

Abstract
Sea spray aerosol (SSA) contributes significantly to natural aerosol particle concentrations globally, in marine areas even dominantly. The potential changes of the omnipresent inorganic fraction of SSA due to atmospheric ageing is largely unexplored. In the atmosphere, SSA may exist as aqueous phase solution droplets or as dried solid or amorphous particles. We demonstrate that ageing of liquid NaCl and artificial sea salt aerosol by exposure to ozone and UV light leads to a substantial decrease in hygroscopicity and cloud activation potential of the dried particles of the same size. The results point towards surface reactions on the liquid aerosols that are more crucial for small particles and the formation of salt structures with water bound within the dried aerosols, termed hydrates. Our findings suggest an increased formation of hydrate forming salts during ageing and the presence of hydrates in dried SSA. Field observations indicate a reduced hygroscopic growth factor of sub-micrometre SSA in the marine atmosphere compared to fresh laboratory generated NaCl or sea salt of the same dry size, which is typically attributed to organic matter or sulphates. Aged inorganic sea salt offers an additional explanation for such a measured reduced hygroscopic growth factor and cloud activation potential.
Subject terms: Atmospheric science, Atmospheric chemistry, Thermodynamics
Introduction
The oceans contribute the largest constant mass of natural aerosol particles to the atmosphere1 in the form of sea spray aerosol (SSA). SSA is produced due to stress applied by winds on the ocean surface, which causes waves to form and break. In this process, air bubbles are entrained in ocean water. When entrained air bubbles rise to the surface they burst, and sea spray droplets are released to the atmosphere. Super-micrometer SSA dominate the emissions by mass but the largest number of SSA is produced in the sub-micrometer size range peaking at dry diameters near 100 nm2–8, which implies the presence of large numbers of particles potentially relevant for cloud formation. The chemical composition of SSA is complex and reflects to some extent the composition of the seawater from which it originates9 although enrichment of inorganic10 as well as organic constituents11 have been reported. While several studies have shown that the organic fraction in SSA is variable depending on location and time of the year12–14, the inorganic fraction is ever-present and a key ingredient across all size ranges13. The hygroscopicity of sea salt has in many years been approximated with that of NaCl but as shown by King et al.15 the CCN forming ability of sea salt particles differs from that of NaCl and e.g. Zieger et al.16 showed that the hygroscopic growth of sea salt is reduced compared to that of NaCl. Rasmussen et al.17 show and Zieger et al.16 conclude that the difference in hygroscopicity for inorganic sea salt and NaCl is likely due to hydrates.
Hydrate forming salts contain water molecules within their structures even after extensive drying and thus fundamentally impact water uptake. and are examples of hydrate forming salts present in sea salt aerosol. A recent study by Rosati et al.18 shows that aerosolized and particles exhibit a substantially reduced water uptake potential compared to predicted values assuming anhydrous salts. Experimental and predicted values could be reconciled when taking into account the hydration states of the dried particles. Thus, measurements of sea salt water uptake at sub- and supersaturated water vapour conditions are profoundly affected by hydrate forming salts. Their presence in dried particles leads to a reduced water uptake which could at least in part be falsely ascribed to other causes, such as the presence of organic matter.
The role of SSA in aerosol-radiation and aerosol-cloud interactions is strongly dictated by its ability to take up water at both sub- and supersaturated water vapour conditions19. Water uptake at sub-saturated conditions, termed hygroscopicity, directly impacts the aerosol optical properties by altering the scattering and absorption potential which is coupled to the ambient relative humidity (RH). Hygroscopicity also influences the SSA’s ability to act as cloud condensation nuclei (CCN) at supersaturated conditions20. The atmospheric lifetime of SSA varies between minutes for the largest particles to weeks for accumulation mode particles1. During its atmospheric transit, SSA is subjected to so-called ageing processes, which are of physico-chemical nature taking place inside the particles as well as at the surface via interactions with other particles, gases or radiation. Ageing may lead to profound changes in aerosol radiation interactions, and the ability to form cloud droplets. At present, accurate representation of SSA in climate models is hindered by incomplete understanding of the physical and chemical properties of SSA leading to large disagreement between model outputs21, where the water uptake is particularly afflicted with large uncertainties22.
Laboratory studies have so far primarily focused on the physical and chemical properties of freshly emitted SSA or its major component, NaCl, while field experiments have by necessity mostly included aged SSA5,7,8,10,15,16,23–31. The hygroscopic growth factor (GF; defined as the ratio of the wet and dry diameter D; GF(RH) = Dwet/Ddry), provides a scale for quantifying hygroscopicity. Based on the hygroscopic growth factor particles are often divided into sub-categories: nearly hydrophobic particles (NHP), less-hygroscopic particles (LHP), more-hygroscopic particles (MHP) and the even more hygroscopic sea-salt particles (SSP)32. As pointed out by Swietlicki et al.32 the MHP, which are significantly less hygroscopic than pure NaCl particles, have been found to be ubiquitous in the marine environment whereas observation of externally mixed sea salt particles with a distinct SSP–group are limited to high wind speeds. This was also highlighted by the first successful eddy covariance flux measurements over the sea, demonstrating a clear GF dependence on wind speed33. Several propositions exist to explain the nature of the reduced hygroscopicity of particles in the marine environment: sea spray may not be present in large enough numbers over the oceans, the particle distribution may be dominated by sulphate containing particles resulting from dimethyl sulphate oxidation, and particles may contain organic species as a result of atmospheric ageing or combinations of these options32. An enrichment in organic matter for particles with diameters smaller than 1 m has been proposed in several studies to cause a reduction in hygroscopicity11,26,34–40, although results from different studies vary quite substantially7,41.
Studies dedicated to effects of ageing processes of SSA are scarce. Oum et al.42 photolysed ozone () in the presence of aqueous pure NaCl particles, finding that this results in the formation of molecular chlorine (). They suggest that this occurs due to generation of hydroxyl radicals (OH) in the aqueous phase which in turn oxidise finally leading to the release of (see reactions 1–15 in42). More recent work has shown that also small amounts of photosensitizers, that are abundant in the marine environment43, could cause the release of dihalogens44. Another study revealed that the key reactions are dominant at the air–water interface45. These studies focused on the gas-phase products of such reactions, neglecting the effects on SSA itself. The experiments by Oum et al.42 were further repeated with pure NaCl, investigating changes in the chemical composition of the aerosols46. For particles with diameters below 1 m it was found that such a reaction gradually depletes the amount of and consequently replaces it by oxygen (O), hence leading to the formation of NaOH inside the liquid particles46. Effects on the physical properties or the use of a complex salt mixture were not investigated.
The current study was designed to explore how ageing by exposure to ozone and UV light affects the hygroscopicity and cloud activation potential of inorganic sea spray aerosol. Through a series of experiments in a well-controlled laboratory environment we examined the hygroscopicity and cloud activation potential of NaCl and artificial sea salt particles of different particle sizes. Additionally, the volatility of the particles was investigated and microscopy analysis was performed to yield insight into the elemental composition of fresh and aged particles.
Results and discussion
Hygroscopicity and cloud activation potential
Sea spray aerosols were generated from aqueous solutions of artificial sea salt (termed sea salt from now on; salinity of 3.5%) or NaCl using a circular plunging jet23 or an atomiser and injected into a 5 m3 Teflon chamber47 at 291 K. Typical particle number concentrations were of the order of 5000 and 15000 #/cm3 at the beginning of the experiments using the plunging jet and atomiser, respectively. The water uptake by the salt particles was examined with a humidified tandem differential mobility analyser, a cloud condensation nucleus counter and a humidified nephelometer. Four different scenarios were investigated: I) particles in the dry chamber exposed to , II) particles in the dry chamber exposed to and UV lights, III) liquid particles in the humid chamber exposed to and IV) liquid particles in the humid chamber exposed to and UV lights. In scenarios I) and II) particles will be either crystalline or amorphous, where NaCl was shown to be crystalline below 70% RH48 while sea salt particles could be amorphous even at very low RH48. Table 1 provides an overview of experimental conditions. Figure 1 shows hygroscopic growth factors at RH = 80% (GF(80%)) for NaCl and sea salt particles as a function of time during the performed experiments using Ægor, i.e. the plunging jet technique, and the atomiser. The data are presented as a mean over separate experiments carried out at the same conditions in the chamber (Exp. 1–19 in Table 1). The uncertainties represent the combined uncertainties of the GF(80%) measurement itself and the standard deviations from averaging the different experiments. Details for each single experiment, including the RH development in the chamber, particulate surface area, as well as humid results from atomiser experiments, are presented in the supplementary information (SI, Figs. S1, S2 and S3).
