

Summary
Translocator protein (TSPO, 18 kDa) levels increase in parallel with the evolution of simple steatosis (SS) to nonalcoholic steatohepatitis (NASH) in nonalcoholic fatty liver disease (NAFLD). However, TSPO function in SS and NASH is unknown. Loss of TSPO in hepatocytes in vitro downregulated acetyl-CoA acetyltransferase 2 and increased free cholesterol (FC). FC accumulation induced endoplasmic reticulum stress via IRE1A and protein kinase RNA-like ER kinase/ATF4/CCAAT-enhancer-binding protein homologous protein pathways and autophagy. TSPO deficiency activated cellular adaptive antioxidant protection; this adaptation was lost upon excessive FC accumulation. A TSPO ligand 19-Atriol blocked cholesterol binding and recapitulated many of the alterations seen in TSPO-deficient cells. These data suggest that TSPO deficiency accelerated the progression of SS. In NASH, however, loss of TSPO ameliorated liver fibrosis through downregulation of bile acid synthesis by reducing CYP7A1 and CYP27A1 levels and increasing farnesoid X receptor expression. These studies indicate a dynamic and complex role for TSPO in the evolution of NAFLD.
Subject areas: Molecular biology, Cell biology, Metabolomics
Graphical abstract
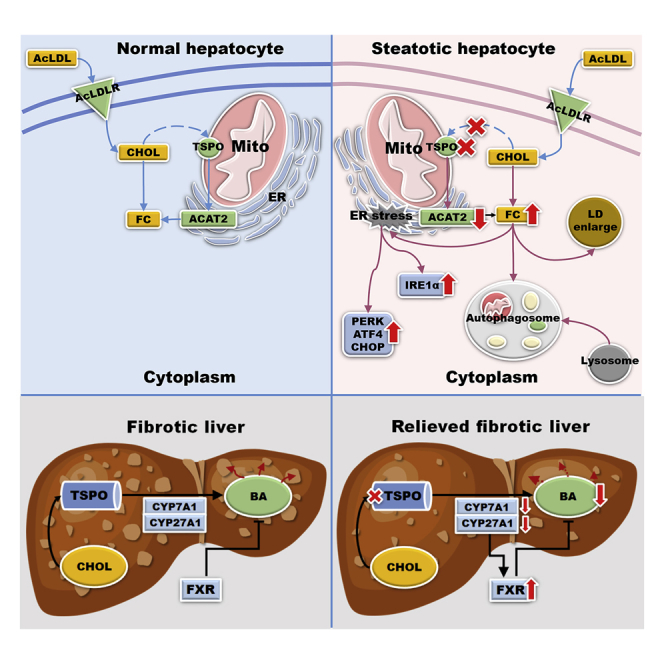
Highlights
-
•
TSPO expression levels correlate with the progression of NAFLD
-
•
TSPO deficiency inhibits ACAT2 leading to FC accumulation
-
•
Loss of TSPO in hepatocytes leads to FC accumulation that promotes simple steatosis
-
•
Loss of TSPO attenuates liver fibrosis via downregulation of bile acid production
Molecular biology; Cell biology; Metabolomics
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a chronic clinicopathological condition associated with significant lipid deposition in hepatocytes and an increased risk of liver injury (Abd El-Kader and El-Den Ashmawy, 2015; Huang et al., 2020). NAFLD encompasses a wide histological spectrum of diseases ranging from simple steatosis (SS) without inflammation or fibrosis to nonalcoholic steatohepatitis (NASH) with inflammation and eventually cirrhosis and ultimately hepatocellular carcinoma (HCC) (Brown and Kleiner, 2016; Kleiner et al., 2005; Takahashi and Fukusato, 2014). In Western countries, NAFLD has become one of the most prevalent chronic liver diseases (Benedict and Zhang, 2017). Therefore, it is of crucial importance to impede SS and NASH progression because these two stages can still be reversed to normal (Kleiner et al., 2005).
Translocator protein (TSPO; 18 kDa), a ubiquitous high-affinity cholesterol- and drug-binding protein located in the outer mitochondrial membrane (OMM), has been well-characterized by its direct or indirect involvement in numerous biological functions, including mitochondrial cholesterol transport, steroid hormone biosynthesis, cell proliferation, apoptosis, porphyrin transport, heme synthesis, and anion transport (Papadopoulos et al., 2006, 2018). In a normal human liver, TSPO expression is low (Papadopoulos et al., 2006; Savino et al., 2016) but increases in NAFLD (Hatori et al., 2015). Recent studies demonstrated serious steatosis aggregates and necroinflammatory infiltration during the progression from NASH to HCC in the higher-uptake regions of 18F-FEDAC, a radiotracer specific for TSPO (Hatori et al., 2014; Xie et al., 2012). Therefore, TSPO has been proposed as a specific molecular imaging biomarker of NAFLD. However, the function of TSPO in the progression of NAFLD is still unknown.
Total cholesterol (TC) is present as unesterified free cholesterol (FC) and cholesteryl ester (CE). Acyl-coenzyme A: cholesterol acyltransferase (ACAT) is an intracellular enzyme that catalyzes the formation of CE from FC and long-chain fatty acyl-CoA (Chang et al., 1998). The ACAT family includes two members, ACAT1 and ACAT2. In the liver, ACAT1 is distributed only in Kupffer cells (Parini et al., 2004), whereas ACAT2 is expressed primarily in hepatocytes (Anderson et al., 1998). Thus, ACAT regulates the balance between FC and CE forms. Any perturbation of this balance will disrupt cholesterol homeostasis and lead to pathological conditions, from hypercholesterolemia (Daniels et al., 2009) to NAFLD (Lee et al., 2009; Tous et al., 2005). Mice fed with a high-fat diet showed FC accumulation in mouse hepatic stellate cells (HSCs), which further sensitized these cells to TGFβ-induced activation, leading to exaggerated liver fibrosis in NASH (Tomita et al., 2014). In addition, after treatment of isolated liver sinusoidal endothelial cells with acetylated LDL (AcLDL) and ACAT inhibitor 58035 (3-[decyldimethylsilyl]-N-[2-{4-methylphenyl}-1-phenylethyl] propenamide), FC but not CE levels were significantly increased in endolysosome-enhanced TLR9-mediated signaling (Teratani et al., 2017), exacerbating the TLR9/inflammasome pathway in the liver. In the HCC cell line Huh7, acute accumulation of FC induced the degradation of perilipin 2 and Rab18-dependent fusion of the endoplasmic reticulum (ER) and lipid droplets (LDs) (Makino et al., 2016), thus perturbing cholesterol metabolism. Increased hepatic FC correlated with disease progression in patients, without CE alteration (Puri et al., 2007). These results indicated that FC accumulation, but not CE, marked NAFLD progression. However, whether TSPO affects NAFLD progression through FC accumulation remains unknown.
In this study, we induced lipid accumulation in vitro by treating Huh7 cells or primary mouse hepatocytes with either AcLDL to increase TC or AcLDL+58035 to induce FC accumulation in the presence or absence of TSPO. We also treated Huh7 cells with 19-Atriol, a ligand binding at the cholesterol recognition amino acid consensus (CRAC) domain of TSPO (Midzak et al., 2011), to inhibit cholesterol binding to assess whether it can reproduce the effects exhibited after loss of TSPO function. We examined FC accumulation, LD alterations, and the TSPO-ACAT2 association. Furthermore, we investigated the effect of FC accumulation on ER stress and autophagy. We also induced in vivo NASH in wild-type and TSPO-knockout (KO) rats using the methionine-choline-deficient (MCD) diet and investigated the role of TSPO in the fibrotic liver.
Results
TSPO levels correlated with the progression for NAFLD
Although TSPO was reported to be a potential indicator for NAFLD progression (Hatori et al., 2014; Xie et al., 2012), there has surprisingly been no comprehensive study of TSPO expression across the spectrum of NAFLD. To that end, we examined TSPO expression at all stages of NAFLD in the human and mouse. The human liver samples were first blind scored using the NAFLD activity score (NAS) system by a pathologist, regardless of gender. A total of 12 samples were randomly picked as representative of different liver stages by NAS grades of normal, steatosis, NASH, and cirrhosis (n = 3 for each stage) for further analysis (Table S1). TSPO immunoreactivity was low in a normal liver but elevated in SS, NASH, and cirrhotic livers (Figure 1A, black arrows). After the removal of areas of blood and vessels by ImageJ software, TSPO signal intensity was significantly higher in patients with steatosis, NASH, and cirrhosis than in controls, and this increase was proportional to the extent of liver damage.
Figure 1.

TSPO is an indicator of the progression of NAFLD
(A) Immunohistochemistry staining showing TSPO levels in human liver paraffin sections (n = 3). The brown color indicates a positive TSPO signal (black arrow, left panel). After the removal of areas of blood and vessels by ImageJ software, quantification showed the TSPO positive signal in each field in normal, steatosis, NASH, and cirrhosis liver (right panel). Scale, 200μm.
(B) Immunofluorescence staining showing TSPO immunoreactivity signal in control (chow diet), steatosis, and NASH mouse models. The upper panels are low magnification, and the lower panels are high magnification of the selected dotted areas. Red colors indicate positive TSPO signal (white arrow). TSPO signals were quantified in control, steatosis, and NASH models. Scale, 40 μm.
(C) qPCR data showing Tspo/Gapdh relative mRNA expression in mouse liver of control, steatosis, and NASH (n = 3). Data are represented as mean ± SD, ∗p < 0.05, ∗∗p < 0.01 by one-way ANOVA.
