus, lacking photoreceptor cells) (de Gooyer et al., 2006b; Liu et al., 2016). Liu et al further showed that photoreceptor cells stressed as a result of the P23H mutation of opsin or deletion of opsin caused retinal vascular damage while the photoreceptor cells were still present (Fig 7), but further damage to the vasculature diminished after the photoreceptor cells had degenerated (Liu et al., 2016). They also showed that the stressed photoreceptor cells stimulated leukocytes to kill retinal endothelial cells (studied ex vivo), and this cytotoxicity against endothelial cells was diminished when leukocytes were collected from older animals in which photoreceptor cells had already degenerated (Liu et al., 2016). Thus, activation of leukocytes by some unidentified soluble product from stressed photoreceptors might contribute to the retinal capillary degeneration observed in patients and animals showing photoreceptor stress or degeneration.
Fig 7.

Two lines of evidence indicate that retinal photoreceptor cells play a role in retinal capillary degeneration. First, molecular alterations that are unique to retinal photoreceptor cells (deletion of opsin (opsin−/−) or knock-in of the P23H mutant opsin (RhoP23H/P23H)) led to the degeneration of retinal capillaries in nondiabetic animals. Second, the diabetes-induced degeneration of retinal capillaries was inhibited if induction of diabetes was delayed until after photoreceptors had degenerated in these models. (a) summarizes retinal capillary degeneration in nondiabetic (N) and diabetic (D) wildtype (wt), opsin−/− and RhoP23H/P23H mice (10 months of age, 8 months of diabetes). (b) shows representative preparations of the isolated retinal vasculature from each of the experimental groups. Arrows illustrate representative degenerate capillaries. Data summarized in (c) show that retinal capillaries actively degenerate in young opsin−/− nondiabetic mice while photoreceptors are present but degenerating due to the opsin deficiency (up to approx. 14 weeks of age), but then the capillary degeneration slows substantially after the photoreceptors have degenerated (after 14 weeks of age). Reprinted from Liu H, Tang J, Du Y, et al. Photoreceptor Cells Influence Retinal Vascular Degeneration in Mouse Models of Retinal Degeneration and Diabetes. Invest Ophthalmol Vis Sci 2016;57:4272–4281. © The Authors. Licensed under a CC BY-NC-ND license.
Photoreceptor activity has been reported to affect the vasculature also in other retinal diseases, including retinopathy of prematurity (ROP) and ischemia/reperfusion. Using oxygen-induced retinopathy as a model of ROP, alterations in structure of both the outer and inner segments of photoreceptors (Fulton et al., 1999) were reported to precede (Reynaud and Dorey, 1994; Reynaud et al., 1995) and predict later vascular changes (Akula et al., 2007; Akula et al., 2008) in retinas of rats. It has been postulated that oxygen demands of the photoreceptors cause the development of the vascular abnormalities that are characteristic of ROP (Akula et al., 2007; Fulton et al., 2009). With regard to retinal ischemia/reperfusion injury, inhibition of the visual cycle by the RPE65 inhibitor, emixustat, or dark adaptation both significantly reduced the ischemia/reperfusion-induced vascular permeability in mice (Dreffs et al., 2020), suggesting that vision-related molecular processes in the outer retina contribute to vascular permeability in this condition (Fig 8). Deletion of Lrat, the enzyme preceding RPE65 on the visual cycle, likewise inhibited the ischemia/reperfusion-induced vascular permeability in mice (Dreffs et al., 2020), but interpretation of those results is confounded by evidence that retinas from those animals were significantly thinner than normal, and thus might have had lower blood flow (and thus, permeability).
Fig 8.

Ocular ischemia-reperfusion injury (IR) increases retinal vascular permeability in normal mice, and this increase is significantly (but partially) inhibited in the absence of visual cycle activity (due to knockout of Lrat (a) or the absence of light (b)). *< 0.05, **< 0.01, ***< 0.001, or ****< 0.0001 Reprinted from Figs 2c and 3c in Dreffs A, Lin CM, Liu X, et al. All-trans-Retinaldehyde Contributes to Retinal Vascular Permeability in Ischemia Reperfusion. Invest Ophthalmol Vis Sci. 2020;61(6):8. © The Authors. Licensed under a CC BY-NC-ND license.
The relationship between photoreceptor cells and degeneration of the retinal microvasculature seems complex. In addition to potential direct release of cytoactive products from the photoreceptor cells that can damage the vasculature either directly or indirectly (such as via activation of leukocytes as described above), diseases in which photoreceptors have already degenerated also could cause vascular changes by retinal remodeling as a result of the decreased metabolic demand by the fewer photoreceptor cells in the remaining retina (Yu and Cringle, 2005). Metabolism of photoreceptor cells and the RPE show strong differences between day and night, and the diabetes-induced increase in retinal superoxide (generated primarily by photoreceptor cells) is greater at night than in the day (Du et al., 2013). Thus, diurnal differences in the effects of the outer retina on health and function of the retinal vasculature also are an important possibility. Unfortunately, there is no data yet available related to diurnal effects of the outer retina on the retinal microvasculature.
5.3. Photoreceptor cells and retinal neovascularization in the absence of diabetes.
Retinal ischemia is a potent stimulus for neovascularization, and the huge metabolic demand by photoreceptor cells has long been recognized to contribute to retinal ischemia, particularly in situations of compromised vascular supply (such as in diabetes). Laser photocoagulation is postulated to exert its documented beneficial effects to inhibit and reverse retinal neovascularization via the destruction of photoreceptor cells (Pournaras et al., 1990) and RPE. Consistent with a role of photoreceptors in ischemia-driven retinal neovascularization, studies of oxygen-induced retinopathy in mice with rapid retinal photoreceptor cell degeneration found that VEGF expression, vascular leakage, nonperfusion and preretinal neovascularization were less severe in mice lacking photoreceptors than that in control mice (Zhang and Zhang, 2014). Likewise, neonatal mice with classic inherited retinal degeneration as a result of the rd1 mutation failed to mount reactive retinal neovascularization in oxygen-induced proliferative retinopathy (Lahdenranta et al., 2001). These data indicate that retinal neovascularization fails to develop or at least is inhibited significantly when the number of photoreceptor cells is markedly reduced.
5.4. Photoreceptor cells contribute to damage of retinal capillaries in diabetes.
Retinitis pigmentosa leads to photoreceptor degeneration, and results of a survey sent to a group of diabetic patients who also had retinitis pigmentosa suggested that diabetics who also had photoreceptor degeneration were protected from the development of DR (Arden, 2001). This study lacked the photographic documentation or systematic quantitation of retinopathy that is found in clinical trials, but it does provide supportive evidence for the hypothesis that photoreceptor cells play an important role in the development of DR. A mechanism postulated by Arden and colleagues for this effect was that the dark current in rods accounts for considerable utilization of energy and oxygen in the dark, making the retina borderline hypoxic at night (Arden and Sivaprasad, 2012; Braun et al., 1995). Therefore, elimination of some rods (as can happen in retinitis pigmentosa or following laser photocoagulation) could reduce this metabolic stress, and thereby reduce the metabolic abnormalities that contribute to the retinopathy. This topic will be covered more in section 6.3. A survey of published manuscripts indicated that the deep vascular layer (adjacent to retinal photoreceptor cells) of diabetic patients with NPDR was affected more severely than was the superficial vascular layer (Dimitrova and Chihara, 2019), thus being consistent with the possibility that photoreceptor cells contribute to the retinal vascular disease of DR. Supplementing with oxygen has been reported to reduce DME and improve aspects of visual function in some diabetic patients (Nguyen et al., 2004). Photoreceptor cells are the major consumer of oxygen in the retina, but there is no direct proof at present that the beneficial effects of oxygen supplementation are due to effects on photoreceptor cells.
Animal studies have directly implicated photoreceptors in the capillary degeneration of DR. As mentioned above, opsin-deficient mice which were diabetic for 5 months were partially protected from the diabetes-induced retinal vascular damage and loss (measured by microvascular density) compared to wild-type diabetic animals (de Gooyer et al., 2006b), whereas if diabetes was not induced until most of the photoreceptors already had degenerated in opsin-deficient or P23H mutant mice, the expected diabetes-induced retinal capillary degeneration failed to develop (Liu et al., 2016) (Fig 7). Mechanisms by which the photoreceptors contribute to the vascular degeneration remain unclear, but degeneration of retinal capillaries in diabetes seems less in the absence of those photoreceptor cells.
The contribution of retinal neurons, particularly photoreceptor cells, to permeability defects in diabetes has been studied less, but recent evidence shows that photoreceptor cells can contribute to enhanced retinal capillary permeability in diabetes (Tonade et al., 2017). In wildtype mice, diabetes increased retinal permeability in all three layers of the retina where the vasculature is located, whereas the permeability defect was inhibited only in some layers of the vasculature in photoreceptor-deficient diabetics (Tonade et al., 2017). In those opsin-deficient diabetics, the permeability defect was significantly inhibited in layers closest to the photoreceptor cells, and inhibited less at distances further from the photoreceptor layer. Photoreceptor cells from diabetic mice have been shown to release soluble inflammatory products that can directly enhance retinal endothelial cell permeability in vitro (Tonade et al., 2017). It seems possible that photoreceptor cells (and neuronal cells in general) release factors that contribute to the permeability defect locally, but have less effect on vessels further away. Likewise, surrounding neurons and glia might release inflammatory mediators that contribute to endothelial cell dropout (Cuenca et al., 2014), which also can increase permeability.
5.5. Can degeneration of the retinal microvasculature in diabetes adversely affect the photoreceptors?
Not all studies have suggested that photoreceptor abnormalities in diabetes lead to damage of the retinal microvasculature. Some clinical investigators came to the opposite conclusion. Using adaptive optics, OCT-A and spectral domain-OCT, investigators observed an association between capillary non-perfusion of the deep retinal capillary plexus and abnormalities in the photoreceptor layer in eyes with DR (Nesper et al., 2017; Scarinci et al., 2016), leading them to consider that the disruption of the photoreceptors was secondary to underlying capillary non-perfusion in DR (Scarinci et al., 2015). Likewise, other investigators interpreted their findings as indicating that retinal vascular degeneration causes the degeneration of photoreceptor cells (van Dijk et al., 2009; van Eeden et al., 2006). Although not involving diabetes, other investigators likewise have provided evidence that cone cell loss in rd10 mice was facilitated by reductions in retinal blood flow and vascular barrier function (Ivanova et al., 2019). Why the retinal vasculature would have such a strong effect on photoreceptor survival seems unclear, since available evidence indicates that the choriocapillaris, and not the retinal vasculature, provides photoreceptors with most of their oxygen and nutrients. Nevertheless, this is a very important area for future investigation.
If damage to the retinal microvasculature contributes to photoreceptor cell disruption, then perhaps maintaining the integrity of the vasculature might help enhance the survival of damaged retinal photoreceptor cells in diabetes. Injection of bone marrow-derived stem cells containing endothelial precursors into eyes of mouse models of retinal degeneration was found to stabilize and rescue retinal blood vessels that ordinarily would have completely degenerated, and led also to a substantial rescue of cone photoreceptors and partial restoration of retinal ERG responses of some photoreceptor cells (Otani et al., 2004). Similar studies have not yet been conducted in diabetes, but the outcome of such studies would be expected to provide valuable perspective on the relation between photoreceptor cells and the retinal vasculature.
5.6. Inter-relationship between abnormalities in photoreceptor cells and choriocapillaris in diabetes
Retinal photoreceptor cells are in close proximity to vascular beds in both the retina and underlying choroid. If photoreceptor cells are releasing soluble factors that adversely affect the retinal vasculature, one would expect that such factors might affect also other vasculatures in the region. Diabetes does affect the choriocapillaris (Lutty, 2017; Muir et al., 2012), and this could have a subsequent, profound impact on RPE and the outer retinal layers. Histopathological studies of diabetic choroids demonstrated thinning of the choroid or its capillary bed with lesions such as vessel drop-out, aneurysms, ischemia, vascular tortuosity, and in some cases, intrachoroidal neovascularization (Berkowitz et al., 2015b; Cao et al., 1998; Fryczkowski et al., 1989; Fryczkowski et al., 1988; Hidayat and Fine, 1985; Hua et al., 2013; Kim et al., 2013). These alterations were interpreted as being due to local inflammation (Lutty, 2017; Lutty et al., 1997). It is not known whether or not photoreceptor cells contribute to the diabetes-induced inflammation and damage to the choriocapillaris, and neither is it known if the opposite possibility (that vascular lesions in choriocapillaris contribute to photoreceptor dysfunction or degeneration in diabetes) occurs.
Conclusions.
Evidence in patients and animals suggests that photoreceptor cells play an important role in the structural and functional integrity of the retinal vasculature, and that the vascular lesions of DR are significantly inhibited or do not develop in the absence of photoreceptor cells. Whether this is a consequence of factors directly released by photoreceptor cells in the abnormal mileau of diabetes, or is caused indirectly by photoreceptor-driven alterations in other
6. Does the outer retina contribute to abnormalities of neuroglial cells in diabetes?
In light of the cell-to-cell connections among neural cells in the retina, it seems likely that altered function of photoreceptor cells or RPE in diabetes will alter the function of downstream cell-types, even if those other cells otherwise were otherwise normal.
Glutamate is regarded as the most important excitatory neurotransmitter in the retina, and it is released by photoreceptors onto horizontal and bipolar cells (and by bipolar cells onto amacrine and ganglion cells) in darkness, whereas the absorption of light decreases its release (Copenhagen and Jahr, 1989). In retinal cells expressing glutamate receptors (especially NMDA receptors (Hahn et al., 1988; Sucher et al., 1991)), however, overstimulation of those receptors by glutamate can lead to neuronal calcium overload and neurotoxicity. In the retina, expression of receptors is found on retinal ganglion cells and subsets of amacrine cells (Brandstätter et al., 1994; Gründer et al., 2000; Hartveit et al., 1994), and a progressive upregulation of these receptors was observed in the ganglion, amacrine and bipolar cells as well as in the inner and outer plexiform layers (IPL and OPL) after 1 and 4 months of diabetes (Ng et al., 2004).
Accumulation of glutamate was found in the vitreous of diabetic patients with PDR (Ambati et al., 1997), and supranormal levels of glutamate have been detected in retinas of rats diabetic for 2 to 3 months (Kowluru et al., 2001a; Lieth et al., 1998; Lieth et al., 2000). The mechanism by which retinal glutamate is elevated in diabetes likely involves changes in glutamate uptake and utilization (Fernandez-Bueno et al., 2017; Hernandez et al., 2013; Zhang et al., 2011), or subnormal glutamine-synthase activity (Lieth et al., 2000; Mizutani et al., 1998) Decreased expression of a glutamate transporter, GLAST, in retina has been reported in diabetes induced by streptozotocin, a genetic model of type 2 diabetes.. Kowluru et al demonstrated that the accumulation of glutamate in the retina of diabetic rats could be inhibited by administering a mixture of antioxidant compounds, indicating that oxidative stress can contribute to the increase in retinal glutamate in diabetes (Kowluru et al., 2001a). Likewise, oxidative stress has been shown to elevate glutamate levels by stimulating the activity of N-methyl-D-aspartate (NMDA) receptors (Agostinho et al., 1996). Since photoreceptor cells are the most populous cell-types in the retina, it is likely that photoreceptor cells are major producers or regulators of increased glutamate in diabetic conditions, but the extent to which accumulation of this neurotransmitter is involved in the pathogenesis of the neural cell dysfunction or degeneration that occurs in diabetes remains to be determined. Products released by photoreceptor cells or RPE also might affect even local non-neuronal cells such as glial cells and retinal neuroglia. This is yet to be investigated.
As described above, deletion of the insulin receptor specifically from RPE cells significantly reduced a- and the inner retina b-wave amplitudes in diabetic animals, as well as reducing the diabetes-induced oxidative stress and greater-than-normal expression of pro-inflammatory cytokines in the retinas of those diabetic mice (Tarchick et al., 2019). Thus, insulin signaling in RPE cells has important effects on other retinal neural cells in diabetes. The contribution of outer retina to abnormalities in glial cells in diabetes remains to be studied.
7. Visual processes and their relation to vascular and neural abnormalities of diabetic retinopathy
The vertebrate visual system relies on two key pathways to detect and transform light into perceived images; phototransduction and the visual (or retinoid) cycle. Phototransduction describes the process of converting light into stimuli that are processed by the brain to form an image. This occurs via a G-protein signaling cascade in photoreceptor outer segments that converts incoming light stimuli into an electrical response, leading to the activation of transducin and cGMP phosphodiesterase, closure of cGMP-gated ion channels, and hyperpolarization of the photoreceptor cells (Arshavsky et al., 2002; Fu and Yau, 2007; Luo et al., 2008) (Fig 9a). The visual cycle is a multistep process that regenerates the light-sensitive photopigments (11-cis-retinal in mammals) so that phototransduction can occur again (Fig 10a). Recently, research has emerged that shows that these processes that underlie vision itself can contribute to the development of early lesions of DR.
Fig 9.

Phototransduction and its role in the development of early stages of diabetic retinopathy. (a) Absorption of a photon of light by rhodopsin results in photoisomerization of the chromophore to all-trans-retinal, which leads to transduction of this signal to cGMP-gated ion channels on photoreceptor cells. Deletion of Gnat1, the gene responsible for expression of transducin1 in rod cells, prevents phototransduction in those photoreceptor cells. Deletion of Gnat1 in diabetic mice did not inhibit the diabetes-induced increase in (b) retinal superoxide, but did significantly inhibit the (c) degeneration of retinal capillaries in mice diabetic for 8 months. This inhibition of phototransduction had variable effects on permeability, significantly inhibiting the leakage of albumin into the neural retina in the inner plexiform layer (c), but not significantly inhibiting it in the outer plexiform layer (d). (N; nondiabetic mice, WT, wildtype mice). Figures (b) and (c) are reprinted from Liu H, Tang J, Du Y, et al. Transducin1, Phototransduction and the Development of Early Diabetic Retinopathy. Invest Ophthalmol Vis Sci. 2019;60:1538–1546. © The Authors. Licensed under a CC BY-NC-ND license.
Fig 10.

Visual cycle and its role in the development of early stages of diabetic retinopathy. (a) The visual cycle refers to the steps to regenerate 11-cis retinal after photoisomerization to all-trans-retinal. Rod photoreceptor cells depend on the output of 11-cis-retinal from adjacent RPE cells, and activity of RPE65 in RPE cells is a critical step in this process. (b) Deletion of RPE65 inhibited the diabetes-induced degeneration of retinal capillaries compared to that in wild type control mice diabetic for ~6 months. Likewise, daily administration of the RPE65 inhibitor, retinylamine (Ret-NH2), to diabetic (D) mice for 8 months inhibited the (c) degeneration of retinal capillaries and (d) leakage of albumin into the neural retina in mice compared to age-matched nondiabetic (N) controls. The leakage of albumin into the neural retina was determined by the leakage of injected FITC-BSA into the neural retina. Figure b was modified from (Thebeau et al., 2020) and available under the Creative Commons CC-BY-NC-ND license, and (c) and (d) are reprinted from (Liu et al., 2015) with permission of the American Society for Biochemistry and Molecular Biology.
7.1. Phototransduction and diabetic retinopathy.
Transducin1 is a subunit of the heterotrimeric G-protein encoded by Gnat1 (Hurley, 1987), and is a key component of the vertebrate phototransduction pathway. It is found only in rod photoreceptor cells in the retina. The absence of transducin1 prevents phototransduction in rods, causing the cGMP-gated ion channels on the rod cells to remain continually open. Diabetes has been reported to cause a reduction in expression of transducin in retinal photoreceptors (Kim et al., 2005), but permanent inhibition of phototransduction in rod cells using diabetic Gnat1−/−mice showed that elimination of phototransduction in rod cells significantly inhibited the diabetes-induced degeneration of retinal capillaries and some molecular processes believed to participate in the pathogenesis of the retinopathy (Liu et al., 2019) (Fig 9). Studies of Gnat1−/− mice by other investigators confirmed that deletion of transducin1 significantly inhibited the diabetes-induced increase in inflammatory mRNAs, ERG defects, and degeneration of retinal capillaries (Thebeau et al., 2020). Thus, elimination of phototransduction in rod cells inhibited diabetes-induced abnormalities in both retinal vasculature structure and neural function. Deletion of Gnat1 did not result in significant degeneration of photoreceptor cells, which would have confounded interpretation of results. Gnat1 deletion had an unexplained effect on the diabetes-induced increase in permeability, in that the leakage of albumin was not uniformly distributed across the retina in those animals (Liu et al., 2019). The abnormal accumulation of albumin in the neural retina was inhibited in diabetic Gnat1−/− mice in the inner plexiform layer, but not in the outer plexiform or inner nuclear layers. In Gnat1-deficient animals, the diabetes-induced increase in inflammatory proteins in the retina and the leukocyte-mediated killing of retinal endothelial cells were inhibited, however, the diabetes-mediated induction of oxidative stress was not inhibited in these aniamls. Since anti-oxidant approaches have been found to inhibit the diabetes-induced degeneration of retinal capillaries in previous long-term rodent studies (Berkowitz et al., 2009a; Du et al., 2015; Du et al., 2003; Kanwar et al., 2007; Kowluru et al., 2001b; Zheng and Kern, 2009), the finding that the diabetes-induced increase in retinal capillary degeneration was inhibited in diabetic Gnat1−/− mice even though the oxidative stress in the retina was not inhibited is unexplained.
Whether or not Gnat2 in cone cells has any similar effect on development of the retinopathy in diabetes has not been explored, but the number of cones in rodents is very small compared to the number of rod cells.