Table 1.
Experimental conditions.
| Exp. # | Date | Compound and generation technique | RH chamber [%] | T chamber [C] | Conditions | conc. [ppb] | Scenario | GF [] |
|---|---|---|---|---|---|---|---|---|
| 1 | 10.04.2018 | NaCl—Jet | 100 | 18 | O3, UV | 150 | IV | – 0.16 |
| 2 | 12.04.2018 | NaCl—Jet | 100 | 18 | O3, UV | 150 | IV | – 0.11 |
| 3 | 17.04.2018 | NaCl—Jet | 100 | 18 | O3 | 150 | III | |
| 4 | 12.06.2018 | NaCl—Atomizer | 0 | 18 | O3 | 150 | I | |
| 5 | 15.06.2018 | NaCl—Atomizer | 0 | 18 | O3, UV | 150 | II | |
| 6 | 18.06.2018 | NaCl—Atomizer | 70 | 18 | O3, UV | 150 | IV | – 0.08 |
| 7 | 17.08.2018 | NaCl—Atomizer | 70 | 18 | O3 | 150 | III | |
| 8 | 21.08.2018 | NaCl—Atomizer | 70 | 18 | O3 | 150 | III | |
| 9 | 19.09.2018 | NaCl—Atomizer | 70 | 18 | O3, UV | 150 | IV | – 0.10 |
| 10 | 09.07.2019 | NaCl—Atomizer | 70 | 18 | O3, UV | 150 | IV | – 0.14 |
| 11 | 03.10.2018 | Sea Salt—Jet | 100 | 18 | O3, UV | 150 | IV | – 0.06 |
| 12 | 07.10.2018 | Sea Salt—Jet | 100 | 18 | O3, UV | 150 | IV | – 0.07 |
| 13 | 10.10.2018 | Sea Salt—Jet | 100 | 18 | O3 | 150 | III | |
| 14 | 22.06.2018 | Sea Salt—Atomizer | 70 | 18 | O3 | 150 | III | |
| 15 | 25.06.2018 | Sea Salt—Atomizer | 70 | 18 | O3, UV | 150 | IV | – 0.07 |
| 16 | 27.06.2018 | Sea Salt—Atomizer | 70 | 18 | O3, UV | 300 | IV | – 0.06 |
| 17 | 25.07.2019 | Sea Salt—Atomizer | 70 | 18 | O3, UV | 150 | IV | – 0.06 |
| 18 | 12.10.2018 | Sea Salt—Atomizer | 0 | 18 | O3, UV | 150 | II | |
| 19 | 08.07.2019 | Sea Salt—Atomizer | 0 | 18 | O3 | 150 | I |
The temperature in the chamber was set to 18C during all experiments, but during experiments with UV lights on it increased to 20C.The GF(80%) decay rates (GF) for particles with Ddry = 50 nm per hour are indicated for experiments with ageing by exposure to and UV lights and are based on a linear regression of the change in GF(80%) during the first three hours of ageing.
Figure 1.

Effect of sea salt ageing on hygroscopicity. Growth factor (GF) measured at RH = 80% for NaCl and sea salt for dry particle sizes of Ddry = 50 nm and 200 nm. (a) Scenarios I (dry particles, dry chamber, ); (b) Scenario II (dry particles, dry chamber, , UV); (c) Scenario III (humid conditions, ); (d) Scenario IV (humid conditions, , UV). The 0-time-point marks the exposure start time to or OH. During t < 0, no oxidant was present in scenario I and III, while particles together with were present in the scenarios II and IV. The uncertainties represent the combined uncertainties of the GF(80%) measurement itself and the standard deviations from averaging the different experiments.
During the first part of the experiments (i.e. t < 0) the GF(80%) for NaCl particles (dry diameters (Ddry) of 50 and 200 nm) are in the range 1.78–1.90 in all experiments and scenarios and somewhat lower, in the range 1.68–1.80 for sea salt particles. The difference in GF between NaCl and sea salt is in agreement with the recent finding that the use of NaCl in experiments or in models overestimates the GF of sea salt16. Our findings coincide well with previously published GF values, recalculated for RH = 80% using -Köhler theory49, of 1.81–1.87 (using a nebulizer to generate the particles and calculated from Köhler theory) and 1.72–1.78 (using a plunging jet and a nebulizer), for NaCl and sea salt, respectively16.
For experiments performed at dry conditions the NaCl and sea salt particles are expected to be crystalline or amorphous in the smog chamber (scenarios I and II, Fig. 1a,b). A slight decrease in GF(80%) with time after exposure to and UV radiation is observed for both salts (max. 7% after 5 hours). This implies chemical and/or physical changes of the particles which at first seem hard to reconcile. They are however consistent with previous findings observing the formation of small amounts of when dry sea salt particles were exposed to and UV light, which were associated with chemical reactions taking place in a film of surface adsorbed water42. Such films can be present on dry NaCl and sea salt even after heating and pumping50,51. Sea salt furthermore contains salts that are hydrated also after drying, e.g. 17,18, providing a liquid layer for surface chemistry52. Exposure of dry NaCl to has also been reported to lead to perchlorate formation, which may have an effect on hygroscopicity53,54.
For particles aged under humid conditions with (no UV light) some decrease in GF(80%) with time is observed (scenario III, Fig. 1c). There is some debate as to the detailed chemistry taking place when aqueous NaCl or sea salt is exposed to in the dark, the formation of chlorates was for example proposed54–56 which might influence hygroscopicity.
A remarkable result in Fig. 1 is the large decrease in GF(80%) when small (50 nm) liquid NaCl and sea salt particles are aged in the presence of and UV lights, which we ascribe to a strong effect of OH chemistry on hygroscopicity (scenario IV, Fig. 1d). After three hours of ageing, the GF(80%) reaches values of 1.38 and 1.46 for NaCl and sea salt, respectively. For the larger particles (200 nm) the GF(80%) after three hours of ageing stays relatively constant reaching values of 1.72 and 1.69 for NaCl and sea salt, respectively. Both particle sizes have been aged in the same way and thus the difference in hygroscopicity shows that the small particles, which have a higher surface to volume ratio, are significantly more aged indicating surface processes being responsible for this finding52.
The weak response of the large particles (200 nm) to chemical ageing was confirmed by measurements of the scattering enhancement factor (f(RH), defined as the ratio between wet and dry scattering coefficient) of the polydisperse sea salt distribution in the chamber (Exp. 15), suggesting no change in the particles’ optical properties with ageing. This optical analysis relies mainly on the particle surface area and is hence dominated by larger particles (mode diameter of surface distribution during experiment: 300 nm, Fig. 2b). The scattering enhancement factor f(RH) in Fig. 2a shows a small decrease over time (f(RH) from 3 to 2.5 at = 635 nm) which can however, be accounted for by the change in RH from about 82% to 75% during the experiment. HTDMA and microscopy results indicate minor amounts of hydrate forming salts for particles of approximately 300 nm and thus a significant change in the particles’ dry index of refraction during ageing for this size seems unlikely. The size and surface area distributions (Fig. 2b) further illustrate that while the mode of the surface distributions remains relatively constant throughout the experiment, a substantial number and surface area of the particles are lost which according to model simulations can be ascribed to wall losses, including sedimentation, and coagulation (more details on the model are presented below). However, this will not affect the qualitative particle properties investigated in this study.
Figure 2.

Hygroscopicity of larger sized sea salt particles (Exp. 15). (a) Scattering enhancement factor (f(RH)) measured at 3 different wavelengths ( = 450 nm, = 525 nm, = 635 nm) and RH in the nephelometer. (b) number (red lines; left y-axis) and surface distributions (blue lines; right y-axis) at the beginning, after 5 hours and mean distributions throughout the experiment as derived from combined scanning mobility particle sizer and optical particle spectrometer measurements.