We assessed TSPO expression in mouse liver sections from different stages of NAFLD generated in mice fed with a diet rich in fructose, palmitate, and cholesterol (Wang et al., 2016). The steatosis group (n = 12) was generated after 20 weeks of feeding with a fructose-palmitate-cholesterol diet (FPC) supplemented with 0.05% cholesterol, and the NASH group (n = 9) was generated after 22 weeks of feeding with a FPC diet supplemented with 1.2% cholesterol. The control group (n = 6) was generated after 20 weeks of feeding with a chow diet. TSPO immunoreactivity was seen by immunofluorescence in control samples (Figure 1B). The TSPO positive signal was significantly higher in steatosis and highest in NASH samples (Figure 1B). qPCR analysis indicated corresponding increases in Tspo mRNA expression in these models, which gradually increased from control (1×) to steatosis (1.99×) to NASH (2.68×) (Figure 1C). Taken together, TSPO expression levels correlated with the progression from control, steatosis, and onto the NASH stage.
FC and TAG accumulation in TSPO-deficient cells after AcLDL+58035 treatment
To examine the role of TSPO in human liver cells, we generated a stable TSPO knockdown (KD) Huh7 hepatocarcinoma cell line by transfection of human TSPO shRNA plasmid or scrambled shRNA plasmid-A (Scram). TSPO was significantly downregulated in TSPO-shRNA-transfected cells compared with Scram-shRNA-transfected cells (Figure 2A). The immunofluorescence staining of TSPO and MitoTracker confirmed reduced TSPO density in mitochondria by shRNA (Figure 2B).
Figure 2.

Total cholesterol or free cholesterol and TAG accumulation in TSPO-deficient cells
(A) Immunoblotting data showing Huh7 TSPO knock down (KD) cells. β-actin is a loading control.
(B) Colocalization of TSPO (green) and MitoTracker (Mito; red) under confocal microscopy. Scale, 10μm.
(C) MTT assay for Scram and KD cells treated with DMSO, AcLDL, or AcLDL+58035 for 24 h.
(D) Determination of TC, FC, and CE content in Scram and TSPO KD Huh7 after treatment with DMSO, AcLDL, and AcLDL+58035 for 24h.
(E) Determination of TC, FC, CE content in WT and TSPO KO primary mouse hepatocytes after treatment with DMSO, AcLDL, and AcLDL+58035 for 24 h.
(F) Determination of TAG in Scram and KD cells after treatment with DMSO, AcLDL, and AcLDL+58035 for 24 h.
(G) Determination of TC, FC, and CE after treatment with DMSO, AcLDL, and AcLDL+19-Atriol for 24 h in Huh7.
(H) MTT assay for Huh7 cells treated with DMSO, AcLDL, and AcLDL+19-Atriol for 24 h.
(I) Measurement of TC in WT and TSPO KO mouse liver by MALDI IMS. Upper: coregistration of MS image with the optical image showing morphological features: The green indicates TC perfusion throughout the liver tissue in the hepatic lobules and around vasculature; the red indicates the distribution of heme molecule from blood cells revealing vasculature. Lower: intensity plots showing cholesterol distribution in the WT and TSPO KO liver. Scale, 5 mm.
(J) Measurement of TAG (50:2) content in the WT and TSPO KO mouse liver by MALDI. Upper: the yellow indicates TAG tissue distribution and the blue shows absence of TAG from respective tissue compartments (blood vessels or biliary ducts). The TAG signal is illustrated as a color gradient from dark blue which represents absence of TAG to yellow which shows strong TAG signal. Lower: intensity plots showing TAG distribution in WT and TSPO KO liver. Scale, 5 mm. Data are represented as mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗P < 0.001 by one-way ANOVA.
Using zinc finger nuclease technology, we previously generated a TSPO null mutant rat (Owen et al., 2017). Serendipitously, we observed abnormal LD accumulation in TSPO homozygous KO rats when compared with WT and heterozygous TSPO rats (Figure S1), implying a liver steatosis-like phenotype in TSPO HO KO rats. This observation prompted us to explore the possible contribution of TSPO to lipid accumulation and NAFLD. First, we demonstrated AcLDL uptake into Huh7 by visualizing the acetylated low-density lipoprotein, DiI complex (Dil-AcLDL) trafficking into the cells (Figure S2A). To assess the effect of TSPO in lipid-loaded Huh7 cells, we treated KD and Scram control Huh7 cells for 24 h with AcLDL to increase TC levels or with AcLDL +58035, the latter being an ACAT inhibitor, to block cholesterol esterification. Cell viability was measured using the MTT assay. Cell viability was significantly decreased in KD cells compared with Scram (Figure 2C). After treatment with AcLDL or AcLDL+58035, cell viability continually declined and decreased more in KD than Scram, suggesting cholesterol-lipid accumulation resulted in cell death and the presence of TSPO was essential for the maintenance of cell viability.
Next, we measured TC, FC, and CE after AcLDL or AcLDL+58035 treatment in Scram and KD cells. The results showed that AcLDL treatment slightly increased TC levels in Scram and KD cells (Figure 2D). However, there was a significant accumulation of FC in KD cells compared with Scram after AcLDL treatment. Surprisingly, TC and FC levels in KD were reduced after treatment with AcLDL+58035 compared with AcLDL (Figure 2D). Considering that cell viability decreased significantly after AcLDL+58035 treatment (Figure 2C), it is likely that excessive FC accumulation caused by 58035 led to cell death. In addition, CE levels did not change in AcLDL+58035 between Scram and KD cells. Meanwhile, we used the same treatments for 24 h on isolated hepatocytes from WT and TSPO global KO mice, previously generated in our laboratory (Fan et al., 2020). We found the treatment of hepatocytes with AcLDL or AcLDL+58035 resulted in significant increase in FC in TSPO KO cells compared with WT (Figure 2E); CE levels did not change with any treatment between WT and KO.
In addition, we isolated human hepatocytes from humanized liver-chimeric mice (Tateno et al., 2015; Yamasaki et al., 2020). After silencing TSPO using human Tspo siRNA (Figure S2B), we examined TC, FC, and CE levels after treatment with DMSO, AcLDL, and AcLDL+58035. The results obtained showed that FC is significantly elevated after silencing TSPO compared with WT cells after AcLDL+58035 (Figure S2C).
Next, we measured triacylglycerol (TAG) levels in KD and Scram cells after AcLDL or AcLDL+58035 treatment. The results showed that TAG was significantly elevated in KD cells versus Scram (Figure 2F). AcLDL or AcLDL+58035 treatment steadily and significantly promoted TAG accumulation in Scram and KD cells, with TAG levels increasing more in KD than in Scram, suggesting that the loss of TSPO enhances TAG accumulation when treated with AcLDL or AcLDL+58035.
To examine whether inhibition of cholesterol binding to TSPO was involved in the observed changes, we treated Huh7 cells with AcLDL or AcLDL+19-Atriol. We previously showed 19-Atriol is a drug ligand inhibiting FC binding at the TSPO CRAC domain (Chung et al., 2013; Midzak et al., 2011). The results showed that TC and FC levels significantly increased with AcLDL+19-Atriol treatment (Figure 2G). Given that FC accumulated after 19-Atriol treatment, it may contribute to reduce cell viability (Figure 2H). Taken together, these results demonstrate that loss of TSPO or inhibition of cholesterol binding to TSPO leads to TC, FC, and TAG accumulation, all hallmarks of hepatic steatosis.
To further examine lipid alteration in vivo after loss of TSPO, we performed matrix-assisted laser desorption/ionization imaging mass spectrometry (MALDI IMS) to assess the levels of cholesterol, TAG, and fatty acids in WT and KO mouse livers. Cholesterol was detected as [cholesterol-H2O + H]+. As seen, cholesterol (green) is evenly distributed through WT and KO livers, while the density is greater in the KO liver (Figure 2I upper). The vessels (red) were revealed by the distribution of heme molecules from blood cells. The intensity plots showed higher levels of cholesterol distribution in the TSPO KO liver compared with WT (Figure 2I lower). TAG was observed both as Na+ and K+ cations, with similar distributions for both. Because of the similarity between ions indicative of TAG (50:2, 52:2, 52:3, 52:4; Figure S3A), we chose TAG (50:2; yellow) for analysis (Figure 2J upper). The intensity plots indicate that TAG (50:2) levels were slightly elevated in the KO liver compared with the WT liver (Figure 2J lower). In addition, neither TAGs nor cholesterol was found in the bile ducts or blood vessels (Figure S3B). Furthermore, some free fatty acids, including palmitoleic/sapienic, linoleic/linoelaidic, oleic/elaidic/vaccenic, eicosapentaenoic, arachidonic, and docosahexaenoic acids were substantially higher in KO livers compared with WT livers (Figure S3C). These observations corroborate the results of cholesterol and TAG elevation in hepatocytes resulting from the loss of TSPO.
Loss of TSPO inhibited ACAT2 expression in mitochondrial-associated ER membranes (MAMs) in a manner independent of TC accumulation
Because FC accumulated in the absence of TSPO, we next determined whether there is an increase in cholesteryl ester hydrolase (CEH), which transforms CE to FC, or a decrease in ACAT2, which transforms FC to CE in TSPO-deficient cells. We detected CEH and ACAT2 levels in Scram and KD cells or WT and KO primary mouse hepatocytes, following the same treatment with AcLDL or AcLDL+58035 for 24 h. In Scram and KD cells, CEH did not change after treatment with DMSO, AcLDL, or AcLDL+58035 (Figure 3A). However, ACAT2 levels were significantly reduced in the absence of TSPO upon AcLDL or AcLDL+58035 treatment compared with their Scram controls (Figure 3B). This was confirmed by confocal imaging microscopy (Figure 3C). In mouse hepatocytes, CEH expression gradually increased with AcLDL or AcLDL+58035 treatment, more so in KO than WT cells (Figure 3D), while ACAT2 expression was downregulated in KO compared with WT, independent of AcLDL or AcLDL+58035 treatment, as shown by immunoblot or immunofluorescence staining (Figures 3E and 3F), respectively. These results indicate that the loss of TSPO inhibited ACAT2 expression in a manner independent of cholesterol accumulation.