A physiologic way to demonstrate the importance of light-initiated processes in the development of DR is to eliminate light. Following 3 or 5 months of hyperglycemia, Thebeau et al (Thebeau et al., 2020) housed 2 different diabetic animal models in total darkness for 1–3 months. Results indicated that the mice housed under conditions of light deprivation developed significantly less diabetes-induced visual dysfunction (a-wave, b-wave, and oscillatory potential), upregulation of pro-inflammatory mRNA in the retina, and retinal neurodegeneration. As the major light-sensing cells in the body, these studies clearly implicate retinal photoreceptors as an early contributor to the retinal dysfunction and pathology in diabetes, and illustrate the importance of light in the diabetes-induced abnormalities (Fig 11).
Fig 11.

Diabetes reduces functional activity of photoreceptors (a-wave; initial downward deflection of ERG line) and inner retina (b-wave; upward curve after a-wave), as determined by scotopic ERG using sub-maximal flashes (a, b). These studies were conducted using db/db mice, a genetic model of type 2 diabetes. After 3 months of continuous darkness, the db/db mice displayed a-wave responses across all stimulus luminances that were similar to those of nondiabetic db/m controls (b). These figures are Figs 3c and e from (Thebeau et al., 2020), and are reprinted under a Under a Creative Commons license.
Exposure to light normally results in expansion of the choroidal space, suggesting that this phenomenon is secondary to phototransduction or its downstream signaling (Longo et al., 2000). The light-induced expansion of the subretinal space becomes abnormal in diabetes (Berkowitz et al., 2015b), and this defect in diabetes can be inhibited with the antioxidant, lipoic acid, suggesting that oxidative stress might underlie that abnormality. Imunoreactivity of rhodopsin kinase has been reported to be increased in the outer limiting membrane and in the outer segments of some rod cells in diabetes (Kim et al., 2005). Rhodopsin kinase activity in retinal rod cells phosphorylates light-activated rhodopsin to terminate the phototransduction signaling cascade, but how this activity in rod cells is affected by diabetes is not known.
7.2. Visual cycle and diabetic retinopathy.
Subnormal rates of adaptation and absolute final threshold sensitivity have been demonstrated in subjects with diabetes (Amemiya, 1977; Greenstein et al., 1993; Henson and North, 1979). Consistent with this, a number of studies have provided biochemical, molecular or functional evidence that diabetes results in subnormal levels of enzymes involved in the visual cycle (Garcia-Ramirez et al., 2009; Ostroy, 1998; Ostroy et al., 1994). After 2–3 months of diabetes, microarray revealed reduced expression of several genes encoding visual cycle proteins, including lecithin/retinol acyltransferase (LRAT), RPE65, and RPE retinal G protein-coupled receptor (RGR) (Kirwin et al., 2011; Zhao et al., 2017). Among visual cycle proteins, interphotoreceptor retinoid-binding protein (IRBP) and Stimulated by Retinoic Acid 6 protein (STRA6) showed significantly lower levels in diabetic rats than those in nondiabetic controls. STRA6 is the receptor for retinol-binding protein 4 (RBP4) which is critical for the transport of vitamin A to the eye from the peripheral blood (Ruiz et al., 2012), and IRBP is an essential protein of the visual cycle which facilitates the transport of retinoids between the RPE and the retina (Crouch et al., 1992). Serum levels of retinol-binding protein 4, a transporter protein for retinol, have been found to be significantly lower in diabetic rats (Malechka et al., 2017). Spontaneously diabetic db/db mice showed reductions in levels of retinoid binding proteins, CRALBP, IRBP, STRA6, LRAT, and retinol dehydrogenase 5 (RDH5) compared to nondiabetic controls, and 10-week treatment with chrysin, a naturally occurring flavonoid, inhibited these defects as well as augmenting levels of RPE65 and rhodopsin (probably by decreasing hyperglycemia) (Kang et al., 2018).
Several rodent studies have demonstrated that diabetes can cause a retinoid deficiency in the retina. Although Western blot analysis revealed no difference in expression of the opsin protein, rhodopsin content of retinas from diabetic animals has been reported to be subnormal (as shown by a difference in absorbance before and after bleaching with light) and levels of 11-cis-retinal were significantly lower in retinas from diabetic rats compared to those in controls (Malechka et al., 2017; Tuitoek et al., 1996). Diabetes has been reported to generate a more acidic environment within rod photoreceptors that causes a delay in rhodopsin regeneration (Ostroy, 1998; Ostroy et al., 1994). Rod cell ion channels, which are normally open in the dark, become partially closed in dark-adapted diabetic mice, and injection of 11-cis-retinaldehyde partially restored the channel-mediated ion movement into rod cells towards normal (Berkowitz et al., 2012a). Likewise, a single administration of the retinoid, 9-cis-retinal, to diabetic Akita mice inhibited diabetes-induced abnormalities in ERG (a- and b-wave amplitudes of scotopic ERG), retinal oxidative stress (as estimated by levels of 3-nitrotyrosine), and apoptosis of retinal cells (Malechka et al., 2020). How long this effect lasted was not investigated. Retinyl esters, which are produced in the visual cycle and stored in the RPE, accumulated to abnormal amounts in diabetes of diabetic mice, suggesting that the visual cycle was disturbed at the level of RPE65 or downstream (Liu et al., 2015). Overall, these results would seem to suggest that retinoid metabolism in the eye can be impaired in diabetes, which can lead to deficient generation of visual pigments and neural retinal dysfunction in early diabetes.
Acute hyperglycemia accelerated rod dark adaptation after bleach in Type 2 diabetic patients who had a subnormal dark adaptation response (compared to historical controls) (Holfort et al., 2010). It is not obvious why acute hyperglycemia would accelerate visual cycle activity and rod recovery rate in patients whose diabetes (or hyperglycemia) initially caused the subnormal rod recovery rate, but other investigators show an increase in retinal blood flow and oxygen consumption during acute hyperglycemia (Tiedeman et al., 1998). A different study found that dark-adapted rod- and cone-sensitivity thresholds were normalized by hyperoxia and moderate hyperglycemia in diabetic subjects, and that cone threshold sensitivity was improved also in non-diabetics during hyperoxia and hyperglycemia (Kurtenbach et al., 2006). Apparently, the disruptions in outer retinal visual processes that develop in diabetes are not irreversible.
In light of evidence that visual cycle activity is subnormal in diabetes, it is very surprising that other evidence indicates that slowing the visual cycle can inhibit the development of lesions of early DR. Retinylamine is a retinoid inhibitor of RPE65, and its administration significantly inhibited both the diabetes-induced increase in retinal vascular permeability and retinal capillary degeneration over an 8 month period (Liu et al., 2015) (Fig 10). Administration of the retinoid, however, had less effect on retinal function; retinylamine partially inhibited the diabetes-induced defect in contrast sensitivity, but had no effect on the defect in spatial frequency threshold. Since RPE65 is a critical component of the visual cycle (regenerating 11-cis retinal after activation of the photoreceptors by light), the beneficial effects of retinylamine on the early vascular lesions of DR are consistent with the possibility that inhibition of the visual cycle inhibits the development of the retinopathy. Retinylamine conceivably might have other effects, but it does not affect the retinoic acid-metabolizing cytochrome P450 enzymes, is not a substrate for cytochrome P450 CYP26 enzymes, does not activate the retinoid-X receptor, and interacts with the retinoic acid receptor only at micromolar concentrations (Golczak et al., 2005b). Retinylamine is acetylated by LRAT and stored in the RPE (Golczak et al., 2005a; Golczak et al., 2005b; Golczak et al., 2008; Liu et al., 2015; Maeda et al., 2006) from which it is then released slowly, thus allowing the compound to be administered only once per week. In addition to implicating the visual cycle in the development of vascular lesions of early DR, this data provides novel evidence also that the RPE contributes in an important way to the pathogenesis of the retinopathy.
Rpe65−/− mice made experimentally diabetic provide further evidence that photoreceptor cells play an important role in the development of molecular, functional and histologic lesions characteristic of DR (Thebeau et al., 2020). Those investigators found that RPE65-deficient mice diabetic for 6 months were protected from the adverse effects of diabetes on expression of inflammatory genes, on visual function, and on the integrity of retinal capillaries (Fig 10).
Other molecular players in the visual cycle also have been implicated in the molecular or physiologic alterations in the retina in diabetes. For example, genetic deletion of Lrat, which catalyzes the esterification of all-trans-retinol into all-trans-retinyl ester during phototransduction, inhibited the diabetes-induced increase in retinal oxidative stress (Liu et al., 2015). Whether or not genetic or pharmacologic manipulation of Lrat or other enzymes in the visual cycle affect the retinal vasculature or neuroglial retina in diabetes has not yet been explored.
7.3. Light as a potential therapy to inhibit diabetic retinopathy.
In contrast to the evidence that light-mediated processes play a role in the development of vascular and neural abnormalities of DR, others have postulated that the absence of light plays a critical role in the development of at least the advanced stages of DR. Photoreceptor cells have very high levels of mitochondria in their inner segment, and consume most of the oxygen in the retina (Ahmed et al., 1993; Linsenmeier and Braun, 1992), especially in the dark (Arden et al., 2005; Arden et al., 1998; Wang et al., 2010), when rod dark currents become maximal (Haugh et al., 1990; Linsenmeier, 1986). Because photoreceptors have an especially high metabolic activity at night, Arden and colleagues postulated that increased photoreceptor metabolic activity would make the retina more hypoxic in the night than during the day, especially in compromised or diseased environments such as in diabetes.
Arden and colleagues reported that application of blue-green (505nm) light all night long could mitigate aspects of DR, presumably by inhibiting the expected increase in metabolic activity of the retina that occurs in darkness (Arden et al., 2010; Arden et al., 2011; Arden et al., 2005; Arden and Sivaprasad, 2011, 2012; Ramsey and Arden, 2015). Their studies were based on evidence that dark-adaptation exacerbates hypoxia in the diabetic retina, acting as a powerful stimulus for the overproduction of VEGF and other less well understood factors. Patients with mild non-proliferative DR and early, untreated non-sight-threatening macular edema slept for 6 months wearing masks that illuminated one closed eye with 505 nm light of sufficient intensity to suppress dark current in that eye (Arden et al., 2011). At 6 months, the number of treated eyes showing intraretinal cysts was reduced by about one third, whereas the number of untreated fellow eyes showing cysts nearly doubled. In the treated eyes, retinal thickness was significantly reduced toward normal, and visual acuity, achromatic contrast sensitivity, and microperimetric thresholds improved significantly. Unfortunately, a larger 2-year Phase 3 clinical trial (CLEOPATRA) of this hypothesis recently was reported (Sivaprasad et al., 2018), and it failed to support the hypothesis that suppression of photoreceptor “dark current” at night would inhibit DME. Review of the results however, suggest that a major factor in the failure of this trial was poor patient compliance to wear the goggles all night long for the duration of the study. An animal study investigating this concept reported that preventing rod cells from dark-adapting did not inhibit retinal neuronal and glial abnormalities in early stages of diabetes (Kur et al., 2016).
Others also have been investigating the use of light (photobiomodulation) to inhibit the development of early DR, but those studies used far-red light (670 nm) for only 3–4 min per day, and do not postulate that the mechanism involves suppression of dark current. Animal studies using albino rats and pigmented mice showed that whole-body photobiomodulation inhibited diabetes-induced abnormalities in retinal oxidative stress, inflammation, and electrophysiology (Saliba et al., 2015; Tang et al., 2014), and very effectively inhibited diabetes-induced increases in capillary degeneration and accumulation of albumin in neural retina (Cheng et al., 2017) (Fig12). Whether or not the beneficial effects of this light therapy involve the outer retina is not clear, because shielding of the head from the light treatment each day nevertheless still had a beneficial effect on molecular abnormalities induced in the retina by diabetes (Saliba et al., 2015). Nevertheless, photobiomodulation is being found to have benefits in a number of disease (Tsai and Hamblin, 2017). Two different research groups (Eells et al., 2017; Tang et al., 2014) have presented pilot studies describing a small number of diabetic patients having retinal edema who were treated with brief sessions of light at 670nm, and both of these reports showed that the light therapy had beneficial effects to reverse the edema in the treated eye. A prospective multi-center pilot study evaluating photobiomodulation therapy for DME currently is being conducted by the Diabetic Retinopathy Clinical Research Network (DRCR).
Fig 12.

Noninvasive application of far-red light (670 nm; 6 joules/cm2) for 4 minutes per day to diabetic (D) mice for 8 months inhibited the diabetes-induced (a) degeneration of retinal capillaries, (b) leakage of FITC-albumin into neural retina at the level of the outer plexiform layer, and reductions in (c) spatial frequency threshold and (d) contrast sensitivity. N; nondiabetic controls. Reproduced from (Cheng et al., 2017) © 2017 by the American Diabetes Association.
Conclusions.
In total, available evidence suggests that the visual cycle and phototransduction play previously unrecognized roles in the development of the microvascular and neural lesions that characterize the early stages of DR, but additional studies will be needed to determine how this occurs. Manipulation of phototransduction or visual cycle activity as a therapeutic approach would need to be approached cautiously to avoid undesirable effects on vision. Application of low intensity light at certain wavelengths is showing interesting potential to inhibit the retinopathy, but again, the mechanisms remain to be clarified.
8. Do clinical therapies for diabetic retinopathy act on the outer retina?
Results of the Diabetic Retinopathy study (DRS) and Early Treatment for Diabetic Retinopathy study (ETDRS) clearly demonstrated the ability of laser photocoagulation to inhibit the progression of clinically significant DR or DME (1981; 1985; 1991), thus significantly reducing the risk of visual loss due to diabetes. Since it is well appreciated that laser photocoagulation leads to focal destruction of the retinal photoreceptors cells and RPE, it seems highly likely that this therapy is mediating its beneficial effects on DR and/or DME via effects on the outer retina. Other therapies used clinically do not have the specificity for the outer retina to judge whether or not they are mediating their beneficial effects via the outer retina.
VEGF is known to be a major contributor to DME and altered vision in diabetic patients, and anti-VEGF agents have become the mainstay of treatment for DME. Evidence in patients and animals implicates VEGF in the pathogenesis in functional defects of the outer retina in diabetes, as illustrated by VEGF-induced modification of RPE tight junction proteins and barrier function (Ablonczy and Crosson, 2007; Ghassemifar et al., 2006a, b). In vivo OCT studies indicated that disruption of photoreceptor layer segments improved after anti-VEGF treatment, at least raising a possibility that the integrity of the inner and outer photoreceptor segments and the OLM might contribute to improvement of functional outcomes in DME patients getting anti-VEGF therapy (Fursova et al., 2017).
9. Molecular mechanisms by which the outer retina might contribute to retinal vascular and neuroglial damage
9.1. Oxidative stress
Diabetes gives rise to increased oxidative stress in the retina (Al-Shabrawey et al., 2008a; Al-Shabrawey et al., 2008b; Berkowitz et al., 2015c; Caldwell et al., 2005; Du et al., 2013; Kanwar et al., 2007; Kowluru et al., 2001b), and the major site of the oxidative stress in diabetes is in photoreceptor cells (Du et al., 2013; Kern and Berkowitz, 2015; Liu et al., 2019). This stress plays a critical role in the development of DR, because vascular lesions such as capillary degeneration are inhibited by administering anti-oxidants (Kowluru et al., 2001b) or over-expression of anti-oxidant enzymes such as superoxide dismutase (Berkowitz et al., 2009a; Kanwar et al., 2007). The extent and mechanisms by which oxidative stress in outer retinal cells affects the retinal neural, glial or vascular cells in diabetes has not yet been clarified.
9.2. Inflammation
Photoreceptor cells contribute to the diabetes-induced increases in inflammatory proteins in the retina (de Gooyer et al., 2006b; Du et al., 2013), and photoreceptor cells themselves are a source of increased inflammatory proteins (e.g. IL-1α, IL-1β, IL-6, IL-12, chemokine C-X-C motif ligand 1 (CXCL1), monocyte chemoattractant protein 1 (MCP-1), CXCL12a, I-309, chemokine ligand 25 (CCL25) and TNF-α) in the retina in diabetes (Scuderi et al., 2015; Tonade et al., 2017). Soluble inflammatory products released from retinal photoreceptors isolated from diabetic mice have been found to induce inflammatory changes in retinal endothelial cells but not to cause endothelial death in vitro (Tonade et al., 2016; Tonade et al., 2017). However, direct evidence that inflammatory mediators released from photoreceptor cells or RPE have adverse effects on the retinal neural, glial or vascular cells in vivo has not been reported to date.
9.3. Glutamate
Glutamate release from photoreceptor cells can alter the function of cells of the inner retina, and excessive release of the neurotransmitter might contribute to degeneration of cells that express the NMDA receptor (such as retinal ganglion cells), but this has not been directly demonstrated to date.
8.4. Ion flux in photoreceptors
The role that ATPases and ion flux in photoreceptor cells may play in retinal vascular damage in diabetes is largely unexplored, but is of interest because of the importance of ion regulation in cell function and integrity, and the strong association between effects of various experimental therapies on retinal ion regulation or ATPase activities in diabetic rodents and the effect of those therapies on the development of vascular lesions of early DR. For example, captopril, anti-oxidant therapies, deletion of iNOS, and photobiomodulation (Berkowitz et al., 2009a; Berkowitz et al., 2007b; Kern et al., 2010; Ottlecz and Bensaoula, 1996; Zhang and Kern, 2003) have been shown to beneficially affect both of these endpoints. The potential roles of ion transport in cells of the outer retina on diabetes-induced injury to other neuronal or glial cells have not been reported.
8.5. Insulin signaling
Insulin clearly lowers blood glucose in diabetes, and that has a strong influence on the development of DR. Direct evidence that insulin acts on cells of the outer retina to affect the response of the retina to diabetes is limited, but diabetic mice with an RPE-specific knockout of the insulin receptor showed lower a- and b-wave electroretinogram amplitudes compared to diabetic mice that expressed the insulin receptor on the RPE (Tarchick et al., 2019). Since photoreceptor cell-specific deletion of the insulin receptor gene results in loss of those photoreceptors (Rajala et al., 2008; Yi et al., 2005), the loss of photoreceptor cells that has been detected in diabetic patients and animals might be due in part to the subnormal insulin and insulin signaling. Several drugs have been reported to treat insulin resistance in retinas of diabetic animals (Fort et al., 2011; Jiang et al., 2015; Jiang et al., 2014a), but whether this effect is mediated also in photoreceptor cells is not known.
8.6. Hypoxia
Photoreceptor cells are known to be major consumers of oxygen, thus resulting in hypoxic conditions at night or when the retinal or choroidal vasculatures are compromised, such as in DR (Wangsa-Wirawan and Linsenmeier, 2003). Retinal oxygen tension in dark-adapted diabetic cats reached a minimum PO2 of nearly 0 mmHg in the outer retina (Linsenmeier, 1986; Wangsa-Wirawan and Linsenmeier, 2003). The resulting hypoxia-induced increase in levels of VEGF in the retina can lead to increased permeability and neovascularization of the retinal vasculature (Miller et al., 2013; Zhang et al., 2009). Effects of hypoxia potentially caused by cells of the outer retina on diabetes-induced abnormalities of other neural or glial cells have not been reported.
10. Future directions
The current clinical definition of DR focuses on microvascular disease, and thus overlooks the potential contribution of neuronal cells, whether direct or indirect, in diabetes-induced vision loss (Simo et al., 2018). Photoreceptor cells are specialized neurons, and as the most numerous and metabolically active cell in the retina, they can have an undue influence on the function and integrity of other retinal cells (Fu et al., 2020). Evidence that photoreceptor cells and RPE contribute to the lesions of DR is accumulating, but is yet incomplete. Additional research is needed.
Patient studies that describe effects of diabetes on the outer retina to date have included relatively small numbers of patients, but available evidence suggests that diabetes can cause thinning or loss of some photoreceptors. The retinal neurodegeneration in diabetes seems to be initiated before gross clinical signs of vasculopathy become apparent, but the importance of that observation needs to be tempered by the modest amount of photoreceptor death that has been detected (which seems minor compared to that in retinal degenerative diseases such as AMD). Thus, the contribution of such minor photoreceptor cell loss on visual impairment in diabetes remains questionable. Cellular dysfunction (as compared to degeneration) of photoreceptor cells, however, is likely to affect many more cells than the number that die in diabetes, and the impact of that photoreceptor cell dysfunction on other retinal cells and vision could be large.
Although Type 1 diabetes and Type 2 diabetes share the common symptom hyperglycemia, they differ in a variety of other ways, including levels of circulating lipids and insulin/insulin signaling. Not enough information is available to know if the type of diabetes affects the involvement of photoreceptor or RPE cells in the pathogenesis of neural, glia or vascular lesions of DR.
Evidence that photoreceptor cells and RPE contribute to the development of DR is strongest with respect to the microvascular disease. The potential contributions of the outer retina to development of diabetes-induced abnormalities of other neuronal cells or glia is less studied. The extent to which photoreceptors affect also non-neuronal cells such as microglia and glial cells in diabetes requires future study.
Evidence is beginning to suggest that the response of the retina to light and dark contributes to its susceptibility to diabetes, so future studies will be needed to identify the mechanisms for how light and dark influence the development of DR. Seemingly contradictory evidence (summarized above in Section 6) suggests that although dark adaptation (and thus visual cycle activity) seem subnormal in diabetes, inhibition of visual cycle activity can inhibit at least the vascular lesions of early DR. The evidence underlying both conclusions seems solid, and so a reasonable interpretation is that both conclusions are correct. Perhaps the new (subnormal) rate of visual cycle activity still causes an adverse effect on the metabolism or integrity of the photoreceptors/RPE in the abnormal mileau of diabetes. Additional work in this area is critical.
Phototransduction and the visual cycle may be novel therapeutic targets to inhibit the retinopathy, but those pathways cannot be casually manipulated due to their importance in visual function. The recent evidence that physiologic levels of light also can be used to inhibit the disease process could open new avenues to treatment of retinal and ocular diseases, but data is yet needed to establish safety and efficacy. The recent failure of the CLEOPATRA trial (testing if chronic suppression of the rod cell “dark current” at night via low intensity light could inhibit DME) is disappointing, but the results obtained suggest that the failure might have been due more to patient non-compliance due to how the light was delivered, rather than a flaw in the scientific hypothesis being tested. Additional research on light and photobiomodulation as a therapeutic approach to inhibit DR, other retinopathies, and other systemic diseases is warranted, especially in light of the uncertainty of whether the outer retina plays a primary role in the effects of the light (as opposed to the light therapy acting systemically).