Simultaneously, the critical supersaturation (SScrit) for dry, monodisperse 50 nm particles was measured (Exp. 2, 14). This diameter falls in the key size range relevant for supersaturations typically experienced in cloud systems in oceanic regions57. The results in Fig. 3 show that fresh NaCl particles (a, blue line) are activated slightly earlier (SScrit = 0.32) than fresh sea salt (b, blue line; SScrit = 0.33). The results for fresh NaCl and sea salt are consistent with previously published data of SScrit,NaCl = 0.32–0.3358,59 and SScrit,sea salt = 0.34–0.3659,60. After three hours of ageing by and UV lights, the SScrit needed to activate the particles is increased for both types of salts reaching values of SScrit = 0.39 (a, red line) and SScrit = 0.41 (b, red line), for NaCl and sea salt, respectively. Hence, the cloud activation potential of such SSA is evidently reduced by the simulated ageing. These findings are in accordance with the hygroscopicity results presented above.
Figure 3.

Effect of ageing on CCN activation potential. Measurement of the critical supersaturation for monodisperse particles of Ddry = 50 nm. Results for NaCl are illustrated in (a) and for sea salt in (b). The blue lines show CCN activation at the initial state of the experiments (fresh NaCl and SSA), while the red lines illustrate CCN activation after 3 hours of ageing in the presence of and UV lights (aged NaCl and SSA).
Although all experiments were performed with particles generated from inorganic salts, we cannot fully exclude a small contamination by organic compounds. We find that it is highly unlikely that organic particle mass can be responsible for the observed changes in hygroscopicity and cloud activation for the following reasons: First, the water uptake potential of dry salt particles was measured with three independent methods yielding values that are well comparable with literature data, indicating negligible contamination. Second, calculations to assess the amount of organics needed to reach the values found in scenario IV for 50 nm particles show that the organic fraction has to be at least 70% to account for a lowering of the growth factor from 1.9 to 1.4 (see Fig. S4), which is very unlikely. Third, a contamination by organic compounds in the initial stages of the experiment by e.g. being present in the used salt solution would yield lower GF values right from the beginning. Organic contamination in later stages would be oxidised through ageing typically leading to an increase in hygroscopicity but it would still be much less hygroscopic compared to the salt particles. Forth, simulations with the Aerosol Dynamics gas- and particle-phase chemistry model for laboratory CHAMber studies (ADCHAM)61,62 revealed that the increase in particle diameter over time, as visible from Fig. 2b, can be fully explained by coagulation and particle wall losses and is not a sign of organic mass condensation (see Figs. S5 and S6 in SI). Specifications on the model settings are described in the SI.
Even though all experiments were carried out at low nitrogen oxide (NOx) levels, a small amount of NOx could lead to the formation of sodium nitrate (NaNO3) inside the particles during high humidity experiments63. The GF(80%) for 50 nm NaNO3 particles is 1.5864, which is much lower compared to that of NaCl and SSA and could lead to a decrease in the overall GF. As it is, however, higher compared to the measured GF(80%,50 nm) of approximately 1.4 after four hours of ageing, even a particle containing only NaNO3 cannot explain the presented results of aged NaCl. Results of the elemental composition also do not suggest the presence of NaNO3 (see discussion below).
Elemental composition
Figure 4a shows microscopy images of fresh NaCl particles appearing relatively cubic with contributions from only Na and Cl (Exp. 2). The sample shows a distribution of particle sizes between approximately Ddry = 50 and 300 nm, all containing about 50% Na and 50% Cl (atomic %). After 3 hours of ageing (Scenario IV), the particles change shape from close to cubic at the beginning of the experiment to more spherical after ageing (Fig. 4b; Exp. 1). Such a change in shape can affect the hygroscopicity results that are based on the comparison of dry and wet volumetric diameters. The diameters are measured with a differential mobility analyser (DMA). For spherical particles the mobility and volumetric diameters coincide while non-spherical particles need to be corrected with a dynamic shape factor to account for differences in shape. According to Zieger et al.16 fresh salt particles generated in the laboratory that appear cubic-like need a dynamic shape correction factor of approximately 1.10 to recalculate volumetric particle diameters from the measured mobility diameters. By applying this correction, the GF values increase by approximately 10%. Therefore, the GF(80%) illustrated in Fig. 1 at t < 0 (fresh particles) present lower estimates. The GF(80%) of the aged particles (t > 0) do not need any correction as aged particles have a more spherical shape.
Figure 4.

Effect of ageing on particle elemental composition. Microscopy images (STEM-EDX) of fresh (a) and aged (b) NaCl particles. Additionally, the elements found in the fresh and aged particles are illustrated. c) overlap of Na and Cl signals in the aged samples.
Additionally, the aged particles exhibit a decrease in Cl content and a concurrent increase in O content. Similar findings were previously published, displaying a replacement of Cl by O mainly for particles below 1 m (46, measurements were carried out above Ddry = 150 nm). Laskin et al.46 attributed the Cl decrease to emissions of molecular chlorine to the gas-phase, while they proposed the formation of NaOH in the particulate phase due to reactions with OH radicals. Another possibility for the appearance of O is the formation of chlorates55,56. By overlapping the signals for Na and Cl a depletion of Cl can be seen primarily at the surface of the particles (Fig. 4c; Exp. 2).
The elemental composition can also be used to infer a possible contamination by e.g. organic matter or NaNO3. Results for fresh NaCl particles (Fig. 4a) do not suggest the presence of either of them at any of the selected sizes as no oxygen was measured. The increased oxygen signal for the aged particles (Fig. 4b) could potentially also originate from organic matter or NaNO3. The concurrent Cl depletion does not support the presence of organics but rather that of NaOH or chlorates as explained above. A detailed microscopy study on NaCl/NaNO3 mixtures by Hoffman et al.65 revealed that NaNO3 is typically found as a shell around the particles producing an oxygen signal concentrated around the particles rather than inside. Although some of NaNO3 could have evaporated during our microscopy measurements, results of aged NaCl in Fig. 4 do not suggest a core–shell structure but an evenly distributed oxygen signal. Finally, Fig. 4b,c shows that aged NaCl particles are still predominately composed of Na and Cl, thus suggestion a very limited potential presence of NaNO3.
The analysis of sea salt reveals that while the fresh particles appear relatively cubic (Fig. 5a; Exp. 11), the aged ones have more round edges (Fig. 5b; Exp. 11). Furthermore, for both the fresh and aged particles, the core mainly consisted of Na and Cl, while S and O are visible in little pockets at the surface and Mg is present around the core of the particles, which coincides with measurements of real sea spray samples7,37. The presence of oxygen in fresh sea salt particles is expected considering the composition of the salt chosen to generate such aerosols (more information can be found under “Materials and methods”). The concentrations of Ca and K were not sufficient for an accurate analysis.
Figure 5.

Effect of ageing on shape and elemental composition. Microscopy images (STEM-EDX) of fresh (a) and aged (b) sea salt particles, illustrating the shape and elemental compositions found in these particles.
Hydrates
Dried NaCl is present in its anhydrous form at 291 K66. During exposure to OH the chemical composition of NaCl in aqueous solution changes, leading to a depletion in Cl and replacement by O46. Therefore, after ageing, the aqueous particles may contain newly formed compounds like NaOH and (chlorates). These compounds are known to exist in hydrated forms upon drying67, implying that even drying to very low RH cannot evaporate all water molecules, keeping a fraction bound to the salt. While NaOH is known to be present as mono- up to heptahydrate68, NaClO is known to mainly exist as pentahydrate69 and as monohydrate69. The GF(80%) for 50 nm NaCl particles was shown to continuously decrease nearly linearly after ageing, indicating a change in the chemical composition within the droplets (Fig. 1). The presence of hydrated salts can explain this phenomenon, as when exposed to high RH a smaller amount of water can be taken up due to the fraction of water molecules already trapped within the crystal. To test this hydrate hypothesis, we dried (RH < 10%) and subsequently heated the NaCl particles with a thermodenuder, set to a temperature of 300C, before exposing them to RH = 80%. The results in Fig. 6a (Exp. 2 and 11) illustrate that this procedure leads to an immediate change in GF(80%) for Ddry = 50 nm. Each time the heating is turned on, aged and heated NaCl (dark red) increases its GF(80%) to values comparable to those for the fresh NaCl particles. Microscopy images of NaCl after the thermodenuder treatment indicate mainly contributions of Na and Cl, while O could not be detected (Fig. S7).