Figure 3.

Loss of TSPO inhibited ACAT2 independent of cholesterol accumulation and TSPO-ACAT2 interacted in MAMs
(A) CEH immunoblots from DMSO-, AcLDL-, and AcLDL+58035-treated Scram and TSPO KD Huh7 cells. The numbers under the lanes represent the CEH/GAPDH.
(B) ACAT2 immunoblot from DMSO-, AcLDL-, and AcLDL+58035-treated Scram and TSPO KD Huh7 cells. The bar graph is the fold change of ACAT2/GAPDH.
(C) Immunofluorescence staining of ACAT2 (green) in DMSO-, AcLDL-, and AcLDL+58035-treated Scram and TSPO KD Huh7 cells. Scale, 10 μm.
(D) CEH immunoblot from DMSO-, AcLDL-, and AcLDL+58035-treated WT and TSPO KO primary mouse hepatocytes. The numbers under the lanes represent the CEH/GAPDH.
(E) ACAT2 immunoblot from DMSO-, AcLDL-, and AcLDL+58035-treated WT, TSPO KO primary mouse hepatocytes. The bar graph is the fold change of ACAT2/GAPDH.
(F) Immunofluorescence staining of ACAT2 (green) in DMSO-, AcLDL-, and AcLDL+58035-treated WT and TSPO KO primary mouse hepatocytes Scale, 10μm.
(G) Coimmunoprecipitation of ACAT2-Turbo-GFP and TSPO-Myc and immunoblot with Anti-GFP or Anti-C-Myc in Huh7 cells.
(H) PLA of ACAT2 and TSPO interactions (red spot) in Huh7 cells. Scale, 10 μm. Data are represented as mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by Student's t-test.
Mitochondria are juxtaposed with the ER, forming MAMs (Vance, 2014). This interorganelle interaction is closely associated with cell stress and/or mitochondrial functions (Rizzuto et al., 1998; Zhou et al., 2020). To address whether TSPO, located at the OMM, and ACAT2, residing in the ER membrane, interact directly, we cotransfected a plasmid containing TSPO-Myc and a plasmid containing ACAT2-Turbo-GFP into Huh7 cells and performed coimmunoprecipitation (CO-IP) studies. We validated the overexpression of ACAT2 and TSPO by immunoblotting with anti-ACAT2 and c-Myc antibodies, respectively (Figures S4A and S4B). After immunoprecipitation of anti-Turbo-GFP or c-Myc antibodies from clarified cell lysates, we performed immunoblot analysis using either anti-rabbit C-Myc or Turbo-GFP antibodies. The results show a band of 18–20 kDa, the approximate size of TSPO-Myc, after immunoprecipitation with anti-Turbo-GFP or a 68-kDa ACAT2-GFP band after immunoprecipitation with anti-C-Myc antibody (Figure 3G), suggesting that TSPO interacts directly with ACAT2. To further examine whether endogenous TSPO and ACAT2 interacts, we performed proximity ligation assay (PLA) in Huh7 cells with anti-TSPO (LifeSpan, # LS-B5755-50) and anti-ACAT2 antibodies. The confocal photomicrographs clearly show that endogenous TSPO and ACAT2 interact as seen by the red fluorescence distributed around the nuclei (Figure 3H). These observations provide strong evidence for the direct interaction between TSPO and ACAT2 at the level of MAMs. D'Eletto et al. reported first that TSPO is present in the MAM fraction and its localization is strictly transglutaminase-type-2-dependent in HEK293 cells (D'Eletto et al., 2018). Herein, we confirmed TSPO presence in MAMs and identified TSPO-ACAT2 interactions in hepatocytes.
To detect whether the TSPO-ACAT2 interaction is associated with cholesterol accumulation, we treated the Huh7 cells with DMSO, AcLDL, or AcLDL+58035 and performed immunoblotting with TSPO-Myc after immunoprecipitation with Turbo-GFP. The results show no change in the intensity of TSPO-Myc (Figures S4C and S4D), suggesting that TSPO and ACAT2 interact in a cholesterol-accumulation-independent manner.
To further explore cholesterol metabolism after the loss of TSPO, we examined the transcriptional levels of Acat2 and liver X receptor α (Lxrα) and Lxrα downstream factors HMG-CoA reductase (Hmgcr) and sterol-regulatory-element-binding transcription factor 1 (Srebp-1c). Acat2 mRNA showed decreased trend and Lxrα levels were significantly reduced after loss of TSPO in control (DMSO), AcLDL, and AcLDL+58035 treatments. Although no significant changes were seen in Hmgcr and Srebp-1c levels, the HMGCR protein was reduced in TSPO KO in all treatments examined (Figures S4E and S4F), suggesting that there was no de novo cholesterol synthesis. It was intriguing, however, that Lxrα and Acat2 levels were both reduced in TSPO KO Huh7 compared with WT cells. This finding is in disagreement with a previous report showing that LXRα may negatively regulate ACAT2 expression (Bonamassa and Moschetta, 2013). To assess whether ACAT2 may be degraded in TSPO KO cells, ACAT2 was immunoprecipitated in extracts of mouse hepatocytes from WT and TSPO KO animals, and the precipitates were immunoblotted with an antiubiquitin antibody. The results obtained showed no changes in the ubiquitination of ACAT2 between WT and TSPO KO cells (Figure S4G), indicative of no ubiquitination-associated degradation of ACAT2 in TSPO-deficient cells.
FC accumulation after loss of TSPO enhanced LD enlargement
LDs are universal cellular organelles for the storage of neutral lipids, such as sterol esters and triacylglycerol synthesis (Farese and Walther, 2009). LDs protect cells from lipotoxic effects associated with various disease states, including obesity (Mardinoglu et al., 2015), type 2 diabetes (Sanjabi et al., 2015), liver steatosis (Gluchowski et al., 2017), and atherosclerosis (Plakkal Ayyappan et al., 2016). To detect LD alteration after loss of TSPO, we extracted LDs from DMSO, AcLDL or AcLDL+58035-treated Scram, and KD Huh7 cells. First, we validated the quality of the extracted LDs by immunoblotting for periplin2 (PLIN2), the LD surface structure protein. The results showed that PLIN2 was abundantly expressed in LDs, while no GAPDH expression was found in these LDs-enriched fractions (Figure 4A, left). In contrast, PLIN2 was weakly and GAPDH highly expressed in non-LD fractions (Figure 4A, right). Next, we stained LDs with BODIPY558/568 C12 (Figure 4B upper) and quantified the frequency distribution of LDs (size and number) by ImageJ. The results show that LDs were enlarged after TC or FC accumulation caused by AcLDL or AcLDL+58035 and this enlargement was enhanced after the loss of TSPO (Figure 4B lower).
Figure 4.

LDs enlargement in TSPO-deficient cells
(A) Immunoblot validation of LDs from Scram and TSPO KD cells treated with DMSO, AcLDL, and AcLDL+58035. The left panel indicates PLIN2 expression but no GAPDH in LD-enriched fraction, the right panel indicates non-LDs fraction by weak PLIN2 but normal GAPDH expression.
(B) LDs staining with BODIPY (red color). LDs size-frequency distribution quantification by ImageJ. Scale, 50 μm.
(C) Nile red staining (red color) of WT and TSPO KO primary mouse hepatocytes treated with DMSO, AcLDL, and AcLDL+58035. Nuclei were stained with DAPI. The LDs were quantified as the % area of LDs/cell. Scale, 50 μm.
(D) qPCR data showing relative mRNA levels of Plin1, Plin2, Ppara, Dgat2, Fasn and Scd1 versus Rps18 in WT and TSPO KO primary mouse hepatocytes treated with DMSO, AcLDL, and AcLDL+58035. Data are represented as mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by student's t-test.
To further confirm the effect of TSPO on neutral lipid storage, we treated WT and KO primary mouse hepatocytes with DMSO, AcLDL, or AcLDL+58035 for 24 h, followed by lipid staining with Nile red (Figure 4C upper). The lipid distribution was characterized by percentage (%) area of the stained LDs normalized by the cell number. Compared with WT, the % area of LDs/cell significantly increased in KO. Upon AcLDL or AcLDL+58035 treatment, the % area of LDs/cell in KO showed significant increase compared with WT (Figure 4C lower). The maximum LD size of each group was as follows: WT DMSO, 34.4 μm; WT AcLDL 57.4 μm; WT AcLDL+58035 71 μm, while KO DMSO, 36.5 μm; KO AcLDL, 127.1 μm, and KO AcLDL+58035, 53 μm. These results suggest that loss of TSPO played a role in the determination of LD size and numbers through TC or FC changes.