Altogether, results suggest that cells of the outer retina play an important role in the pathogenesis of retinal vascular and neuronal damage or dysfunction in diabetes (either directly or indirectly) (Fig 13). Abundant evidence has been developed regarding the participation also of other cell-types in the development of the retinopathy, so evaluation of the relative importance of all of these various cell-types can be expected to provide useful insight into the best therapeutic targets at which to inhibit the retinopathy.
Fig 13.

Postulated contribution of the outer retina to the development of early stages of DR. Grey lines represent areas that are outside of the topic of this review.
Acknowledgements
This work was supported by NIH grants EY022938 and R24 EY024864, and BX003604 from the Department of Veterans Affairs. Dr. Kern is the recipient of a Research Career Scientist award from the Department of Veterans Affairs. The authors acknowledge an RPB unrestricted grant to the Department of Ophthalmology from Research to Prevent Blindness, New York, NY). Clinical photos in Figs 2 and 6 were generously provided by Dr. Andrew Browne (University of California-Irvine), the 2-photon microscope image (Fig 2) and description were provided by Dr. Grazyna Palczewska (University of California-Irvine), and the OCT-A image (Fig 6) was provided by Dr. Amani Fawzi (Northwestern University, Chicago, IL).
Neither of the authors have any financial and personal relationships that could inappropriately influence or bias this work.
Glossary
- 5-Lox
5-lipoxygenase
- AMD
age-related macular degeneration
- AO
adaptive optics
- ATP
adenosine triphosphate
- BRB
blood-retinal barrier
- CCL
chemokine ligand
- cGMP
cyclic guanosine monophosphate
- CXCL
chemokine C-X-C motif ligand
- DM
diabetes mellitus
- DME
diabetic macular edema
- DR
diabetic retinopathy
- DRIL
Disorganization of the retinal inner layers
- ER
endoplasmic reticulum
- ERG
electroretinogram
- FA
fluorescein angiography
- FGF21
fibroblast growth factor 21
- Glut1
glucose transporter 1
- GMP
guanosine monophosphate
- HIF-1α
Hypoxia-inducible factor 1 alpha
- IKK
I kappa B kinase
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- IR
Infra-red
- IS
inner segment
- L
long wavelength
- logMAR
Logarithm of the Minimum Angle of Resolution
- LRAT
lecithin/retinol acyltransferase
- M
mid-wavelength opsin
- MAPK
Mitogen-activated protein kinase
- MCP-1
monocyte chemoattractant protein 1
- MEMRI
Manganese-enhanced magnetic resonance imaging
- MRI
magnetic resonance imaging
- MyD88
Innate immune signal transduction adapter
- Na/K-ATPase
sodium potassium ATPase
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NF-κB
nuclear factor kappa light chain enhancer of activated B cells
- NMDA
N-methyl-D-aspartate
- NPDR
non-proliferative DR
- OCT
optical coherence tomography
- OCT-A
OCT-angiography
- OLM
outer limiting membrane
- ONL
outer nuclear layer
- OS
outer segment
- PACAP
pituitary adenylate cyclase activating polypeptide
- PARP1
Poly(ADP-ribose) polymerase 1
- PDR
proliferative diabetic retinopathy
- PKC
protein kinase C
- PLGF-1
Placental Growth Factor-1
- RBP3
retinol binding protein 3
- rd
retinal degeneration
- ROP
retinopathy of prematurity
- RPE
retinal pigment epithelium
- RPE65
Retinal pigment epithelium-specific 65 kDa protein (retinoid isomerohydrolase)
- S
short wavelength opsin
- STRA6
stimulated by retinoic acid 6 protein
- TAK1
transforming growth factor β-activated kinase 1
- TLR
toll-like receptor
- TNF-α
tumor necrosis factor alpha
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- VA
visual acuity
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ablonczy Z, Crosson CE, 2007. Vegf Modulation of Retinal Pigment Epithelium Resistance. Experimental eye research 85, 762–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams AJ, Zisman F, Ai E, Bresnick G, 1987. Macular Edema Reduces B Cone Sensitivity in Diabetics. Applied optics 26, 1455–1457. [DOI] [PubMed] [Google Scholar]
- Adamus G, Wang S, Kyger M, Worley A, Lu B, Burrows GG, 2012. Systemic Immunotherapy Delays Photoreceptor Cell Loss and Prevents Vascular Pathology in Royal College of Surgeons Rats. Molecular vision 18, 2323–2337. [PMC free article] [PubMed] [Google Scholar]
- Adler AJ, Southwick RE, 1992. Distribution of Glucose and Lactate in the Interphotoreceptor Matrix. Ophthalmic research 24, 243–252. [DOI] [PubMed] [Google Scholar]
- Agostinho P, Duarte CB, Oliveira CR, 1996. Activity of Ionotropic Glutamate Receptors in Retinal Cells: Effect of Ascorbate/Fe(2+)-Induced Oxidative Stress. J Neurochem 67, 1153–1163. [DOI] [PubMed] [Google Scholar]
- Ahmed J, Braun RD, Dunn R Jr., Linsenmeier RA, 1993. Oxygen Distribution in the Macaque Retina. Investigative ophthalmology & visual science 34, 516–521. [PubMed] [Google Scholar]
- Aizu Y, Oyanagi K, Hu J, Nakagawa H, 2002. Degeneration of Retinal Neuronal Processes and Pigment Epithelium in the Early Stage of the Streptozotocin-Diabetic Rats. Neuropathology 22, 161–170. [DOI] [PubMed] [Google Scholar]
- Akula JD, Hansen RM, Martinez-Perez ME, Fulton AB, 2007. Rod Photoreceptor Function Predicts Blood Vessel Abnormality in Retinopathy of Prematurity. Investigative ophthalmology & visual science 48, 4351–4359. [DOI] [PubMed] [Google Scholar]
- Akula JD, Mocko JA, Benador IY, Hansen RM, Favazza TL, Vyhovsky TC, Fulton AB, 2008. The Neurovascular Relation in Oxygen-Induced Retinopathy. Molecular vision 14, 2499–2508. [PMC free article] [PubMed] [Google Scholar]
- Al-Shabrawey M, Bartoli M, El-Remessy A, Ma G, Matragoon S, Lemtalsi T, Caldwell RW, Caldwell RB, 2008a. Role of Nadph Oxidase and Stat3 in Statin-Mediated Protection against Diabetic Retinopathy. Investigative ophthalmology & visual science 49, 3231–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shabrawey M, Rojas M, Sanders T, Behzadian A, El-Remessy A, Bartoli M, Parpia AK, Liou G, Caldwell RB, 2008b. Role of Nadph Oxidase in Retinal Vascular Inflammation. Investigative ophthalmology & visual science 49, 3239–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert-Fort M, Hombrebueno JR, Pons-Vazquez S, Sanz-Gonzalez S, Diaz-Llopis M, Pinazo-Duran MD, 2014. Retinal Neurodegenerative Changes in the Adult Insulin Receptor Substrate-2 Deficient Mouse. Experimental eye research 124, 1–10. [DOI] [PubMed] [Google Scholar]
- Alm A, Bill A, 1972. The Oxygen Supply to the Retina. Ii. Effects of High Intraocular Pressure and of Increased Arterial Carbon Dioxide Tension on Uveal and Retinal Blood Flow in Cats. A Study with Radioactively Labelled Microspheres Including Flow Determinations in Brain and Some Other Tissues. Acta Physiol Scand 84, 306–319. [DOI] [PubMed] [Google Scholar]
- Alvarez Y, Chen K, Reynolds AL, Waghorne N, O’Connor JJ, Kennedy BN, 2010. Predominant Cone Photoreceptor Dysfunction in a Hyperglycaemic Model of Non-Proliferative Diabetic Retinopathy. Dis Model Mech 3, 236–245. [DOI] [PubMed] [Google Scholar]
- Ambati J, Chalam KV, Chawla DK, D’Angio CT, Guillet EG, Rose SJ, Vanderlinde RE, Ambati BK, 1997. Elevated Gamma-Aminobutyric Acid, Glutamate, and Vascular Endothelial Growth Factor Levels in the Vitreous of Patients with Proliferative Diabetic Retinopathy. Archives of ophthalmology 115, 1161–1166. [DOI] [PubMed] [Google Scholar]
- Amemiya T, 1977. Dark Adaptation in Diabetics. Ophthalmologica. Journal international d’ophtalmologie. International journal of ophthalmology. Zeitschrift fur Augenheilkunde 174, 322–326. [DOI] [PubMed] [Google Scholar]
- Ames A 3rd, 1992. Energy Requirements of Cns Cells as Related to Their Function and to Their Vulnerability to Ischemia: A Commentary Based on Studies on Retina. Canadian journal of physiology and pharmacology 70 Suppl, S158–164. [DOI] [PubMed] [Google Scholar]
- Ames A 3rd, Li YY, Heher EC, Kimble CR, 1992. Energy Metabolism of Rabbit Retina as Related to Function: High Cost of Na+ Transport. The Journal of neuroscience : the official journal of the Society for Neuroscience 12, 840–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonetti DA, Klein R, Gardner TW, 2012. Diabetic Retinopathy. The New England journal of medicine 366, 1227–1239. [DOI] [PubMed] [Google Scholar]
- Antonetti DA, VanGuilder HD, Mao-Lin C, 2008. Vascular Permeability in Diabetic Retinopathy, in: Duh E. (Ed.), Diabetic Retinopathy. Humana Press, Totowa, NJ, pp. 333–352. [Google Scholar]
- Applebury ML, Antoch MP, Baxter LC, Chun LL, Falk JD, Farhangfar F, Kage K, Krzystolik MG, Lyass LA, Robbins JT, 2000. The Murine Cone Photoreceptor: A Single Cone Type Expresses Both S and M Opsins with Retinal Spatial Patterning. Neuron 27, 513–523. [DOI] [PubMed] [Google Scholar]
- Arden GB, 2001. The Absence of Diabetic Retinopathy in Patients with Retinitis Pigmentosa: Implications for Pathophysiology and Possible Treatment. Br. J. Ophthalmol. 85, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden GB, Gunduz MK, Kurtenbach A, Volker M, Zrenner E, Gunduz SB, Kamis U, Ozturk BT, Okudan S, 2010. A Preliminary Trial to Determine Whether Prevention of Dark Adaptation Affects the Course of Early Diabetic Retinopathy. Eye 24, 1149–1155. [DOI] [PubMed] [Google Scholar]
- Arden GB, Jyothi S, Hogg CH, Lee YF, Sivaprasad S, 2011. Regression of Early Diabetic Macular Oedema Is Associated with Prevention of Dark Adaptation. Eye 25, 1546–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden GB, Sidman RL, Arap W, Schlingemann RO, 2005. Spare the Rod and Spoil the Eye. The British journal of ophthalmology 89, 764–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden GB, Sivaprasad S, 2011. Hypoxia and Oxidative Stress in the Causation of Diabetic Retinopathy. Curr Diabetes Rev 7, 291–304. [DOI] [PubMed] [Google Scholar]
- Arden GB, Sivaprasad S, 2012. The Pathogenesis of Early Retinal Changes of Diabetic Retinopathy. Documenta ophthalmologica. Advances in ophthalmology 124, 15–26. [DOI] [PubMed] [Google Scholar]
- Arden GB, Wolf JE, Tsang Y, 1998. Does Dark Adaptation Exacerbate Diabetic Retinopathy? Evidence and a Linking Hypothesis. Vision research 38, 1723–1729. [DOI] [PubMed] [Google Scholar]
- Arshavsky VY, Lamb TD, Pugh EN Jr., 2002. G Proteins and Phototransduction. Annual review of physiology 64, 153–187. [DOI] [PubMed] [Google Scholar]
- Badr GA, Tang J, Ismail-Beigi F, Kern TS, 2000. Diabetes Down-Regulates Glut1 Expression in Retina and Its Microvessels, but Not in Cerebral Cortex or Its Microvessels. Diabetes 49, 1016–1021. [DOI] [PubMed] [Google Scholar]
- Bailey TA, Kanuga N, Romero IA, Greenwood J, Luthert PJ, Cheetham ME, 2004. Oxidative Stress Affects the Junctional Integrity of Retinal Pigment Epithelial Cells. Investigative ophthalmology & visual science 45, 675–684. [DOI] [PubMed] [Google Scholar]
- Bandah-Rozenfeld D, Collin RW, Banin E, van den Born LI, Coene KL, Siemiatkowska AM, Zelinger L, Khan MI, Lefeber DJ, Erdinest I, Testa F, Simonelli F, Voesenek K, Blokland EA, Strom TM, Klaver CC, Qamar R, Banfi S, Cremers FP, Sharon D, den Hollander AI, 2010. Mutations in Impg2, Encoding Interphotoreceptor Matrix Proteoglycan 2, Cause Autosomal-Recessive Retinitis Pigmentosa. Am J Hum Genet 87, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banford D, North RV, Dolben J, Butler G, Owens DR, 1994. Longitudinal Study of Visual Functions in Young Insulin Dependent Diabetics. Ophthalmic & physiological optics : the journal of the British College of Ophthalmic Opticians 14, 339–346. [PubMed] [Google Scholar]
- Barber AJ, Antonetti DA, Gardner TW, 2000. Altered Expression of Retinal Occludin and Glial Fibrillary Acidic Protein in Experimental Diabetes. The Penn State Retina Research Group. Investigative ophthalmology & visual science 41, 3561–3568. [PubMed] [Google Scholar]
- Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW, 1998. Neural Apoptosis in the Retina During Experimental and Human Diabetes. Early Onset and Effect of Insulin. The Journal of clinical investigation 102, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barham R, El Rami H, Sun JK, Silva PS, 2017. Evidence-Based Treatment of Diabetic Macular Edema. Semin Ophthalmol 32, 56–66. [DOI] [PubMed] [Google Scholar]
- Bavinger JC, Dunbar GE, Stem MS, Blachley TS, Kwark L, Farsiu S, Jackson GR, Gardner TW, 2016. The Effects of Diabetic Retinopathy and Pan-Retinal Photocoagulation on Photoreceptor Cell Function as Assessed by Dark Adaptometry. Investigative ophthalmology & visual science 57, 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearse MA Jr., Adams AJ, Han Y, Schneck ME, Ng J, Bronson-Castain K, Barez S, 2006. A Multifocal Electroretinogram Model Predicting the Development of Diabetic Retinopathy. Progress in retinal and eye research 25, 425–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearse MA Jr., Ozawa GY, 2014. Multifocal Electroretinography in Diabetic Retinopathy and Diabetic Macular Edema. Current diabetes reports 14, 526. [DOI] [PubMed] [Google Scholar]
- Beasley S, El-Sherbiny M, Megyerdi S, El-Shafey S, Choksi K, Kaddour-Djebbar I, Sheibani N, Hsu S, Al-Shabrawey M, 2014. Caspase-14 Expression Impairs Retinal Pigment Epithelium Barrier Function: Potential Role in Diabetic Macular Edema. Biomed Res Int 2014, 417986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S, Carroll LS, Vinberg F, 2020. Rod Phototransduction and Light Signal Transmission During Type 2 Diabetes. BMJ Open Diabetes Res Care 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bek T, 1994. Transretinal Histopathological Changes in Capillary-Free Areas of Diabetic Retinopathy. Acta Ophthalmol. 72, 409–415. [DOI] [PubMed] [Google Scholar]
- Bensaoula T, Ottlecz A, 1995. Decreased Activity of Retinal Atpases and Na+, K+-Atpase Isozymes in the Microvascular and Neural Compartments of the Experimentally Diabetic Retina. Arvo Abstracts. Invest. Ophthalmol. Vis. Sci. 36, S897. [Google Scholar]
- Bensaoula T, Ottlecz A, 2001. Biochemical and Ultrastructural Studies in the Neural Retina and Retinal Pigment Epithelium of Stz-Diabetic Rats: Effect of Captopril. J Ocul Pharmacol Ther 17, 573–586. [DOI] [PubMed] [Google Scholar]
- Berkowitz BA, 2020. Preventing Diabetic Retinopathy by Mitigating Subretinal Space Oxidative Stress in Vivo. Visual neuroscience 37, E002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Bissig D, Patel P, Bhatia A, Roberts R, 2012a. Acute Systemic 11-Cis-Retinal Intervention Improves Abnormal Outer Retinal Ion Channel Closure in Diabetic Mice. Molecular vision 18, 372–376. [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Bissig D, Roberts R, 2016a. Mri of Rod Cell Compartment-Specific Function in Disease and Treatment in Vivo. Progress in retinal and eye research 51, 90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Bissig D, Ye Y, Valsadia P, Kern TS, Roberts R, 2012b. Evidence for Diffuse Central Retinal Edema in Vivo in Diabetic Male Sprague Dawley Rats. PloS one 7, e29619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Bredell BX, Davis C, Samardzija M, Grimm C, Roberts R, 2015a. Measuring in Vivo Free Radical Production by the Outer Retina. Investigative ophthalmology & visual science 56, 7931–7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Gradianu M, Bissig D, Kern TS, Roberts R, 2009a. Retinal Ion Regulation in a Mouse Model of Diabetic Retinopathy: Natural History and the Effect of Cu/Zn Superoxide Dismutase Overexpression. Investigative ophthalmology & visual science 50, 2351–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Grady EM, Khetarpal N, Patel A, Roberts R, 2015b. Oxidative Stress and Light-Evoked Responses of the Posterior Segment in a Mouse Model of Diabetic Retinopathy. Investigative ophthalmology & visual science 56, 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Grady EM, Roberts R, 2014. Confirming a Prediction of the Calcium Hypothesis of Photoreceptor Aging in Mice. Neurobiology of aging 35, 1883–1891. [DOI] [PubMed] [Google Scholar]
- Berkowitz BA, Kern TS, Bissig D, Patel P, Bhatia A, Kefalov VJ, Roberts R, 2015c. Systemic Retinaldehyde Treatment Corrects Retinal Oxidative Stress, Rod Dysfunction, and Impaired Visual Performance in Diabetic Mice. Investigative ophthalmology & visual science 56, 6294–6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Lewin AS, Biswal MR, Bredell BX, Davis C, Roberts R, 2016b. Mri of Retinal Free Radical Production with Laminar Resolution in Vivo. Investigative ophthalmology & visual science 57, 577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Podolsky RH, Farrell B, Lee H, Trepanier C, Berri AM, Dernay K, Graffice E, Shafie-Khorassani F, Kern TS, Roberts R, 2018. D-Cis-Diltiazem Can Produce Oxidative Stress in Healthy Depolarized Rods in Vivo. Investigative ophthalmology & visual science 59, 2999–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Podolsky RH, Lins-Childers KM, Li Y, Qian H, 2019. Outer Retinal Oxidative Stress Measured in Vivo Using Quench-Assisted (Quest) Oct. Investigative ophthalmology & visual science 60, 1566–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Roberts R, Goebel DJ, Luan H, 2006. Noninvasive and Simultaneous Imaging of Layer-Specific Retinal Functional Adaptation by Manganese-Enhanced Mri. Investigative ophthalmology & visual science 47, 2668–2674. [DOI] [PubMed] [Google Scholar]
- Berkowitz BA, Roberts R, Luan H, Bissig D, Bui BV, Gradianu M, Calkins DJ, Vingrys AJ, 2007a. Manganese-Enhanced Mri Studies of Alterations of Intraretinal Ion Demand in Models of Ocular Injury. Investigative ophthalmology & visual science 48, 3796–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Roberts R, Oleske DA, Chang M, Schafer S, Bissig D, Gradianu M, 2009b. Quantitative Mapping of Ion Channel Regulation by Visual Cycle Activity in Rodent Photoreceptors in Vivo. Investigative ophthalmology & visual science 50, 1880–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz BA, Roberts R, Stemmler A, Luan H, Gradianu M, 2007b. Impaired Apparent Ion Demand in Experimental Diabetic Retinopathy: Correction by Lipoic Acid. Investigative ophthalmology & visual science 48, 4753–4758. [DOI] [PubMed] [Google Scholar]
- Berkowitz BA, Wen X, Thoreson WB, Kern TS, Roberts R, 2015d. Abnormal Rod Calcium Homeostasis and the Development of Retinal Oxidative Stress in Diabetes (Arvo Abstract). Investigative ophthalmology & visual science 56, 4280. [Google Scholar]
- Beutelspacher SC, Serbecic N, Barash H, Burgansky-Eliash Z, Grinvald A, Krastel H, Jonas JB, 2011. Retinal Blood Flow Velocity Measured by Retinal Function Imaging in Retinitis Pigmentosa. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 249, 1855–1858. [DOI] [PubMed] [Google Scholar]
- Bhutto IA, Amemiya T, 2001. Retinal Vascular Architecture Is Maintained in Retinal Degeneration: Corrosion Cast and Electron Microscope Study. Eye 15, 531–538. [DOI] [PubMed] [Google Scholar]
- Blair NP, Tso MO, Dodge JT, 1984. Pathologic Studies of the Blood--Retinal Barrier in the Spontaneously Diabetic Bb Rat. Investigative ophthalmology & visual science 25, 302–311. [PubMed] [Google Scholar]
- Blanks JC, Johnson LV, 1986. Vascular Atrophy in the Retinal Degenerative Rd Mouse. The Journal of comparative neurology 254, 543–553. [DOI] [PubMed] [Google Scholar]
- Boettner EA, Wolter JR, 1962. Transmission of the Ocular Media Investigative ophthalmology & visual science 1, 776–7873. [Google Scholar]