Figure 6.
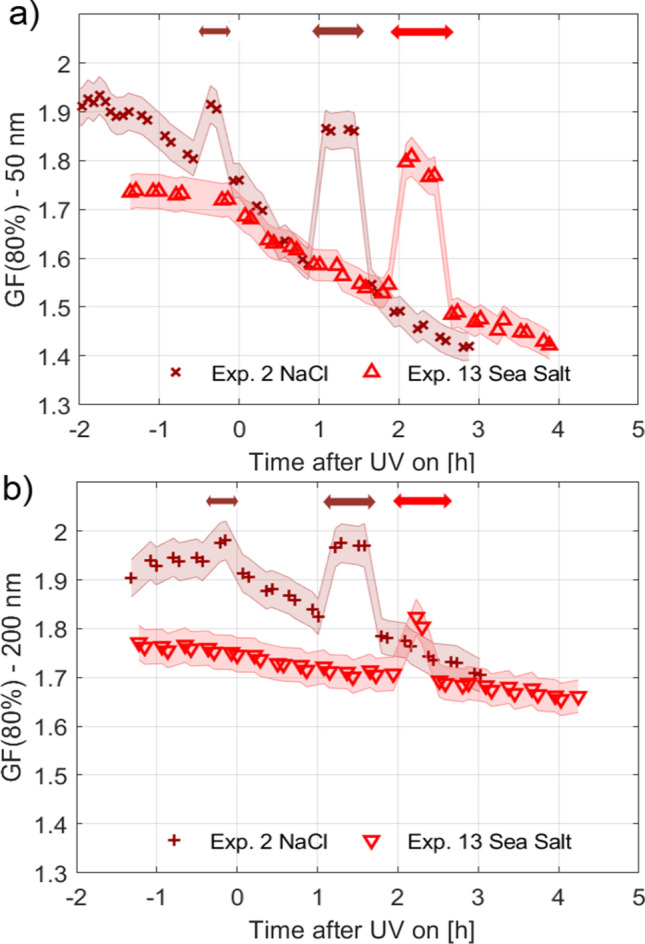
Effect of heating the aerosol particles. Growth factors at RH = 80% measured for NaCl and sea salt for the two different sizes of Ddry = 50 nm (a) and 200 nm (b) are shown. The influence of heating the particles to 300C, additionally to drying them, is illustrated. The arrows indicate the time period when the heating was switched on.
Sea salt is a complex inorganic mixture containing compounds like and , which are known to exist in hydrated forms under various conditions67. This was recently suggested to influence the interpretation of thermodenuder data17 and to be the reason for the slightly lower hygroscopicity of fresh sea salt particles compared to pure NaCl particles16. This small difference is also visible in this study (Fig. 6). When sea salt is dried (RH < 10%) and additionally heated to 300C, the GF(80%) increases rapidly to values slightly higher than those for fresh sea salt particles. This implies that at 300C, water bound as hydrates in the compounds formed during ageing, but also hydrates present even in the freshly generated sea salt particles are evaporated.
The GF(80%) of particles with Ddry = 200 nm (Fig. 6b) is also affected by the heating process, reaching values comparable to those at the beginning of the experiment in the case of NaCl and slightly higher values for sea salt. In the marine atmosphere, SSA may exist as aqueous phase solution droplets and to investigate its hygroscopicity, for example using an HTDMA, the particles have to be dried most certainly leading to formation of several hydrated salts18. The decreasing hygroscopic growth factors with time for the same dry particle size observed in our study are ascribed to ageing reactions in the aqueous droplets leading to formation of hydrate forming salts and as a result hydrates in the dried particles. There is no real reason to assume that formation of hydrate forming salts would not take place also in the marine boundary layer with time. Since SSA may undergo several RH cycles during their atmospheric transit70, hydrate forming salts are expected to have an effect on the aerosols’ physical and chemical properties and ultimately their role in climate.
Conclusion and implications
The unique setup combining an environmental chamber with a sea spray simulation tank directly targets the evolution of SSA affected by different ageing mechanisms. We follow the common practice of laboratory and field studies and report hygroscopic growth and cloud forming potential based on dried size-selected particles. This study reports key findings on inorganic SSA ageing by exposure to and UV lights, yielding OH radicals, that are crucial in daytime chemical reactions. Our results suggest that ageing processes in aqueous sea spray solution droplets induce surface reactions, which significantly alter the hygroscopic growth and cloud activation potential of the dried particles. In particular we observed an effect on the smaller particles (Ddry = 50 nm), while the effect on larger ones (Ddry = 200 nm) is minimal. The lowering of the growth factor and CCN activation potential after ageing is explained by a change in the amount of water retained under dry conditions as a result of hydrate formation. To relate these results to ageing of SSA under atmospheric conditions four key parameters have to be compared: the prevailing RH, , OH and aerosol number concentrations. During our simulated ageing, the RH was kept at values that are representative for the marine boundary layer (> 60%), while the and aerosol number concentrations were somewhat higher: 150 ppb and 2000–20000 #/ in our experiments vs. typically 10–60 ppb 71 and < 1000 #/72 in the clean marine boundary layer. Although the particle number concentrations and hence particle surface in the chamber was substantially higher compared to the real atmosphere, the water uptake properties of sea salt and NaCl at different concentrations are comparable, suggesting that this did not considerably affect the herein presented results. Based on an experiment monitoring the decay of 1-butanol at elevated RH (60%), 150 ppb and using UV lamps to initiate the photolysis of we estimate the OH concentration to be in the range of molecules/ or lower (for more information see "Materials and methods") and hence values comparable to those found in the troposphere1. This agreement suggests that the measured GF(80%) decay rates are representative of changes occurring over the oceans due to ageing processes and do not need to be scaled to the degree the elevated levels would suggest, to represent in-situ conditions. The GF(80%) decay rates for NaCl and sea salt are presented in Table 1 (see also Fig. S8), as calculated from the linear decrease of each experiment during the first three hours of ageing. The results for sea salt are comparable between the repetitions and the two generation techniques amounting to on average, while a more pronounced and more variable decrease is found for NaCl reaching on average. General Circulation Models (GCM) assuming that sea salt aerosols behave like NaCl will thus overestimate their GF as previously suggested16, but our data suggests in addition that the assumption of similarity to NaCl will be more problematic with time if the models do not also include the ageing processes of sea salt: losses, oxidation and hydration.
The significant change in hygroscopicity, demonstrated in this work, and the formation of hydrate forming salts during atmospheric ageing has not been previously reported but could be of major importance for interpretation of the hygroscopic and cloud activation properties of marine SSA measured over the oceans, since such measurements are typically performed with dried particles as the baseline. The observations in the marine boundary layer state a prevalent MHP GF-group described as GF(90%,100nm) > 1.33, while the SSP group with GF(90%,100nm) > 1.85 occurred only at high wind speeds32,33. By using -Köhler theory49 to recalculate the GFs measured in this laboratory study for sea salt at RH = 90%, values of GFfresh(90%,50nm) = 2.18 ( = 1.20) and GFaged(90%,50nm) = 1.73 ( = 0.59) (after 3 hours of ageing) are found. These results are also in good agreement with -values calculated from SScrit yielding fresh = 0.99 and aged = 0.65 for sea salt particles with Ddry = 50 nm.
When applying the above mentioned GF-categories, the fresh particles would hence be classified as SSP and the aged ones as MHP. Therefore, our results suggest that the dominant MHP-group over the oceans could be explained by the presence of aged inorganic SSA leading to an increased amount of hydrate forming salts within the particles and that these observations are compatible with sea spray being a major aerosol source in marine regions. It has to be noted however, that real SSA is not solely composed of inorganics (like sea salt used in this study) and thus other suggested alternatives comprising the presence of sulphate and organic matter might also play a role. On the other hand the argument that GFs in the MHP-range disprove the presence of sea salt in favour of biogenic sulphates and organics is invalid. Indeed, these compounds could contribute to a lower GF, but the amount of biogenic sulphate and organics change with the biological production and thus with season and location, while sea salt is always present as long as there is some wind33, pointing to aged sea salt being a key explanation for the lower GFs.