PLIN1 and PLIN2 levels are increased in steatotic hepatocytes (Fujii et al., 2009; Straub et al., 2008) and in rodents and humans with NAFLD (Straub et al., 2008), promoting triglyceride accumulation (Imai et al., 2007). After AcLDL treatment, we found that there was a significant increase in Plin2 but not Plin1 expression in TSPO KO hepatocytes compared with WT cells (Figure 4D), suggesting the changes seen in LD morphology are due to changes in expression of structural LD proteins. Interestingly, we noticed that Lxrα levels may be positively correlated with Plin2 at the transcriptional level (Figure S4E). Expression of Pparα, responsible for fatty acid β-oxidation, and the lipogenesis genes Dgat2, Fasn, and Scd1 was not significantly changed after cholesterol accumulation (Figure 4D). Together, these results suggest that loss of TSPO augmented LD size partly through changes in genes important for LD structure, which agrees with previous studies (Makino et al., 2016).
We also investigated the effect of inhibiting Niemann-Pick type C1 (NPC1) on LDs in Scram and KD cells. NPC1 is a protein critical for late endosomes and lipid transport, and disruption of NPC1 by the inhibitor U18666A has been shown to result in cholesterol accumulation in lysosomes. We found that LD size increased more significantly in TSPO KD cells versus Scram in response to U18666A treatment (Figure S5A). Likewise, we found LD size is larger in KD cells than Scram after treatment with MβCD (methyl-β-cyclodextrin)/cholesterol, which depletes cholesterol, followed by loading of cholesterol in the presence of 58035 (Figure S5B). However, after treatment with the fatty acid principle monounsaturated oleic acid, we did not see changes in LD size in KD compared with Scram cells (Figure S5C), suggesting the presence of different regulatory mechanisms for TSPO in cholesterol and free fatty acid accumulation.
TSPO deficiency aggravated ER stress via excessive FC
To determine whether loss of TSPO initiated ER stress, we examined the levels of the master regulator of ER stress GRP-78 (glucose-regulated protein 78) and ER stress markers, IRE1α (inositol requiring kinase enzyme 1 alpha), PERK (protein-kinase-RNA-like ER kinase), and ATF (activating transcription factor) in primary mouse hepatocytes after treatment. With DMSO treatment, GRP-78, PERK, ATF4, and its downstream target CCAAT-enhancer-binding protein homologous protein (CHOP) showed higher levels in KO compared with WT cells, indicating ER stress induction after loss of TSPO. After AcLDL or AcLDL+58035 treatment, GRP-78, PERK, ATF4, and CHOP continuously increased in KO compared with WT cells. GRP78 and CHOP levels were quantified in the PERK/ATF4/CHOP cascade (Figure 5A). IRE1α levels increased in KO compared with WT in DMSO. After AcLDL or AcLDL+58035 treatment, IRE1α levels decreased continuously, although they remained higher in KO than those seen in WT, suggesting dynamic regulation. ATF6, another ER-stress-signal-transduction cascade marker, did not change under these conditions (Figure S6). The CHOP levels (green) in WT and KO mouse hepatocytes were confirmed by immunostaining in these treatments (Figure 5B). Given the presence of greater amounts of FC accumulation in TSPO-deficient cells, our data suggest that ER stress induced IRE1A and the PERK/ATF4/CHOP pathway after loss of TSPO and this stress was aggravated by excessive FC accumulation mainly through the PERK/ATF4/CHOP signaling pathway.
Figure 5.

Loss of TSPO enhanced ER stress and adaption mechanism owing to FC accumulation
(A) Immunoblot of GRP-78, PERK, ATF4, CHOP, and IRE1A in WT and TSPO KO primary mouse hepatocytes treated with DMSO, AcLDL, and AcLDL+58035. GRP-78, CHOP, and IRE1A were quantified after DMSO, AcLDL, and AcLDL+58035 treatments.
(B) CHOP immunofluorescence staining (green) in WT and TSPO KO primary mouse hepatocytes treated with DMSO, AcLDL, and AcLDL+58035. Scale, 20 μm.
(C) Immunoblot of catalase and TRX in DMSO-, AcLDL-, and AcLDL+58035-treated WT and TSPO KO primary mouse hepatocytes.
(D) Immunoblot of ACOX1 and PPARα in WT and TSPO KO primary mouse hepatocytes treated with DMSO, AcLDL, and AcLDL+58035.
(E) Immunoblot of GRP-78, IRE1A, CHOP, catalase, SOD1, and TRX in Huh7 cells treated with DMSO, AcLDL, and AcLDL+19-Atriol. The ratio in A, C, and D indicates the densitometric analysis of the tested protein versus the corresponding GAPDH. Data are represented as mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by student's t-test.
The adaptation of antioxidant defense protection was lost upon excessive FC accumulation in TSPO-deficient cells
Hepatic catalase levels indicate antioxidant defense mechanisms. To assess whether TSPO plays a role in this protective system upon cholesterol accumulation, we treated primary mouse hepatocytes with DMSO, AcLDL, or AcLDL+58035. With DMSO, catalase in KO markedly increased compared with the WT, suggesting TSPO plays a protective role in the antioxidant defense process. Moreover, the catalase levels increased more in KO cells compared with WT in response to AcLDL or AcLDL+58035 (Figure 5C). The ratio of KO/WT (numbers under the lanes) after AcLDL treatment reached the highest level (4.3:1). Then, the ratio of KO/WT upon AcLDL+58035 treatment became less compared with the ratio from AcLDL treatment (3.0:1). Thioredoxin (TRX), another antioxidant marker, is playing a key role in redox signaling by removal of reactive oxygen species and nitrogen species (Matsuzawa, 2017). Here, TRX levels increased in KO compared with WT and kept a consistent ratio (1.4:1) in DMSO and AcLDL treatments, but the ratio became less apparent (1.2:1) after AcLDL+58035 treatment (Figure 5C). These data suggest that the antioxidant defense was gradually lost upon excessive FC accumulation caused by AcLDL+58035 after loss of TSPO.
Next, we examined the levels of ACOX1 (acyl-coenzyme A oxidase 1) and PPARα, responsible for fatty acid β-oxidation in mouse hepatocytes. After AcLDL or AcLDL+58035 treatment, the ACOX1 ratio of KO/WT is 2.0:1.0:0.4 and the PPARα ratio of KO/WT is 1.4:1.7:1.3, respectively (Figure 5D). These results imply that increased fatty acid β-oxidation allowed cells to consume accumulated lipid upon DMSO or AcLDL treatment in KO. However, this consumption was compromised upon excessive FC accumulation with AcLDL+58035 treatment in KO. In agreement with this, decreased fatty acid β-oxidation led to accumulation of triglycerides in cells after the loss of TSPO (Figure 2G), which may contribute to the known insulin resistance in NAFLD.
Furthermore, we examined whether 19-Atriol treatment in Huh 7 cells could initiate ER stress or cell stress adaptation mechanisms. The results show that GRP-78, IRE1A, and CHOP levels substantially increased in response to AcLDL+19-Atriol (Figure 5E). Considering the level of FC accumulation after AcLDL+19-Atriol treatment, these results suggest that FC accumulation with 19-Atriol initiated ER stress via the IRE1A and PERK/ATF4/CHOP signaling pathways, the same pathway as loss of TSPO. Meanwhile, catalase, SOD1, and TRX levels were also elevated after AcLDL+19-Atriol treatment (Figure 5E), indicative of cell adaptation to cell stress caused by FC. Taken together, these results suggest that FC accumulation derived from either TSPO deficiency or TSPO inhibition initiated an adaptive antioxidant defense process and fatty acid β-oxidation. However, this adaptation was gradually lost upon excessive FC accumulation as seen with 58035 treatment, which also marks the transition from SS to late SS or early NASH.
TSPO deficiency causes autophagy
Transmission electron microscopy (TEM) was performed on Huh7 Scram and TSPO KD cells. Compared with Scram, the mitochondrial structure in KD cells was disrupted, showing that the inner mitochondrial matrix content disappeared or showed abnormal cristae (Figure 6A red arrows). LD size was enlarged in Scram but more prominent in KD after AcLDL treatment (middle, Figures 6A and S7 red arrow), consistent with our previous observations (Figure 4B). LDs were wrapped tightly by ER in AcLDL-treated Scram cells (Figure 6A black arrowhead, upper middle), suggesting active lipid mobilization at this contact site. Cholesterol accumulation with AcLDL treatment after loss of TSPO caused more autophagosome formation (Figure 6A black arrow, lower middle panel). After treatment with AcLDL+58035, Scram LD numbers increased and sizes became larger (Figure 6A upper right). The treatment of AcLDL+58035 in TSPO KD showed more autophagolysosomes formation (Figure 6A double black arrows, lower right).
Figure 6.

Loss of TSPO caused autophagy owing to FC accumulation
(A) Representative electron micrographs showing the ultrastructure of Scram and TSPO KD cells. Mitochondria (red arrow), autophagosome (black arrow) in TSPO KD after AcLDL treatment and autophagolysosome formation (double black arrows) in TSPO KD after AcLDL+58035 treatment as compared with the corresponding control. Mit, mitochondria; ER, endoplasmic reticulum (black arrowhead); LD, lipid droplet; Pero, peroxisome. Scale, 200 nm.
(B) Immunoblot of LC3 (upper) and LAMP1 (lower) in Scram and TSPO KD cells treated with DMSO, AcLDL, and AcLDL+58035. The ratio indicates the densitometric analysis of LC3-II/GAPDH.
(C and D) Immunoblot of LC3 (C), ATG12 and LAMP2 (D) in WT, and TSPO KO mouse hepatocytes treated with DMSO, AcLDL, and AcLDL+58035. LC3-II was quantified with GAPDH in (C). The ratio indicates the densitometric analysis of LC3-II/GAPDH, ATG12/GAPDH, and LAMP2/GAPDH. Data are represented as mean ± SD, ∗p < 0.05, ∗∗p < 0.01 by one-way ANOVA.