- Bogdanov P, Corraliza L, A Villena J, Carvalho AR, Garcia-Arumi J, Ramos D, Ruberte J, Simo R, Hernandez C, 2014. The Db/Db Mouse: A Useful Model for the Study of Diabetic Retinal Neurodegeneration. PloS one 9, e97302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth AA, Khalifah RG, Hudson BG, 1996. Thiamine Pyrophosphate and Pyridoxamine Inhibit the Formation of Antigenic Advanced Glycation End-Products: Comparison with Aminoguanidine. Biochem Biophys Res Commun 220, 113–119. [DOI] [PubMed] [Google Scholar]
- Boynton GE, Stem MS, Kwark L, Jackson GR, Farsiu S, Gardner TW, 2015. Multimodal Characterization of Proliferative Diabetic Retinopathy Reveals Alterations in Outer Retinal Function and Structure. Ophthalmology 122, 957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramall AN, Wright AF, Jacobson SG, McInnes RR, 2010. The Genomic, Biochemical, and Cellular Responses of the Retina in Inherited Photoreceptor Degenerations and Prospects for the Treatment of These Disorders. Annual review of neuroscience 33, 441–472. [DOI] [PubMed] [Google Scholar]
- Brandstätter JH, Hartveit E, Sassoè-Pognetto M, Wässle H, 1994. Expression of Nmda and High-Affinity Kainate Receptor Subunit Mrnas in the Adult Rat Retina. European Journal of Neuroscience 6, 1100–1112. [DOI] [PubMed] [Google Scholar]
- Braun RD, Linsenmeier RA, Goldstick TK, 1995. Oxygen Consumption in the Inner and Outer Retina of the Cat. Investigative ophthalmology & visual science 36, 542–554. [PubMed] [Google Scholar]
- Bresnick GH, Palta M, 1987. Temporal Aspects of the Electroretinogram in Diabetic Retinopathy. Archives of ophthalmology 105, 660–664. [DOI] [PubMed] [Google Scholar]
- Bueno JM, Cruz-Castillo R, Aviles-Trigueros M, Bautista-Elivar N, 2020. Arrangement of the Photoreceptor Mosaic in a Diabetic Rat Model Imaged with Multiphoton Microscopy. Biomedical optics express 11, 4901–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui BV, Armitage JA, Tolcos M, Cooper ME, Vingrys AJ, 2003. Ace Inhibition Salvages the Visual Loss Caused by Diabetes. Diabetologia 46, 401–408. [DOI] [PubMed] [Google Scholar]
- Bui BV, Loeliger M, Thomas M, Vingrys AJ, Rees SM, Nguyen CT, He Z, Tolcos M, 2009. Investigating Structural and Biochemical Correlates of Ganglion Cell Dysfunction in Streptozotocin-Induced Diabetic Rats. Experimental eye research 88, 1076–1083. [DOI] [PubMed] [Google Scholar]
- Burns SA, Elsner AE, Chui TY, Vannasdale DA Jr., Clark CA, Gast TJ, Malinovsky VE, Phan AD, 2014.In Vivo Adaptive Optics Microvascular Imaging in Diabetic Patients without Clinically Severe Diabetic Retinopathy. Biomedical optics express 5, 961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busik JV, Olson LK, Grant MB, Henry DN, 2002. Glucose-Induced Activation of Glucose Uptake in Cells from the Inner and Outer Blood-Retinal Barrier. Investigative ophthalmology & visual science 43, 2356–2363. [PubMed] [Google Scholar]
- Caldwell RB, Bartoli M, Behzadian MA, El-Remessy AE, Al-Shabrawey M, Platt DH, Liou GI, Caldwell RW, 2005. Vascular Endothelial Growth Factor and Diabetic Retinopathy: Role of Oxidative Stress. Curr Drug Targets 6, 511–524. [DOI] [PubMed] [Google Scholar]
- Caldwell RB, Roque RS, Solomon SW, 1989. Increased Vascular Density and Vitreo-Retinal Membranes Accompany Vascularization of the Pigment Epithelium in the Dystrophic Rat Retina. Current eye research 8, 923–937. [PubMed] [Google Scholar]
- Campochiaro PA, Aiello LP, Rosenfeld PJ, 2016. Anti-Vascular Endothelial Growth Factor Agents in the Treatment of Retinal Disease: From Bench to Bedside. Ophthalmology 123, S78–S88. [DOI] [PubMed] [Google Scholar]
- Cao J, McLeod S, Merges CA, Lutty GA, 1998. Choriocapillaris Degeneration and Related Pathologic Changes in Human Diabetic Eyes. Archives of ophthalmology 116, 589–597. [DOI] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM, 1979. Rods and Cones in the Mouse Retina. I. Structural Analysis Using Light and Electron Microscopy. The Journal of comparative neurology 188, 245–262. [DOI] [PubMed] [Google Scholar]
- Chalam KV, Sambhav K, 2016. Optical Coherence Tomography Angiography in Retinal Diseases. J Ophthalmic Vis Res 11, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WJ, Wu C, Xu Z, Kuse Y, Hara H, Duh EJ, 2017. Nrf2 Protects Photoreceptor Cells from Photo-Oxidative Stress Induced by Blue Light. Experimental eye research 154, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Li J, Yan Y, Shen X, 2016. Diabetic Macular Morphology Changes May Occur in the Early Stage of Diabetes. BMC ophthalmology 16, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Du Y, Liu H, Tang J, Veenstra A, Kern TS, 2017. Photobiodulation Inhibits Long-Term Structural and Functional Lesions of Diabetic Retinopathy. Diabetes 67, 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchore Y, Begaj T, Wu D, Drokhlyansky E, Cepko CL, 2017. Glycolytic Reliance Promotes Anabolism in Photoreceptors. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho NC, Poulsen GL, Ver Hoeve JN, Nork TM, 2000. Selective Loss of S-Cones in Diabetic Retinopathy. Archives of ophthalmology 118, 1393–1400. [DOI] [PubMed] [Google Scholar]
- Cogan DG, Toussaint D, Kuwabara T, 1961. Retinal Vascular Patterns. Iv. Diabetic Retinopathy. Archives of ophthalmology 66, 366–378. [DOI] [PubMed] [Google Scholar]
- Copenhagen DR, Jahr CE, 1989. Release of Endogenous Excitatory Amino Acids from Turtle Photoreceptors. Nature 341, 536–539. [DOI] [PubMed] [Google Scholar]
- Crider JY, Yorio T, Sharif NA, Griffin BW, 1997. The Effects of Elevated Glucose on Na+/K(+)-Atpase of Cultured Bovine Retinal Pigment Epithelial Cells Measured by a New Nonradioactive Rubidium Uptake Assay. J Ocul Pharmacol Ther 13, 337–352. [DOI] [PubMed] [Google Scholar]
- Crouch RK, Hazard ES, Lind T, Wiggert B, Chader G, Corson DW, 1992. Interphotoreceptor Retinoid-Binding Protein and Alpha-Tocopherol Preserve the Isomeric and Oxidation State of Retinol. Photochemistry and photobiology 56, 251–255. [DOI] [PubMed] [Google Scholar]
- Cuenca N, Fernandez-Sanchez L, Campello L, Maneu V, De la Villa P, Lax P, Pinilla I, 2014. Cellular Responses Following Retinal Injuries and Therapeutic Approaches for Neurodegenerative Diseases. Progress in retinal and eye research 43, 17–75. [DOI] [PubMed] [Google Scholar]
- Cunha-Vaz J, Ribeiro L, Lobo C, 2014. Phenotypes and Biomarkers of Diabetic Retinopathy. Progress in retinal and eye research 41, 90–111. [DOI] [PubMed] [Google Scholar]
- D’Amico AG, Maugeri G, Reitano R, Bucolo C, Saccone S, Drago F, D’Agata V, 2015. Pacap Modulates Expression of Hypoxia-Inducible Factors in Streptozotocin-Induced Diabetic Rat Retina. Journal of molecular neuroscience : MN 57, 501–509. [DOI] [PubMed] [Google Scholar]
- Dahrouj M, Alsarraf O, Liu Y, Crosson CE, Ablonczy Z, 2013. C-Type Natriuretic Peptide Protects the Retinal Pigment Epithelium against Advanced Glycation End Product-Induced Barrier Dysfunction. The Journal of pharmacology and experimental therapeutics 344, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahrouj M, Alsarraf O, McMillin JC, Liu Y, Crosson CE, Ablonczy Z, 2014. Vascular Endothelial Growth Factor Modulates the Function of the Retinal Pigment Epithelium in Vivo. Investigative ophthalmology & visual science 55, 2269–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilova I, Medvedeva S, Shmakova S, Chereshneva M, Sarapultsev A, Sarapultsev P, 2018. Pathological Changes in the Cellular Structures of Retina and Choroidea in the Early Stages of Alloxan-Induced Diabetes. World J Diabetes 9, 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daruich A, Matet A, Moulin A, Kowalczuk L, Nicolas M, Sellam A, Rothschild PR, Omri S, Gelize E, Jonet L, Delaunay K, De Kozak Y, Berdugo M, Zhao M, Crisanti P, Behar-Cohen F, 2018. Mechanisms of Macular Edema: Beyond the Surface. Progress in retinal and eye research 63, 20–68. [DOI] [PubMed] [Google Scholar]
- de Gooyer TE, Stevenson KA, Humphries P, Simpson DA, Curtis TM, Gardiner TA, Stitt AW, 2006a. Rod Photoreceptor Loss in Rho−/− Mice Reduces Retinal Hypoxia and Hypoxia-Regulated Gene Expression. Invest. Ophthalmol. Vis. Sci.. 47, 5553–5560. [DOI] [PubMed] [Google Scholar]
- de Gooyer TE, Stevenson KA, Humphries P, Simpson DA, Gardiner TA, Stitt AW, 2006b. Retinopathy Is Reduced During Experimental Diabetes in a Mouse Model of Outer Retinal Degeneration. Invest. Ophthalmol. Vis. Sci.. 47, 5561–5568. [DOI] [PubMed] [Google Scholar]
- Dean FM, Arden GB, Dornhorst A, 1997. Partial Reversal of Protan and Tritan Colour Defects with Inhaled Oxygen in Insulin Dependent Diabetic Subjects. The British journal of ophthalmology 81, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins D, Liu Y, Crosson CE, Ablonczy Z, 2016a. Histone Deacetylase Inhibition Restores Retinal Pigment Epithelium Function in Hyperglycemia. PloS one 11, e0162596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins DM, Yates PW, Dahrouj M, Liu Y, Crosson CE, Ablonczy Z, 2016b. Progressive Early Breakdown of Retinal Pigment Epithelium Function in Hyperglycemic Rats. Investigative ophthalmology & visual science 57, 2706–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diabetes Control and Complications Trial Research Group, 1993. The Effect of Intensive Treatment of Diabetes on the Development of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. The New England journal of medicine 329, 977–986. [DOI] [PubMed] [Google Scholar]
- Diabetic Retinopathy Study Research Group, 1981. Photocoagulation Treatment of Proliferative Diabetic Retinopathy. Clinical Application of Diabetic Retinopathy Study (Drs) Findings, Drs Report Number 8.. Ophthalmology 88, 583–600. [PubMed] [Google Scholar]
- Dimitrova G, Chihara E, 2019. Implication of Deep-Vascular-Layer Alteration Detected by Optical Coherence Tomography Angiography for the Pathogenesis of Diabetic Retinopathy. Ophthalmologica. Journal international d’ophtalmologie. International journal of ophthalmology. Zeitschrift fur Augenheilkunde, 1–4. [DOI] [PubMed] [Google Scholar]
- Dmitriev AV, Mangel SC, 2001. Circadian Clock Regulation of Ph in the Rabbit Retina. The Journal of neuroscience : the official journal of the Society for Neuroscience 21, 2897–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreffs A, Lin CM, Liu X, Shanmugam S, Abcouwer SF, Kern TS, Antonetti DA, 2020. All-Trans-Retinaldehyde Contributes to Retinal Vascular Permeability in Ischemia Reperfusion. Investigative ophthalmology & visual science 61, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Rountree A, Cleghorn WM, Contreras L, Lindsay KJ, Sadilek M, Gu H, Djukovic D, Raftery D, Satrustegui J, Kanow M, Chan L, Tsang SH, Sweet IR, Hurley JB, 2016. Phototransduction Influences Metabolic Flux and Nucleotide Metabolism in Mouse Retina. The Journal of biological chemistry 291, 4698–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Cramer M, Lee CA, Tang J, Muthusamy A, Antonetti DA, Jin H, Palczewski K, Kern TS, 2015. Adrenergic and Serotonin Receptors Affect Retinal Superoxide Generation in Diabetic Mice: Relationship to Capillary Degeneration and Permeability. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 29, 2194–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Miller CM, Kern TS, 2003. Hyperglycemia Increases Mitochondrial Superoxide in Retina and Retinal Cells. Free radical biology & medicine 35, 1491–1499. [DOI] [PubMed] [Google Scholar]
- Du Y, Veenstra A, Palczewski K, Kern TS, 2013. Photoreceptor Cells Are Major Contributors to Diabetes-Induced Oxidative Stress and Local Inflammation in the Retina. Proceedings of the National Academy of Sciences of the United States of America 110, 16586–16591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early Treatment Diabetic Retinopathy Study Research Group, 1985. Photocoagulation Therapy for Diabetic Eye Disease. Jama 254, 3086. [PubMed] [Google Scholar]
- Eells JT, Gopalakrishnan S, Connor TB, Stepien K, Carroll J, Williams V, Packard K, Kim JE, 2017. 670 Nm Photobiomodulation as a Therapy for Diabetic Macular Edema: A Pilot Study. Investigative ophthalmology & visual science 58, 932; ARVO E-Abstract A0111. [Google Scholar]
- Eliwa TF, Hussein MA, Zaki MA, Raslan OA, 2018. Outer Retinal Layer Thickness as Good Visual Predictor in Patients with Diabetic Macular Edema. Retina 38, 805–811. [DOI] [PubMed] [Google Scholar]
- Engelgau MM, Geiss LS, Saaddine JB, Boyle JP, Benjamin SM, Gregg EW, Tierney EF, Rios-Burrows N, Mokdad AH, Ford ES, Imperatore G, Narayan KM, 2004. The Evolving Diabetes Burden in the United States. Annals of internal medicine 140, 945–950. [DOI] [PubMed] [Google Scholar]
- Engerman RL, Bloodworth JMB Jr, Nelson S, 1977. Relationship of Microvascular Disease in Diabetes to Metabolic Control. Diabetes 26, 760–769. [DOI] [PubMed] [Google Scholar]
- Enzsoly A, Szabo A, Kantor O, David C, Szalay P, Szabo K, Szel A, Nemeth J, Lukats A, 2014. Pathologic Alterations of the Outer Retina in Streptozotocin-Induced Diabetes. Investigative ophthalmology & visual science 55, 3686–3699. [DOI] [PubMed] [Google Scholar]
- Fain GL, Hardie R, Laughlin SB, 2010. Phototransduction and the Evolution of Photoreceptors. Current biology : CB 20, R114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan N, Silverman SM, Liu Y, Wang X, Kim BJ, Tang L, Clark AF, Liu X, Pang IH, 2017. Rapid Repeatable in Vivo Detection of Retinal Reactive Oxygen Species. Experimental eye research 161, 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felius J, Thompson DA, Khan NW, Bingham EL, Jamison JA, Kemp JA, Sieving PA, 2002. Clinical Course and Visual Function in a Family with Mutations in the Rpe65 Gene. Archives of ophthalmology 120, 55–61. [DOI] [PubMed] [Google Scholar]
- Feng Y, Wang Y, Stock O, Pfister F, Tanimoto N, Seeliger MW, Hillebrands JL, Hoffmann S, Wolburg H, Gretz N, Hammes HP, 2009. Vasoregression Linked to Neuronal Damage in the Rat with Defect of Polycystin-2. PloS one 4, e7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Bueno I, Jones R, Soriano-Romaní L, López-García A, Galvin O, Cheetham S, Diebold Y, 2017. Histologic Characterization of Retina Neuroglia Modifications in Diabetic Zucker Diabetic Fatty Rats. Investigative ophthalmology & visual science 58, 4925–4933. [DOI] [PubMed] [Google Scholar]
- Fernandez-Sanchez L, Lax P, Campello L, Pinilla I, Cuenca N, 2015a. Astrocytes and Muller Cell Alterations During Retinal Degeneration in a Transgenic Rat Model of Retinitis Pigmentosa. Frontiers in cellular neuroscience 9, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Sanchez L, Lax P, Esquiva G, Martin-Nieto J, Pinilla I, Cuenca N, 2012. Safranal, a Saffron Constituent, Attenuates Retinal Degeneration in P23h Rats. PloS one 7, e43074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Sanchez L, Lax P, Noailles A, Angulo A, Maneu V, Cuenca N, 2015b. Natural Compounds from Saffron and Bear Bile Prevent Vision Loss and Retinal Degeneration. Molecules 20, 13875–13893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipek S, Stenkamp RE, Teller DC, Palczewski K, 2003. G Protein-Coupled Receptor Rhodopsin: A Prospectus. Annual review of physiology 65, 851–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman GA, Birch DG, Holder GE, Brigell MG, 2001. Electrophysiologic Testing in Disorders of the Retina, Optic Nerve, and Visual Pathway Second ed. American Academy of Ophthalmology, San Francisco. [Google Scholar]
- Flores M, Debellemaniere G, Bully A, Meillat M, Tumahai P, Delbosc B, Saleh M, 2015. Reflectivity of the Outer Retina on Spectral-Domain Optical Coherence Tomography as a Predictor of Photoreceptor Cone Density. American journal of ophthalmology 160, 588–595 e582. [DOI] [PubMed] [Google Scholar]
- Fort PE, Losiewicz MK, Reiter CE, Singh RS, Nakamura M, Abcouwer SF, Barber AJ, Gardner TW, 2011. Differential Roles of Hyperglycemia and Hypoinsulinemia in Diabetes Induced Retinal Cell Death: Evidence for Retinal Insulin Resistance. PloS one 6, e26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryczkowski AW, Hodes BL, Walker J, 1989. Diabetic Choroidal and Iris Vasculature Scanning Electron Microscopy Findings. Int Ophthalmol 13, 269–279. [DOI] [PubMed] [Google Scholar]
- Fryczkowski AW, Sato SE, Hodes BL, 1988. Changes in the Diabetic Choroidal Vasculature: Scanning Electron Microscopy Findings. Annals of ophthalmology 20, 299–305. [PubMed] [Google Scholar]
- Fu Y, Yau KW, 2007. Phototransduction in Mouse Rods and Cones. Pflugers Arch 454, 805–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Lofqvist CA, Liegl R, Wang Z, Sun Y, Gong Y, Liu CH, Meng SS, Burnim SB, Arellano I, Chouinard MT, Duran R, Poblete A, Cho SS, Akula JD, Kinter M, Ley D, Pupp IH, Talukdar S, Hellstrom A, Smith LE, 2017. Photoreceptor Glucose Metabolism Determines Normal Retinal Vascular Growth. EMBO molecular medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Sun Y, Cakir B, Tomita Y, Huang S, Wang Z, Liu CH, S SC, Britton W, T.S. K, Antonetti DA, Hellstrom A, L.E. S, 2020. Targeting Neurovascular Interaction in Retinal Disorders. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Wang Z, Liu CH, Gong Y, Cakir B, Liegl R, Sun Y, Meng SS, Burnim SB, Arellano I, Moran E, Duran R, Poblete A, Cho SS, Talukdar S, Akula JD, Hellstrom A, Smith LEH, 2018. Fibroblast Growth Factor 21 Protects Photoreceptor Function in Type 1 Diabetic Mice. Diabetes 67, 974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton AB, Akula JD, Mocko JA, Hansen RM, Benador IY, Beck SC, Fahl E, Seeliger MW, Moskowitz A, Harris ME, 2009. Retinal Degenerative and Hypoxic Ischemic Disease. Documenta ophthalmologica. Advances in ophthalmology 118, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton AB, Reynaud X, Hansen RM, Lemere CA, Parker C, Williams TP, 1999. Rod Photoreceptors in Infant Rats with a History of Oxygen Exposure. Investigative ophthalmology & visual science 40, 168–174. [PubMed] [Google Scholar]
- Fursova AZ, Chubar NV, Tarasov MS, Saifullina IF, Pustovaya GG, 2017. Clinical Associations between Photoreceptor Status and Visual Outcomes in Diabetic Macular Edema. Vestnik oftalmologii 133, 11–18. [DOI] [PubMed] [Google Scholar]
- Garcia-Ramirez M, Hernandez C, Villarroel M, Canals F, Alonso MA, Fortuny R, Masmiquel L, Navarro A, Garcia-Arumi J, Simo R, 2009. Interphotoreceptor Retinoid-Binding Protein (Irbp) Is Downregulated at Early Stages of Diabetic Retinopathy. Diabetologia 52, 2633–2641. [DOI] [PubMed] [Google Scholar]
- Gastinger MJ, Barber AJ, Khin SA, McRill CS, Gardner TW, Marshak DW, 2001. Abnormal Centrifugal Axons in Streptozotocin-Diabetic Rat Retinas. Investigative ophthalmology & visual science 42, 2679–2685. [PMC free article] [PubMed] [Google Scholar]
- Gastinger MJ, Kunselman AR, Conboy EE, Bronson SK, Barber AJ, 2008. Dendrite Remodeling and Other Abnormalities in the Retinal Ganglion Cells of Ins2akita Diabetic Mice. Investigative ophthalmology & visual science 49, 2635–2642. [DOI] [PubMed] [Google Scholar]
- Gastinger MJ, Singh RS, Barber AJ, 2006. Loss of Cholinergic and Dopaminergic Amacrine Cells in Streptozotocin-Diabetic Rat and Ins2akita-Diabetic Mouse Retinas. Investigative ophthalmology & visual science 47, 3143–3150. [DOI] [PubMed] [Google Scholar]
- Gaucher D, Chiappore JA, Paques M, Simonutti M, Boitard C, Sahel JA, Massin P, Picaud S, 2007. Microglial Changes Occur without Neural Cell Death in Diabetic Retinopathy. Vision research 47, 612–623. [DOI] [PubMed] [Google Scholar]
- Gerhardinger C, Brown LF, Roy S, Mizutani M, Zucker CL, Lorenzi M, 1998. Expression of Vascular Endothelial Growth Factor in the Human Retina and in Nonproliferative Diabetic Retinopathy. The American journal of pathology 152, 1453–1462. [PMC free article] [PubMed] [Google Scholar]