We also show that slightly larger particles (Ddry = 200 nm) are not equally affected by the same amount of ageing. The analysis of the chemical composition using microscopy techniques accentuates processes occurring mainly at the surface of the particles, which are more decisive for smaller sizes and could explain the differences found for the two dry particle sizes after ageing, as particles with Ddry = 50 nm are four times as easily changed in bulk composition due to surface processes than 200 nm particles. The new oxygen containing compounds formed in aged particles initially containing only NaCl, are most likely NaOH and Na-chlorates. Both are known to form hydrates at low RH, which strongly influence the hygroscopic properties of particles. By additionally heating the particles to 300C, the initial GF(80%) was restored, suggesting that this temperature was sufficient to decompose the hydrated compounds leaving a particle predominately containing NaCl behind. Hydrates hinder the water uptake as they already contain water molecules in their crystalline structure reducing the amount of water that can be taken up at elevated RH. In the case of the more complex (inorganic) sea salt mixture some of the compounds, especially and , are known to exist in their hydrated form in the atmosphere. The ageing effects on the complex sea salt mixture are comparable to those of NaCl exhibiting a substantial decrease in hygroscopicity and CCN activity and can, again, be reversed by drying and additionally heating the particles before exposing them to high RH. Thus, there is a need to further investigate atmospheric ageing of sea spray aerosol and examine uncertainties arising from likely overestimations of the particles’ hygroscopicity and ability to form CCN in climate models and adapt the parametrizations for sea salt aerosols accordingly.
Materials and methods
Experimental procedure
All experiments were performed at the AURA smog chamber facility47. Before each experiment, the Teflon chamber was filled to approximately 5 m3 with purified dry air provided by passing compressed air through first an active carbon and HEPA filter and second a zero-air generator (Aadeco model 737-14) to clean for volatile organic compounds, particles and residual gases. The chamber was kept at atmospheric pressure and a temperature of 293 K. Two techniques were used to generate SSA: a temperature controlled sea spray simulation tank Ægor23 and an atomiser (TSI model 3076). Ægor was operated with a plunging jet (nozzle diameter 4 mm) using a jet water flow rate of 5 l/min. Sweep air was pushed through the head-space with a flow rate of 10 l/min and transferred to the AURA smog chamber via stainless steel tubing. Transfer continued until the RH inside the Teflon chamber reached RH > 90%. This is above the deliquescence point of the chosen salts and thus it was ensured that the SSA existed in the chamber as aqueous solution droplets. SSA generation with the atomiser was very efficient and hence an external custom-made humidifier was employed to pre-condition the chamber to RH > 70%. Then SSA droplets from the atomiser were injected into the chamber for one minute without drying. In this set-up liquid SSA was ensured by keeping RH above the efflorescence point of the salts (47% for NaCl16,28,73, 50% for sea salt16). For the dry experiments the RH in the chamber was kept close to 0% and SSA was injected using the atomiser. The salts chosen for the experiments were pure NaCl (Sigma Aldrich; purity > 99.5%) and artificial sea salt (Sigma Aldrich S9883; mass fraction: 55% , 31% , 8% , 4% , 1% , 1% , < 1% other; termed sea salt throughout the manuscript). In all experiments the salts were dissolved in MilliQ water (EMD Millipore, 18.2 Mcm at 25C resistivity, 2 ppb TOC) achieving a salinity of 3.5% (by mass).
The experiments proceeded through three stages: In the first stage the smog chamber contained the SSA in either a humid or dry atmosphere. In the second stage approximately 150 ppb of (ozone generator Model 610, Jetlight Company, Inc.) were injected and dark chemistry was allowed to occur. In the third stage, daytime chemistry was simulated by turning on UV lamps photolysing (wavelengths from 300–400 nm) thereby producing OH radicals. and concentrations were continuously monitored using a UV absorption ozone analyser (O342 Module, Environment S.A) and a chemiluminescent monitor (AC32M, Environnement S.A), respectively. The levels were always < 5 ppb. Particle size distributions (Ddry = 10–450 nm) were measured using a scanning mobility particle sizer (SMPS) comprising a Kr-85 neutraliser (TSI 3077A) and an electrostatic classifier (TSI 3082) coupled with a nano water-based condensation particle counter (CPC; TSI 3788), while sizes between D = 0.35–10 m were measured with an optical particle spectrometer (OPS; TSI 3330). A silica gel diffusion dryer was placed in front of each instrument to record dry size distributions. To reconcile SMPS and OPS measurements, the optical diameters determined from the OPS were recalculated for an index of refraction of m = 1.54 representative for NaCl74 and assumed to be the same for the artificial sea salt mixture75.
Hygroscopicity and cloud activation instruments
The hygroscopic growth of particles with Ddry = 50 and 200 nm was measured with a humidified tandem differential mobility analyser (HTDMA; Brechtel, Model 300276). The RH for dry particle measurement was constantly monitored and always kept < 10%, while humid experiments were all carried out at 80% RH. The accuracy of the HTDMA was verified with measurements using ammonium sulphate (Sigma Aldrich, purity > 99.95%). The combination of instrumental uncertainty (estimated to be 2% sizing accuracy and 1% RH uncertainty) and deviation from theoretical values calculated with Köhler theory (max. 2% for the chosen size range) lead to an overall measurement uncertainty of less than 3% in the GF(80%) values. A reliable correction for the shape of the particles was not possible in this analysis as microscopy results revealed an evolution of the particles’ shape with time. Thus, a different correction would be necessary for fresh (more cubic) and aged (more spherical) particles. It should be pointed out that we did not measure the amount of water in the aqueous solution droplets during ageing. Additionally, a thermodenuder (TD; Brechtel, Model 3105) was utilised in combination with the HTDMA. In this set-up the TD was set to a temperature of 300C and placed downstream of the HTDMA drier but upstream of the first DMA. In this way the hygroscopic behaviour of not only dried but also thermally treated particles was analysed.
Hygroscopicity was additionally probed with a commercially available humidified nephelometer system employing a three wavelength nephelometer (Ecotech Pty Ltd., Aurora 4000) together with an especially designed humidification system (aerosol conditioning system (ACS1000) by Ecotech Pty Ltd.). The original set-up was altered to contain solely one nephelometer and RH in the humidifier was alternating between 30% (dry) and 80% (wet) every 20 minutes to measure once dry and once humidified scattering coefficients. The scattering enhancement factor f(RH) can be calculated from the scattering coefficient at dry and elevated RH: f(RH) = /. Measurements using the nephelometer were carried out using the polydisperse size distribution directly obtained from the smog chamber. The nephelometer was calibrated using particle-free air and as a span gas and the system was additionally calibrated with ammonium sulphate (Sigma Aldrich, purity > 99.95%). The observed deliquescence RH coincided with theoretical values within 5%.
The CCN activity of the particles was monitored with a CCN counter (CCN 100, Droplet Measurement Technologies). Measurements were performed using a constant monodisperse diameter of Ddry = 50 nm, selected by a DMA (TSI, model 3081) keeping at sheath to aerosol flow of 1:10. After the DMA the aerosol flow was split between the CCN counter (0.5 l/min) and a CPC (0.3 l/min TSI, Model 3776). The supersaturation of the CCN counter was calibrated using dried ammonium sulphate particles (Sigma Aldrich, purity > 99.9999%) generated by an atomiser according to77, with theoretical values calculated using the E-AIM model (Extended-Aerosol Inorganics Model, http://www.aim.env.uea.ac.uk/aim/aim.php). The calibration curve obtained with ammonium sulphate was further used to infer the uncertainty of SScrit. For this purpose the method described in Harris et al.78 was applied, which is based on the uncertainty of the fit values, i.e. the slope and intercept. Overall, a mean uncertainty of ±0.02% was determined for the measured SScrit values. In the data analysis the first 2 minutes at each supersaturation were omitted to ensure that temperatures in the column had stabilised. For the lowest supersaturation setting, the initial 5 minutes were omitted. The data was averaged on a per minute basis, the activation ratio calculated and normalised to unity to account for different particle losses and counting efficiencies of the CPC and CCN counter. The SScrit was found by fitting a sigmoidal curve in MATLAB, where 50% had activated to CCN. We accounted for multiple charged particles by fitting from the level of the lower plateau in the spectrum following77.