(E) Immunoblot of LC3 in Huh7 cells treated with DMSO, AcLDL, and AcLDL+19-Atriol.
To determine if cholesterol accumulation affected autophagy as seen under TEM, we detected autophagy-specific markers microtubule-associated protein 1 light chain 3 (LC3) and lysosome-associated membrane protein 1 and 2 (LAMP1 and LAMP2, respectively) in Scram and KD cells and in WT and KO primary mouse hepatocytes. LC3-II was upregulated in KD cells and increased with AcLDL and AcLDL+58035 treatment compared to Scram (Figure 6B). LAMP1 did not change with DMSO or AcLDL treatment between Scram and KD but was slightly upregulated with AcLDL+58035 in KD (Figure 6B). The upregulated LC3-II and LAMP1 confirmed elevated autophagolysosome formation in TSPO KD compared with Scram under AcLDL+58035 treatment as observed under TEM (double black arrows, lower right Figure 6A). In mouse hepatocytes, LC3B-II gradually increased in TSPO KO compared with WT upon DMSO, AcLDL, and AcLDL+58035 treatment (Figure 6C). In addition, Atg12, critical for the ubiquitin-like conjugation system of autophagy and targeted to autophagosomes, was upregulated in KO cells compared with WT. Upon AcLDL or AcLDL+58035 treatment, the ratio of ATG12 between KO and WT increased (Figure 6D). In addition, LAMP2 was upregulated 1.5 times in KO primary hepatocytes compared with WT after treatment with DMSO and about 2 times after treatment with AcLDL or AcLDL+58035. These data also indicate that TC and FC accumulation from AcLDL or AcLDL+58035 treatment augmented autophagosome and autophagolysosome formation in TSPO-deficient hepatocytes (Figure 6D).
Moreover, we treated Huh7 cells with AcLDL+19-Atriol and found LC3-II substantially increased compared with Scram and AcLDL-treated cells (Figure 6E). Because AcLDL+19-Atriol led to FC accumulation (Figure 2G), this result may also suggest that FC accumulation derived from TSPO inhibition caused autophagy but need further investigation. Thus, we concluded that loss or inhibition of TSPO altered mitochondrial morphology and enhanced interactions between the ER and LDs. TC accumulation after loss of TSPO resulted in autophagy, and excessive FC accumulation led to more autophagolysosomes.
Loss of TSPO ameliorated liver fibrosis and inflammation in MCD-Induced NASH
To assess whether changes in TSPO expression regulated liver fibrogenesis, we fed WT and Tspo KO rats (Owen et al., 2017) an MCD diet for 2 months, which is enough to generate NASH associated with fibrosis. Serum analysis showed that the circulating cholesterol levels tended to increase, while serum alanine transaminase (ALT) was reduced in Tspo KO MCD as compared with WT MCD rats (Figure 7A). Bile acid (BA) levels were significantly reduced in Tspo KO MCD compared with WT MCD. To compare the transcriptome profiles between Tspo WT and KO rats after a chow diet or MCD, we performed RNA sequencing (RNA-seq) on samples from 4 groups: WT chow, Tspo KO chow, WT MCD, and Tspo KO MCD. Principal component analysis (PCA) highlighted the differences (Figure 7B). Hierarchical clustering analysis further confirmed that each group displayed a distinct gene expression pattern (Figure 7C). The transcriptome changes between these 4 groups were analyzed as scatterplots to display the changes (Figure S8). Ingenuity Pathway Analysis was performed to identify potential canonical pathways, diseases and functions, and gene networks associated with RNA-seq readouts, and the results are listed in Table S2. Comparisons of NASH markers Col1a1 and Acta2 and markers for cholesterol metabolism Cyp27a1, Cyp7a1, and Fgf21 between WT chow and WT MCD and between KO chow and KO MCD were analyzed (Figures S9A and S9B). Tspo levels were increased in MCD compared with a chow diet, suggesting Tspo expression paralleled liver fibrosis development (Figures S9C–S9E). qPCR and immunoblot analyses validated Tspo depletion in KO rats (Figures 7D and 7E). The inflammation markers Lgals1, Tnfα, F4/80, Tlr4, and Slc20a1 (also known as PiT1) were decreased (Figure 7D), suggesting less inflammation in KO than WT after MCD. TNFα (Tumor necrosis factor alpha)was again shown downregulated in Tspo KO MCD compared with WT MCD by immunoblot analysis (Figure 7E). Farnesoid X receptor (Fxr or Nr1h4), a negative regulator of BA formation, was significantly increased in Tspo KO versus WT with MCD (Figure 7D), suggesting less BA synthesis in Tspo KO. Increased levels of FGF21 have been shown to correlate with NAFLD progression (Su et al., 2019), which was seen in WT MCD compared to WT chow (Figure 7D). Immunoblots also showed that actin alpha 2 (ACTA2) was decreased in Tspo KO versus WT with MCD. Responsible for BA synthesis, cholesterol 7 alpha-hydroxylase A1 (CYP7A1) showed a trend to decrease, while CYP27A1 significantly decreased in Tspo KO MCD as compared with WT MCD. However, FXR levels significantly increased in Tspo KO versus WT after MCD (Figure 7E), suggesting reduced BA production in Tspo KO rats compared to WT. Furthermore, Sirius red staining showed significantly reduced collagen fibers in Tspo KO in comparison to WT MCD rats (Figure 7F), indicating less fibrosis in KO MCD compared with WT MCD. Taken together, our results suggest that the loss of TSPO ameliorated liver fibrosis and inflammation through downregulation of BA synthesis.
Figure 7.

Loss of TSPO reduced serum BAs and inflammation and ameliorated liver fibrosis
(A) Serum test of cholesterol, triglyceride, ALT, and total BAs in WT and TSPO KO after MCD feeding.
(B) Schematic summary of PCA in WT and TSPO KO with chow or MCD feeding.
(C) Heatmap of cluster marker genes in WT and TSPO KO with chow or MCD feeding.
(D) qPCR data showing relative mRNA levels of Tspo, Lgals1, Slc20a1, F4/80, Tlr4, Tnfa, Fxr, and Fgf21 versus Rps18 in WT and TSPO KO after MCD feeding.
(E) Immunoblot showing TSPO, ACTA2, CYP71A, CYP27A1, FXR, and TNFα after MCD feeding between WT and TSPO KO. Immunoblots were quantified by ImageJ.
(F) Sirius red staining showing collagen fiber (red color) distribution between WT and TSPO KO after chow and MCD feeding. Quantification of Sirius red staining. Scale, 400 μM. Data are represented as mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by Student's t-test.
Discussion
Herein, we demonstrated several essential roles for TSPO in the progression of NAFLD. Loss of TSPO in hepatocytes triggers the onset of steatosis by TC, FC, and TAG accumulation. Mechanistically, TSPO physically interacts with ACAT2 at MAMs, and loss of TSPO inhibited ACAT2, resulting in FC accumulation in a manner independent of TC accumulation. Moreover, TSPO deficiency induced IRE1A and PERK/ATF4/CHOP pathways in ER stress and autophagy formation and FC accumulation aggravated these events. Meanwhile, TC and FC accumulation after the loss of TSPO led to LD enlargement. Mild TC accumulation after the loss of TSPO initially triggered a cell adaptation mechanism, but excessive FC accumulation caused the loss of this adaptive response. 19-Atriol can specifically block TSPO functions leading to FC accumulation, which phenocopies many biological events in ER stress, autophagy, and antioxidant reaction. In the NASH model, the loss of TSPO ameliorated the liver fibrosis and inflammation by reducing BA production through upregulating FXR and downregulating CYP27A1 and CYP7A1. Collectively, our results suggest that TSPO plays an essential role in all stages of NAFLD progression. The loss of TSPO may initiate SS, but in vivo it also ameliorates liver fibrogenesis and inflammation, halting progression in NAFLD.
TSPO is ubiquitously expressed (Papadopoulos et al., 2006, Papadopoulos et al., 2018; Wyatt et al., 2012). In normal livers, TSPO expression is relatively low but becomes elevated upon liver injuries, such as those in response to carbon tetrachloride (CCl4) or cycloheximide injection (Hatori et al., 2014, 2015; Xie et al., 2012). In this study, we confirmed that TSPO levels were indicative of the progression of NAFLD from patients and rodent samples.
To determine TSPO levels in different cell subtypes of the liver, we isolated hepatocytes, HSCs, and Kupffer cells from rats. Tspo mRNA is mainly found in HSCs and Kupffer cells and in low levels in hepatocytes (Figure S10). However, considering hepatocytes occupy more than 70% of the liver, we specifically investigated TSPO role in the hepatocyte cell line Huh7 cell and in primary mouse hepatocytes after lipid loading to mimic NAFLD.
Under chow diet feeding, we observed lipid accumulation in the TSPO KO rat liver by LDs staining and the TSPO KO mouse liver by MALDI IMS as compared with the controls. Thus, we specifically examined the role of TSPO in hepatocytes for SS progression because there is no (or less) activation of HSCs and Kupffer cells at this stage. Given the role of TSPO in binding and transporting cholesterol into mitochondria, we expected TC accumulation in the absence of TSPO. Surprisingly, we noticed FC elevation in TSPO-deficient cells compared with the control, even in the absence of AcLDL treatment. To gain further insight into the mechanism of FC accumulation in TSPO-deficient cells, we looked at levels of the convertases, CEH, and ACAT2, which ultimately determine the levels of free and esterified cholesterol in hepatocytes (Anderson et al., 1998; Chang et al., 1998; Parini et al., 2004). The loss of TSPO suppressed ACAT2 activity independent of cholesterol accumulation, implying that TSPO deficiency can induce FC accumulation by decreasing ACAT2 conversion of FC to CE. Furthermore, we performed Co-IP and PLA and revealed that the mitochondrial TSPO directly interacts with the ER ACAT2 independent of cholesterol accumulation. These findings provide evidence that the TSPO-ACAT2 interaction in MAMs may be a potential modulator for the TC and FC homeostasis and, furthermore, should have an impact on NAFLD progression.