- Ghassemifar R, Lai CM, Rakoczy PE, 2006a. Regulation of Tight Junction Proteins in Cultured Retinal Pigment Epithelial Cells and in Vegf Overexpressing Transgenic Mouse Retinas. Advances in experimental medicine and biology 572, 179–185. [DOI] [PubMed] [Google Scholar]
- Ghassemifar R, Lai CM, Rakoczy PE, 2006b. Vegf Differentially Regulates Transcription and Translation of Zo-1alpha+ and Zo-1alpha- and Mediates Trans-Epithelial Resistance in Cultured Endothelial and Epithelial Cells. Cell and tissue research 323, 117–125. [DOI] [PubMed] [Google Scholar]
- Giordano CR, Roberts R, Krentz KA, Bissig D, Talreja D, Kumar A, Terlecky SR, Berkowitz BA, 2015. Catalase Therapy Corrects Oxidative Stress-Induced Pathophysiology in Incipient Diabetic Retinopathy. Investigative ophthalmology & visual science 56, 3095–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golczak M, Imanishi Y, Kuksa V, Maeda T, Kubota R, Palczewski K, 2005a. Lecithin:Retinol Acyltransferase Is Responsible for Amidation of Retinylamine, a Potent Inhibitor of the Retinoid Cycle. The Journal of biological chemistry 280, 42263–42273. [DOI] [PubMed] [Google Scholar]
- Golczak M, Kuksa V, Maeda T, Moise AR, Palczewski K, 2005b. Positively Charged Retinoids Are Potent and Selective Inhibitors of the Trans-Cis Isomerization in the Retinoid (Visual) Cycle. Proceedings of the National Academy of Sciences of the United States of America 102, 8162–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golczak M, Maeda A, Bereta G, Maeda T, Kiser PD, Hunzelmann S, von Lintig J, Blaner WS, Palczewski K, 2008. Metabolic Basis of Visual Cycle Inhibition by Retinoid and Nonretinoid Compounds in the Vertebrate Retina. The Journal of biological chemistry 283, 9543–9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Fernandez F, Ghosh D, 2008. Focus on Molecules: Interphotoreceptor Retinoid-Binding Protein (Irbp). Experimental eye research 86, 169–170. [DOI] [PubMed] [Google Scholar]
- Green FD, Ghafour IM, Allan D, Barrie T, McClure E, Foulds WS, 1985. Colour Vision of Diabetics. The British journal of ophthalmology 69, 533–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein V, Sarter B, Hood D, Noble K, Carr R, 1990. Hue Discrimination and S Cone Pathway Sensitivity in Early Diabetic Retinopathy. Investigative ophthalmology & visual science 31, 1008–1014. [PubMed] [Google Scholar]
- Greenstein VC, Thomas SR, Blaustein H, Koenig K, Carr RE, 1993. Effects of Early Diabetic Retinopathy on Rod System Sensitivity. Optom Vis Sci 70, 18–23. [DOI] [PubMed] [Google Scholar]
- Grimes PA, Laties AM, 1980. Early Morphological Alteration of the Pigment Epithelium in Streptozotocin-Induced Diabetes: Increased Surface Area of the Basal Cell Membrane. Experimental eye research 30, 631–639. [DOI] [PubMed] [Google Scholar]
- Group ETDRSR, 1991. Early Photocoagulation for Diabetic Retinopathy. Etdrs Report Number 9.. Ophthalmology 98, 766–785. [PubMed] [Google Scholar]
- Gründer T, Kohler K, Kaletta A, Guenther E, 2000. The Distribution and Developmental Regulation of Nmda Receptor Subunit Proteins in the Outer and Inner Retina of the Rat. Journal of Neurobiology 44, 333–342. [PubMed] [Google Scholar]
- Gubitosi-Klug RA, Talahalli R, Du Y, Nadler JL, Kern TS, 2008. 5-Lipoxygenase, but Not 12/15-Lipoxygenase, Contributes to Degeneration of Retinal Capillaries in a Mouse Model of Diabetic Retinopathy. Diabetes 57, 1387–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon B, Elphege E, Flores M, Gauthier AS, Delbosc B, Saleh M, 2017. Retinal Reflectivity Measurement for Cone Impairment Estimation and Visual Assessment after Diabetic Macular Edema Resolution (Recover-Dme). Investigative ophthalmology & visual science 58, 6241–6247. [DOI] [PubMed] [Google Scholar]
- Hagins WA, Penn RD, Yoshikami S, 1970. Dark Current and Photocurrent in Retinal Rods. Biophys J 10, 380–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn JS, Aizenman E, Lipton SA, 1988. Central Mammalian Neurons Normally Resistant to Glutamate Toxicity Are Made Sensitive by Elevated Extracellular Ca2+: Toxicity Is Blocked by the N-Methyl-D-Aspartate Antagonist Mk-801. Proceedings of the National Academy of Sciences of the United States of America 85, 6556–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer M, Roggan A, Schweitzer D, Muller G, 1995. Optical Properties of Ocular Fundus Tissues--an in Vitro Study Using the Double-Integrating-Sphere Technique and Inverse Monte Carlo Simulation. Phys Med Biol 40, 963–978. [DOI] [PubMed] [Google Scholar]
- Hammes HP, Feng Y, Pfister F, Brownlee M, 2011. Diabetic Retinopathy: Targeting Vasoregression. Diabetes 60, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoum I, Benlarbi M, Dellaa A, Szabo K, Dekany B, Csaba D, Almasi Z, Hajdu RI, Azaiz R, Charfeddine R, Lukats A, Ben Chaouacha-Chekir R, 2017. Study of Retinal Neurodegeneration and Maculopathy in Diabetic Meriones Shawi: A Particular Animal Model with Human-Like Macula. The Journal of comparative neurology 525, 2890–2914. [DOI] [PubMed] [Google Scholar]
- Hancock HA, Kraft TW, 2004. Oscillatory Potential Analysis and Ergs of Normal and Diabetic Rats. Investigative ophthalmology & visual science 45, 1002–1008. [DOI] [PubMed] [Google Scholar]
- Hanna J, Yucel YH, Zhou X, Mathieu E, Paczka-Giorgi LA, Gupta N, 2018. Progressive Loss of Retinal Blood Vessels in a Live Model of Retinitis Pigmentosa. Can J Ophthalmol 53, 391–401. [DOI] [PubMed] [Google Scholar]
- Hardy KJ, Fisher C, Heath P, Foster DH, Scarpello JH, 1995. Comparison of Colour Discrimination and Electroretinography in Evaluation of Visual Pathway Dysfunction in Aretinopathic Iddm Patients. The British journal of ophthalmology 79, 35–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy KJ, Lipton J, Scase MO, Foster DH, Scarpello JH, 1992. Detection of Colour Vision Abnormalities in Uncomplicated Type 1 Diabetic Patients with Angiographically Normal Retinas. The British journal of ophthalmology 76, 461–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison WW, Bearse MA Jr., Ng JS, Jewell NP, Barez S, Burger D, Schneck ME, Adams AJ, 2011. Multifocal Electroretinograms Predict Onset of Diabetic Retinopathy in Adult Patients with Diabetes. Investigative ophthalmology & visual science 52, 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartveit E, Brandstätter JH, Sassoè-Pognetto M, Laurie DJ, Seeburg PH, Wässle H, 1994. Localization and Developmental Expression of the Nmda Receptor Subunit Nr2a in the Mammalian Retina. Journal of Comparative Neurology 348, 570–582. [DOI] [PubMed] [Google Scholar]
- Haugh LM, Linsenmeier RA, Goldstick TK, 1990. Mathematical Models of the Spatial Distribution of Retinal Oxygen Tension and Consumption, Including Changes Upon Illumination. Annals of biomedical engineering 18, 19–36. [DOI] [PubMed] [Google Scholar]
- Heegaard S, Rosenberg T, Preising M, Prause JU, Bek T, 2003. An Unusual Retinal Vascular Morphology in Connection with a Novel Aipl1 Mutation in Leber’s Congenital Amaurosis. The British journal of ophthalmology 87, 980–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson DB, North RV, 1979. Dark Adaptation in Diabetes Mellitus. The British journal of ophthalmology 63, 539–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez C, Garcia-Ramirez M, Corraliza L, Fernandez-Carneado J, Farrera-Sinfreu J, Ponsati B, Gonzalez-Rodriguez A, Valverde AM, Simo R, 2013. Topical Administration of Somatostatin Prevents Retinal Neurodegeneration in Experimental Diabetes. Diabetes 62, 2569–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidayat AA, Fine BS, 1985. Diabetic Choroidopathy. Light and Electron Microscopic Observations of Seven Cases. Ophthalmology 92, 512–522. [PubMed] [Google Scholar]
- Hoang QV, Linsenmeier RA, Chung CK, Curcio CA, 2002. Photoreceptor Inner Segments in Monkey and Human Retina: Mitochondrial Density, Optics, and Regional Variation. Visual neuroscience 19, 395–407. [DOI] [PubMed] [Google Scholar]
- Holfort SK, Jackson GR, Larsen M, 2010. Dark Adaptation During Transient Hyperglycemia in Type 2 Diabetes. Experimental eye research 91, 710–714. [DOI] [PubMed] [Google Scholar]
- Holopigian K, Greenstein VC, Seiple W, Hood DC, Carr RE, 1997. Evidence for Photoreceptor Changes in Patients with Diabetic Retinopathy. Investigative ophthalmology & visual science 38, 2355–2365. [PubMed] [Google Scholar]
- Holopigian K, Seiple W, Lorenzo M, Carr R, 1992. A Comparison of Photopic and Scotopic Electroretinographic Changes in Early Diabetic Retinopathy. Investigative ophthalmology & visual science 33, 2773–2780. [PubMed] [Google Scholar]
- Hombrebueno JR, Ali IH, Xu H, Chen M, 2015. Sustained Intraocular Vegf Neutralization Results in Retinal Neurodegeneration in the Ins2(Akita) Diabetic Mouse. Scientific reports 5, 18316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombrebueno JR, Chen M, Penalva RG, Xu H, 2014. Loss of Synaptic Connectivity, Particularly in Second Order Neurons Is a Key Feature of Diabetic Retinal Neuropathy in the Ins2akita Mouse. PloS one 9, e97970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood DC, Bach M, Brigell M, Keating D, Kondo M, Lyons JS, Marmor MF, McCulloch DL, Palmowski-Wolfe AM, 2012. Iscev Standard for Clinical Multifocal Electroretinography (Mferg) (2011 Edition). Documenta ophthalmologica. Advances in ophthalmology 124, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta N, Koh N, Sakakibara F, Nakamura J, Hamada Y, Hara T, Takeuchi N, Inukai S, Kasama N, Fukasawa H, et al. , 1995. An Aldose Reductase Inhibitor, Tat, Prevents Electroretinographic Abnormalities and Adp-Induced Hyperaggregability in Streptozotocin-Induced Diabetic Rats. Eur J Clin Invest 25, 948–954. [DOI] [PubMed] [Google Scholar]
- Hotta N, Koh N, Sakakibara F, Nakamura J, Hara T, Hamada Y, Fukasawa H, Kakuta H, Sakamoto N, 1997. Effect of an Aldose Reductase Inhibitor on Abnormalities of Electroretinogram and Vascular Factors in Diabetic Rats. Eur J Pharmacol 326, 45–51. https://clinicaltrials.gov/ct2/show/NCT03866473, A Pilot Study Evaluating Photobiomodulation Therapy for Diabetic Macular Edema [DOI] [PubMed] [Google Scholar]
- Hua R, Liu L, Wang X, Chen L, 2013. Imaging Evidence of Diabetic Choroidopathy in Vivo: Angiographic Pathoanatomy and Choroidal-Enhanced Depth Imaging. PloS one 8, e83494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Gandhi JK, Zhong X, Wei Y, Gong J, Duh EJ, Vinores SA, 2011. Tnfalpha Is Required for Late Brb Breakdown in Diabetic Retinopathy, and Its Inhibition Prevents Leukostasis and Protects Vessels and Neurons from Apoptosis. Investigative ophthalmology & visual science 52, 1336–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JB, 1987. Molecular Properties of the Cgmp Cascade of Vertebrate Photoreceptors. Annual review of physiology 49, 793–812. [DOI] [PubMed] [Google Scholar]
- Imai H, Misra GP, Wu L, Janagam DR, Gardner TW, Lowe TL, 2015. Subconjunctivally Implanted Hydrogels for Sustained Insulin Release to Reduce Retinal Cell Apoptosis in Diabetic Rats. Invest. Ophthalmol. Vis. Sci.. 56, 7839–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi Y, Batten ML, Piston DW, Baehr W, Palczewski K, 2004a. Noninvasive Two-Photon Imaging Reveals Retinyl Ester Storage Structures in the Eye. J Cell Biol 164, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi Y, Gerke V, Palczewski K, 2004b. Retinosomes: New Insights into Intracellular Managing of Hydrophobic Substances in Lipid Bodies. J Cell Biol 166, 447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M, Sawada Y, Yoshitomi T, 2015. Structure and Function of the Interphotoreceptor Matrix Surrounding Retinal Photoreceptor Cells. Experimental eye research 133, 3–18. [DOI] [PubMed] [Google Scholar]
- Ismail GM, Whitaker D, 1998. Early Detection of Changes in Visual Function in Diabetes Mellitus. Ophthalmic & physiological optics : the journal of the British College of Ophthalmic Opticians 18, 3–12. [PubMed] [Google Scholar]
- Ivanova E, Alam NM, Prusky GT, Sagdullaev BT, 2019. Blood-Retina Barrier Failure and Vision Loss in Neuron-Specific Degeneration. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe GJ, Roberts WL, Wong HL, Yurochko AD, Cianciolo GJ, 1995. Monocyte-Induced Cytokine Expression in Cultured Human Retinal Pigment Epithelial Cells. Experimental eye research 60, 533–543. [DOI] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH, 1998. The Major Cell Populations of the Mouse Retina. The Journal of neuroscience : the official journal of the Society for Neuroscience 18, 8936–8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Liu L, Steinle JJ, 2018. Mirna15a Regulates Insulin Signal Transduction in the Retinal Vasculature. Cell Signal 44, 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Thakran S, Bheemreddy R, Coppess W, Walker RJ, Steinle JJ, 2015. Sodium Salicylate Reduced Insulin Resistance in the Retina of a Type 2 Diabetic Rat Model. PloS one 10, e0125505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Thakran S, Bheemreddy R, Ye EA, He H, Walker RJ, Steinle JJ, 2014a. Pioglitazone Normalizes Insulin Signaling in the Diabetic Rat Retina through Reduction in Tumor Necrosis Factor Alpha and Suppressor of Cytokine Signaling 3. The Journal of biological chemistry 289, 26395–26405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Zhang Q, Steinle JJ, 2014b. Intravitreal Injection of Igfbp-3 Restores Normal Insulin Signaling in Diabetic Rat Retina. PloS one 9, e93788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Zhang Q, Ye EA, Steinle JJ, 2014c. Beta1-Adrenergic Receptor Stimulation by Agonist Compound 49b Restores Insulin Receptor Signal Transduction in Vivo. Molecular vision 20, 872–880. [PMC free article] [PubMed] [Google Scholar]
- Jin M, Li S, Nusinowitz S, Lloyd M, Hu J, Radu RA, Bok D, Travis GH, 2009. The Role of Interphotoreceptor Retinoid-Binding Protein on the Translocation of Visual Retinoids and Function of Cone Photoreceptors. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 1486–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LE, Larsen M, Perez MT, 2013. Retinal Adaptation to Changing Glycemic Levels in a Rat Model of Type 2 Diabetes. PloS one 8, e55456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MA, Lutty GA, McLeod DS, Otsuji T, Flower RW, Sandagar G, Alexander T, Steidl SM, Hansen BC, 2005. Ocular Structure and Function in an Aged Monkey with Spontaneous Diabetes Mellitus. Exp. Eye Res. 80, 37–42. [DOI] [PubMed] [Google Scholar]
- Joussen AM, Doehmen S, Le ML, Koizumi K, Radetzky S, Krohne TU, Poulaki V, Semkova I, Kociok N, 2009. Tnf-Alpha Mediated Apoptosis Plays an Important Role in the Development of Early Diabetic Retinopathy and Long-Term Histopathological Alterations. Molecular vision 15, 1418–1428. [PMC free article] [PubMed] [Google Scholar]
- Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, Schraermeyer U, Kociok N, Fauser S, Kirchhof B, Kern TS, Adamis AP, 2004. A Central Role for Inflammation in the Pathogenesis of Diabetic Retinopathy. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 18, 1450–1452. [DOI] [PubMed] [Google Scholar]
- Joyal JS, Gantner M, Smith LEH, 2017. Retinal Lipid and Glucose Metabolism and Angiogenesis in the Retina. Progress in retinal and eye research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyal JS, Gantner ML, Smith LEH, 2018. Retinal Energy Demands Control Vascular Supply of the Retina in Development and Disease: The Role of Neuronal Lipid and Glucose Metabolism. Progress in retinal and eye research 64, 131–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang EC, Seo Y, Byeon SH, 2016. Diabetic Retinal Pigment Epitheliopathy: Fundus Autofluorescence and Spectral-Domain Optical Coherence Tomography Findings. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 254, 1931–1940. [DOI] [PubMed] [Google Scholar]
- Kang MK, Lee EJ, Kim YH, Kim DY, Oh H, Kim SI, Kang YH, 2018. Chrysin Ameliorates Malfunction of Retinoid Visual Cycle through Blocking Activation of Age-Rage-Er Stress in Glucose-Stimulated Retinal Pigment Epithelial Cells and Diabetic Eyes. Nutrients 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanow MA, Giarmarco MM, Jankowski CS, Tsantilas K, Engel AL, Du J, Linton JD, Farnsworth CC, Sloat SR, Rountree A, Sweet IR, Lindsay KJ, Parker ED, Brockerhoff SE, Sadilek M, Chao JR, Hurley JB, 2017. Biochemical Adaptations of the Retina and Retinal Pigment Epithelium Support a Metabolic Ecosystem in the Vertebrate Eye. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanwar M, Chan PS, Kern TS, Kowluru RA, 2007. Oxidative Damage in the Retinal Mitochondria of Diabetic Mice: Possible Protection by Superoxide Dismutase. Invest. Ophthalmol. Vis. Sci.. 48, 3805–3811. [DOI] [PubMed] [Google Scholar]
- Karaca C, Karaca Z, 2018. Beyond Hyperglycemia, Evidence for Retinal Neurodegeneration in Metabolic Syndrome. Investigative ophthalmology & visual science 59, 1360–1367. [DOI] [PubMed] [Google Scholar]
- Kern TS, Berkowitz BA, 2015. Photoreceptors in Diabetic Retinopathy. J Diabetes Investig 6, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern TS, Kowluru R, Engerman RL, 1994. Abnormalities of Retinal Metabolism in Diabetes or Galactosemia. Atpases and Glutathione. Investigative ophthalmology & visual science 35, 2962–2967. [PubMed] [Google Scholar]
- Kern TS, Tang J, Berkowitz BA, 2010. Validation of Structural and Functional Lesions of Diabetic Retinopathy in Mice. Molecular vision 16, 2121–2131. [PMC free article] [PubMed] [Google Scholar]
- Kim DI, Lim SK, Park MJ, Han HJ, Kim GY, Park SH, 2007. The Involvement of Phosphatidylinositol 3-Kinase /Akt Signaling in High Glucose-Induced Downregulation of Glut-1 Expression in Arpe Cells. Life Sci 80, 626–632. [DOI] [PubMed] [Google Scholar]
- Kim JT, Lee DH, Joe SG, Kim JG, Yoon YH, 2013. Changes in Choroidal Thickness in Relation to the Severity of Retinopathy and Macular Edema in Type 2 Diabetic Patients. Investigative ophthalmology & visual science 54, 3378–3384. [DOI] [PubMed] [Google Scholar]
- Kim YH, Kim YS, Noh HS, Kang SS, Cheon EW, Park SK, Lee BJ, Choi WS, Cho GJ, 2005. Changes in Rhodopsin Kinase and Transducin in the Rat Retina in Early-Stage Diabetes. Experimental eye research 80, 753–760. [DOI] [PubMed] [Google Scholar]
- Kirber WM, Nichols CW, Grimes PA, Winegrad AI, Laties AM, 1980. A Permeability Defect of the Retinal Pigment Epithelium. Occurrence in Early Streptozocin Diabetes. Archives of ophthalmology 98, 725–728. [DOI] [PubMed] [Google Scholar]
- Kirwin SJ, Kanaly ST, Hansen CR, Cairns BJ, Ren M, Edelman JL, 2011. Retinal Gene Expression and Visually Evoked Behavior in Diabetic Long Evans Rats. Investigative ophthalmology & visual science 52, 7654–7663. [DOI] [PubMed] [Google Scholar]
- Klaassen I, Van Noorden CJ, Schlingemann RO, 2013. Molecular Basis of the Inner Blood-Retinal Barrier and Its Breakdown in Diabetic Macular Edema and Other Pathological Conditions. Progress in retinal and eye research 34, 19–48. [DOI] [PubMed] [Google Scholar]
- Kohner EM, Dollery CT, 1970. Fluorescein Angiography of the Fundus in Diabetic Retinopathy. British medical bulletin 26, 166–170. [DOI] [PubMed] [Google Scholar]
- Kohner EM, Henkind P, 1970. Correlation of Fluorescein Angiogram and Retinal Digest in Diabetic Retinopathy. American journal of ophthalmology 69, 403–414. [DOI] [PubMed] [Google Scholar]
- Kohzaki K, Vingrys AJ, Bui BV, 2008. Early Inner Retinal Dysfunction in Streptozotocin-Induced Diabetic Rats. Investigative ophthalmology & visual science 49, 3595–3604. [DOI] [PubMed] [Google Scholar]