Microscopy
Samples for microscopy analysis were collected on pure carbon coated Ni-pure grids (CF400-NI-UL, EMS) with a Micro INertial Impactor (MINI;79) using one sampling stage with an aerodynamic cut-off diameter of either 50 or 60 nm. A flow of 0.5 l/min was utilised and the sampling time was approximately four minutes. Two samples were taken twice per experiment: two samples before any oxidant was added (fresh aerosol) and two after approximately 3 hours of exposure to OH radicals (aged aerosol). Scanning Transmission Electron Microscopy (STEM; FEI TALOS F200A, Thermo Fischer Scientific) was performed in combination with Energy-Dispersive X-Ray (EDX) detection to retrieve the elemental composition of the SSA and their shape. Carbon was not measured as it was already present on the used TEM grid. The STEM images were obtained using a high angle annular dark field detector (HAADF) and energy dispersive X-ray spectroscopy elemental maps were obtained using a Super-X EDS detector. The measurement of the elemental composition of 50 nm particles was not possible as such particles were not stable enough for this analysis and evaporated under the electron beam.
Estimation of OH radical concentration during the experiments
We performed an additional experiment (3.3.2019) monitoring the decay of 1-butanol at elevated RH (60%), 150 ppb and using the UV lamps in the AURA chamber to initiate the photolysis of leading to the formation of OH. Based on the reaction rate constant for 1-butanol and OH, known to be at 298 K80, we estimate OH concentrations in the range of molecules/ and hence values comparable to those found in the troposphere1. Although the chamber was carefully cleaned after each experiment, a possible influence by Cl radicals generated from leftover salt particles on the walls on the 1-butanol decrease rate cannot be fully excluded which makes the estimate an upper limit. The UV intensity might have varied between experiments due to failing of some of the lamps in the AURA chamber and this might influence the concentration of OH radicals available for reactions. It is, however, difficult to know the UV intensity in each experiment, as they were carried out over several months (April until October 2018). To ensure that this did not have a significant effect on the results, two ageing experiments were repeated with a complete set of new UV lamps (Exp. 9 and 17 in Table 1. An experiment without lamps was also repeated, Exp. 19). The data presented in Figs. S2 and S3, suggest a negligible influence as the results fall in the same range as those from previous experiments. This is also clearly visible in Fig. S8 where the GF decrease rates for particles of Ddry = 50 nm (also presented in Table 1) are displayed and compare well to those recorded in the other experiments.
Supplementary information
Acknowledgements
This research was supported by the Austrian Science Fund (FWF: J 3970-N36), Aarhus University and the Swedish Research Council Formas Project No. 2018-01745-COBACCA, Swedish Research Council VR Project No. 2019-05006. The authors thank Jacques Chevalier and Mohammad Aref Hasen Mamakhel for their help with the STEM-EDX analysis. We are also grateful to Anders Brostrøm for assistance with the MINI sampler and Ecotech, in particular Felicity Sharp, for providing us with the Aurora 4000 nephelometer for this study. We thank the Villum Foundation for support for instrumentation (HTDMA and TD) involved in this study.
Author contributions
B.R., E.D.N. and M.B. designed the research; B.R. performed the experiments and analysed data; A.D. performed a part of the microscopy analysis; A.M. provided the HTDMA instrument; S.C. contributed to the sea spray tank employment and microscopy analysis; P.R. performed the ADCHAM model runs; and B.R., D.N. and M.B. wrote the paper with contributions from S.C., A.D., P.R. and A.M.
Data availability
The data obtained in this study and the codes for the analysis, written in Matlab, are available on request from the first author B.R. The ADCHAM model source code, which was used to analyse the impact of aerosol dynamics, wall losses and dilution effects, can be obtained upon request from P.R.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Bernadette Rosati, Email: bernadette.rosati@chem.au.dk.
Merete Bilde, Email: bilde@chem.au.dk.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-89346-6.
References
- 1.Seinfeld J, Pandis S. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. 3. Wiley; 2016. [Google Scholar]
- 2.de Leeuw G, et al. Production flux of sea spray aerosol. Rev. Geophys. 2011;49:1–39. doi: 10.1029/2010RG000349. [DOI] [Google Scholar]
- 3.Forestieri SD, et al. Temperature and composition dependence of sea spray aerosol production. Geophys. Res. Lett. 2018;45:7218–7225. doi: 10.1029/2018GL078193. [DOI] [Google Scholar]
- 4.Gong SL. A parameterization of sea-salt aerosol source function for sub- and super-micron particles. Glob. Biogeochem. Cycles. 2003 doi: 10.1029/2003GB002079. [DOI] [Google Scholar]
- 5.Mårtensson EM, Nilsson ED, de Leeuw G, Cohen LH, Hansson H-C. Laboratory simulations and parameterization of the primary marine aerosol production. J. Geophys. Res. Atmos. 2003;108:1–12. doi: 10.1029/2002JD002263. [DOI] [Google Scholar]
- 6.Ovadnevaite J, et al. A sea spray aerosol flux parameterization encapsulating wave state. Atmos. Chem. Phys. 2014;14:1837–1852. doi: 10.5194/acp-14-1837-2014. [DOI] [Google Scholar]
- 7.Prather K, et al. Bringing the ocean into the laboratory to probe the chemical complexity of sea spray aerosol. Proc. Natl. Acad. Sci. U.S.A. 2013;110:7550–7555. doi: 10.1073/pnas.1300262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salter ME, Nilsson ED, Butcher A, Bilde M. On the seawater temperature dependence of the sea spray aerosol generated by a continuous plunging jet. J. Geophys. Res. Atmos. 2014;119:9052–9072. doi: 10.1002/2013JD021376. [DOI] [Google Scholar]
- 9.Bertram TH, Cochran RE, Grassian VH, Stone EA. Sea spray aerosol chemical composition: Elemental and molecular mimics for laboratory studies of heterogeneous and multiphase reactions. Chem. Soc. Rev. 2018;47:2374–2400. doi: 10.1039/C7CS00008A. [DOI] [PubMed] [Google Scholar]
- 10.Salter M, et al. Calcium enrichment in sea spray aerosol particles. Geophys. Res. Lett. 2016;43:8277–8285. doi: 10.1002/2016GL070275. [DOI] [Google Scholar]
- 11.Facchini M, et al. Primary submicron marine aerosol dominated by insoluble organic colloids and aggregates. Geophys. Res. Lett. 2008;35:1–5. doi: 10.1029/2008GL034210. [DOI] [Google Scholar]
- 12.Gantt B, Meskhidze N. The physical and chemical characteristics of marine primary organic aerosol: A review. Atmos. Chem. Phys. 2013;13:3979–3996. doi: 10.5194/acp-13-3979-2013. [DOI] [Google Scholar]
- 13.Vaishya A, et al. Bistable effect of organic enrichment on sea spray radiative properties. Geophys. Res. Lett. 2013;40:6395–6398. doi: 10.1002/2013GL058452. [DOI] [Google Scholar]
- 14.Murphy DM, et al. The distribution of sea-salt aerosol in the global troposphere. Atmos. Chem. Phys. 2019;19:4093–4104. doi: 10.5194/acp-19-4093-2019. [DOI] [Google Scholar]
- 15.King S, et al. Investigating primary marine aerosol properties: CCN activity of sea salt and mixed inorganic–organic particles. Environ. Sci. Technol. 2012;46:10405–10412. doi: 10.1021/es300574u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zieger P, et al. Revising the hygroscopicity of inorganic sea salt particles. Nat. Commun. 2017 doi: 10.1038/ncomms15883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasmussen BB, Nguyen QT, Kristensen K, Nielsen LS, Bilde M. What controls volatility of sea spray aerosol? Results from laboratory studies using artificial and real seawater samples. J. Aerosol Sci. 2017;107:134–141. doi: 10.1016/j.jaerosci.2017.02.002. [DOI] [Google Scholar]
- 18.Rosati B, Paul A, Iversen EM, Massling A, Bilde M. Reconciling atmospheric water uptake by hydrate forming salts. Environ. Sci. Process. Impacts. 2020;22:1759–1767. doi: 10.1039/D0EM00179A. [DOI] [PubMed] [Google Scholar]
- 19.IPCC. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. (Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013). Stocker, T.F. and Qin, D. and Plattner, G.-K. and Tignor, M. and Allen, S.K. and Boschung, J. and Nauels, A. and Xia, Y. and Bex, V. and Midgley, P.M.