An inhibitor of ACAT 58035 enhances FC accumulation after AcLDL treatment by blocking FC conversion to CE. However, we did not see a continuous increase in FC in response to AcLDL+58035 compared with AcLDL treatment in Huh7. Instead, both TC and FC content decreased in TSPO KD after AcLDL+58035 treatment compared with AcLDL, which might be due to the decreased cell viability in AcLDL+58035. However, in isolated mouse hepatocytes, TC and FC continuously increased after AcLDL or AcLDL+58035 treatment, which was more striking in TSPO KO cells compared with WT, implying that mouse primary hepatocytes are less susceptible to the impairment caused by FC accumulation than the human Huh7 cell line.
The catalase antioxidant system was triggered further after AcLDL treatment in mouse TSPO KO hepatocytes compared with DMSO, suggesting that the cells adapt to maintain cellular homeostasis in some extent. However, this adaptation was lost under additional stress as seen with FC accumulation after treatment with AcLDL+58035 in TSPO-deficient hepatocytes. Previous studies reported that β-oxidation, mitochondrial respiration, antioxidant reactions, and TCA cycle flux were induced in mouse models with nutritional overload, as well as in human subjects with obesity and SS (Iozzo et al., 2010; Koliaki et al., 2015; Miele et al., 2003; Patterson et al., 2016; Satapati et al., 2012; Sunny et al., 2011). ACOX1 is a rate-limiting enzyme in the peroxisomal β-oxidation pathway (Moreno-Fernandez et al., 2018), and PPARα regulates lipid transport and metabolism chiefly through the activation of the mitochondrial and peroxisomal fatty acid β-oxidation pathways (Tyagi et al., 2011). Mitochondrial fatty acid oxidation stimulation is enhanced upon the upregulation of ACOX1 and PPARα by a high-fat diet or the dysregulation of lipid metabolism (Eccleston et al., 2011; Sunny et al., 2011). We showed increased ACOX1 and PPARα levels in TSPO KO cells compared with controls after AcLDL treatment. However, this ratio did not increase more but rather decreased after AcLDL+58035 treatment compared with AcLDL. Taken all these observations into account, our results suggest that mild lipid accumulation primed antioxidant defense and free fatty acid oxidation, but excessive lipid overload is deleterious to these protective systems. Thus, FC accumulation resulting from a disturbed cholesterol balance in TSPO-deficient cells promotes the progression of SS to a later stage.
TSPO was identified as a novel element in the regulation of mitochondrial quality control by autophagy. Gatliff et al. demonstrated that this process depended upon the level of voltage-dependent anion channel 1 (VDAC1) expression; a higher TSPO/VDAC1 ratio leads to the reduction of the autophagic removal of mitochondria by limiting PINK1/PARK2-mediated mitochondrial ubiquitination via periorganelle accumulation of reactive oxygen species (ROS). On the other hand, a reduced TSPO/VDAC1 ratio leads to the induction of mitophagy (a specific autophagy with mitochondria) by reducing ROS production to promote PARK2-mediated ubiquitination of proteins and, thus, removal of dysfunctional mitochondria, allowing for the maintenance of cellular functions (Gatliff et al., 2014). Herein, we found that lipid accumulation (cholesterol and TAG) in TSPO-deficient cells promoted autophagy. In agreement with this, Kim et al. reported that ablation of TSPO in mouse hypothalamic glial cells elicited AMPK-dependent lipophagy (autophagic degradation of intracellular LDs), breaking down LDs into free fatty acids, thereby elevating ATP in a lipid stimulus (Kim et al., 2020). Whether there is an induction of lipophagy as a consequence of lipid accumulation in TSPO-deficient cells remains to be investigated.
BAs are the final steroid metabolites of cholesterol utilization in the liver (Hanukoglu, 1992). Many BAs are highly cytotoxic (Li and Apte, 2015), and therefore, BA synthesis is under tight regulatory control. In patients with NAFLD, BA homeostasis is disrupted and worsens with progression of the disease (Aranha et al., 2008). In contrast, stabilization of BA levels to normal significantly improved NALFD and type 2 diabetes mellitus (Huang et al., 2019). In our MCD NASH study, serum BA levels were suppressed in TSPO KO compared with WT rats. CYP7A1 and CYP27A1, a cholesterol 7α-hydroxylase responsible for the classic pathway and a steroid 27-hydroxylase responsible for the alternative pathway for BA synthesis, were both suppressed in the KO MCD liver compared with the WT MCD liver. The BA level was reduced by 66.7% and 73% in Cyp7a1−/− and Cyp27a1−/− mice and even more in double-knockout (Cyp7a1−/−; Cyp27a1−/−) mice (Rizzolo et al., 2019), indicative of the critical roles of these two hydroxylases in the BA metabolism. In addition, levels of FXR, a negative regulator of BA production and a BA receptor, were significantly increased in KO MCD rats compared with WT MCD. Recently, many emerging and promising therapies for NASH are designed as FXR agonists, for instance, LJN452 (Tropifexor, Novartis) (Tully et al., 2017), GS-9674 (Gilead) (Trauner et al., 2019), and Ocaliva (Intercept) (Samur et al., 2017). Ocaliva (obeticholic acid) is in phase III clinical trials for its ability to improve cholestasis liver disease and NASH by suppressing BA production in the liver, thus reducing the exposure of the liver to toxic levels of BAs (Donkers et al., 2019). Our data also showed that loss of TSPO ameliorated liver fibrosis through downregulation of BA synthesis by reducing CYP7A1 and CYP27A1 and increasing FXR expression, part of the same signaling pathway affected by Ocaliva. These data suggest that TSPO might be a potential therapeutic target for the suppression of NASH.
Despite the unequivocal evidence for the essential role of TSPO in NAFLD progression, several puzzling features remain. TSPO is a ubiquitous protein expressed in hepatocytes, HSCs, and Kupffer cells. Our study demonstrated that loss of TSPO in hepatocytes facilitated SS progression owing to FC and TAG accumulation. This model was simulated by the in vitro hepatocyte study in two aspects. First, in the SS stage, HSCs and Kupffer cells are not or less activated. Second, hepatocytes occupy most of liver parenchyma. However, it is still uncertain whether the results obtained from the hepatocyte studies depleted in TSPO can reflect the reality of SS in the liver. Therefore, to assess the liver phenotype in an SS animal model caused by high-fat diet feeding after depletion of TSPO to recapitulate the TSPO deficiency in hepatocytes is essential. In the MCD NASH model, the loss of TSPO function improved liver fibrogenesis by reducing BA formation. This discrepancy of the TSPO role between SS and NASH suggests distinct roles for TSPO dependent on liver cell type and stages of NAFLD. In the MCD NASH model, the reduced fibrosis and inflammation after the loss of TSPO is presumably mediated by TSPO deficiency in HSCs and Kupffer cells, but not hepatocytes. Thus, using HSCs or cocultures of hepatocytes and HSCs or Kupffer cells may be alternative strategies to tease out the cellular basis for TSPO effects in NASH.
Limitations of the study
Although the small cohort of patient samples used is a limitation in our study, the results obtained showed statistical significance between each stage of NAFLD. Moreover, the results obtained are in line with the observations made using labeled TSPO ligands to image TSPO expression in patients with NAFLD (Hatori et al., 2014; Xie et al., 2012). Thus, these human data provide the link between the published imaging studies and the animal models explored herein.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Vassilios Papadopoulos: Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, Los Angeles, CA 90089, USA; vpapadop@usc.edu.
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA-seq data were deposited in the Gene Expression Omnibus under accession number GSE138666. All data supporting the present study are available from the corresponding author on request.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Cheng Ji at USC (University of Southern California) for the stimulating discussions on cell stress and autophagy, Dr. Junji Watanabe (USC) for help with confocal microscopy and MALDI coordination, Mr. Anthony Rodriguez (USC) for technical assistance with transmission electron microscopy and the Liver Histology Core of the USC Research Center for Liver Diseases for histology services (NIH grant No. P30 DK048522). We also thank Ms. Melanie Galano for critical proofreading of the manuscript. This work was supported by grants from the Canadian Institutes of Health Research (MOP125983 and PJT148659), funds from the USC School of Pharmacy and the John Stauffer Dean's Chair in Pharmaceutical Sciences at USC, and the National Institutes of Health (R21AA027222).
Author contributions
Conceptualization, L.Y., and P.V.; Methodology, L.Y., W,J., S.C., and P.V.; Investigation, L.Y., C,L., L.L., S.C., P.S., L,A., M,P., W.H., W,J., S.C., G.S., C.G., and H.T.; Writing – Original Draft, L.Y., C.L., L.L., and P.V.; Writing –Review & Editing, L.Y., and P.V.; Funding Acquisition, P.V.; Resources, P.S., L,A., M,P., I,Y., S.T., G,L., and R.H.; Supervision, F,J., C.M., B,S., A,K., and P.V. All authors revised the article critically for important intellectual content. All authors have read and approved the final version of the manuscript.