- Kolb H, 1995. Photoreceptors, in: Kolb H, Fernandez E, Nelson R. (Eds.), Webvision: The Organization of the Retina and Visual System, Salt Lake City (UT). [PubMed] [Google Scholar]
- Kowluru RA, 2002. Retinal Metabolic Abnormalities in Diabetic Mouse: Comparison with Diabetic Rat. Current eye research 24, 123–128. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, 2013. Mitochondria Damage in the Pathogenesis of Diabetic Retinopathy and in the Metabolic Memory Associated with Its Continued Progression. Curr Med Chem 20, 3226–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA, Engerman RL, Case GL, Kern TS, 2001a. Retinal Glutamate in Diabetes and Effect of Antioxidants. Neurochem Int 38, 385–390. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Engerman RL, Kern TS, 1999. Abnormalities of Retinal Metabolism in Diabetes or Experimental Galactosemia. Vi. Comparison of Retinal and Cerebral Cortex Metabolism, and Effects of Antioxidant Therapy. Free Radic. Biol. Med. 26, 371–378. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Jirousek MR, Stramm LE, Farid NA, Engerman RL, Kern TS, 1998. Abnormalities of Retinal Metabolism in Diabetes or Experimental Galactosemia. V. Relationship between Protein Kinase C and Atpases. Diabetes 47, 464–469. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Kowluru A, Mishra M, Kumar B, 2015. Oxidative Stress and Epigenetic Modifications in the Pathogenesis of Diabetic Retinopathy. Progress in retinal and eye research 48, 40–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA, Mishra M, 2015. Oxidative Stress, Mitochondrial Damage and Diabetic Retinopathy. Biochimica et biophysica acta. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Tang J, Kern TS, 2001b. Abnormalities of Retinal Metabolism in Diabetes and Experimental Galactosemia. Vii. Effect of Long-Term Administration of Antioxidants on the Development of Retinopathy. Diabetes 50, 1938–1942. [DOI] [PubMed] [Google Scholar]
- Krebs HA, 1927. On the Metabolism of the Retina. Biochemische Zeitschrift 189, 57–59. [Google Scholar]
- Kubota R, Calkins DJ, Henry SH, Linsenmeier RA, 2019. Emixustat Reduces Metabolic Demand of Dark Activity in the Retina. Investigative ophthalmology & visual science 60, 4924–4930. [DOI] [PubMed] [Google Scholar]
- Kumagai AK, Glasgow BJ, Pardridge WM, 1994. Glut1 Glucose Transporter Expression in the Diabetic and Nondiabetic Human Eye. Investigative ophthalmology & visual science 35, 2887–2894. [PubMed] [Google Scholar]
- Kumar U, Ramkumar H, McAnany J, 2013. Electroretinogram, EyeWiki. American Academy of Ophthalmology. [Google Scholar]
- Kur J, Burian MA, Newman EA, 2016. Light Adaptation Does Not Prevent Early Retinal Abnormalities in Diabetic Rats. Scientific reports 6, 21075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtenbach A, Flogel W, Erb C, 2002. Anomaloscope Matches in Patients with Diabetes Mellitus. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 240, 79–84. [DOI] [PubMed] [Google Scholar]
- Kurtenbach A, Mayser HM, Jagle H, Fritsche A, Zrenner E, 2006. Hyperoxia, Hyperglycemia, and Photoreceptor Sensitivity in Normal and Diabetic Subjects. Visual neuroscience 23, 651–661. [DOI] [PubMed] [Google Scholar]
- Kurtenbach A, Wagner U, Neu A, Schiefer U, Ranke MB, Zrenner E, 1994. Brightness Matching and Colour Discrimination in Young Diabetics without Retinopathy. Vision research 34, 115–122. [DOI] [PubMed] [Google Scholar]
- Kusari J, Zhou S, Padillo E, Clarke KG, Gil DW, 2007. Effect of Memantine on Neuroretinal Function and Retinal Vascular Changes of Streptozotocin-Induced Diabetic Rats. Investigative ophthalmology & visual science 48, 5152–5159. [DOI] [PubMed] [Google Scholar]
- Kuwabara T, Cogan DG, 1960. Studies of Retinal Vascular Patterns: I. Normal Architecture. Archives of ophthalmology 64, 904–911. [DOI] [PubMed] [Google Scholar]
- Lagnado L, Baylor D, 1992. Signal Flow in Visual Transduction. Neuron 8, 995–1002. [DOI] [PubMed] [Google Scholar]
- Lahdenranta J, Pasqualini R, Schlingemann RO, Hagedorn M, Stallcup WB, Bucana CD, Sidman RL, Arap W, 2001. An Anti-Angiogenic State in Mice and Humans with Retinal Photoreceptor Cell Degeneration. PNAS 98, 10368–10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammer J, Prager SG, Cheney MC, Ahmed A, Radwan SH, Burns SA, Silva PS, Sun JK, 2016. Cone Photoreceptor Irregularity on Adaptive Optics Scanning Laser Ophthalmoscopy Correlates with Severity of Diabetic Retinopathy and Macular Edema. Investigative ophthalmology & visual science 57, 6624–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau JC, Linsenmeier RA, 2012. Oxygen Consumption and Distribution in the Long-Evans Rat Retina. Experimental eye research 102, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau JC, Linsenmeier RA, 2014. Increased Intraretinal Po2 in Short-Term Diabetic Rats. Diabetes 63, 4338–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver NM, Robison WG Jr., Pfeffer BA, 1993. Novel Procedures for Isolating Intact Retinal Vascular Beds from Diabetic Humans and Animal Models. Investigative ophthalmology & visual science 34, 2097–2104. [PubMed] [Google Scholar]
- Lee H, Chung H, Arnouk H, Lamoke F, Hunt RC, Hrushesky WJ, Wood PA, Lee SH, Jahng WJ, 2010. Cleavage of the Retinal Pigment Epithelium-Specific Protein Rpe65 under Oxidative Stress. Int J Biol Macromol 47, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SG, Lee CG, Yun IH, Hur DY, Yang JW, Kim HW, 2012. Effect of Lipoic Acid on Expression of Angiogenic Factors in Diabetic Rat Retina. Clinical & experimental ophthalmology 40, e47–57. [DOI] [PubMed] [Google Scholar]
- Lee VK, Hosking BM, Holeniewska J, Kubala EC, Lundh von Leithner P, Gardner PJ, Foxton RH, Shima DT, 2018. Btbr Ob/Ob Mouse Model of Type 2 Diabetes Exhibits Early Loss of Retinal Function and Retinal Inflammation Followed by Late Vascular Changes. Diabetologia 61, 2422–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Veenstra AA, Talahalli RR, Wang X, Gubitosi-Klug RA, Sheibani N, Kern TS, 2012a. Marrow-Derived Cells Regulate the Development of Early Diabetic Retinopathy and Tactile Allodynia in Mice. Diabetes 61, 3294–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Cai Y, Wang YS, Shi YY, Hou W, Xu CS, Wang HY, Ye Z, Yao LB, Zhang J, 2012b. Hyperglycaemia Exacerbates Choroidal Neovascularisation in Mice Via the Oxidative Stress-Induced Activation of Stat3 Signalling in Rpe Cells. PloS one 7, e47600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieth E, Barber AJ, Xu B, Dice C, Ratz MJ, Tanase D, Strother JM, 1998. Glial Reactivity and Impaired Glutamate Metabolism in Short-Term Experimental Diabetic Retinopathy. Penn State Retina Research Group. Diabetes 47, 815–820. [DOI] [PubMed] [Google Scholar]
- Lieth E, LaNoue KF, Antonetti DA, Ratz M, 2000. Diabetes Reduces Glutamate Oxidation and Glutamine Synthesis in the Retina. The Penn State Retina Research Group. Experimental eye research 70, 723–730. [DOI] [PubMed] [Google Scholar]
- Lindsay KJ, Du J, Sloat SR, Contreras L, Linton JD, Turner SJ, Sadilek M, Satrustegui J, Hurley JB, 2014. Pyruvate Kinase and Aspartate-Glutamate Carrier Distributions Reveal Key Metabolic Links between Neurons and Glia in Retina. Proceedings of the National Academy of Sciences of the United States of America 111, 15579–15584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsenmeier RA, 1986. Effects of Light and Darkness on Oxygen Distribution and Consumption in the Cat Retina. The Journal of general physiology 88, 521–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsenmeier RA, Braun RD, 1992. Oxygen Distribution and Consumption in the Cat Retina During Normoxia and Hypoxemia. The Journal of general physiology 99, 177–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Tang J, Du Y, Lee CA, Golczak M, Muthusamy A, Antonetti DA, Veenstra AA, Amengual J, von Lintig J, Palczewski K, Kern TS, 2015. Retinylamine Benefits Early Diabetic Retinopathy in Mice. J. Biol. Chem. 290, 21568–21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Tang J, Du Y, Saadane A, Samuels I, Veenstra A, Kiser JZ, Palczewski K, Kern TS, 2019. Transducin1, Phototransduction and the Development of Early Diabetic Retinopathy. Investigative ophthalmology & visual science 60, 1538–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Tang J, Du Y, Saadane A, Tonade D, Samuels I, Veenstra A, Palczewski K, Kern TS, 2016. Photoreceptor Cells Influence Retinal Vascular Degeneration in Mouse Models of Retinal Degeneration and Diabetes. Invest. Ophthalmol. Vis. Sci.. 57, 4272–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Deng Y, 2001. The Analysis of Electroretinography of Diabetes Mellitus. Yan Ke Xue Bao 17, 173–175, 179. [PubMed] [Google Scholar]
- Lombardo M, Parravano M, Lombardo G, Varano M, Boccassini B, Stirpe M, Serrao S, 2014. Adaptive Optics Imaging of Parafoveal Cones in Type 1 Diabetes. Retina 34, 546–557. [DOI] [PubMed] [Google Scholar]
- Lombardo M, Parravano M, Serrao S, Ziccardi L, Giannini D, Lombardo G, 2016. Investigation of Adaptive Optics Imaging Biomarkers for Detecting Pathological Changes of the Cone Mosaic in Patients with Type 1 Diabetes Mellitus. PloS one 11, e0151380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhin E, Tormene AP, Olivato E, Convento E, Vujosevic S, Pilotto E, Kotsafti O, Midena E, 2016. Rod Function in Diabetic Patients without and with Early Diabetic Retinopathy. European journal of ophthalmology 26, 418–424. [DOI] [PubMed] [Google Scholar]
- Longo A, Geiser M, Riva CE, 2000. Subfoveal Choroidal Blood Flow in Response to Light-Dark Exposure. Investigative ophthalmology & visual science 41, 2678–2683. [PubMed] [Google Scholar]
- Lovasik JV, Spafford MM, 1988. An Electrophysiological Investigation of Visual Function in Juvenile Insulin-Dependent Diabetes Mellitus. Am J Optom Physiol Opt 65, 236–253. [DOI] [PubMed] [Google Scholar]
- Lu ZY, Bhutto IA, Amemiya T, 2003. Retinal Changes in Otsuka Long-Evans Tokushima Fatty Rats (Spontaneously Diabetic Rat)--Possibility of a New Experimental Model for Diabetic Retinopathy. Japanese journal of ophthalmology 47, 28–35. [DOI] [PubMed] [Google Scholar]
- Luo DG, Xue T, Yau KW, 2008. How Vision Begins: An Odyssey. Proceedings of the National Academy of Sciences of the United States of America 105, 9855–9862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutty GA, 2017. Diabetic Choroidopathy. Vision research 139, 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutty GA, Cao J, McLeod DS, 1997. Relationship of Polymorphonuclear Leukocytes to Capillary Dropout in the Human Diabetic Choroid. The American journal of pathology 151, 707–714. [PMC free article] [PubMed] [Google Scholar]
- MacGregor LC, Matschinsky FM, 1985. Treatment with Aldose Reductase Inhibitor or with Myo-Inositol Arrests Deterioration of the Electroretinogram of Diabetic Rats. The Journal of clinical investigation 76, 887–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGregor LC, Matschinsky FM, 1986a. Altered Retinal Metabolism in Diabetes Ii. Measurement of Sodium-Potassium Atpase and Total Sodium and Potassium in Individual Retinal Layers. The Journal of biological chemistry 261, 4052–4058. [PubMed] [Google Scholar]
- MacGregor LC, Matschinsky FM, 1986b. Experimental Diabetes Mellitus Impairs the Function of the Retinal Pigmented Epithelium. Metabolism 35, 28–34. [DOI] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Golczak M, Imanishi Y, Leahy P, Kubota R, Palczewski K, 2006. Effects of Potent Inhibitors of the Retinoid Cycle on Visual Function and Photoreceptor Protection from Light Damage in Mice. Mol Pharmacol 70, 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshwary AS, Oster SF, Yuson RM, Cheng L, Mojana F, Freeman WR, 2010. The Association between Percent Disruption of the Photoreceptor Inner Segment-Outer Segment Junction and Visual Acuity in Diabetic Macular Edema. American journal of ophthalmology 150, 63–67 e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malechka VV, Chen J, Cheng R, Ma JX, Moiseyev G, 2020. The Single Administration of a Chromophore Alleviates Neural Defects in Diabetic Retinopathy. The American journal of pathology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malechka VV, Moiseyev G, Takahashi Y, Shin Y, Ma JX, 2017. Impaired Rhodopsin Generation in the Rat Model of Diabetic Retinopathy. The American journal of pathology 187, 2222–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes G, Meunier I, Avila-Fernandez A, Banfi S, Le Meur G, Zanlonghi X, Corton M, Simonelli F, Brabet P, Labesse G, Audo I, Mohand-Said S, Zeitz C, Sahel JA, Weber M, Dollfus H, Dhaenens CM, Allorge D, De Baere E, Koenekoop RK, Kohl S, Cremers FP, Hollyfield JG, Senechal A, Hebrard M, Bocquet B, Ayuso Garcia C, Hamel CP, 2013. Mutations in Impg1 Cause Vitelliform Macular Dystrophies. Am J Hum Genet 93, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein AD, 2001. The Polarity of the Retinal Pigment Epithelium. Traffic 2, 867–872. [DOI] [PubMed] [Google Scholar]
- Marmorstein LY, Munier FL, Arsenijevic Y, Schorderet DF, McLaughlin PJ, Chung D, Traboulsi E, Marmorstein AD, 2002. Aberrant Accumulation of Efemp1 Underlies Drusen Formation in Malattia Leventinese and Age-Related Macular Degeneration. Proceedings of the National Academy of Sciences of the United States of America 99, 13067–13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PM, Roon P, Van Ells TK, Ganapathy V, Smith SB, 2004. Death of Retinal Neurons in Streptozotocin-Induced Diabetic Mice. Investigative ophthalmology & visual science 45, 3330–3336. [DOI] [PubMed] [Google Scholar]
- May CA, Narfstrom K, 2012. Retinal Capillary Morphology in the Abyssinian Cat with Hereditary Retinal Degeneration. Experimental eye research 99, 45–47. [DOI] [PubMed] [Google Scholar]
- McAnany JJ, Liu K, Park JC, 2020. Electrophysiological Measures of Dysfunction in Early-Stage Diabetic Retinopathy: No Correlation between Cone Phototransduction and Oscillatory Potential Abnormalities. Documenta ophthalmologica. Advances in ophthalmology 140, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAnany JJ, Park JC, 2019. Cone Photoreceptor Dysfunction in Early-Stage Diabetic Retinopathy: Association between the Activation Phase of Cone Phototransduction and the Flicker Electroretinogram. Investigative ophthalmology & visual science 60, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod DS, Lefer DJ, Merges C, Lutty GA, 1995. Enhanced Expression of Intercellular Adhesion Molecule-1 and P-Selectin in the Diabetic Human Retina and Choroid. Am. J. Pathol. 147, 642–653. [PMC free article] [PubMed] [Google Scholar]
- McLeod DS, Lutty GA, 1994. High-Resolution Histologic Analysis of the Human Choroidal Vasculature. Invest. Ophthalmol. Vis. Sci. 35, 3799–3811. [PubMed] [Google Scholar]
- Medrano CJ, Fox DA, 1995. Oxygen Consumption in the Rat Outer and Inner Retina: Light- and Pharmacologically-Induced Inhibition. Experimental eye research 61, 273–284. [DOI] [PubMed] [Google Scholar]
- Miceli MV, Newsome DA, 1991. Cultured Retinal Pigment Epithelium Cells from Donors with Type I Diabetes Show an Altered Insulin Response. Investigative ophthalmology & visual science 32, 2847–2853. [PubMed] [Google Scholar]
- Miller JW, Le Couter J, Strauss EC, Ferrara N, 2013. Vascular Endothelial Growth Factor a in Intraocular Vascular Disease. Ophthalmology 120, 106–114. [DOI] [PubMed] [Google Scholar]
- Miller SS, Steinberg RH, 1977. Active Transport of Ions across Frog Retinal Pigment Epithelium. Experimental eye research 25, 235–248. [DOI] [PubMed] [Google Scholar]
- Miyamoto N, de Kozak Y, Jeanny JC, Glotin A, Mascarelli F, Massin P, BenEzra D, Behar-Cohen F, 2007. Placental Growth Factor-1 and Epithelial Haemato-Retinal Barrier Breakdown: Potential Implication in the Pathogenesis of Diabetic Retinopathy. Diabetologia 50, 461–470. [DOI] [PubMed] [Google Scholar]
- Mizutani M, Gerhardinger C, Lorenzi M, 1998. Muller Cell Changes in Human Diabetic Retinopathy. Diabetes 47, 445–449. [DOI] [PubMed] [Google Scholar]
- Mizutani M, Kern TS, Lorenzi M, 1996. Accelerated Death of Retinal Microvascular Cells in Human and Experimental Diabetic Retinopathy. The Journal of clinical investigation 97, 2883–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney JB, Drury MI, 1982. The Effect of Pregnancy on the Natural Course of Diabetic Retinopathy. American journal of ophthalmology 93, 745–756. [DOI] [PubMed] [Google Scholar]
- Moore-Dotson JM, Beckman JJ, Mazade RE, Hoon M, Bernstein AS, Romero-Aleshire MJ, Brooks HL, Eggers ED, 2016. Early Retinal Neuronal Dysfunction in Diabetic Mice: Reduced Light-Evoked Inhibition Increases Rod Pathway Signaling. Investigative ophthalmology & visual science 57, 1418–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortlock KE, Chiti Z, Drasdo N, Owens DR, North RV, 2005. Silent Substitution S-Cone Electroretinogram in Subjects with Diabetes Mellitus. Ophthalmic & physiological optics : the journal of the British College of Ophthalmic Opticians 25, 392–399. [DOI] [PubMed] [Google Scholar]
- Motohashi Y, Kemmochi Y, Maekawa T, Tadaki H, Sasase T, Tanaka Y, Kakehashi A, Yamada T, Ohta T, 2018. Diabetic Macular Edema-Like Ocular Lesions in Male Spontaneously Diabetic Torii Fatty Rats. Physiol Res 67, 423–432. [DOI] [PubMed] [Google Scholar]
- Muftuoglu IK, Mendoza N, Gaber R, Alam M, You Q, Freeman WR, 2017a. Integrity of Outer Retinal Layers after Resolution of Central Involved Diabetic Macular Edema. Retina 37, 2015–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu IK, Unsal E, Ozturker ZK, 2017b. Restoration of Photoreceptors in Eyes with Diabetic Macular Edema. European journal of ophthalmology 27, 585–590. [DOI] [PubMed] [Google Scholar]
- Muir ER, Renteria RC, Duong TQ, 2012. Reduced Ocular Blood Flow as an Early Indicator of Diabetic Retinopathy in a Mouse Model of Diabetes. Investigative ophthalmology & visual science 53, 6488–6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Yoshimura N, 2013. Structural Changes in Individual Retinal Layers in Diabetic Macular Edema. Journal of diabetes research 2013, 920713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi M, Tanimoto M, Gohda T, Hagiwara S, Ohara I, Toyoda H, Ishikawa Y, Horikoshi S, Tomino Y, 2009. Pleiotropic Effect of Pyridoxamine on Diabetic Complications Via Cd36 Expression in Kk-Ay/Ta Mice. Diabetes Res Clin Pract 83, 183–189. [DOI] [PubMed] [Google Scholar]
- Naeser P, 1997. Insulin Receptors in Human Ocular Tissues. Immunohistochemical Demonstration in Normal and Diabetic Eyes. Ups J Med Sci 102, 35–40. [DOI] [PubMed] [Google Scholar]
- Nagesh BN, Takkar B, Azad S, Azad R, 2016. Optical Coherence Tomography and Multifocal Electroretinography in Diabetic Macular Edema: A Neurovascular Relation with Vision. Ophthalmic Surg Lasers Imaging Retina 47, 626–631. [DOI] [PubMed] [Google Scholar]
- Nair G, Tanaka Y, Kim M, Olson DE, Thule PM, Pardue MT, Duong TQ, 2011. Mri Reveals Differential Regulation of Retinal and Choroidal Blood Volumes in Rat Retina. Neuroimage 54, 1063–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natalini PM, Mateos MV, Ilincheta de Boschero MG, Giusto NM, 2016. Insulin-Related Signaling Pathways Elicited by Light in Photoreceptor Nuclei from Bovine Retina. Experimental eye research 145, 36–47. [DOI] [PubMed] [Google Scholar]
- Nathans J, Thomas D, Hogness DS, 1986. Molecular Genetics of Human Color Vision: The Genes Encoding Blue, Green, and Red Pigments. Science 232, 193–202. [DOI] [PubMed] [Google Scholar]