- 20.Väisänen O, et al. In-cloud measurements highlight the role of aerosol hygroscopicity in cloud droplet formation. Atmos. Chem. Phys. 2016;16:10385–10398. doi: 10.5194/acp-16-10385-2016. [DOI] [Google Scholar]
- 21.Lewis E, Schwartz S. Sea Salt Aerosol Production: Mechanisms, Methods, Measurements and Models. American Geophysical Union; 2004. [Google Scholar]
- 22.Textor C, et al. Analysis and quantification of the diversities of aerosol life cycles within aerocom. Atmos. Chem. Phys. 2006;6:1777–1813. doi: 10.5194/acp-6-1777-2006. [DOI] [Google Scholar]
- 23.Christiansen S, Salter ME, Gorokhova E, Nguyen QT, Bilde M. Sea spray aerosol formation: Laboratory results on the role of air entrainment, water temperature and phytoplankton biomass. Environ. Sci. Technol. 2019 doi: 10.1021/acs.est.9b04078. [DOI] [PubMed] [Google Scholar]
- 24.Hultin KAH, et al. In situ laboratory sea spray production during the Marine Aerosol Production 2006 cruise on the northeastern Atlantic ocean. J. Geophys. Res. Atmos. 2006;115:1–19. [Google Scholar]
- 25.Hultin KA, et al. Aerosol and bacterial emissions from baltic seawater. Atmos. Res. 2011;99:1–14. doi: 10.1016/j.atmosres.2010.08.018. [DOI] [Google Scholar]
- 26.Keene WC, et al. Chemical and physical characteristics of nascent aerosols produced by bursting bubbles at a model air–sea interface. J. Geophys. Res. Atmos. 2007 doi: 10.1029/2007JD008464. [DOI] [Google Scholar]
- 27.Salter ME, et al. An empirically derived inorganic sea spray source function incorporating sea surface temperature. Atmos. Chem. Phys. 2015;15:11047–11066. doi: 10.5194/acp-15-11047-2015. [DOI] [Google Scholar]
- 28.Tang I, Tridico A, Fung K. Thermodynamic and optical properties of sea salt aerosols. J. Geophys. Res. Atmos. 1997;102:23269–23275. doi: 10.1029/97JD01806. [DOI] [Google Scholar]
- 29.Zábori J, et al. Wintertime Arctic ocean sea water properties and primary marine aerosol concentrations. Atmos. Chem. Phys. 2012;12:10405–10421. doi: 10.5194/acp-12-10405-2012. [DOI] [Google Scholar]
- 30.Zábori J, Matisäns M, Krejci R, Nilsson ED, Ström J. Artificial primary marine aerosol production: A laboratory study with varying water temperature, salinity, and succinic acid concentration. Atmos. Chem. Phys. 2012;12:10709–10724. doi: 10.5194/acp-12-10709-2012. [DOI] [Google Scholar]
- 31.Zábori J, et al. Comparison between summertime and wintertime Arctic ocean primary marine aerosol properties. Atmos. Chem. Phys. 2013;13:4783–4799. doi: 10.5194/acp-13-4783-2013. [DOI] [Google Scholar]
- 32.Swietlicki E, et al. Hygroscopic properties of submicrometer atmospheric aerosol particles measured with H-TDMA instruments in various environments—A review. Tellus Ser. B Chem. Phys. Meteorol. 2008;60B:432–469. doi: 10.1111/j.1600-0889.2008.00350.x. [DOI] [Google Scholar]
- 33.Nilsson ED, et al. Turbulent aerosol fluxes over the arctic ocean: 2. Wind-driven sources from the sea. J. Geophys. Res. Atmos. 2001;106:32139–32154. doi: 10.1029/2000JD900747. [DOI] [Google Scholar]
- 34.Gershey RM. Characterization of seawater organic matter carried by bubble-generated aerosols. Limnol. Oceanogr. 1983;28:309–319. doi: 10.4319/lo.1983.28.2.0309. [DOI] [Google Scholar]
- 35.Middlebrook AM, Murphy DM, Thomson DS. Observations of organic material in individual marine particles at cape grim during the first aerosol characterization experiment (ACE 1) J. Geophys. Res. Atmos. 1998;103:16475–16483. doi: 10.1029/97JD03719. [DOI] [Google Scholar]
- 36.O’Dowd C, et al. Biogenically driven organic contribution to marine aerosol. Nature. 2004;431:676–680. doi: 10.1038/nature02959. [DOI] [PubMed] [Google Scholar]
- 37.Ault AP, et al. Size-dependent changes in sea spray aerosol composition and properties with different seawater conditions. Environ. Sci. Technol. 2013;47:5603–5612. doi: 10.1021/es400416g. [DOI] [PubMed] [Google Scholar]
- 38.Cavalli F, et al. Advances in characterization of size-resolved organic matter in marine aerosol over the North Atlantic. J. Geophys. Res. Atmos. 2004 doi: 10.1029/2004JD005137. [DOI] [Google Scholar]
- 39.Orellana MV, et al. Marine microgels as a source of cloud condensation nuclei in the high arctic. Proc. Natl. Acad. Sci. 2011;108:13612–13617. doi: 10.1073/pnas.1102457108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quinn P, et al. Contribution of sea surface carbon pool to organic matter enrichment in sea spray aerosol. Nat. Geosci. 2014;7:228–232. doi: 10.1038/ngeo2092. [DOI] [Google Scholar]
- 41.Fuentes E, Coe H, Green D, McFiggans G. On the impacts of phytoplankton-derived organic matter on the properties of the primary marine aerosol part 2: Composition, hygroscopicity and cloud condensation activity. Atmos. Chem. Phys. 2011;11:2585–2602. doi: 10.5194/acp-11-2585-2011. [DOI] [Google Scholar]
- 42.Oum K, Lakin M, DeHaan D, Brauers T, Finlayson-Pitts B. Formation of molecular chlorine from the photolysis of ozone and aqueous sea-salt particles. Science. 1998;279:74–77. doi: 10.1126/science.279.5347.74. [DOI] [PubMed] [Google Scholar]
- 43.Trueblood JV, et al. Shedding light on photosensitized reactions within marine-relevant organic thin films. ACS Earth Space Chem. 2019;3:1614–1623. doi: 10.1021/acsearthspacechem.9b00066. [DOI] [Google Scholar]
- 44.Jammoul A, Dumas S, D’Anna B, George C. Photoinduced oxidation of sea salt halides by aromatic ketones: A source of halogenated radicals. Atmos. Chem. Phys. 2009;9:4229–4237. doi: 10.5194/acp-9-4229-2009. [DOI] [Google Scholar]
- 45.Knipping EM, et al. Experiments and simulations of ion-enhanced interfacial chemistry on aqueous NaCl aerosols. Science. 2000;288:301–306. doi: 10.1126/science.288.5464.301. [DOI] [PubMed] [Google Scholar]
- 46.Laskin A, et al. Reactions at interfaces as a source of sulfate formation in sea-salt particles. Science. 2003;301:340–344. doi: 10.1126/science.1085374. [DOI] [PubMed] [Google Scholar]
- 47.Kristensen K, Jensen L, Glasius M, Bilde M. The effect of sub-zero temperature on the formation and composition of secondary organic aerosol from ozonolysis of alpha-pinene. Environ. Sci. Process. Impacts. 2017;19:1220–1234. doi: 10.1039/c7em00231a. [DOI] [PubMed] [Google Scholar]
- 48.Wise ME, Martin ST, Russell LM, Buseck PR. Water uptake by NaCl particles prior to deliquescence and the phase rule. Aerosol Sci. Technol. 2008;42:281–294. doi: 10.1080/02786820802047115. [DOI] [Google Scholar]
- 49.Petters MD, Kreidenweis SM. A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos. Chem. Phys. 2007;7:1961–1971. doi: 10.5194/acp-7-1961-2007. [DOI] [Google Scholar]
- 50.De Haan DO, Finlayson-Pitts BJ. Knudsen cell studies of the reaction of gaseous nitric acid with synthetic sea salt at 298 K. J. Phys. Chem. A. 1997;101:9993–9999. doi: 10.1021/jp972450s. [DOI] [Google Scholar]
- 51.Beichert P, Finlayson-Pitts BJ. Knudsen cell studies of the uptake of gaseous HNO and other oxides of nitrogen on solid NaCl: The role of surface-adsorbed water. J. Phys. Chem. 1996;100:15218–15228. doi: 10.1021/jp960925u. [DOI] [Google Scholar]