Declaration of interests
Dr. Yuji Ichida is an employee of PhoenixBio, Co., Ltd, Higashi-Hiroshima, Hiroshima, Japan; Dr. Cristina I. Silvescu is an employee of Bruker Daltonics, Billerica, MA 01821, USA; Dr. Jeremy J. Wolff was an employee of Bruker Daltonics, Billerica, MA 01821, USA, when this work was done.
Published: May 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102457.
Supplemental information
References
- Abd El-Kader S.M., El-Den Ashmawy E.M. Non-alcoholic fatty liver disease: the diagnosis and management. World J. Hepatol. 2015;7:846–858. doi: 10.4254/wjh.v7.i6.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R.A., Joyce C., Davis M., Reagan J.W., Clark M., Shelness G.S., Rudel L.L. Identification of a form of acyl-CoA:cholesterol acyltransferase specific to liver and intestine in nonhuman primates. J. Biol. Chem. 1998;273:26747–26754. doi: 10.1074/jbc.273.41.26747. [DOI] [PubMed] [Google Scholar]
- Aranha M.M., Cortez-Pinto H., Costa A., da Silva I.B., Camilo M.E., de Moura M.C., Rodrigues C.M. Bile acid levels are increased in the liver of patients with steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2008;20:519–525. doi: 10.1097/MEG.0b013e3282f4710a. [DOI] [PubMed] [Google Scholar]
- Benedict M., Zhang X. Non-alcoholic fatty liver disease: an expanded review. World J. Hepatol. 2017;9:715–732. doi: 10.4254/wjh.v9.i16.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonamassa B., Moschetta A. Atherosclerosis: lessons from LXR and the intestine. Trends Endocrinol. Metab. 2013;24:120–128. doi: 10.1016/j.tem.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Brown G.T., Kleiner D.E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism. 2016;65:1080–1086. doi: 10.1016/j.metabol.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.C., Lee C.Y., Chang E.T., Cruz J.C., Levesque M.C., Chang T.Y. Recombinant acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) purified to essential homogeneity utilizes cholesterol in mixed micelles or in vesicles in a highly cooperative manner. J. Biol. Chem. 1998;273:35132–35141. doi: 10.1074/jbc.273.52.35132. [DOI] [PubMed] [Google Scholar]
- Chung J.Y., Chen H., Midzak A., Burnett A.L., Papadopoulos V., Zirkin B.R. Drug ligand-induced activation of translocator protein (TSPO) stimulates steroid production by aged brown Norway rat Leydig cells. Endocrinology. 2013;154:2156–2165. doi: 10.1210/en.2012-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Eletto M., Rossin F., Occhigrossi L., Farrace M.G., Faccenda D., Desai R., Marchi S., Refolo G., Falasca L., Antonioli M. Transglutaminase type 2 regulates ER-mitochondria contact sites by interacting with GRP75. Cell Rep. 2018;25:3573–3581 e3574. doi: 10.1016/j.celrep.2018.11.094. [DOI] [PubMed] [Google Scholar]
- Daniels T.F., Killinger K.M., Michal J.J., Wright R.W., Jr., Jiang Z. Lipoproteins, cholesterol homeostasis and cardiac health. Int. J. Biol. Sci. 2009;5:474–488. doi: 10.7150/ijbs.5.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donkers J.M., Roscam Abbing R.L.P., van de Graaf S.F.J. Developments in bile salt based therapies: a critical overview. Biochem. Pharmacol. 2019;161:1–13. doi: 10.1016/j.bcp.2018.12.018. [DOI] [PubMed] [Google Scholar]
- Eccleston H.B., Andringa K.K., Betancourt A.M., King A.L., Mantena S.K., Swain T.M., Tinsley H.N., Nolte R.N., Nagy T.R., Abrams G.A. Chronic exposure to a high-fat diet induces hepatic steatosis, impairs nitric oxide bioavailability, and modifies the mitochondrial proteome in mice. Antioxid. Redox Signal. 2011;15:447–459. doi: 10.1089/ars.2010.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Campioli E., Sottas C., Zirkin B., Papadopoulos V. Amhr2-Cre-Mediated global Tspo knockout. J. Endocr. Soc. 2020;4 doi: 10.1210/jendso/bvaa001. bvaa001–029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farese R.V., Jr., Walther T.C. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell. 2009;139:855–860. doi: 10.1016/j.cell.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H., Ikura Y., Arimoto J., Sugioka K., Iezzoni J.C., Park S.H., Naruko T., Itabe H., Kawada N., Caldwell S.H. Expression of perilipin and adipophilin in nonalcoholic fatty liver disease; relevance to oxidative injury and hepatocyte ballooning. J. Atheroscler. Thromb. 2009;16:893–901. doi: 10.5551/jat.2055. [DOI] [PubMed] [Google Scholar]
- Gatliff J., East D., Crosby J., Abeti R., Harvey R., Craigen W., Parker P., Campanella M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy. 2014;10:2279–2296. doi: 10.4161/15548627.2014.991665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluchowski N.L., Becuwe M., Walther T.C., Farese R.V., Jr. Lipid droplets and liver disease: from basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017;14:343–355. doi: 10.1038/nrgastro.2017.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanukoglu I. Steroidogenic enzymes: structure, function, and role in regulation of steroid hormone biosynthesis. J. Steroid Biochem. Mol. Biol. 1992;43:779–804. doi: 10.1016/0960-0760(92)90307-5. [DOI] [PubMed] [Google Scholar]
- Hatori A., Yui J., Xie L., Kumata K., Yamasaki T., Fujinaga M., Wakizaka H., Ogawa M., Nengaki N., Kawamura K. Utility of translocator protein (18 kDa) as a molecular imaging biomarker to monitor the progression of liver fibrosis. Sci. Rep. 2015;5:17327. doi: 10.1038/srep17327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatori A., Yui J., Xie L., Yamasaki T., Kumata K., Fujinaga M., Wakizaka H., Ogawa M., Nengaki N., Kawamura K. Visualization of acute liver damage induced by cycloheximide in rats using PET with [(18)F]FEDAC, a radiotracer for translocator protein (18 kDa) PLoS One. 2014;9:e86625. doi: 10.1371/journal.pone.0086625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.H., Lee W.J., Chen S.C., Chen T.F., Lee S.D., Chen C.Y. Bile acid and fibroblast growth factor 19 regulation in obese diabetics, and non-alcoholic fatty liver disease after sleeve gastrectomy. J. Clin. Med. 2019;8:815. doi: 10.3390/jcm8060815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T.D., Behary J., Zekry A. Non-alcoholic fatty liver disease: a review of epidemiology, risk factors, diagnosis and management. Intern. Med. J. 2020;50:1038–1047. doi: 10.1111/imj.14709. [DOI] [PubMed] [Google Scholar]
- Imai Y., Varela G.M., Jackson M.B., Graham M.J., Crooke R.M., Ahima R.S. Reduction of hepatosteatosis and lipid levels by an adipose differentiation-related protein antisense oligonucleotide. Gastroenterology. 2007;132:1947–1954. doi: 10.1053/j.gastro.2007.02.046. [DOI] [PubMed] [Google Scholar]
- Iozzo P., Bucci M., Roivainen A., Nagren K., Jarvisalo M.J., Kiss J., Guiducci L., Fielding B., Naum A.G., Borra R. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology. 2010;139:846–856, 856 e841-846. doi: 10.1053/j.gastro.2010.05.039. [DOI] [PubMed] [Google Scholar]
- Kim S., Kim N., Park S., Jeon Y., Lee J., Yoo S.J., Lee J.W., Moon C., Yu S.W., Kim E.K. Tanycytic TSPO inhibition induces lipophagy to regulate lipid metabolism and improve energy balance. Autophagy. 2020;16:1200–1220. doi: 10.1080/15548627.2019.1659616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W., Ferrell L.D., Liu Y.C., Torbenson M.S., Unalp-Arida A. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- Koliaki C., Szendroedi J., Kaul K., Jelenik T., Nowotny P., Jankowiak F., Herder C., Carstensen M., Krausch M., Knoefel W.T. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–746. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Lee L., Alloosh M., Saxena R., Van Alstine W., Watkins B.A., Klaunig J.E., Sturek M., Chalasani N. Nutritional model of steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Hepatology. 2009;50:56–67. doi: 10.1002/hep.22904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Apte U. Bile acid metabolism and signaling in cholestasis, inflammation, and cancer. Adv. Pharmacol. 2015;74:263–302. doi: 10.1016/bs.apha.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino A., Hullin-Matsuda F., Murate M., Abe M., Tomishige N., Fukuda M., Yamashita S., Fujimoto T., Vidal H., Lagarde M. Acute accumulation of free cholesterol induces the degradation of perilipin 2 and Rab18-dependent fusion of ER and lipid droplets in cultured human hepatocytes. Mol. Biol. Cell. 2016;27:3293–3304. doi: 10.1091/mbc.E15-10-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinoglu A., Heiker J.T., Gartner D., Bjornson E., Schon M.R., Flehmig G., Kloting N., Krohn K., Fasshauer M., Stumvoll M. Extensive weight loss reveals distinct gene expression changes in human subcutaneous and visceral adipose tissue. Sci. Rep. 2015;5:14841. doi: 10.1038/srep14841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa A. Thioredoxin and redox signaling: roles of the thioredoxin system in control of cell fate. Arch. Biochem. Biophys. 2017;617:101–105. doi: 10.1016/j.abb.2016.09.011. [DOI] [PubMed] [Google Scholar]