- Nesper PL, Scarinci F, Fawzi AA, 2017. Adaptive Optics Reveals Photoreceptor Abnormalities in Diabetic Macular Ischemia. PloS one 12, e0169926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SK, Wood JP, Chidlow G, Han G, Kittipassorn T, Peet DJ, Casson RJ, 2015. Cancer-Like Metabolism of the Mammalian Retina. Clin Exp Ophthalmol 43, 367–376. [DOI] [PubMed] [Google Scholar]
- Ng YK, Zeng XX, Ling EA, 2004. Expression of Glutamate Receptors and Calcium-Binding Proteins in the Retina of Streptozotocin-Induced Diabetic Rats. Brain research 1018, 66–72. [DOI] [PubMed] [Google Scholar]
- Nguyen QD, Shah SM, Van Anden E, Sung JU, Vitale S, Campochiaro PA, 2004. Supplemental Oxygen Improves Diabetic Macular Edema: A Pilot Study. Investigative ophthalmology & visual science 45, 617–624. [DOI] [PubMed] [Google Scholar]
- Nikonov SS, Kholodenko R, Lem J, Pugh EN Jr., 2006. Physiological Features of the S- and M-Cone Photoreceptors of Wild-Type Mice from Single-Cell Recordings. The Journal of general physiology 127, 359–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RV, Cooney O, Chambers D, Dolben J, Owens DR, 1997a. Does Hyperglycaemia Have an Influence Upon Colour Vision of Patients with Diabetes Mellitus? Ophthalmic & physiological optics : the journal of the British College of Ophthalmic Opticians 17, 95–101. [PubMed] [Google Scholar]
- North RV, Farrell U, Banford D, Jones C, Gregory JW, Butler G, Owens DR, 1997b. Visual Function in Young Iddm Patients over 8 Years of Age. A 4-Year Longitudinal Study. Diabetes care 20, 1724–1730. [DOI] [PubMed] [Google Scholar]
- Okawa H, Sampath AP, Laughlin SB, Fain GL, 2008. Atp Consumption by Mammalian Rod Photoreceptors in Darkness and in Light. Current biology : CB 18, 1917–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omri S, Behar-Cohen F, de Kozak Y, Sennlaub F, Verissimo LM, Jonet L, Savoldelli M, Omri B, Crisanti P, 2011. Microglia/Macrophages Migrate through Retinal Epithelium Barrier by a Transcellular Route in Diabetic Retinopathy: Role of Pkczeta in the Goto Kakizaki Rat Model. The American journal of pathology 179, 942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omri S, Behar-Cohen F, Rothschild PR, Gelize E, Jonet L, Jeanny JC, Omri B, Crisanti P, 2013. Pkczeta Mediates Breakdown of Outer Blood-Retinal Barriers in Diabetic Retinopathy. PloS one 8, e81600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omri S, Omri B, Savoldelli M, Jonet L, Thillaye-Goldenberg B, Thuret G, Gain P, Jeanny JC, Crisanti P, Behar-Cohen F, 2010. The Outer Limiting Membrane (Olm) Revisited: Clinical Implications. Clin Ophthalmol 4, 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortin-Martinez A, Nadal-Nicolas FM, Jimenez-Lopez M, Alburquerque-Bejar JJ, Nieto-Lopez L, Garcia-Ayuso D, Villegas-Perez MP, Vidal-Sanz M, Agudo-Barriuso M, 2014. Number and Distribution of Mouse Retinal Cone Photoreceptors: Differences between an Albino (Swiss) and a Pigmented (C57/Bl6) Strain. PloS one 9, e102392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster SF, Mojana F, Brar M, Yuson RM, Cheng L, Freeman WR, 2010. Disruption of the Photoreceptor Inner Segment/Outer Segment Layer on Spectral Domain-Optical Coherence Tomography Is a Predictor of Poor Visual Acuity in Patients with Epiretinal Membranes. Retina 30, 713–718. [DOI] [PubMed] [Google Scholar]
- Ostroy SE, 1998. Altered Rhodopsin Regeneration in Diabetic Mice Caused by Acid Conditions within the Rod Photoreceptors. Current eye research 17, 979–985. [DOI] [PubMed] [Google Scholar]
- Ostroy SE, Frede SM, Wagner EF, Gaitatzes CG, Janle EM, 1994. Decreased Rhodopsin Regeneration in Diabetic Mouse Eyes. Investigative ophthalmology & visual science 35, 3905–3909. [PubMed] [Google Scholar]
- Otani A, Dorrell MI, Kinder K, Moreno SK, Nusinowitz S, Banin E, Heckenlively J, Friedlander M, 2004. Rescue of Retinal Degeneration by Intravitreally Injected Adult Bone Marrow-Derived Lineage-Negative Hematopoietic Stem Cells. The Journal of clinical investigation 114, 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani T, Yamaguchi Y, Kishi S, 2010. Correlation between Visual Acuity and Foveal Microstructural Changes in Diabetic Macular Edema. Retina 30, 774–780. [DOI] [PubMed] [Google Scholar]
- Ottlecz A, Bensaoula T, 1996. Captopril Ameliorates the Decreased Na+, K+-Atpase Activity in the Retina of Streptozotocin-Induced Diabetic Rats. Invest. Ophthalmol. Vis. Sci. 37, 1633–1641. [PubMed] [Google Scholar]
- Ottlecz A, Garcia CA, Eichberg J, Fox DA, 1993. Alterations in Retinal Na+, K(+)-Atpase in Diabetes: Streptozotocin-Induced and Zucker Diabetic Fatty Rats. Current eye research 12, 1111–1121. [DOI] [PubMed] [Google Scholar]
- Ozkaya A, Alkin Z, Karakucuk Y, Karatas G, Fazil K, Gurkan Erdogan M, Perente I, Taskapili M, 2017. Thickness of the Retinal Photoreceptor Outer Segment Layer in Healthy Volunteers and in Patients with Diabetes Mellitus without Retinopathy, Diabetic Retinopathy, or Diabetic Macular Edema. Saudi journal of ophthalmology : official journal of the Saudi Ophthalmological Society 31, 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewska G, Boguslawski J, Stremplewski P, Kornaszewski L, Zhang J, Dong Z, Liang XX, Gratton E, Vogel A, Wojtkowski M, Palczewski K, 2020. Noninvasive Two-Photon Optical Biopsy of Retinal Fluorophores. Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewska G, Stremplewski P, Suh S, Alexander N, Salom D, Dong Z, Ruminski D, Choi EH, Sears AE, Kern TS, Wojtkowski M, Palczewski K, 2018. Two-Photon Imaging of the Mammalian Retina with Ultrafast Pulsing Laser. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, 2006. G Protein-Coupled Receptor Rhodopsin. Annual review of biochemistry 75, 743–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kiser PD, 2020. Shedding New Light on the Generation of the Visual Chromophore. Proceedings of the National Academy of Sciences of the United States of America 117, 19629–19638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh JG, Saraswathy S, Rao NA, 2008. Photoreceptor Oxidative Damage in Sympathetic Ophthalmia. American journal of ophthalmology 146, 866–875 e862. [DOI] [PubMed] [Google Scholar]
- Park SH, Park JW, Park SJ, Kim KY, Chung JW, Chun MH, Oh SJ, 2003. Apoptotic Death of Photoreceptors in the Streptozotocin-Induced Diabetic Rat Retina. Diabetologia 46, 1260–1268. [DOI] [PubMed] [Google Scholar]
- Penn JS, Li S, Naash MI, 2000. Ambient Hypoxia Reverses Retinal Vascular Attenuation in a Transgenic Mouse Model of Autosomal Dominant Retinitis Pigmentosa. Investigative ophthalmology & visual science 41, 4007–4013. [PubMed] [Google Scholar]
- Penn RD, Hagins WA, 1969. Signal Transmission Along Retinal Rods and the Origin of the Electroretinographic a-Wave. Nature 223, 201–204. [DOI] [PubMed] [Google Scholar]
- Phipps JA, Fletcher EL, Vingrys AJ, 2004. Paired-Flash Identification of Rod and Cone Dysfunction in the Diabetic Rat. Investigative ophthalmology & visual science 45, 4592–4600. [DOI] [PubMed] [Google Scholar]
- Piano I, Novelli E, Della Santina L, Strettoi E, Cervetto L, Gargini C, 2016. Involvement of Autophagic Pathway in the Progression of Retinal Degeneration in a Mouse Model of Diabetes. Frontiers in cellular neuroscience 10, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poitry-Yamate CL, Poitry S, Tsacopoulos M, 1995. Lactate Released by Muller Glial Cells Is Metabolized by Photoreceptors from Mammalian Retina. The Journal of neuroscience : the official journal of the Society for Neuroscience 15, 5179–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnalagu M, Subramani M, Jayadev C, Shetty R, Das D, 2017. Retinal Pigment Epithelium-Secretome: A Diabetic Retinopathy Perspective. Cytokine 95, 126–135. [DOI] [PubMed] [Google Scholar]
- Pournaras CJ, Tsacopoulos M, Strommer K, Gilodi N, Leuenberger PM, 1990. Scatter Photocoagulation Restores Tissue Hypoxia in Experimental Vasoproliferative Microangiopathy in Miniature Pigs. Ophthalmology 97, 1329–1333. [DOI] [PubMed] [Google Scholar]
- Prunty MC, Aung MH, Hanif AM, Allen RS, Chrenek MA, Boatright JH, Thule PM, Kundu K, Murthy N, Pardue MT, 2015. In Vivo Imaging of Retinal Oxidative Stress Using a Reactive Oxygen Species-Activated Fluorescent Probe. Investigative ophthalmology & visual science 56, 5862–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puro DG, 2002. Diabetes-Induced Dysfunction of Retinal Muller Cells. Transactions of the American Ophthalmological Society 100, 339–352. [PMC free article] [PubMed] [Google Scholar]
- Rajala A, Anderson RE, Ma JX, Lem J, Al-Ubaidi MR, Rajala RV, 2007. G-Protein-Coupled Receptor Rhodopsin Regulates the Phosphorylation of Retinal Insulin Receptor. The Journal of biological chemistry 282, 9865–9873. [DOI] [PubMed] [Google Scholar]
- Rajala A, Dighe R, Agbaga MP, Anderson RE, Rajala RV, 2013. Insulin Receptor Signaling in Cones. The Journal of biological chemistry 288, 19503–19515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala A, Tanito M, Le YZ, Kahn CR, Rajala RV, 2008. Loss of Neuroprotective Survival Signal in Mice Lacking Insulin Receptor Gene in Rod Photoreceptor Cells. The Journal of biological chemistry 283, 19781–19792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala RV, Anderson RE, 2010. Rhodopsin-Regulated Insulin Receptor Signaling Pathway in Rod Photoreceptor Neurons. Molecular neurobiology 42, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala RV, Elliott MH, McClellan ME, Anderson RE, 2006. Localization of the Insulin Receptor and Phosphoinositide 3-Kinase in Detergent-Resistant Membrane Rafts of Rod Photoreceptor Outer Segments. Advances in experimental medicine and biology 572, 491–497. [DOI] [PubMed] [Google Scholar]
- Rajala RV, Rajala A, Kooker C, Wang Y, Anderson RE, 2016. The Warburg Effect Mediator Pyruvate Kinase M2 Expression and Regulation in the Retina. Scientific reports 6, 37727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendram R, Saraswathy S, Rao NA, 2007. Photoreceptor Mitochondrial Oxidative Stress in Early Experimental Autoimmune Uveoretinitis. The British journal of ophthalmology 91, 531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey DJ, Arden GB, 2015. Hypoxia and Dark Adaptation in Diabetic Retinopathy: Interactions, Consequences, and Therapy. Current diabetes reports 15, 118. [DOI] [PubMed] [Google Scholar]
- Ramsey DJ, Ripps H, Qian H, 2006. An Electrophysiological Study of Retinal Function in the Diabetic Female Rat. Investigative ophthalmology & visual science 47, 5116–5124. [DOI] [PubMed] [Google Scholar]
- Reichenbach A, Bringmann A, 2013. New Functions of Muller Cells. Glia 61, 651–678. [DOI] [PubMed] [Google Scholar]
- Reichhart N, Crespo-Garcia S, Haase N, Golic M, Skosyrski S, Rubsam A, Herrspiegel C, Kociok N, Alenina N, Bader M, Dechend R, Strauss O, Joussen AM, 2017. The Teto Rat as a New Translational Model for Type 2 Diabetic Retinopathy by Inducible Insulin Receptor Knockdown. Diabetologia 60, 202–211. [DOI] [PubMed] [Google Scholar]
- Reis A, Mateus C, Melo P, Figueira J, Cunha-Vaz J, Castelo-Branco M, 2014. Neuroretinal Dysfunction with Intact Blood-Retinal Barrier and Absent Vasculopathy in Type 1 Diabetes. Diabetes 63, 3926–3937. [DOI] [PubMed] [Google Scholar]
- Reiter CE, Sandirasegarane L, Wolpert EB, Klinger M, Simpson IA, Barber AJ, Antonetti DA, Kester M, Gardner TW, 2003. Characterization of Insulin Signaling in Rat Retina in Vivo and Ex Vivo. Am J Physiol Endocrinol Metab 285, E763–774. [DOI] [PubMed] [Google Scholar]
- Reiter CE, Wu X, Sandirasegarane L, Nakamura M, Gilbert KA, Singh RS, Fort PE, Antonetti DA, Gardner TW, 2006. Diabetes Reduces Basal Retinal Insulin Receptor Signaling: Reversal with Systemic and Local Insulin. Diabetes 55, 1148–1156. [DOI] [PubMed] [Google Scholar]
- Ren X, Sun H, Zhang C, Li C, Wang J, Shen J, Yu D, Kong L, 2016. Protective Function of Pyridoxamine on Retinal Photoreceptor Cells Via Activation of the Perk1/2/Nrf2/Trx/Ask1 Signalling Pathway in Diabetic Mice. Molecular medicine reports 14, 420–424. [DOI] [PubMed] [Google Scholar]
- Reynaud X, Dorey CK, 1994. Extraretinal Neovascularization Induced by Hypoxic Episodes in the Neonatal Rat. Investigative ophthalmology & visual science 35, 3169–3177. [PubMed] [Google Scholar]
- Reynaud X, Hansen RM, Fulton AB, 1995. Effect of Prior Oxygen Exposure on the Electroretinographic Responses of Infant Rats. Investigative ophthalmology & visual science 36, 2071–2079. [PubMed] [Google Scholar]
- Rizzolo LJ, 1997. Polarity and the Development of the Outer Blood-Retinal Barrier. Histology and histopathology 12, 1057–1067. [PubMed] [Google Scholar]
- Roy MS, Gunkel RD, Podgor MJ, 1986. Color Vision Defects in Early Diabetic Retinopathy. Archives of ophthalmology 104, 225–228. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Mark M, Jacobs H, Klopfenstein M, Hu J, Lloyd M, Habib S, Tosha C, Radu RA, Ghyselinck NB, Nusinowitz S, Bok D, 2012. Retinoid Content, Visual Responses, and Ocular Morphology Are Compromised in the Retinas of Mice Lacking the Retinol-Binding Protein Receptor, Stra6. Investigative ophthalmology & visual science 53, 3027–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungger-Brandle E, Dosso AA, Leuenberger PM, 2000. Glial Reactivity, an Early Feature of Diabetic Retinopathy. Investigative ophthalmology & visual science 41, 1971–1980. [PubMed] [Google Scholar]
- Saari JC, 2000. Biochemistry of Visual Pigment Regeneration: The Friedenwald Lecture. Investigative ophthalmology & visual science 41, 337–348. [PubMed] [Google Scholar]
- Sahajpal N, Kowluru A, Kowluru RA, 2019. The Regulatory Role of Rac1, a Small Molecular Weight Gtpase, in the Development of Diabetic Retinopathy. J Clin Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto A, Nishijima K, Kita M, Oh H, Tsujikawa A, Yoshimura N, 2009. Association between Foveal Photoreceptor Status and Visual Acuity after Resolution of Diabetic Macular Edema by Pars Plana Vitrectomy. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 247, 1325–1330. [DOI] [PubMed] [Google Scholar]
- Saleh M, Flores M, Gauthier AS, Elphege E, Delbosc B, 2017. Quantitative Analysis of Photoreceptor Layer Reflectivity on En-Face Optical Coherence Tomography as an Estimator of Cone Density. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 255, 2119–2126. [DOI] [PubMed] [Google Scholar]
- Saliba A., Du Y., Liu H., Patel S., Roberts R., Berkowitz BA., Kern TS., 2015. Photobiomodulation Mitigates Diabetes-Induced Retinopathy by Direct and Indirect Mechanisms: Evidence from Intervention Studies in Pigmented Mice. PloS one 10, e0139003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samardzija M, von Lintig J, Tanimoto N, Oberhauser V, Thiersch M, Remé CE, Seeliger M, Grimm C, Wenzel A, 2008. R91w Mutation in Rpe65 Leads to Milder Early-Onset Retinal Dystrophy Due to the Generation of Low Levels of 11-Cis-Retinal. Hum Mol Genet 17, 281–292. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Bell BA, Pereira A, Saxon J, Peachey NS, 2015. Early Retinal Pigment Epithelium Dysfunction Is Concomitant with Hyperglycemia in Mouse Models of Type 1 and Type 2 Diabetes. Journal of neurophysiology 113, 1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels IS, Lee CA, Petrash JM, Peachey NS, Kern TS, 2012. Exclusion of Aldose Reductase as a Mediator of Erg Deficits in a Mouse Model of Diabetic Eye Disease. Visual neuroscience 29, 267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Chavez G, Pena-Rangel MT, Riesgo-Escovar JR, Martinez-Martinez A, Salceda R, 2012. Insulin Stimulated-Glucose Transporter Glut 4 Is Expressed in the Retina. PloS one 7, e52959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JM, Tewari S, Lin JY, Kowluru RA, 2013. Interrelationship between Activation of Matrix Metalloproteinases and Mitochondrial Dysfunction in the Development of Diabetic Retinopathy. Biochem Biophys Res Commun 438, 760–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawides L, Sapoznik KA, de Castro A, Walker BR, Gast TJ, Elsner AE, Burns SA, 2017. Alterations to the Foveal Cone Mosaic of Diabetic Patients. Investigative ophthalmology & visual science 58, 3395–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyad Z, Sirohi K, Radha V, Swarup G, 2017. 661w Is a Retinal Ganglion Precursor-Like Cell Line in Which Glaucoma-Associated Optineurin Mutants Induce Cell Death Selectively. Scientific reports 7, 16855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarinci F, Jampol LM, Linsenmeier RA, Fawzi AA, 2015. Association of Diabetic Macular Nonperfusion with Outer Retinal Disruption on Optical Coherence Tomography. JAMA ophthalmology 133, 1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarinci F, Nesper PL, Fawzi AA, 2016. Deep Retinal Capillary Nonperfusion Is Associated with Photoreceptor Disruption in Diabetic Macular Ischemia. American journal of ophthalmology 168, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuderi S, D’Amico A,G, Federico C, Saccone S, Magro G, Bucolo C, Drago F, D’Agata V, 2015. Different Retinal Expression Patterns of Il-1alpha, Il-1beta, and Their Receptors in a Rat Model of Type 1 Stz-Induced Diabetes. Journal of molecular neuroscience : MN 56, 431–439. [DOI] [PubMed] [Google Scholar]
- Sharma R, Schwarz C, Hunter JJ, Palczewska G, Palczewski K, Williams DR, 2017. Formation and Clearance of All-Trans-Retinol in Rods Investigated in the Living Primate Eye with Two-Photon Ophthalmoscopy. Investigative ophthalmology & visual science 58, 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Liu K, Xu X, 2016. Correlation between Visual Function and Photoreceptor Integrity in Diabetic Macular Edema: Spectral-Domain Optical Coherence Tomography. Current eye research 41, 391–399. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Kobayashi Y, Muraoka K, 1981. Midperipheral Fundus Involvement in Diabetic Retinopathy. Ophthalmology 88, 601–612. [DOI] [PubMed] [Google Scholar]
- Simo R, Stitt AW, Gardner TW, 2018. Neurodegeneration in Diabetic Retinopathy: Does It Really Matter? Diabetologia 61, 1902–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simo R, Sundstrom JM, Antonetti DA, 2014. Ocular Anti-Vegf Therapy for Diabetic Retinopathy: The Role of Vegf in the Pathogenesis of Diabetic Retinopathy. Diabetes care 37, 893–899. [DOI] [PubMed] [Google Scholar]
- Simo R, Villarroel M, Corraliza L, Hernandez C, Garcia-Ramirez M, 2010. The Retinal Pigment Epithelium: Something More Than a Constituent of the Blood-Retinal Barrier--Implications for the Pathogenesis of Diabetic Retinopathy. J Biomed Biotechnol 2010, 190724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaprasad S, Vasconcelos JC, Prevost AT, Holmes H, Hykin P, George S, Murphy C, Kelly J, Arden GB, Group, C.S., 2018. Clinical Efficacy and Safety of a Light Mask for Prevention of Dark Adaptation in Treating and Preventing Progression of Early Diabetic Macular Oedema at 24 Months (Cleopatra): A Multicentre, Phase 3, Randomised Controlled Trial. The lancet. Diabetes & endocrinology 6, 382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleightholm MA, Aldington SJ, Arnold J, Kohner EM, 1988. Diabetic Retinopathy: Ii. Assessment of Severity and Progression from Fluorescein Angiograms. J Diabet Complications 2, 117–120. [DOI] [PubMed] [Google Scholar]