- 52.Finlayson-Pitts BJ. Reactions at surfaces in the atmosphere: Integration of experiments and theory as necessary (but not necessarily sufficient) for predicting the physical chemistry of aerosols. Phys. Chem. Chem. Phys. 2009;11:7760–7779. doi: 10.1039/B906540G. [DOI] [PubMed] [Google Scholar]
- 53.Kang N, Jackson WA, Dasgupta PK, Anderson TA. Perchlorate production by ozone oxidation of chloride in aqueous and dry systems. Sci. Total Environ. 2008;405:301–309. doi: 10.1016/j.scitotenv.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 54.Jackson WA, Wang S, Rao B, Anderson T, Estrada NL. Heterogeneous production of perchlorate and chlorate by ozone oxidation of chloride: Implications on the source of (per)chlorate in the solar system. ACS Earth Space Chem. 2018;2:87–94. doi: 10.1021/acsearthspacechem.7b00087. [DOI] [Google Scholar]
- 55.Rao B, Anderson T, Redder A, Jackson W. Perchlorate formation by ozone oxidation of aqueous chlorine/oxy-chlorine species: Role of clxoy radicals. Environ. Sci. Technol. 2010;44:2961–2967. doi: 10.1021/es903065f. [DOI] [PubMed] [Google Scholar]
- 56.Rao B, et al. Perchlorate production by photodecomposition of aqueous chlorine solutions. Environ. Sci. Technol. 2012;46:11635–11643. doi: 10.1021/es3015277. [DOI] [PubMed] [Google Scholar]
- 57.Fossum KN, et al. Sea-spray regulates sulfate cloud droplet activation over oceans. npj Clim. Atmos. Sci. 2020;3:1–6. doi: 10.1038/s41612-020-0116-2. [DOI] [Google Scholar]
- 58.Kreidenweis SM, et al. Water activity and activation diameters from hygroscopicity data—Part I: Theory and application to inorganic salts. Atmos. Chem. Phys. 2005;5:1357–1370. doi: 10.5194/acp-5-1357-2005. [DOI] [Google Scholar]
- 59.Christiansen S, et al. Influence of arctic microlayers and algal cultures on sea spray hygroscopicity and the possible implications for mixed-phase clouds. J. Geophys. Res. Atmos. 2020;125:1–16. doi: 10.1029/2020JD032808. [DOI] [Google Scholar]
- 60.Nguyen QT, Kjær KH, Kling KI, Boesen T, Bilde M. Impact of fatty acid coating on the CCN activity of sea salt particles. Tellus B Chem. Phys. Meteorol. 2017;69:1304064. doi: 10.1080/16000889.2017.1304064. [DOI] [Google Scholar]
- 61.Roldin P, et al. Modelling non-equilibrium secondary organic aerosol formation and evaporation with the aerosol dynamics, gas- and particle-phase chemistry kinetic multilayer model adcham. Atmos. Chem. Phys. 2014;14:7953–7993. doi: 10.5194/acp-14-7953-2014. [DOI] [Google Scholar]
- 62.Roldin P, et al. The role of highly oxygenated organic molecules in the boreal aerosol-cloud-climate system. Nat. Commun. 2019;10:4370. doi: 10.1038/s41467-019-12338-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.ten Brink HM. Reactive uptake of HNO and HSO in sea-salt (NaCl) particles. J. Aerosol Sci. 1998;29:57–64. doi: 10.1016/S0021-8502(97)00460-6. [DOI] [Google Scholar]
- 64.Gysel M , Weingartner E, Baltensperger U. Hygroscopicity of aerosol particles at low temperatures. 2. Theoretical and experimental hygroscopic properties of laboratory generated aerosols. Environ. Sci. Technol. 2002;36:63–68. doi: 10.1021/es010055g. [DOI] [PubMed] [Google Scholar]
- 65.Hoffman RC, Laskin A, Finlayson-Pitts BJ. Sodium nitrate particles: Physical and chemical properties during hydration and dehydration, and implications for aged sea salt aerosols. J. Aerosol Sci. 2004;35:869–887. doi: 10.1016/j.jaerosci.2004.02.003. [DOI] [Google Scholar]
- 66.Braitsch O. Salt Deposits Their Origin and Composition. Minerals, Rocks and Inorganic Materials. Springer; 1971. [Google Scholar]
- 67.Hardie LA, Eugster HP. Evaporation of seawater: Calculated mineral sequences. Science. 1980;208:498–500. doi: 10.1126/science.208.4443.498. [DOI] [PubMed] [Google Scholar]
- 68.Siemens PR, Giauque WF. Entropies of the hydrates of sodium hydroxide. II. Low-temperature heat capacities and heats of fusion of NaOH.2HO and NaOH.3.5HO. J. Phys. Chem. 1969;73:149–157. doi: 10.1021/j100721a024. [DOI] [Google Scholar]
- 69.Lide D. CRC Handbook of Chemistry and Physics. 85. CRC Press; 2005. [Google Scholar]
- 70.Estillore AD, et al. Linking hygroscopicity and the surface microstructure of model inorganic salts, simple and complex carbohydrates, and authentic sea spray aerosol particles. Phys. Chem. Chem. Phys. 2017;19:21101–21111. doi: 10.1039/C7CP04051B. [DOI] [PubMed] [Google Scholar]
- 71.Boylan P , Helmig D, Oltmans S. Ozone in the Atlantic ocean marine boundary layer. Elementa. 2015 doi: 10.12952/journal.elementa.000045. [DOI] [Google Scholar]
- 72.Koponen IK, Virkkula A, Hillamo R, Kerminen V-M, Kulmala M. Number size distributions and concentrations of marine aerosols: Observations during a cruise between the english channel and the coast of antarctica. J. Geophys. Res. Atmos. 2002;107:AAC 6–1–AAC 6–8. doi: 10.1029/2002JD002533. [DOI] [Google Scholar]
- 73.Cheng Y, Su H, Koop T, Mikhailov E, Pöschl U. Size dependence of phase transitions in aerosol nanoparticles. Nat. Commun. 2015;6:1–7. doi: 10.1038/ncomms6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Polyanskiy, M. N. Refractive index database. https://refractiveindex.info (2019, accessed 25 January 2019).
- 75.Irshad R, et al. Laboratory measurements of the optical properties of sea salt aerosol. Atmos. Chem. Phys. 2009;9:221–230. doi: 10.5194/acp-9-221-2009. [DOI] [Google Scholar]
- 76.Lopez-Yglesias XF, Yeung MC, Dey SE, Brechtel FJ, Chan CK. Performance evaluation of the Brechtel Mfg. humidified tandem differential mobility analyzer (BMI HTDMA) for studying hygroscopic properties of aerosol particles. Aerosol Sci. Technol. 2014;48:969–980. doi: 10.1080/02786826.2014.952366. [DOI] [Google Scholar]
- 77.Rose D, et al. Calibration and measurement uncertainties of a continuous-flow cloud condensation nuclei counter (DMT-CCNC): CCN activation of ammonium sulfate and sodium chloride aerosol particles in theory and experiment. Atmos. Chem. Phys. 2008;8:1153–1179. doi: 10.5194/acp-8-1153-2008. [DOI] [Google Scholar]
- 78.Harris DC, Lucy CA. Quantitative Chemical Analysis. 9. Freeman Custom Publishing; 2016. [Google Scholar]
- 79.Kandler, K. A miniature impactor for aerosol collection with emphasis on single particle analysis. European Aerosol Conference, Karlsruhe, Germany (T092A04) (2009).
- 80.Yujing M, Mellouki A. Temperature dependence for the rate constants of the reaction of OH radicals with selected alcohols. Chem. Phys. Lett. 2001;333:63–68. doi: 10.1016/S0009-2614(00)01346-4. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data obtained in this study and the codes for the analysis, written in Matlab, are available on request from the first author B.R. The ADCHAM model source code, which was used to analyse the impact of aerosol dynamics, wall losses and dilution effects, can be obtained upon request from P.R.