- Midzak A., Akula N., Lecanu L., Papadopoulos V. Novel androstenetriol interacts with the mitochondrial translocator protein and controls steroidogenesis. J. Biol. Chem. 2011;286:9875–9887. doi: 10.1074/jbc.M110.203216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele L., Grieco A., Armuzzi A., Candelli M., Forgione A., Gasbarrini A., Gasbarrini G. Hepatic mitochondrial beta-oxidation in patients with nonalcoholic steatohepatitis assessed by 13C-octanoate breath test. Am. J. Gastroenterol. 2003;98:2335–2336. doi: 10.1111/j.1572-0241.2003.07725.x. [DOI] [PubMed] [Google Scholar]
- Moreno-Fernandez M.E., Giles D.A., Stankiewicz T.E., Sheridan R., Karns R., Cappelletti M., Lampe K., Mukherjee R., Sina C., Sallese A. Peroxisomal beta-oxidation regulates whole body metabolism, inflammatory vigor, and pathogenesis of nonalcoholic fatty liver disease. JCI Insight. 2018;3:e93626. doi: 10.1172/jci.insight.93626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D.R., Fan J., Campioli E., Venugopal S., Midzak A., Daly E., Harlay A., Issop L., Libri V., Kalogiannopoulou D. TSPO mutations in rats and a human polymorphism impair the rate of steroid synthesis. Biochem. J. 2017;474:3985–3999. doi: 10.1042/BCJ20170648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos V., Baraldi M., Guilarte T.R., Knudsen T.B., Lacapere J.J., Lindemann P., Norenberg M.D., Nutt D., Weizman A., Zhang M.R. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006;27:402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V., Fan J., Zirkin B. Translocator protein (18 kDa): an update on its function in steroidogenesis. J. Neuroendocrinol. 2018;30:e12500. doi: 10.1111/jne.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parini P., Davis M., Lada A.T., Erickson S.K., Wright T.L., Gustafsson U., Sahlin S., Einarsson C., Eriksson M., Angelin B. ACAT2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation. 2004;110:2017–2023. doi: 10.1161/01.CIR.0000143163.76212.0B. [DOI] [PubMed] [Google Scholar]
- Patterson R.E., Kalavalapalli S., Williams C.M., Nautiyal M., Mathew J.T., Martinez J., Reinhard M.K., McDougall D.J., Rocca J.R., Yost R.A. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol. Endocrinol. Metab. 2016;310:E484–E494. doi: 10.1152/ajpendo.00492.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plakkal Ayyappan J., Paul A., Goo Y.H. Lipid droplet-associated proteins in atherosclerosis (Review) Mol. Med. Rep. 2016;13:4527–4534. doi: 10.3892/mmr.2016.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri P., Baillie R.A., Wiest M.M., Mirshahi F., Choudhury J., Cheung O., Sargeant C., Contos M.J., Sanyal A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081–1090. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- Rizzolo D., Buckley K., Kong B., Zhan L., Shen J., Stofan M., Brinker A., Goedken M., Buckley B., Guo G.L. Bile acid homeostasis in a cholesterol 7alpha-hydroxylase and sterol 27-hydroxylase double knockout mouse model. Hepatology. 2019;70:389–402. doi: 10.1002/hep.30612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R., Pinton P., Carrington W., Fay F.S., Fogarty K.E., Lifshitz L.M., Tuft R.A., Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Samur S., Klebanoff M., Banken R., Pratt D.S., Chapman R., Ollendorf D.A., Loos A.M., Corey K., Hur C., Chhatwal J. Long-term clinical impact and cost-effectiveness of obeticholic acid for the treatment of primary biliary cholangitis. Hepatology. 2017;65:920–928. doi: 10.1002/hep.28932. [DOI] [PubMed] [Google Scholar]
- Sanjabi B., Dashty M., Ozcan B., Akbarkhanzadeh V., Rahimi M., Vinciguerra M., van Rooij F., Al-Lahham S., Sheedfar F., van Kooten T.G. Lipid droplets hypertrophy: a crucial determining factor in insulin regulation by adipocytes. Sci. Rep. 2015;5:8816. doi: 10.1038/srep08816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satapati S., Sunny N.E., Kucejova B., Fu X., He T.T., Mendez-Lucas A., Shelton J.M., Perales J.C., Browning J.D., Burgess S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012;53:1080–1092. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savino S., Denora N., Iacobazzi R.M., Porcelli L., Azzariti A., Natile G., Margiotta N. Synthesis, characterization, and cytotoxicity of the first oxaliplatin Pt(IV) derivative having a TSPO ligand in the axial position. Int. J. Mol. Sci. 2016;17:1010. doi: 10.3390/ijms17071010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub B.K., Stoeffel P., Heid H., Zimbelmann R., Schirmacher P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology. 2008;47:1936–1946. doi: 10.1002/hep.22268. [DOI] [PubMed] [Google Scholar]
- Su X., Kong Y., Peng D. Fibroblast growth factor 21 in lipid metabolism and non-alcoholic fatty liver disease. Clin. Chim. Acta. 2019;498:30–37. doi: 10.1016/j.cca.2019.08.005. [DOI] [PubMed] [Google Scholar]
- Sunny N.E., Parks E.J., Browning J.D., Burgess S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–810. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y., Fukusato T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2014;20:15539–15548. doi: 10.3748/wjg.v20.i42.15539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno C., Kawase Y., Tobita Y., Hamamura S., Ohshita H., Yokomichi H., Sanada H., Kakuni M., Shiota A., Kojima Y. Generation of novel chimeric mice with humanized livers by using hemizygous cDNA-uPA/SCID mice. PLoS One. 2015;10:e0142145. doi: 10.1371/journal.pone.0142145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teratani T., Tomita K., Suzuki T., Furuhashi H., Irie R., Hida S., Okada Y., Kurihara C., Ebinuma H., Nakamoto N. Free cholesterol accumulation in liver sinusoidal endothelial cells exacerbates acetaminophen hepatotoxicity via TLR9 signaling. J. Hepatol. 2017;67:780–790. doi: 10.1016/j.jhep.2017.05.020. [DOI] [PubMed] [Google Scholar]
- Tomita K., Teratani T., Suzuki T., Shimizu M., Sato H., Narimatsu K., Okada Y., Kurihara C., Irie R., Yokoyama H. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology. 2014;59:154–169. doi: 10.1002/hep.26604. [DOI] [PubMed] [Google Scholar]
- Tous M., Ferre N., Camps J., Riu F., Joven J. Feeding apolipoprotein E-knockout mice with cholesterol and fat enriched diets may be a model of non-alcoholic steatohepatitis. Mol. Cell. Biochem. 2005;268:53–58. doi: 10.1007/s11010-005-2997-0. [DOI] [PubMed] [Google Scholar]
- Trauner M., Gulamhusein A., Hameed B., Caldwell S., Shiffman M.L., Landis C., Eksteen B., Agarwal K., Muir A., Rushbrook S. The nonsteroidal farnesoid X receptor agonist cilofexor (GS-9674) improves markers of cholestasis and liver injury in patients with primary sclerosing cholangitis. Hepatology. 2019;70:788–801. doi: 10.1002/hep.30509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully D.C., Rucker P.V., Chianelli D., Williams J., Vidal A., Alper P.B., Mutnick D., Bursulaya B., Schmeits J., Wu X. Discovery of tropifexor (LJN452), a highly potent non-bile acid FXR agonist for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH) J. Med. Chem. 2017;60:9960–9973. doi: 10.1021/acs.jmedchem.7b00907. [DOI] [PubMed] [Google Scholar]
- Tyagi S., Gupta P., Saini A.S., Kaushal C., Sharma S. The peroxisome proliferator-activated receptor: a family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011;2:236–240. doi: 10.4103/2231-4040.90879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance J.E. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta. 2014;1841:595–609. doi: 10.1016/j.bbalip.2013.11.014. [DOI] [PubMed] [Google Scholar]
- Wang X., Zheng Z., Caviglia J.M., Corey K.E., Herfel T.M., Cai B., Masia R., Chung R.T., Lefkowitch J.H., Schwabe R.F. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2016;24:848–862. doi: 10.1016/j.cmet.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt S.K., Manning H.C., Bai M., Ehtesham M., Mapara K.Y., Thompson R.C., Bornhop D.J. Preclinical molecular imaging of the translocator protein (TSPO) in a metastases model based on breast cancer xenografts propagated in the murine brain. Curr. Mol. Med. 2012;12:458–466. doi: 10.2174/156652412800163361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L., Yui J., Hatori A., Yamasaki T., Kumata K., Wakizaka H., Yoshida Y., Fujinaga M., Kawamura K., Zhang M.R. Translocator protein (18 kDa), a potential molecular imaging biomarker for non-invasively distinguishing non-alcoholic fatty liver disease. J. Hepatol. 2012;57:1076–1082. doi: 10.1016/j.jhep.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Yamasaki C., Ishida Y., Yanagi A., Yoshizane Y., Kojima Y., Ogawa Y., Kageyama Y., Iwasaki Y., Ishida S., Chayama K. Culture density contributes to hepatic functions of fresh human hepatocytes isolated from chimeric mice with humanized livers: novel, long-term, functional two-dimensional in vitro tool for developing new drugs. PLoS One. 2020;15:e0237809. doi: 10.1371/journal.pone.0237809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Torres M., Sha H., Halbrook C.J., Van den Bergh F., Reinert R.B., Yamada T., Wang S., Luo Y., Hunter A.H. Endoplasmic reticulum-associated degradation regulates mitochondrial dynamics in brown adipocytes. Science. 2020;368:54–60. doi: 10.1126/science.aay2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data were deposited in the Gene Expression Omnibus under accession number GSE138666. All data supporting the present study are available from the corresponding author on request.