- Soliman MK, Sadiq MA, Agarwal A, Sarwar S, Hassan M, Hanout M, Graf F, High R, Do DV, Nguyen QD, Sepah YJ, 2016. High-Resolution Imaging of Parafoveal Cones in Different Stages of Diabetic Retinopathy Using Adaptive Optics Fundus Camera. PloS one 11, e0152788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt A, Gardiner TA, Alderson NL, Canning P, Frizzell N, Duffy N, Boyle C, Januszewski AS, Chachich M, Baynes JW, Thorpe SR, Anderson NL, 2002. The Age Inhibitor Pyridoxamine Inhibits Development of Retinopathy in Experimental Diabetes. Diabetes 51, 2826–2832. [DOI] [PubMed] [Google Scholar]
- Stitt AW, Curtis TM, Chen M, Medina RJ, McKay GJ, Jenkins A, Gardiner TA, Lyons TJ, Hammes HP, Simo R, Lois N, 2016. The Progress in Understanding and Treatment of Diabetic Retinopathy. Progress in retinal and eye research 51, 156–186. [DOI] [PubMed] [Google Scholar]
- Strauss O, 2005. The Retinal Pigment Epithelium in Visual Function. Physiol Rev 85, 845–881. [DOI] [PubMed] [Google Scholar]
- Sucher NJ, Aizenman E, Lipton SA, 1991. N-Methyl-D-Aspartate Antagonists Prevent Kainate Neurotoxicity in Rat Retinal Ganglion Cells in Vitro. The Journal of neuroscience : the official journal of the Society for Neuroscience 11, 966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M, Cutler A, Shen B, Moss SE, Iyengar SK, Klein R, Folkman J, Anand-Apte B, 2013. Inhibition of Egf Signaling Protects the Diabetic Retina from Insulin-Induced Vascular Leakage. The American journal of pathology 183, 987–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadfi K, Atlasz T, Kiss P, Reglodi D, Szabo A, Kovacs K, Szalontai B, Setalo G Jr., Banki E, Csanaky K, Tamas A, Gabriel R, 2012. Protective Effects of the Neuropeptide Pacap in Diabetic Retinopathy. Cell and tissue research 348, 37–46. [DOI] [PubMed] [Google Scholar]
- Talukdar S, Zhou Y, Li D, Rossulek M, Dong J, Somayaji V, Weng Y, Clark R, Lanba A, Owen BM, Brenner MB, Trimmer JK, Gropp KE, Chabot JR, Erion DM, Rolph TP, Goodwin B, Calle RA, 2016. A Long-Acting Fgf21 Molecule, Pf-05231023, Decreases Body Weight and Improves Lipid Profile in Non-Human Primates and Type 2 Diabetic Subjects. Cell Metab 23, 427–440. [DOI] [PubMed] [Google Scholar]
- Tan W, Wright T, Rajendran D, Garcia-Sanchez Y, Finkelberg L, Kisilak M, Campbell M, Westall CA, 2015. Cone-Photoreceptor Density in Adolescents with Type 1 Diabetes. Investigative ophthalmology & visual science 56, 6339–6343. [DOI] [PubMed] [Google Scholar]
- Tang J, Herda AA, Kern TS, 2014. Photobiomodulation in the Treatment of Patients with Non-Center-Involving Diabetic Macular Oedema. The British journal of ophthalmology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Kern TS, 2011. Inflammation in Diabetic Retinopathy. Progress in retinal and eye research 30, 343–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Lee CA, Du Y, Sun Y, Pearlman E, Sheibani N, Kern T, 2013. Myd88-Dependent Pathways in Leukocytes Affect the Retina in Diabetes. PloS one 8, e68871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Zhang Y, Jiang Y, Willard L, Ortiz E, Wark L, Medeiros D, Lin D, 2011. Dietary Wolfberry Ameliorates Retinal Structure Abnormalities in Db/Db Mice at the Early Stage of Diabetes. Exp Biol Med (Maywood) 236, 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarchick MJ, Bassiri P, Rohwer RM, Samuels IS, 2016. Early Functional and Morphologic Abnormalities in the Diabetic Nyxnob Mouse Retina. Investigative ophthalmology & visual science 57, 3496–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarchick MJ, Cutler AH, Trobenter TD, Kozlowski MR, Makowski ER, Holoman N, Shao J, Shen B, Anand-Apte B, Samuels IS, 2019. Endogenous Insulin Signaling in the Rpe Contributes to the Maintenance of Rod Photoreceptor Function in Diabetes. Experimental eye research 180, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares Ferreira J, Alves M, Dias-Santos A, Costa L, Santos BO, Cunha JP, Papoila AL, Abegao Pinto L, 2016. Retinal Neurodegeneration in Diabetic Patients without Diabetic Retinopathy. Investigative ophthalmology & visual science 57, 6455–6460. [DOI] [PubMed] [Google Scholar]
- Tavares Ferreira J, Proenca R, Alves M, Dias-Santos A, Santos BO, Cunha JP, Papoila AL, Abegao Pinto L, 2017. Retina and Choroid of Diabetic Patients without Observed Retinal Vascular Changes: A Longitudinal Study. American journal of ophthalmology 176, 15–25. [DOI] [PubMed] [Google Scholar]
- Tehrani NM, Riazi-Esfahani H, Jafarzadehpur E, Mirzajani A, Talebi H, Amini A, Mazloumi M, Roohipoor R, Riazi-Esfahani M, 2015. Multifocal Electroretinogram in Diabetic Macular Edema; Correlation with Visual Acuity and Optical Coherence Tomography. J Ophthalmic Vis Res 10, 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thebeau C, Zhang S, Kolesnikov AV, Kefalov VJ, Semenkovich CF, Rajagopal R, 2020. Light Deprivation Reduces the Severity of Experimental Diabetic Retinopathy. Neurobiol Dis 137, 104754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedeman JS, Kirk SE, Srinivas S, Beach JM, 1998. Retinal Oxygen Consumption During Hyperglycemia in Patients with Diabetes without Retinopathy. Ophthalmology 105, 31–36. [DOI] [PubMed] [Google Scholar]
- Tonade D, Kern TS, 2017. Diabetes of 5 Years Duration Does Not Lead to Photoreceptor Degeneration in the Canine Non-Tapetal Inferior-Nasal Retina. Experimental eye research 162, 126–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonade D, Liu H, Kern TS, 2016. Photoreceptor Cells Produce Inflammatory Mediators That Contribute to Endothelial Cell Death in Diabetes. Investigative ophthalmology & visual science 57, 4264–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonade D, Liu H, Palczewski K, Kern TS, 2017. Photoreceptor Cells Produce Inflammatory Products That Contribute to Retinal Vascular Permeability in a Mouse Model of Diabetes. Diabetologia 60, 2111–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toprak I, Yildirim C, Yaylali V, 2015. Impaired Photoreceptor Inner Segment Ellipsoid Layer Reflectivity in Mild Diabetic Retinopathy. Can J Ophthalmol 50, 438–441. [DOI] [PubMed] [Google Scholar]
- Tornquist P, Alm A, 1979. Retinal and Choroidal Contribution to Retinal Metabolism in Vivo. A Study in Pigs. Acta Physiol. Scand. 106, 351–357. [DOI] [PubMed] [Google Scholar]
- Toyoda F, Tanaka Y, Shimmura M, Kinoshita N, Takano H, Kakehashi A, 2016. Diabetic Retinal and Choroidal Edema in Sdt Rats. Journal of diabetes research 2016, 2345141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tregear SJ, Knowles PJ, Ripley LG, Casswell AG, 1997. Chromatic-Contrast Threshold Impairment in Diabetes. Eye 11 ( Pt 4), 537–546. [DOI] [PubMed] [Google Scholar]
- Trick GL, Burde RM, Gordon MO, Santiago JV, Kilo C, 1988. The Relationship between Hue Discrimination and Contrast Sensitivity Deficits in Patients with Diabetes Mellitus. Ophthalmology 95, 693–698. [DOI] [PubMed] [Google Scholar]
- Tsai SR, Hamblin MR, 2017. Biological Effects and Medical Applications of Infrared Radiation. Journal of photochemistry and photobiology. B, Biology 170, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tso MO, Cunha-Vaz JG, Shih CY, Jones CW, 1980. Clinicopathologic Study of Blood-Retinal Barrier in Experimental Diabetes Mellitus. Archives of ophthalmology 98, 2032–2040. [DOI] [PubMed] [Google Scholar]
- Tuitoek PJ, Ziari S, Tsin AT, Rajotte RV, Suh M, Basu TK, 1996. Streptozotocin-Induced Diabetes in Rats Is Associated with Impaired Metabolic Availability of Vitamin a (Retinol). Br J Nutr 75, 615–622. [DOI] [PubMed] [Google Scholar]
- Uji A, Murakami T, Nishijima K, Akagi T, Horii T, Arakawa N, Muraoka Y, Ellabban AA, Yoshimura N, 2012. Association between Hyperreflective Foci in the Outer Retina, Status of Photoreceptor Layer, and Visual Acuity in Diabetic Macular Edema. American journal of ophthalmology 153, 710–717, 717 e711. [DOI] [PubMed] [Google Scholar]
- United Kingdom Prospective Diabetes Study, 1998. Intensive Blood-Glucose Control with Sulphonylureas or Insulin Compared with Conventional Treatment and Risk of Complications in Patients with Type 2 Diabetes. Lancet 352, 837–853. [PubMed] [Google Scholar]
- Unoki N, Nishijima K, Sakamoto A, Kita M, Watanabe D, Hangai M, Kimura T, Kawagoe N, Ohta M, Yoshimura N, 2007. Retinal Sensitivity Loss and Structural Disturbance in Areas of Capillary Nonperfusion of Eyes with Diabetic Retinopathy. American journal of ophthalmology 144, 755–760. [DOI] [PubMed] [Google Scholar]
- van Dijk HW, Kok PH, Garvin M, Sonka M, Devries JH, Michels RP, van Velthoven ME, Schlingemann RO, Verbraak FD, Abramoff MD, 2009. Selective Loss of Inner Retinal Layer Thickness in Type 1 Diabetic Patients with Minimal Diabetic Retinopathy. Investigative ophthalmology & visual science 50, 3404–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eeden PE, Tee LB, Lukehurst S, Lai CM, Rakoczy EP, Beazley LD, Dunlop SA, 2006. Early Vascular and Neuronal Changes in a Vegf Transgenic Mouse Model of Retinal Neovascularization. Investigative ophthalmology & visual science 47, 4638–4645. [DOI] [PubMed] [Google Scholar]
- Veenstra A, Liu H, Lee CA, Du Y, Tang J, Kern TS, 2015. Diabetic Retinopathy: Retina-Specific Methods for Maintenance of Diabetic Rodents and Evaluation of Vascular Histopathology and Molecular Abnormalities. Curr Protoc Mouse Biol 5, 247–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey KA, Greferath U, Aplin FP, Jobling AI, Phipps JA, Ho T, De Iongh RU, Fletcher EL, 2014. Adenosine Triphosphate-Induced Photoreceptor Death and Retinal Remodeling in Rats. The Journal of comparative neurology 522, 2928–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinores SA, Campochiaro PA, Williams EH, May EE, Green WR, Sorenson RL, 1988. Aldose Reductase Expression in Human Diabetic Retina and Retinal Pigment Epithelium. Diabetes 37, 1658–1664. [DOI] [PubMed] [Google Scholar]
- Vinores SA, Gadegbeku C, Campochiaro PA, Green WR, 1989. Immunohistochemical Localization of Blood-Retinal Barrier Breakdown in Human Diabetics. The American journal of pathology 134, 231–235. [PMC free article] [PubMed] [Google Scholar]
- Vujosevic S, Midena E, 2013. Retinal Layers Changes in Human Preclinical and Early Clinical Diabetic Retinopathy Support Early Retinal Neuronal and Muller Cells Alterations. Journal of diabetes research 2013, 905058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallow IH, 1983. Posterior and Anterior Permeability Defects? Morphologic Observations on Streptozotocin-Treated Rats. Investigative ophthalmology & visual science 24, 1259–1268. [PubMed] [Google Scholar]
- Wanek J, Blair NP, Chau FY, Lim JI, Leiderman YI, Shahidi M, 2016. Alterations in Retinal Layer Thickness and Reflectance at Different Stages of Diabetic Retinopathy by En Face Optical Coherence Tomography. Investigative ophthalmology & visual science 57, OCT341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JS, Kefalov VJ, 2011. The Cone-Specific Visual Cycle. Progress in retinal and eye research 30, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Kondo M, Bill A, 1997. Glucose Metabolism in Cat Outer Retina. Effects of Light and Hyperoxia. Investigative ophthalmology & visual science 38, 48–55. [PubMed] [Google Scholar]
- Wang S, Birol G, Budzynski E, Flynn R, Linsenmeier RA, 2010. Metabolic Responses to Light in Monkey Photoreceptors. Current eye research 35, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Liu Y, Tan JW, Hu T, Zhang HF, Sorenson CM, Smith JA, Sheibani N, 2019. Tunicamycin-Induced Photoreceptor Atrophy Precedes Degeneration of Retinal Capillaries with Minimal Effects on Retinal Ganglion and Pigment Epithelium Cells. Experimental eye research 187, 107756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XN, Li ST, Li W, Hua YJ, Wu Q, 2018. The Thickness and Volume of the Choroid, Outer Retinal Layers and Retinal Pigment Epithelium Layer Changes in Patients with Diabetic Retinopathy. Int J Ophthalmol 11, 1957–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangsa-Wirawan ND, Linsenmeier RA, 2003. Retinal Oxygen: Fundamental and Clinical Aspects. Archives of ophthalmology 121, 547–557. [DOI] [PubMed] [Google Scholar]
- Warburg O, Posener K, Negrelein E, 1924. On the Metabolism of Carcinoma Cells. Journal of Cancer Research 152, 309–344. [Google Scholar]
- Weber BH, Vogt G, Pruett RC, Stohr H, Felbor U, 1994. Mutations in the Tissue Inhibitor of Metalloproteinases-3 (Timp3) in Patients with Sorsby’s Fundus Dystrophy. Nature genetics 8, 352–356. [DOI] [PubMed] [Google Scholar]
- Weinberger D, Fink-Cohen S, Gaton DD, Priel E, Yassur Y, 1995. Non-Retinovascular Leakage in Diabetic Maculopathy. The British journal of ophthalmology 79, 728–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger D, Kramer M, Priel E, Gaton DD, Axer-Siegel R, Yassur Y, 1998. Indocyanine Green Angiographic Findings in Nonproliferative Diabetic Retinopathy. American journal of ophthalmology 126, 238–247. [DOI] [PubMed] [Google Scholar]
- Winkler BS, 1981. Glycolytic and Oxidative Metabolism in Relation to Retinal Function. The Journal of general physiology 77, 667–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley MT, 2010. Energy Metabolism of the Visual System. Eye Brain 2, 99–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia HH, Chen JL, Chen HY, Lin HJ, 2020. Correlation between Optical Coherence Tomography, Multifocal Electroretinogram Findings and Visual Acuity in Diabetic Macular Edema. Int J Ophthalmol 13, 1592–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Rizzolo LJ, 2017. Effects of Diabetic Retinopathy on the Barrier Functions of the Retinal Pigment Epithelium. Vision research 139, 72–81. [DOI] [PubMed] [Google Scholar]
- Xiao C, He M, Nan Y, Zhang D, Chen B, Guan Y, Pu M, 2012. Physiological Effects of Superoxide Dismutase on Altered Visual Function of Retinal Ganglion Cells in Db/Db Mice. PloS one 7, e30343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HZ, Le YZ, 2011. Significance of Outer Blood-Retina Barrier Breakdown in Diabetes and Ischemia.Investigative ophthalmology & visual science 52, 2160–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HZ, Song Z, Fu S, Zhu M, Le YZ, 2011. Rpe Barrier Breakdown in Diabetic Retinopathy: Seeing Is Believing. J Ocul Biol Dis Infor 4, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wei Y, Gong J, Cho H, Park JK, Sung ER, Huang H, Wu L, Eberhart C, Handa JT, Du Y, Kern TS, Thimmulappa R, Barber AJ, Biswal S, Duh EJ, 2014. Nrf2 Plays a Protective Role in Diabetic Retinopathy in Mice. Diabetologia 57, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Macleish PR, 1994. Ionic Conductances of Monkey Solitary Cone Inner Segments. Journal of neurophysiology 71, 656–665. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Kamiyama M, Nitta K, Yamada T, Hayasaka S, 1996. Selective Reduction of the S Cone Electroretinogram in Diabetes. The British journal of ophthalmology 80, 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Takeuchi S, Kamiyama M, 1997. The Short Wavelength-Sensitive Cone Electroretinogram in Diabetes: Relationship to Systemic Factors. Documenta ophthalmologica. Advances in ophthalmology 94, 193–200. [DOI] [PubMed] [Google Scholar]
- Yanyali A, Bozkurt KT, Macin A, Horozoglu F, Nohutcu AF, 2011. Quantitative Assessment of Photoreceptor Layer in Eyes with Resolved Edema after Pars Plana Vitrectomy with Internal Limiting Membrane Removal for Diabetic Macular Edema. Ophthalmologica. Journal international d’ophtalmologie. International journal of ophthalmology. Zeitschrift fur Augenheilkunde 226, 57–63. [DOI] [PubMed] [Google Scholar]
- Yi X, Schubert M, Peachey NS, Suzuma K, Burks DJ, Kushner JA, Suzuma I, Cahill C, Flint CL, Dow MA, Leshan RL, King GL, White MF, 2005. Insulin Receptor Substrate 2 Is Essential for Maturation and Survival of Photoreceptor Cells. The Journal of neuroscience : the official journal of the Society for Neuroscience 25, 1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomizo H, Maeda Y, Park K, Clermont AC, Hernandez SL, Fickweiler W, Li Q, Wang CH, Paniagua SM, Simao F, Ishikado A, Sun B, Wu IH, Katagiri S, Pober DM, Tinsley LJ, Avery RL, Feener EP, Kern TS, Keenan HA, Aiello LP, Sun JK, King GL, 2019. Retinol Binding Protein 3 Is Increased in the Retina of Patients with Diabetes Resistant to Diabetic Retinopathy. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DY, Cringle SJ, 2005. Retinal Degeneration and Local Oxygen Metabolism. Experimental eye research 80, 745–751. [DOI] [PubMed] [Google Scholar]
- Yu M, Zhang X, Zhong X, Yu Q, Jiang F, Ma J, Wu D, 2002. Multifocal Electroretinograms in the Early Stages of Diabetic Retinopathy. Chin Med J (Engl) 115, 563–566. [PubMed] [Google Scholar]
- Zaleska-Zmijewska A, Piatkiewicz P, Smigielska B, Sokolowska-Oracz A, Wawrzyniak ZM, Romaniuk D, Szaflik J, Szaflik JP, 2017. Retinal Photoreceptors and Microvascular Changes in Prediabetes Measured with Adaptive Optics (Rtx1): A Case-Control Study. Journal of diabetes research 2017, 4174292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaleska-Zmijewska A, Wawrzyniak ZM, Dabrowska A, Szaflik JP, 2019. Adaptive Optics (Rtx1) High-Resolution Imaging of Photoreceptors and Retinal Arteries in Patients with Diabetic Retinopathy. Journal of diabetes research 2019, 9548324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng HY, Green WR, Tso MO, 2008. Microglial Activation in Human Diabetic Retinopathy. Archives of ophthalmology 126, 227–232. [DOI] [PubMed] [Google Scholar]
- Zeng XX, Ng YK, Ling EA, 2000. Neuronal and Microglial Response in the Retina of Streptozotocin-Induced Diabetic Rats. Visual neuroscience 17, 463–471. [DOI] [PubMed] [Google Scholar]
- Zhang J-Z, Kern TS, 2003. Captopril Inhibits Intracellular Glucose Accumulation in Retinal Cells in Diabetes. Investigative ophthalmology & visual science 44, 4001–4005. [DOI] [PubMed] [Google Scholar]
- Zhang J, Kiser PD, Badiee M, Palczewska G, Dong Z, Golczak M, Tochtrop GP, Palczewski K, 2015. Molecular Pharmacodynamics of Emixustat in Protection against Retinal Degeneration. The Journal of clinical investigation 125, 2781–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhang ZM, 2014. Oxygen-Induced Retinopathy in Mice with Retinal Photoreceptor Cell Degeneration. Life Sci 102, 28–35. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bao S, Hambly BD, Gillies MC, 2009. Vascular Endothelial Growth Factor-A: A Multifunctional Molecular Player in Diabetic Retinopathy. The international journal of biochemistry & cell biology 41, 2368–2371. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang J, Wang Q, Lei X, Chu Q, Xu GT, Ye W, 2011. Intravitreal Injection of Exendin-4 Analogue Protects Retinal Cells in Early Diabetic Rats. Investigative ophthalmology & visual science 52, 278–285. [DOI] [PubMed] [Google Scholar]
- Zhao W, Wang D, Zhao J, Zhao W, 2017. Bioinformatic Analysis of Retinal Gene Function and Expression in Diabetic Rats. Experimental and therapeutic medicine 14, 2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Du Y, Miller C, Gubitosi-Klug RA, Ball S, Berkowitz BA, Kern TS, 2007. Critical Role of Inducible Nitric Oxide Synthase in Degeneration of Retinal Capillaries in Mice with Streptozotocin-Induced Diabetes. Diabetologia 50, 1987–1996. [DOI] [PubMed] [Google Scholar]
- Zheng L, Kern T, 2009. Role of Nitric Oxide, Superoxide, Peroxynitrite and Poly(Adp-Ribose) Polymerase in Diabetic Retinopathy. Front Biosci. 14, 3974–3987. [DOI] [PubMed] [Google Scholar]
- Ziccard L., Paris V., Piccon F., Di Renz A., Lombard M., Fronton S., Parravan M., 2018. Early and Localized Retinal Dysfunction in Patients with Type 1 Diabetes Mellitus Studied by Multifocal Electroretinogram. Acta Diabetol 55, 1191–1200. [DOI] [PubMed] [Google Scholar]